

Abstract
Supported single atom catalysts on defected graphene show great potential for electrochemical reduction of CO2 to CO. In this study, we perform a computational screening of single and di-atom catalysts (MNCs and FeMNC respectively) with M varying from Sc to Zn on nitrogen-doped graphene for CO2 reduction using hybrid-density functional theory and potential dependent micro-kinetic modeling. The formation energy calculations reveal several stable single and di-atom doping site motifs. We consider the kinetics of CO2 using the binding energies of CO2* and COOH* intermediates as the descriptors to analyze the activity of these catalysts. In comparison to (211) transition metal (TM) surfaces, both MNCs and FeMNCs show a variety of binding motifs of the reaction intermediates on different metal dopants. We find four MNCs as CrNC, MnNC, FeNC, and CoNC with high catalytic efficiency for CO2R. Among the different FeMNCs with varying doping geometry and surrounding N-coordination, we have identified 11 candidates having high TOF for CO production and lower selectivity for the hydrogen evolution reaction. FeMnNC shows the highest activity for CO2R. Large CO2* dipole–field interactions in both the MNCs and FeMNCs give rise to deviations in scaling from TM surfaces.
Keywords: single and di-atom catalysts, electrocatalysis, CO2 reduction, density functional theory, microkinetic modeling
Introduction
Electrochemical carbon dioxide reduction (CO2R) is one of the promising approaches for the sustainable production of carbon-based fuels and chemicals.1,2 In this process, CO2 is electrochemically reduced to organic products containing one or more carbon atoms (C1 or C2 products). CO is the simplest of these products, which can be used as feedstock for the production of hydrocarbons in the Fischer–Tropsch reaction.3 Nanostructured gold (Au) surfaces are presently the most efficient catalysts for CO2 to CO conversion.4 Nevertheless, they are expensive, and the hydrogen evolution reaction (HER) at reductive potentials reduces the selectivity for CO production on these transition metal (TM) surfaces.5 For industrial-scale applications, earth-abundant and stable electro-catalysts with high activity and selectivity for CO2R are needed.2
Two-dimensional (2D) materials have in the past decade emerged as a promising class of catalysts with unique geometries.6−9 However, for electrocatalytic conversion of CO2 to C1 and C2 products, pristine surfaces of some of the 2D materials such as graphene and graphitic-C3N4 are found to be inert and the introduction of dopants, defect states, or edge sites are needed to activate for electrocatalysis.10−17 Embedding single transition metal atoms on nitrogen-doped defected graphene (MNC) surfaces has been found as efficient electrocatalysts for CO2R.18−21 While CO2* adsorption is the limiting step for transition and coinage metal surfaces over relevant potentials, on the single atom catalysts (SACs), either the CO2* adsorption or COOH* formation is identified as the rate limiting step.21 Furthermore, the single metal dopants show greater surface-adsorbate dipole–field interactions, due to their narrow d-bands. This enhances the binding affinity of reactive intermediates on the catalytic centers under applied potential and provides a greater tunability of catalytic activity beyond the TM scaling.21
Inspired by the bimetallic alloys and transition metal complexes,4,22,23 we hypothesize that enhancing the number of active sites by introducing two adjacently placed metal dopants will open up new possibilities for coordination of reaction intermediates. Recent literature has reported the synthesis of a few di-atom catalysts (DACs).23−32 For example, a DAC consisting of Ni and Fe pair supported on a nitrogen-doped graphene surface was reported to show a high Faradaic efficiency (FE) of 98% for CO formation within the potential window −0.4 to −1.0 V vs RHE. X-ray absorption spectroscopy and HAADF-STEM images of the catalyst surface gives clear evidence for the presence of Ni–Fe metal pairs. We expect that screening of other metal dimers and analysis of their activity will allow us to identify a greater number of promising electrocatalysts for CO2R.
In this work, we perform a computational screening of single and di-atom dopants supported on a nitrogen-doped graphene surface (MNCs and M′MNCs respectively, where M and M′ are transition metal dopants) based on the stability and activity for CO2R. The metal dopants, M, range from Sc to Zn. We consider Fe as the candidate for M′, given that recent experimental studies have investigated synthetic pathways for FeMNC di-atom catalysts.24−32 We evaluate the formation energies of single and di-atom metal dopants on different defected sites of a nitrogen-doped graphene sheet. Based on hybrid DFT functional HSE06 and potential dependent free-energy calculations,33−36 we determine how the binding geometries of reactive intermediates on metallic dopants in SACs are influenced by the introduction of another metal dopant at an adjacent position in DACs. Using a kinetic volcano with the adsorption energies of CO2* and COOH* as the activity descriptors,21 we have identified several single and di-atom candidates as potential candidates with a good catalytic activity. Among the eight FeM dimers, we find that FeMn has both higher stability and TOF for CO2R in comparison to the earlier reported FeNi dimer. We show that the adsorbate dipole–field interactions influence the binding affinity of the adsorbates, giving rise to different binding motifs of the intermediates on the metal dopants and deviation of the binding energies beyond the transition metal scaling.
Results and Discussion
Computations of Formation Energies Suggest a Variety of Stable Single- and Di-Atom Site Motifs
We have calculated the formation energies (ΔEform) of the single and di-atom metal dopants on different vacancy sites using eq 1 to understand the stability of the site motifs in SACs and DACs.
| 1 |
Here, EM1/M2 – NC denotes the energy of the SACs and DACs. The reference energies for metal dopants denoted as EM1/M2 are obtained from the DFT calculated energies of the bulk metal. n is the total number of dopant sites in SACs and DACs, and ENC denotes the DFT calculated energy of the defected graphene surface without the metal dopants.
The ΔEform values for the SACs with single and double carbon atom vacancy sites on N-doped graphene show that the double carbon atom vacancy is the most favorable doping site. Figures S1 and S2 in the Supporting Information show the structural possibilities and formation energies of SACs, respectively, considered for the different metal dopants with the single and double carbon atom vacancy sites. However, for two metal atoms, the possible doping sites remain debated in the recent works. Therefore, to understand the preferred structure for DACs, we calculate the formation energies of dual metal dopants on different vacancy sites, varying from two to six carbon atoms.
Figure 1 shows the structural arrangement of the vacancy defects and metal atom arrangements. The vacancy defects are denoted by a two-letter symbol based on the vacancy site. For example, DV corresponds to the di-carbon atom vacancy sites, TV for tri-carbon atom vacancy, and so on. The di- and quad-carbon atom vacancies show three different atomic arrangements of the defect sites, whereas for the other vacancy types, we identify only one possible arrangement. The metal dopants are coordinated to the 2D material support through the N-atoms.
Figure 1.
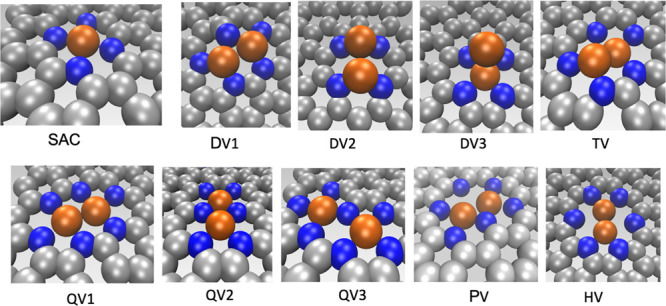
Single and di-atom doping sites on the defected monolayered graphene sheet. Metal dopants are coordinated to the graphene atoms through N atoms. The metal dopants are represented in orange, carbon atoms in gray, and N atoms in blue. SAC corresponds to the doping site for the single metal dopants. For DACs, the dopant sites are named by two-letter symbols as derived from the initials of the vacancy sites. Here, DV denotes the divacancy site, TV for trivacancy, QV for quadvacancy, PV for pentavacancy, and HV for hexavacancy. In the case of DV and QV, three stable structural geometries are obtained, whereas for the other vacancy sites, only one configuration is found to be stable.
Figure 2 shows the formation energies (ΔEform) of the di-atom catalysts (FeMNC). Based on the formation energy values, we find that the stability of the DACs depends on the defect structure of the graphene surface. The quad-atom vacancy sites (QV) are seen to be the most preferable binding sites for di-atom dopants. Among the three QVs, QV2 and/or QV3 sites show slightly higher stability compared to the QV1 site. Higher exothermic binding energies for the di-atom dopants on the quad-atom vacancy sites suggest stronger binding affinities and therefore would not allow the formation of metal nanoclusters on the graphene surfaces.
Figure 2.
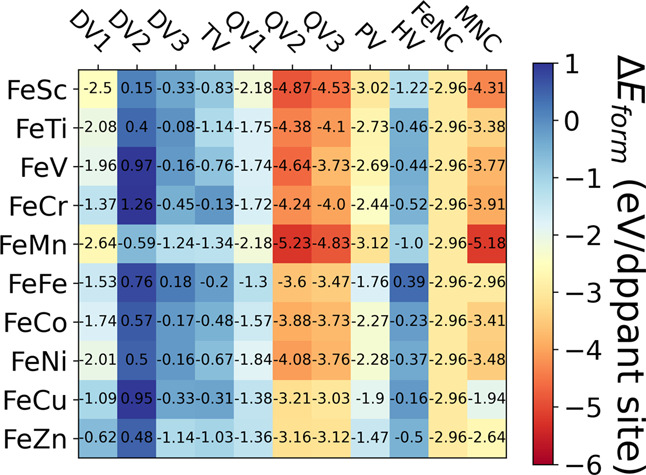
Formation energy (ΔEform) heatmap of the different metal dimer combinations on different vacancy sites as shown in Figure 1. The formation energy values for the corresponding single metal dopants are also shown here. The calculation details for the formation energies are given in Section S-II of the SI. The color map shows the relative stability in eV/dopant site. The metal combinations are denoted as FeM, where M corresponds to the first-row transition metal atoms from Sc to Zn. FeNC and MNC corresponds to the respective SACs.
Along with the configuration of the doping sites, the stability of the DACs is found to be influenced by the surrounding N atom coordination. The effect of N atom coordination on the stability of DACs is shown in Figure S3 for the three QV sites with 6, 4, and 2N atom coordination.
Comparing the formation energies (ΔEform) of the di-atom catalysts in Figure 2 with the corresponding single atom catalysts, we find that the most stable di-atom doping sites for some of the dimers have comparable stability with single doping sites. This suggests that the single and di-atom configurations could be formed simultaneously during the thermodynamic synthetic process. However, recently suitable methods have been reported for synthesizing the preferred di-atom catalysts under carefully controlled conditions inside metal–organic frameworks as the templates.24−32,37
Computational Catalyst Screening Shows Several di-Atom Catalysts with Good Activity
Using potential dependent DFT calculations, we study the activity of the SACs and DACs with QV1, QV2, and QV3 geometries. As in ref (21), we consider the following reaction mechanism for CO formation on the catalyst surfaces:
The corresponding mean-field microkinetic model was developed using the binding energies of CO2* and COOH* as the activity descriptors in refs (20 and 21).
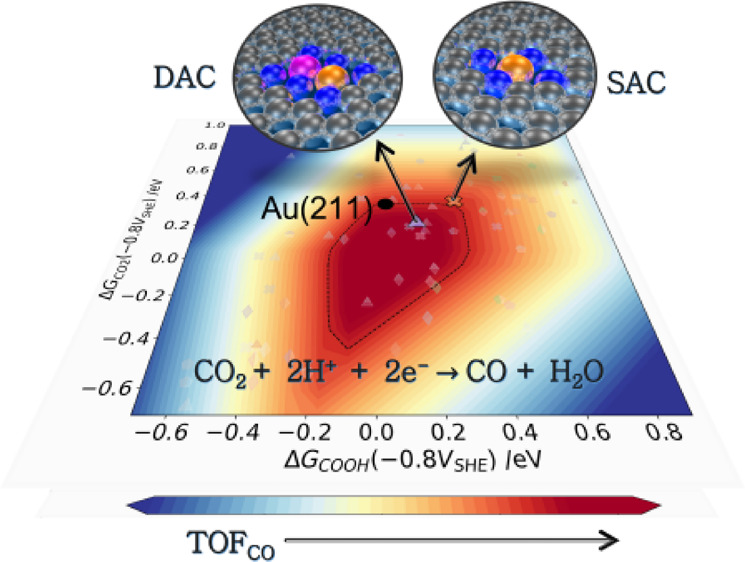
In Figure 3a,b, we show the activities for CO2 to CO reduction on SACs and DACs, respectively, at an applied potential of −0.8 V vs SHE and a buffer pH of 6.8. The choice of pH value is motivated by the recent experimental reports on di-atom catalysts, where CO TOF is measured in a CO2-saturated KHCO3 electrolyte.24,27,30,31Section S-III in the SI shows the potential dependence of the rate map at −0.6, −1.0, and −1.2 V vs SHE for the SACs and DACs. The activity is determined for different numbers of N-atom coordination surrounding the metal dopants in both SACs and DACs. To compare the relative activity with the TM surfaces, the corresponding activity for (211) surfaces of Pt, Pd, Ag, Au, and Cu are also shown in this figure through the black dots. The black solid line in Figure 3a,b represents the parity line for COOH* vs CO2* binding energies. Deviation from the parity line would lead either of the two reaction steps, CO2(g) → CO2* or CO2 → COOH* to be the rate limiting step as indicated in the figures.
Figure 3.

Activity volcano plots for SACs and DACs plotted using the binding energies of CO2* and COOH* as the activity descriptors at an applied potential −0.8 V vs SHE and pH of 6.8. (a) Unified activity of SACs with first row transition metals doped on the defected graphene layer with 2 and 4 surrounding coordinating N atoms. The early and late transition metal dopants prefer different binding motifs as shown in the inset of the figure. (b) The activity of FeM DACs doped on the three quad-atom vacancy sites (QV1, QV2, and QV3) for 2, 4, and 6/7 coordinating N atoms is shown. The metal dimers with TOF comparable to or greater than the Au (211) surface and lower surface coverage for CO are mainly highlighted in solid markers. The less active materials are shown in faded markers. The top-left panel of Figure 3b shows the chemical symbols of the metal dimers activity comparable to or greater than the Au (211) surface and those reported in recent experimental studies. The black dotted curve denotes the rate map for the Au (211) surface.
The activity volcano plot in Figure 3a shows considerable deviation in the binding affinity for both COOH* and CO2* intermediates in SACs with respect to the (211) TM surfaces. The two descriptors follow a linear scaling relationship denoted by the black dotted line on the TM (211) surfaces. The SACs, on the other hand, show a large scatter, which in part arises from variations in binding motifs for the intermediates depending on the metal dopants. The early transition SACs with Sc, Ti, and V metal dopants (denoted by solid circles in blue) prefer a bidentate binding motif for both CO2* and COOH* intermediates through the C and O atoms attaching to the metal. This binding geometry tends to increase the binding strengths in early TM dopants and results into the CO* desorption as the reaction limiting step (see coverages in Figure S4a). The later TM dopants from Cr to Zn (solid circle in purple) show a monodentate binding motif of CO2* and COOH* intermediates attaching through the C atom. This motif reduces the binding affinities of the intermediates resulting into either of the CO2* adsorption or COOH* formation as the rate determining step in late TM SACs.
In addition to the variation in the binding structures, the larger dipole moments associated with the CO2* on SACs leads to the deviation in the binding energies from the (211) TM scaling line.21 The adsorbate dipole moments for all the CO2R intermediates on the SACs are shown in Figure S5 of the SI. The narrow d-bands of all the metal dopants result in large dipoles for CO2*.21 The enhancement in the electrostatic stabilization for CO2* increases the binding energies under a field relative to COOH* and therefore results in greater deviation from the 211 scaling line.
The DACs considered here show an even larger scatter in the binding energies for the CO2* and COOH* intermediates. Figure 3b shows the activity volcano plot for the different FeM dimers doped on the three quad-atom vacancy (QV) sites at −0.8 V and buffer pH of 6.8 and their representative binding motifs for CO2*. The metal dopants on the QV1 site prefer a two-site binding motif for CO2* and COOH*, where both the metal atoms bind to the intermediates through C and O atoms as shown in the inset (1) of Figure 3b. However, on the QV2 and QV3 sites, the single-site binding motifs are mainly preferred as observed for the SACs. Most of the metal dopants on QV2 and QV3 show the monodentate binding of CO2* and COOH* through the C atom shown in the insets (2) and (3) of Figure 3b.
We observe that the adsorbate dipole moments of the intermediates on the active DACs show a similar trend as seen for the SACs. CO2* shows greater dipoles compared to both COOH* and CO* (Figure S5 in SI). Differences in the preferential binding motifs of the CO2R intermediates depending on the dopant geometry of DACs (QV1, QV2, and QV3) result in greater CO2* dipoles and greater preferential stabilization of CO2* relative to COOH*. This results in large scattering of the binding energies on these catalysts.
The variation in the binding geometries on SACs and DACs considerably influences the catalytic activity for CO2R. We compare here the activity of the SACs and DACs with the Au (211) surface. The single and di-atom candidates with TOF for CO production comparable to or greater than the Au (211) surface and CO coverage (θCO) lower than 0.4 as seen from the CO coverage plot in Figure S4) are shown in solid markers in Figure 3a,b. The ones with lower activity compared to the Au (211) surface are shown by the faded markers.
Screening the 3d metal dopants on single doping sites, we find only three SACs, CrNC, FeNC, and CoNC, with high TOF for CO production. FeNC shows an activity comparable to the Au surface, whereas we have seen greater TOF for CrNC and CoNC SACs than Au (211) surface. At a more reducing potential (Section S-III in the SI), we find that in addition to these three SACs, MnNC shows a comparable activity to the Au (211) surface. Earlier studies have identified some of these metal dopants as favorable catalysts for CO2R.11,18,20,21
With the di-atom doping sites, we have identified more candidates within the high activity region of the volcano plot. Five dimer candidates show comparable activity as the Au (211) surface, while eight di-atom pairs are identified with slightly greater TOF than the Au (211) surface. These DACs with higher activity are labeled in the left top inset of Figure 3b. The QV1 and QV2 DACs (denoted by the diamond and triangular markers, respectively, in Figure 3b) show the maximum number of the candidates within this high activity region. Comparatively, the QV3 site shows only one metal dimer (FeCr) with higher TOF. In addition to the catalyst doping sites, we see that lowering the N atom coordination surrounding the metal dimers enhances the catalytic activity for most of the FeM dimers. With 6N atom coordination, only three candidates (FeNi on the QV1 site, and FeMn and FeCo on the QV2 site) are identified with TOF comparable to the Au (211) surface. The other metal dimers identified with higher activity are surrounded by 4 or 2 coordinating N atoms.
Among the 10 FeM dimers, FeV, FeMn, FeCo, and FeCr are found to have higher activity on more than one QV doping sites. FeCr show higher activity on QV2 and QV3 sites, whereas both QV1 and QV2 doping sites are found to be active for FeV, FeMn, and FeCo dimers. On the other hand, FeSc, FeTi, and FeNi dimers show specific selectivity of the doping sites for the CO2R activity. While FeSc and FeNi DACs are found to have greater activity with QV1 geometry only, FeTi show greater TOF with the QV2 doping site. For FeFe and FeCu, all the QV doping sites show lower activity than the Au (211) surface. With FeZn, we have identified the QV1 site with 4N atom coordination as the most active for CO2 to CO reduction. However, the CO coverage plot in Figure S4 suggests higher binding affinity for CO*; thereby, the catalytic site would be poisoned by CO at this reducing potential.
Focusing on the di-atom catalysts that have been synthetically reported in recent studies, we find three possible candidates as FeCo, FeMn, and FeNi with catalytic activity comparable to the Au (211) surface.24,25,27,28 While experimental studies have also reported FeNi to be a good catalyst for CO2 to CO production,24,28 for both FeMn and FeCo, experimental characterization of their catalytic activity for CO2R have not been performed. TOF of FeMn is found to be higher than the recently identified FeNi dimer for CO2R. The other two di-atom candidates that have been recently reported in experiments as a good catalyst for CO2 to CO reduction are FeFe and FeCu.30,31 In our screening study, we have also identified these two dimer candidates as viable; however, the activity is found to be lower than the Au (211) surface. The activity volcano plot for DACs in Figure 3b shows that the activity for both FeFe and FeCu is comparable to the Ag(211) surface. For FeFe, we have obtained both the QV1 site with 6N and the QV2 site with 4N atom coordination as the most active catalytic site. However, the QV1 (6N) site has higher binding affinity for H+ ions (Figure S8 in the SI). Therefore, we expect that the QV2 site with 4N should be the active site for FeFe. On the other hand, for FeCu, we have found that the QV1 site with 4N atom coordination has the highest activity among the other doping sites.
Along with the rate of CO production, probing the rate of the competing hydrogen evolution reaction (HER) at the reduction potentials is essential for determining the catalytic efficiency of the catalysts. Therefore, to understand the selectivity of the best performing single and di-atom catalysts for CO vs H2 evolution,38 we classify the binding energies of CO* and H* intermediates on these catalysts with (111) TM surfaces as shown in the binding-energy plot in Figure S8 of the Supporting Information. We found that the MNCs with higher activity for CO2R screened in this study show lower selectivity for H2 evolution. The binding-energy plot shows corresponding similarity with Cu, Ag, and Au surfaces. Following a similar classification for the FeMNCs identified with higher TOF for CO, we find two metal dimers FeV (QV1) and FeTi (QV2) with 2N coordination to show higher selectivity for HER. The other active DACs show greater selectivity for CO production.
We also consider the pathway to the formation of formic acid for the 11 best di-atom catalysts identified with the comparable CO2 to CO reduction activity to the Au (211) surface in the activity volcano plot (Figure 3). The CO2 to CO reduction mechanism has been studied in detail; however, there is no clear indication for the initial catalytic intermediate for the electroreduction of CO2 to formate or formic acid. In recent studies, the selectivity of formic acid or CO on the transition metal surfaces and p-block elements has been speculated to depend upon the binding mode of CO2.39−42 The CO formation should involve the binding of CO2 through the C atom, allowing the protonation on one of the O-atoms to form the COOH* intermediate. On the other hand, for formate formation, the CO2 intermediate should bind through the O atoms for allowing the hydrogenation of the C atom to form the first protonated intermediate *OCHO. Therefore, to find out the possibility for the formation of *OCHO intermediate on the di-atom catalysts, we optimized different O binding motifs of CO2 (*OCO) on the metal dopant sites.
We find that at zero surface charge, CO2 does not prefer to bind to the metal centers through the O atoms. However, with an applied negative surface charge, a bidentate binding motif of CO2 through the two O atoms is found to be stable for some di-atom catalysts. We have not found a minimum energy structure for *OCO on FeNi (QV1), FeMn (QV2), and FeCo (QV2) with 6N atom coordination, FeTi (QV2) with 2N atom coordination, and FeCr (QV3) with 4N atom coordination. Meanwhile, all five remaining di-atom candidates able to stabilize *OCO still prefer the CO2* motif at potential of −0.8 V vs SHE (cf. Table S2 in the SI). Therefore, at the reducing potential of −0.8 V vs SHE, the selectivity toward formation of CO dominates.
The other CO2R reaction pathways leading to the formation of CH4 or CH3OH have not been considered in this study. It is because the potential range we have considered to screen the single and di-atom catalysts shows small CO* coverage for the 11 most active candidates. When coverages of adsorbed CO are very small, the production of CH4 or CH3OH would be less likely.
It is important to note that the systematic errors in describing the O–C–O backbone structure using DFT functionals results in a constant energy shift in the gas phase reference values.43−45 These uncertainties yield unrealistic errors in the estimation of adsorbate binding energy calculations and approximation of the kinetic barriers, particularly for CO2 adsorption. It is observed that due to these systematic errors, an uncertainty of 5 to 7 orders of magnitude is obtained in the computed absolute TOF values. Therefore, in this study to reduce these empirical errors, the binding energies of the reaction intermediates on single and di-atom catalysts are corrected with single-point energy calculations at hybrid HSE06 functional using the relaxed geometries obtained with the RPBE functional. Furthermore, instead of using the absolute TOF values, we have focused on relative TOF values with respect to Au (211) surfaces for screening the best candidates of the single and di-atom catalysts for CO2 reduction. The single and di-atom catalysts with TOFs comparable or greater than Au (211) surface have been denoted as the active catalysts. We also note that the solvation effects are critical for accurate evaluation of the catalytic activity.46 By referring to the relative activity with respect to the Au (211) surface, we also effectively remove the systematic errors associated with the implicit solvation scheme.
Conclusions
We study the relative stability and catalytic activity of single and di-atom catalysts denoted as MNC and FeMNC with M corresponding to the 3d transition metal from Sc to Ni doped on defected graphene support for electrochemical CO2R. We found that two C-atom vacancies are the most stable doping site for MNCs. On the other hand, for di-atoms, the formation energy calculations show that the quad-atom carbon vacancies denoted as QV1, QV2, and QV3 are the favorable doping sites.
We used a potential dependent micro-kinetic model based on CO2* and COOH* binding energies as the activity descriptors to understand the catalytic activity and rate of CO production on these catalysts. Compared to transition metal (211) surfaces, both MNCs and FeMNCs show greater extent of scattering for ΔGCO2* and ΔGCOOH*, bound to the variations in their binding geometry and larger adsorbate dipole–field interactions. The early TM MNCs prefer a bidentate binding motif for CO2* and COOH* through C and O atoms, whereas the late TM MNCs show a monodentate motif through the C atom. We have screened four MNCs as CrNC, MnNC, FeNC, and CoNC with higher TOF for CO2R under lower overpotential for CO2R.
Among the three quad-atom vacancy sites, QV1 allows a two-site bidentate motif, where both the metal atoms bind to CO2* and COOH* intermediates. The metal dopants on QV2 and QV3 sites show similar binding motifs as the corresponding MNCs, where only one of the metal atoms act as the active site for the reaction. The unified activity on QV sites is found to depend on the site geometry of catalysts and the surrounding coordinating N-atoms. Analyzing the CO2R activity, we have identified 11 stable FeM dimers having TOF for CO production comparable to the Au (211) surface at −0.8 V vs SHE and buffer pH of 6.8. Most of these dimers show lower selectivity for HER. Among these DACs, both FeMn and FeNi dimers have been synthesized in the recent studies.
The single or di-atom catalysts lead to larger dipoles for CO2* because of their localized d-states on the metal dopants.21 The larger dipole enhances the stabilization of the adsorbed CO2* upon interacting with the interfacial electric field under an applied potential and electrolyte. This electrostatic stabilization has been identified as one defining source of the deviation from the transition metal scaling line at working potentials and tunable field–dipole interactions in catalyst design. In contrast to the SACs, the di-atom metal dopants create a larger variety of binding sites for CO2*, resulting in a wider spread of the created surface dipoles. We interpret this new degree of freedom as the source of the increased scattering in the activity plot shown in Figure 3.
In addition to the site geometries, the electrocatalytic activity could be effectively tuned by the identity of the alkali metal ions in the electrolyte. Several theoretical and experimental studies have elucidated the effect of the electrolyte cation size on the intrinsic activity of metal catalysts for CO2 electro-reduction.3,47 We expect that careful evaluation of the cation effects on the metal doped graphene catalysts will open new possibilities to tune the catalytic activity of SACs and DACs for renewable energy generation technologies.
Acknowledgments
The research leading to these results has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement no. 851441. We also acknowledge the VILLUM Centre for the Science of Sustainable Fuels and Chemicals (#9455) from VILLUM FONDEN. We are grateful to Prof. Jens K Nørskov for valuable discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.1c05750.
Computational detail for the simulation (Section S-I); (Figure S1) structural possibilities of SACs with various N atom coordination; formation energy calculations and relative stability of the SACs and DACs (Figures S2 and S3, Section S-II); Figures S4 and S5 CO coverage plot and adsorbate dipole moment plots for best performing SACs and DACs; (Figures S6 and S7, Section S-III) potential dependent activity rate map for SACs and DACs; CO2R vs HER selectivity (Figure S8); (Section S-IV) formation free energies of the best di-atom candidates; comparison of *CO2 and *OCO binding motifs on the best di-atom catalysts (Section S-V); (Figure S9, Section S-VI) comparison of the binding energies and magnetic moment values for FeNi DACs calculated as RPBE and HSE06 functional with and without structural relaxation; and free energy diagrams for the CO2 to CO reduction pathway for the best di-atom candidates (Figure S10) (PDF)
The authors declare no competing financial interest.
Notes
The structural and calculation details and for all the stable di-atom candidates identified in this study under the following weblink: https://github.com/CatTheoryDTU/SAC_DAC_screening
Supplementary Material
References
- Seh Z. W.; Kibsgaard J.; Dickens C. F.; Chorkendorff I.; Nørskov J. K.; Jaramillo T. F. Combining Theory and Experiment in Electrocatalysis: Insights into Materials Design. Science 2017, 355, eaad4998. 10.1126/science.aad4998. [DOI] [PubMed] [Google Scholar]
- Jouny M.; Luc W.; Jiao F. General Techno-Economic Analysis of CO2 Electrolysis Systems. Ind. Eng. Chem. Res. 2018, 57, 2165–2177. 10.1021/acs.iecr.7b03514. [DOI] [Google Scholar]
- Nitopi S.; Bertheussen E.; Scott S. B.; Liu X.; Engstfeld A. K.; Horch S.; Seger B.; Stephens I. E.; Chan K.; Hahn C.; Nørskov J. K.; Jaramillo T. F.; Chorkendorff I. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119, 7610–7672. 10.1021/acs.chemrev.8b00705. [DOI] [PubMed] [Google Scholar]
- Hansen H. A.; Varley J. B.; Peterson A. A.; Nørskov J. K. Understanding Trends in the Electrocatalytic Activity of Metals and Enzymes for CO2 Reduction to CO. J. Phys. Chem. Lett. 2013, 4, 388–392. 10.1021/jz3021155. [DOI] [PubMed] [Google Scholar]
- Hori Y.Electrochemical CO2 Reduction on Metal Electrodes. In Modern Aspects of Electrochemistry; Vayenas C. G., White R. E., Gamboa-Aldeco M. E., Eds.; Springer New York: New York, NY, 2008; 89–189. [Google Scholar]
- Bonaccorso F.; Colombo L.; Yu G.; Stoller M.; Tozzini V.; Ferrari A. C.; Ruoff R. S.; Pellegrini V. Graphene, Related Two-Dimensional Crystals, and Hybrid Systems for Energy Conversion and Storage. Science 2015, 347, 1246501 10.1126/science.1246501. [DOI] [PubMed] [Google Scholar]
- Hinnemann B.; Moses P. G.; Bonde J.; Jørgensen K. P.; Nielsen J. H.; Horch S.; Chorkendorff I.; Nørskov J. K. Biomimetic Hydrogen Evolution: MoS2 Nanoparticles as Catalyst for Hydrogen Evolution. J. Am. Chem. Soc. 2005, 127, 5308–5309. 10.1021/ja0504690. [DOI] [PubMed] [Google Scholar]
- Karmodak N.; Andreussi O. Catalytic Activity and Stability of Two-Dimensional Materials for the Hydrogen Evolution Reaction. ACS Energy Lett. 2020, 5, 885–891. 10.1021/acsenergylett.9b02689. [DOI] [Google Scholar]
- Karmodak N.; Bursi L.; Andreussi O. Oxygen Evolution and Reduction on Two-Dimensional Transition Metal Dichalcogenides. J. Phys. Chem. Lett. 2022, 58–65. 10.1021/acs.jpclett.1c03431. [DOI] [PubMed] [Google Scholar]
- Asadi M.; Kumar B.; Behranginia A.; Rosen B. A.; Baskin A.; Repnin N.; Pisasale D.; Phillips P.; Zhu W.; Haasch R.; Klie R. F.; Král P.; Abiade J.; Salehi-Khojin A. Robust Carbon Dioxide Reduction on Molybdenum Disulphide Edges. Nat. Commun. 2014, 5, 1–8. 10.1038/ncomms5470. [DOI] [PubMed] [Google Scholar]
- He Y.; Liu S.; Priest C.; Shi Q.; Wu G. Atomically Dispersed Metal–Nitrogen–Carbon Catalysts for Fuel Cells: Advances in Catalyst Design, Electrode Performance, and Durability Improvement. Chem. Soc. Rev. 2020, 49, 3484–3524. 10.1039/C9CS00903E. [DOI] [PubMed] [Google Scholar]
- Li H.; Tsai C.; Koh A. L.; Cai L.; Contryman A. W.; Fragapane A. H.; Zhao J.; Han H. S.; Manoharan H. C.; Abild-Pedersen F.; Nørskov J. K.; Zheng X. Activating and Optimizing MoS2 Basal Planes for Hydrogen Evolution through the Formation of Strained Sulphur Vacancies. Nature Mater. 2016, 15, 48–53. 10.1038/nmat4465. [DOI] [PubMed] [Google Scholar]
- Karmodak N.; Andreussi O. Oxygen Evolution on MoS2 Edges: Activation through Surface Oxidation. J. Phys. Chem. C 2021, 125, 10397–10405. 10.1021/acs.jpcc.1c02210. [DOI] [Google Scholar]
- Wang C.; Zhu C.; Zhang M.; Geng Y.; Su Z. Copper Dimer Anchored in G-CN Monolayer as an Efficient Electrocatalyst for CO2 Reduction Reaction: A Computational Study. Adv. Theory Simul. 2020, 3, 2000218. 10.1002/adts.202000218. [DOI] [Google Scholar]
- Ouyang Y.; Shi L.; Bai X.; Li Q.; Wang J. Breaking Scaling Relations for Efficient CO2 Electrochemical Reduction through Dual-Atom Catalysts. Chem. Sci. 2020, 11, 1807–1813. 10.1039/C9SC05236D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K.; Tsai C.; Hansen H. A.; Nørskov J. K. Molybdenum Sulfides and Selenides as Possible Electrocatalysts for CO2 Reduction. ChemCatChem 2014, 6, 1899–1905. 10.1002/cctc.201402128. [DOI] [Google Scholar]
- Siahrostami S.; Jiang K.; Karamad M.; Chan K.; Wang H.; Nørskov J. Theoretical Investigations into Defected Graphene for Electrochemical Reduction of CO2. ACS Sustainable Chem. Eng. 2017, 5, 11080–11085. 10.1021/acssuschemeng.7b03031. [DOI] [Google Scholar]
- Varela A. S.; Kroschel M.; Leonard N. D.; Ju W.; Steinberg J.; Bagger A.; Rossmeisl J.; Strasser P. PH Effects on the Selectivity of the Electrocatalytic CO2 Reduction on Graphene-Embedded Fe–N–C Motifs: Bridging Concepts between Molecular Homogeneous and Solid-State Heterogeneous Catalysis. ACS Energy Lett. 2018, 3, 812–817. 10.1021/acsenergylett.8b00273. [DOI] [Google Scholar]
- Prslja P.; López N. Stability and Redispersion of Ni Nanoparticles Supported on N-Doped Carbons for the CO2 Electrochemical Reduction. ACS Catal. 2020, 11, 88–94. [Google Scholar]
- Vijay S.; Gauthier J. A.; Heenen H. H.; Bukas V. J.; Kristoffersen H. H.; Chan K. Dipole-Field Interactions Determine the CO2 Reduction Activity of 2D Fe–N–C Single-Atom Catalysts. ACS Catal. 2020, 10, 7826–7835. 10.1021/acscatal.0c01375. [DOI] [Google Scholar]
- Vijay S.; Ju W.; Brückner S.; Tsang S.-C.; Strasser P.; Chan K. Unified Mechanistic Understanding of CO2 Reduction to CO on Transition Metal and Single Atom Catalysts. Nat. Catal. 2021, 4, 1024–1031. 10.1038/s41929-021-00705-y. [DOI] [Google Scholar]
- Kim D.; Xie C.; Becknell N.; Yu Y.; Karamad M.; Chan K.; Crumlin E. J.; Nørskov J. K.; Yang P. Electrochemical Activation of CO2 through Atomic Ordering Transformations of AuCu Nanoparticles. J. Am. Chem. Soc. 2017, 139, 8329–8336. 10.1021/jacs.7b03516. [DOI] [PubMed] [Google Scholar]
- Ma S.; Sadakiyo M.; Heima M.; Luo R.; Haasch R. T.; Gold J. I.; Yamauchi M.; Kenis P. J. A. Electroreduction of Carbon Dioxide to Hydrocarbons Using Bimetallic Cu–Pd Catalysts with Different Mixing Patterns. J. Am. Chem. Soc. 2017, 139, 47–50. 10.1021/jacs.6b10740. [DOI] [PubMed] [Google Scholar]
- Ren W.; Tan X.; Yang W.; Jia C.; Xu S.; Wang K.; Smith S. C.; Zhao C. Isolated Diatomic Ni-Fe Metal–Nitrogen Sites for Synergistic Electroreduction of CO2. Angew. Chem., Int. Ed. 2019, 58, 6972–6976. 10.1002/anie.201901575. [DOI] [PubMed] [Google Scholar]
- Wang J.; Huang Z.; Liu W.; Chang C.; Tang H.; Li Z.; Chen W.; Jia C.; Yao T.; Wei S.; Wu Y.; Li Y. Design of N-Coordinated Dual-Metal Sites: A Stable and Active Pt-Free Catalyst for Acidic Oxygen Reduction Reaction. J. Am. Chem. Soc. 2017, 139, 17281–17284. 10.1021/jacs.7b10385. [DOI] [PubMed] [Google Scholar]
- Wei Y.-S.; Sun L.; Wang M.; Hong J.; Zou L.; Liu H.; Wang Y.; Zhang M.; Liu Z.; Li Y.; Horike S.; Suenaga K.; Xu Q. Fabricating Dual-Atom Iron Catalysts for Efficient Oxygen Evolution Reaction: A Heteroatom Modulator Approach. Angew. Chem., Int. Ed. 2020, 59, 16013–16022. 10.1002/anie.202007221. [DOI] [PubMed] [Google Scholar]
- Yang G.; Zhu J.; Yuan P.; Hu Y.; Qu G.; Lu B.-A.; Xue X.; Yin H.; Cheng W.; Cheng J.; Xu W.; Li J.; Hu J.; Mu S.; Zhang J.-N. Regulating Fe-Spin State by Atomically Dispersed Mn-N in Fe-NC Catalysts with High Oxygen Reduction Activity. Nat. Commun. 2021, 12, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Z.; Gan L. Y.; Yang H. B.; Su X.; Gao J.; Liu W.; Matsumoto H.; Gong J.; Zhang J.; Cai W.; Zhang Z.; Yan Y.; Liu B.; Chen P. Orbital Coupling of Hetero-Diatomic Nickel-Iron Site for Bifunctional Electrocatalysis of CO2 Reduction and Oxygen Evolution. Nat. Commun. 2021, 12, 1–11. 10.1038/s41467-021-24052-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X.; Ling X.; Yu D.; Xie D.; Li L.; Peng S.; Zhong C.; Zhao N.; Deng Y.; Hu W. Atomically Dispersed Binary Co-Ni Sites in Nitrogen-Doped Hollow Carbon Nanocubes for Reversible Oxygen Reduction and Evolution. Adv. Mater. 2019, 31, 1905622. 10.1002/adma.201905622. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Park B. J.; Paidi V. K.; Huang R.; Lee Y.; Noh K.-J.; Lee K.-S.; Han J. W. Precisely Constructing Orbital Coupling-Modulated Dual-Atom Fe Pair Sites for Synergistic CO2 Electroreduction. ACS Energy Lett. 2022, 640–649. 10.1021/acsenergylett.1c02446. [DOI] [Google Scholar]
- Feng M.; Wu X.; Cheng H.; Fan Z.; Li X.; Cui F.; Fan S.; Dai Y.; Lei G.; He G. Well-Defined Fe–Cu Diatomic Sites for Efficient Catalysis of CO2 Electroreduction. J. Mater. Chem. A 2021, 9, 23817–23827. 10.1039/D1TA02833B. [DOI] [Google Scholar]
- Li F.; Ding X.-B.; Cao Q.-C.; Qin Y.-H.; Wang C. A ZIF-Derived Hierarchically Porous Fe–Zn–N–C Catalyst Synthesized via a Two-Stage Pyrolysis for the Highly Efficient Oxygen Reduction Reaction in Both Acidic and Alkaline Media. Chem. Commun. 2019, 55, 13979–13982. 10.1039/C9CC07489A. [DOI] [PubMed] [Google Scholar]
- Hammer B.; Hansen L. B.; Nørskov J. K. Improved Adsorption Energetics within Density-Functional Theory Using Revised Perdew-Burke-Ernzerhof Functionals. Phys. Rev. B 1999, 59, 7413. 10.1103/PhysRevB.59.7413. [DOI] [Google Scholar]
- Heyd J.; Scuseria G. E.; Ernzerhof M. Hybrid Functionals Based on a Screened Coulomb Potential. J. Chem. Phys. 2003, 118, 8207–8215. 10.1063/1.1564060. [DOI] [Google Scholar]
- Ge H. J. S.; Ernzerhof M. Erratum:“Hybrid Functionals Based on a Screened Coulomb Potential”[J. Chem. Phys. 118, 8207 (2003)]. J Chem Phys 2006, 124, 219906. [Google Scholar]
- Gauthier J. A.; Dickens C. F.; Heenen H. H.; Vijay S.; Ringe S.; Chan K. Unified Approach to Implicit and Explicit Solvent Simulations of Electrochemical Reaction Energetics. J. Chem. Theory Comput. 2019, 15, 6895–6906. 10.1021/acs.jctc.9b00717. [DOI] [PubMed] [Google Scholar]
- Pedersen A.; Barrio J.; Li A.; Jervis R.; Brett D. J. L.; Titirici M. M.; Stephens I. E. L. Dual- Metal Atom Electrocatalysts: Theory, Synthesis, Characterization, and Applications. Adv. Energy Mater. 2022, 12, 2102715. 10.1002/aenm.202102715. [DOI] [Google Scholar]
- Bagger A.; Ju W.; Varela A. S.; Strasser P.; Rossmeisl J. Electrochemical CO2 Reduction: A Classification Problem. ChemPhysChem 2017, 18, 3266–3273. 10.1002/cphc.201700736. [DOI] [PubMed] [Google Scholar]
- Kortlever R.; Shen J.; Schouten K. J. P.; Calle-Vallejo F.; Koper M. T. M. Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. 10.1021/acs.jpclett.5b01559. [DOI] [PubMed] [Google Scholar]
- Chaplin R. P. S.; Wragg A. A. Effects of Process Conditions and Electrode Material on Reaction Pathways for Carbon Dioxide Electroreduction with Particular Reference to Formate Formation. J. Appl. Electrochem. 2003, 33, 1107–1123. 10.1023/B:JACH.0000004018.57792.b8. [DOI] [Google Scholar]
- Feaster J. T.; Shi C.; Cave E. R.; Hatsukade T.; Abram D. N.; Kuhl K. P.; Hahn C.; Nørskov J. K.; Jaramillo T. F. Understanding Selectivity for the Electrochemical Reduction of Carbon Dioxide to Formic Acid and Carbon Monoxide on Metal Electrodes. ACS Catal. 2017, 7, 4822–4827. 10.1021/acscatal.7b00687. [DOI] [Google Scholar]
- Hatsukade T.; Kuhl K. P.; Cave E. R.; Abram D. N.; Jaramillo T. F. Insights into the Electrocatalytic Reduction of CO2 on Metallic Silver Surfaces. Phys. Chem. Chem. Phys. 2014, 16, 13814–13819. 10.1039/C4CP00692E. [DOI] [PubMed] [Google Scholar]
- Christensen R.; Hansen H. A.; Vegge T. Identifying Systematic DFT Errors in Catalytic Reactions. Catal. Sci. Technol. 2015, 5, 4946–4949. 10.1039/C5CY01332A. [DOI] [Google Scholar]
- Studt F.; Abild-Pedersen F.; Varley J. B.; Nørskov J. K. CO and CO2 Hydrogenation to Methanol Calculated Using the BEEF-VdW Functional. Catal. Lett. 2013, 143, 71–73. 10.1007/s10562-012-0947-5. [DOI] [Google Scholar]
- Peterson A. A.; Abild-Pedersen F.; Studt F.; Rossmeisl J.; Nørskov J. K. How Copper Catalyzes the Electroreduction of Carbon Dioxide into Hydrocarbon Fuels. Energy Environ. Sci. 2010, 3, 1311–1315. 10.1039/c0ee00071j. [DOI] [Google Scholar]
- Gray C. M.; Saravanan K.; Wang G.; Keith J. A. Quantifying Solvation Energies at Solid/Liquid Interfaces Using Continuum Solvation Methods. Mol. Simul. 2017, 43, 420–427. 10.1080/08927022.2016.1273525. [DOI] [Google Scholar]
- Ringe S.; Clark E. L.; Resasco J.; Walton A.; Seger B.; Bell A. T.; Chan K. Understanding Cation Effects in Electrochemical CO2 Reduction. Energy Environ. Sci. 2019, 12, 3001–3014. 10.1039/C9EE01341E. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.