

Abstract
Two heterobimetallic Mo,M’ complexes (M’ = IrIII, RhIII) were synthesized and fully characterized. Their catalytic activity in homogeneous carbon dioxide hydrogenation to formate was studied. A pronounced synergistic effect between the two metals was found, most notably between Mo and Ir, leading to a fourfold increase in activity compared with a binary mixture of the two monometallic counterparts. This synergism can be attributed to spatial proximity of the two metals rather than electronic interactions. To further understand the nature of this interaction, the mechanism of the CO2 hydrogenation to formate by a monometallic IrIII catalyst was studied using computational and spectroscopic methods. The resting state of the reaction was found to be the metal-base adduct, whereas the rate-determining step is the inner-sphere hydride transfer to CO2. Based on these findings, the synergism in the heterobimetallic complex is beneficial in this key step, most likely by further activating the CO2.
Keywords: bimetallic complex, carbon dioxide, hydrogenation, synergistic catalysis
1. Introduction
As society is faced with ever-increasing atmospheric CO2 levels and its impact on the world climate, chemists strive to develop novel catalysts that can create value-added products from abundant and environmentally benign substrates and reagents [1,2]. A common approach is taking inspiration from nature—more specifically, enzymes which have fast conversion rates, high selectivity, and low activation energies [3,4]. Several me-talloproteins, capable of redox transformations of small molecules such as H2, CO2, N2 and CO, employ multi- and bimetallic assemblies [5]. One example is the metalloenzyme carbon monoxide dehydrogenase (Ni-CODH, Figure 1), in which the cooperation of the Fe and the Ni center enable a fast and selective reduction of CO2 to CO [6].
Figure 1.
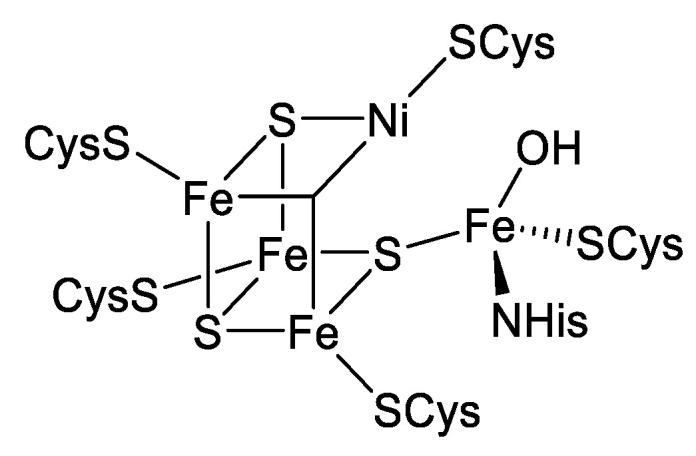
Active site of Ni-CODH [6].
Inspired by this approach, a plethora of heterobimetallic complexes have been developed, which show superior catalytic behavior compared with their monometallic counterparts. In most of these cases, the increase in activity can be attributed to the beneficial electronic interaction between the two metals, which means that exclusively one metal center performs all substrate transformations [7,8,9,10,11,12,13]. A different approach is to utilize the reactivity of each metal to facilitate tandem or one-pot reactions, meaning both metals interact directly with substrates [14,15,16,17,18,19,20]. However, in biological systems such as Ni-CODH, both metals are used to perform a single reaction [6]. Synergistic effects within a heterobimetallic complex have only recently been recognized and utilized. For example, heterobimetallic MgII–ZnII complexes are very active catalysts for ring-opening copolymerization reactions between CO2 and epoxides [21,22,23]. Synergistic effects are also vital in the activity of heterobimetallic catalysts for olefin hydrogenation and formate dehydrogenation [24].
Herein, we report the utilization of heterobimetallic Mo0,M’ complexes (M’ = RhIII (C2), IrIII (C1)) for homogeneous CO2 hydrogenation to formate salts. Homogeneous hydrogenation of CO2 to formic acid and formate salts is a fast-developing field, ever since the pioneering works of Inoue [25]. While formic acid is widely used as silage acid, it is also interesting for potential application in chemical H2 storage [25,26]. Currently, there are multiple examples of precious metal catalysts with very high turnover numbers (TON) and turnover frequency (TOF), as well as several base metal catalysts with similarly high TON and TOF [27,28,29,30]. Recently, heterobimetallic Ni0–GaIII and Co-I–GaIII complexes with outstanding performance have been reported. However, in both examples, the second metal (Ga) is used to impart nobility on the first (Co/Ni) through a metal–metal bond [31,32,33].
2. Results and Discussion
2.1. Complex Synthesis
In the synthesis of heterobimetallic complexes, two main challenges arise: selectivity and stability. For a complex with two different catalytic sites, the metals must be selectively bound and kinetically stable within each respective coordination sphere. In the ditopic ligand 1 (Scheme 1), two distinctively different coordination environments are available. Through a stepwise coordination, complexes C1 and C2 can be obtained in good to excellent yields. [Mo0(CO)3(NCMe)3] was chosen as a precursor as the resulting complex 2 (Scheme 1) is stable enough to undergo subsequent cyclometallation with [MIIICp*Cl2]2 (M = Ir or Rh, Cp* = C5Me5). The resulting complexes C1 and C2 were fully characterized, including single crystal structure determinations. Additionally, the kinetic stability was tested using solutions of each complex in acetonitrile-d3, which showed no change over the course of a week in the 1H- and 31P{1H}-NMR spectra. Furthermore, a 1:1 mixture of the monometallic complexes M1 and M3 (see Table 1) was studied, showing no reaction. This indicates a high kinetic stability of the metals in their respective coordinative environments.
Scheme 1.

Synthesis of heterobimetallic complexes C1 and C2 (Cp* = C5Me5).
Table 1.
Catalytic CO2 hydrogenation to formate using different complexes and bases (Cp* = C5Me5).(a)
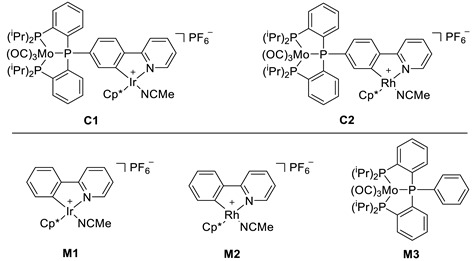
| |||||
| Entry | Catalyst | Catalyst Loading (mol%) (b) | Base | TON (c) | Yield (%) (d) |
| 1 | C1 | 0.6 | DBU | 128 | 77 |
| 2 | M1 | 0.6 | DBU | 23 | 14 |
| 3 | M3 | 0.6 | DBU | traces | <1 |
| 4 | M1 + M3 (e) | 0.6 | DBU | 34 | 21 |
| 5 | C2 | 0.6 | DBU | 17 | 10 |
| 6 | M2 | 0.6 | DBU | 5 | 3 |
| 7 | M2 + M3 (e) | 0.6 | DBU | 3 | 2 |
| 8 | C1 | 0.6 | TMG | 168 | Quant. |
| 9 | C1 | 0.6 | NEt3 | 45 | 27 |
| 10 | C1 | 0.6 | - | - | 0 |
| 11 | C1 (f) | 0.6 | DBU | 12 | 7 |
(a) Reaction conditions: catalyst (3 µmol), base (500 µmol), 2 mL total volume of MeCN solution, 50 °C, H2 (30 bar), 30 min; CO2 (30 bar, total pressure 60 bar), 22 h; DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene, TMG = 1,1,3,3-tetramethylguanidine. (b) In relation to amount of base. (c) Based on 1H-NMR signal of HCO2− relative to the internal standard (benzene in C6D6 in a capillary). (d) Determined by n(formate)/n(base) by 1H-NMR spectroscopy. (e) 3 µmol of each complex. (f) H2 (15 bar), CO2 (15 bar, total pressure 30 bar).
2.2. Catalysis
All complexes were tested for catalytic activity towards CO2 hydrogenation (Table 1). It is essential to activate the complexes, by first applying H2 for 30 min and then adding CO2. If both CO2 and H2 are added simultaneously, no catalytic activity can be observed. Different conditions (solvents, base, H2/CO2 pressure, and reaction time) were investigated (for a comprehensive list of the tested conditions see electronic supporting information). Since C1 outperformed C2 significantly, the focus of this study is on the Mo,Ir complex C1. The system performed best with acetonitrile as solvent and a strong base, such as 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) or 1,1,3,3-tetramethylguanidine (TMG) (Table 1, entries 1, 8). Using a weaker base such as triethylamine led to a significant decrease in TON (Table 1, entry 9). Strong inorganic bases such as LiOH or Cs2CO3 led to no observable formate production, probably due to their insolubility in acetonitrile. The necessity of using strong organic bases can be understood by comparing the pKaH of the organic base and formic acid in acetonitrile [34,35]. While DBU (pKaH = 24.31) and TMG (pKaH = 23.35) are sufficiently strong bases to quantitively deprotonate formic acid (pKaH = 20.7), triethylamine (pKaH = 18.83) and diisopropylethylamine (DIPEA, pKaH = 18.2) are not [34,35]. Since the hydrogenation of CO2 to formic acid is an inherently endergonic reaction, the base must be strong enough to provide a thermodynamic driving force. In the absence of base, no reaction occurs (Table 1, entry 10).
After 70 h, with TMG as base, more than the expected stoichiometric amounts of formic acid were formed, namely two equivalents in relation to the amount of base employed. Increasing the reaction time to 140 h did not result in even higher yields (see electronic supporting information, Table S4), indicating that the reaction stops when two equivalents of formic acid have been formed. While unexpected, higher than the expected stoichiometric amounts of formic acid in relation to the base have been reported before and are due to the formation of a formate-formic acid dimer [36]. Counterion effects in M1 and complexes similar to M1 have been studied; while higher catalytic activity could most likely be achieved employing an even more weakly coordinating anion, studying these effects in depth is beyond the scope of the present article [37,38].
Using more polar solvents such as DMSO, water or methanol led to a significantly lower TON. Reducing the total pressure from 60 bar to 30 bar led to a tenfold drop in reactivity (entry 11). The TOF for complex C1, using TMG as base (see electronic supporting information, Table S4), was determined by monitoring the reaction and is 5.0 h−1.
The performance of the monometallic species M1 or M3 is poorer than the 1:1 mixture of M1 and M3, especially considering that M3 is catalytically inactive. This shows that there is a synergistic effect between the two complexes. This synergistic effect is much more pronounced in the heterobimetallic complex C1, leading to a reactivity that is ~4 times as high as the mixture of M1 and M3 (entry 1 vs. entry 4, Table 1). Furthermore, there is no observable synergistic effect between M2 and M3 (entry 6 vs. entry 7, Table 1) but in the heterobimetallic complex C2, leading to a threefold increase in reactivity compared to M2 (Table 1, entry 5 vs. entry 6).
2.3. Comparing Heterobimetallic and Monometallic Systems
The spectroscopic data of the heterobimetallic complexes C1 and C2 were compared to M3 to elucidate whether the increased reactivity in the heterobimetallic complexes stems from an actual synergistic effect or is due to a beneficial electronic interaction. The chemical shift of the central phosphorus atom in the ligand of all three complexes is almost identical (79.7 ppm in M3 vs. 80.2 and 80.5 ppm in C1 and C2, respectively; see supporting information). The CO stretching vibrations of M3, C2 and C1 are almost identical (two signals at 1930 cm−1 and 1845 cm−1).
Most notably, however, the experimental E1/2 values for Mo0/I are only about 30 mV apart (Figure 2; E1/2(M3) = 5 mV, E1/2(C1) = 25 mV, E1/2(C2) = 35 mV, vs. [FcH]/[FcH]+). When measuring the E1/2 values of Mo0/I for M3 while M1 or M2 is present in the same concentration, one can see a very small shift (E1/2(M3 and M2) = 12 mV, E1/2(M3 and M1) = 15 mV, see Sections 8.2 and 8.3 in the supporting information) The very similar electrochemical potential as well as the spectroscopic data suggest that the Mo centers in C1, C2 and M3 are electronically very similar, which in turn means that the Ir center of C1 and M1 and the Rh center of C2 and M2 must also be comparable. It further indicates that any increase in reactivity must be from synergistic effects between the two metal centers and cannot be attributed to electronic interactions. This is in contrast to other synergistic heterobimetallic systems, where an electronic interaction between the metals is very likely and the increase in reactivity is probably caused by a combination of synergism and electronic interaction [21,22,23,24,39].
Figure 2.

Cyclic voltammogram of M3 (1 mM), C1 (1 mM) and, C2 (3 mM) in MeCN at room temperature, with 0.10 M [N(nBu)4]PF6 as supporting electrolyte, a glassy carbon electrode (surface area 0.001 cm2), a Ag/AgNO3 (0.01 mol∙l−1) reference electrode, a Pt wire (99.9%) counter electrode, and a voltage sweep rate ν = 100 mV∙s−1.
Similar to natural enzymes, the synergism in C1 and C2 is likely caused by the close spatial proximity of the two metals. While C1 does not crystallize readily, single crystals were obtained of a complex in which the labile MeCN ligand was exchanged with N2 (Figure 3). The intermetallic distance Mo···Ir is 6.731(1) Å is longer than the proposed ideal intermetallic distance of 3.5–6 Å, but the metals are not ideally aligned to achieve the shortest possible distance [40]. Additionally, the closest approach of a CO ligand at the Mo center to the N2 at the Ir atom is 3.7589(3) Å. This suggests that substrates bound at one metal can readily interact with a ligand and/or substrates bound at the second metal.
Figure 3.

Molecular structure of a derivative of complex C1 in which the coordinated MeCN has been replaced by N2 during crystallization. The non-coordinating PF6− counterion, all non-coordinating solvent molecules and hydrogen atoms have been omitted for clarity. All non-coordinating carbon atoms and Cp* are drawn as wireframe.
2.4. Understanding the CO2 Hydrogenation Using M1 as Catalyst
Considering that the two metals in C1 and C2 are not electronically communicating, the monometallic complexes M1 and M2 can be used as models for the Ir and Rh center in the heterobimetallic complexes, reducing the complexity of the system considerably. Furthermore, studying the heterobimetallic system directly has several disadvantages. The main challenge is that any changes at either metal lead to small shifts in the 1H- and 31P-NMR spectra, which renders interpreting the spectra difficult, or even impossible. Since NMR spectroscopy is the only analytical technique available to us, which can be performed under an H2 or CO2 atmosphere, without conclusive NMR measurement no mechanistic insight could be gained into the heterobimetallic system.
During catalytic trials M3 showed no catalytic activity as the complex is unable to form the necessary hydride species to perform the reaction; thus, we focused the subsequent mechanistic investigations on M1, as the Mo center in C1 only has an auxiliary role in the mechanism. Furthermore, a thorough understanding how M1 performs CO2 hydrogenation reactions can provide a basis for including synergistic interactions later. As the two metals are not in electronic communication, one can deduce that the two metals will be electronically isolated in the catalytically active species as well. Therefore, the catalytic cycle of M1 is expected to bear strong resemblance to that of C1, the main difference being that the Mo center has a beneficial interaction during the rate-determining step (RDS). While additional interactions are possible, they will only have an influence if the activation energy of the RDS is lowered so significantly that a new RDS arises. Nevertheless, this means that by identifying the RDS in the CO2 hydrogenation, using M1 as catalyst the nature of the synergistic interaction becomes more apparent, as an energetically lower transition state of the RDS is essential for higher TON and TOF.
Even before the catalytic cycle (Figure 4) begins, there are two parameters to be considered. The first is the coordinative strength of the solvent, quantified by the free binding enthalpy (ΔG0bind). These values were calculated for acetonitrile (−50.2 kJ mol−1) and for DMSO (−158.9 kJ mol−1, coordination through the oxygen atom). The strong coordination of DMSO explains the low activity when the latter is used as a solvent. The resulting [IrIII(DMSO)]+ complex (Figure 4) would act as a thermodynamic sink. While DMSO binds too strongly, THF, methanol and water show only weak coordination (ΔG0bind = −36.9 kJ mol−1, −29.9 kJ mol−1, and −32.1 kJ mol−1, respectively). The lack of stabilization by the solvent leads to instability of [IrIII(solv)]+. Accordingly, when M1 is subjected to 1 atm of H2 atmosphere in THF-d8, signals similar to reported Rh nanoclusters can be observed in the 1H-NMR spectra [41]. Thus, MeCN is an ideal solvent as it provides good stabilization of the Ir complex, while not binding too strongly.
Figure 4.

Proposed catalytic scheme for CO2 hydrogenation using a monometallic iridium catalyst (Cp* = C5Me5).
Both DBU and TMG are sufficiently strong bases to provide a driving force for the reaction by deprotonating the product formic acid; they also bind strongly to the metal center [42]. For the coordination of DBU to M1, the calculated free binding enthalpy in different solvents is about −100 kJ mol−1 (ΔG0bind = −98.6 kJ mol−1 (MeCN), −98.3 kJ mol−1 (DMSO, coordination through the oxygen atom), −96.5 kJ mol−1 (H2O), −98.7 kJ mol−1 (MeOH), and −103.0 kJ mol−1 (THF)). It is reasonable to assume that the resting state of the reaction is [IrIII(Base)]+ (Figure 4); thus, the effective concentration of [IrIII(solv)]+ in the reaction is very low, contributing to the low TOF. When M1 is subjected to 1 atm of H2 in the presence of TMG (see Sections 5.1 and 5.2 in the electronic supporting information), TMG binds quantitatively at Ir; hence, no [IrIIIH] formation can be observed.
[IrIII(solv)]+ reacts with H2 to give the intermediate [IrIII(η2-H2)]+ (Figure 4). The calculated H-H bond length (92.7 pm) in the side-on coordinated dihydrogen indicates a significant activation considering an H-H bond length of 76.1 pm in molecular hydrogen [43]. The formation of the key intermediate [IrIIIH] proceeds through a heterolytic cleavage of the η2-bound H2. In the presence of the strong base DBU, [IrIII(η2-H2)]+ seems to be deprotonated instantaneously, as it was not possible to detect the transition state (TS) of the heterolytic cleavage using DFT calculations. Furthermore, the structure of [IrIII(η2-H2)]+ was found to be only stable on the potential energy hypersurface in the absence of base, supporting the idea of a barrierless reaction. Interestingly, a TS for the heterolytic cleavage of H2—with a very small barrier—was found when the D3 dispersion correction was omitted, suggesting that the non-covalent interactions between the large complex [IrIII(η2-H2)]+ and a bulky base could decrease and remove the barrier. Furthermore, the counterion PF6− was neglected in the DFT calculations, but could also have an effect on the stability of [IrIII(η2-H2)]+ as well as on [TS1].
The next step in the catalytic cycle is the hydride transfer from [IrIIIH] to CO2. In similar catalytic processes, this is often the overall RDS [27,28,29,30]. However, not only the kinetics of this step bear remarkable consequences for the overall reaction, but also its thermodynamics. By determining the thermodynamic hydricity (ΔG0H−), the Gibbs free energy change of the hydride transfer from the metal hydride to carbon dioxide can be calculated [44]. Since both hydride complexes [RhIIIH] and [IrIIIH] are isostructural, the hydride transfer has to undergo the same transition state with both metals and thus the Bell–Evans–Polanyi principle can be applied, i.e., the more endergonic reaction will also have a higher transition state [45,46]. The thermodynamic hydricity of [RhIIIH] has been determined by 1H-NMR spectroscopy (Scheme 2); using a similar procedure, ΔG0H− for [IrIIIH] was calculated [42].
Scheme 2.

Equilibrium between hydrido complex [IrIIIH] and cationic [IrIII(NCCD3)]+ used to determine the thermodynamic hydricity [42].
While the hydricity of [RhIIIH] could be assessed using a triethylammonium salt, [IrIIIH] was too reactive and reacted quantitatively. Using a less acidic phosphorimidic triamide salt a ΔGH−([IrIIIH]) of 197.1 kJ mol−1 was determined (Figure 5).
Figure 5.

Hydrido complexes [IrIIIH] and [RhIIIH] (Cp* = C5Me5) with their respective thermodynamic hydricity [42].
The relatively high ΔGH− indicates that the hydride transfer from both Ir and Rh to CO2 is thermodynamically unfavorable (Scheme 3), and the catalytic activity is most likely due to the basicity of the base (in case of the sufficiently strong bases DBU or TMG), high pressure and temperature [44]. This would also explain the strong pressure dependency of the catalytic activity (Table 1, entry 1 and entry 11). When subjecting [IrIIIH] to 1 atm of CO2 in CD3CN, no reaction was observed over the course of one week. Furthermore, the hydride transfer from Rh is 10.0 kJ mol−1 less favorable than the hydride transfer from Ir, which explains the much higher reactivity of C1 and M1.
Scheme 3.

Calculated reaction enthalpies for hydride transfer from [MIIIH] to CO2.
The hydride transfer can follow an inner-sphere or outer-sphere mechanism. The outer-sphere reaction would proceed through [TS2] to form the H-bound formate adduct [IrIIIHCO2]*. This adduct can either release the formate anion followed by recoordination via oxygen, or [IrIIIHCO2] can be formed through an intramolecular isomerization. An inner-sphere mechanism would involve direct binding of CO2 to form [IrIIIHCO2] directly through [TS3]. Both pathways result in the formation of [IrIIIHCO2], which then releases the formate to close the catalytic cycle. When M1 is mixed in a 1:1 ratio with [N(nBu)4]HCO2, [IrIIIHCO2] is formed exclusively (see Section 5.3 in the electronic supporting information). To further assess whether the liberation of formate could be the overall rate-determining step, phosphorimidic triamide and H2 (1 atm) were added to this mixture. While the formation of [IrIIIH] is slower from [IrIIIHCO2] than staring directly from M1, it can be observed. This shows that the liberation of formate is unfavorable but not rate determining. While the hydrogen activation is only slowed down in the presence of formate, no reaction was observed when [IrIIIH] was subjected to a CO2 atmosphere at room temperature. This shows that the hydride transfer to CO2 is indeed the RDS of the catalysis.
The complex [MIIIHCO2]* and transition state [TS2] could not be optimized through DFT calculations, as the structures always spontaneously rearranged into [MIIIHCO2] and [TS3], respectively. Thus, it is reasonable to assume that the reaction follows an inner-sphere mechanism.
Calculating accurate solution phase entropies is a challenge for quantum chemistry, and for the hydrogenation of CO2 to formate, two components of the reaction must enter the solution from gas phase at elevated pressures. To avoid discussing this elusive term, we will here consider only the more reliable calculated relative enthalpies; the corresponding Gibbs free energies are given in the electronic supporting information. The reaction enthalpy profile is presented in Figure 6 for M1 in MeCN and with DBU as base; detailed numerical data for all other variations are summarized in Table 2. The counterion PF6− was neglected in all calculations, assuming its influence on the catalyst to be only minor. As discussed above, the coordination of the base or the solvent result in a highly stable resting state. Taking [IrIII(solv)]+ as reference, the formation of the complex with an η2-coordinated dihydrogen ligand is endothermic for all solvents (MeCN, DMSO, H2O and MeOH). Splitting the H-H bond in the before mentioned barrierless step is thermodynamically more favorable with DBU than NEt3 as base yielding [IrIIIH]. In agreement with the discussion above, the reaction of the base with the formed formic acid is necessary if the overall catalytic cycle is to be thermodynamically favorable. Accordingly, the formation of a salt consisting of the protonated base and a formate anion was observed experimentally.
Figure 6.

Reaction enthalpy profile for the catalyst M1 in MeCN using DBU as a base.
Table 2.
Calculated relative enthalpies (DFT, B3LYP-D3/def2-SVP level of theory in kJ mol−1) for the catalytic cycle of monometallic Ir complexes for different solvents and bases.
| Intermediate. | Base | MeCN (M1) | DMSO | H2O | MeOH |
|---|---|---|---|---|---|
| [IrIII(solv)]+ | DBU | 0.0 | 0.0 | 0.0 | 0.0 |
| NEt3 | 0.0 | 0.0 | 0.0 | 0.0 | |
| [IrIII(base)]+ | DBU | −66.7 | −34.2 | −86.2 | −84.0 |
| NEt3 | −9.0 | 22.5 | −28.7 | −26.2 | |
| [IrIII(η2-H2)]+ | DBU | 56.4 | 87.7 | 36.3 | 39.3 |
| NEt3 | 56.4 | 87.7 | 36.3 | 39.3 | |
| [IrIIIH] | DBU | −56.8 | −25.8 | −77.6 | −73.8 |
| NEt3 | −9.0 | 21.7 | −30.4 | −25.8 | |
| [TS3] | DBU | 28.4 | 59.1 | 7.8 | 11.6 |
| NEt3 | 76.3 | 106.7 | 55.0 | 59.6 | |
| [IrIIIHCO2] | DBU | −57.4 | −26.6 | −78.7 | −74.3 |
| NEt3 | −9.6 | 21.0 | −31.5 | −26.3 | |
| [IrIII(solv)]+ (a) | DBU | −73.4 | −73.3 | −73.3 | −73.4 |
| NEt3 | −59.6 | −59.7 | −59.7 | −59.5 |
(a) With considering the formation of the [DBU(H)]HCO2 salt.
Clearly, the values discussed above would be different if entropic effects were considered, since the reaction of H2 with [IrIII(solv)]+ and of CO2 with [IrIIIH] involve the association of molecules. However, since these effects are similar for all the systems that were compared in this study, the trends observed in enthalpies are valid for Gibbs free energies as well.
It should further be noted that all shown solution phase enthalpies were calculated using an implicit continuum solvation model. This approach, while acting as a good foundation for the comparison of different solvents, allows for some inaccuracy for not including explicit solvation.
2.5. Identifying the Synergistic Interaction
As the hydride transfer from [IrIIIH] to CO2 is the overall RDS, the synergism that enhances the reactivity in C1 needs to assist in this step. There are several examples of Mo-phosphine complexes capable of binding and activating CO2, e.g., for coupling with ethylene forming acrylate. However, all these complexes contain at least one labile ligand [47,48,49,50,51,52]. It is possible that during the activation period, before the addition of CO2, one or more CO ligands are cleaved from the Mo center of C1. However, considering that no CO ligands were eliminated at high temperature (>110 °C) in the presence of trimethylamine N-oxide or under irradiation with UV light, we infer that the Mo complex is stable under the reaction conditions.
In a first attempt to study the activation period of the catalyst (30 bar H2 for 30 min), a solution containing M1 and M3 was subjected to 1 atm of H2 in the presence of excess NEt3 (see Section 5.4 in the electronic supporting information). While no transformation on the Mo center of M3 could be observed in either the 1H- or 31P-NMR spectra over the course of one week, formation of [IrIIIH] from M1 occurred. This indicates an interaction in which M3 assists in the formation of [IrIIIH] and stabilizes the latter. As mentioned before, in the absence of M3, HNEt3+ is too acidic and quantitatively reacts with [IrIIIH]. The exact nature of the interaction between M1 and M3 is not yet understood as M3 does not interact with HNEt3+, at least not in a manner that is observable by IR or NMR spectroscopy.
When C1 is subjected to 1 atm of H2 in the presence of excess NEt3, the formation of an iridium hydride species can be observed as well using 1H-NMR spectroscopy. Additionally, a shift in the 31P{1H}-NMR spectrum takes place supporting the formation of a corresponding hydrido complex, which, however, decomposed over the course of several days. The fact that a transformation occurred while no reaction was observed at the molybdenum center in the mixture of the two monometallic species further confirms that the synergistic effect is much more pronounced in the heterobimetallic complex. Future work is now focused on identifying the intermediate and understanding the synergistic interaction.
3. Computational Methods
All calculations were carried out using ORCA (version 3.0) [53] for the complexes M1 and M2. Density functional theory with the exact exchange functional B3LYP and the def2-SVP basis set was used [53,54,55,56]. All calculations utilize the atom-pairwise dispersion correction with the Becke-Johnson damping scheme (D3BJ) [57,58]. The settings TightSCF, grid5, and TightOpt were employed. After each optimization, a numerical frequency calculation was carried out in order to confirm minimum structures on the potential energy surface. For the transition state search the same settings with unrestricted calculations were used, but with the OptTS and the SlowConv keywords. A single-point calculation on each of the structures optimized in the gas phase was performed, using the conductor-like polarizable continuum model (CPCM) for each solvent with the default settings [59].
4. Conclusions
Two heterobimetallic complexes and their monometallic counterparts were synthesized, fully characterized, and their catalytic activity was studied in homogeneous CO2 hydrogenation. Best results in terms of catalytic activity were obtained with the heterobimetallic Mo,Ir complex C1 (TON 168, TOF 5 h−1). A synergistic effect was observed in all heterobimetallic complexes, leading to a significant increase in reactivity, as compared with the 1:1 mixture of their monometallic counterparts. Electrochemical analysis indicates that this synergism cannot be attributed to an electronic interaction, which is further supported by spectroscopic data.
As the two metal centers in the heterobimetallic complex do not communicate electronically, the monometallic Ir complex M1 was studied extensively as a model for the Ir center in C1. A mechanism for the CO2 hydrogenation using M1 was proposed and is supported by extensive calculations and NMR spectroscopic data. The overall rate-determining step is the hydride transfer from the Ir catalyst to CO2. The resting state of the reaction is the base adduct of M1, thus removing it from the reaction and leading to low TOF. Since the mechanism of the CO2 hydrogenation using C1 must be similar to the mechanism using M1, one can deduce that a synergism assists in the hydride transfer to CO2. For further understanding of the heterobimetallic system, the active catalyst must be identified, and the catalytic reaction studied by in situ spectroscopy.
Acknowledgments
Many thanks to Wieland Körber for developing the synthetic protocol for ligand 1.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28062574/s1. Crystallographic data for M3, 2, the starting materials [Mo(CO)3;RhCl] (3) and [Mo(CO)3;IrCl] (4), and the side products [Mo(CO)3;Rh(CO)]PF6 and [Mo(CO)3;Ir(N2)]PF6. Additional data for synthesis, spectroscopic characterizations, catalysis studies, computational studies, hydricity measurements, mechanistic NMR experiments and electrochemical data. References [60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78] are cited in the Supplementary Materials.
Author Contributions
Conceptualization, Z.B.G.F. and E.H.-H.; methodology, Z.B.G.F.; formal analysis (spectroscopy), Z.B.G.F.; formal analysis (diffractometry), P.L.; calculation A.K.M., O.H. and B.K.; formal analysis (calculations), A.K.M., O.H. and B.K.; writing—original draft preparation, Z.B.G.F. and A.K.M.; writing—review and editing, Z.B.G.F., E.H.-H., P.L., A.K.M., O.H. and B.K.; supervision, E.H.-H. and B.K.; project administration, E.H.-H.; funding acquisition, E.H.-H., O.H., B.K. and Z.B.G.F. All authors have read and agreed to the published version of the manuscript.
Data Availability Statement
X-ray crystallography data can be accessed through the CCDC (2210067-2210072). All other data are given in the Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
Financial support from the Studienstiftung des deutschen Volkes (doctoral fellowships for Z.B.G.F) and the Graduate School BuildMoNa is gratefully acknowledged. O.H. acknowledges the financial support from the National Research, Development and Innovation Office through the project OTKA-FK 138823, the János Bolyai Research Scholarship of the Hungarian Academy of Sciences, and the ÚNKP-22-5 New National Excellence Program from the National Research, Development and Innovation Fund.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Rockström J., Steffen W., Noone K., Persson A., Chapin F.S., Lambin E.F., Lenton T.M., Scheffer M., Folke C., Schellnhuber H.J., et al. A safe operating space for humanity. Nature. 2009;461:472–475. doi: 10.1038/461472a. [DOI] [PubMed] [Google Scholar]
- 2.Beller M., Bolm C. Transition Metals for Organic Synthesis: Building Blocks and Fine Chemicals. 2nd ed. Wiley VCH; Weinheim, Germany: 2004. [Google Scholar]
- 3.Steinhagen H., Helmchen G. Asymmetric Two-Center Catalysis—Learning from Nature. Angew. Chem. Int. Ed. 1996;35:2339–2342. doi: 10.1002/anie.199623391. [DOI] [Google Scholar]
- 4.Steinhagen H., Helmchen G. Asymmetrische Zweizentren-Katalyse—Von der Natur lernen. Angew. Chem. 1996;108:2489–2492. doi: 10.1002/ange.19961082005. [DOI] [Google Scholar]
- 5.Appel A.M., Bercaw J.E., Bocarsly A.B., Dobbek H., DuBois D.L., Dupuis M., Ferry J.G., Fujita E., Hille R., Kenis P.J.A., et al. Frontiers, opportunities, and challenges in biochemical and chemical catalysis of CO2 fixation. Chem. Rev. 2013;113:6621–6658. doi: 10.1021/cr300463y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Can M., Armstrong F.A., Ragsdale S.W. Structure, function, and mechanism of the nickel metalloenzymes, CO dehydrogenase, and acetyl-CoA synthase. Chem. Rev. 2014;114:4149–4174. doi: 10.1021/cr400461p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buchwalter P., Rosé J., Braunstein P. Multimetallic catalysis based on heterometallic complexes and clusters. Chem. Rev. 2015;115:28–126. doi: 10.1021/cr500208k. [DOI] [PubMed] [Google Scholar]
- 8.Chaudhary A., Singh A., Kamboj R.C. Heterobimetallic Complexes as Promising Catalysts. Chem. Sci. Rev. Lett. 2016;5:170–192. [Google Scholar]
- 9.Cooper B.G., Napoline J.W., Thomas C.M. Catalytic Applications of Early/Late Heterobimetallic Complexes. Catal. Rev. 2012;54:1–40. doi: 10.1080/01614940.2012.619931. [DOI] [Google Scholar]
- 10.Kalck P. Homo-and Heterobimetallic Complexes in Catalysis. Springer International Publishing; Basel, Switzerland: 2016. [Google Scholar]
- 11.Mankad N.P. Catalysis with Multinuclear Complexes. In: Moret M.-E., Gebbink R.J.M., editors. Non-Noble Metal Catalysis: Molecular Approaches and Reactions. Wiley-VCH; Weinheim, Germany: 2019. pp. 49–68. [Google Scholar]
- 12.Bratko I., Gómez M. Polymetallic complexes linked to a single-frame ligand: Cooperative effects in catalysis. Dalton Trans. 2013;42:10664–10681. doi: 10.1039/c3dt50963j. [DOI] [PubMed] [Google Scholar]
- 13.Mata J.A., Hahn F.E., Peris E. Heterometallic complexes, tandem catalysis and catalytic cooperativity. Chem. Sci. 2014;5:1723–1732. doi: 10.1039/C3SC53126K. [DOI] [Google Scholar]
- 14.Bitzer M.J., Kühn F.E., Baratta W. Tandem Suzuki–Miyaura/transfer hydrogenation reaction catalyzed by a Pd–Ru complex bearing an anionic dicarbene. J. Catal. 2016;338:222–226. doi: 10.1016/j.jcat.2016.02.031. [DOI] [Google Scholar]
- 15.Gu S., Xu D., Chen W. Heterobimetallic complexes containing an N-heterocyclic carbene based multidentate ligand and catalyzed tandem click/Sonogashira reactions. Dalton Trans. 2011;40:1576–1583. doi: 10.1039/c0dt01211d. [DOI] [PubMed] [Google Scholar]
- 16.Sabater S., Mata J.A., Peris E. Heterobimetallic Iridium–Ruthenium Assemblies through an Ambidentate Triazole-Diylidene Ligand: Electrochemical Properties and Catalytic Behavior in a Cascade Reaction. Organometallics. 2012;31:6450–6456. doi: 10.1021/om300675p. [DOI] [Google Scholar]
- 17.Wei L., Zhu Q., Xu S.-M., Chang X., Wang C.-J. Stereodivergent Synthesis of α,α-Disubstituted α-Amino Acids via Synergistic Cu/Ir Catalysis. J. Am. Chem. Soc. 2018;140:1508–1513. doi: 10.1021/jacs.7b12174. [DOI] [PubMed] [Google Scholar]
- 18.Zanardi A., Mata J.A., Peris E. Well-defined Ir/Pd complexes with a triazolyl-diylidene bridge as catalysts for multiple tandem reactions. J. Am. Chem. Soc. 2009;131:14531–14537. doi: 10.1021/ja906028g. [DOI] [PubMed] [Google Scholar]
- 19.Zanardi A., Mata J.A., Peris E. An Ir-Pt catalyst for the multistep preparation of functionalized indoles from the reaction of amino alcohols and alkynyl alcohols. Chemistry. 2010;16:13109–13115. doi: 10.1002/chem.201001180. [DOI] [PubMed] [Google Scholar]
- 20.Brereton K.R., Pitman C.L., Cundari T.R., Miller A.J.M. Solvent-Dependent Thermochemistry of an Iridium/Ruthenium H2 Evolution Catalyst. Inorg. Chem. 2016;55:12042–12051. doi: 10.1021/acs.inorgchem.6b02223. [DOI] [PubMed] [Google Scholar]
- 21.Garden J.A., Saini P.K., Williams C.K. Greater than the Sum of Its Parts: A Heterodinuclear Polymerization Catalyst. J. Am. Chem. Soc. 2015;137:15078–15081. doi: 10.1021/jacs.5b09913. [DOI] [PubMed] [Google Scholar]
- 22.Trott G., Garden J.A., Williams C.K. Heterodinuclear zinc and magnesium catalysts for epoxide/CO2 ring opening copolymerizations. Chem. Sci. 2019;10:4618–4627. doi: 10.1039/C9SC00385A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deacy A.C., Durr C.B., Garden J.A., White A.J.P., Williams C.K. Groups 1, 2 and Zn(II) Heterodinuclear Catalysts for Epoxide/CO2 Ring-Opening Copolymerization. Inorg. Chem. 2018;57:15575–15583. doi: 10.1021/acs.inorgchem.8b02923. [DOI] [PubMed] [Google Scholar]
- 24.Hong D., Shimoyama Y., Ohgomori Y., Kanega R., Kotani H., Ishizuka T., Kon Y., Himeda Y., Kojima T. Cooperative Effects of Heterodinuclear IrIII-MII Complexes on Catalytic H2 Evolution from Formic Acid Dehydrogenation in Water. Inorg. Chem. 2020;59:11976–11985. doi: 10.1021/acs.inorgchem.0c00812. [DOI] [PubMed] [Google Scholar]
- 25.Inoue Y., Izumida H., Sasaki Y., Hashimoto H. Catalytic Fixation of Carbon Dioxide to Formic Acid by Transition-Metal Complexes under Mild Conditions. Chem. Lett. 1976;5:863–864. doi: 10.1246/cl.1976.863. [DOI] [Google Scholar]
- 26.Yu X., Pickup P.G. Recent advances in direct formic acid fuel cells (DFAFC) J. Power Sources. 2008;182:124–132. doi: 10.1016/j.jpowsour.2008.03.075. [DOI] [Google Scholar]
- 27.Klankermayer J., Wesselbaum S., Beydoun K., Leitner W. Selective Catalytic Synthesis Using the Combination of Carbon Dioxide and Hydrogen: Catalytic Chess at the Interface of Energy and Chemistry. Angew. Chem. Int. Ed. 2016;55:7296–7343. doi: 10.1002/anie.201507458. [DOI] [PubMed] [Google Scholar]
- 28.Klankermayer J., Wesselbaum S., Beydoun K., Leitner W. Selektive katalytische Synthesen mit Kohlendioxid und Wasserstoff: Katalyse-Schach an der Nahtstelle zwischen Energie und Chemie. Angew. Chem. 2016;128:7416–7467. doi: 10.1002/ange.201507458. [DOI] [Google Scholar]
- 29.Dong K., Razzaq R., Hu Y., Ding K. Homogeneous Reduction of Carbon Dioxide with Hydrogen. Top. Curr. Chem. 2017;375:23. doi: 10.1007/s41061-017-0107-x. [DOI] [PubMed] [Google Scholar]
- 30.Wang W.-H., Himeda Y., Muckerman J.T., Manbeck G.F., Fujita E. CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction. Chem. Rev. 2015;115:12936–12973. doi: 10.1021/acs.chemrev.5b00197. [DOI] [PubMed] [Google Scholar]
- 31.Cammarota R.C., Vollmer M.V., Xie J., Ye J., Linehan J.C., Burgess S.A., Appel A.M., Gagliardi L., Lu C.C. A Bimetallic Nickel-Gallium Complex Catalyzes CO2 Hydrogenation via the Intermediacy of an Anionic d10 Nickel Hydride. J. Am. Chem. Soc. 2017;139:14244–14250. doi: 10.1021/jacs.7b07911. [DOI] [PubMed] [Google Scholar]
- 32.Ye J., Cammarota R.C., Xie J., Vollmer M.V., Truhlar D.G., Cramer C.J., Lu C.C., Gagliardi L. Rationalizing the Reactivity of Bimetallic Molecular Catalysts for CO2 Hydrogenation. ACS Catal. 2018;8:4955–4968. doi: 10.1021/acscatal.8b00803. [DOI] [Google Scholar]
- 33.Vollmer M.V., Ye J., Linehan J.C., Graziano B.J., Preston A., Wiedner E.S., Lu C.C. Cobalt-Group 13 Complexes Catalyze CO2 Hydrogenation via a Co(−I)/Co(I) Redox Cycle. ACS Catal. 2020;10:2459–2470. doi: 10.1021/acscatal.9b03534. [DOI] [Google Scholar]
- 34.Tshepelevitsh S., Kütt A., Lõkov M., Kaljurand I., Saame J., Heering A., Plieger P.G., Vianello R., Leito I. On the Basicity of Organic Bases in Different Media. Eur. J. Org. Chem. 2019;2019:6735–6748. doi: 10.1002/ejoc.201900956. [DOI] [Google Scholar]
- 35.Matsubara Y., Grills D.C., Koide Y. Thermodynamic Cycles Relevant to Hydrogenation of CO2 to Formic Acid in Water and Acetonitrile. Chem. Lett. 2019;48:627–629. doi: 10.1246/cl.190180. [DOI] [Google Scholar]
- 36.Hansen P.E., Lund T., Krake J., Spanget-Larsen J., Hvidt S. A Reinvestigation of the Ionic Liquid Diisopropylethylammonium Formate by NMR and DFT Methods. J. Phys Chem. B. 2016;120:11279–11286. doi: 10.1021/acs.jpcb.6b08561. [DOI] [PubMed] [Google Scholar]
- 37.Corre Y., Iali W., Hamdaoui M., Trivelli X., Djukic J.-P., Agbossou-Niedercorn F., Michon C. Efficient hydrosilylation of imines using catalysts based on iridium(III) metallacycles. Catal. Sci. Technol. 2015;5:1452–1458. doi: 10.1039/C4CY01233J. [DOI] [Google Scholar]
- 38.Hamdaoui M., Desrousseaux C., Habbita H., Djukic J.-P. Iridacycles as Catalysts for the Autotandem Conversion of Nitriles into Amines by Hydrosilylation: Experimental Investigation and Scope. Organometallics. 2017;36:4864–4882. doi: 10.1021/acs.organomet.7b00749. [DOI] [Google Scholar]
- 39.Hong D., Ohgomori Y., Shimoyama Y., Kotani H., Ishizuka T., Kon Y., Kojima T. Mechanistic Insight into Synergistic Catalysis of Olefin Hydrogenation by a Hetero-Dinuclear RuII-CoII Complex with Adjacent Reaction Sites. Inorg. Chem. 2019;58:11284–11288. doi: 10.1021/acs.inorgchem.9b02104. [DOI] [PubMed] [Google Scholar]
- 40.van den Beuken E.K., Feringa B.L. Bimetallic catalysis by late transition metal complexes. Tetrahedron. 1998;54:12985–13011. doi: 10.1016/S0040-4020(98)00319-6. [DOI] [Google Scholar]
- 41.Kim S., Loose F., Bezdek M.J., Wang X., Chirik P.J. Hydrogenation of N-Heteroarenes Using Rhodium Precatalysts: Reductive Elimination Leads to Formation of Multimetallic Clusters. J. Am. Chem. Soc. 2019;141:17900–17908. doi: 10.1021/jacs.9b09540. [DOI] [PubMed] [Google Scholar]
- 42.Hu Y., Norton J.R. Kinetics and thermodynamics of H-/H•/H+ transfer from a rhodium(III) hydride. J. Am. Chem. Soc. 2014;136:5938–5948. doi: 10.1021/ja412309j. [DOI] [PubMed] [Google Scholar]
- 43.Huber K.-P., Herzberg G. Molecular Spectra and Molecular Structure: IV. Constants of Diatomic Molecules. Springer; Boston, MA, USA: 1979. Springer US; Imprint. [Google Scholar]
- 44.DuBois D.L., Berning D.E. Hydricity of transition-metal hydrides and its role in CO2 reduction. Appl. Organometal. Chem. 2000;14:860–862. doi: 10.1002/1099-0739(200012)14:12<860::AID-AOC87>3.0.CO;2-A. [DOI] [Google Scholar]
- 45.Hu Y., Li L., Shaw A.P., Norton J.R., Sattler W., Rong Y. Synthesis, Electrochemistry, and Reactivity of New Iridium(III) and Rhodium(III) Hydrides. Organometallics. 2012;31:5058–5064. doi: 10.1021/om300398r. [DOI] [Google Scholar]
- 46.Evans M.G., Polanyi M. Inertia and driving force of chemical reactions. Trans. Faraday Soc. 1938;34:11. doi: 10.1039/tf9383400011. [DOI] [Google Scholar]
- 47.Alvarez R., Carmona E., Marin J.M., Poveda M.L., Gutierrez-Puebla E., Monge A. Carbon dioxide chemistry. Synthesis, properties, and structural characterization of stable bis(carbon dioxide) adducts of molybdenum. J. Am. Chem. Soc. 1986;108:2286–2294. doi: 10.1021/ja00269a026. [DOI] [PubMed] [Google Scholar]
- 48.Bernskoetter W.H., Tyler B.T. Kinetics and Mechanism of Molybdenum-Mediated Acrylate Formation from Carbon Dioxide and Ethylene. Organometallics. 2011;30:520–527. doi: 10.1021/om100891m. [DOI] [Google Scholar]
- 49.Carmona E., Munoz M.A., Perez P.J., Poveda M.L. Rotational isomerism in bis(carbon dioxide) complexes of molybdenum generated by conrotatory motion of the CO2 ligands. Organometallics. 1990;9:1337–1339. doi: 10.1021/om00118a077. [DOI] [Google Scholar]
- 50.Hanna B.S., MacIntosh A.D., Ahn S., Tyler B.T., Palmore G.T.R., Williard P.G., Bernskoetter W.H. Ancillary Ligand Effects on Carbon Dioxide-Ethylene Coupling at Zerovalent Molybdenum. Organometallics. 2014;33:3425–3432. doi: 10.1021/om500324h. [DOI] [Google Scholar]
- 51.Zhang Y., Hanna B.S., Dineen A., Williard P.G., Bernskoetter W.H. Functionalization of Carbon Dioxide with Ethylene at Molybdenum Hydride Complexes. Organometallics. 2013;32:3969–3979. doi: 10.1021/om400448m. [DOI] [Google Scholar]
- 52.Ziegler W., Nicholas K.M. Photochemistry of (Me3P)4Mo(η-CO2)2: Deoxygenation of coordinated carbon dioxide and phosphine oxidation. J. Organomet. Chem. 1992;423:C35–C37. doi: 10.1016/0022-328X(92)83136-6. [DOI] [Google Scholar]
- 53.Neese F. The ORCA program system. WIREs Comput. Mol. Sci. 2012;2:73–78. doi: 10.1002/wcms.81. [DOI] [Google Scholar]
- 54.Becke A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A Gen. Phys. 1988;38:3098–3100. doi: 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- 55.Lee C., Yang W., Parr R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter. 1988;37:785–789. doi: 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- 56.Weigend F., Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005;7:3297–3305. doi: 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- 57.Grimme S., Ehrlich S., Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011;32:1456–1465. doi: 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- 58.Grimme S., Antony J., Ehrlich S., Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010;132:154104. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 59.Barone V., Cossi M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A. 1998;102:1995–2001. doi: 10.1021/jp9716997. [DOI] [Google Scholar]
- 60.Bellan J., Marre M.R., Sanchez M., Wolf R. Nouveaux ylures du phosphore a partir e tris(amino)iminophosphanes et d’esters acetyleniques. etude structurale par RMN de 13C. Phosphorus Sulfur Silicon Relat. Elem. 1981;12:11–18. doi: 10.1080/03086648108078285. [DOI] [Google Scholar]
- 61.Buscagan T.M., Oyala P.H., Peters J.C. N2 -to-NH3 Conversion by a triphos-Iron Catalyst and Enhanced Turnover under Photolysis. Angew. Chem. Int. Ed. 2017;56:6921–6926. doi: 10.1002/anie.201703244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mankad N.P., Rivard E., Harkins S.B., Peters J.C. Structural snapshots of a flexible Cu2P2 core that accommodates the oxidation states CuICuI, Cu1.5Cu1.5, and CuIICuII. J. Am. Chem. Soc. 2005;127:16032–16033. doi: 10.1021/ja056071l. [DOI] [PubMed] [Google Scholar]
- 63.Li L., Brennessel W.W., Jones W.D. C−H Activation of Phenyl Imines and 2-Phenylpyridines with [Cp*MCl2]2 (M = Ir, Rh): Regioselectivity, Kinetics, and Mechanism. Organometallics. 2009;28:3492–3500. doi: 10.1021/om9000742. [DOI] [Google Scholar]
- 64.Kang J.W., Moseley K., Maitlis P.M. Pentamethylcyclopentadienylrhodium and -iridium halides. I. Synthesis and properties. J. Am. Chem. Soc. 1969;91:5970–5977. doi: 10.1021/ja01050a008. [DOI] [Google Scholar]
- 65.Harris R.K., Becker E.D., Cabral De Menezes S.M., Goodfellow R., Granger P. NMR nomenclature: Nuclear spin properties and conventions for chemical shifts (IUPAC recommendations 2001) Concepts Magn. Reson. 2002;14:326–346. doi: 10.1002/cmr.10035. [DOI] [PubMed] [Google Scholar]
- 66.Clark R.C., Reid J.S. The analytical calculation of absorption in multifaceted crystals. Acta. Crystallogr. A. 1995;51:887–897. doi: 10.1107/S0108767395007367. [DOI] [Google Scholar]
- 67.Rigaku Oxford Diffraction CrysAlisPro Software Systems, Rigaku Corporation, Oxford, UK 201-2020. [(accessed on 17 February 2023)]. Available online: https://www.rigaku.com/products/crystallography/crysalis.
- 68.Sheldrick G.M. SHELXT - integrated space-group and crystal-structure determination. Acta. Crystallogr. A. 2015;71:3–8. doi: 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gritzner G., Kuta J. Recommendations on reporting electrode potentials in nonaqueous solvents (Recommendations 1983) Pure Appl. Chem. 1984;56:461–466. doi: 10.1351/pac198456040461. [DOI] [Google Scholar]
- 70.Noviandri I., Brown K.N., Fleming D.S., Gulyas P.T., Lay P.A. The Decamethylferrocenium/Decamethylferrocene Redox Couple: A Superior Redox Standard to the Ferrocenium/Ferrocene Redox Couple for Studying Solvent Effects on the Thermodynamics of Electron Transfer. J Phys Chem B. 1999;103:6713–6722. doi: 10.1021/jp991381+. [DOI] [Google Scholar]
- 71.Burchat A.F., Chong J., Nielsen N. Titration of alkyllithiums with a simple reagent to a blue endpoint. J. Organomet. Chem. 1997;542:281–283. doi: 10.1016/S0022-328X(97)00143-5. [DOI] [Google Scholar]
- 72.Krasovskiy A., Knochel P. Convenient Titration Method for Organometallic Zinc, Magnesium, and Lanthanide Reagents. Synthesis. 2006;2006:890–891. doi: 10.1055/s-2006-926345. [DOI] [Google Scholar]
- 73.Albinati A., Bakhmutov V.I., Caulton K.G., Clot E., Eckert J., Eisenstein O., Gusev D.G., Grushin V.V., Hauger B.E. Reaction of molecular hydrogen (H2) with chlorohydridoiridium phosphines IrHCl2P2 (P = PiPr3 or PtBu2Ph): Stereoelectronic control of the stability of molecular H2 transition metal complexes. J. Am. Chem. Soc. 1993;115:7300–7312. doi: 10.1021/ja00069a032. [DOI] [Google Scholar]
- 74.Desrosiers P.J., Cai L., Lin Z., Richards R., Halpern J. Assessment of the "T1 criterion" for distinguishing between classical and nonclassical transition-metal hydrides: Hydride relaxation rates in tris(triarylphosphine)osmium tetrahydrides and related polyhydrides. J. Am. Chem. Soc. 1991;113:4173–4184. doi: 10.1021/ja00011a019. [DOI] [Google Scholar]
- 75.Kubas G.J. Dihydrogen complexes as prototypes for the coordination chemistry of saturated molecules. Proc. Natl. Acad. Sci. USA. 2007;104:6901–6907. doi: 10.1073/pnas.0609707104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kubas G.J. Activation of dihydrogen and coordination of molecular H2 on transition metals. J. Organomet. Chem. 2014;751:33–49. doi: 10.1016/j.jorganchem.2013.07.041. [DOI] [Google Scholar]
- 77.Spek A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. C. 2015;71:9–18. doi: 10.1107/S2053229614024929. [DOI] [PubMed] [Google Scholar]
- 78.Sheldrick G.M. A short history of SHELX. Acta. Crystallogr. A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
X-ray crystallography data can be accessed through the CCDC (2210067-2210072). All other data are given in the Supplementary Materials.