

Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia, with associated morbidity and mortality including stroke, dementia, congestive heart failure, and death. Despite the burden of disease, therapeutic advances are lagging. Animal models present an opportunity to gain better insight into the mechanistic basis for AF. Mouse models are attractive owing due to low cost, accessibility, and ease of genetic manipulation. However, their role in the study of AF has been limited, as mice typically do not develop spontaneous AF, and most require time-dependent cardiac dysfunction and remodeling.1 Here we present a model of sepsis-related atrial fibrillation in wild type mice with a characteristic electrophysiologic (EP) phenotype within 1 week of insult.
Studies were approved by the Institutional Animal Care and Use Committee at Massachusetts General Hospital. Cecal ligation and puncture (CLP), a commonly used animal model of polymicrobial sepsis, can be easily performed in mice.2 We subjected 10–20 week old male C57/Bl6 mice to either mild CLP or sham operation; a separate batch of mice were used as naïve controls (A). After one week, ~89% of the CLP-treated mice were alive, as compared to 100% of controls. There were no significant differences in tail-cuff blood pressure, nor cardiac structure/function determined by transthoracic echocardiography.
One week post-CLP, invasive EP studies were performed on the three groups of mice to determine electrophysiologic characteristics and arrhythmia inducibility. Inducible AF was defined by rapid atrial cycle length and concomitant irregular R-R intervals for >5 seconds in duration (B). There were no significant differences in baseline EP parameters except for a higher HR in CLP-treated mice as compared to controls. CLP-treated mice had an increased incidence (C, left)), frequency (C, middle) and burden (C, right) of AF compared to the controls. Optical mapping revealed a CLP-related increase in action potential duration (APD) in both atrial chambers (D) as well as a trend towards slowed action potential upstroke (dV/dt) (E). We next investigated the effects of sepsis on impulse propagation and found a CLP-induced slowing in atrial conduction velocity (CV) (F).
We utilized quantitative PCR and Western blotting to explore possible differences in atrial gene/protein expression that may underlie the CLP-related phenotype. As connexin expression is critical to impulse propagation, we were interested to find CLP-related decreases in mRNA encoding Cx40 and Cx43, as well as the protein expression of Cx40 (G,H). We identified a CLP-related decrease in atrial expression of several genes that encode ion channel subunits responsible for the cardiac action potential (I). Considering the effects on CV and dV/dt, we were interested to note CLP-related decrease in protein expression of the voltage-gated sodium channel, NaV1.5 (J).
Calcium/calmodulin-dependent protein kinase II (CaMKII) is integral to calcium homeostasis and can centrally affect APD through its effects on multiple ion channels. We were interested to find a CLP-induced increase in phosphorylation of CaMKII (K). To investigate the role of CaMKII further, we treated mice with CLP followed by intra-peritoneal injections of either a CaMKII inhibitor, KN-93, or vehicle. KN-93 treatment significantly reduced AF inducibility in CLP treated mice, suggesting that CaMKII activation is necessary for sepsis-related atrial arrhythmogenesis (L).
As systemic inflammation is evident during sepsis, we investigated local leukocyte recruitment to the left atrium. Flow cytometry of CLP- and sham-treated left atria revealed a significant increase in inflammatory leukocytes and fibroblasts (M). Inflammasome activity has been posited as involved in AF pathogenesis, so we assayed the components of the NLRP3 inflammasome via western blotting. CLP significantly increased atrial expression of inflammasome components procaspase 1, cleaved caspase p20, and ASC without a change in expression of NLRP3 itself (N). In addition, CLP resulted in increased expression of the phosphorylated form of the NF-kB p65 subunit, consistent with transactivation of this pathway. These data demonstrate an atrial inflammatory milieu, as would be expected in the setting of sepsis.
Here we introduce a mouse model of sepsis-related AF caused by CLP and mediated by altered atrial electrophysiology associated with dysregulated expression of conduction-related genes and atrial inflammation. AF is the most commonly encountered arrhythmia in the setting of severe sepsis, which increases the odds of new-onset AF by nearly ~7-fold.3 Sepsis is independently associated with QT prolongation,4 and our data suggests that a similar effect on repolarization reserve may underlie atrial pro-arrhythmia. Interestingly, CLP-treated mice demonstrate an atrial inflammatory infiltrate resulting in both “priming” and “triggering” of the NLRP3 inflammasome, which is associated with AF.5 Commonly utilized mouse models of AF require transgenic mice or extensive remodeling from a chronic exposure; these would increase the cost and time required for use. Thus, this clinically relevant model provides an opportunity to perform high throughput experimentation to better understand the role of inflammation in AF pathogenesis. The data supporting this study’s findings are available from the corresponding author on reasonable request, according to Transparency and Openness Promotion guidelines.
Figure. Cecal Ligation and Puncture Produces a Mouse Model of Atrial Fibrillation.
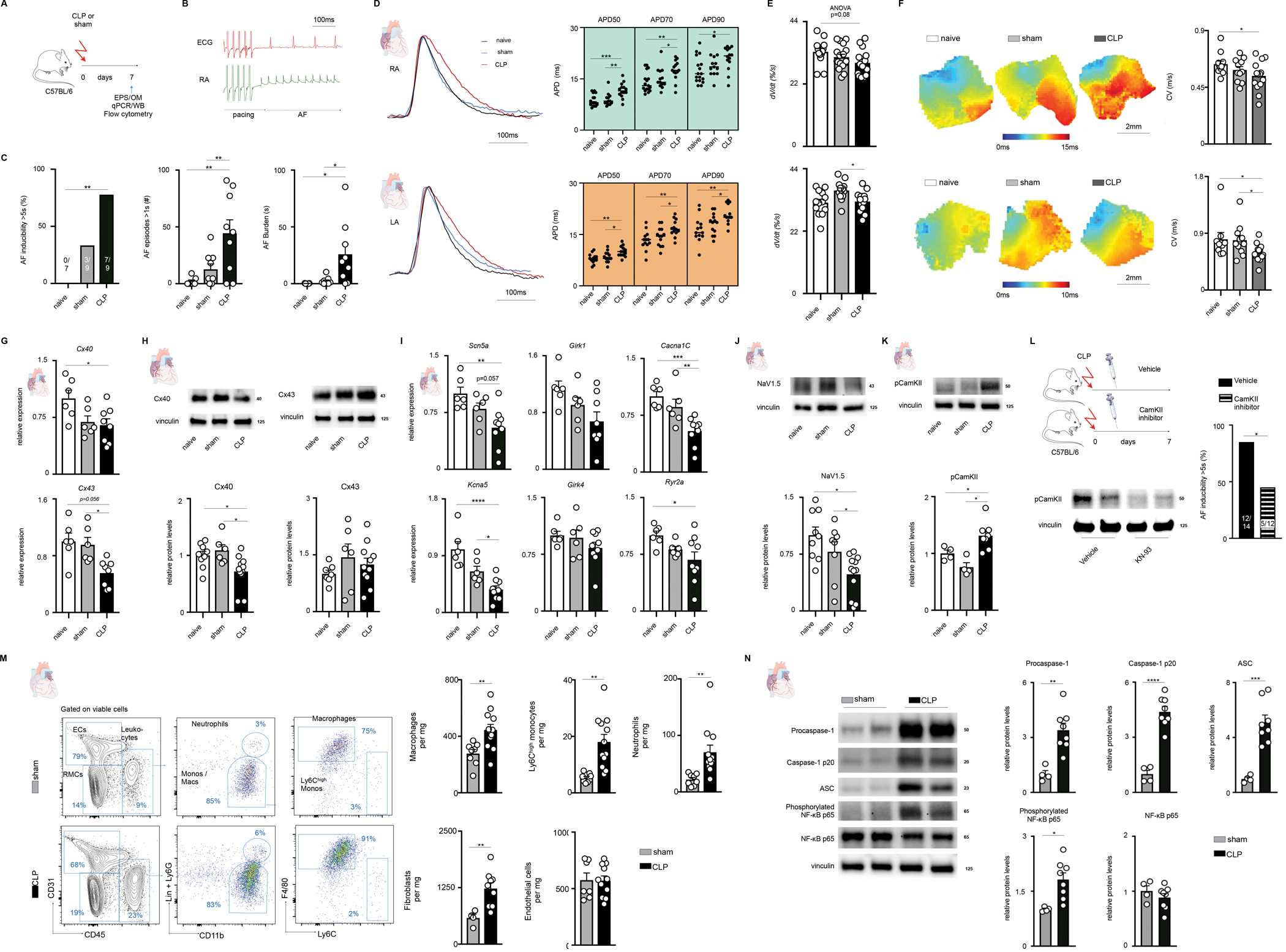
A, 57/Bl6 mice underwent mild cecal ligation and punction (CLP) or sham operation and were studied 7 days later. Naïve C57/Bl6 mice were used as additional controls.
B, Representative surface ECG and right atrial intracardiac electrogram acquired during an invasive EP study and showing pacing-induced AF.
C, Left, percentage of mice with an episode of atrial fibrillation (AF) longer than 5 seconds. Two-sided Fisher’s exact test. Middle, number of AF episodes longer than 1 second during the entirety of each EP study. One-way ANOVA, post-hoc Sidak test. Right, AF burden assessed per mouse during the entire EP study. One-way ANOVA, post-hoc Sidak test.
D, Left, representative action potential tracings derived from optical mapping in the right (upper) and left (lower) atrium 7 days after CLP. Right, quantification of the APD at 50%, 70%, and 90% repolarization; obtained at a pacing cycle length of 100ms. One-way ANOVA, post-hoc Sidak test.
E, Action potential upstroke velocity, (dV/dt) in the right (upper) and left (lower) atrium; obtained at a pacing cycle length of 100ms. One-way ANOVA, post-hoc Sidak test.
F, Optical mapping derived representative activation maps from right (upper) and left (lower) atria of mice with quantification of conduction velocity to the right; obtained at a pacing cycle length of 100ms. One-way ANOVA, post-hoc Sidak test.
G, Expression of atrial connexin genes measured by qPCR. One-way ANOVA, post-hoc Sidak test.
H, Representative blots of connexin 40 (left) and connexin 43 (right), with quantification below. One-way ANOVA, post-hoc Sidak test.
I, Atrial expression of ion channel genes measured by qPCR. One-way ANOVA, post-hoc Sidak test.
J, Representative blots of NaV1.5 sodium channel subunit with quantification shown below. One-way ANOVA, post-hoc Sidak test.
K, Representative blots of phosphorylated CaMKII with quantification shown below. One-way ANOVA, post-hoc Sidak test.
L, C57/Bl6 mice underwent CLP and then were treated with either vehicle or a CaMKII inhibitor, KN-93, every other day (total of 4 doses) until study (left upper). Left lower, representative blot showing decreased CaMKII phosphorylation in KN-93 treated mice. Right, percentage of mice with an episode of AF longer than 5 seconds in vehicle treated or KN-93 treated mice. Two-sided Fisher’s exact test.
M, Left, flow cytometry plots of the left atrium. Right, quantification of leukocytes, endothelial cells and fibroblasts in the left atrium. Up to 2 independent experiments. Two-tailed Student’s t-test.
N, Left, representative blots of protein expression in the atria. Right, quantification of relative protein expression in the atria. Two-tailed Student’s t-test.
Data are mean ± SEM with individual values for data distribution, *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001.
Acknowledgements
We acknowledge Servier Medical Art (smart.servier.com) for cartoon components.
A.B. and M.J.S. conceived the study, designed, performed, and analyzed experiments. A.B. and M.J.S. interpreted data and made the figures. M.Y., J.G. and M.H. performed and analyzed experiments. M.N. and D.M. discussed results and strategy. P.T.E. provided funding and discussed results and strategy. A.B. and M.J.S. wrote the manuscript with input from all authors.
Funding Sources
This work was funded in part by U.S. federal funds from the National Institutes of Health (T32HL076136, 1RO1HL092577, 1R01HL157635, 5R01HL139731). A.B was supported by the NIH grant T32HL007604. M.J.S. was funded by Deutsche Forschungsgemeinschaft (SCHL 2221/1–1). J.G. was supported by funding from the German Research Foundation (GR 5261/1–1, SFB-1470-A04), German Society for Cardiology, German Center for Cardiovascular Research and Corona-Stiftung. M.H. was supported by an American Heart Association Career Development Award (19CDA34490005) and NIH grant HL155097. M.N. was supported by NIH grant HL139598. P.T.E. was supported by a grant from the American Heart Association Strategically Focused Research Networks (18SFRN34110082), and by a grant from the European Union (MAESTRIA 965286).
Footnotes
Conflict of Interest Disclosures
M.N. has received funds or material research support from Alnylam, Biotronik, CSL Behring, GlycoMimetics, GSK, Medtronic, Novartis and Pfizer, as well as consulting fees from Biogen, Gimv, IFM Therapeutics, Molecular Imaging, Sigilon and Verseau Therapeutics. P.T.E has received sponsored research support from Bayer AG, IBM Research, Bristol Myers Squibb and Pfizer; he has also served on advisory boards or consulted for Bayer AG, MyoKardia and Novartis
REFERENCES:
- 1.Schüttler D, Bapat A, Kääb S, Lee K, Tomsits P, Clauss S, Hucker WJ. Animal Models of Atrial Fibrillation. Circ Res. 2020;127:91–110. [DOI] [PubMed] [Google Scholar]
- 2.Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nature Protocols. 2009;4:31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walkey AJ, Wiener RS, Ghobrial JM, Curtis LH, Benjamin EJ. Incident stroke and mortality associated with new-onset atrial fibrillation in patients hospitalized with severe sepsis. JAMA - Journal of the American Medical Association. 2011;306:2248–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tisdale JE, Jaynes HA, Kingery JR, Mourad NA, Trujillo TN, Overholser BR, Kovacs RJ. Development and validation of a risk score to predict QT interval prolongation in hospitalized patients. Circ Cardiovasc Qual Outcomes. 2013;6:479–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yao C, Veleva T, Scott L, Cao S, Luge L, Chen G, Jeyabal P, Pan X, Alsina KM, Abu-Taha I, Ghezelbash S, Reynolds CL, Shen YH, LeMaire SA, Schmitz W, Müller FU, El-Armouche A, Eissa NT, Beeton C, Nattel S, Wehrens XHT, Dobrev D, Li N. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation [Internet]. 2018;1. Available from: http://insights.ovid.com/crossref?an=00003017-900000000-95079 [DOI] [PMC free article] [PubMed] [Google Scholar]