

Abstract
Chlamydia trachomatis (CT) causes the most prevalent sexually transmitted bacterial disease in the United States. The lack of drug selectivity is one of the main challenges of the current antichlamydial pharmacotherapy. The metabolic needs of CT are controlled, among others, by cylindrical proteases and their chaperones (e.g., ClpX). It has been shown that dihydrothiazepines can disrupt CT-ClpXP. Based on this precedent, we synthesized a dihydrothiazepine library and characterized its antichlamydial activity using a modified semi-high-throughput screening assay. Then, we demonstrated their ability to inhibit ClpX ATPase activity in vitro, supporting ClpX as a target. Further, our lead compound displayed a promising selectivity profile against CT, acceptable cytotoxicity, no mutagenic potential, and good in vitro stability. A two-dimensional quantitative structure–activity relationship (2D QSAR) model was generated as a support tool in the identification of more potent antichlamydial molecules. This study suggests dihydrothiazepines are a promising starting point for the development of new and selective antichlamydial drugs.
Graphical Abstract
INTRODUCTION
Chlamydia trachomatis (CT) is an obligate intracellular Gram-negative bacterium1 and the cause of the most prevalent sexually transmitted bacterial disease in the United States.2 More than 1.8 million cases were reported in 2019, representing a 19% increase since 2015.3 The medical cost of CT infection treatments in the U.S. is estimated to be ~$700 million.4 CT transmission occurs almost exclusively through sexual contact, although there are reported cases of infections during labor, which can lead to newborn conjunctivitis.5 CT infection is especially problematic in reproductive-aged women because it can result in pelvic inflammatory disease (PID), tubal factor infertility, ectopic pregnancy, and chronic pelvic pain.6 CT can result in trachoma7 (the leading cause of infectious blindness worldwide),8 facilitate human immunodeficiency virus (HIV) infection,9 and is associated with preterm labor, low birth weight, and perinatal mortality.10 Further, the reported infection rate for chlamydia of African Americans/Blacks is six times that of Whites.3
CT displays a biphasic developmental cycle.11 The cycle begins when the elementary body (EB), the infectious but nondividing form of the bacterium, interacts with membranes of host cells, leading to its uptake.12 After entering the host cell, the EB remains within a host-derived vesicle, known as an inclusion, in which the EB differentiates into the reticulate body (RB) form.11 RBs are the replicative but noninfectious form of CT that, after repeated cycles of bacterial cell division, undergo secondary differentiation to EBs. Later, EBs are released by lysis or inclusion extrusion and a new infective cycle ensues.13
Broad-spectrum antibiotics such as azithromycin (AZM) and doxycycline (Doxy) are the drugs of choice to eradicate this infection.14 However, accumulating evidence suggests that AZM presents failure rates; i.e., 16% for the treatment of urethritis15 and 14% for female urogenital chlamydial infections.16 These levels are higher than the desired World Health Organization’s target chlamydia treatment failure rate of <5%.17 Further, broad-spectrum drugs affect commensal flora,18 altering their normal function19 and both AZM and Doxy may compromise the microbiome.20,21 Of note, studies have shown that gut microbial dysbiosis can contribute to the development of diabetes,22 obesity,23 asthma,24 and other conditions.25 Further, these drugs can foster the development of general antibiotic resistance.26,27 For example, treatment of CT with broad-spectrum antibiotics, including AZM, can lead to nasopharyngeal carriage of macrolide-resistant Staphylococcus aureus,28 macrolide-resistant gonorrhea,29 and Mycoplasma genitalium.30 Additionally, treatment of trachoma with AZM has led to induction of resistance in Streptococcus pneumoniae.28,31 Thus, narrow-spectrum/selective drugs can prevent damage or alterations in the patient’s microbiome32 and limit the development of general resistance. Unfortunately, there are few reports of molecules that are specific for Chlamydia, and none of those molecules has received U.S. Food and Drug Administration (FDA) approval.26,33–36 In this context, novel scaffolds or targets are needed to advance antibacterial research.
Proteolytic enzymes can play an important role as new drug targets.37 Affecting the degradation machinery is an attractive, yet barely explored, strategy to control pathogens.38 The ClpXP protease system is well conserved in bacteria and relevant for protein turnover.39 This degradation complex is composed of a serine protease subunit, known as ClpP, and an unfoldase chaperone, e.g., ClpX (which has ATPase activity). The chaperone component recognizes, unfolds, and transfers target proteins to ClpP for degradation.40 The degraded proteins include damaged or nonfunctional proteins as well as transcriptional regulators and other specific targets.41 The ClpP protease component of the ClpXP complex has been more extensively studied as a drug target than the unfoldases.42 Some of the structures that target the ClpP are (i) acyldepsipeptide (ADEP) derivatives;43 (ii) activators of self-compartmentalizing proteases (ACP);44 (iii) sclerotiamide;45 (iv) β-lactones;46 (v) phenyl esters;47 or (vi) bortezomib48 (Figure 1). However, only a few reports describe small molecules targeting the ClpX ATPase.49 The dihydrothiazepine scaffold reported by Fetzer and co-workers50 is the first reversible inhibitor of ClpX. Their most potent dihydrothiazepine derivative (334—Figure 1) disrupts the hexameric state of Methicillin-resistant S. aureus (MRSA) ClpX and blocks its ATPase activity with an IC50 value of 0.8 μM. Additionally, according to the authors, the whole ClpXP proteolytic complex collapses in response to dihydrothiazepine derivative action. Recently, it has been demonstrated that the ClpXP proteolytic system is critical to CT development51–53 and that 334 (Figure 1) can disrupt chlamydial ClpX function affecting Chlamydia growth in cell culture.39 Thus, targeting the ClpX component of the CT proteolytic system with dihydrothiazepine derivatives could produce anti-CT agents.
Figure 1.

Examples of structures that target the ClpXP protease system. The rings A, B, and C of 334 are color coded as indicated above. (aBortezomib can exert an activating effect on ClpP depending on its concentration.54).
In the present work, we report the synthesis and biological evaluation of dihydrothiazepines with antichlamydial activity. Some of the synthesized derivatives showed substantial reduction in chlamydial inclusion number and size in infected HEp-2 cells. To investigate the selectivity of our most promising structure, we tested its antimicrobial activity against clinically relevant Gram-positive/Gram-negative bacteria, yeast, and bacteria from the human gastrointestinal (GI) and genital tract microbiota. Inhibition of ClpX ATPase activity was assessed to acquire initial insights on the mechanism of action responsible for chlamydial eradication. Further, we tested the toxicity of our most promising analogues toward human cells, their stability in serum and simulated gastric and intestinal fluids. Moreover, the potential mutagenicity was investigated using an Ames test. To understand the activity and develop more potent structures, we generated a two-dimensional (2D) quantitative structure–activity relationship (QSAR) model. We used this model to guide the design of compounds with superior potency than the parent molecules. Our results indicate that our best dihydrothiazepine derivative is selective for CT and represents a starting point for the development of new antichlamydial drugs.
RESULTS AND DISCUSSION
Chemistry.
First, we explored modifications on the ortho, meta, and para positions of ring A. The synthesis consisted of Knoevenagel condensation of indan-1,3-dione (1) with the corresponding benzaldehyde (2) following the method of Fetzer et al.55 Then, dihydrothiazepines (5a–y) were obtained by the cyclization of 2-benzylideneindan-1,3-diones (3a–y) with 2-aminothiophenol (4) (Scheme 1).
Scheme 1. Synthesis of Dihydrothiazepines Containing Modifications on Ring Aa.

a(a) l-Proline, MeOH, room temperature (RT), molecular sieve (MS) 4 Å, 16–20 h; (b) isopropanol (iPrOH)/HOAc or p-toluenesulfonic acid (pTsOH)/iPrOH, RT, MS 4 Å, 18–24 h.
To place an amine on ring A, we performed catalytic hydrogenation of the nitro derivative 5n (Scheme 2). To investigate the importance of the thioether on the seven-member ring heterocycle, we oxidized this group to a sulfone with meta-chloroperoxybenzoic acid (mCPBA).
Scheme 2. Synthesis of Dihydrothiazepines Containing Amine and Sulfone Groupsa.

a (a) Pd/C (10%), H2(g), tetrahydrofuran (THF)/MeOH, RT, 24 h; (b) mCPBA, THF, RT, 5 h.
To determine the importance of ring B on the antichlamydial activity, we removed it by reacting selected 2-benzylideneindan-1,3-dione intermediates (3) with 2-amino-ethanethiol (8) using the same reaction conditions (Scheme 3).
Scheme 3. Synthesis of Dihydrothiazepines without Ring Ba.

a (a) iPrOH/HOAc or pTsOH/iPrOH, RT, MS 4 Å, 18–24 h
Since ring B proved to be key for activity, we explored how modifications on this ring affect the activity. First, we placed a trifluoromethyl group on the meta position. Further, we replaced the thioether by a secondary amine on the seven-member ring heterocycle (diazepine derivatives). The synthetic route for these modifications is presented in Scheme 4.
Scheme 4. Synthesis of Dihydrothiazepines Containing Modification on Ring B and Diazepine Derivativesa.

a (a) iPrOH, RT, MS 4 Å, 18–24 h; (b) iPrOH/HOAc or pTsOH/iPrOH, N2(g), RT, MS 4 Å, 18–24 h.
Biological Evaluation.
Determination of Antichlamydial Activity.
We tested our compounds against C. trachomatis serovar L2. As mentioned, CT has a biphasic developmental cycle.56 Once the EB is internalized into the cell, it resides in it as an inclusion where it differentiates into the replicative RB form.57 Since the inclusions are linked to the growth and replication of the pathogen,58 we quatitatively determined the antichlamydial activity by analyzing the number of inclusions after treatment and comparing them to untreated (UTD) samples and drug controls. To do so, we adapted an immunofluorescence assay (IFA) described previously36,52,59 to develop a semi-high-throughput screening platform (see full description in the Experimental Procedures section). This allowed us to determine the IC50 (the concentration to induce 50% of inclusion inhibition) of all analogues using a screening approach that is faster than other methods described in the literature.52,60–62
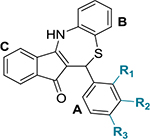
We observed that substitutions at ortho and meta positions on ring A had a positive effect on the antichlamydial activity (Table 1). These positions allow substituents with diverse electronic characteristic (both electron-withdrawing and electron-donating groups maintained the activity). In accordance, our top five most active compounds (5a–e) presented substitutions at one or both of these positions in ring A. On the contrary, groups in the para position caused a reduction in the IC50 (Table 1). In particular, bulky groups at the para position had the most significant drop in potency (a benzyloxy group (5s) led to a 14-fold decrease in the IC50 and a phenyl (5v) group completely abolished the activity). Of note, derivatives displaying a thiophene instead of a benzene in ring A (11a and 5y, Table 4) retained activity.
Table 1.
Chemical Structures of Dihydrothiazepine Derivatives 5a–v and Their IC50 against C. trachomatis Serovar L2a
| ||||
|---|---|---|---|---|
| compound | R1 | R2 | R3 | IC50 (μg/mL)b,c |
| 5a | CN | H | H | 1.61 ± 0.11 |
| 5b | OCH3 | H | H | 3.32 ± 0.51 |
| 5c | F | CH3 | H | 4.69 ± 1.20 |
| 5d | H | Cl | H | 4.79 ± 0.19 |
| 5e | H | CF3 | H | 5.18 ± 1.67 |
| 5f | H | NO2 | H | 6.35 ± 0.70 |
| 5g | Cl | H | Cl | 6.37 ± 0.67 |
| 5h | H | H | Cl | 6.59 ± 0.18 |
| H | H | CH3 | 6.72 ± 0.58 | |
| 5j | H | H | OC(O)CH3 | 7.18 ± 0.50 |
| 5k | H | H | OCF3 | 7.44 ± 0.30 |
| 5l | H | H | Br | 7.50 ± 1.01 |
| 5m | H | CN | H | 9.00 ± 1.15 |
| 5n | H | H | NO2 | 9.01 ± 0.25 |
| 5o | H | H | CN | 10.21 ± 1.47 |
| 5P | H | H | OH | 14.41 ± 2.76 |
| 5q | H | H | OCH3 | 14.69 ± 1.26 |
| 334 | H | OH | H | 16.53 ± 0.65 |
| 6a | H | H | NH2 | 17.63 ± 0.43 |
| 5s | H | H | OCH2Ph | 22.76 ± 0.34 |
| 5t | H | H | CO2H | NI |
| 5u | CO2H | H | H | NI |
| 5v | H | H | Ph | NI |
IC50 indicates 50% inhibition of control Chlamydia inclusions. NI: no inhibition seen at the tested concentration range (25–0.39 μg/ mL).
All assays were performed in triplicate.
IC50 values are also presented in μM and pIC50 units in Table S1. The presented data corresponds to mean ± standard deviation (SD).
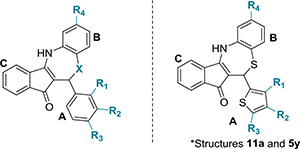
Table 4.
Chemical Structures of Dihydrothiazepine Derivatives 11a−13c and Their IC50 against C. trachomatis Serovar L2a,c
| ||||||
|---|---|---|---|---|---|---|
| compound | R1 | R2 | R3 | R4 | X | IC50 (μg/mL)b |
| 11a | H | H | H | CF3 | S | 6.53 ± 0.45 |
| 11b | H | H | Br | CF3 | S | 6.93 ± 0.37 |
| 5y | H | H | H | H | S | 7.36 ± 0.78 |
| 7a | H | OCH3 | H | H | SO2 | 10.00 ± 3.69 |
| 11c | H | H | OCH3 | CF3 | S | 12.30 ± 0.87 |
| 13a | H | H | CH3 | H | NH | 15.00 ± 3.23 |
| 13b | H | H | CN | H | NH | 20.88 ± 1.94 |
| 13c | H | H | OCH3 | H | NH | 22.61 ± 0.32 |
IC50 indicates 50% inhibition of control Chlamydia inclusions. NA: not applicable. Compounds 11a and 5y possess a 2-thiophene ring instead of the benzyl ring A.
All assays were performed in triplicate.
IC50 values are also presented in μM and pIC50 units in Table S1.
The presented data corresponds to mean ± SD.
Further, we observed that acidic groups abolished the antichlamydial activity. This can be seen with derivatives 5t and 5u, which present carboxylic acid in ring A at ortho and para positions, respectively. This observation can be partially explained by the ionization of these two molecules at the pH used in the assays (~7.4) (calculated pKa: 4.10 and 3.48 for 5t and 5u, respectively), which can diminish their permeability into the host cell preventing them from displaying antichlamydial activity. This theory is supported by the fact that the esterified version of 5t, compound 5j, displays antichlamydial activity (Table 1), supporting the notion of permeability and activity. However, the polar derivatives 5f and 5n, with a nitro group at meta and para positions, respectively, possess antichlamydial activity (IC50 of 6.35 and 9.01 μg/mL).
To analyze these results, we also calculated in silico permeability parameters (values for all of the compounds can be found in File S1; Table 2 shows selected compounds). Among the evaluated parameters, two descriptors indicated the derivatives having a nitro group should have better permeability than the carboxylic acids: (i) the predicted apparent Caco-2 cell permeability (QPPCaco) and (ii) the predicted apparent Madin–Darby Canine Kidney (MDCK) cell permeability (QPPMDC). First, QPPCaco values for the acid derivatives are lower than the ones predicted for the nitro analogues (101.98 and 345.75 nm/s for 5t and 5u against 429.39 and 605.69 nm/s for 5n and 5f, respectively). The same pattern is seen on the second permeability prediction: 82.60 and 298.59 for 5t and 5u against 307.34 and 444.67 for 5n and 5f, respectively. Thus, the predicted permeability for the nitro derivatives is considerably better than that for the carboxylic acid analogues (the ester analogue, 5j, also has higher predicted permeability than the acid—Table 2). Meanwhile, the predicted octanol/water partition coefficient (QPlogPo/w (pH 7.4)) and solvent accessible surface area (SASA) were similar between the analogues. Of course, electronic effects might also contribute to the distinct antichlamydial activities. Resonance forms, position of the substituents, and electronegativity cannot be discarded.63
Table 2.
Detailing of In Silico Permeability Parameters for Some Selected Compounds and Comparison with IC50 Valúes
| compound | IC50 (μg/mL) | QPlogPo/wa | QPPCacob | QPPMDCKc | SASAd |
|---|---|---|---|---|---|
| 5a | 1.61 ± 0.11 | 4.08 | 1794.2 | 1465.8 | 563.86 |
| 5b | 3.32 ± 0.51 | 4.63 | 3707.8 | 3212.5 | 569.57 |
| 5c | 4.69 ± 1.20 | 5.06 | 3639.2 | 3188.8 | 589.42 |
| 5d | 4.79 ± 0.19 | 5.21 | 3600.4 | 6855.0 | 582.84 |
| 5e | 5.18 ± 1.67 | 5.68 | 3448.6 | 10 000.0 | 605.77 |
| 5f | 6.35 ± 0.70 | 4.47 | 605.7 | 444.7 | 624.51 |
| 5j | 6.72 ± 0.58 | 4.40 | 1155.8 | 900.70 | 661.59 |
| 11a | 6.53 ± 0.45 | 5.75 | 3352.8 | 10 000.0 | 608.64 |
| 11b | 6.93 ± 0.37 | 6.45 | 3384.2 | 10 000.0 | 654.63 |
| 5y | 7.36 ± 0.78 | 4.80 | 3269.7 | 4617.9 | 563.87 |
| 5l | 7.50 ± 1.01 | 5.42 | 3588.0 | 8068.2 | 595.47 |
| 5n | 9.01 ± 0.25 | 4.16 | 429.4 | 307.3 | 605.51 |
| 11c | 12.30 ± 0.87 | 5.97 | 3910.5 | 10 000.0 | 653.20 |
| 5q | 14.69 ± 1.26 | 4.95 | 3500.7 | 2967.1 | 603.41 |
| 5s | 22.76 ± 0.34 | 6.82 | 4104.2 | 3532.7 | 734.32 |
| 5t | NI | 4.14 | 102.0 | 82.6 | 604.58 |
| 5u | NI | 4.36 | 345.8 | 298.6 | 575.65 |
| 9d | NI | 2.44 | 149.7 | 112.6 | 561.09 |
QPlogPo/w—predicted octanol/water partition coefficient (−2.0 to 6.5).
QPPCaco—predicted apparent Caco-2 cell permeability in nm/s (<25 poor, >500 great).
QPPMDCK—predicted apparent MDCK cell permeability in nm/s (<25 poor, >500 great).
Total solvent accessible surface area (SASA) in square angstroms (300–1000). Range values are related to 95% of known drugs.64 Predictions relate to drug absorption as oral dosage form. Values for the entire library of compounds can be found in File S1.
Further, the removal of ring B led to a significant drop in the IC50 values (Table 3) when compared to the molecules displaying the same substitution pattern but presenting such a ring (for derivative 9d, due to no inhibition seen at the concentration range tested, we could not calculate the IC50). Structural studies need to be done to assess if there is a π-stacking interaction that is critical for activity or if this is related to cell permeability/lipophilicity.
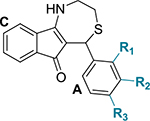
Table 3.
Chemical Structures of Dihydrothiazepine Derivatives 9a−d and Their IC50 against C. trachomatis Serovar L2a,c
| ||||
|---|---|---|---|---|
| compound | R1 | R2 | R3 | IC50 (μg/mL)b |
| 9a | H | H | OCH3 | 16.08 ± 0.54 |
| 9b | H | H | Br | 21.35 ± 2.88 |
| 9c | H | H | CH3 | 21.45 ± 0.22 |
| 9d | H | H | CN | NI |
IC50 indicates 50% inhibition of control Chlamydia inclusions. NI: no inhibition seen at the tested concentration range (25−0.39 μg/mL).
All assays were performed in triplicate.
IC50 values are also presented in μM and pIC50 units in Table S1. The presented data corresponds to mean ± SD.
Interestingly, the introduction of a trifluoromethyl at the meta position on ring B (Table 4) led to a slight increase in the potency of all of the derivatives (when compared to analogues possessing the same substitution on the rest of the structure—Table 1). For instance, the IC50 of compound 5q improved from 14.69 to 12.30 μg/mL (11c), compound 5y from 7.36 to 6.53 μg/mL (11a), and compound 5l from 7.50 to 6.93 μg/mL (11b).
A potential explanation for the slight improvement in the potency of 11a–c may be a result of increased permeability into host cells. The in silico permeability parameters (Table 2) analyzed for these molecules support this hypothesis. Molecules lacking the trifluoromethyl (5y, 5l, 5q) group present lower values for the predicted QPPCaco and QPPMDC cell permeabilities as well as lower predicted octanol/water partition coefficient values (QPlogPo/w—pH 7.4) when compared to analogues that display this functional group (see File S1 and Table 2).
Electronic effects might also contribute to the beneficial activity of the trifluoromethyl group at the meta position on ring B. Figure 2a displays the calculated quantum mechanics molecular electrostatic potential (MEP) for various analogues. The MEP surface suggests an electron density deficiency on ring B for the CF3 analogues when compared to their unsubstituted counterparts. A potential explanation (assuming aromatic amino acids are nearby and keeping in mind the binding pocket of the ClpX is unknown at this moment) might be improved π-stacking with Tyr, Phe, or Trp in the binding pocket. In fact, the MEP for these amino acids shows the electron-rich nature (dark red color) of the aromatic ring (especially Trp and Try) side chains (Figure 2b). Thus, electronic effects complementary to the CF3 substituted B-ring (Figure 2a) would be favored. We understand that the existence of such interactions depends also on other factors like the orientation of the interacting groups and distance. However, due to the absence of a crystal structure and the binding pocket identification, our current investigation tools are limited. Another beneficial interaction mediated by the CF3 might be the establishment of hydrogen bonds between the fluorine atoms and residues in the binding pocket. Altogether, the data suggest that the B-ring is crucial for the antichlamydial activity and that both rings A and B are permissive to modifications while maintaining activity against CT.
Figure 2.

Molecular electrostatic potential (MEP) surface. (a) Top: compounds with a H atom on ring B. Bottom: compounds containing the trifluoromethyl on ring B. The rest of the molecules possess the same substitution pattern. (b) Tryptophan, tyrosine, and phenylalanine. Detailing of the MEP surface of ring B and the aromatic side chain for Trp, Try, and Phe. The most positive potential regions are displayed in deepest blue color and the most negative potential regions in deepest red color.
Next, we assessed the SAR for the dihydrothiazepine seven-member ring. We noticed the exchange of sulfur for a nitrogen increased the IC50 values, but it did not abolish activity (Table 4, compounds 13a–c). Further, oxidation of the thioether to a sulfone also reduced the activity but still the derivative containing this modification (7a) showed intermediate activity (10 μg/mL).
We found the permeability parameters could be reasonably correlated to better antichlamydial activity for specific comparison cases, for instance, carboxylic acids versus nitro structures, or molecules with CF3 groups (described above), or compound 13a–c analogues (S replaced by NH). Due to the intracellular nature of CT, it is expected that permeability will affect the activity (improved membrane permeability leads to higher accumulation of drug inside the cell65). In fact, the most potent molecules (5a–d) present high-permeability-related parameters (Table 2) and not active compounds like 9d display very low predicted permeability. However, not all highly lipophilic molecules presented good activity. For instance, low active molecule 5s (22.76 μg/mL) presents high predicted permeability (QPPCaco: 4104.20; QPPMDCK: 3532.70 nm/s). Further, the most active compound (5a) displays lower values for the predicted permeability (QPPCaco: 1794.20 and QPPMDCK: 1465.80) than the second or third most active structures (5b: 3707.80, 3212.50 and 5c: 3639.20, 3188.80 for QPPCaco and QPPMDCK, respectively). Further, the acetyl ester (para position, 5j) of the phenol (5p) is more active than the methyl ether (5q) of the same molecule, although its permeability is lower. This may suggest electronic effects are as important as the permeability parameter (although the occurrence of intracellular ester hydrolysis may not be discarded since the similar acid derivative—5u, presenting a carboxylic acid at the meta position—displays ClpX anti-ATPase activity but no antichlamydial activity in the cellular assay). These illustrate the multifactorial nature of drug activity.66 In accordance, small structural changes will affect the biological activity67 (especially when comparing a cellular assay directly to an enzymatic experiment68).
Fingerprint-Based Two-Dimensional (2D) QSAR Studies.
Although it is common to infer a SAR through the qualitative evaluation of a library of molecules, some intricacies of the relationships are better elucidated using a visual projection of the quantitative contributions of different chemical groups.69 Accordingly, to analyze the key structural features of the dihydrothiazepine library, a 2D quantitative structure–activity relationship model (QSAR) was generated. This ligand-based tool is necessary for this study because there is no published chlamydial ClpX crystal structure, and the binding pocket has not been identified. Further, the use of a three-dimensional (3D) QSAR model (based on 3D descriptors) will be inadequate since the antichlamydial activity reported is based on racemic mixtures. Additionally, the biologically active conformation is unknown at the moment. Although the active conformation of the ligand is not absolutely indispensable for all 3D QSAR methods,70 it increases the reliability of the models and is a requirement for most of the 3D QSAR approaches.71
To develop our model, we used 2D fingerprint descriptors and AutoQSAR, a machine-learning software that builds and applies QSAR models through automation.72 The program computes fingerprints and uses machine-learning statistical methods to create a predictive QSAR model that can describe the studied biological activity. The kernel-based partial least-square (KPLS) regression method using 2D fingerprint radial descriptors produced a statistically relevant QSAR model (Figures S1, 3, and 4). The best KPLS model generated using radial descriptors (model KPLS—radial 13) possesses good predictive accuracy in the training as well as in the test set (Table S3). The obtained model displays satisfactory statistical results as evaluated by the correlation coefficient (R2—a measure of the data explained by the model73); the coefficient of determination between the observed and predicted biological activities for the test set (Q2—a measure of predictability74); low standard deviation (SD) of the regression calculated based on the training set; and low root mean square error (RMSE) in the test set predictions (RMSE—expresses the variability not explained by the model75). Further details regarding the predictive accuracy in the test and training set and statistical parameters/reference values for the QSAR model validation are presented in Tables S2 and S3 and Figure S1.
Figure 3.

Color maps for the 2D fingerprint (radial 13) QSAR model. (a) Most active molecules. (b) Molecules with less antichlamydial activity. Atoms displaying a favorable contribution to the antichlamydial activity are colored red and unfavorable are colored blue. Color intensity reflects the strength of the effect. Highly active molecules are shown in predominant red color and less active molecules are presented in predominant blue. (Note: stereochemistry is displayed because the molecules are drawn in the 3D form, which is required for Maestro software to display the QSAR color maps. However, stereochemistry is not considered in the generation of the 2D descriptors used to build the model).
Figure 4.

Color maps for the 2D fingerprint (radial 13) QSAR model. (a) Representative molecules of the favorable effect of lipophilic groups on the antichlamydial activity. (b) Representative molecules of the unfavorable effect of H bond donors at ring A on the antichlamydial activity. Atoms displaying a favorable contribution to the antichlamydial activity are colored red and unfavorable are colored blue. Color intensity reflects the strength of the effect. (Note: stereochemistry is displayed because the molecules are drawn in the 3D form, which is required for Maestro software to display the QSAR color maps. However, stereochemistry is not considered in the generation of the 2D descriptors used to build the model).
The used fingerprints descriptors allow the KPLS model to be decomposed into individually atomic effects, allowing a convenient way of visualizing favorable and unfavorable aspects of the chemical structures. The evaluation of the individual atomic contribution to the model is illustrated in Figure 3a using active and Figure 3b using molecules presenting low antichlamydial activity (color maps for the entire dataset can be found in Figure S2). Groups that contribute positively to activity are highlighted in red and the detrimental ones in blue. Color intensity reflects the strength of the effect. The 2D KPLS-QSAR model provides an efficient approach to visualize molecules (Figures 3 and 4). The compounds in panel “a” are more active than the ones presented in panel “b”.
Other relevant features are highlighted by this QSAR model. For instance, for molecule 5s, the darkest blue color is centered at the methylene group at the para position (ring A), indicating that this substitution pattern is highly unsuited for modifications (Figure 3b). The same effect is seen in 5v (phenyl group at the para position—ring A), which does not present antichlamydial activity at the tested concentration. These observations could relate to the size and shape of the binding pocket, which probably does not accommodate bulky groups at the para position. Further, molecules 6a, 9c, and 13a presenting smaller electron-donating groups at the para position of ring A (amine and methyl groups, respectively) are also detrimental for the activity.
The QSAR model indicates the favorable effect of lipophilic groups regardless of the position within the dihydrothiazepine core (Figure 4a). As mentioned above, substituents at the para position have a detrimental effect on the biological activity (ring A is highlighted in blue when such positions are substituted). It is possible that the presence of lipophilic and electronegative substituents (e.g., trifluoromethyl or chloro at ring A) may counterbalance this unfavorable effect (note the red sphere around these groups in Figure 4a, molecules 5h and 5k). This could also be associated to the relationship between increasing lipophilicity and a superior ability to cross the membrane of the host cell and of the inclusions to access the target.
Another important feature is the unfavorable effect of hydrogen bond (H bond) donor groups at the A ring as shown in Figure 4b as demonstrated by the more intense spherical blue color at those groups (Figure 4b, molecules 5p, 6a, and 334). Their effects are more evident when the group is placed at the para position versus the meta position (for 334, the A ring displays more faded blue color than 5p and 6a). Interestingly, even though 334 possessed an inhibitory effect against ClpX of S. aureus,55 it only showed moderate activity (IC50 ~ 16 μg/mL) against C. trachomatis. This might be a result of differences in the composition of the binding pocket between ClpX orthologues in these different microorganisms (52.09% of protein similarity according to Uniprot databank alignment tool76). Further, the differences in membrane constitution may influence permeability, and consequently, the biological activity of 334 across bacterial species. To be able to inhibit ClpX (or other targets) in CT, 334 needs to cross the host cell membrane, the inclusion membrane, and the double membrane of CT (a Gram-negative bacteria). On the opposite side, for S. aureus, a single membrane needs to be crossed by the drug. The impact of such differences might be clarified once the binding pocket of dihydrothiazepines are identified.
EB Progeny Assay.
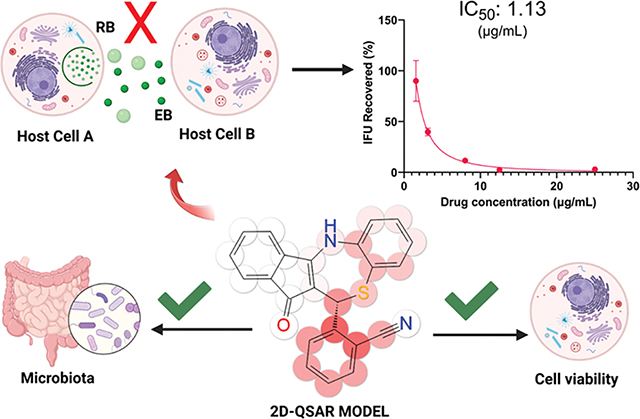
To determine the ability of the compounds to inhibit infectious (EB) progeny production and to validate the IC50 obtained by our semi-high-throughput IFA screening, we performed an inclusion forming units (IFU) reinfection assay77 using the three most potent structures. Here, we quantified the number of C. trachomatis infectious forms (EB) from previously treated cultures at five concentrations (as well as untreated and AZM controls) following infection of a new cell monolayer. The chlamydial infectivity, reported as IFUs, was determined by counting the number of green-fluorescent inclusions after immunofluorescent staining at 24 h post-secondary infection (Figure 5). In addition to the number of inclusions, we also evaluated the size and appearance of the chlamydial inclusions after treatment with the studied drugs. The most promising compound (5a) displayed inhibitory activity against CT with an IC50 of 1.13 μg/mL (similar to the IC50 value determined in the semi-high-throughput screening—1.61 μg/mL, Figure 5a). The dose–response curve displays a concentration-dependent profile for the reduction in the chlamydial progeny, as seen by the reduction in the number of chlamydial inclusions (Figure 5a). Importantly, we noticed a remarkable reduction in the inclusion size compared to the control (UTD) even in lower doses, for instance, 3.12 and 1.56 μg/mL (Figure 5c). Moreover, even at 3.12 μg/mL, compound 5a achieved an inclusion recovery ~60% lower than the untreated samples (Figure 5c). When compared against the drug control AZM (Figure 5b), our tested structure displayed lower potency. However, the superior antichlamydial activity of AZM is also followed by a remarkable lower selectivity when tested against different bacteria, including different species from the human microbiota (Table 5). Moreover, AZM is a commercialized drug that had its potency optimized throughout its preclinical research phase, while the dihydrothiazepine scaffold has not been fully investigated. The obtained IC50 values for 5b and 5c in the progeny assay also display the same pattern as for the semi-high-throughput screening. The first molecule has superior activity (6.41 μg/mL) compared to the latter (7.13 μg/mL). The dose–response curves and immunofluorescence images for compounds 5b and 5c can be found in Figure S3.
Figure 5.

Dose–response curves for the effect of 5a (a) and AZM (b) reported as the percentage of recovered IFU. Error bars report standard error of the mean (SEM) (N = 3). (c) Immunofluorescence images for the inhibitory effect of 5a and AZM. Chlamydial inclusions are displayed in green (MOMP - Ctr L2) and HEp-2 cell nuclei in blue (4′,6-diamidino-2-phenylindole (DAPI)). Images were acquired on 20× magnification using an Olympus CKX53 fluorescence microscope with EP50 camera. Channel merge was processed using ImageJ software. UTD: untreated; AZM: azithromycin. Omitted error bars indicate a SD value shorter than the size of the symbol presented in the plots.
Table 5.
Antimicrobial Activity Panel of Compound 5a
| MICp (μg/mL) |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Saa | Abb | Ecc | Pad | Ece | Lrf | Lpg | BPh | Cri | Btj | Ack | Cal | Ccm | Ckn | Cto | |
| 5a | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >1000 | >1000 | >1000 | >1000 |
| AZM q | 2 | <1 | <1 | 125 | 2 | 1 | 2 | <0.06 | <0.06 | 0.5 | >125 | ner | ne | ne | ne |
| FCZ s | ne | ne | ne | ne | ne | ne | ne | ne | ne | ne | ne | 1 | 2 | 16 | 0.5 |
S. aureus USA 300 JE2.
Acinetobacter baumannii 2208.
Escherichia coli K12.
Pseudomonas aeruginosa PA01.
E. coli nonpathogenic ATCC MG1655.
Lactobacillus rhamnosus CGMCC 1.3724.
Lactobacillus plantarum DSM 20174.
Blautia producta DSM 2950.
Clostridium ramosum DSM 1402.
Bacteroides thetaiotaomicron DSM 2079.
Anaerostipes caccae DSM 14662.
Candida albicans 90028.
Candida glabrata 2001.
Candida krusei 6558.
Candida tropicalis 750.
MIC: minimum inhibitory concentration (μg/mL).
AZM: azithromycin.
ne: not evaluated.
FCZ: fluconazole.
Cytotoxicity.
Following the determination of the antibacterial activity of our library, we evaluated the in vitro cytotoxicity of the three most active molecules (5a–c) against epithelial type 2 (HEp-2, host cell in the antichlamydial assays), epithelial endometrium adenocarcinoma (HEC-1-A), and immortalized human embryonic kidney (HEK-293) cells. HEC-1-A and HEK-293 cells showed a very good viability profile, over ≥75% survival at the highest concentration for all tested compounds: 256 μg/mL (Figure 6b,c). HEp-2 cells proved to be more sensitive to the tested molecules. At the highest tested concentration, around 50% cell viability was seen (Figure 6a). Importantly, in the highest concentration used in the antichlamydial assays (25 μg/mL), molecules 5a and 5c displayed high cell viability values (84 and 78%, respectively). Compound 5b shows a slightly increased cytotoxicity against HEp-2 cells at this concentration, displaying a viability value of 73%. Compound 5a was selected for further investigations since it presents higher potency (IC50 for the infectious progeny inhibition: 1.13 μg/mL versus 6.41 and 7.13 for 5b and 5c, respectively–Figure S3) and a similar cytotoxicity profile when compared to the other derivatives analyzed.
Figure 6.

Viability analysis of the most active compounds against HEp-2 (a), HEC-1-A (b), and HEK-293 (c) cell lines. Error bars report SEM (N = 3). Each experiment included technical replicates. Omitted error bars indicate an SD value shorter than the size of the symbol presented in the plots.
Selectivity.
One major challenge of current antichlamydial treatments is their lack of selectivity. To determine the spectrum of antibacterial effect of 5a, we tested it against a broad panel of microorganisms spanning from pathogenic bacteria and fungi to bacteria from the human microbiota (Table 5). Molecule 5a proved to be inactive against Methicillin-resistant S. aureus USA 300 JE2, P. aeruginosa PA01, A. baumannii 2208, and E. coli K12 up to the highest concentration analyzed.
Additionally, 5a also showed no antibacterial activity against L. rhamnosus (CGMCC 1.3724) and L. plantarum (DSM 20174). This result is encouraging since L. rhamnosus is found in the human gut and vaginal microbiota.78 Further, both species are widely used as probiotic strains.78,79 To further determine the antibacterial activity against bacteria that comprise the human gut microbiota, we tested our compound against B. producta,80 C. ramosum,81 B. thetaiotaomicron,82 A. caccae,83 and E. coli MG1655. Compound 5a was completely inactive against all species tested up to the highest concentration evaluated. On the other hand, AZM showed very potent antibacterial activity against most tested bacteria. In particular, for the gut microbiota tested strains, AZM displays MIC values as low as <0.06 μg/mL for B. producta DSM 2950 and C. ramosum DSM 1402, 0.5 μg/mL for B. thetaiotaomicron DSM 2079, and 2 μg/mL for E. coli MG1655. Regarding the lactobacillus strains, AZM also presented low MIC values that ranged around 2 to <1 μg/mL. The only gut microbiota bacteria that showed no susceptibility to AZM in the concentration range tested was A. caccae DSM 14662. The fact that compound 5a did not affect the tested gut microbiota strains is very interesting, since, as pointed out, gut microbiota symbiosis is linked to the development of numerous diseases25 and to increased rates of antibiotic resistance to other pathogens.
Molecule 5a also proved to be inactive (MIC > 1000 μg/mL) against C. albicans (90028), C. glabrata (2001), C. krusei (6558), and C. tropicalis (750). The lack of activity against Candida is expected given that it is a eukaryotic organism. These species are commonly found in the gastrointestinal (GI) and genital tract microbiota of healthy humans. However, during host immunosuppression or alterations in the bacterial microbiota, these microorganisms can disseminate and cause life-threatening illness.84–87
Overall, these results suggest that normal microbiota in humans might be unaffected by this class of compounds, which in turn suggests that the development of widespread resistance or alterations in the bacterial composition of the microbiota is unlikely. We hypothesize that the lack of activity against other classes of microorganisms might be a result of the unique cell wall structure of CT88 when compared to other bacteria and/or the unique developmental cycle of Chlamydia.11 In fact, the ClpXP complex51 is essential for CT development,39,51,53 whereas it is dispensable for other types of bacteria like E. coli.89 These factors might play a role in the selectivity of this compound.
ClpX Anti-ATPase Activity.
To investigate the mechanism of action, we studied the inhibition of CT ClpX ATPase activity in vitro. We assessed the ability of 5a, 5c, 5u, and 9d to block ATP depletion over time using recombinant CT ClpX and a Kinase-Glo endpoint assay (Figure 7). All tested derivatives at 25 μg/mL significantly reduced Ct ClpX ATP consumption compared to the dimethyl sulfoxide (DMSO) solvent control by ~20%. While in vitro recombinant ClpX inhibition was expected for compounds 5a and 5c based on the antichlamydial assays, the in vitro activity of 5u and 9d contrasted with those experiments where no inhibition was observed. It is conceivable that membrane permeability negatively impacted in vivo activity for 5u and 9d. Compound 5u, as discussed above, is ionized in the pH of the cell culture assay. This fact reinforces the highly influential effect of permeability in the activity of this scaffold and explains the difference in the cell culture assay versus the in vitro ClpX inhibition. Furthermore, the same rationale can be applied to explain the in vitro ClpX inhibition of 9d. This structure presents very low predicted permeability compared to other highly active molecules of our library according to Caco-2 cell permeability and MDCK cell permeability parameters (File S1). For instance, 5a displays high potence in the cell culture assay and high-predicted-permeability Caco-2 cell permeability of 1794.16 nm/s and MDCK cell permeability of 1465.85 nm/s. On the contrary, 9d displays 149.74 and 112.61 nm/s for the same parameters, respectively (File S1).
Figure 7.

Inhibition of ClpX ATPase activity. Recombinant ClpX was incubated with either the inhibitor or DMSO as a solvent control. Reactions were halted by the addition of Kinase-Glo at 60 min and the remaining levels of ATP were determined by measuring luminescence as a marker for ClpX ATPase activity. The columns represent the amount of ATP depleted over time versus the ATP only control. Error bars report SEM. Each experiment included technical replicates and at least three biological replicates. **P ≤ 0.01; ****P ≤ 0.0001; by one-way analysis of variance (ANOVA), Tukey correction.
The activities of 5a and 5c against ClpX in vitro support that CT inhibition in the cell culture assays by these compounds is due, at least in part, to reduced ClpX activity. However, the results from the Sieber group, using an S. aureus ClpX knockout strain, indicate that the dihydrothiazepine scaffold could also have other targets within the bacterium.55 Thus, a second target cannot be discarded.
Stability Studies.
To analyze the drug-like properties of 5a to a greater extent, we evaluated its stability in human serum (Figure 8a), simulated gastric fluid (SGF, pH 1.40, Figure 8b), and simulated intestinal fluid (SIF, pH 7.60, Figure 8c). As shown in Figure 8, all of the tested compounds exhibited a satisfactory stability profile for the serum, SGF, and SIF assays during the study. For the stability in serum, at 180 min, the percentage of drug remaining in the solution was around 100%, and at 24 h showed a decrease to 80%. In SGF, the percentage of drug remaining at 180 min was around 90% with a decrease to 70% after 24 h of exposure. Furthermore, for SIF, the same stable pattern was visualized with the percentage of drug remaining around 85% at 24 h.
Figure 8.

Stability profile of 5a in human serum (a), simulated gastric fluid (b), and simulated intestinal fluid (c). The studied compound (2 mg/mL) was incubated in human serum; simulated gastric fluid–pH 1.4 containing 3.2 mg/mL of pepsin; or simulated intestinal fluid–pH 7.40 containing 10 mg/mL of pancreatin over 24 h in a shaking incubator (37 °C and 75 rpm). Relative percentage of drug remaining is presented as a function of incubation time. Error bars represent SD of three independent experiments. Omitted error bars indicate an SD value shorter than the size of the symbol presented in the plots.
Ames Test.
Analysis of the mutagenic potential in drug discovery intends to rapidly recognize unpromising candidates, removing them from development.90 Particularly, for antibacterial drugs, the evaluation of the potential mutagenic effect is highly significant since certain antibiotics induce elevated rates of mutation in bacteria.91 The elevated rate of mutagenesis expands the genetic diversity of bacterial populations, which might increase the rate at which bacteria develop antibiotic resistance.91,92 To further characterize the potential of 5a, we evaluated its mutagenic activity using a bacterial reverse mutation assay (Ames test). The basis of this assay is the detection of substances, which by induction of mutations, can revert previous mutations present in the tester strains.93
Assays were performed with Salmonella typhimurium strains TA98 and TA100. Since molecules can be converted to mutagenic compounds after metabolism, we performed the assay in the presence and absence of metabolic activation from the microsomal fraction (S9). The results are displayed in Table 6. The mutagenicity index (MI) was calculated according to the formula: MI = number of induced revertants/number of spontaneous revertants. The sample is considered to have mutagenic potential when the mutagenicity index was ≥2 in at least one of the concentrations tested and when there was a dose–response relationship between the concentrations tested and the number of induced revertants.94
Table 6.
Mutagenic Activity and Mutagenic Index (Displayed in Parenthesis) of 5a against S. typhimurium TA98 and TA100 Strainsa
| number of revertants/plate and (MI) |
||||
|---|---|---|---|---|
| TA98 |
TA100 |
|||
| concentration (μg/plate) | S9− | S9+ | S9− | S9+ |
| 0b | 25.67c ± 5.51 | 24.33 ± 9.50 | 206.00 ± 7.07 | 201.00 ± 9.90 |
| 50 | 26.00 ± 1.41 (1.01) | 32.00 ± 11.31 (1.31) | 190.50 ± 4.95 (0.92) | 185.00 ± 7.07 (0.92) |
| 150 | 34.50 ± 0.71 (1.34) | 35.00 ± 1.41 (1.43) | 187.50 ± 3.54 (0.91) | 187.00 ± 5.66 (0.93) |
| 500 | 38.00 ± 2.83 (1.48) | 43.50 ± 6.36 (1.78) | 233.00 ± 7.07 (1.13) | 191.50 ± 9.19 (0.95) |
| 1500 | 39.00 ± 9.90 (1.51) | 30.50 ± 3.54 (1.25) | 241.00 ± 2.83* (1.16) | 198.50 ± 4.95 (0.98) |
| 5000 | 37.50 ± 9.19 (1.46) | 43.50 ± 0.71 (1.78) | 241.00 ± 1.41* (1.16) | 227.00 ± 9.90 (1.12) |
| control | 34.50 ± 2.12d | 137.00 ± 7.07f | 260.70 ± 15.30e | 1256.00 ± 102.10f |
P < 0.05 (ANOVA) concentratons of 5a compared with negative control.
The results displayed represent the data for the assays in the absence (S9−) and presence (S9+) of an exogenous metabolism system.
Negative control (DMSO).
Number of revertant colonies.
4-Nitrophenylenediamine (NPD, 10 μg/plate).
Sodium azide (2.5 μg/plate).
2-Aminoanthracene (AA, 2.5 μg/plate).
The results are reported as means ± SD.
Analysis of the data presented in Table 6 suggests the studied molecule does not display mutagenic activity against S. typhimurium TA98 or TA100 strains. The calculated MI is less than 2 at all tested concentrations with or without metabolic activation. Further, a positive dose–response relationship between concentrations and number of revertant colonies was not observed for the tested samples. Some concentrations showed a significant increase in the number of revertant colonies per plate (*P < 0.05, 1500, and 5000 μg/mL for S9- and TA100 strain) when compared against the negative control, but overall, based on these data, the molecule can be considered nonmutagenic. Lastly, strain TA98 is used to detect frameshift mutations and strain TA100 detects substitution of DNA bases.93 Compound 5a was considered nonmutagenic to both TA98 and TA100 strains, suggesting that this dihydrothiazepine does not have the potential to cause gene mutations by frameshift or substitution of DNA bases. Of note, caveats remain for this assay because the compound may not penetrate into S. typhimurium. However, if this is the case, it reinforces the potential selectivity of this compound for Chlamydia.
Experimental Validation of the QSAR Model (KPLS—Radial 13).
The reliability of a QSAR model is conditioned to its ability to result in confident predictions, which can aid in the design of new molecules not used in the generation of the model.75 Therefore, we designed three new antichlamydial dihydrothiazepines (11d, 11e, 5x, Figure 9a) to experimentally validate our QSAR model. In our new molecular design, we focused on modifications of the most active compounds from the initial dataset (5a and 5b). To these initial structures, a trifluoromethyl group was inserted at ring B (structures 11d and 11e). Furthermore, since the presence of a sp3 carbon from the methoxy in 5b is detrimental for activity (marked in blue in the QSAR model representation in Figure 3a), we exchanged this group for a methyl substituent to get derivative 5x. From this, we used our QSAR model to predict the antichlamydial activity of the proposed new molecules. Next, we synthesized and determined the antichlamydial activity of the designed new molecules using our semi-high-throughput screening assay. Details of the new compounds and predicted versus experimental activity comparisons can be found in Figure 9a,b. These simple modifications, as predicted by the QSAR model, resulted in more potent structures. Compound 11d presented an IC50 value of 0.70 μg/mL against 1.61 μg/mL for the parent molecule 5a. The same pattern is observed for structures 11e (IC50 1.08 μg/mL) and 5x (IC50 2.78 μg/mL) versus 3.32 μg/mL for 5b (parent molecule). The viability profile of the new most potent compounds (11d and 11e) against HEp-2 cells indicated a very promising cytotoxic activity for these two structures (viability at 256 μg/mL: ~100%, Figure S4).
Figure 9.

(a) Predictions color maps for the 2D fingerprint (radial 13) QSAR for molecules 11d, 11e, and 5x. (b) Experimental versus KPLS-radial 2D QSAR model (radial 13) predicted values of antichlamydial activity (pIC50). Atoms displaying a favorable contribution to the antichlamydial activity are colored red and unfavorable are colored blue. The color intensity reflects the strength of the effect. (Note: stereochemistry is displayed because the molecules are drawn in the 3D form, which is required for Maestro software to display the QSAR color maps. However, stereochemistry is not considered in the generation of the 2D descriptors used to build the model).
As Figure 9 shows, our KPLS-QSAR model predicted the antichlamydial activity for the new derivatives with a low prediction error (Figure 9b). Noteworthy, the model can effectively predict the activity trend of the molecules in a satisfactory manner. In addition, predictions of biological activities of new molecules using a QSAR model are conditioned to such molecules falling within the applicability domain (AD) of the model.95 The AD corresponds to the chemical space covered by the nature of compounds in the training set.96 We tested our new molecules 11d, 11e, and 5x using the AutoQsar generated domain label alert,97 which suggested that these compounds are within the applicability domain of the KPLS model. These results indicate that the presented QSAR model can be a support tool in the design and guided synthesis of new dihydrothiazepines. Therefore, the model’s prediction power grants us the ability to rationalize our process of finding new highly potent molecules by focusing our synthesis on the molecules with superior predicted activity. Further, this model can have its prediction power improved even more as we expand our library to cover a larger chemical space.
We understand our model (due to the nature of the intracellular assays discussed here) is not able to distinguish between variations in the IC50 caused by differences in the permeability of the compounds or in their potency. There is a possibility that other targets in addition to the ClpX are affected, which will result in the antichlamydial activity being a result of multiple factors. However, we theorize the use of a QSAR model based on the overall antichlamydial activity is a better representation of potency than using different QSAR models (each of them based on one specific biological measurement). Experiments that seek to increase the permeability of dihydrothiazepines are currently underway.
CONCLUSIONS
The lack of drug selectivity is one of the main challenges of current therapies against CT infection owing to disruption of the microbiome and risk for the development of antibiotic resistance in other (non-CT) bacteria. In this study, we synthesized a new library of dihydrothiazepine derivatives and characterized their antichlamydial activity using a newly developed semi-high-throughput screening assay. Some of the synthesized derivatives presented substantial reduction in chlamydial inclusion number and size in infected HEp-2 cells. Further, our generated 2D fingerprint (radial 13) QSAR model was used to better elucidate the SAR using a visual projection of the quantitative contributions of different groups onto the chemical structures. Interestingly, our lead compound 5a displayed a very promising selectivity profile in our initial evaluation when tested against a panel of both pathogenic bacterial species and microorganisms from the human microbiota. Additionally, this compound showed a good cytotoxicity profile against the tested human cell lines and no mutagenic potential was seen in the Ames test. Furthermore, 5a proved to be stable in human serum and simulated gastric and intestinal fluids. A set of our dihydrothiazepines inhibited the ClpX ATPase activity in vitro, supporting ClpX as a target of this class of compounds. Our results indicate that our lead derivative 5a is selective for CT and represents a starting point for the development of new selective antichlamydial drugs. In addition, we were able to use a validated QSAR model to guide the design of compounds with superior potency when compared to the parent molecules. This tool provides us the ability to acquire more potent molecules in the future in a quicker and rationalized manner.
EXPERIMENTAL PROCEDURES
Chemistry.
The solvents and reagents were from Fisher Scientific and used with no further purification. All chemical reactions were performed using oven-dried glassware. Qualitative analysis of reactions was performed by thin-layer chromatography (TLC) with silica gel as the adsorbent (Merck silica gel IB2-F plates 0.25 mm thickness). The visualization techniques employed to analyze TLC results were either ultraviolet light (254 nm) or iodine on silica. Chromatographic separations were performed on silica gel columns (normal phase silica columns, 20–40 μm, RediSep Rf Gold high-performance columns) by a flash chromatography method on a RF 200i Flash Chromatography System from Teledyne ISCO. The solvent system used was hexanes/ethyl acetate (Hex/EtOAc) with an increasing strength of the polar component from 0 to 100% during 25 min (unless otherwise stated). To perform structural characterization of the resulting molecules we used proton nuclear magnetic resonance (1H NMR, Bruker instrument, 500 or 600 MHz) with deuterated dimethyl sulfoxide (DMSO-d6), methanol (CD3OD-d4), or chloroform (CDCl3) as solvents. 1H NMR chemical shifts are reported in parts per million (δ ppm). Chemical purities of the final compounds were determined by high-performance liquid chromatography (HPLC)-UV, which confirmed >95% purity for all biologically tested compounds. The purity determination assay was performed using the following protocol: Agilent Technologies 1220 infinity LC instrument coupled with a multiple wavelength UV detector (acquisition 254 nm), Kinetex column from Phenomenex: 5 μm (C18 reversed phase), 250 × 4.6 mm2 with a mobile phase of methanol/water, 1 mL/min flow, gradient from 0 to 100% methanol from 0 to 23 min, and maintained it until 25 min. All reported yields refer to final tested molecules. The compounds were named using the naming algorithm employed by ChemDraw Professional 15.0 (https://perkinelmerinformatics.com/products/research/chemdraw/).
General Procedure for the Knoevenagel Condensation Reaction (A).
Knoevenagel condensation was performed to obtain 2-benzylidene-1H-indene-1,3(2H)-dione intermediates (3a–y, Scheme 1) by adapting the procedure reported by Fetzer and co-workers.55 A suspension of ~1 mmol of indan-1,3-dione (1) in ethanol or methanol (see individual descriptions below, 10 mL) was added to the appropriate benzaldehyde (2) (1.10 equiv with respect to indan-1,3-dione) along with L-proline (0.30 equiv) and molecular sieves 4 Å. Specifically for derivative 3r, 1 mmol of starting material 1 was reacted with 3-hydroxybenzaldehyde in 10 mL of water (no catalyst).98 The reaction mixture was stirred at room temperature (RT) for 16–24 h (depending on the intermediate to be synthesized). Next, the reaction content was filtered using vacuum and washed with methanol (3 × 10 mL) to obtain a solid mass. These intermediates were used in the following reaction without further purification.
General Procedure for Cyclization in Acidic Media (B).
To obtain final compounds 5a–v, 2-benzylidene-1H-indene-1,3(2H)-dione intermediates (3) were cyclized by adapting the method of Fetzer and co-workers (Scheme 1).55 To a solution of a given benzylidene-1H-indene-1,3(2H)-dione (3) intermediate (amount indicated below according to different compounds) in isopropanol (iPrOH, 6 mL) were added acetic acid (AcOH, 4 mL), 2-aminothiophenol (4) (1.5 equiv with respect to benzylidene intermediate), and molecular sieves 4 Å. For some final compounds, this reaction was performed using p-toluenesulfonic acid (0.5 equiv amount indicated below according to the specific final compound), instead of acetic acid. The reaction mixture was stirred at RT or using higher temperature for 18–24 h depending on the intermediate to be synthesized (see individual descriptions below). The solvent was removed under vacuum, and the crude product was absorbed onto silica gel followed by purification using flash column chromatography eluting with a 0–100% gradient of EtOAc in hexanes. The physical characters and the spectral data of the obtained products are listed below.
General Procedure for Nitro Reduction (C).
To a mixture of methanol (MeOH, 10 mL) and tetrahydrofuran (THF, 10 mL) was added 11-(4-nitrophenyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e]-[1,4]thiazepin-12-one (5n—0.362 mmol, Scheme 2). To this solution was added 20 mg of palladium on carbon (Pd/C, 10%). This mixture was stirred under a hydrogen gas atmosphere (with a balloon apparatus) for 24 h at room temperature. Next, the solution was filtered, the solvent was removed under vacuum, and the crude product was absorbed onto silica gel followed by purification using flash column chromatography eluting with a 0–100% gradient of EtOAc in hexanes. The physical characters and the spectral data of the obtained products are listed below.
General Procedure for Thioester Oxidation (D).
To a solution of 5w (68 mg, 0.18 mmol) in dry dichloromethane (DCM, 8 mL) was added mCPBA (107 mg, 0.620 mmol, Scheme 2), and the mixture was stirred for 5 h at room temperature. The mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulfate and filtered. The solvent was removed under vacuum, and the crude product was absorbed onto silica gel followed by purification using flash column chromatography eluting with a 0–100% gradient of EtOAc in hexanes. The physical characters and the spectral data of the obtained product are listed below.
General Procedure for Cyclization of Aminoethanethiol Derivatives (E).
Procedure to obtain final derivatives without ring B (9a–d, Scheme 3): To a solution of benzylidene-1H-indene-1,3(2H)-dione (3) intermediate (amount indicated below according to different compounds) in isopropanol (iPrOH, 6 mL) were added acetic acid (AcOH, 4 mL), 2-aminoethanethiol (8) (1.5 equiv with respect to benzylidene intermediate), and molecular sieves 4 Å. For some final compounds, this reaction was performed using p-toluenesulfonic acid (0.5 equiv amount indicated below according to the specific final compound), instead of acetic acid. The reaction mixture was stirred at RT or using higher temperature for 18–24 h depending on the intermediate to be synthesized. The solvent was removed under vacuum, and the crude product was absorbed onto silica gel followed by purification using flash column chromatography eluting with a 0–70% gradient of EtOAc in hexanes. The physical characters and the spectral data of the obtained products are listed below.
General Procedure for Cyclization of Ring B Substituted Derivatives (F).
Procedure to obtain final derivatives containing a trifluoromethyl group on ring B (11a–e, Scheme 4): To a solution of benzylidene-1H-indene-1,3(2H)-dione intermediate (3) (amount indicated below according to different compounds) in isopropanol (iPrOH, 15 mL) were added 2-amino-4-(trifluoromethyl) benzenethiol hydrochloride (10) (1.5 equiv with respect to benzylidene intermediate) and molecular sieves 4 Å. The reaction mixture was stirred at room temperature for 18–24 h depending on the intermediate to be synthesized (see individual descriptions below). The solvent was removed under vacuum, and the crude product was absorbed onto silica gel followed by purification using flash column chromatography eluting with a 0–70% gradient of EtOAc in hexanes. The physical characters and the spectral data of the obtained products are listed below.
General Procedure for Cyclization of Diazepine Derivatives (G).
Procedure to obtain final derivatives containing the diazepine group (13a–c, Scheme 4): In a three-neck round-bottom flask, to a solution of benzylidene-1H-indene-1,3(2H)-dione intermediate (3) (amount indicated below according to different compounds) in isopropanol (iPrOH, 6 mL) were added acetic acid (AcOH, 4 mL), ortho-phenylenediamine (12) (1.5 equiv with respect to benzylidene intermediate), and molecular sieves 4 Å. For some final compounds, this reaction was performed using p-toluenesulfonic acid (0.5 equiv amount indicated below according to the specific final compound) instead of acetic acid. The reaction mixture was stirred at room temperature for 18–24 h under a nitrogen gas atmosphere. The solvent was removed under vacuum, and the crude product was absorbed onto silica gel followed by purification using flash column chromatography eluting with a 0–70% gradient of EtOAc in hexanes. The physical characters and the spectral data of the obtained products are listed below.
Characterization of the Compounds.
2-((1,3-Dioxo-1,3-dihydro-2H-inden-2-ylidene)methyl)benzonitrile (3a).
According to procedure A, 150 mg of indan-1,3-dione (1, 1.03 mmol) was added in 10 mL of MeOH. To this solution was added 148 mg of 2-cyanobenzaldehyde (1.13 mmol) and 28 mg of l-proline (0.25 mmol). The reaction was stirred for 16 h at RT. Orange solid, 258.00 mg (96.7%). 1H NMR (600 MHz, DMSO-d6): δ 8.69 (s, 1H), 8.01 (br s, 6H), 7.89–7.76 (m, 2H). 13C NMR (150 MHz, DMSO-d6): 188.52, 187.62, 142.14, 139.88, 138.21, 136.50, 136.36, 134.57, 133.47, 132.98, 132.90, 132.25, 132.17, 23.46, 117.25, 113.85.
2-(2-Methoxybenzylidene)-1H-indene-1,3(2H)-dione (3b).
According to procedure A, 112 mg of indan-1,3-dione (1, 0.770 mmol) was added in 10 mL of MeOH. To this solution was added 115 mg of 2-methoxy-benzaldehyde (0.847 mmol) and 28 mg of l-proline (0.25 mmol). The reaction was stirred for 18 h at RT. Yellow solid, 131.60 mg (64.7%). 1H NMR (500 MHz, CDCl3): δ 8.90 (dd, J = 7.90, 1.65 Hz, 1H), 8.51 (s, 1H), 8.02–7.97 (m, 2H), 7.81–7.78 (m, 2H), 7.52 (td, J = 7.50, 1.70 Hz, 1H), 7.08 (t, J = 7.58 Hz, 1H), 6.95 (d, J = 8.29 Hz, 1H), 3.94 (s, 3H). 13C NMR (125 MHz, CDCl3): 190.75, 189.41, 160.67, 142.53, 141.53, 140.23, 135.50, 135.29, 135.13, 134.10, 128.42, 123.32, 123.31, 122.18, 120.53, 110.80, 55.90.
2-(2-Fluoro-3-methylbenzylidene)-1H-indene-1,3(2H)-dione (3c).
According to procedure A, 112 mg of indan-1,3-dione (1, 0.770 mmol) was added in 10 mL of EtOH. To this solution was added 103 μL of 2-fluoro-3-methylbenzaldehyde (0.847 mmol) and 28 mg of l-proline (0.25 mmol). The reaction was stirred for 16 h at RT. Yellow solid, 118.50 mg (58.7%). 1H NMR (500 MHz, CDCl3): δ 8.26 (t, J = 6.95 Hz, 1H), 8.25 (s, 1H), 8.04–7.99 (m, 2H), 7.84–7.80 (m, 2H), 7.38 (t, J = 7.18 Hz, 1H), 7.18 (t, J = 7.73 Hz, 1H), 2.33 (s, 3H). 13C NMR (125 MHz, CDCl3): 190.12, 189.14, 161.82 (d, JCF = 256.01 Hz), 142.84, 140.51, 138.37 (d, JCF = 8.03 Hz), 137.15 (d, JCF = 6.40 Hz), 135.73 (d, JCF = 15.20 Hz), 131.54, 130.40, 125.45 (d, JCF = 17.59 Hz), 124.03 (d, JCF = 4.51 Hz), 123.74 (d, JCF = 2.82 Hz), 121.27 (d, JCF = 2.82 Hz), 14.86 (d, JCF = 4.64 Hz).
2-(3-Chlorobenzylidene)-1H-indene-1,3(2H)-dione (3d).
According to procedure A, 112 mg of indan-1,3-dione (1, 0.770 mmol) was added in 10 mL of MeOH. To this solution was added 113 μL of 3-chlorobenzaldehyde (0.814 mmol) and 28 mg of l-proline (0.25 mmol). The reaction was stirred for 18 h at RT. Yellow solid, 154.30 mg (74.8%). 1H NMR (500 MHz, CDCl3): δ 8.56 (t, J = 1.85 Hz, 1H), 8.25 (d, J = 7.75 Hz, 1H), 8.05–8.01 (m, 2H), 7.86–7.82 (m, 2H), 7.81 (s, 1H), 7.53–7.51 (m, 1H), 7.45 (t, J = 7.87 Hz, 1H). 13C NMR (125 MHz, CDCl3): 189.90, 188.91. 145.04, 142.71, 140.27, 135.81, 135.63, 134.89, 134.72, 133.42, 132.96, 132.24, 130.42, 130.10, 123.69, 123.64.
2-(3-(Trifluoromethyl)benzylidene)-1H-indene-1,3(2H)-dione (3e).
According to procedure A, 112 mg of indan-1,3-dione (1, 0.770 mmol) was added in 10 mL of MeOH. To this solution was added 147 mg of 3-(trifluoromethyl)benzaldehyde (0.847 mmol) and 28 mg of L-proline (0.25 mmol). The reaction was stirred for 16 h at RT. White solid, 146.5 mg (62.9%). 1H NMR (500 MHz, CDCl3): δ 8.80 (s, 1H), 8.57 (d, J = 7.84 Hz, 1H), 8.07–8.02 (m, 2H), 7.90 (s, 1H), 7.87–7.84 (m, 2H), 7.80 (d, J = 7.85 Hz, 1H), 7.65 (d, J = 7.85 Hz, 1H). 19F NMR δ −62.86 (s). 13C NMR (125 MHz, CDCl3): 189.75, 188.89, 144.62, 142.71, 140.33, 136.88, 135.91, 135.75, 133.66, 131.62, 131.36, 130.85, 130.47 (q, JCF = 3.81 Hz), 129.46, 129.26 (q, JCF = 3.53 Hz), 123.78, 123.71.
2-(3-Nitrobenzylidene)-1H-indene-1,3(2H)-dione (3f).
According to procedure A, 112 mg of indan-1,3-dione (1, 0.770 mmol) was added in 10 mL of MeOH. To this solution was added 123 mg of 3-nitrobenzaldehyde (0.814 mmol) and 28 mg of l-proline (0.25 mmol). The reaction was stirred for 16 h at RT. White solid, 146.0 mg (67.9%). 1H NMR (600 MHz, CDCl3): δ 9.57 (s, 1H), 8.68 (d, J = 7.68 Hz, 1H), 8.43 (d, J = 8.11 Hz, 1H), 8.07–8.04 (m, 2H), 8.00–7.99 (m, 3H), 7.85 (d, J = 7.97 Hz, 1H). 13C NMR (150 MHz, CDCl3): 188.91, 188.56, 147.85, 142.26, 142.10, 139.79, 139.77, 136.34, 136.25, 134.05, 131.62, 130.31, 127.05, 126.74, 123.48, 123.32.
2-(2,4-Dichlorobenzylidene)-1H-indene-1,3(2H)-dione (3g).
According to procedure A, 112 mg of indan-1,3-dione (1, 0.770 mmol) was added in 10 mL of MeOH. To this solution was added 148 mg of 2,4-dichlorobenzaldehyde (0.847 mmol) and 28 mg of L-proline (0.25 mmol). The reaction was stirred for 18 h at RT. Yellow solid, 208.80 mg (89.5%). Spectral data are identical to those published in the literature.55
2-(4-Chlorobenzylidene)-1H-indene-1,3(2H)-dione (3h).
According to procedure A, 112 mg of indan-1,3-dione (1, 0.770 mmol) was added in 10 mL of MeOH. To this solution was added 114 mg of 4-chlorobenzaldehyde (0.814 mmol) and 28 mg of l-proline (0.25 mmol). The reaction was stirred for 16 h at RT. Yellow solid, 150.0 mg (72.7%). 1H NMR (600 MHz, CDCl3): δ 8.42 (d, J = 8.26 Hz, 2H), 8.03–7.99 (m, 2H), 7.84–7.81 (m, 3H), 7.48 (d, J = 8.28 Hz, 2H). 13C NMR (150 MHz, CDCl3): 190.15, 189.18, 145.34, 142.66, 140.23, 139.67, 135.69, 135.53, 135.51, 131.69, 129.60, 129.30, 123.59, 123.57.
2-(4-Methylbenzylidene)-1H-indene-1,3(2H)-dione (3i).
According to procedure A, 112 mg of indan-1,3-dione (1, 0.770 mmol) was added in 10 mL of MeOH. To this solution was added 100 μL of 4-methylbenzaldehyde (0.847 mmol) and 28 mg of l-proline (0.25 mmol). The reaction was stirred for 20 h at RT. Yellow solid, 131.0 mg (68.6%). 1H NMR (500 MHz, CD3OD-d4): δ 8.25 (d, J = 6.98 Hz, 2H), 7.87 (s, 2H), 7.75–7.69 (m, 3H), 7.20 (d, J = 7.07 Hz, 2H), 2.32 (s, 3H). 13C NMR (125 MHz, CDCl3, some drops of CD3OD-d4): 190.94, 189.39, 147.40, 144.87, 139.87, 135.46, 135.21, 134.48, 130.51, 129.61, 128.01, 124.52, 123.22, 21.87.
4-((1,3-Dioxo-1,3-dihydro-2H-inden-2-ylidene)methyl)phenyl Acetate (3j).
According to procedure A, 146 mg of indan-1,3-dione (1, 1.00 mmol) was added in 10 mL of MeOH. To this solution was added 154 μL of 2-acetoxy-benzaldehyde (1.10 mmol) and 32 mg of l-proline (0.28 mmol). The reaction was stirred for 16 h at RT. Yellow solid, 241.7 mg (82.8%). 1H NMR (500 MHz, CDCl3): δ 8.55 (d, J = 8.74 Hz, 2H), 8.04–8.00 (m, 2H), 7.88 (s, 1H), 7.85–7.81 (m, 2H), 7.28–7.26 (m, 2H), 2.35 (s, 3H). 13C NMR (125 MHz, CDCl3): 190.33, 189.22, 168.92, 154.44, 145.79, 142.64, 140.22, 136.01, 135.59, 135.42, 130.93, 129.12, 123.53, 123.50, 122.16, 21.36.
2-(4-(Trifluoromethoxy)benzylidene)-1H-indene-1,3(2H)-dione (3k).
According to procedure A, 112 mg of indan-1,3-dione (1, 0.770 mmol) was added in 10 mL of MeOH. To this solution was added 120 μL of 4-(trifluoromethoxy)benzaldehyde (0.847 mmol) and 28 mg of l-proline (0.25 mmol). The reaction was stirred for 16 h at RT. Yellow solid, 136.5 mg (55.7%). 1H NMR (500 MHz, CDCl3): δ 8.52 (s, 2H), 8.04–7.99 (m, 2H), 7.84–7.81 (m, 3H), 7.80 (d, J = 2.04 Hz, 2H). 13C NMR (125 MHz, CDCl3): 190.24, 189.29, 152.65 (q, JCF = 1.55 Hz), 145.07, 142.83, 140.44, 136.34, 135.92, 135.77, 131.70, 129.94, 123.81, 123.78, 120.77 (q, JCF = 257.84 Hz), 120.71.
2-(4-Bromobenzylidene)-1H-indene-1,3(2H)-dione (3l).
According to procedure A, 112 mg of indan-1,3-dione (1, 0.770 mmol) was added in 10 mL of MeOH. To this solution was added 156 mg of 4-bromobenzaldehyde (0.847 mmol) and 28 mg of L-proline (0.25 mmol). The reaction was stirred for 17 h at RT. Yellow solid, 205.3 mg (85.2%). Spectral data identical to the literature.99
3-((1,3-Dioxo-1,3-dihydro-2H-inden-2-ylidene)methyl)benzonitrile (3m).
According to procedure A, 112 mg of indan-1,3-dione (1, 0.770 mmol) was added in 10 mL of MeOH. To this solution was added 111 mg of 3-cyanobenzaldehyde (0.847 mmol) and 28 mg of l-proline (0.25 mmol). The reaction was stirred for 22 h at RT. White solid, 179.7 mg (90.3%). 1H NMR (500 MHz, DMSO-d6): δ 8.94 (s, 1H), 8.68 (d, J = 9.48 Hz, 1H), 8.07–7.98 (m, 5H), 7.90 (s, 1H), 7.78 (t, J = 9.42 Hz, 1H). 13C NMR (125 MHz, DMSO-d6): 188.95, 188.49, 142.32, 142.07, 139.72, 137.64, 136.34, 136.22, 135.47, 133.64, 131.37, 129.94, 123.42, 123.30, 118.29, 111.80.
2-(4-Nitrobenzylidene)-1H-indene-1,3(2H)-dione (3n).
According to procedure A, 112 mg of indan-1,3-dione (1, 0.770 mmol) was added in 10 mL of MeOH. To this solution was added 128 mg of 4-nitrobenzaldehyde (0.847 mmol) and 28 mg of l-proline (0.25 mmol). The reaction was stirred for 24 h at RT. Yellow solid, 174.90 mg (81.4%). 1H NMR (500 MHz, CDCl3): δ 8.55 (d, J = 8.80 Hz, 2H), 8.33 (d, J = 8.86 Hz, 2H), 8.08–8.04 (m, 2H), 7.90–7.87 (m, 3H). 13C NMR (125 MHz, CDCl3): 189.26, 188.54, 149.60, 142.80, 142.77, 140.42, 138.57, 136.16, 136.06, 134.40, 132.39, 123.93, 123.88, 123.83.
4-((1,3-Dioxo-1,3-dihydro-2H-inden-2-ylidene)methyl)benzonitrile (3o).
According to procedure A, 112 mg of indan-1,3-dione (1, 0.770 mmol) was added in 10 mL of MeOH. To this solution was added 128 mg of 4-cyanobenzaldehyde (0.847 mmol) and 28 mg of L-proline (0.25 mmol). The reaction was stirred for 16 h at RT. Yellow solid, 162.6 mg (81.7%). 1H NMR (500 MHz, CDCl3, with some drops of CD3OD-d4): δ 8.40 (d, J = 7.60 Hz, 2H), 7.80 (s, 2H), 7.80–7.77 (m, 3H), 7.71 (d, J = 7.60 Hz, 2H). 13C NMR (125 MHz, CDCl3, with some drops of CD3OD-d4): 189.54, 188.60, 143.47, 142.53, 140.15, 136.76, 136.11, 135.97, 133.72, 132.26, 131.81, 123.71, 123.66, 118.19, 115.15.
2-(4-Hydroxybenzylidene)-1H-indene-1,3(2H)-dione (3p).
According to procedure A, 146 mg of indan-1,3-dione (1, 1 mmol) was added in 10 mL of MeOH. To this solution was added 134 mg of 4-hydroxybenzaldehyde (1.100 mmol) and 56 mg of L-proline (0.50 mmol). The reaction was stirred for 16 h at RT. Yellow solid, 97.4 mg (39.0%). 1H NMR (600 MHz, DMSO-d6): δ 10.88 (s br, 1H), 8.54 (d, J = 8.79 Hz, 2H), 7.96–7.90 (m, 4H), 7.76 (s, 1H), 6.95 (d, J = 8.82 Hz, 2H). 13C NMR (150 MHz, DMSO-d6): 190.00, 189.04, 163.30, 146.29, 141.65, 139.22, 137.61, 135.58, 135.41, 125.19, 124.60, 122.76, 122.71, 116.00.
2-(4-Methoxybenzylidene)-1H-indene-1,3(2H)-dione (3q).
According to procedure A, 112 mg of indan-1,3-dione (1, 0.770 mmol) was added in 10 mL of MeOH. To this solution was added 102.5 μL of 4-methoxybenzaldehyde (0.847 mmol) and 28 mg of l-proline (0.25 mmol). The reaction was stirred for 20 h at RT. Yellow solid, 152.3 mg (75.0%). Spectral data identical to the literature.99
2-(3-Hydroxybenzylidene)-1H-indene-1,3(2H)-dione (3r).
According to procedure A, 146 mg of indan-1,3-dione (1, 1.0 mmol) was added in 10 mL of water. To this solution was added 134 mg of 3-hydroxybenzaldehyde (1.1 mmol). The reaction was stirred for 19 h at RT. Yellow solid, 199.2 mg (79.7%). Spectral data identical to the literature.100
2-(4-(Benzyloxy)benzylidene)-1H-indene-1,3(2H)-dione (3s).
According to procedure A, 112 mg of indan-1,3-dione (1, 0.770 mmol) was added in 10 mL of EtOH. To this solution was added 179 mg of 4-(benzyloxy)benzaldehyde (0.847 mmol) and 28 mg of L-proline (0.25 mmol). The reaction was stirred for 19 h at RT. Yellow solid, 160.8 mg (61.1%). 1H NMR (500 MHz, CDCl3): δ 8.57 (d, J = 8.86 Hz, 2H), 8.00–7.97 (m, 2H), 7.84 (s, 1H), 7.81–7.77 (m, 2H), 7.45–7.40 (m, 4H), 7.36 (t, J = 7.25 Hz, 1H), 7.09 (d, J = 8.95 Hz, 2H), 5.18 (s, 2H). 13C NMR (125 MHz, CDCl3): 190.95, 189.67, 163.33, 146.91, 142.53, 140.12, 137.33, 136.12, 135.23, 135.02, 128.88, 128.47, 127.67, 127.62, 126.82, 126.80, 123.23, 115.33, 70.41.
4-((1,3-Dioxo-1,3-dihydro-2H-inden-2-ylidene)methyl)benzoic Acid (3t).
According to procedure A, 112 mg of indan-1,3-dione (1, 0.770 mmol) was added in 10 mL of MeOH. To this solution was added 127 mg of 4-carboxaldehyde (1.1 mmol) and 28 mg of l-proline (0.25 mmol). The reaction was stirred for 18 h at RT. Yellow solid, 181.5 mg (84.7%). Spectral data identical to the literature.55
2-((1,3-Dioxo-1,3-dihydro-2H-inden-2-ylidene)methyl)benzoic Acid (3u).
According to procedure A, 112 mg of indan-1,3-dione (1, 0.770 mmol) was added in 10 mL of MeOH. To this solution was added 127 mg of 2-carboxaldehyde (0.847 mmol) and 28 mg of l-proline (0.25 mmol). The reaction was stirred for 23 h at RT. White solid, 155.6 mg (72.7%). Spectral data are identical to those published in the literature.99
2-([1,1′-Biphenyl]-4-ylmethylene)-1H-indene-1,3(2H)-dione (3v).
According to procedure A, 112 mg of indan-1,3-dione (1, 0.770 mmol) was added in 10 mL of MeOH. To this solution was added 150 mg of [1,1′-biphenyl]-4-carbaldehyde (0.824 mmol) and 28 mg of l-proline (0.25 mmol). The reaction was stirred for 16 h at RT. Yellow solid, 206.0 mg (86.2%). Spectral data are identical to those published in the literature.101
2-(3-Methoxybenzylidene)-1H-indene-1,3(2H)-dione (3w).
According to procedure A, 146 mg of indan-1,3-dione (1, 0.770 mmol) was added in 10 mL of MeOH. To this solution was added 118 mg of 3-methoxybenzaldehyde (0.847 mmol) and 28 mg of l-proline (0.25 mmol). The reaction was stirred for 16 h at RT. Yellow solid, 190.0 mg (84.9%). 1H NMR (600 MHz, CDCl3): δ 8.46 (s, 1H), 8.04–8.01 (m, 2H), 7.89 (s, 1H), 7.84–7.81 (m, 2H), 7.79 (t, J = 7.62 Hz, 1H), 7.41 (t, J = 7.92 Hz, 1H), (dd, J = 8.28, 2.46 Hz, 1H). 13C NMR (150 MHz, CDCl3): 190.44, 189.32, 159.87, 147.34, 142.73, 140.16, 135.59, 135.40, 134.51, 129.79, 129.30, 127.89, 123.52, 123.51, 120.97, 117.02, 55.68.
2-(2-Methylbenzylidene)-1H-indene-1,3(2H)-dione (3x).
According to procedure A, 112 mg of indan-1,3-dione (1, 0.770 mmol) was added in 10 mL of MeOH. To this solution was added 98.2 μL of 2-methylbenzaldehyde (0.847 mmol) and 28 mg of l-proline (0.25 mmol). The reaction was stirred for 18 h at RT. Yellow solid, 132.6 mg (64.7%). 1H NMR (600 MHz, CDCl3): δ 8.54 (d, J = 7.76 Hz, 1H), 8.24 (s, 1H), 8.04–7.99 (m, 2H), 7.84–7.81 (m, 2H), 7.42 (td, J = 7.44, 1.02 Hz, 1H), 7.33 (t, J = 7.57 Hz, 1H), 7.28 (d, J = 7.56 Hz, 1H), 2.55 (s, 3H). 13C NMR (150 MHz, CDCl3): 190.41, 188.94, 144.49, 142.65, 140.98, 140.20, 135.57, 135.38, 132.91, 132.63, 131.60, 130.70, 129.33, 126.05, 123.50, 123.47, 20.50.
2-(Thiophen-2-ylmethylene)-1H-indene-1,3(2H)-dione (3y).
According to procedure A, 300 mg of indan-1,3-dione (1, 2.05 mmol) was added in 20 mL of MeOH. To this solution was added 210 μL of 2-thiophenecarboxaldehyde (2.26 mmol) and 56 mg of l-proline (0.50 mmol). The reaction was stirred for 16 h at RT. Orange solid, 464.4 mg (94.2%). Spectral data are identical to those published in the literature.99
2-(12-Oxo-11,12-dihydro-5H-benzo[b]indeno[1,2-e][1,4]thiazepin-11-yl)benzonitrile (5a).
According to general procedure B, 100 mg of 3a (0.386 mmol), 61.9 μL of 2-aminothiophenol (0.579 mmol), 4 mL of acetic acid, 6 mL of iPrOH, and molecular sieves 4 Å were reacted for 20 h at RT. Orange solid, 79.73 mg (56.4%). 1H NMR (600 MHz, DMSO-d6): δ 10.26 (s, 1H), 8.15 (d, J = 7.37 Hz, 1H), 7.80 (dd, J = 7.56, 1.14 Hz, 1H), 7.63 (d, J = 8.22 Hz, 1H), 7.58 (td, J = 7.56, 1.02 Hz, 1H), 7.44 (t, J = 7.35 Hz, 1H), 7.41–7.38 (m, 2H), 7.25 (td, J = 7.56, 0.94 Hz, 1H), 7.18 (td, J = 7.74, 1.26 Hz, 1H), 6.94–6.91 (m, 2H), 6.62 (d, J = 7.84 Hz, 1H), 5.63 (s, 1H). 13C NMR (150 MHz, DMSO-d6): 189.17, 157.25, 145.17, 143.50, 139.19, 136.05, 133.43, 132.54, 132.26, 131.90, 130.34, 129.71, 127.62, 126.78, 125.14, 123.81, 122.93, 120.69, 119.54, 117.46, 110.69, 106.89, 44.17. High-resolution mass spectrometry-electrospray ionization (HRMS-ESI) (m/z) calcd for C23H14N2OS [M + H]+ 367.0900, found 367.0914. HPLC purity: 95.4%.
11-(2-Methoxyphenyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (5b).
According to general procedure B, 50 mg of 3b (0.203 mmol), 32.6 μL of 2-aminothiophenol (0.305 mmol), 4 mL of acetic acid, 6 mL of iPrOH, and molecular sieves 4 Å were reacted for 19 h at 70 °C. Red solid, 15.05 mg (19.8%). 1H NMR (600 MHz, DMSO-d6): δ 10.04 (s, 1H), 8.11 (d, J = 7.44 Hz, 1H), 7.60 (d, J = 7.97 Hz, 1H), 7.55 (td, J = 7.56, 0.96 Hz, 1H), 7.40 (d, J = 7.33 Hz, 1H), 7.35–7.32 (m, 2H), 7.03 (td, J = 8.28, 1.62 Hz, 1H), 6.94 (d, J = 8.10 Hz, 1H), 6.90–6.87 (m, 2H), 6.40 (t, J = 7.47 Hz, 1H), 6.28 (t, J = 7.59, 1.47 Hz, 1H), 5.62 (s, 1H), 3.91 (s, 3H). 13C NMR (150 MHz, DMSO-d6): 189.16, 156.87, 155.75, 143.21, 139.36, 136.11, 132.65, 131.61, 130.09, 129.95, 128.91, 127.95, 126.36, 124.55, 124.33, 123.20, 120.37, 119.05, 119.03, 110.75, 109.23, 55.68, 40.03. HRMS-ESI (m/z) calcd for C23H17NO2S [M + Na]+ 394.0878, found 394.2260. HPLC purity: 98.3%.
11-(2-Fluoro-3-methylphenyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (5c).
According to procedure B, 50 mg of 3c (0.188 mmol), 30.20 μL of 2-aminothiophenol (0.282 mmol), 4 mL of acetic acid, and 6 mL of iPrOH were reacted for 18 h at RT. Red solid, 48.80 mg (69.1%). 1H NMR (500 MHz, DMSO-d6): δ 10.12 (s, 1H), 8.13 (d, J = 7.38 Hz, 1H), 7.61 (d, J = 8.06 Hz, 1H), 7.56 (t, J = 7.44 Hz, 1H), 7.42 (t, J = 7.45 Hz, 1H), 7.39–7.36 (m, 2H), 6.99–6.91 (m, 3H), 6.56 (t, J = 7.62 Hz, 1H), 6.24 (t, J = 7.25 Hz, 1H), 5.56 (s, 1H), 2.23 (s, 3H). 19F NMR δ −121.87 (s). 13C NMR (150 MHz, DMSO-d6): 189.12, 157.69 (d, JCF = 244.66 Hz), 157.01, 143.33, 139.25, 136.05, 132.56, 131.73, 130.16 (d, JCF = 7.86 Hz), 129.29, 129.07 (d, JCF = 13.71 Hz), 125.16 (d, JCF = 2.15 Hz), 124.86, 123.87 (d, JCF = 17.15 Hz), 123.73, 123.47, 122.65 (d, JCF = 3.86 Hz), 120.52, 119.27, 108.07, 40.05, 14.14 (d, JCF = 3.92 Hz). HRMS-ESI (m/z) calcd for C23H16FNOS [M + H]+ 374.1009, found 374.1008. HPLC purity: 95.8%.
11-(3-Chlorophenyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (5d).
According to procedure B, 50 mg of 3d (0.186 mmol), 20.55 μL of 2-aminothiophenol (0.219 mmol), 4 mL of acetic acid, and 6 mL of iPrOH were reacted for 18 h at RT. Red solid, 43.00 mg (61.8%). 1H NMR (500 MHz, DMSO-d6): δ 10.11 (s, 1H), 8.11 (d, J = 7.39 Hz, 1H), 7.60–7.55 (m, 2H), 7.43 (t, J = 7.39 Hz, 1H), 7.39–7.34 (m, 2H), 7.09–7.06 (m, 3H), 7.00–6.94 (m, 3H), 5.55 (s, 1H). 13C NMR (125 MHz, DMSO-d6): 189.25, 156.68, 144.81, 143.35, 139.18, 136.03, 132.60, 132.46, 131.70, 130.13, 129.51, 129.24, 127.04, 126.36, 125.74, 124.88, 123.86, 123.50, 120.50, 119.28, 108.20, 45.16. HRMS-ESI (m/z) calcd for C22H14ClNOS [M + H]+ 376.0557, found 376.0559. HPLC purity: 97.5%.
11-(3-(Trifluoromethyl)phenyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (5e).
According to procedure B, 50 mg of 3e (0.166 mmol), 18.7 μL of 2-aminothiophenol (0.199 mmol), 4 mL of acetic acid, and 6 mL of iPrOH were reacted for 18 h at RT. Red solid, 18.26 mg (26.9%). 1H NMR (600 MHz, DMSO-d6): δ 10.15 (s, 1H), 8.12 (d, J = 7.40 Hz, 1H), 7.59–7.55 (m, 2H), 7.43 (d, J = 7.55 Hz, 1H), 7.39–7.34 (m, 3H), 7.32–7.28 (m, 3H). 6.92–6.91 (m, 2H), 5.68 (s, 1H). 19F NMR δ −61.18 (s). 13C NMR (150 MHz, DMSO-d6): 189.27, 156.83, 143.84, 143.38, 139.15, 136.04, 132.60, 131.76, 131.11, 130.20, 129.29, 128.84, 128.40 (q, JCF = 31.51 Hz), 124.95, 123.80, 123.57, 123.40 (q, JCF = 4.01 Hz), 123.11 (q, JCF = 3.89 Hz), 120.56, 119.34, 107.97, 45.29. HRMS-matrix-assisted laser desorption ionization (MALDI) (m/z) calcd for C23H14F3NOS [M + H]+ 410.0820, found 410.0726. HPLC purity: 96.7%.
11-(3-Nitrophenyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (5f).
According to procedure B, 50 mg of 3f (0.179 mmol), 20.2 μL of 2-aminothiophenol (0.214 mmol), 4 mL of acetic acid, and 6 mL of iPrOH were reacted for 18 h at RT. Red solid, 18.3 mg (26.9%). 1H NMR (600 MHz, DMSO-d6): δ 10.29 (s, 1H), 8.14 (d, J = 7.38 Hz, 1H), 7.90 (dd, J = 8.16, 1.59 Hz, 1H), 7.83 (t, J = 1.73 Hz, 1H), 7.61–7.56 (m, 2H), 7.51–7.49 (m, 1H), 7.45–7.43 (m, 1H), 7.40–7.34 (m, 3H), 6.95–6.90 (m, 2H), 5.76 (s, 1H). 13C NMR (150 MHz, DMSO-d6): 189.26, 156.96, 147.20, 144.78, 143.45, 139.11, 136.08, 133.68, 132.58, 131.81, 130.27, 129.45, 129.35, 125.06, 123.68, 123.59, 121.44, 121.42, 120.61, 119.42, 107.64, 45.06. HRMS-MALDI (m/z) calcd for C22H14N2O3S [M + Na]+ 409.0623, found 409.0959. HPLC purity: 98.13%.
11-(2,4-Dichlorophenyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (5g).
According to procedure B, 50 mg of 3g (0.16 mmol), 26.5 μL of 2-aminothiophenol (0.247 mmol), 4 mL of acetic acid, and 6 mL of iPrOH were reacted for 18 h at RT. Orange solid, 27.2 mg (40.0%). 1H NMR (500 MHz, DMSO-d6): δ 10.22 (s, 1H), 8.14 (d, J = 7.38 Hz, 1H), 7.63 (d, J = 8.14 Hz, 1H), 7.59–7.56 (m, 2H), 7.45–7.37 (m, 3H), 6.97–6.89 (m, 2H), 6.90 (dd, J = 8.45 Hz, 1H), 6.48 (d, J = 8,43 Hz, 1H), 5.58 (s, 1H). 13C NMR (125 MHz, DMSO-d6): 189.06, 157.22, 143.38, 139.18, 138.18, 136.15, 133.21, 132.48, 132.00, 131.80, 130.24, 129.58, 128.93, 128.75, 126.17, 125.10, 123.61, 123.10, 120.60, 119.41, 107.53, 42.86. HRMS-MALDI (m/z) calcd for C22H13Cl2NOS [M + H]+ 410.0167, found 410.0040. HPLC purity: 97.0%.
11-(4-Chlorophenyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (5h).
According to procedure B, 50 mg of 3h (0.19 mmol), 23.9 μL of 2-aminothiophenol (0.22 mmol), 4 mL of acetic acid, and 6 mL of iPrOH were reacted for 18 h at RT. Red solid, 25 mg (35%). 1H NMR (600 MHz, DMSO-d6): δ 10.07 (s, 1H), 8.11 (d, J = 7.38 Hz, 1H), 7.57–7.54 (m, 2H), 7.42 (t, J = 7.31 Hz, 1H), 7.38–7.33 (m, 2H), 7.12 (d, J = 8.53 Hz, 2H), 7.03 (d, J = 8.51 Hz, 2H), 6.98 (dd, J = 7.68, 1.44 Hz, 1H), 6.94 (td, J = 7.74, 1.01 Hz, 1H), 5.53 (s, 1H). 13C NMR (150 MHz, DMSO-d6): 189.25, 156.64, 143.38, 141.34, 139.24, 136.07, 132.64, 131.70, 130.87, 130.11, 129.21, 128.99, 127.69, 124.83, 123.94, 123.51, 120.49, 119.22, 108.55, 45.05. HRMS-MALDI (m/z) calcd for C22H14ClNOS [M + H]+ 376.0557, found 376.0560. HPLC purity: 99.4%.
11-(p-Tolyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (5i).
According to procedure B, 50 mg of 3i (0.20 mmol), 25.9 μL of 2-aminothiophenol (0.242 mmol), 4 mL of acetic acid, and 6 mL of iPrOH were reacted for 24 h at RT. Orange solid, 42.83 mg (59.4%). 1H NMR (500 MHz, DMSO-d6): δ 9.99 (s, 1H), 8.10 (d, J = 7.38 Hz, 1H), 7.57–7.54 (m, 2H), 7.42 (t, J = 7.29 Hz, 1H), 7.38 (d, J = 6.65 Hz, 1H), 7.35–7.32 (m, 1H), 6.98 (dd, J = 7.70, 1.45 Hz, 1H), 6.94–6.86 (m, 5H), 5.45 (s, 1H), 2.13 (s, 3H). 13C NMR (125 MHz, DMSO-d6): δ 189.23, 156.34, 143.37, 139.32, 139.26, 136.05, 135.33, 132.69, 131.61, 129.97, 128.95, 128.32, 127.09, 124.60, 124.27, 123.30, 120.39, 119.08, 109.39, 45.46, 20.48. HRMS-ESI (m/z) calcd for C23H17NOS [M + H]+ 356.1104, found 356.1102. HPLC purity: 98.9%.
4-(12-Oxo-11,12-dihydro-5H-benzo[b]indeno[1,2-e][1,4]thiazepin-11-yl)phenyl Acetate (5j).
According to procedure B, 150 mg of 3j (0.51 mmol), 2-aminothiophenol (0.77 mmol), 8 mL of acetic acid, and 12 mL of iPrOH were reacted for 24 h at RT. Orange solid, 102 mg (50.0%). 1H NMR (500 MHz, DMSO-d6): δ 10.04 (s, 1H), 8.11 (d, J = 7.39 Hz, 1H), 7.57–7.54 (m, 2H), 7.42 (t, J = 7.30 Hz, 1H), 7.37 (d, J = 6.55 Hz, 1H), 7.36–7.32 (m, 1H), 7.04 (d, J = 8.45 Hz, 2H), 6.99 (dd, J = 7.70 Hz, 1.45 Hz,1H), 6.92 (td, J = 7.65, 1.13, 1.45 Hz, 1H), 6.82 (d, J = 8.60 Hz, 2H), 5.52 (s, 1H), 2.17 (s, 3H). 13C NMR (125 MHz, DMSO-d6): 189.26, 168.95, 156.48, 148.73, 143.37, 139.70, 139.28, 136.05, 132.66, 131.65, 130.04, 129.08, 128.41, 124.70, 124.06, 123.43, 121.00, 120.44, 119.16, 109.00, 45.16, 20.77. HRMS-MALDI (m/z) calcd for C24H17NO3S [M + Na]+ 422.0827, found 422.2483. HPLC purity: 98.2%.
11-(4-(Trifluoromethoxy)phenyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (5k).
According to procedure B, 50 mg of 3k (0.16 mmol), 17.7 μL of 2-aminothiophenol (0.199 mmol), 4 mL of acetic acid, and 6 mL of iPrOH were reacted for 18 h at RT. Red solid, 37.6 mg (56.4%). 1H NMR (500 MHz, DMSO-d6): δ 10.08 (s, 1H), 8.11 (d, J = 7.39 Hz, 1H), 7.58–7.54 (m, 2H), 7.43 (t, J = 7.29 Hz, 1H), 7.38–7.33 (m, 2H), 7.14 (d, J = 8.64 Hz, 2H), 7.05 (d, J = 8.34 Hz, 2H), 6.97 (dd, J = 7.75, 1.5 Hz, 1H), 6.94–6.91 (m, 1H), 5.58 (s, 1H). 19F NMR δ −56.89 (s). 13C NMR (125 MHz, DMSO-d6): 189.26, 156.65, 146.10, 143.38, 141.70, 139.23, 136.02, 132.63, 131.70, 130.10, 129.21, 128.96, 124.73, 123.89, 123.53, 120.48, 120.23, 119.23, 108.51, 44.96. HRMS-MALDI (m/z) calcd for C23H14F3NO2S [M + H]+ 426.0769, found 426.0709. HPLC purity: 96.7%.
11-(4-Bromophenyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (5l).
According to procedure B, 50 mg of 3l (0.16 mmol), 18.1 μL of 2-aminothiophenol (0.168 mmol), 4 mL of acetic acid, and 6 mL of iPrOH were reacted for 18 h at RT. Orange solid, 33.7 mg (50.2%). 1H NMR (500 MHz, DMSO-d6): δ 10.06 (s, 1H), 8.11 (d, J = 7.35 Hz, 1H), 7.57–7.54 (m, 2H), 7.42 (t, J = 7.25 Hz, 1H), 7.38–7.34 (m, 2H), 7.26 (d, J = 8.50 Hz, 2H), 7.00–6.93 (m, 4H), 5.51 (s, 1H). 13C NMR (125 MHz, DMSO-d6): 189.24, 156.62, 143.37, 141.76, 139.23, 136.06, 132.62, 131.69, 130.60, 130.09, 129.35, 129.21, 124.82, 123.91, 123.50, 120.48, 119.43, 119.21, 108.48, 45.10. HRMS-MALDI (m/z) calcd for C22H14BrNOS [M + H]+ 420.0051, found 420.0066. HPLC purity: 97.2%.
3-(12-Oxo-11,12-dihydro-5H-benzo[b]indeno[1,2-e][1,4]thiazepin-11-yl)benzonitrile (5m).
According to procedure B, 50 mg of 3m (0.19 mmol), 31 μL of 2-aminothiophenol (0.29 mmol), 4 mL of acetic acid, and 6 mL of iPrOH were reacted for 18 h at 75 °C. Orange solid, 17.58 mg (22.2%). 1H NMR (500 MHz, DMSO-d6): δ 10.13 (s, 1H), 8.12 (d, J = 7.35 Hz, 1H), 7.60–7.55 (m, 2H), 7.50 (d, J = 7.62 Hz, 1H), 7.45–7.42 (m, 2H), 7.39–7.35 (m, 3H), 7.29 (t, J = 7.77 Hz, 1H), 6.96–6.91 (m, 2H), 5.64 (s, 1H). 13C NMR (125 MHz, DMSO-d6): 189.73, 157.47, 144.52, 143.91, 139.70, 136.52, 133.12, 132.42, 132.18, 130.93, 130.71, 130.62, 129.84, 129.50, 125.43, 124.12, 124.11, 120.99, 119.82, 119.04, 111.11, 108.06, 45.52. HRMS-ESI (m/z) calcd for C23H14N2OS [M + H]+ 367.0900, found 367.0899. HPLC purity: 96.3%.
11-(4-Nitrophenyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (5n).
According to procedure B, 50 mg of 3n (0.58 mmol), 92.3 μL of 2-aminothiophenol (0.868 mmol), 50 mg of p-toluenesulfonic acid monohydrate (0.260 mmol), and 20 mL of iPrOH were reacted for 24 h at RT. Orange solid, 213.25 mg (95.3%). 1H NMR (500 MHz, DMSO-d6): δ 10.16 (s, 1H), 8.14 (d, J = 7.39 Hz, 1H), 7.94 (d, J = 8.77 Hz, 2H), 7.60–7.56 (m, 2H), 7.43 (t, J = 7.31 Hz, 1H), 7.40–7.35 (m, 2H), 7.30 (d, J = 8.72 Hz, 2H), 6.97 (dd, J = 7.70, 1.45 Hz, 1H), 6.92 (t, J = 7.41 Hz, 1H), 5.71 (s, 1H). 13C NMR (125 MHz, DMSO-d6): 189.29, 156.84, 150.07, 145.78, 143.35, 139.15, 135.98, 132.58, 131.78, 130.21, 129.44, 128.39, 124.95, 123.66, 123.46, 122.99, 120.57, 119.36, 107.58, 45.19. HRMS-ESI (m/z) calcd for C22H14N2O3S [M + H]+ 387.0798, found 387.0796. HPLC purity: 97.1%.
4-(12-Oxo-11,12-dihydro-5H-benzo[b]indeno[1,2-e][1,4]thiazepin-11-yl)benzonitrile (5o).
According to procedure B, 50 mg of 3o (0.19 mmol), 31 μL of 2-aminothiophenol (0.29 mmol), 4 mL of acetic acid, and 6 mL of iPrOH were reacted for 24 h at RT. Orange solid, 67.6 mg (95.3%). 1H NMR (500 MHz, CD3OD-d4): δ 7.92 (d, J = 7.35 Hz, 1H), 7.55–7.51 (m, 1H), 7.49 (dd, J = 8.10, 0.93 Hz, 1H), 7.45–7.42 (m, 4H), 7.33 (dd, J = 8.35, 1.47 Hz, 1H), 7.24 (d, J = 8.25 Hz, 2H), 7.01 (dd, J = 7.75, 1.45 Hz, 1H), 6.94 (td, J = 7.50, 1.10 Hz, 1H), 5.57 (s, 1H). 13C NMR (125 MHz, DMSO-d6): δ 189.28, 156.83, 147.95, 143.35, 139.19, 135.97, 132.59, 131.75, 130.16, 129.37, 128.11, 124.89, 123.62, 123.54, 120.53, 119.31, 118.65, 109.00, 107.61, 45.40. HRMS-ESI (m/z) calcd for C23H14N2OS [M + H]+ 367.0900, found 367.0898. HPLC purity: 99.2%.
11-(4-Hydroxyphenyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (5p).
According to procedure B, 97.4 mg of 3p (0.39 mmol), 2-aminothiophenol (0.58 mmol), 8 mL of acetic acid, 12 mL of iPrOH, and molecular sieves were reacted for 24 h at RT. Red solid, 13.00 mg (9.3%). Spectral data are identical to those published in the literature.55 HPLC purity: 97.4%.
11-(4-Methoxyphenyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (5q).
According to procedure B, 50 mg of 3q (0.22 mmol), 25.3 μL of 2-aminothiophenol (0.265 mmol), 4 mL of acetic acid, and 6 mL of iPrOH were reacted for 18 h at RT. Red solid, 14.3 mg (17.4%). 1H NMR (500 MHz, DMSO-d6): δ 9.98 (s, 1H), 8.06 (d, J = 7.38 Hz, 1H), 7.56–7.53 (m, 2H), 7.41 (t, J = 7.28 Hz, 1H), 7.37–7.31 (m, 2H), 6.99 (dd, J = 7.70, 1.45 Hz, 1H), 6.94–6.91 (m, 3H), 6.62 (d, J = 8.75 Hz, 2H), 5.44 (s, 1H), 3.60 (s, 3H). 13C NMR (125 MHz, DMSO-d6): 189.20, 157.59, 156.31, 143.38, 139.32, 136.09, 134.25, 132.69, 131.61, 129.97, 128.94, 128.30, 124.61, 124.37, 123.32, 120.38, 119.07, 113.06, 109.61, 54.88, 45.27. HRMS-ESI (m/z) calcd for C23H17NO2S [M + H]+ 372.1053, found 372.1051. HPLC purity: 97.8%.
11-(3-Hydroxyphenyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (334).
According to procedure B, 60 mg of 3r (0.17 mmol), 2-aminoethanethiol (19.4 mg, 0.252 mmol), 4 mL of acetic acid, and 6 mL of iPrOH were reacted for 19 h at RT. Red solid, 44.4 mg (52.0%). Spectral data are identical to those published in the literature.55 HPLC purity: 95.9%.
11-(4-Aminophenyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (6a).
According to procedure C, 140 mg of 5n (0.362 mmol), 10 mL of dried MeOH, and 10 mL of dried THF were added in a three-neck round-bottom flask under a nitrogen atmosphere. To this solution was added 20 mg of palladium on carbon (Pd/C, 10%) and the solution was stirred for 24 h at RT. Red solid, 65.7 mg (52.3%). 1H NMR (600 MHz, DMSO-d6): δ 9.92 (s, 1H), 8.07 (d, J = 7.36 Hz, 1H), 7.53 (td, J = 7.38, 1.02 Hz, 2H), 7.40 (t, J = 7.31 Hz, 1H), 7.35 (d, J = 6.93 Hz, 1H), 7.31 (td, J = 7.38, 1.46 Hz, 1H), 6.99 (dd, J = 7.74, 1.50 Hz, 1H), 6.92 (td, J = 7.50, 1.20 Hz, 1H), 6.65 (d, J = 8.40 Hz, 2H), 6.22 (d, J = 4.25 Hz, 2H), 5.32 (s, 1H), 4.84 (s, 2H). 13C NMR (150 MHz, DMSO-d6): 189.15, 156.17, 146.90, 143.39, 139.40, 136.21, 132.80, 131.54, 129.88, 129.14, 128.71, 127.83, 124.84, 124.52, 123.18, 120.32, 118.94, 113.17, 110.32, 45.83. HRMS-ESI (m/z) calcd for C22H16N2OS [M + H]+ 357.1056, found 357.1059. HPLC purity: 95.9%.
11-(4-(Benzyloxy)phenyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (5s).
According to procedure B, 50 mg of 3s (0.15 mmol), 23.5 μL of 2-aminothiophenol (0.220 mmol), 4 mL of acetic acid, and 6 mL of iPrOH were reacted for 18 h at 80 °C. Orange solid, 13.7 mg (20.0%). 1H NMR (500 MHz, DMSO-d6): δ 9.99 (s, 1H), 8.10 (d, J = 7.35 Hz, 1H), 7.56–7.53 (m, 2H), 7.41 (t, J = 7.25 Hz, 1H), 7.37–7.29 (m, 7H), 7.00 (d, J = 7.62 Hz, 1H), 6.94–6.91 (m, 3H), 6.70 (d, J = 8.65 Hz, 2H), 5.45 (s, 1H), 4.94 (s, 2H). 13C NMR (125 MHz, DMSO-d6): 189.20, 156.71, 156.31, 143.38, 139.32, 136.95, 136.08, 134.51, 132.68, 131.60, 129.96, 128.94, 128.33, 127.74, 127.65, 124.62, 124.35, 123.34, 120.38, 119.07, 113.94, 109.57, 69.04, 45.26. HRMS-MALDI (m/z) calcd for C29H21NO2S [M + H]+ 448.1365, found 448.0505. HPLC purity: 96.4%.
4-(12-Oxo-11,12-dihydro-5H-benzo[b]indeno[1,2-e][1,4]thiazepin-11-yl)benzoic Acid (5t).
According to procedure B, 50 mg of 3t (0.18 mmol), 28.8 μL of 2-aminothiophenol (0.269 mmol), 25 mg of p-toluenesulfonic acid monohydrate (0.130 mmol), and 10 mL of iPrOH were reacted for 20 h at RT. Orange solid, 30.8 mg (44.7%). 1H NMR (500 MHz, DMSO-d6): δ 12.80 (s, 1H), 10.10 (s, 1H), 8.12 (d, J = 7.40 Hz, 1H), 7.64 (d, J = 8.35 Hz, 2H), 7.58–7.54 (m, 2H), 7.42 (t, J = 7.30 Hz, 1H), 7.38 (d, J = 7.55 Hz, 1H), 7.36–7.32 (m, 1H), 7.13 (d, J = 8.25 Hz, 2H), 6.96 (dd, J = 7.65 Hz, 1H), 6.92–6.89 (m, 1H), 5.59 (s, 1H). 13C NMR (125 MHz, DMSO-d6): 189.29, 166.97, 156.69, 147.20, 143.37, 139.25, 136.01, 132.67, 131.70, 130.10, 129.19, 128.86, 128.69, 127.33, 124.77, 123.86, 123.51, 120.50, 119.25, 108.32, 45.49. HRMS-MALDI (m/z) calcd for C23H15NO3S [M + Na]+ 408.0671, found 408.0961. HPLC purity: 96.2%.
2-(12-Oxo-11,12-dihydro-5H-benzo[b]indeno[1,2-e][1,4]thiazepin-11-yl)benzoic Acid (5u).
According to procedure B, 50 mg of 3u (0.179 mmol), 28.8 μL of 2-aminothiophenol (0.269 mmol), 25 mg of p-toluenesulfonic acid monohydrate (0.13 mmol), and 10 mL of iPrOH were reacted for 19 h at RT. Orange solid, 56.7 mg (82.1%). 1H NMR (600 MHz, DMSO-d6): δ 13.11 (s, 1H), 10.09 (s, 1H), 8.12 (d, J = 7.41 Hz, 1H), 7.75 (dd, J = 7.68, 1.32 Hz, 1H), 7.57–7.54 (m, 2H), 7.42 (t, J = 7.33 Hz, 1H), 7.37 (d, J = 6.84 Hz, 1H), 7.30 (td, J = 7.14, 1.58 Hz, 1H), 7.10 (td, J = 7.50, 0.92 Hz, 1H), 6.98 (td, J = 7.68, 1.32 Hz, 1H), 6.88–6.82 (m, 2H), 6.60 (s, 1H), 6.53 (d, J = 7.81 Hz, 1H). 13C NMR (125 MHz, DMSO-d6): 189.34, 168.90, 157.15, 143.21, 142.69, 139.34, 135.98, 132.71, 131.66, 130.58, 130.38, 130.05, 129.45, 129.06, 126.72, 126.52, 124.64, 124.02, 123.19, 120.45, 119.11, 108.67, 42.85. HRMS-MALDI (m/z) calcd for C23H15NO3S [M + Na]+ 408.0671, found 408.0302. HPLC purity: 97.9%.
11-([1,1′-Biphenyl]-4-yl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (5v).
According to procedure B, 50 mg of 3v (0.16 mmol), 21 μL of 2-aminothiophenol (0.19 mmol), 4 mL of acetic acid, and 6 mL of iPrOH were reacted for 18 h at RT. Red solid, 43.3 mg (64.4%). 1H NMR (600 MHz, DMSO-d6): δ 10.05 (s, 1H), 8.13 (d, J = 7.41 Hz, 1H), 7.59 (dd, J = 8.22, 1.02 Hz, 1H), 7.56 (td, J = 7.56, 1.14 Hz, 1H), 7.54–7.52 (m, 2H), 7.43 (t, J = 7.32 Hz, 1H), 7.39–7.37 (m, 5H), 7.34 (td, J = 7.44, 1.38 Hz, 1H), 7.30 (t, J = 7.36 Hz, 1H), 7.11 (d, J = 8.26 Hz, 2H), 7.03 (dd, J = 7.72, 1.42 Hz, 1H), 6.92 (dd, J = 7.50, 1.12 Hz, 1H), 5.54 (s, 1H). 13C NMR (150 MHz, DMSO-d6): 189.30, 156.42, 143.39, 141.53, 139.40, 139.30, 137.92, 136.06, 132.66, 131.66, 130.03, 129.06, 128.83, 127.80, 127.30, 126.35, 125.93, 124.66, 124.13, 123.41, 120.43, 119.15, 109.05, 45.43. HRMS-MALDI (m/z) calcd for C28H19NOS [M + H]+ 418.1259, found 418.1215. HPLC purity: 97.8%.
11-(3-Methoxyphenyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (5w).
According to procedure B, 100 mg of 3w (0.381 mmol), 49 μL of 2-aminothiophenol (0.53 mmol), 4 mL of acetic acid, and 6 mL of iPrOH were reacted for 18 h at RT. Orange solid, 68.00 mg (48.03%). Spectral data are identical to those published in the literature.55
5-(4-Methoxyphenyl)-1,5-dihydro-6H-indeno[1,2-e][1,4]thiazepin-6-one (9a).
According to procedure E, 50 mg of 3q (0.22 mmol), 2-aminoethanethiol (0.332 mmol), 4 mL of acetic acid, and 6 mL of iPrOH were reacted for 18 h at RT. Orange solid, 18.1 mg (30.0%). 1H NMR (500 MHz, DMSO-d6): δ 8.36 (s, 1H), 7.65 (d, J = 7.30 Hz, 1H), 7.44 (td, J = 7.50, 0.80 Hz, 1H), 7.35 (t, J = 7.25 Hz, 1H), 7.25 (d, J = 6.95 Hz, 1H), 7.20 (d, J = 8.55 Hz, 2H), 6.84 (d, J = 8.72 Hz, 2H), 5.00 (s, 1H), 3.94 (m, 1H), 3.70 (s, 3H), 3.51 (m, 1H), 2.87 (m, 1H), 2.65 (m, 1H). 13C NMR (150 MHz, DMSO-d6): 189.97, 161.65, 157.70, 139.18, 134.87, 133.72, 131.03, 129.82, 128.64, 119.42, 118.17, 113.49, 106.88, 55.01, 48.31, 40.46, 27.55. HRMS-ESI (m/z) calcd for C19H17NO2S [M + H]+ 324.1053, found 324.1051. HPLC purity: 95.4%
5-(4-Bromophenyl)-1,5-dihydro-6H-indeno[1,2-e][1,4]thiazepin-6-one (9b).
According to procedure E, 100 mg of 3l (0.27 mmol), 2-aminoethanethiol (0.48 mmol), 4 mL of acetic acid, and 6 mL of iPrOH were reacted for 24 h at RT. Orange solid, 61.8 mg (52.0%). 1H NMR (500 MHz, DMSO-d6): δ 8.43 (s, 1H), 7.66 (d, J = 7.30 Hz, 1H), 7.48–7.44 (m, 3H), 7.36 (t, J = 7.35 Hz, 1H), 7.25 (t, J = 8.30 Hz, 3H), 5.04 (s, 1H), 3.95–3.90 (m, 1H), 3.57–3.52 (m, 1H), 2.91–2.87 (m, 1H), 2.65–2.59 (m, 1H). 13C NMR (125 MHz, DMSO-d6): 190.01, 161.65, 142.77, 139.14, 133.63, 131.12, 131.01, 129.94, 129.80, 119.52, 119.32, 118.33, 105.83, 48.56, 40.64, 27.55. HRMS-ESI (m/z) calcd for C18H14BrNOS [M + H]+ 372.0051, found 372.0050. HPLC purity: 95.5%.
5-(p-Tolyl)-1,5-dihydro-6H-indeno[1,2-e][1,4]thiazepin-6-one (9c).
According to procedure E, 68 mg of 3i (0.27 mmol), 2-aminoethanethiol (0.41 mmol), 4 mL of acetic acid, and 6 mL of iPrOH were reacted for 24 h at RT. Orange solid, 24.5 mg (29.0%). 1H NMR (500 MHz, DMSO-d6): δ 8.36 (s, 1H), 7.65 (d, J = 7.28 Hz, 1H), 7.44 (t, J = 7.42 Hz, 1H), 7.35 (t, J = 7.34 Hz, 1H), 7.25 (d, J = 6.94 Hz, 1H), 7.17 (d, J = 8.00 Hz, 2H), 7.09 (d, J = 7.99 Hz, 2H), 5.00 (s, 1H), 3.95–3.91 (m, 1H), 3.54–3.49 (m, 1H), 2.88–2.84 (m, 1H), 2.68–2.63 (m, 1H), 2.25 (s, 3H). 13C NMR (125 MHz, DMSO-d6): 190.00, 161.73, 140.11, 139.19, 135.35, 133.75, 131.04, 129.84, 128.71, 127.49, 119.44, 118.19, 106.69, 48.30, 40.75, 27.63, 20.55. HRMS-MALDI (m/z) calcd for C19H17NOS [M + H]+ 308.1103, found 308.1102. HPLC purity: 98.5%
4-(6-Oxo-2,3,5,6-tetrahydroindeno[1,2-e][1,4]thiazepin-5-yl)benzonitrile (9d).
According to general procedure E, 100 mg of 3o (0.386 mmol), 44.6 mg of 2-aminoethanethiol (0.579 mmol), 10 mL of iPrOH, 4 mL of acetic acid, and molecular sieves were reacted for 24 h at RT. Orange solid, 53.18 mg (43.3%). 1H NMR (500 MHz, DMSO-d6): δ 8.48 (br s, 1H), 7.75 (d, J = 8.30 Hz, 2H), 7.69 (d, J = 7.30 Hz, 1H), 7.46 (t, J = 7.76 Hz, 3H), 7.37 (t, J = 7.35 Hz, 1H), 7.27 (d, J = 6.97 Hz, 1H), 5.16 (s, 1H), 3.96–3.91 (m, 1H), 3.59–3.24 (m, 1H), 2.91–2.87 (m, 1H), 2.60–2.56 (m, 1H). 13C NMR (125 MHz, DMSO-d6): δ 190.08, 161.63, 149.21, 139.15, 133.54, 132.20, 131.18, 130.00, 128.52, 119.58, 118.87, 118.43, 109.03, 105.03, 48.87, 41.19, 27.50. HRMS-ESI (m/z) calcd for C19H14N2OS [M + H]+ 319.0899, found 319.0900. HPLC purity: 96.6%
11-(Thiophen-2-yl)-7-(trifluoromethyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (11a).
According to general procedure F, 150 mg of 3y (0.624 mmol), 224 mg of 2-amino-4-(trifluoromethyl) benzenethiol hydrochloride (0.749 mmol), 15 mL of iPrOH, and molecular sieves were reacted for 24 h at RT. Orange solid, 139 mg (53.8%). 1H NMR (500 MHz, CD3OD-d4): δ 7.87 (d, J = 7.34 Hz, 1H), 7.79 (s, 1H), 7.51 (td, J = 7.35, 1.35 Hz, 1H), 7.45–7.40 (m, 2H), 7.37 (d, J = 8.08 Hz, 1H), 7.22 (dd, J = 8.35, 1.40 Hz, 1H), 7.07 (d, J = 5.00 Hz, 1H), 6.65–6.63 (m, 1H), 6.57 (d, J = 3.51 Hz, 1H), 5.72 (s, 1H). 13C NMR (150 MHz, DMSO-d6): δ 189.16, 155.61, 146.17, 143.79, 138.95, 137.40, 132.14, 132.01, 130.32, 129.29 (q, JCF = 32.14 Hz), 126.39, 125.69, 125.60, 123.74 (q, JCF = 270.90 Hz), 120.86, 120.54 (q, JCF = 3.47 Hz), 119. 81 (q, JCF = 3.83 Hz), 119.45, 110.69, 41.51. 19F NMR (CD3OD-d4) δ −64.37(s). HRMS-MALDI (m/z) calcd for C21H12F3NOS2 [M + H]+ 416.0384, found 415.9460. HPLC purity: 97.6%
11-(4-Bromophenyl)-7-(trifluoromethyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (11b).
According to general procedure F, 100 mg of 3l (0.319 mmol), 144 mg of 2-amino-4-(trifluoromethyl) benzenethiol hydrochloride (0.479 mmol), 10 mL of iPrOH, and molecular sieves were reacted for 24 h at RT. Orange solid, 62.1 mg (39.8%). 1H NMR (500 MHz, DMSO-d6): δ 10.18 (br s, 1H), 8.08 (d, J = 7.38 Hz, 1H), 7.98 (s, 1H), 7.59 (td, J = 7.40, 1.20 Hz, 1H), 7.45–7.40 (m, 2H), 7.30–7.26 (m, 3H), 7.21 (d, J = 8.06 Hz 1H), 7.02 (d, J = 8.45 Hz 2H), 5.62 (s, 1H). 19F NMR δ −63.37 (s). 13C NMR (125 MHz, DMSO-d6): δ 189.46, 156.22, 144.14, 141.52, 139.14, 137.07, 132.19, 131.99, 130.82, 130.22, 129.32 (q, JCF = 32.13 Hz), 129.29, 128.78, 123.72 (q, JCF = 271.25 Hz), 120.79, 120.56 (q, JCF = 3.77 Hz), 119. 85 (q, JCF = 4.33 Hz), 119.75, 119.31, 109.12, 45.21. HRMS-MALDI (m/z) calcd for C23H13BrF3NOS [M + H]+ 487.9925, found 487.9705. HPLC purity: 98.5%
11-(Thiophen-2-yl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (5y).
According to procedure B, 50 mg of 3y (0.21 mmol), 26.5 μL of 2-aminothiophenol (0.248 mmol), 4 mL of acetic acid, and 6 mL of iPrOH were reacted for 18 h at RT. Red solid, 39.6 mg (55.0%). 1H NMR (600 MHz, DMSO-d6): δ 10.05 (s, 1H), 8.10 (d, J = 7.38 Hz, 1H), 7.56–7.54 (m, 2H), 7.43 (t, J = 7.28 Hz, 1H), 7.39 (d, J = 6.50 Hz, 1H), 7.35 (td, J = 7.26, 1.56 Hz, 1H), 7.18 (td, J = 8.04, 1.44 Hz, 2H), 7.00 (td, J = 7.41, 1.17 Hz, 1H), 6.67–7.66 (m, 1H), 6.54–7.53 (m, 1H), 5.67 (s, 1H). 13C NMR (150 MHz, DMSO-d6): 188.94, 155.94, 146.53, 143.01, 139.03, 136.36, 132.54, 131.69, 130.15, 129.11, 126.23, 125.35, 125.24, 124.75, 124.24, 123.44, 120.53, 119.32, 110.05, 41.32. HRMS-ESI (m/z) calcd for C20H13NOS2 [M + Na]+ 370.0337, found 370.1523. HPLC purity: 98.3%.
11-(3-Methoxyphenyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one 10,10-Dioxide (7a).
According to general procedure D, to a solution of 5w (68 mg, 0.18 mmol) in dry dichloromethane (DCM, 8 mL) was added mCPBA (107 mg, 0.620 mmol), and the mixture was stirred for 5 h at room temperature. Yellow solid, 45.3 mg (62.2%). 1H NMR (600 MHz, DMSO-d6): δ 10.05 (s, 1H), 8.18 (d, J = 7.38 Hz, 1H), 7.85 (d, J = 8.15 Hz, 1H), 7.73 (td, J = 7.38, 1.44 Hz, 1H), 7.64 (td, J = 7.26, 1.62 Hz, 1H), 7.50–7.46 (m, 3H), 7.20 (d, J = 7.60 Hz, 1H), 7.04 (d, J = 7.92 Hz, 1H), 6.74 (dd, J = 8.04, 2.04 Hz, 1H), 6.65–6.63 (m, 2H), 5.68 (s, 1H), 3.55 (s, 3H). 13C NMR (150 MHz, DMSO-d6): 190.64, 158.79, 155.50, 138.96, 136.41, 135.54, 134.82, 132.36, 132.13, 130.56, 129.15, 129.08, 127.17, 124.28, 123.32, 121.46, 121.03, 120.11, 114.85, 114.10, 100.15, 66.36, 54.96. HRMS-MALDI (m/z) calcd for C23H17NO4S [M + Na]+ 426.0776, found 426.0566. HPLC purity: 96.8%.
11-(4-Methoxyphenyl)-7-(trifluoromethyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (11c).
According to general procedure F, 150 mg of 3q (0.378 mmol), 195 mg of 2-amino-4-(trifluoromethyl) benzenethiol hydrochloride (0.568 mmol), 15 mL of iPrOH, and molecular sieves were reacted for 24 h at RT. Orange solid, 83 mg (33%). 1H NMR (600 MHz, DMSO-d6): δ 10.12 (s, 1H), 8.06 (d, J = 7.40 Hz, 1H), 7.97 (s, 1H), 7.58 (td, J = 7.50, 1.20 Hz, 1H), 7.44–7.39 (m, 2H), 7.25 (dd, J = 8.16, 1.53 Hz, 1H), 7.20 (d, J = 8.07 Hz, 1H), 6.97 (d, J = 8.72 Hz, 2H), 6.65 (d, J = 8.80 Hz, 2H), 5.55 (s, 1H), 3.60 (s, 3H). 19F NMR δ −61.32 (s). 13C NMR (125 MHz, DMSO-d6): δ 189.43, 157.78, 155.95, 144.14, 139.22, 137.08, 133.97, 132.26, 131.91, 130.10, 129.31, 129.10 (q, JCF = 32.17 Hz), 128.27, 123.74 (q, JCF = 271.13 Hz), 120.70, 120.36 (q, JCF = 3.50 Hz), 119. 66 (q, JCF = 3.98 Hz), 119.18, 113.24, 110.23, 54.91, 45.45. HRMS-MALDI (m/z) calcd for C24H16F3NO2S [M + H]+ 440.0926, found 440.0735. HPLC purity: 98.4%.
11-(p-Tolyl)-10,11-dihydrobenzo[b]indeno[1,2-e][1,4]diazepin-12(5H)-one (13a).
According to general procedure G, 150 mg of 3i (0.605 mmol), 15 mL of isopropanol, 98.1 mg of ortho-phenylenediamine (0.646 mmol), 3 mL of acetic acid, and molecular sieves were added in a three-neck round-bottom flask and reacted for 24 h at RT under nitrogen gas. Red solid, 54.2 mg (26.4%). 1H NMR (500 MHz, DMSO-d6): δ 9.83 (s, 1H), 7.98 (d, J = 7.29 Hz, 1H), 7.48 (t, J = 7.45 Hz, 1H), 7.36–7.34 (m, 2H), 7.31 (d, J = 6.84 Hz, 1H), 7.02 (d, J = 8.09 Hz, 2H), 6.95 (d, J = 7.98 Hz, 2H), 6.98–6.76 (m, 2H), 6.67–6.65 (m, 1H), 6.22 (d, J = 4.69 Hz, 1H), 5.28 (d, J = 4.69 Hz, 1H), 2.15 (s, 3H). 13C NMR (125 MHz, DMSO-d6): δ 188.76, 156.01, 141.38, 138.71, 138.37, 135.29, 134.47, 130.92, 130.22, 128.57, 126.91, 124.37, 122.60, 121.51, 120.26, 119.83, 118.78, 107.78, 107.75, 55.94, 20.51. HRMS-ESI (m/z) calcd for C23H18N2O [M + H]+ 339.1492, found 339.1497. HPLC purity: 95.1%
11-(p-Tolyl)-10,11-dihydrobenzo[b]indeno[1,2-e][1,4]diazepin-12(5H)-one (13b).
According to general procedure G, 150 mg of 3o (0.579 mmol), 93.1 mg of ortho-phenylenediamine (0.868 mmol), 3 mL of acetic acid, 15 mL of iPrOH, and molecular sieves were added in a three-neck round-bottom flask and reacted for 24 h at RT. Red solid, 62.16 mg (37.7%). 1H NMR (500 MHz, DMSO-d6): δ 9.99 (br s, 1H), 8.01 (d, J = 7.32 Hz, 1H), 7.65 (d, J = 8.30 Hz, 2H), 7.50 (t, J = 7.47 Hz 1H), 7.40–7.37 (m, 2H), 7.33 (d, J = 8.30 Hz, 3H), 6.84–6.81 (m, 2H), 6.69–6.67 (m, 1H), 6.40 (d, J = 4.69 Hz, 1H), 5.38 (d, J = 4.66 Hz, 1H). 13C NMR (125 MHz, DMSO-d6): δ 188.79, 156.32, 149.84, 138.20, 138.06, 134.28, 132.11, 131.10, 130.19, 129.88, 127.95, 124.71, 122.55, 121.81, 120.77, 120.01, 119.09, 118.78, 109.21, 106.20, 56.13. HRMS-ESI (m/z) calcd for C23H15N3O [M + H]+ 350.1288, found 350.1291. HPLC purity: 96.5%.
11-(4-Methoxyphenyl)-10,11-dihydrobenzo[b]indeno[1,2-e][1,4]diazepin-12(5H)-one (13c).
According to general procedure G, 73 mg of 3q (0.323 mmol), ortho-phenylenediamine (69.8 mg, 0.646 mmol), 4 mL of acetic acid, 10 mL of iPrOH, and molecular sieves were added in a three-neck round-bottom flask and reacted for 24 h at RT. Red solid, 40.25 mg (41.0%). 1H NMR (600 MHz, DMSO-d6): δ 9.83 (s, 1H), 7.97 (d, J = 7.32 Hz, 1H), 7.48 (td, J = 7.56, 1.02 Hz, 1H), 7.38–7.33 (m, 2H), 7.30 (d, J = 6.94 Hz, 1H), 7.05 (d, J = 8.72 Hz, 2H), 6.80–6.76 (m, 2H), 6.72–6.69 (m, 2H), 6.68–6.66 (m, 1H), 6.19 (d, J = 4.28 Hz, 1H), 5.26 (d, J = 4.14 Hz, 1H), 3.63 (s, 3H). 13C NMR (150 MHz, DMSO-d6): 188.72, 157.65, 155.97, 138.76, 138.34, 136.42, 134.44, 130.91, 130.25, 129.64, 128.05, 124.36, 122.59, 121.50, 120.25, 119.81, 118.75, 113.29, 107.95, 55.60, 54.84. HRMS-ESI (m/z) calcd for C23H18N2O2 [M + Na]+ 377.1266, found 377.2529. HPLC purity: 95.1%.
11-(o-Tolyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (5x).
According to procedure B, 50 mg of 3x (0.201 mmol), 32.2 μL of 2-aminothiophenol (0.302 mmol), 4 mL of acetic acid, and 6 mL of iPrOH were reacted for 18 h at 80 °C. Orange solid, 5.53 mg (7.4%). 1H NMR (500 MHz, DMSO-d6): δ 10.08 (s, 1H), 8.12 (d, J = 7.38 Hz, 1H), 7.60 (d, J = 8.08 Hz, 1H), 7.55 (t, J = 7.51 Hz, 1H), 7.41 (t, J = 7.31 Hz, 1H), 7.36–7.32 (m, 2H), 7.11 (d, J = 7.39 Hz, 1H), 6.92 (t, J = 7.38 Hz, 1H), 6.87 (d, J = 4.24 Hz, 2H), 6.64 (t, J = 7.46 Hz, 1H), 6.31 (d, J = 7.64 Hz, 1H), 5.47 (s, 1H), 2.54 (s, 3H). 13C NMR (125 MHz, DMSO-d6): δ 189.14, 156.82, 143.44, 139.72, 139.40, 136.10, 135.09, 132.68, 131.68, 130.25, 130.02, 129.07, 126.64, 125.71, 124.81, 124.65, 123.86, 123.21, 120.43, 119.11, 109.66, 43.16, 19.16. HRMS-ESI (m/z) calcd for C23H17NOS [M + Na]+ 378.0929, found 378.3829. HPLC purity: 97.7%.
2-(12-Oxo-7-(trifluoromethyl)-11,12-dihydro-5H-benzo[b]indeno[1,2-e][1,4]thiazepin-11-yl)benzonitrile (11d).
According to general procedure F, 100 mg of 3a (0.386 mmol), 133 mg of 2-amino-4-(trifluoromethyl) benzenethiol hydrochloride (0.579 mmol), 10 mL of iPrOH, and molecular sieves were reacted for 24 h at RT. Orange solid, 28.3 mg (17.0%). 1H NMR (500 MHz, CD3OD): δ 10.37 (s, 1H), 8.12 (d, J = 7.34 Hz, 1H), 8.04 (s, 1H), 7.83 (d, J = 7.52 Hz, 1H), 7.62 (td, J = 7.50, 1.05 Hz, 1H), 7.48–7.42 (m, 2H), 7.30–7.22 (m, 3H), 7.16 (d, J = 8.08 Hz 1H), 6.71 (d, J = 7.72 Hz, 1H), 5.70 (s, 1H). 19F NMR (CD3OD) δ −61.45 (s). 13C NMR (150 MHz, DMSO-d6): δ 189.36, 156.90, 144.76, 144.29, 139.11, 137.15, 133.65, 132.50, 132.18, 132.12, 130.47, 129.76 (q, J = 32.84 Hz), 127.96, 127.72, 126.74, 124.15 (q, J = 260.63 Hz), 121.00, 120.86 (q, J = 3.66 Hz), 120.22 (q, J = 3.59 Hz), 119.63, 117.33, 110.73, 107.53, 44.29. HRMS-ESI (m/z) calcd for C24H13F3N2OS [M + CH3OH + H]+ 467.1035, found 467.1649. HPLC purity: 99.6%.
11-(2-Methoxyphenyl)-7-(trifluoromethyl)-5,11-dihydro-12H-benzo[b]indeno[1,2-e][1,4]thiazepin-12-one (11e).
According to general procedure F, 100 mg of 3b (0.378 mmol), 130 mg of 2-amino-4-(trifluoromethyl) benzenethiol hydrochloride (0.568 mmol), 10 mL of iPrOH, and molecular sieves were reacted for 24 h at RT. Orange solid, 85.6 mg (51.5%). 1H NMR (600 MHz, DMSO-d6): δ 10.18 (s, 1H), 8.08 (d, J = 7.40 Hz, 1H), 8.00 (s, 1H), 7.58 (td, J = 7.50, 1.08 Hz, 1H), 7.42 (t, J = 7.33 Hz, 1H), 7.38 (d, J = 6.90 Hz 1H), 7.20 (dd, J = 8.10, 1.50 Hz, 1H), 7.11 (d, J = 8.05 Hz, 1H), 7.06 (td, J = 7.74, 1.62 Hz, 1H), 6.96 (d, J = 8.25 Hz, 1H), 6.45 (td, J = 7.50, 0.78 Hz, 1H), 6.35 (dd, J = 7.56, 1.44 Hz, 1H), 5.68 (s, 1H), 3.92 (s, 3H). 19F NMR (DMSO-d6): δ −61.32 (s). 13C NMR (150 MHz, DMSO-d6): δ 189.36, 156.53, 155.76, 143.92, 139.24, 137.10, 132.23, 131.91, 130.09, 129.75, 129.20, 129.06 (q, J = 32.16 Hz), 128.34, 126.20, 123.77 (q, J = 270.99 Hz), 120.69, 120.28 (q, J = 3.40 Hz), 119.59, (q, J = 3.74 Hz), 119.17, 119.16, 110.93, 109.78, 55.70, 40.17. HRMS-ESI (m/z) calcd for C24H16F3NO2S [M + Na]+ 462.0762, found 462.4431. HPLC purity: 99.5%.
Quantitative Structure–Activity Relationship (QSAR) Model.
Molecular modeling was performed using Maestro interface102 (Schrödinger package, version 2020–4). Molecules were modeled from a structure retrieved from the ChemSpider database (ID 2117364) followed by standard molecular mechanics geometry optimization using Maestro standard settings. The experimental antichlamydial IC50 values obtained from the semi-high-throughput imaging inclusion forming units (IFU) screen were converted to the corresponding pIC50 using the pIC50 value calculator from the Sanjeev’s Lab103 (https://www.sanjeevslab.org/tools.html). Only molecules with a determined value of antichlamydial activity were used to generate the QSAR models. Therefore, molecules with no inhibition seen within the concentration range tested (25–0.39 μg/mL) do not compose the dataset used in the QSAR study. The dataset was automatically divided into training (21 compounds–77% of the total dataset) and test set (compounds 5c, 5d, 5h, 5n, 7a, 13c–23% of the total dataset) using autoQSAR tool72 from the Schrödinger package, version 2020–4. AutoQSAR, is a machine-learning tool to build, validate, and deploy quantitative structure–activity relationship models in an automated manner.72 For the descriptor generation, the following set of binary fingerprints were calculated: radial, linear, dendritic, and molprint2D.104 For feature selection, the maximum allowed correlation between any pair of independent variables was set to 0.80. The maximum number of generated models was set to 50 and all of the AutoQSAR available methods to generate models were employed.72 The model selection was based on the predictive power (Q2 values) and the internal ranking score from the software (based on the performance).105 Model visualization and preparation of the color maps was performed using Maestro interface and AutoQSAR tools.
In Silico Drug-Likeness Properties.
The entire library of dihydrothiazepines (including the molecules with no defined value for the antichlamydial IC50) were modeled according to the description in the QSAR section above. Such dataset was used to calculate drug-likeness properties using standard settings of Qikprop tool106 (Schrödinger package, version 2020–4). Predictions are related to drug absorption as oral dosage form.
Quantum Mechanics Molecular Electrostatic Potential Surface.
All calculations were performed using Jaguar software107 incorporated in the Schrödinger package, version 2020–4 and using the computer resources of the Holland Computing Center from the University of Nebraska—Lincoln.108 The selected molecules (5q, 5y, 5l, 11a, 11b, 11c) were modeled according to the description in the QSAR section. Tryptophan, tyrosine, and phenylalanine were modeled using Maestro build tool by direct insertion of these amino acids from the internal software database and optimization using the same protocol as described for dihydrothiazepines. Next, the molecules were optimized using density functional theory (DFT) level using the functional Becke, three-parameter, Lee–Yang–Parr (B3LYP), with the basis 6–31G(d,p). Structures were optimized using water as the solvent (Poisson–Boltzmann continuum solvation as model) and the accuracy of the calculations was set to accurate. All of the other parameters were used as the standard settings of Jaguar. The calculated quantum mechanics molecular electrostatic potential (MEP) surfaces were displayed using Maestro interface102 (Schrödinger package, version 2020–4).
Biology.
Antichlamydial Semi-High-Throughput Imaging Screen. HEp-2 Cell Culture.
Human epithelial cell line HEp-2 was routinely cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco ref 11965–092) supplemented with 10% fetal bovine serum (FBS) and 10 μg/mL gentamicin at 37 °C in a humidified environment with 5% CO2. The same culture conditions were used for both infected and uninfected cell culture samples.
Semi-High-Throughput Imaging Inclusion Forming Units (IFU) Screen.
Antichlamydial activity can be analyzed by the effect of the drugs on the number of inclusions using an immunofluorescence assay (IFA). Therefore, HEp-2 cells were first seeded in a 96-well plate (PerkinElmer Cell carrier plate, black well walls, and optically clear bottom) compatible with the Operetta microscope. After incubation for 24 h at 37 °C in a humidified environment with 5% CO2, cells were infected with C. trachomatis serovar L2 at a multiplicity of infection (MOI) of 0.6, and the test molecules were added at seven concentration points: 25, 12.5, 6.25, 3.12, 1.56, 0.78, and 0.39 μg/mL. The drug solutions were prepared by serial dilution in a separated 96-well plate and carefully transferred to the cell plate after 8 h post-infection (hpi). After 16 h (total incubation of the cells for 24 h), the cells were washed with 100 μL of phosphate-buffered saline/azide (PBS-azide—0.025%) and fixed with methanol (100 μL) for 10 min. Next, the samples were stained using primary goat anti-MOMP antibody (Meridian Biosciences, ref B65266G) and a secondary donkey anti-goat antibody labeled with Alexa488 (Invitrogen, ref A32814). Finally, the samples were stained with 4′,6-diamidino-2-phenylindole (DAPI). All staining solutions were used in 1:1000 concentration diluted in PBS. The green-fluorescent inclusions (number) were automatically identified, counted, and compared against the vehicle DMSO control (untreated) and positive control azithromycin (1 μg/mL) using the microplate imager Operetta High Content Analysis System (PerkinElmer, Inc.). The following parameters were considered by the system to identify the inclusions: green fluorescence intensity (>600), area (>40 μm2), and roundness (>0.85). All of the samples were tested in triplicate. The IC50 was determine using GraphPad Prism software, version 9.3.1 for Windows.109
EB Progeny Assay.
Aiming to further evaluate the effect of the most active compounds against C. trachomatis L2 (Ctr L2), HEp-2 cells were seeded in a 24-well plate and infected with Ctr L2 at a multiplicity of infection of 1. The samples were treated with drugs in triplicate at 8 hpi. Next, after 16 h (total incubation of the cells for 24 h), the cells were lysed to release C. trachomatis elementary bodies, which were collected in sucrose storage medium (2SP) and stored at −80 °C. Next, the collected cell lysates were used to infect a fresh HEp-2 cell monolayer in a series of 10-fold dilutions. Samples were incubated for 24 h at 37 °C in a humidified environment with 5% CO2 in media containing cycloheximide (2 μg/mL). After 24 hpi, the cells were fixed with methanol for 15 min and the recoverable EBs from the initial infection were stained using primary goat anti-MOMP antibody (Meridian Biosciences, ref B65266G) and a secondary donkey anti-goat antibody labeled with Alexa488 (Invitrogen, ref A32814). The fluorescent inclusions were counted from 15 fields of view (FOV) at 20× magnification and images were acquired using an Olympus CKX53 fluorescence microscope equipped with an EP50 camera. Channel merge was processed using ImageJ software. Images were assembled with Adobe Photoshop 26.0.2 (Adobe Systems Inc., San Jose, CA). The experiment was performed in triplicate. The IC50 was determine using GraphPad Prism software, version 9.3.1 for Windows.109 Infectious progeny was calculated using the the same procedure as described before for a volume of 0.25 mL (volume used per well).52,110
Cytotoxicity Assay. Cell Culture.
Epithelial endometrium adenocarcinoma cells (HEC-1-A) were cultured in McCoy’s 5A media (Gibco, ref 16600082) containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Gibco, 15140–122) at 37 °C in a humidified environment with 5% CO2. Human embryonic kidney 293 cells (HEK-293) were cultured in DMEM media (Gibco, ref 11965–092) containing 10% fetal bovine serum (FBS), 1% penicillin/streptomycin (Gibco, 15140–122), and 200 mmol L-glutamine at 37 °C in a humidified environment with 5% CO2. HEp-2 well cultured as described above.
XTT Assay.
The cells were seeded at a density of 5 × 103 cells per well in 96-well plates. After 24 h, he cells were treated within a concentration range from 256 to 2 μg/mL (in triplicate). As control, DMSO was employed at a concentration equivalent to the ones used in drug-treated cells. Next, the plates were incubated for an additional 20 h. After the incubation, the wells were washed (2×) with 100 μL of PBS to remove any residual compound precipitation and 100 μL of new media was added to all samples. Next, 70 μL of the XTT assay reagent (CyQUANT XTT Cell Viability Assay, Invitrogen, ref X12223) was added followed by incubation of the plates for an additional 4 h (total 24 h), accordingly to the manufacturer’s instructions. Corrected absorbance readings were measured with a 450 nm filter using a multiscan FC microplate photometer (Thermo Fisher Scientific).
Antibacterial Assay against Clinically Relevant Bacterial Strains. Minimum Inhibitory Concentration (MIC) Determination.
For both tested compounds and controls drugs, the MIC was determined using the broth microdilution method, according to the Clinical and Laboratory Standards Institute (CLSI) reference M07-A9.111 The compounds were serially diluted in Mueller–Hinton broth (MHB, Difco, ref 275730) at a concentration range of 125–1.9 μg/mL in 96-well microtiter plates. S. aureus USA 300 JE2, A. baumannii 2208, E. coli K12, and P. aeruginosa PA01 were cultivated in trypticase soy agar (TSA—Difco, ref 236940) at 37 °C for 18 h and standardized to 1.5 × 108 colony forming units (CFU)/mL (0.5 McFarland) on saline (0.85%). Afterward, the bacteria solutions were diluted to 1.5 × 106 CFU/mL in MHB and added to the microtiter plates containing 100 μL of drug media (final bacterial concentration: 1.5 × 105 CFU/mL). The plates were incubated for 24 h at 37 °C. A 0.1% triphenyl tetrazolium chloride (TTC) solution was added to all wells before reading the results. The MIC was defined as the lowest drug concentration that showed no visible bacterial growth. The experiment was performed in triplicate in at least two different moments.
Antibacterial Assay against L. rhamnosus. Minimum Inhibitory Concentration (MIC) Determination.
L. rhamnosus was isolated from the commercially available probiotic product INESSA ADVANCED DAILY BIOTIC (INESSA Wellness, London W1W 7FA, U.K.). For this, the bacterium was aseptically isolated from the respective product on De Man, Rogosa, and Sharpe (MRS) broth (HiMedia, Mumbai, India), incubated at 37 °C for 72 h with 5% CO2. Next, an isolate typical colony of L. rhamnosus was grown in MRS agar (Weber Scientific, NJ). The compounds were serially diluted in MRS broth at a concentration range of 125–1.9 μg/mL in 96-well microtiter plates. L. rhamnosus were cultivated in MRS agar at 37 °C with 5% CO2 for 24 h and standardized to 1.5 × 108 colony forming units (CFU)/mL (0.5 McFarland) on saline (0.85%). Afterward, the bacterial solution was diluted to 1.5 × 106 CFU/mL in MRS broth and added to the microtiter plates containing 100 μL of drug media (final bacterial concentration: 1.5 × 105 CFU/mL). The plates were incubated for 24 h at 37 °C, with 5% CO2. A 0.1% triphenyl tetrazolium chloride (TTC) solution was added to all wells of the plates to read the results. The MIC was defined as the lowest drug concentration that showed no visible bacterial growth. The experiment was performed in triplicate in at least two different moments.
Antibacterial Assay against Anaerobic Bacteria. Minimum Inhibitory Concentration (MIC) Determination.
Drugs were serially diluted in Brucella broth (BRU, Anaerobe systems, ref AS-105) at a concentration range of 125–0.06 μg/mL in 96-well microtiter plates. B. producta DSM 2950, C. ramosum DSM 1402, B. thetaiotaomicron DSM 2079, and A. caccae DSM 14662 were cultivated in brain heart infusion (BHI) agar (Anaerobe systems, ref AS-6426) at 37 °C for 24 h followed by another round of growing in Brucella broth (Anaerobe systems, ref AS-105). From the Brucella broth, a sample was centrifuged at 5000 rpm for 5 min. Next, media was replaced by saline (0.85%) and the sample was standardized to 1.5 × 108 CFU/mL (0.5 McFarland). Afterward, bacterial solutions were diluted to 1.5 × 107 CFU/mL in Brucella broth and added to the microtiter plates containing 100 μL of drug media (final bacterial concentration: 1.5 × 106 CFU/mL). The plates were incubated for 48 h at 37 °C under anaerobic conditions using an anaerobic laminar flow workstation. The environment inside the workstation was routinely checked for maintenance of anaerobiosis using anaerobic strip indicators (Thermo Scientific, ref BR0055B). The MIC was defined as the lowest drug concentration that showed no visible bacterial growth. The experiment was performed in triplicate. L. plantarum DSM 20174 was cultivated in MRS agar (Difco, ref 288130) at 30 °C for 24 h followed by another round of growing in MRS broth (HiMedia, ref M369) at 30 °C for 24 h. From this point, the same protocol described above was followed, except using MRS broth for bacterial cultivation/compounds dilution and incubation at 30 °C for 24 h.
Antifungal Susceptibility Testing. Minimum Inhibitory Concentration (MIC) Determination.
For both the tested compound and fluconazole drug control, the MIC was determined using the broth microdilution method, according to the Clinical and Laboratory Standards Institute (CLSI) reference M27-A3.112 Drugs were serially diluted in Roswell Park Memorial Institute 1640 (RPMI 1640—Gibco, ref 11875093) at a concentration range of 1000–1.95 μg/mL in 96-well microtiter plates. C. albicans (90028), C. glabrata (2001), C. Krusei (6558), and C. tropicalis (750) (American Type Culture Collection—ATCC) were suspended in sterile saline (0.9%) and adjusted to a concentration of 2.5 × 106 UFC/mL using a spectrophotometer (Quimis, Brazil) with 90% (±2) transmittance and 530 nm wavelength. Next, the microorganism suspension was diluted 1:20 and 1:50 in RPMI 1640 medium to obtain (2–3) × 103 UFC/mL. Drugs were serially diluted in RPMI 1640 medium at a concentration range of 1000–1.9 μg/mL in 96-well microtiter plates. Next, 100 μL of the microorganism suspension was added to the microtiter plates containing 100 μL of drug media. The MIC was defined as the lowest drug concentration that showed no visible microorganism growth. The experiment was performed in triplicate.
Stability Studies. Human Serum Stability.
The studied compound (2 mg/mL) was incubated in human serum AB (50% v/v—Vally Biomedical Inc.) over 24 h in a shaking incubator (37 °C and 75 rpm). Next, 50 μL aliquots were taken at selected time intervals of 0, 30, 60, 90, 120, 150, 180, and 1440 min. To the aliquots was added 50 μL of HPLC-grade acetonitrile and the solutions were heated to 80 °C for 1 min followed by centrifugation at 12,000 rpm (Eppendorf Centrifuge) for 5 min. Next, a 50 μL sample of the resulting supernatant was taken from each time point sample and mixed with 50 μL of 5-methoxy indole (1 mg/mL—standard control) in acetonitrile solution and centrifuged again. An aliquot of the final supernatant (70 μL) was removed for analysis using HPLC. The stability results were expressed as the percentage of drug remaining versus time. The calculations were performed as percentage parent drug remaining at select time points relative to 0 min (100% parent drug).
Simulated Gastric Fluid Stability.
The studied drug (2 mg/mL) was incubated in simulated gastric fluid—pH 1.4 (Ricca Chemical Company, ref 7108–16) containing 3.2 mg/mL of pepsin (MP biomedicals LLC, ref 195367) over 24 h in a shaking incubator (37 °C and 75 rpm). At this point, the same experimental procedure described above for serum stability was followed.
Simulated Intestinal Fluid Stability.
The studied drug (2 mg/mL) was incubated in simulated Intestinal fluid—pH 7.40 (Ricca Chemical Company, ref 7109.75–16) containing 10 mg/mL of pancreatin (MP biomedicals LLC, ref 102557) over 24 h in a shaking incubator (37 °C and 75 rpm). At this point, the same experimental procedure described above for serum stability was followed.
ClpX ATPase Inhibition Assay.
CT ClpX containing a C-terminal 6x-HIS tag was purified from E. coli BL21(DE3) ΔPAX as previously described by Wood et al.39 using cobalt-based immobilized metal affinity chromatography. Protein purity was assessed by running 1 μg samples on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels following by staining with Coomassie Brilliant Blue. CT ClpX sourced from three independent purifications was used for ATPase assays. The assays were performed using Kinase-Glo (ATP) from Promega with 1 μM ATP and 1.5 μg of CT ClpX.39 The reactions were incubated at 30 °C for 60 min and terminated via addition of the Kinase-Glo reagent. Luminescence (ATP not consumed by ClpX) was measured on a BioTek Synergy HT plate reader. Data are reported as the amount of ATP depleted versus an ATP only control. Experiments were run in at least triplicate with replicates used for each individual experiment.
Mutagenic Potential Determination. Ames Test.
The mutagenic potential of the studied molecule was determined by the Ames test using a microsuspension method described by Kado et al.113 and according to the OCDE guidelines, test no. 471.114 The assays were conducted using S. typhimurium strains TA98 and TA100 (Moltox, Molecular Toxicology, Inc.) in the presence and absence of metabolic activation from the microsomal fraction. To prepare the inoculum, a bacterial suspension was incubated in nutrient broth no. 2 at 37 °C for 16 h. Next, this suspension was concentrated by centrifugation (10,000g/10 min, 4 °C) to obtain a bacterial density of (1–2) × 109 UFC/mL in PBS 0.2 mM (pH 7,4). In the assay without metabolic activation, 5 μL of the studied compound diluted in DMSO was mixture with 50 μL of bacterial inoculum and 50 μL of PBS 0.2 mM (pH 7.4) to obtain different drug concentrations (50, 150, 500, 1500, and 5000 μg/mL). For the assay with metabolic activation, 50 μL of microsomal fraction S9 was added instead of PBS. Next, the samples were preincubated for 90 min at 37 °C. Next, 2 mL of top agar (sodium chloride 0.6% (m/v); agar–agar 0.6% (m/v); 0.5 mM l-histidine, d-biotin 0.5% (w/v)) was added to the prepared samples, homogenized, and poured onto Petri dishes containing minimal glucose agar (MGA): agar–agar 1.5% (m/v); glucose solution 10% (v/v); glucose dextrose (20% (w/v); 0.02% (v/v) of a Vogel–Boner solution 50×; magnesium sulfate heptahydrate 0.02% (v/v); citric acid monohydrate 0.2% (v/v); potassium phosphate dibasic 1% (m/v); and sodium phosphate 0.35% (v/v)). Following 66 h of incubation at 37 °C, the number of revertant colonies per plate (his+) was determined. The experiment was performed in triplicate. The positive controls used in the assays without metabolic activation were 4-nitrophenylenediamine (NPD, 10 μg/plate) for TA98 strain and sodium azide (2.5 μg/plate) for TA100 strain. In the assays with metabolic activation, 2-aminoanthracene (2-AA, 0.625 μg/plate) was used for TA98 and TA100 strains. Negative controls were performed with the sample solvent, DMSO (5 μL/plate).
The results were analyzed using the Salanal Statistical Software (U.S. Environmental Protection Agency, Monitoring Systems Laboratory, version 1.0, from the Research Triangle Institute, RTP), adopting the model by Bernstein et al.115 The mutagenicity index (MI) was calculated according to the formula: MI = number of induced revertants/number of spontaneous revertants. The sample was considered to have mutagenic potential when the mutagenicity index was ≥2 in at least one of the concentrations tested and when there was a dose–response relationship between the concentrations tested and the number of induced revertants.94 Concentrations that had an MI of less than 0.7 were considered cytotoxic.116
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge the University of Nebraska Medical Center and the office of graduate studies for the fellowship awarded to L.J.d.C. in support of this work. In addition, L.J.d.C., S.P.O., D.J.F., and M.C.-S. were supported in part by an NIH/NIAID award to S.P.O. (1R56AI146062). Further, J.B.C. was supported by the Office of the Director, National Institutes of Health of the National Institutes of Health under Award Number K01OD030514. The authors also express gratitude to Drs. Jered Garrison and Audifas Salvador for providing some mass spectrometry (MS) analysis presented in this paper. The authors thank Drs. Elizabeth A. Rucks and Camille Riffaud for providing the epithelial endometrium adenocarcinoma cell line (HEC-1-A). Also, the authors are grateful to Huihua Xing for the help in the stability studies presented here. Finally, the authors acknowledge the computing resources provided by the University of Nebraska—Lincoln’s Holland Computing Center.
ABBREVIATIONS USED
- ACP
activators of self-compartmentalizing proteases
- AD
applicability domain of the model
- ADEP
acyldepsipeptide
- AZM
azithromycin
- CT
Chlamydia trachomatis
- DMSO
dimethyl sulfoxide
- Doxy
doxycycline
- EB
elementary body
- HEC-1-A
epithelial endometrium adenocarcinoma cell line
- HEK-293
immortalized human embryonic kidney cell line
- HEp-2
epithelial type 2 cell line
- IFA
immunofluorescence assay
- KPLS
kernel-based partial least-square regression method
- MRSA
methicillin-resistant Staphylococcus aureus
- OECD
organization for economic cooperation and development
- PID
pelvic inflammatory disease
- Q 2
coefficient of determination between the observed and predicted biological activities for the test set
- QPlogPo/w
predicted octanol/water partition coefficient
- QPPCaco
predicted apparent Caco-2 cell permeability
- QPPMDC
predicted apparent Madin–Darby Canine Kidney (MDCK) cell permeability
- R 2
correlation coefficient
- RB
reticulate body
- RMSE
root mean square error
- SAR
structure–activity relationships
- SD
standard deviation of the regression
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01894.
Detailing of the antichlamydial potency in different unit measures; detailing of the prediction accuracy of KPLS-radial 2D QSAR Model (radial 13); experimental versus KPLS-radial 2D QSAR model (radial 13) predicted values of antichlamydial activity (pIC50); color maps for the 2D fingerprint (radial 13) QSAR model for all of the molecules used to generate the QSAR model; dose–response curves for the effects of 5b and 5c reported as percentage of recovered IFU; viability analysis for molecules 11d and 11e against the HEp-2 cell line; and spectral information of the studied molecules (PDF) Calculated in silico permeability parameters (CSV) Molecular formula strings and biological data (CSV)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.2c01894
Contributor Information
Luana Janaína de Campos, Department of Pharmaceutical Sciences, College of Pharmacy, University of Nebraska Medical Center, Omaha, Nebraska 68198, United States.
Mohamed A. Seleem, Department of Pharmaceutical Sciences, College of Pharmacy, University of Nebraska Medical Center, Omaha, Nebraska 68198, United States
Jiachen Feng, Department of Pharmaceutical Sciences, College of Pharmacy, University of Nebraska Medical Center, Omaha, Nebraska 68198, United States.
Kelly Mari Pires de Oliveira, Faculty of Biological and Environmental Science, Federal University of Grande Dourados, Dourados, MS 79804-970, Brazil.
João Víctor de Andrade dos Santos, Faculty of Health Sciences, Federal University of Grande Dourados, Dourados, MS 79804-970, Brazil.
Shivdeep Hayer, Department of Biology, University of Nebraska at Omaha, Omaha, Nebraska 68182, United States.
Jonathan B. Clayton, Department of Biology, University of Nebraska at Omaha, Omaha, Nebraska 68182, United States; Department of Food Science and Technology, University of Nebraska—Lincoln, Lincoln, Nebraska 68588, United States; Department of Pathology and Microbiology, University of Nebraska Medical Center, Omaha, Nebraska 68198, United States; Nebraska Food for Health Center, University of Nebraska—Lincoln, Lincoln, Nebraska 68508, United States
Sharvath Kathi, School of Biological Sciences, Southern Illinois University, Carbondale, Illinois 62901, United States.
Derek J. Fisher, School of Biological Sciences, Southern Illinois University, Carbondale, Illinois 62901, United States
Scot P. Ouellette, Department of Pathology and Microbiology, College of Medicine, University of Nebraska Medical Center, Omaha, Nebraska 68198, United States
Martin Conda-Sheridan, Department of Pharmaceutical Sciences, College of Pharmacy, University of Nebraska Medical Center, Omaha, Nebraska 68198, United States.
REFERENCES
- (1).Witkin SS; Minis E; Athanasiou A; Leizer J; Linhares IM Chlamydia trachomatis: the Persistent Pathogen. Clin. Vaccine Immunol. 2017, 24, No. e00203–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).CDC. New CDC Analysis Shows Steep and Sustained Increases in STDs in Recent Years, 2018. https://www.cdc.gov/media/releases/2018/p0828-increases-in-stds.html (accessed Dec 07, 2021).
- (3).CDC. Sexually Transmitted Disease Surveillance, 2019, 2019. https://www.cdc.gov/std/statistics/2019/default.htm (accessed Nov 29, 2021).
- (4).Kumar S; Chesson H; Gift TL Estimating the Direct Medical Costs and Productivity Loss of Outpatient Chlamydia and Gonorrhea Treatment. Sex. Transm. Dis. 2021, 48, e18–e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Satpathy G; Behera HS; Ahmed NH Chlamydial eye infections: Current perspectives. Indian J Ophthalmol. 2017, 65, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Adachi K; Nielsen-Saines K; Klausner JD Chlamydia trachomatis Infection in Pregnancy: The Global Challenge of Preventing Adverse Pregnancy and Infant Outcomes in Sub-Saharan Africa and Asia. BioMed Res. Int. 2016, 2016, No. 9315757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).O’Connell CM; Ferone ME Chlamydia trachomatis Genital Infections. Microb. Cell 2016, 3, 390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Mohammadpour M; Abrishami M; Masoumi A; Hashemi H Trachoma: Past, present and future. J. Curr. Ophthalmol. 2016, 28, 165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).CDC. Trachoma, 2021. https://www.who.int/news-room/fact-sheets/detail/trachoma#:~:text=Trachoma%20is%20hyperendemic%20in%20many,1.4%25%20of%20all%20blindness%20worldwide (accessed Feb 14, 2022).
- (9).Silva G. A. R. d.; Motta H. L. S. á. N.; de Souza EFA; Cardoso PANM; Pilotto JH; Eyer-Silva WA; Ribeiro LCP; Dos Santos MS; de Azevedo MCVM; Pinto JFDC; et al. Chlamydia trachomatis asymptomatic urethritis recurrence among males living with HIV-1. Rev. Inst. Med. Trop. Sao Paulo 2018, 60, No. e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Keegan MB; Diedrich JT; Peipert JF Chlamydia trachomatis Infection: Screening and Management. J. Clin. Outcomes Manage. 2014, 21, 30–38. [PMC free article] [PubMed] [Google Scholar]
- (11).AbdelRahman YM; Belland RJ The chlamydial developmental cycle. FEMS Microbiol. Rev. 2005, 29, 949–959. [DOI] [PubMed] [Google Scholar]
- (12).Bastidas RJ; Elwell CA; Engel JN; Valdivia RH Chlamydial intracellular survival strategies. Cold Spring Harbor Perspect. Med. 2013, 3, No. a010256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hybiske K; Stephens RS Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 11430–11435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Tuddenham S; Hamill MM; Ghanem KG Diagnosis and Treatment of Sexually Transmitted Infections: A Review. JAMA 2022, 327, 161–172. [DOI] [PubMed] [Google Scholar]
- (15).Mohammadzadeh F; Dolatian M; Jorjani M; Afrakhteh M; Majd HA; Abdi F; Pakzad R Urogenital Chlamydia trachomatis treatment failure with azithromycin: A meta-analysis. Int. J. Reprod. Biomed. 2019, 17, 603–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Batteiger BE; Tu W; Ofner S; Van Der Pol B; Stothard DR; Orr DP; Katz BP; Fortenberry JD Repeated Chlamydia trachomatis genital infections in adolescent women. J. Infect. Dis. 2010, 201, 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kissinger PJ; White S; Manhart LE; Schwebke J; Taylor SN; Mena L; Khosropour CM; Wilcox L; Schmidt N; Martin DH Azithromycin Treatment Failure for Chlamydia trachomatis Among Heterosexual Men With Nongonococcal Urethritis. Sex. Transm. Dis. 2016, 43, 599–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wei S; Mortensen MS; Stokholm J; Brejnrod AD; Thorsen J; Rasmussen MA; Trivedi U; Bisgaard H; Sørensen SJ Short- and long-term impacts of azithromycin treatment on the gut microbiota in children: A double-blind, randomized, placebo-controlled trial. EBioMedicine 2018, 38, 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Raju SC; Viljakainen H; Figueiredo RAO; Neuvonen PJ; Eriksson JG; Weiderpass E; Rounge TB Antimicrobial drug use in the first decade of life influences saliva microbiota diversity and composition. Microbiome 2020, 8, No. 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Tamarelle J; Ma B; Gajer P; Humphrys MS; Terplan M; Mark KS; Thiébaut ACM; Forney LJ; Brotman RM; Delarocque-Astagneau E; et al. Nonoptimal Vaginal Microbiota After Azithromycin Treatment for Chlamydia trachomatis Infection. J. Infect. Dis. 2020, 221, 627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Edwards VL; Smith SB; McComb EJ; Tamarelle J; Ma B; Humphrys MS; Gajer P; Gwilliam K; Schaefer AM; Lai SK; et al. The Cervicovaginal Microbiota-Host Interaction Modulates Chlamydia trachomatis Infection. mBio 2019, 10, No. e01548–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chen Z; Radjabzadeh D; Chen L; Kurilshikov A; Kavousi M; Ahmadizar F; Ikram MA; Uitterlinden AG; Zhernakova A; Fu J; et al. Association of Insulin Resistance and Type 2 Diabetes With Gut Microbial Diversity: A Microbiome-Wide Analysis From Population Studies. JAMA Network Open 2021, 4, No. e2118811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ley RE; Bäckhed F; Turnbaugh P; Lozupone CA; Knight RD; Gordon JI Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 11070–11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hufnagl K; Pali-Schöll I; Roth-Walter F; Jensen-Jarolim E Dysbiosis of the gut and lung microbiome has a role in asthma. Semin. Immunopathol. 2020, 42, 75–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).DeGruttola AK; Low D; Mizoguchi A; Mizoguchi E Current Understanding of Dysbiosis in Disease in Human and Animal Models. Inflammatory Bowel Dis. 2016, 22, 1137–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Good JAD; Kulén M; Silver J; Krishnan KS; Bahnan W; Núñez-Otero C; Nilsson I; Wede E; de Groot E; Gylfe Å; et al. Thiazolino 2-Pyridone Amide Isosteres As Inhibitors of Chlamydia trachomatis Infectivity. J. Med. Chem. 2017, 60, 9393–9399. [DOI] [PubMed] [Google Scholar]
- (27).Pickering H; Hart JD; Burr S; Stabler R; Maleta K; Kalua K; Bailey RL; Holland MJ Impact of azithromycin mass drug administration on the antibiotic-resistant gut microbiome in children: a randomized, controlled trial. Gut Pathog. 2022, 14, No. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).O’Brien KS; Emerson P; Hooper PJ; Reingold AL; Dennis EG; Keenan JD; Lietman TM; Oldenburg CE Antimicrobial resistance following mass azithromycin distribution for trachoma: a systematic review. Lancet. Infect. Dis. 2019, 19, e14–e25. [DOI] [PubMed] [Google Scholar]
- (29).Wetten S; Mohammed H; Yung M; Mercer CH; Cassell JA; Hughes G Diagnosis and treatment of chlamydia and gonorrhoea in general practice in England 2000–2011: a population-based study using data from the UK Clinical Practice Research Datalink. BMJ Open 2015, No. e007776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).(a) Jensen JS; Bradshaw CS; Tabrizi SN; Fairley CK; Hamasuna R Azithromycin Treatment Failure in Mycoplasma genitalium–Positive Patients with Nongonococcal Urethritis Is Associated with Induced Macrolide Resistance. Clin. Infect. Dis. 2008, 47, 1546–1553. [DOI] [PubMed] [Google Scholar]; (b) Pond MJ; Nori AV; Witney AA; Lopeman RC; Butcher PD; Sadiq ST High prevalence of antibiotic-resistant Mycoplasma genitalium in nongonococcal urethritis: the need for routine testing and the inadequacy of current treatment options. Clin. Infect. Dis. 2014, 58, 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Skalet AH; Cevallos V; Ayele B; Gebre T; Zhou Z; Jorgensen JH; Zerihun M; Habte D; Assefa Y; Emerson PM; et al. Antibiotic selection pressure and macrolide resistance in nasopharyngeal Streptococcus pneumoniae: a cluster-randomized clinical trial. PLoS Med. 2010, 7, No. e1000377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Cao X; Boyaci H; Chen J; Bao Y; Landick R; Campbell EA Basis of narrow-spectrum activity of fidaxomicin on Clostridioides difficile. Nature 2022, 604, 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Balakrishnan A; Patel B; Sieber SA; Chen D; Pachikara N; Zhong G; Cravatt BF; Fan H Metalloprotease inhibitors GM6001 and TAPI-0 inhibit the obligate intracellular human pathogen Chlamydia trachomatis by targeting peptide deformylase of the bacterium. J. Biol. Chem. 2006, 281, 16691–16699. [DOI] [PubMed] [Google Scholar]
- (34).Mojica SA; Salin O; Bastidas RJ; Sunduru N; Hedenström M; Andersson CD; Núñez-Otero C; Engström P; Valdivia RH; Elofsson M; Gylfe Å N-Acylated Derivatives of Sulfamethoxazole Block Chlamydia Fatty Acid Synthesis and Interact with FabF. Antimicrob. Agents Chemother. 2017, 61, No. e00716–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Keurulainen L; Salin O; Siiskonen A; Kern JM; Alvesalo J; Kiuru P; Maass M; Yli-Kauhaluoma J; Vuorela P Design and Synthesis of 2-Arylbenzimidazoles and Evaluation of Their Inhibitory Effect against Chlamydia pneumoniae. J. Med. Chem. 2010, 53, 7664–7674. [DOI] [PubMed] [Google Scholar]
- (36).Marwaha S; Uvell H; Salin O; Lindgren AEG; Silver J; Elofsson M; Gylfe A N-acylated derivatives of sulfamethoxazole and sulfafurazole inhibit intracellular growth of Chlamydia trachomatis. Antimicrob. Agents Chemother. 2014, 58, 2968–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Cromm PM; Crews CM The Proteasome in Modern Drug Discovery: Second Life of a Highly Valuable Drug Target. ACS Cent. Sci. 2017, 3, 830–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Culp E; Wright GD Bacterial proteases, untapped antimicrobial drug targets. J. Antibiot. 2017, 70, 366. [DOI] [PubMed] [Google Scholar]
- (39).Wood NA; Blocker AM; Seleem MA; Conda-Sheridan M; Fisher DJ; Ouellette SP; Msadek T The ClpX and ClpP2 Orthologs of Chlamydia trachomatis Perform Discrete and Essential Functions in Organism Growth and Development. mBio 2020, 11, No. e02016–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Baker TA; Sauer RT ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim. Biophys. Acta, Mol. Cell Res. 2012, 1823, 15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Baker TA; Sauer RT ATP-dependent proteases of bacteria: recognition logic and operating principles. Trends Biochem. Sci. 2006, 31, 647–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ye F; Li J; Yang C-G The development of small-molecule modulators for ClpP protease activity. Mol. BioSyst. 2017, 13, 23–31. [DOI] [PubMed] [Google Scholar]
- (43).Brötz-Oesterhelt H; Sass P Bacterial caseinolytic proteases as novel targets for antibacterial treatment. Int. J. Med. Microbiol. 2014, 304, 23–30. [DOI] [PubMed] [Google Scholar]
- (44).Leung E; Datti A; Cossette M; Goodreid J; McCaw SE; Mah M; Nakhamchik A; Ogata K; El Bakkouri M; Cheng YQ; et al. Activators of cylindrical proteases as antimicrobials: identification and development of small molecule activators of ClpP protease. Chem. Biol. 2011, 18, 1167–1178. [DOI] [PubMed] [Google Scholar]
- (45).Lavey NP; Coker JA; Ruben EA; Duerfeldt AS Sclerotiamide: The First Non-Peptide-Based Natural Product Activator of Bacterial Caseinolytic Protease P. J. Nat. Prod. 2016, 79, 1193–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Böttcher T; Sieber SA β-Lactones as Specific Inhibitors of ClpP Attenuate the Production of Extracellular Virulence Factors of Staphylococcus aureus. J. Am. Chem. Soc. 2008, 130, 14400–14401. [DOI] [PubMed] [Google Scholar]
- (47).Hackl MW; Lakemeyer M; Dahmen M; Glaser M; Pahl A; Lorenz-Baath K; Menzel T; Sievers S; Böttcher T; Antes I; et al. Phenyl Esters Are Potent Inhibitors of Caseinolytic Protease P and Reveal a Stereogenic Switch for Deoligomerization. J. Am. Chem. Soc. 2015, 137, 8475–8483. [DOI] [PubMed] [Google Scholar]
- (48).Moreira W; Ngan GJ; Low JL; Poulsen A; Chia BC; Ang MJ; Yap A; Fulwood J; Lakshmanan U; Lim J; et al. Target mechanism-based whole-cell screening identifies bortezomib as an inhibitor of caseinolytic protease in mycobacteria. mBio 2015, 6, No. e00253–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Ju Y; An Q; Zhang Y; Sun K; Bai L; Luo Y Recent advances in Clp protease modulation to address virulence, resistance and persistence of MRSA infection. Drug Discovery Today 2021, 26, 2190–2197. [DOI] [PubMed] [Google Scholar]
- (50).Fetzer C; Korotkov VS; Thänert R; Lee KM; Neuenschwander M; von Kries JP; Medina E; Sieber SA A Chemical Disruptor of the ClpX Chaperone Complex Attenuates the Virulence of Multidrug-Resistant Staphylococcus aureus. Angew. Chem., Int. Ed. 2017, 56, 15746–15750. [DOI] [PubMed] [Google Scholar]
- (51).Wood NA; Chung KY; Blocker AM; Rodrigues de Almeida N; Conda-Sheridan M; Fisher DJ; Ouellette SP Initial Characterization of the Two ClpP Paralogs of Chlamydia trachomatis Suggests Unique Functionality for Each. J. Bacteriol. 2019, 201, No. e00635–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Seleem MA; Rodrigues de Almeida N; Chhonker YS; Murry DJ; Guterres Z. d. R.; Blocker AM; Kuwabara S; Fisher DJ; Leal ES; Martinefski MR; et al. Synthesis and Antichlamydial Activity of Molecules Based on Dysregulators of Cylindrical Proteases. J. Med. Chem. 2020, 63, 4370–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Wood NA; Swoboda AR; Blocker AM; Fisher DJ; Ouellette SP Tag-Dependent Substrate Selection of ClpX Underlies Secondary Differentiation of Chlamydia trachomatis. mBio 2022, 13, No. e0185822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Felix J; Weinhäupl K; Chipot C; Dehez F; Hessel A; Gauto DF; Morlot C; Abian O; Gutsche I; Velazquez-Campoy A; et al. Mechanism of the allosteric activation of the ClpP protease machinery by substrates and active-site inhibitors. Sci. Adv. 2019, 5, No. eaaw3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Fetzer C; Korotkov VS; Thänert R; Lee KM; Neuenschwander M; von Kries JP; Medina E; Sieber SA A Chemical Disruptor of the ClpX Chaperone Complex Attenuates the Virulence of Multidrug-Resistant Staphylococcus aureus. Angew. Chem., Int. Ed. 2017, 56, 15746–15750. [DOI] [PubMed] [Google Scholar]
- (56).Abdelrahman YM; Belland RJ The chlamydial developmental cycle. FEMS Microbiol. Rev. 2005, 29, 949–959. [DOI] [PubMed] [Google Scholar]
- (57).Fields KA; Hackstadt T The chlamydial inclusion: escape from the endocytic pathway. Annu. Rev. Cell Dev. Biol. 2002, 18, 221–245. [DOI] [PubMed] [Google Scholar]
- (58).Volceanov L; Herbst K; Biniossek M; Schilling O; Haller D; Nölke T; Subbarayal P; Rudel T; Zieger B; Häcker G Septins arrange F-actin-containing fibers on the Chlamydia trachomatis inclusion and are required for normal release of the inclusion by extrusion. mBio 2014, 5, No. e01802–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Hakala E; Hanski L; Uvell H; Yrjönen T; Vuorela H; Elofsson M; Vuorela PM Dibenzocyclooctadiene lignans from Schisandra spp. selectively inhibit the growth of the intracellular bacteria Chlamydia pneumoniae and Chlamydia trachomatis. J. Antibiot. 2015, 68, 609–614. [DOI] [PubMed] [Google Scholar]
- (60).Lawrence A; Fraser T; Gillett A; Tyndall JDA; Timms P; Polkinghorne A; Huston WM Chlamydia Serine Protease Inhibitor, targeting HtrA, as a New Treatment for Koala Chlamydia infection. Sci. Rep. 2016, 6, No. 31466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Patel P; De Boer L; Timms P; Huston WM Evidence of a conserved role for Chlamydia HtrA in the replication phase of the chlamydial developmental cycle. Microbes Infect. 2014, 16, 690–694. [DOI] [PubMed] [Google Scholar]
- (62).Gloeckl S; Ong VA; Patel P; Tyndall JD; Timms P; Beagley KW; Allan JA; Armitage CW; Turnbull L; Whitchurch CB; et al. Identification of a serine protease inhibitor which causes inclusion vacuole reduction and is lethal to Chlamydia trachomatis. Mol. Microbiol. 2013, 89, 676–689. [DOI] [PubMed] [Google Scholar]
- (63).Libby RD Advanced Organic Chemistry, Part A: Structure and Mechanism, 4th Edition (Carey Francis A.; Sundberg Richard J.). J. Chem. Educ. 2001, 78, 314. [Google Scholar]
- (64).Schrödinger. QikProp User Manual 2015. http://gohom.win/ManualHom/Schrodinger/Schrodinger_2015-2_docs/qikprop/qikprop_user_manual.pdf (accessed May 02, 2022).
- (65).Lee Y; Choi SQ Quantitative analysis for lipophilic drug transport through a model lipid membrane with membrane retention. Eur. J. Pharm. Sci. 2019, 134, 176–184. [DOI] [PubMed] [Google Scholar]
- (66).Feng Y; Mitchison TJ; Bender A; Young DW; Tallarico JA Multi-parameter phenotypic profiling: using cellular effects to characterize small-molecule compounds. Nat. Rev. Drug Discovery 2009, 8, 567–578. [DOI] [PubMed] [Google Scholar]
- (67).Kenakin T Quantifying Biological Activity in Chemical Terms: A Pharmacology Primer To Describe Drug Effect. ACS Chem. Biol. 2009, 4, 249–260. [DOI] [PubMed] [Google Scholar]
- (68).Nierode G; Kwon PS; Dordick JS; Kwon SJ Cell-Based Assay Design for High-Content Screening of Drug Candidates. J. Microbiol. Biotechnol. 2016, 26, 213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).An Y; Sherman W; Dixon SL Kernel-Based Partial Least Squares: Application to Fingerprint-Based QSAR with Model Visualization. J. Chem. Inf. Model. 2013, 53, 2312–2321. [DOI] [PubMed] [Google Scholar]
- (70).Asakawa N; Kobayashi S; Goto J; Hirayama N AutoGPA: An Automated 3D-QSAR Method Based on Pharmacophore Alignment and Grid Potential Analysis. Int. J. Med. Chem. 2012, 2012, No. 498931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Verma J; Khedkar VM; Coutinho EC 3D-QSAR in drug design–a review. Curr. Top. Med. Chem. 2010, 10, 95–115. [DOI] [PubMed] [Google Scholar]
- (72).Dixon SL; Duan J; Smith E; Bargen CDV; Sherman W; Repasky MP AutoQSAR: an automated machine learning tool for best-practice quantitative structure–activity relationship modeling. Future Med. Chem. 2016, 8, 1825–1839. [DOI] [PubMed] [Google Scholar]
- (73).Alexander DLJ; Tropsha A; Winkler DA Beware of R2: Simple, Unambiguous Assessment of the Prediction Accuracy of QSAR and QSPR Models. J. Chem. Inf. Model. 2015, 55, 1316–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Golbraikh A; Tropsha A Beware of q2! J. Mol. Graphics Modell. 2002, 20, 269–276. [DOI] [PubMed] [Google Scholar]
- (75).Gramatica P Principles of QSAR models validation: internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar]
- (76).Uniprot Databank. Uniprot Aligment Tool, 2022. https://www.uniprot.org/align/A20220503A084FC58F6BBA219896F365D15F2EB4401E091V (accessed May 02, 2022).
- (77).Ouellette SP; Karimova G; Subtil A; Ladant D Chlamydia co-opts the rod shape-determining proteins MreB and Pbp2 for cell division. Mol. Microbiol. 2012, 85, 164–178. [DOI] [PubMed] [Google Scholar]
- (78).Segers ME; Lebeer S Towards a better understanding of Lactobacillus rhamnosus GG–host interactions. Microb. Cell Fact. 2014, 13, S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Kaźmierczak-Siedlecka K; Daca A; Folwarski M; Witkowski JM; Bryl E; Makarewicz W The role of Lactobacillus plantarum 299v in supporting treatment of selected diseases. Cent. Eur. J. Immunol. 2020, 45, 488–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Maturana JL; Cárdenas JP Insights on the Evolutionary Genomics of the Blautia Genus: Potential New Species and Genetic Content Among Lineages. Front. Microbiol. 2021, 12, No. 660920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Senda S; Fujiyama Y; Ushijima T; Hodohara K; Bamba T; Hosoda S; Kobayashi K Clostridium ramosum, an IgA protease-producing species and its ecology in the human intestinal tract. Microbiol. Immunol. 1985, 29, 1019–1028. [DOI] [PubMed] [Google Scholar]
- (82).Liu H; Shiver AL; Price MN; Carlson HK; Trotter VV; Chen Y; Escalante V; Ray J; Hern KE; Petzold CJ; et al. Functional genetics of human gut commensal Bacteroides thetaiotaomicron reveals metabolic requirements for growth across environments. Cell Rep. 2021, 34, No. 108789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Morinaga K; Kusada H; Watanabe M; Tamaki H Complete Genome Sequence of Anaerostipes caccae Strain L1–92(T), a Butyrate-Producing Bacterium Isolated from Human Feces. Microbiol. Resour. Announce. 2021, 10, No. e00056–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Mason KL; Erb Downward JR; Mason KD; Falkowski NR; Eaton KA; Kao JY; Young VB; Huffnagle GB Candida albicans and bacterial microbiota interactions in the cecum during recolonization following broad-spectrum antibiotic therapy. Infect. Immun. 2012, 80, 3371–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Ho H -l.; Haynes, K. Candida glabrata: new tools and technologies–expanding the toolkit. FEMS Yeast Res. 2015, 15, No. fov066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Di Martino L; De Salvo C; Buela K-A; Hager C; Ghannoum M; Osme A; Buttò L; Bamias G; Pizarro TT; Cominelli F Candida tropicalis Infection Modulates the Gut Microbiome and Confers Enhanced Susceptibility to Colitis in Mice. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 901–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Pérez JC Fungi of the human gut microbiota: Roles and significance. Int. J. Med. Microbiol. 2021, 311, No. 151490. [DOI] [PubMed] [Google Scholar]
- (88).Pilhofer M; Aistleitner K; Biboy J; Gray J; Kuru E; Hall E; Brun YV; VanNieuwenhze MS; Vollmer W; Horn M; Jensen GJ Discovery of chlamydial peptidoglycan reveals bacteria with murein sacculi but without FtsZ. Nat. Commun. 2013, 4, No. 2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Gottesman S Proteases and Their Targets in Escherichia coli. Annu. Rev. Genet. 1996, 30, 465–506. [DOI] [PubMed] [Google Scholar]
- (90).McCarren P; Springer C; Whitehead L An investigation into pharmaceutically relevant mutagenicity data and the influence on Ames predictive potential. J. Cheminf. 2011, 3, No. 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Revitt-Mills SA; Robinson A Antibiotic-Induced Mutagenesis: Under the Microscope. Front. Microbiol. 2020, 11, No. 585175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Woodford N; Ellington MJ The emergence of antibiotic resistance by mutation. Clin. Microbiol. Infect. 2007, 13, 5–18. [DOI] [PubMed] [Google Scholar]
- (93).Ames BN; McCann J; Yamasaki E Methods for detecting carcinogens and mutagens with the salmonella/mammalian-microsome mutagenicity test. Mutat. Res., Environ. Mutagen. Relat. Subj. 1975, 31, 347–363. [DOI] [PubMed] [Google Scholar]
- (94).Valente-Campos S; Dias CL; Barbour ED; de Souza Nascimento E; de Aragão Umbuzeiro G The introduction of the Salmonella/microsome mutagenicity assay in a groundwater monitoring program. Mutat. Res., Genet. Toxicol. Environ. Mutagen. 2009, 675, 17–22. [DOI] [PubMed] [Google Scholar]
- (95).Roy K; Kar S; Ambure P On a simple approach for determining applicability domain of QSAR models. Chemom. Intell. Lab. Syst. 2015, 145, 22–29. [Google Scholar]
- (96).Ruiz IL; Gómez-Nieto MÁ Study of the Applicability Domain of the QSAR Classification Models by Means of the Rivality and Modelability Indexes. Molecules 2018, 23, No. 2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Schrödinger. AutoQSAR; Schrödinger, 2021. https://www.schrodinger.com/products/autoqsar (accessed Oct 28, 2022). [Google Scholar]
- (98).Yang PH; Zhang QZ; Sun W Clean synthesis of 2-arylideneindan-1,3-diones in water. Res. Chem. Intermed. 2012, 38, 1063–1068. [Google Scholar]
- (99).Lee C-J; Sheu C-N; Tsai C-C; Wu Z-Z; Lin W Direct β-acylation of 2-arylidene-1,3-indandiones with acyl chlorides catalyzed by organophosphanes. Chem. Commun. 2014, 50, 5304–5306. [DOI] [PubMed] [Google Scholar]
- (100).Adibi H; Mehrabi M; Amiri K; Balalaie S; Khodarahmi R Synthesis and characterization of 2-benzylidene-1,3-indandione derivatives as in vitro quantification of amyloid fibrils. J. Iran. Chem. Soc. 2020, 17, 423–432. [Google Scholar]
- (101).Meyer T; Krug R; Müller TJJ Three-Component Suzuki–Knoevenagel Synthesis of Merocyanine Libraries and Correlation Analyses of Their Oxidation Potentials and Optical Band Gaps. Molecules 2021, 26, No. 5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Maestro; Schrödinger: New York, NY, 2020. [Google Scholar]
- (103).Selvaraj C; Tripathi SK; Reddy KK; Singh SK Tool development for Prediction of pIC50 values from the IC50 values - A pIC50 value calculator. Curr. Trends Biotechnol. Pharm. 2011, 5, 1104–1109. [Google Scholar]
- (104).Duan J; Dixon SL; Lowrie JF; Sherman W Analysis and comparison of 2D fingerprints: Insights into database screening performance using eight fingerprint methods. J. Mol. Graphics Modell. 2010, 29, 157–170. [DOI] [PubMed] [Google Scholar]
- (105).Schrödinger. AutoQSAR. Automated Creation and Application of Predictive QSAR Models Following Best Practices; Schrödinger, 2021. https://www.schrodinger.com/products/autoqsar (accessed Sept 26, 2022). [Google Scholar]
- (106).QikProp; Schrödinger: New York, NY, 2020. [Google Scholar]
- (107).Bochevarov AD; Harder E; Hughes TF; Greenwood JR; Braden DA; Philipp DM; Rinaldo D; Halls MD; Zhang J; Friesner RA Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar]
- (108).Holland Computing Center. https://hcc.unl.edu/ (accessed April 11, 2022).
- (109).GraphPad Prism; GraphPad Software: San Diego, CA, 2022. www.graphpad.com. [Google Scholar]
- (110).Seleem MA; Wood NA; Brinkworth AJ; Manam S; Carabeo RA; Murthy AK; Ouellette SP; Conda-Sheridan M In Vitro and In Vivo Activity of (Trifluoromethyl)pyridines as Anti-Chlamydia trachomatis Agents. ACS Infect. Dis. 2022, 8, 227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).CLSI. M7-A9 Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Wayne: PA, 2008. [Google Scholar]
- (112).CLSI. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts. Approved Standard. CLSI Document M27-A3; Clinical and Laboratory Standards Institute: Wayne, PA, 2008. [Google Scholar]
- (113).Kado NY; Langley D; Eisenstadt E A simple modification of the Salmonella liquid-incubation assay. Increased sensitivity for detecting mutagens in human urine. Mutat. Res. Lett. 1983, 121, 25–32. [DOI] [PubMed] [Google Scholar]
- (114).OECD. Test No. 471: Bacterial Reverse Mutation Test. OECD Guidelines for the Testing of Chemicals, Section 4; OECD, 2020. [Google Scholar]
- (115).Bernstein L; Kaldor J; McCann J; Pike MC An empirical approach to the statistical analysis of mutagenesis data from the Salmonella test. Mutat. Res., Environ. Mutagen. Relat. Subj. 1982, 97, 267–281. [DOI] [PubMed] [Google Scholar]
- (116).Kummrow F; Magalhães D; Franco A; de Aragão Umbuzeiro G Blue rayon and Salmonella/microsome assay in the evaluation of coastal water quality. Rev. Saúde Pública 2006, 40, 890. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.