

Abstract
Naturally-derived cell membranes have shown great promise in functionalizing nanoparticles to enhance biointerfacing functions for drug delivery applications. However, its potential for functionalizing macroporous scaffolds to enhance tissue regeneration in vivo remains unexplored. Engineering scaffolds with immunomodulatory functions represents an exciting strategy for tissue regeneration but is largely limited to soft tissues. Critical sized bone defects cannot heal on its own, and the role of adaptive immune cells in scaffold-mediated healing of cranial bone defects remain largely unknown. Here we report mensenchymal stem cell membrane (MSCM)-coated microribbon (μRB) scaffolds for treating critical size cranial bone defects via targeting immunomodulation. Confocal imaging and proteomic analyses were used to confirm successful coating and characterize the compositions of cell membrane coating. We demonstrate MSCM coating promotes Mφ polarization towards regenerative phenotype, induces CD8+ T cell apoptosis, and enhances regulatory T cell differentiation in vitro and in vivo. MSCM primed with pro-inflammatory cytokines enhances regenerative immune response and promotes MSC osteogenesis. When combined with a low dosage of BMP-2, primed MSCM coating further accelerates bone regeneration and suppresses inflammation. These results establish cell membrane-coated microribbon scaffolds as a promising strategy for treating critical size bone defects via immunomodulation. The platform may be broadly used with different cell membranes and scaffolds to enhance regeneration of multiple tissue types.
Keywords: Immunomodulation, bone regeneration, biomaterials, critical-sized bone defect, cell membrane coating
Graphical Abstract
Cell membrane coating represents a biomimetic strategy for modifying biomaterials surface. Coating macroporous microribbon scaffolds with mesenchymal stem cell membrane (MSCM) enhances cranial bone healing via immunomodulation. Primed MSCM-coating contains ligands that promote Mφ polarization towards M2, induces Treg differentiation and TCD8+ apoptosis, and enhances MSC osteogenesis. MSCM coating synergizes with BMP-2 to further accelerate bone regeneration while suppressing inflammation.
1. Introduction
Previous research on harnessing biomaterials to aid in repairing critical size bone defects has largely focused on promoting stem cell osteogenesis and vasculature [1, 2]. Immune cells are early responders during bone injury, which impact inflammation, stem cell recruitment, differentiation, and tissue repair[3]. Macrophages (Mφ) with pro-inflammatory M1-like phenotype respond early to bone injury, and are the dominant players in acute inflammation phase.[4] While pro-inflammatory, M1-like Mφ is essential for progenitor cell recruitment [5], prolonged presence of M1 Mφ can lead to chronic inflammation and stem cell apoptosis[6]. As such, timely transition from pro-inflammatory M1 Mφ to pro-regenerative M2-like Mφ is a critical step for normal bone healing.[7] Furthermore, majority of previous studies in bone injury only focused on Mφ,[8] yet the role of adaptive T cells in the repair of critical size bone defects remains unclear. Recently, adaptive immune cell have been shown to play an important role in bone fracture healing, with increased CD8+ T cell and decreased regulatory T cells (Tregs) in peripheral blood of patients with delayed fracture healing.[9, 10] However, it remains unclear how different subpopulation of T cells affect bone healing within a critical-sized bone defect. Engineering biomaterials with immunomodulatory function that target both innate and adaptive immune cells represent a great opportunity to enhance critical size bone defect regeneration, which remains largely unexplored.
Recently, cell membrane-coated nanoparticles have emerged as promising biomimetic therapeutics for drug delivery applications. [11, 12] Cell membrane coating is advantageous due to its ability to mimic sophisticated cellular function by directly borrowing complex cues contained in native cell membrane. For example, red blood cell membrane-coated nanoparticles can evade immune clearance and exhibited prolonged circulating time for drug delivery.[13] Cancer cell membrane-coated nanoparticles have shown promise for cancer vaccine applications by presenting complex antigens derived from cancer cell membrane.[11] Emerging studies have shown the promise of coating nanofibrous scaffolds with cell membrane for promoting pancreatic beta cell function[14] or keratinocyte proliferation[15]. However, these studies were limited to nanoporous scaffolds and in vitro cell studies, and potential of cell membrane coating for macroporous scaffolds to enhance bone regeneration has not been investigated before. Macroporosity has been shown to be critical for bone regeneration in vivo[16]. Combining cell membrane coating with macroporous scaffolds offers a unique opportunity for innovation for promoting bone regeneration in vivo.
In this study, we report a mesenchymal stem cell (MSC) membrane (MSCM)-coated macroporous microribbon scaffold for repairing critical-sized cranial bone defect through targeting both innate and adaptive immune systems. MSCs were chosen due to their well-known immunomodulatory functions, [17] and MSC cell membrane contains immunomodulatory ligands such as programmed death-ligand 1 (PD-L1) and tumor necrosis factor ligand superfamily member 6 (TNFL6). [18] Previous research has shown the promise of MSC-derived exosomes for enhancing bone regeneration by targeting stem cell differentiation or angiogenesis.[19] Another previous study has used MSCM-coated nanoparticles for target drug delivery to inflammatory cartilage.[20] However, the potential of using MSC-derived cell membrane for promoting bone tissue regeneration has never been explored. Gelatin microribbon (μRB) scaffolds were chosen because its demonstrated efficacy in supporting stem cell-based bone regeneration in vivo.[21] Its inherent macroporosity facilitate rapid cell infiltration without the need for degradation, offering additional advantage as a scaffold for immunomodulation. Previous studies have shown hydrogels containing macroporosity enhance cell infiltration and tissue regeneration in both a skin wound healing model and the cranial defect model.[21, 22] Our previous studies showed bone regeneration in critical size cranial bone defects decreased substantially when switching from immunocompromised mouse model to immunocompetent mouse model [21, 23], further highlighting the need for immunomodulation strategy in repairing critical size bone defects. We hypothesize that MSCM-coated μRB scaffolds would enhance critical-sized cranial bone regeneration through promoting regenerative phenotype of both Mφ and T cells. Given that interferon (IFN)-γ primed MSCs exhibit enhanced immunomodulatory function [24, 25], cell membrane from both naïve and IFN-γ primed MSCs were tested. Bone morphogenetic proteins-2 (BMP-2) has been widely used for boosting bone regeneration, but often requires high concentration (i.e. 1000 ng/defect), which leads to side effects of uncontrolled bone formation and severe inflammation[26, 27]. To address this limitation, we further assess the potential of using MSCM-coated scaffolds to support bone regeneration in vivo using reduced dosage of BMP-2 (200 ng per defect). Our results showed successful cell membrane coating on microribbon scaffolds. We demonstrate MSCM coating, especially primed MSCM, induced regenerative immune response of both innate and adaptive immune cells in vitro and in vivo. Furthermore, pMSCM-coating synergized with a low dose of BMP-2 to induce robust endogenous bone regeneration in a critical sized mouse cranial defect model.
Results and Discussion
We first established a method that achieve efficient cell membrane coating on the macroporous μRB scaffolds by electrostatic interaction (Figure 1a). In brief, μRBs were covalently modified with positively charged poly-lysine, which allows binding to negatively charged MSCM through electrostatic interaction. Dynamic light scattering (DLS) showed MSCM vesicles exhibit an average diameter of 264 nm and zeta potential of −29.8 mV (Figure S2a and S2b, Supporting Information). Transmission electron microscopy (TEM) confirmed MSCM forming nanosize vesicle-like morphologies after isolation (Figure S2c, Supporting Information). Given MSCM coating blocked methacrylate groups on μRB surface needed for photo-crosslinking, the MSCM-coated μRBs were mixed with uncoated μRBs at 1:1 ratio to allow sufficient crosslinking. Enhanced MSCM retention in the μRB scaffolds were observed with increasing poly-lysine concentration (Figure 1b). After 2-week incubation in vitro, the retention rate of MSCM was stabilized around 82% (0.4% poly-lysine) and 71% (0.1% poly-lysine), while minimal MSCM remained in the scaffolds in 0.01% poly-lysine group (Figure 1b). μRB scaffolds with best MSCM retention rate (0.4% poly-lysine) were chosen for the following experiments. To evaluate the distribution of MSCM on μRBs, MSCM were fluorescently labelled. Uniform MSCM coating on the μRB surface was observed before crosslinking into scaffolds (Figure S2d, Supporting Information). SEM also confirmed the successful coating of MSCM on μRBs, with the scaffold showing a spongy surface morphology after MSCM coating (Figure S2e, Supporting Information). After mixing with uncoated μRBs (1:1 ratio) and crosslinking, the MSCM (red) coating remained stable on μRBs and can be seen mixed with uncoated μRBs (green) throughout the scaffolds (Figure 1c).
Figure 1. Characterization of MSCM-coated μRB scaffolds.
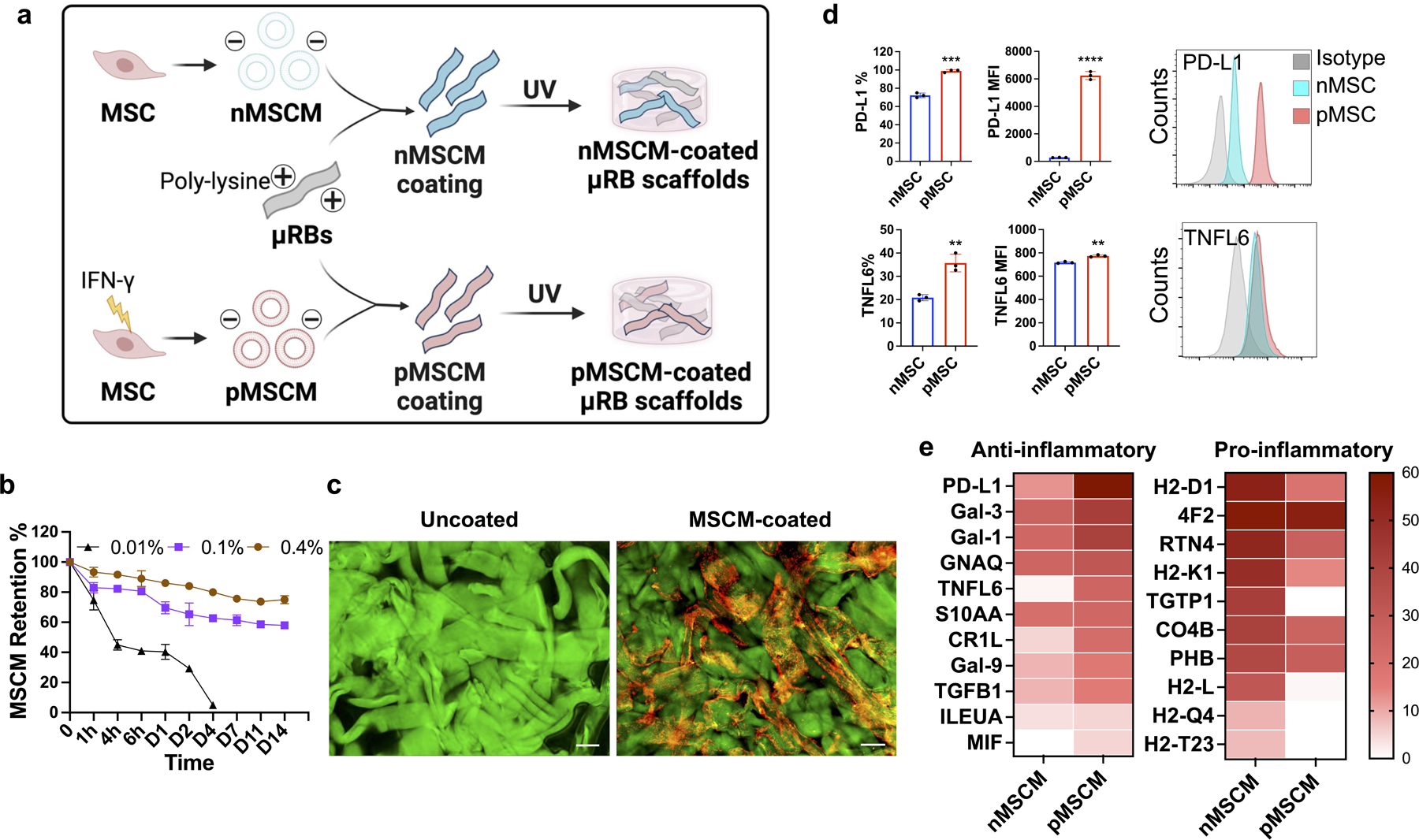
(a) Coating μRB scaffolds with mesenchymal stem cell membrane derived from naïve MSCs (nMSCM) or primed MSCs (pMSCM) treated with inflammatory cytokines. (b) Optimizing the retention of MSCM coating on μRB scaffolds by tuning poly-lysine concentration (n=4/group). (c) Visualizing MSCM coating using confocal imaging. Uncoated μRBs: green, coated μRBs: red; MSCM-coated scaffold is formed using 1:1 mixture of MSCM-coated μRBs and uncoated μRBs to allow efficient photocrosslinking. Scale bar: 100 μm. (d) Flow cytometry confirmed the presence of PD-L1 and TNFL6 on the surface of intact MSCs (n=3/group). (e) Proteomics characterization of naïve or primed MSCM showed abundant ligands related to anti-inflammatory and pro-inflammatory signaling. Data are represented as mean ± S.D. **P < 0.01, ***P < 0.001, ****P < 0.0001.
Next, we characterized the protein contents of MSCM, with a focus on analyzing ligands with immunomodulatory functions. MSCM from both naïve MSCs and IFN-γ primed MSCs were collected and compared. PD-L1 and TNFL6 are two well-known immunomodulatory ligands on MSCs that are known to inhibit adaptive T cell immune response through cell-cell contact.[18, 28] We first confirmed the presence of PD-L1 and TNFL6 on intact MSCs using flow cytometry (Figure 1d). Consistent with our hypothesis, IFN-γ priming significantly increased the expression level of PD-L1 and TNFL6, compared to naïve MSCs. Proteomics was also performed to characterize protein content of naive MSCM (nMSCM)and primed MSCM (pMSCM) in a high-throughput and comprehensive manner. Consistent with flow cytometry results, both PD-L1 and TNFL6 were detected in MSCM, with primed MSCM exhibiting higher expression level than naïve MSCM (Figure 1e). In addition to PD-L1 and TNFL6, other immunomodulatory molecules were detected including galectin (Gal)-1/Gal-3/Gal-9 [29, 30], macrophage migration inhibitory factor (MIF) [31] and transforming growth factor beta-1 (TGFB1).[32] These signals are known for inducing M2 Mφ polarization, Treg differentiation and cytotoxic T cell apoptosis. Similar to PD-L1 and TNFL6, the expression level of the observed anti-inflammatory molecules was also upregulated in primed MSCM compared to naive MSCM. Pro-inflammatory components were also found in MSCM such as histocompatibility molecules, and were downregulated in primed MSCM. In addition to immunomodulation, other protein motifs with biological function of adhesion, such as angiogenesis and osteogenesis, were also detected, but exhibited similar expression level between naive MSCM and primed MSCM (Figure S2f, Supporting Information). The above results indicate both naive MSCM and primed MSCM contain protein compositions that could modulate immune responses on Mφ and T cells. Given IFN-γ priming increased the expression level of anti-inflammatory molecules, we further hypothesize that primed MSCM will induce more regenerative immune response compared to naive MSCM.
To assess the function of MSCM-coating on innate immune cells, Mφ polarization and their crosstalk with stem cells were first evaluated in vitro. To mimic the acute inflammation after injury, Mφ were primed to M1 phenotype with IFN-γ/LPS and then seeded on μRB scaffolds with or without MSCM coating (Figure 2a). Compared to 2D tissue culture plate (TCP), all 3D μRB scaffold groups significantly decreased M1 marker gene expression, including CD86, iNOS and tumor necrosis factor (TNF)-α (Figure 2b), and TNF-α secretion (Figure 2d). These results suggest the benefit of 3D μRB scaffolds over 2D culture in quenching pro-inflammatory cytokine production. A previous study has shown growing macrophages on soft substrates (~kPa) reduced M1 inflammatory phenotype compared to growing cells on glass or stiff substrates (~GPa).[33] The stiffness of μRBs used in our study is ~28 kPa,[34] which is several orders of magnitude softer than 2D tissue culture plastic (~GPa). Our observation of decreased M1 in μRB vs. 2D is consistent with the previous literature. Compared to the uncoated μRB scaffolds, MSCM-coating further significantly decreased M1 polarization, with primed MSCM-coating exhibiting the lowest pro-inflammatory signals. Overall, an opposite trend was observed on anti-inflammatory M2 marker expression and interleukin (IL)-10 secretion. Primed MSCM-μRB showed significantly higher M2 marker gene expression (CD206, Egr2, IL-10) (Figure 2c) and IL-10 secretion (Figure 2e), indicating the primed MSCM-coating facilitated transition of Mφ from pro-inflammatory M1 to regenerative M2 phenotype. It is worth noting naïve MSCM showed no significant changes for most of the M2 markers except for CD206, compared to uncoated μRB scaffolds. Next, how MSCM-coating induced Mφ phenotypic change affect MSC bone formation through secretome was assessed. Conditioned medium (CM) from M1 Mφ cultured in different conditions was collected, mixed with osteogenic medium (20% CM and 80% OM), and used to treat MSCs cultured in 2D (Figure 2f). Compared to the control group without the CM treatment, all groups treated with M1 Mφ CM showed significantly decreased MSC bone formation and mineralization, as indicated by alizarin red staining (ARS) staining (Figure 2g). Importantly, MSCM-coating partially rescued this M1 Mφ CM-induced inhibition on MSC-based bone formation compared to uncoated μRB scaffolds, with primed MSCM-μRB showing the highest amount of bone formation. ARS+ area was quantified in figure S3a. These results validated our hypothesis that MSCM-coating can reduce undesirable prolonged pro-inflammatory response, facilitated macrophage phenotype transition toward pro-regenerative state and rescued the inhibited MSC bone formation. Furthermore, our results also validated the hypothesis that primed MSCM further enhance the regenerative immune response than naïve MSCM.
Figure 2. MSCM-coated μRB scaffolds significantly inhibited M1 Mφ polarization, and partially rescued MSC osteogenesis in the presence of M1 Mφ-secreted cytokines in vitro.
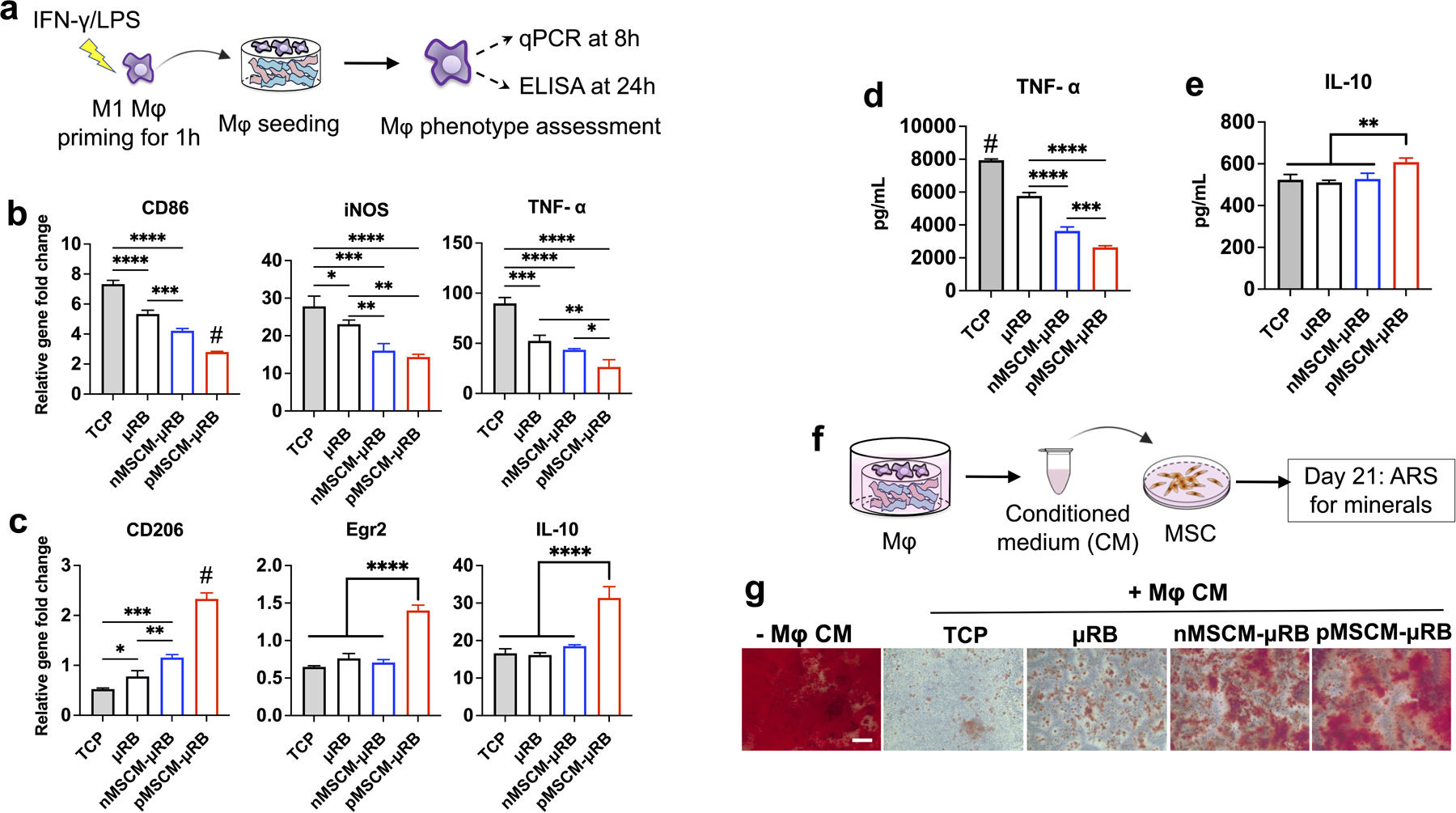
(a) Schematic of experimental design. Four groups were evaluated including 2D tissue culture plastic (TCP), uncoated μRB scaffolds, and μRB scaffolds coated with nMSCM or pMSCM. (b, c) Normalized gene expressions of M1 Mφ markers (b) and M2 Mφ markers (c) at 8 hours after seeding; (d, e) ELISA measurement of TNF-α (d) and IL-10 (e) released by Mφ after seeding on TCP or different μRB scaffolds for 24 hours. (f) Evaluating the effects of Mφ condition medium and MSCM coating on MSC osteogenesis in vitro. (g) Alizarin red S staining showed conditioned medium (CM) from Mφ in TCP and μRB alone inhibited MSC mineralization, whereas MSCM coating rescued MSC mineralization. Scale bar: 200 μm. Data are represented as mean ± S.D. (n = 4/group). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
While emerging studies have shown the promise of using biomaterials to modulate Mφ response to facilitate bone regeneration, [35, 36] the role of T cell subtypes in regenerating critical-sized cranial bone defects remains largely unclear. Using alginate hydrogels and a rat critical-sized femoral defect model, a recent study showed bone regeneration was more robust in T cell-deficient animals than immunocompetent animals.[37] These results highlighted the importance to better understand the role of T cell subtypes in scaffold-mediated bone regeneration. Furthermore, previous studies were limited to long bone niche, yet how biomaterials mediate T cell immunomodulation in cranial bone regeneration remains unknown. To fill in these critical unmet needs, we next assess the effect of MSCM-coating on T cells in vitro and in a mouse critical sized cranial defect model. Quantitative gene expression analyses of cells infiltrated into the scaffold reveal that macroporous μRB scaffolds recruited CD4+, Tregs and CD8+ T cells, with an increasing trend from day 7 to day 14 (Figure S4, Supporting Information). These results provide the rationale to evaluate MSCM-coating on T cell immunomodulation.
The effect of MSCM-coating on T cell response were first evaluated in vitro. Given PD-L1 and TNFL6 were identified on MSCM (Figure 1e) and their known role in inducing apoptosis of activated T cells [18], CD8+ T cell apoptosis was first evaluated. CD8+ T cells were isolated from mouse spleen and activated by CD3/CD28 Dynabeads for 3 days prior cell seeding. Apoptosis was evaluated by flow cytometry after 5 days (Figure 3a). Compared to 2D TCP, uncoated μRB scaffolds significantly increased CD8+ T cell apoptosis from 20% to 35% (Figure 3b, 3c). Recently, substrate stiffness has been reported to modulate T cell fates, and increasing stiffness enhances T-cell activation, proliferation, and migration [38]. These suggest mechanotransduction likely contributes to increased CD8+T cell apoptosis in μRB scaffolds compared to 2D. Compared to uncoated μRB scaffolds, naive MSCM coating and primed MSCM coating further significantly increased CD8+ T cell apoptosis to 50% and 73%, respectively. These are likely due to the changes in biochemical cues associated with various ligands on MSCM coating. The apoptotic cells exhibited double positive of PI and Annexin V, indicating a late stage of apoptosis. A previous study showed CD8+ T cells inhibited MSC osteogenesis in vitro [39], suggesting excessive CD8+ T cell may inhibit bone formation. Consistent with the previous report, we found CM from CD8+ T cells reduced MSC mineralization, indicating secretome of CD8+ T cells has an inhibitory effect on MSC bone formation (Figure 3d and 3e). Compared to uncoated μRB scaffolds, naïve MSCM-coating partially rescued the MSC mineral deposition. Strikingly, CM from primed MSCM-μRB scaffolds restored the bone formation to the level comparable to the no CM control. ARS+ area was quantified in figure S3b. This result suggests primed MSCM coating was sufficient to minimize the negative effect of CD8+ T cells on MSC-mediated bone regeneration.
Figure 3. MSCM-coating rescued MSC osteogenesis and mineral deposition in vitro by inducing CD8+ T cell apoptosis while enhancing Treg differentiation.
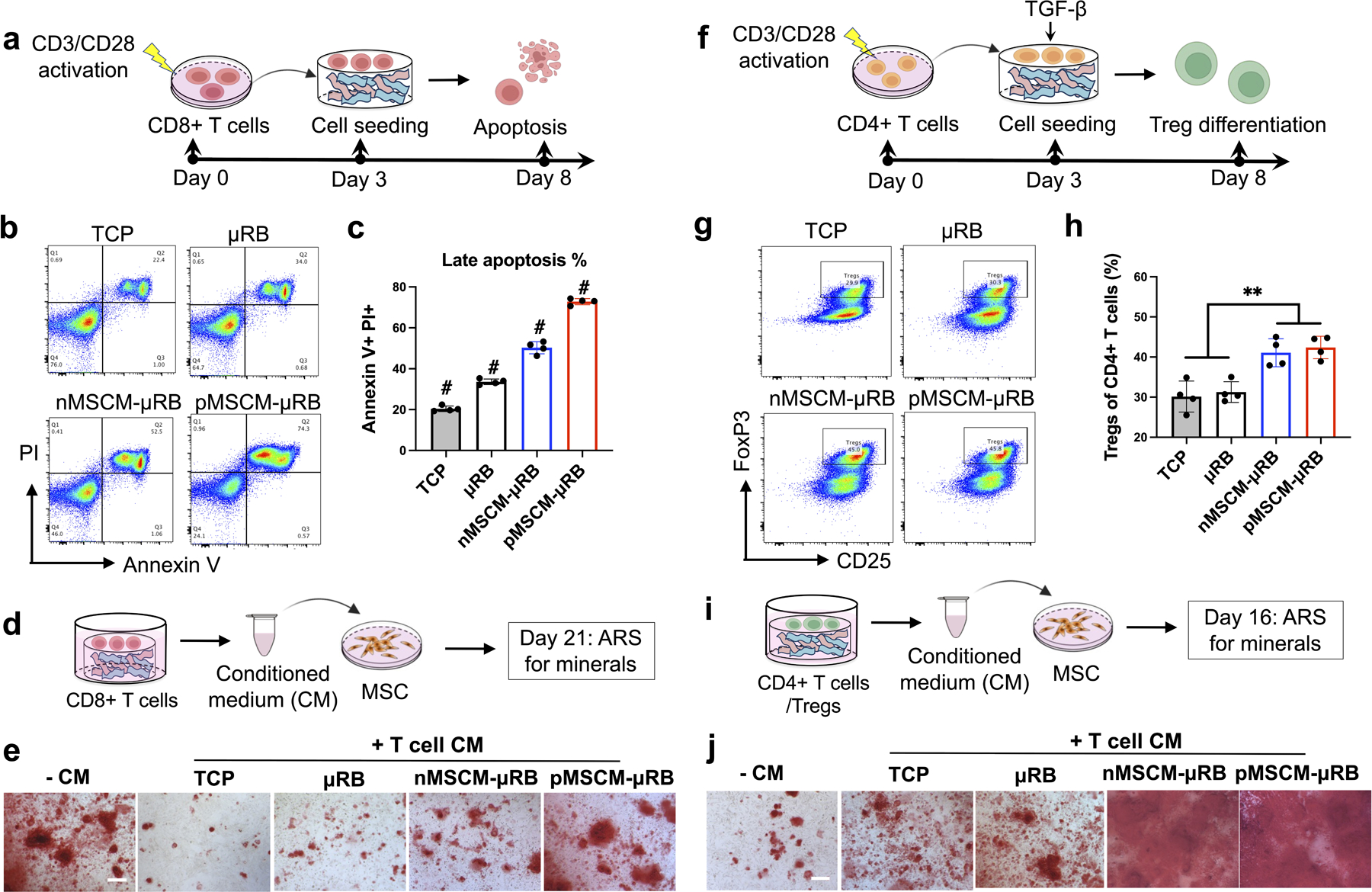
(a-c) Evaluating the effects of MSCM coating on TCD8+ apoptosis using flow cytometry; (b) representative flow cytometry plots and (c) quantification of apoptotic CD8+ cells, (d-e) Evaluating the effects of conditioned medium (CM) from TCD8+ cultured under different conditions on MSC mineralization using ARS staining; (f-h) Evaluating the effects of MSCM coating on Treg differentiation; (g) Representative flow cytometry plots and (h) quantification of Tregs (of CD25+ FoxP3+); (i-j) Evaluating the effects of CM from Treg cultured under different conditions on MSC mineralization; Data are represented as mean ± S.D. (n = 4/group). Scale bars: 200 μm. **P < 0.01, # indicates P < 0.001 when compared with all other groups.
Next, we assessed the MSCM-coating on Tregs differentiation in vitro. A previous study showed intravenous delivery of Tregs accelerated bone healing in a mouse critical-sized cranial defect model [40], suggesting a potential beneficial role of Tregs on bone regeneration. Here, CD4+ T cells were isolated from mouse spleen and activated by CD3/CD28 Dynabeads for 3 days. For Treg differentiation, the expanded CD4+ T cells were seeded to μRB scaffolds or TCP, supplemented with TGF-β. Treg differentiation was analyzed by flow cytometry after 5 days (Figure 3f). MSCM-coating significantly enhanced Treg differentiation compared to the uncoated scaffold, with no differences observed between naïve MSCM- and primed MSCM-μRB scaffolds (Figure 3g and 3h). The MSCM-induced Treg differentiation is supported by the proteomic characterization, where Gal-1/Gal-3/Gal-9 were detected on both naïve and primed MSCM (Figure 1e). The expression level of Gal-1/Gal-3/Gal-9 are comparable between naïve and primed MSCM, which might explain their similar capacity in inducing Treg differentiation. To further assess the effect of enhanced differentiation of Tregs on MSC-based bone formation, CM from Tregs cultured under different culture conditions were collected, mixed with osteogenic medium, and applied to MSC. Compared to no CM control, CM from Treg differentiation culture increased MSC mineral deposition (Figure 3i and 3j), indicating the beneficial role of secretome from Tregs in MSC bone formation. Impressively, Treg CM from naive MSCM- and primed MSCM-μRB scaffolds substantially accelerated MSC-based bone formation, with mineralization reaching a saturated level as early as day 16. ARS+ area was quantified in figure S3c. Consistent with our finding, a recent study demonstrated that exosomes secreted by Tregs can enhance MSC osteogenesis. [41] Taken together, these results validate that MSCM-coating enhances T-reg differentiation, which further accelerate MSC-based bone regeneration through secretomes.
The effect of MSCM-coating on immunomodulation was further evaluated in vivo using a mouse critical-sized cranial bone defect model (Figure 4a). The potential synergy between MSCM-coating and BMP-2 delivery was also explored using low dosage of BMP-2 (200 ng per defect). BMP-2 was loaded using dopamine-coated mesoporous silica nanoparticles (MSNs) as previously reported by our lab.[42] One advantage of μRB scaffold is its macroporosity, which facilitate rapid immune cell infiltration in vivo. Flow cytometry analysis showed ~ 1 million cells have infiltrated into the scaffold by day 7, with comparable level of Mφ (CD11b+ F4/80+) and T cells (CD3+) infiltrated to the implanted scaffolds across all groups. Mφ consisted about 15% of total infiltrated cells at day 7. Compared to uncoated μRB or BMP-2 alone controls, both naïve and primed MSCM-coating significantly decreased M1 Mφ infiltration to a comparable level (Figure 4b). BMP-2 synergized with MSCM-coating to further decrease M1 Mφ population, with primed MSCM+BMP exhibited the lowest number of M1 Mφ among all groups. While MSCM-coating alone decreased proinflammatory response by suppressing M1 Mφ and CD8+ T cell activity (Figure 4b and 4d), it was not sufficient to increase regenerative immune response of M2 Mφ and Tregs (Figure 4c and 4e) in vivo. Importantly, co-delivery with BMP-2 significantly increased M2 Mφ population compared to μRB control and BMP-2 alone group. Co-delivery of BMP-2 with MSCM-coating also increased Tregs population, with primed MSCM+BMP inducing highest number of Tregs (Figure 4e). It is worth noting MSCM-coating alone without BMP-2 did not induce significantly change in Treg number compared to control, suggesting BMP-2 and MSCM-coating synergy is required for modulating Treg population. Together, these results showed both MSCM-coating and BMP-2 are required to elicit regenerative immunomodulation of both innate and adaptive immune cells in vivo.
Figure 4. Primed-MSCM coating synergizes with BMP-2 to modulate both innate and adaptive immune responses and stem cell recruitment in a mouse critical-sized cranial defect model.
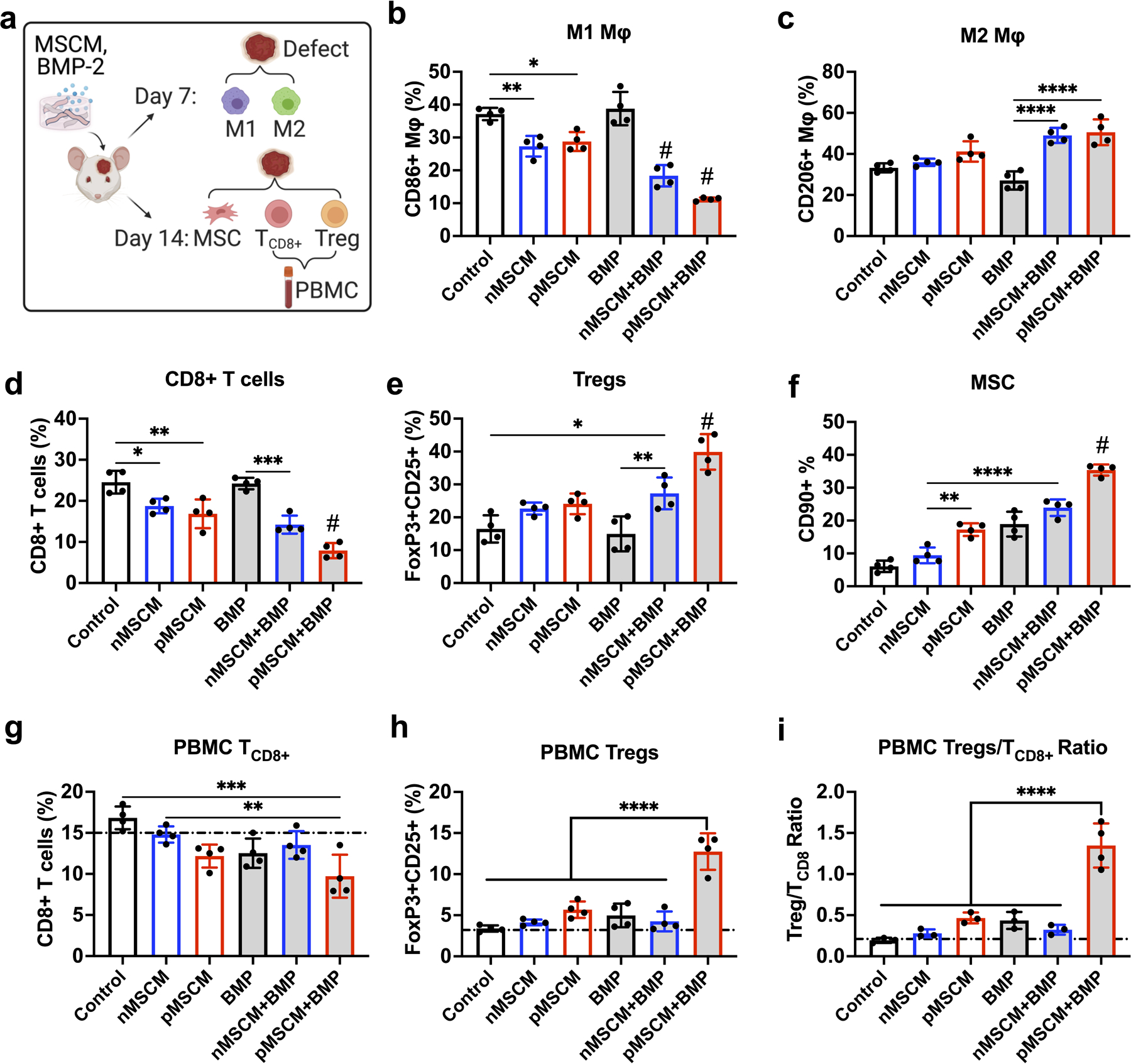
(a) In vivo experimental design for characterizing infiltrated immune cells and MSCs into μRB scaffolds in the cranial defect using flow cytometry. (b-e) Percentage of CD86+ M1 Mφ (b) and CD206+ M2 Mφ (c) at day 7; Percentage of CD8+ T cells (d) and FoxP3+CD25+ Tregs (e) at day 14; (f) Percentage of CD90+ MSCs at day 14. (g-i) Percentage of CD8+ T cells (g), FoxP3+CD25+ Tregs (h) and the ratio of T CD8+/Tregs (i) in peripheral blood at day 14. Dashed line indicates baseline from mice without surgery. Data are represented as mean ± S.D. (n = 4). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. # indicates P < 0.001 compared to all other groups.
Immune response can directly modulate endogenous stem cell recruitment to implanted scaffolds in vivo.[43] We next assessed early recruitment of MSCs using flow cytometry. The flow cytometry gating and representative dot plots for characterizing immune cell and MSC population were shown in Figure S5. Similar to the trend observed with Treg modulation, primed MSCM+BMP induced the highest percentage of endogenous MSC recruitment (Figure 4f). Compared to uncoated μRB control and naïve MSCM group, primed MSCM, BMP and naïve MSCM+BMP groups also increased endogenous MSC recruitment with significance. These results suggest BMP-2 delivery can directly increase endogenous MSC recruitment, which is further enhanced by crosstalk with immune system through primed MSCM coating.
Delayed fracture healing in patients has been shown to correlate with decreased Tregs and CD8+ T cell ratio in blood [44] [45], suggesting bone healing may be predicted by monitoring adaptive immune response at a systemic level. Unlike samples from the cranial defect, one advantage of using peripheral blood to monitor immunomodulation is that it is a minimal invasive procedure that does not require harvesting samples from the defect. To assess if peripheral blood may be used for predicting biomaterials-mediated immunomodulation in the cranial bone niche, Tregs and CD8+ T cell ratio were collected from the peripheral blood at day 14 and analyzed (Figure 4a). Similar to the trend observed in the cranial bone niche, primed MSCM+BMP group was the leading group with significantly decreased CD8+ T cells, increased Tregs, and increased Tregs/TCD8+ ratio in peripheral blood compared to other groups (Figure 4g–i). These results suggest it is possible to use peripheral blood to predict adaptive immune response induced by biomaterials implanted in cranial bone defects, although the sensitivity in detecting differences among groups is still higher in the local niche than peripheral blood.
Next, we assess the effect of MSCM-coating and BMP-2 delivery on μRB scaffold-mediated endogenous bone regeneration using a mouse critical size cranial defect model. Impressively, both MSCM-coated groups with BMP-2 induced early mineralized bone regeneration at week 2 (Figure 5a and 5b). This trend continued to increase. By week 6, 90% of bone defect was filled in pMSCM+BMP group, followed by 60% filling by nMSCM+BMP group. In contrast, BMP-2 alone led to only 37% of bone filled by week 6. Group without BMP-2 showed minimal bone formation, indicating that while MSCM coating induced regenerative immune response, a low dosage of BMP-2 is still needed to achieve robust bone regeneration. The trend in bone regeneration correlates with the trend observed in immunomodulation in the bone niche (Figure 4b–e), indicating the enhanced bone regeneration is, at least partly, contributed by immunomodulation. Previous studies suggested highly inflammatory environment can desensitize endogenous stem cells response to growth factor,[46, 47] which may explain why MSCM coating enhances BMP-2-induced bone regeneration via immunomodulation. The group that showed most robust bone formation (pMSCM+BMP) also showed the significantly higher Tregs/TCD8+ ratio in peripheral blood (Figure 4i). This data suggest Tregs/TCD8+ ratio in peripheral blood could be used as a biomarker to predict the healing outcomes in patients with critical sized cranial defects.
Figure 5. MSCM-coating synergized with BMP-2 to enhance new bone formation, angiogenesis and stem cell recruitment in vivo.
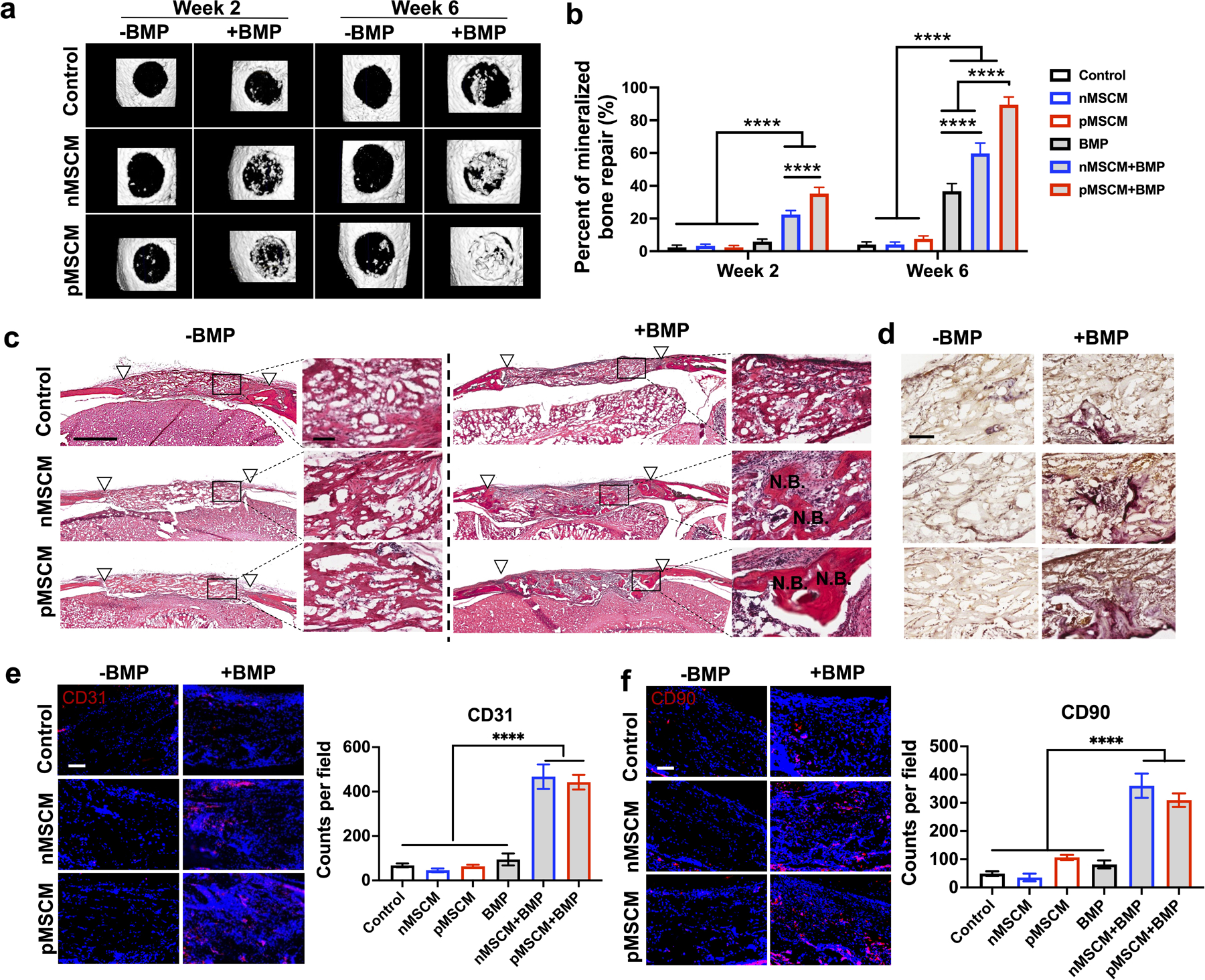
(a) Representative μCT images and percentage of newly formed bone volume in the mouse cranial defects at week 2 and week 6. (c) Representative morphology of newly formed bone at week 6, as shown by H&E staining. White triangles mark the edges of the defect. Scale bar: 1 mm in low magnification images, 100 μm in high magnification images. (d) Representative TRAP staining images for osteoclasts at week 6. (e, f) Representative immunofluorescent staining images and quantification of CD31+ endothelial cells (e) and CD90+ MSCs (f) in the cranial defects at week 6. Scale bar in (e, f): 100 μm. Data are represented as mean ± S.D. (n = 5). ****P < 0.0001.
To further assess newly formed bone morphology, remodeling and vascularization, histology and immunostainings were performed. Newly formed bone was highest in pMSCM+BMP group, followed by nMSCM+BMP (Figure 5c). Similar trend was observed in osteoclast activity, as shown by TRAP staining, indicating active bone remodeling (Figure 5d). Osteoclasts are indispensable for replacing biomaterial implants with newly regenerated bone, and suppression of osteoclastogenesis has been shown to inhibit biomaterial-mediated bone regeneration.[48, 49] Osteoclasts can also directly promote osteogenesis and angiogenesis through secretome,[50, 51] and increasing osteoclast population promoted vascularization and bone regeneration in a mouse critical-sized cranial defect model.[52] Both MSCM-coated groups with BMP-2 showed significantly higher vascularization, as shown by CD31 staining (Figure 5e). Proteomic analysis of MSCM identified ligands related to angiogenesis including myeloid-derived growth factor (MYDGF)[53], cell surface hyaluronidase 2 (CEIP2)[54], and CCN1[55] (Figure S2f, Supporting Information). These ligands may directly promote angiogenesis, which contributes to the observed enhanced bone regeneration. Finally, immunostaining of MSC marker (CD90+) showed both MSCM coated group with BMP-2 led to significantly enhanced recruitment of endogenous MSCs (Figure 5f). This is consistent with the trend of flow cytometry result (Figure 4f). Interestingly, pMSCM+BMP group showed comparable vessel density and MSC number as nMSCM+BMP group (Figure 5e, f), but resulted in significantly higher bone formation (Figure 5b). These results further highlight the importance of immunomodulation for bone healing, given pMSCM coating induces a more robust regenerative immune response than nMSCM coating (Figure 4) in vivo.
To further elucidate how BMP-2 and pMSCM-coating synergize to enhance on bone regeneration, we conducted in vitro studies to assess macrophage phenotype and MSC osteogenesis in response to singular or combination treatments (Figure 6a). BMP-2 alone induced polarization of macrophage towards inflammatory phenotype, as shown by increasing CD86 expression and TNF-α secretion (Figure 6b, c). This recapitulates the inflammatory responses of BMP-2 that are typically associated with high dosage use in clinical setting.[56]. High dosage of BMP-2 can lead to undesirable swelling, associated with increased recruitment of monocytes, macrophages and lymphocytes.[57] While high dosage of BMP2 is reported to induce inflammation and uncontrolled bone formation, low dosage of BMP2 has also been shown to induce M2 polarization and enhanced osteogenesis in vitro.[58, 59] These results suggest the effect of BMP2 on inflammation is dosage-dependent.Importantly, when BMP-2 is combined with pMSCM coating, the undesirable M1 Mφ activation induced by BMP-2 was minimized. Instead, Mφ exhibited pro-regenerative phenotype, as shown by significantly upregulated CD206 expression and increased IL-10 secretion. These results indicate that pMSCM coating can not only quench BMP-2 induced inflammation, but also promote macrophage polarization towards regenerative phenotype. There are several possible mechanisms through which MSCM suppress BMP2-induced inflammatory responses. First, MSCM contains rich immunomodulatory ligands, which could direct bind to corresponding receptors on both innate and adaptive immune cells to modulate their signaling and phenotype. Second, the immunomodulatory ligands present on MSCM can also directly bind to free inflammatory cytokines, thus reducing the concentration of undesirable inflammatory signals. Future research could conduct inhibition studies to identify the specific role of certain ligands on MSCM that are responsible for suppressing BMP-induced inflammation.
Figure 6. Primed MSCM coating mitigated BMP-induced inflammation, promoted macrophage polarization towards M2 phenotype, and enhanced MSC osteogenesis in vitro.
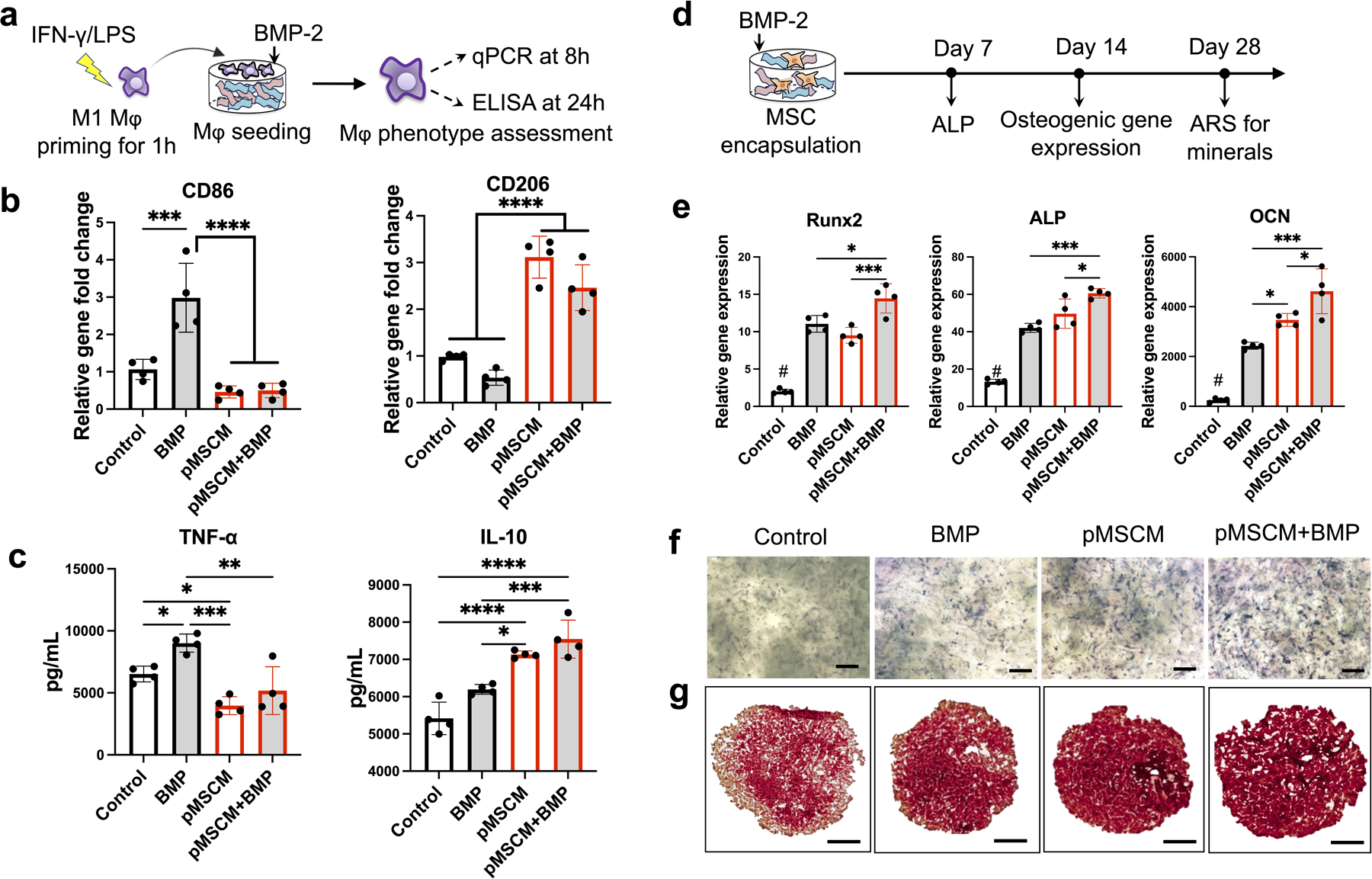
(a) Schematics and timeline for evaluating Mφ polarization in response to BMP-2 and pMSCM coating. (b-c) Gene expressions of CD86, CD206 (b) and cytokine secretion (TNF-α and IL-10) (c). (d) Schematics and timeline for evaluating MSC osteogenesis in response to primed MSCM-coating and BMP-2 treatment. (e) Osteogenic gene expression of MSCs at week 2 in uncoated μRB scaffolds or MSCM-μRB scaffolds. (f, g) Representative images of ALP staining at week 1 (f) and ARS staining at week 4 (g). Scale bar: 1 mm. Data are represented as mean ± S.D. (n = 4/group). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Furthermore, pMSCM-coating alone also showed beneficial effect on MSC osteogenesis, as shown by significantly upregulated bone marker expressions including runt-related transcription factor 2 (Runx2), alkaline phosphatase (ALP) and osteocalcin (OCN) (Figure 6e). This trend was further validated using ALP staining and ARS staining for mineralization (Figure 6f and 6g). The beneficial effect of pMSCM-coating on MSC osteogenesis may be explained by proteomic analysis of pMSCM, where identified pro-osteogenic ligands were identified including ALP [60], Annexin A4 (ANXA4)[61], and stem cell recruiting molecules (SDF-2) (Figure S2f, Supporting Information). Combining pMSCM coating and BMP-2 led to the highest level of bone marker expressions (Fig. 6e), further validating their synergistic effect on enhancing bone formation.
In summary, here we report stem cell membrane-coated macroporous μRB scaffolds for immunomodulation and enhancing endogenous bone regeneration in a critical size cranial defect model (Fig. 7). We found MSC membrane coating elicited regenerative immune responses of both Mφ and T cells in vitro and in vivo. Cell membrane from MSCs primed with pro-inflammatory cytokines further enhances regenerative immune response and promotes MSC osteogenesis. When combined with a low dosage of BMP-2, primed MSC membrane coating further accelerated endogenous cranial bone regeneration, while suppressing BMP-2 induced inflammation. The present study filled the gap of knowledge on the role of adaptive immune cells in the regeneration of critical-sized bone defects. Our findings provide strong evidence to support future research in applying cell membrane coating for functionalizing macroporous scaffolds for immunomodulation and tissue regeneration applications. Previous studies have shown cell membrane bound secretory factors, such as TGFβ and galectins, could modulate immune cell responses via cell-cell contact.[62] Using proteomic analysis, we also identified MSCM contains transmembrane ligands such as FASL and PD-L1 with known immunomodulatory functions (Fig. 1).[18] We speculate that MSCM coating-induced immune responses are contributed collectively by a combination of these ligands from MSCM, instead of just one or two ligands. Compared to strategies that modify scaffold surface with a few specific ligands, cell membrane coating has the advantage of targeting multiple biological processes and ease of application. Here we chose MSC membrane due to their known immunomodulatory functions. Future studies may explore cell membrane derived from other cell types for targeting different biological processes associated with tissue regeneration. While the present study focuses on microribbon scaffolds and bone regeneration, the concept of integrating cell membrane coating with scaffolds for immunomodulation can be broadly applied for functionalizing other scaffolds and regenerating other tissue types.
Figure 7. A schematic summary of primed MSCM-coated μRB scaffolds enhance bone regeneration through inducing regenerative immune response of both innate and adaptive immune cells, modulating the crosstalk between immune cells and MSCs, and synergizing with BMP-2.
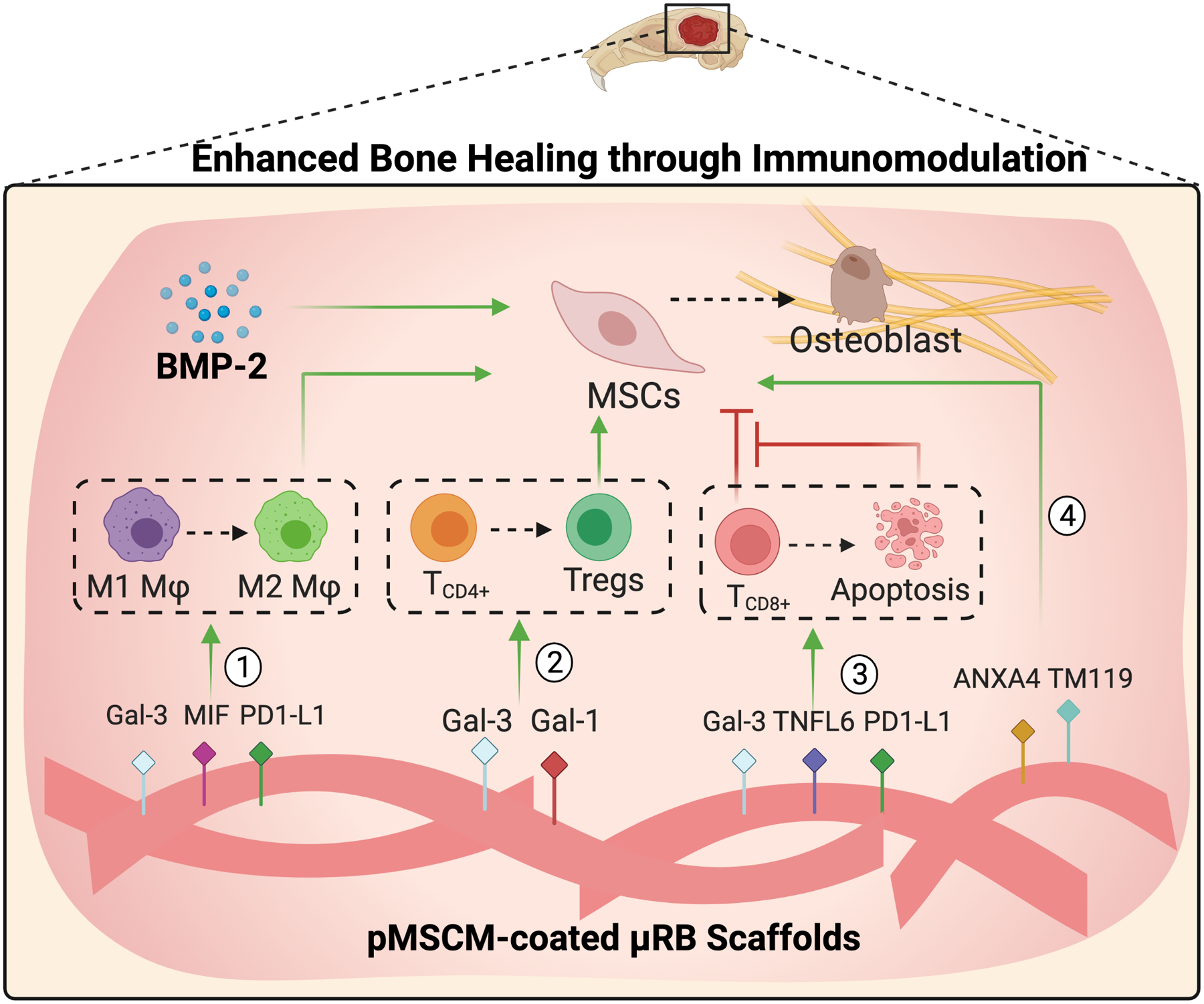
Primed MSCM-coating contains ligands that promote Mφ polarization towards M2-like phenotype, induces Treg differentiation and TCD8+ apoptosis, and directly enhances MSC osteogenesis.
Supplementary Material
Acknowledgements
The authors would like to thank NIH R01DE024772 (F. Y.), R01AR074502 (FY), Stanford Maternal and Children’s Health Institute Postdoctoral Fellowship (N.S.), National Science Foundation Predoctoral fellowship (C. V.) and EDGE Fellowship (C.V.) for support. The authors would also like to thank Dr. Liangfang Zhang and Dr. Weiwei Gao from University of California San Diego for kindly sharing their experiences with cell membrane coating for nanoparticles and helpful discussions.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Yin S, Zhang W, Zhang Z, Jiang X, Adv Healthc Mater 2019, 8, e1801433. [DOI] [PubMed] [Google Scholar]
- [2].Shang F, Yu Y, Liu S, Ming L, Zhang Y, Zhou Z, Zhao J, Jin Y, Bioact Mater 2021, 6, 666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Su N, Villicana C, Yang F, Biomaterials 2022, 121604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wu AC, Raggatt LJ, Alexander KA, Pettit AR, Bonekey Rep 2013, 2, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gerstenfeld LC, Cullinane DM, Barnes GL, Graves DT, Einhorn TA, J Cell Biochem 2003, 88, 873. [DOI] [PubMed] [Google Scholar]
- [6].Liu H, Li D, Zhang Y, Li M, Histochem Cell Biol 2018, 149, 393. [DOI] [PubMed] [Google Scholar]
- [7].Pajarinen J, Lin T, Gibon E, Kohno Y, Maruyama M, Nathan K, Lu L, Yao Z, Goodman SB, Biomaterials 2019, 196, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Niu Y, Wang Z, Shi Y, Dong L, Wang C, Bioact Mater 2021, 6, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lei H, Schmidt-Bleek K, Dienelt A, Reinke P, Volk HD, Front Pharmacol 2015, 6, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schlundt C, Reinke S, Geissler S, Bucher CH, Giannini C, Mardian S, Dahne M, Kleber C, Samans B, Baron U, Duda GN, Volk HD, Schmidt-Bleek K, Front Immunol 2019, 10, 1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jiang Y, Krishnan N, Zhou J, Chekuri S, Wei X, Kroll AV, Yu CL, Duan Y, Gao W, Fang RH, Zhang L, Adv Mater 2020, 32, e2001808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ai X, Wang S, Duan Y, Zhang Q, Chen MS, Gao W, Zhang L, Biochemistry 2021, 60, 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dehaini D, Wei X, Fang RH, Masson S, Angsantikul P, Luk BT, Zhang Y, Ying M, Jiang Y, Kroll AV, Gao W, Zhang L, Adv Mater 2017, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen W, Zhang Q, Luk BT, Fang RH, Liu Y, Gao W, Zhang L, Nanoscale 2016, 8, 10364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chooi WH, Dong Q, Low JZY, Yuen C, Chin JS, Lin J, Ong W, Liu Q, Chew SY, ACS Appl Bio Mater 2021, 4, 4079. [DOI] [PubMed] [Google Scholar]
- [16].Perez RA, Mestres G, Mater Sci Eng C Mater Biol Appl 2016, 61, 922. [DOI] [PubMed] [Google Scholar]
- [17].Wu X, Jiang J, Gu Z, Zhang J, Chen Y, Liu X, Stem Cell Res Ther 2020, 11, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gu YZ, Xue Q, Chen YJ, Yu GH, Qing MD, Shen Y, Wang MY, Shi Q, Zhang XG, Hum Immunol 2013, 74, 267. [DOI] [PubMed] [Google Scholar]
- [19].Brennan MÁ, Layrolle P, Mooney DJ, Advanced Functional Materials 2020. [DOI] [PMC free article] [PubMed]
- [20].Zhang X, Sun Y, Chen W, Yang J, Chen J, Chen S, Biomater Adv 2022, 136, 212802. [DOI] [PubMed] [Google Scholar]
- [21].Han LH, Conrad B, Chung MT, Deveza L, Jiang X, Wang A, Butte MJ, Longaker MT, Wan D, Yang F, J Biomed Mater Res A 2016, 104, 1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Griffin DR, Archang MM, Kuan CH, Weaver WM, Weinstein JS, Feng AC, Ruccia A, Sideris E, Ragkousis V, Koh J, Plikus MV, Di Carlo D, Segura T, Scumpia PO, Nat Mater 2021, 20, 560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tang Y, Tong X, Conrad B, Yang F, Theranostics 2020, 10, 6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lin T, Pajarinen J, Nabeshima A, Lu L, Nathan K, Jamsen E, Yao Z, Goodman SB, Stem Cell Res Ther 2017, 8, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Noronha NC, Mizukami A, Caliari-Oliveira C, Cominal JG, Rocha JLM, Covas DT, Swiech K, Malmegrim KCR, Stem Cell Res Ther 2019, 10, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zara JN, Siu RK, Zhang X, Shen J, Ngo R, Lee M, Li W, Chiang M, Chung J, Kwak J, Wu BM, Ting K, Soo C, Tissue Eng Part A 2011, 17, 1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shields LB, Raque GH, Glassman SD, Campbell M, Vitaz T, Harpring J, Shields CB, Spine 2006, 31, 542. [DOI] [PubMed] [Google Scholar]
- [28].da Costa Goncalves F, Paz AH, World J Stem Cells 2019, 11, 618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yaseen H, Butenko S, Polishuk-Zotkin I, Schif-Zuck S, Perez-Saez JM, Rabinovich GA, Ariel A, Front Pharmacol 2020, 11, 901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Reesink HL, Sutton RM, Shurer CR, Peterson RP, Tan JS, Su J, Paszek MJ, Nixon AJ, Stem Cell Res Ther 2017, 8, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Deng M, Tan J, Dai Q, Luo F, Xu J, Front Cell Dev Biol 2021, 9, 714011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kim H, Lee MJ, Bae E-H, Ryu JS, Kaur G, Kim HJ, Kim JY, Barreda H, Jung SY, Choi JM, Molecular Therapy 2020, 28, 1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Blakney AK, Swartzlander MD, Bryant SJ, Journal of biomedical materials research. Part A 2012, 100, 1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Conrad B, Hayashi C, Yang F, ACS Biomaterials Science & Engineering 2020, 6, 3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Qiu P, Li M, Chen K, Fang B, Chen P, Tang Z, Lin X, Fan S, Biomaterials 2020, 227, 119552. [DOI] [PubMed] [Google Scholar]
- [36].Won JE, Lee YS, Park JH, Lee JH, Shin YS, Kim CH, Knowles JC, Kim HW, Biomaterials 2020, 227, 119548. [DOI] [PubMed] [Google Scholar]
- [37].Garske DS, Schmidt-Bleek K, Ellinghaus A, Dienelt A, Gu L, Mooney DJ, Duda GN, Cipitria A, Tissue Eng Part A 2020, 26, 852. [DOI] [PubMed] [Google Scholar]
- [38].Majedi FS, Hasani-Sadrabadi MM, Thauland TJ, Li S, Bouchard L-S, Butte MJ, Biomaterials 2020, 252, 120058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wendler S, Schlundt C, Bucher CH, Birkigt J, Schipp CJ, Volk HD, Duda GN, Schmidt-Bleek K, Front Immunol 2019, 10, 713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Liu Y, Wang L, Kikuiri T, Akiyama K, Chen C, Xu X, Yang R, Chen W, Wang S, Shi S, Nat Med 2011, 17, 1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chen L, Xiong Y, Hu Y, Yu C, Panayi AC, Zhou W, Cao F, Sun Y, Liu M, Liu G, Xue H, Hu L, Mi B, Liu G, Chemical Engineering Journal 2022, 427. [Google Scholar]
- [42].Barati D, Gegg C, Yang F, Ann Biomed Eng 2020, 48, 1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Alshoubaki YK, Nayer B, Das S, Martino MM, Stem Cells Transl Med 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Reinke S, Geissler S, Taylor WR, Schmidt-Bleek K, Juelke K, Schwachmeyer V, Dahne M, Hartwig T, Akyüz L, Meisel C, Unterwalder N, Singh NB, Reinke P, Haas NP, Volk HD, Duda GN, Sci Transl Med 2013, 5, 177ra36. [DOI] [PubMed] [Google Scholar]
- [45].Jiang H, Ti Y, Wang Y, Wang J, Chang M, Zhao J, Sun G, Clin Exp Pharmacol Physiol 2018, 45, 430. [DOI] [PubMed] [Google Scholar]
- [46].Huang R-L, Yuan Y, Zou G-M, Liu G, Tu J, Li Q, Stem cells and development 2014, 23, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Julier Z, Karami R, Nayer B, Lu YZ, Park AJ, Maruyama K, Kuhn GA, Müller R, Akira S, Martino MM, Sci Adv 2020, 6, eaba7602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gómez-Cerezo N, Casarrubios L, Saiz-Pardo M, Ortega L, de Pablo D, Díaz-Güemes I, Fernández-Tomé B, Enciso S, Sánchez-Margallo FM, Portolés MT, Arcos D, Vallet-Regí M, Acta Biomater 2019, 90, 393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hauser M, Siegrist M, Denzer A, Saulacic N, Grosjean J, Bohner M, Hofstetter W, J Orthop Surg (Hong Kong) 2018, 26, 2309499018802487. [DOI] [PubMed] [Google Scholar]
- [50].Pederson L, Ruan M, Westendorf JJ, Khosla S, Oursler MJ, Proc Natl Acad Sci U S A 2008, 105, 20764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Xie H, Cui Z, Wang L, Xia Z, Hu Y, Xian L, Li C, Xie L, Crane J, Wan M, Zhen G, Bian Q, Yu B, Chang W, Qiu T, Pickarski M, Duong LT, Windle JJ, Luo X, Liao E, Cao X, Nat Med 2014, 20, 1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sun JL, Jiao K, Niu LN, Jiao Y, Song Q, Shen LJ, Tay FR, Chen JH, Biomaterials 2017, 113, 203. [DOI] [PubMed] [Google Scholar]
- [53].Korf-Klingebiel M, Reboll MR, Klede S, Brod T, Pich A, Polten F, Napp LC, Bauersachs J, Ganser A, Brinkmann E, Nature medicine 2015, 21, 140. [DOI] [PubMed] [Google Scholar]
- [54].De Angelis JE, Lagendijk AK, Chen H, Tromp A, Bower NI, Tunny KA, Brooks AJ, Bakkers J, Francois M, Yap AS, Simons C, Wicking C, Hogan BM, Smith KA, Dev Cell 2017, 40, 123. [DOI] [PubMed] [Google Scholar]
- [55].Yeger H, Perbal B, J Cell Commun Signal 2007, 1, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].James AW, LaChaud G, Shen J, Asatrian G, Nguyen V, Zhang X, Ting K, Soo C, Tissue Eng Part B Rev 2016, 22, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Pardali E, Makowski LM, Leffers M, Borgscheiper A, Waltenberger J, J Cell Mol Med 2018, 22, 5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wei F, Zhou Y, Wang J, Liu C, Xiao Y, Tissue Eng Part A 2018, 24, 584. [DOI] [PubMed] [Google Scholar]
- [59].Munoz J, Akhavan NS, Mullins AP, Arjmandi BH, Nutrients 2020, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Vimalraj S, Gene 2020, 754, 144855. [DOI] [PubMed] [Google Scholar]
- [61].Davies OG, Cox SC, Williams RL, Tsaroucha D, Dorrepaal RM, Lewis MP, Grover LM, Sci Rep 2017, 7, 12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hofer HR, Tuan RS, Stem Cell Research & Therapy 2016, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.