

Abstract
Single-cell proteomics is growing rapidly and has made several technological advancements. As most research has been focused on improving instrumentation and sample preparation methods, very little attention has been given to algorithms responsible for identifying and quantifying proteins. Given the inherent difference between bulk data and single-cell data, it is necessary to realize that current algorithms being employed on single-cell data were designed for bulk data and have underlying assumptions that may not hold true for single-cell data. In order to develop and optimize algorithms for single-cell data, we need to characterize the differences between single-cell data and bulk data and assess how current algorithms perform on single-cell data. Here, we present a review of algorithms responsible for identifying and quantifying peptides and proteins. We will give a review of how each type of algorithm works, assumptions it relies on, how it performs on single-cell data, and possible optimizations and solutions that could be used to address the differences in single-cell data.
Keywords: bioinformatics, peptide identification, quantification, algorithm, single-cell proteomics
Graphical Abstract
Highlights
-
•
Single cell proteomics (SCP) data is rapidly becoming available.
-
•
Proteomics workflows and algorithms were optimized for bulk datasets.
-
•
The low signal of SCP data challenges assumptions of computational workflows.
-
•
Tradeoff between identification and quantification in experimental designs.
-
•
Uncertainty in protein quant from high percentage of missing peptide abundances.
In Brief
Single-cell proteomics is growing rapidly, but most research has focused on improving instrumentation and sample preparation. Current algorithms being employed on single-cell data were designed for bulk data and have underlying assumptions that may not hold true for single-cell data. Here, we present a review of algorithms for identifying and quantifying peptides and proteins, including how each type of algorithm works, assumptions it relies on, and possible optimizations for single-cell data.
Complex biological systems consist of numerous individual cells sensing and responding to their environment. Most systems include a variety of heterogeneous cells and therefore our understanding of the system compels us to know these individual and diverse cells through single-cell measurements. Quantifying the proteins within individual cells is essential to characterize the cell, since proteins perform most cellular functions. Mass spectrometry–based proteomics enables high throughput profiling of proteins within a sample and has proved to be valuable in cellular biology and biomedical research (1, 2, 3, 4, 5). Traditional proteomics workflows analyze all cells in a sample together and therefore measure population averages. This not only confounds differences that exist between individual cells but also makes it difficult to detect cell-type–specific proteins which are low-abundance in the bulk homogenate (6, 7). To address these problems, researchers are rapidly improving the capabilities for single-cell proteomics (SCP).
Current progress in SCP has been heavily focused on developing methods for sample preparation, chromatography, and data acquisition. These advancements have been previously reviewed (8, 9, 10). It is important to understand that changes to data acquisition parameters can affect algorithm performance in unexpected ways—specifically, they may improve data quality for peptide identification but detract from quantitative accuracy. This tradeoff is described in detail below, but a motivating example here will suffice. Various methods have been used to increase the number of ions collected for an MS2 spectrum. In a label-free experiment, the chromatography may be shortened and/or the ion acquisition time increased, thus delivering more peptide ions for fragmentation which improves the likelihood of achieving a confident peptide spectrum match (PSM). However, these actions also significantly reduce the number of MS1 spectra acquired during a peptide’s elution and could potentially harm quantitative accuracy. Thus, as experimental researchers are varying data acquisition parameters, they should be aware of how this might affect algorithms for both identification and quantification.
After significant progress in overcoming major technical hurdles for generating SCP data, attention is now turning to the algorithms used to identify and quantify proteins (11). Although a diverse set of data acquisition strategies are used in SCP, data analysis for all of these methodologies generally involves four steps: 1) peptide identification, 2) peptide quantitation, 3) protein identification, and 4) protein quantitation (12, 13). As no dedicated software yet exists for SCP analysis, researchers are currently employing algorithms that were developed for bulk data. However, single-cell data is fundamentally different from bulk data and algorithms may need to adapt for optimal performance (14). To assist researchers in developing and optimizing algorithms for single-cell data, this review analyzes common algorithms and workflows used in each of these four fundamental steps of mass spectrometry data processing. The adaptation of downstream analysis of quantitative SCP data has also started (15, 16). However, the focus of this review is to explain important algorithmic assumptions in each of the four processing steps used in quantitative proteomics, their challenges, and potential solutions for SCP.
Due to the recent emergence of single-cell proteomics and its strong focus on the development of experimental methods, the published literature on computational methods for data analysis is very limited. Therefore, this manuscript is a hybrid review of sorts. Our goal is not to provide a review of general computational methods and software developed for proteomics, as many quality reviews already exist (17, 18, 19, 20, 21); but rather our goal is to point out where common assumptions used in software for bulk proteomics will be problematic for SCP. Thus, we review the published peer-reviewed manuscripts but also provide context for unaddressed challenges.
Peptide Identification
This review is limited to bottom-up proteomics, where proteins are digested into peptides and the peptides are analyzed in the mass spectrometer. Moreover, we are primarily concerned with the aspects of peptide identification that are problematic in SCP. Two common methods exist for data acquisition in bottom-up proteomics: data-dependent analysis (DDA) and data-independent analysis (DIA). For both DIA and DDA, the first data analysis step is to identify peptides from fragmentation spectra. The data generation and analysis algorithms are different for DDA and DIA methods, and we will address each one separately. After scoring PSMs, both DDA and DIA methods assign statistical probability to the match, often using the target/decoy model (22) or a mixture modeling approach (23). The data from SCP does not change the assumptions of these approaches and so will not be discussed here.
DDA Peptide Identification
Data-dependent acquisition identifies highly abundant ions in MS1 spectra and isolates and fragments them for measurement in an MS2 spectrum (24). The most common analysis method for this data type is a database search where algorithms identify the most likely candidate peptide for each spectrum. Database search algorithms began in the 1990s (25) and have continued to evolve with new advances for novel search methods (26, 27), improved speed (28), rigorous statistical confidence (23, 29), identification of protein modifications (30, 31), combining search engines (32), open source software (33), compatibility for multiple fragmentation methods (34, 35), and user friendly and robust tools (36). The specific implementation of scoring to judge PSMs is unique to each algorithm, however, they share common fundamental assumptions. In general, these tools generate a theoretical spectrum for each candidate peptide which is compared to the observed spectrum and scored to reflect the quality of the match (reviewed in (37)). The highest scoring peptide is assigned to the spectra. Although each software tool may use slightly different metrics, there are two main features which are fundamental and common to all: the number of annotated fragment ions is expected to be high for the correct PSM and annotated ions are expected to be more intense than background/noise ions.
In previous work, we characterized these two essential scoring metrics by comparing single-cell spectra to bulk spectra (14). Figure 1 illustrates the differences between single-cell spectra and bulk spectra. We found the number of annotated fragment ion peaks to be reduced in single-cell spectra. This result is expected, as the lower ion signal diminishes the probability of observing low intensity fragment ions. Since most algorithms rely heavily on the number matching peaks between the observed and theoretical spectrum, single-cell PSMs will get a lower score and therefore be more likely to be excluded by false-discovery rate (FDR) cutoffs. We also investigated the difference in intensity between annotated peaks and background noise. In single-cell data, we found a compressed ratio of annotated peaks to background noise (S/N ratio). This compression makes it more difficult for algorithms to distinguish true fragment ion peaks. The reduced number of ions and the compressed S/N ratio make it more challenging to confidently assign a peptide to a spectrum, resulting in fewer PSMs passing FDR cutoffs. While this work was done on label-free data, it is reasonable to expect similar results in tandem mass tag (TMT) data. In the SCoPE protocol, many cells are combined into a ‘carrier channel’ to increase the number of ions and therefore improve MS2 signal. The increase in ions improves peptide identification yet substantially affects quantitative accuracy as discussed below in the section on quantification.
Fig. 1.

Comparison of single-cell and bulk MS2. For the peptide GFAFVTFDDHDSVDK, the top spectrum is from single-cell data, and the bottom spectrum is from bulk data. The bulk spectrum has 17 annotated y-ions and single-cell only has five annotated y-ions. A similar fraction of b-ions are lost in single cell spectra. The lost peaks are mostly from medium and low intensity peaks.
DIA Peptide Identification
In DIA, an MS2 spectrum is acquired by isolating and fragmenting a broad m/z range which typically contains multiple peptides (38). This m/z range is fragmented repeatedly throughout the experiment, including many times during the chromatographic elution time of a single peptide. Thus, DIA spectra require a different method to identify peptides in a complex multiplexed spectrum (39, 40, 41). As with the previous section, our goal is not to exhaustively review all experimental or computational DIA methodologies; there are numerous variants of DIA which have been developed and optimized for specific needs, and these have been reviewed and benchmarked elsewhere (42, 43). Rather, our focus is to analyze their commonalities and how the basic parameters of DIA might be challenged by the data from SCP.
One common method for DIA data analysis compares observed spectra and chromatographic traces of ions to data from a spectral library (44, 45). Library optimization is a significant computational and experimental topic for the analysis of bulk samples with DIA (46, 47). Thus, it is natural to ask how library optimization can improve the results of SCP-DIA. In single-cell DIA data, we can expect the loss of input ions to create spectra that are noticeably different from bulk DIA data, due to the loss of numerous medium and low intensity fragment ions. This suggests that building spectral libraries from single-cell data would improve peptide identification. Siyal et al. rigorously explored the effect of input sample size for library creation on the effectiveness of DIA peptide identification. They created three libraries of varying sizes: a small library of 1.5 ng (∼10 cells), a medium library of 15 ng (∼100 cells), and a large library of 1 μg (∼5000 cells). A comprehensive spectral library created from multiple bulk experiments with cell lines and human samples was created as a control. They benchmarked the number of proteins identified using a digest of PC-9 cells of various amounts. DIA data was created with 0.75 ng (∼5 cells) and analyzed using the four different libraries. Two thousand three hundred eighty protein groups were identified with the small library, 1908 with the medium library, and 1749 with the large library. Using the comprehensive library, only 107 protein groups were identified. Thus, the protein identifications dramatically increased when using single-cell data to create the library as compared to a library created from ‘small input’. Comparing single-cell data against a typical comprehensive library was completely unsuccessful. Their results were consistent with multiple software tools (Spectronaut and DIA-NN) (48). Other optimizations of DIA for SCP have been mostly experimental (not computational), including adaptations to the chromatography gradient to concentrate ion signals and help them rise above noise (49).
In addition to library-based methods, there are also library-free DIA methods. These tools associate chromatograms for peptides and fragment ions without relying on a library to describe observed fragment ions. Scoring can happen through a pseudospectrum, as with DIA-umpire (40) or directly on chromatograms as with DIA-NN (50). These tools would likely be affected by single-cell data in the same way that label-free quantification (LFQ) chromatogram detection would be due to weaker signal, poor correlation between isotopes, and rough peak elution edges (see Section Label-free quantification).
To help improve DIA accuracy, Demichev et al. (51) incorporated both technical and computational methods in their pipeline that leverage ion-mobility separation. This improved sensitivity by making it easier to associate the chromatograms of precursors with their fragment ions using a multidimensional peak-picking algorithm for ion mobility data. An optimized spectral library and neural network algorithm are used to score precursor-spectrum matches. Their tool was assessed at low sample amounts (10 ng or ∼100 cells). We anticipate that these improvements will be beneficial at the single-cell level.
Possible Solutions to Improve Peptide Identification
Given the recent emergence of SCP, there are no peptide identification algorithms that have been developed specifically for single-cell data. However, with the observations above, there are few obvious optimizations that could be made. 1) All database search algorithms currently rely on the number of annotated peaks that are matched between the observed and theoretical spectrum. The limited number of observed peaks suggests that algorithms should compensate and incorporate expected fragment ion abundances into part of their PSM score. As spectral libraries provide a realistic spectrum that contains intensity differences between peaks, this should become a more significant part of the algorithm. 2) Some database search tools mimic a ‘library search’ by leveraging a MS2 prediction in their score (52). However, this is typically used very late in the algorithm workflow at the stage of p-value assignment. Putting this earlier in the workflow would significantly improve sensitivity. 3) Since there are significant differences between bulk spectra and single-cell spectra, a spectral library should be created from single-cell data, rather than bulk data. If using computational predictions of libraries (52), this should also be trained on the ultra low input of single-cell data to ensure it accurately predicts the substantial loss of fragment ions, as was done in DeepSCP (53). This will improve the accuracy of matches being made between the observed spectrum and the theoretical spectrum. 4) It has been recently shown that PSM identifications significantly improve when using a rescoring tool like Percolator or MokaPot (54, 55). The general advantage of this method is that it uses a rich set of features to calculate a score, instead of the simplistic ‘peak count’ often used in database search tools. The Dart-ID method corroborates this improvement with the inclusion of retention time as a PSM feature (56). Additionally MS2rescore (57), while developed for nontryptic immunopeptidome data, also demonstrates improved scoring with an expanded feature set. Thus, we suggest that peptide identification algorithms for SCP expand their feature space to include a diverse set of metrics and leverage these in semisupervised post hoc scoring. We also note that the differences between single-cell MS2 data and bulk may be instrument-specific and vary based on the physics of fragmentation and ion detection (58).
Peptide Quantitation
After peptides have been identified, they need to be quantified. The method of quantitation is different for label-free and labeled (multiplexed) experimental designs. LFQ methods calculate abundances based on the ion intensity in the MS1 chromatogram (or potentially MS2 in DIA). Multiplexed experiments that use isobaric labeling (TMT) quantitation instead measure the intensity of the reporter ions that appear in MS2 spectra. In this section, we will review work being done for both LFQ and TMT peptide quantitation algorithms.
Label-free Quantification
A peptide’s abundance value is most often calculated by the area of the extracted ion chromatogram (XIC), which reflects the number of ions detected for a peptide. An example XIC for several peptides from single-cell data is shown in Figure 2. Computing the abundance metric varies between software tools. Some tools simply sum intensities, some smooth the data prior to summing (36, 59), and some perform more computationally intense signal processing, e.g., Savitzky-Golay (60). Alternatively, some software tools do not use XICs but rather report the maximal abundance of an ion’s elution (61).
Fig. 2.

Example XIC of peptide ions for single-cell data. Four chromatograms were selected to represent challenging, but not uncommon, scenarios that occur in single-cell data. They demonstrate three challenges with peptide quantification: peak boundary detection, poor correlation within the isotopic peaks, and sporadic and low number of MS1 measurements. These were sampled to represent a range of intensities. Panel A demonstrates a good looking XIC, but the +2 isotope does not correlate well. Panel B shows a much worse chromatogram, where there is coelution interference, and the peak boundaries are difficult to define. Panel D also struggled to define peak boundaries. Panel C and D both have a nonsmooth XIC. XIC, extracted ion chromatogram.
In single-cell proteomics, there are fewer ions for each peptide. Although there are no currently published manuscripts exploring how this affects quantitative accuracy, we recognize several potential problems. Figure 2 demonstrates these problems and shows examples across the intensity range of MS1 peaks. At higher abundances, chromatograms generally look better, but few peptides in SCP have high abundance relative to the norms on which most algorithms are trained and evaluated.
The first challenge is that an ion’s signal is less intense and is closer to the level of noise. This can impact boundary identification and other aspects of peak detection, which require high signal-to-noise. Some algorithms might face a second challenge because of a loss in correlation between the XICs within the isotopic profile. As seen in Figure 2, the most common problem is that correlations between the isotope chromatograms are often poor. If algorithms require strict correlation in the peak detection methods, it is likely that many peptide ions will not be discovered. The next challenge we anticipate with LFQ peptide quantification arises because the ion accumulation time for MS2 spectra is dramatically longer for single-cell data, increasing the spacing between sequential MS1 and yielding fewer measurements across an ion’s elution profile. In SCP, some researchers are shortening the LC gradient in an attempt to tighten a peak’s elution and increase signal. However, this exacerbates the problems associated with having too few measurements across an elution profile (62, 63). A final subtle challenge for single-cell peptide quantification is that the time interval between successive MS1 scans can be highly irregular. This comes from both a variable number of MS2 spectra acquired as well as the widely varying time for ion accumulation. To our knowledge, only the Skyline algorithm accounts for this type of temporal irregularity in calculating ion quantity, by resampling the chromatogram (60). Other algorithms which simply sum MS1 scans or do a kernel smoothing would be significantly affected in quantitative accuracy.
Match Between Runs
Match between runs (MBR) was developed to address situations where ions were measured in MS1 spectra but not selected for MS2 identification (36, 64). To rescue these potential peptides, MBR transfers the peptide identification from another run based on the ion’s mass and aligned retention time, thus improving peptide identification coverage within an experiment. This technique is part of many software tools as an intermediate step between peptide identification and peptide quantification. One of the most important advances to be made to MBR was the development of a statistical confidence measure of each transfer, facilitating FDR-based cutoffs and improving accuracy (65). As MS2 identifications are generally more stochastic with single-cell data, MBR plays a relatively larger role in peptide identification/quantification.
Two recent papers have adapted the MBR technique for the unique challenges of SCP. Woo et al. (66) recently developed TIFF to improve sensitivity and accuracy. The TIFF experimental workflow uses FAIMS to filter out background and +1 ions to increase signal for peptide ions. Moreover, the TIFF algorithm incorporates the FAIMS CV into the MBR match to supplement the traditional m/z and retention time. By including a third metric into the matching criteria, they report a significant increase in sensitivity and accuracy. A second tool, IceR, seeks to improve MBR for SCP by improving ion detection in ultra-low input samples (67). As MBR was developed for bulk data with rich ion features, most peak detection methods require molecular ion peaks in MS1 spectra to have peaks that correspond to the isotopic envelope. However, ion abundances in single-cell data are so low that isotopic peaks sometimes are not detected or have poor correlation (see Fig. 2 and discussion above). Thus, the IceR implementation of MBR does not require an isotopic envelope for peak detection, which significantly reduces missing data for peptide quantification.
Tandem Mass Tag
In multiplexed isobaric labeling with tandem mass tags (TMTs), each sample is labeled with an isobaric tag. After labeling each individual sample, all samples are combined and analyzed via LC-MS/MS. Due to the unique placement of the isotopes, each tag produces a unique fragment ion in MS2 spectra which is used to measure abundance (i.e., reporter ion) (68). Multiplexed experiments are especially valuable in single-cell proteomics, as combining samples increase signal intensity and yield a better MS2 spectrum. To further increase signal, the SCoPE-MS method utilizes a carrier proteome containing dozens to hundreds of cells to boost the signal (69). However, caution must be used when implementing the carrier proteome. While the increase in ions improves peptide identification, there is a tradeoff with protein quantitation accuracy (70, 71). Cheung et al. (70) recommend a carrier limit of ∼20 to 50× along with SNR filtering, but note that higher carrier might be used if instrument setting and postanalysis filtering is carefully optimized. They found that high carrier amounts significantly under samples the number of ions for other reporter channels, making it more difficult to distinguish true differences between single-cells and limiting quantitative accuracy. Similarly, Ye et al. (71) suggest the most accurate quantitation is achieved with a carrier limit of 42×, but 98× could achieve an acceptable balance between identification and quantification. Thus, both manuscripts recommend a carrier proteome limit of ∼50×, while allowing up to ∼100×.
Beyond undersampling of the reporter ion channels, a high carrier proteome also distorts the quantitation of nearby channels due to impurities in the reagent. To demonstrate the impact of this bleedover of TMT signal from carrier channel to other channels, we analyzed a recently published unique TMT experiment (72). While this experiment was not designed specifically to study the effects of the carrier proteome, it omits a large number of reporter ion channels and thus provides important insight into the magnitude of bleedover. Figure 3 shows the intensity of all reporter ion channels across 6451 high confidence PSMs. Channel 126 is the boost of 10 ng (∼100 cells), and 127N is a reference of 0.5 ng (∼5 cells). Nine channels were used for 0.1 ng (single-cell amounts) of three different cell types: channels 130N, 131C, and 133N are epithelial cells, channels 130C, 132N, and 133C are immune cells, and channels 131N, 132C, and 134N are endothelial cells. Importantly, channels 127C, 128N, 128C, 129N, and 129C were not used and thus any signal detected in them is noise. When using a large number of cells in the 126 channel, researchers typically leave 127C blank, as a 13C impurity in the 126 reagent would show up as signal in 127C. As shown in Figure 3, channel 127C has a high signal, similar to the abundance of the reference channel. Because this dataset left four other channels blank, we are able to see whether the bleedover from the carrier channel and reference channel extend beyond the expected 127C. Channel 128C has high intensity that is most likely to be attributed to a double 13C impurity in the 126 carrier channel. The intensity in 128N is likely to be a 13C impurity from the 127N reference channel. Indeed, both 128N and 128C have intensity similar to 130N, which is data for a single epithelial cell. This bleedover beyond the one standard blank, 127C, was also noted by Ye et al. (71). Thus, with a 100× carrier channel, we see significant bleedover into many channels that would typically be used for experiments.
Fig. 3.

Measured signal intensity in blank TMT channels. The intensity of each TMT reporter ion is plotted for 6451 PSMs filtered to 1% FDR. Shown in rose colored boxes are two channels which are not a single cell: channel 126 is used for the 100× carrier, and channel 127N is used for the 5× reference sample. Shown in gray boxes are the blank channels. Channels 127C, 128N, 128C, 129N, and 129C were omitted from the experimental design, yet they contain noticeable measured signal. Shown in yellow, blue, and red boxes are channels for the single cells from three different cell lines, as described in reference (72). FDR, false-discovery rate; PSM, peptide spectrum match.
Several methods are currently used to improve quantitative accuracy in TMT-labeled SCP, however, we note that they are experimental and not computational improvements. First, in an attempt to remove the effect of coisolated precursor ions, researchers have utilized the SPS-MS3 method developed in bulk studies (73, 74). Here multiple intense fragment ions in MS2 are chosen for MS3. This second round of isolation and fragmentation removes the contaminating signal from potential cofragmented precursor ions, avoiding the quantitative inaccuracies caused by ‘ratio compression’ (75). Although this has shown to be very impactful for quantitative accuracy in bulk studies, there are several challenges with regards to SCP. First, creating an MS2 and MS3 spectrum for each peptide ion results in significantly reduced peptide/protein coverage (74). In an attempt to overcome this time cost, researchers have explored using the faster linear ion trap (76). Although this improves peptide identification, there are two drawbacks: 1) the inherently lower mass accuracy of the ion trap limits the number of TMT channels that can be used in an experiment and 2) the protein quantification in the ion trap had a substantially higher %CV than an Orbitrap. A second challenge for using MS3 with SCP is that the extremely limited number of ions present in single cell data means that secondary selection for MS3 spectra will potentially not have enough ions for meaningful quantitation, especially for experiments using a large carrier proteome. Third, as shown in Figure 3, quantitative accuracy of SCP is significantly impacted by the bleedover of the carrier channel, which is not resolved by SPS-MS3. Therefore, a second essential experimental design consideration for multiplexed SCP is the inclusion of a blank channel within the multiplexed design. The intensity in this blank channel is a reliable measure of noise, which may arise for myriad cases. Using the blank channel to quantify noise allows for a simple calculation of whether specific PSMs have sufficient signal/noise to be considered quantifiable.
Possible Solutions to Improve Peptide Quantification
While there have been various implementations of MBR designed to account for a specific difference in single-cell data (66, 67), we lack an in-depth analysis of how MBR is performing on single-cell data. One fundamental difference that has not been accounted for yet is that MBR was developed for bulk data, where ions are plentiful and time is limited. This contrast leads to data acquisition where it is not possible to select every peptide for MS2 fragmentation. Single-cell data is the opposite. The limiting factor is the paucity of ions. MBR could be greatly improved for single-cell data by accounting for base assumptions that were made for bulk data that are not true for single-cell data.
In proteomics, there is a long history of separately defining the limit of detection from the limit of quantification, especially in targeted proteomics (77, 78). Some analytes are detectable, but not reliably quantifiable. We believe that this distinction will be very important for SCP, as the ultra low sample input often means that analytes are between the limit of detection and limit of quantification. Thus, researchers may need to be more obvious and direct in the reporting of SCP results, bluntly stating that many peptides are detectable but not quantifiable. This may be unpopular as it would reduce the overall depth of proteome coverage reported in a manuscript, but we believe it is an essential part of research integrity. Towards this end, the SCPCompanion tool is designed for TMT multiplexed single-cell data and calculates a signal/noise ratios for all PSMs based on the instrument’s native S/N calculation (70). SCPCompanion suggests a cutoff used on filtering data which is nonquantifiable. Additionally, we strongly suggest that ‘blanks’ be more heavily relied upon for experimental design; questionably quantified analytes are easy to find for both TMT and LFQ studies that include blanks to explicitly measure noise. Although blanks are often mentioned in the experimental design section, many manuscripts are curiously silent about how these are utilized in data analysis.
Protein Identification and Quantitation
In bottom-up proteomics, the measured analyte is a peptide. Yet, the ultimate goal of proteomics is often to identify and quantify proteins. In proteolytic digestion, the direct connection between peptides and protein is lost. Inferring which peptides belong to which proteins and determining a protein’s quantitative value has both theoretical and algorithmic challenges (79, 80). We will give a brief overview of how protein identification and quantitation work—pointing the reader to more thorough reviews and benchmarks (77, 81, 82, 83)—and then discuss the challenges for single-cell proteomics.
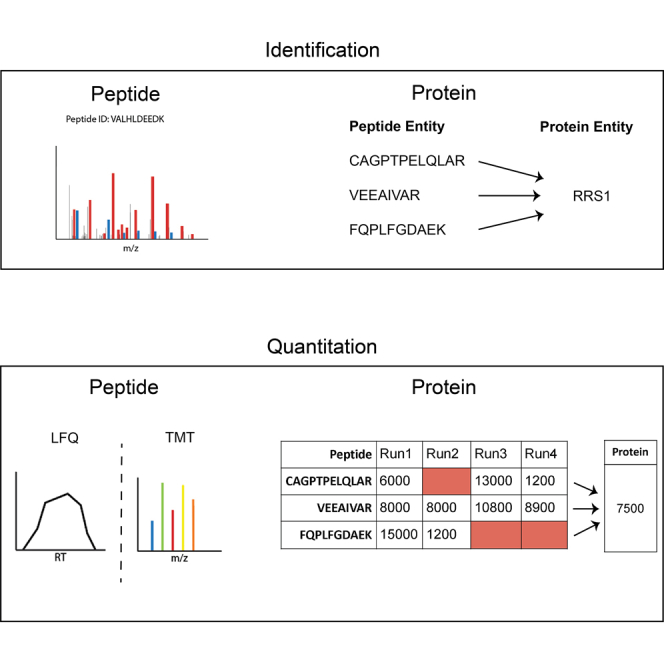
Protein identification algorithms group peptide sequences into a protein. If each peptide uniquely mapped to a single protein, this would be a trivial step. Since peptides often belong to multiple proteins, assigning peptides to proteins becomes a much more complex problem (84, 85). The most common method used for protein identification is parsimony, which derives the minimal protein list that is sufficient to account for all identified peptides (86). Two other solutions are to completely filter out peptides that map to multiple proteins or to assign shared peptides to the protein group with the greatest number of matching peptides (the razor solution). However, condensing peptide measurements into a single protein report may potentially obscure biologically relevant differences between related protein isoforms (87, 88). To avoid these problems with protein-based quantification, some researchers have advocated for working more directly with peptide abundances to identify significant differences between samples (89, 90, 91, 92). As peptide-centric methods have yet to be used in SCP, this review will focus on protein-level quantification.
Proteins are quantified by summarizing peptide level abundances into a single protein value. The most common types of methods are sum_all, topN, and median value. As suggested by their names, these methods combine peptide abundances into a single, protein-level abundance using basic mathematical operations. For example, the ‘sum_all’ method calculates the protein abundance by summing all peptide abundances; the ‘median value’ method calculates the protein abundance from the median abundance value of constituent peptides. There are many differences in specific implementations of each of these general types, e.g., simple median versus Tukey’s median polish; sum all versus iBAQ. The quantitative accuracy of various methods and implementations for bulk proteomics has been reviewed elsewhere (93). Currently, there is no consistently used method within the SCP community (11).
Challenges
In SCP, the sparseness of the peptide quantitative data matrix is likely to make protein quantification a significant challenge. Regardless of the details of algorithmic implementation, these methods implicitly require a large number of consistently quantified peptides per protein for stable and reliable summarization (87). Although algorithmic details of protein summarization are poorly described for many software tools, we review below how the common methods for sum_all, topN, and median might be challenged with SCP data. The problems outlined below are not unique to SCP. But they are much more prominent and should be directly addressed as they are no longer simply an ‘edge case.’
One of the most significant adjustments that algorithms will need to make is the expectation on how many peptides can contribute to the summarization formula and the level of sparsity in peptide quantification tables. For example, the topN method is most often implemented as Top3. However, this begs the question for how often proteins in SCP have at least three peptides, and is there consistent data for these three peptides? If choosing a Top3 method and not doing an explicit peptide abundance imputation, there is an implicit imputation of zero for any missing data.
To demonstrate challenges with quantitation, we use six LC-MS/MS technical replicates of a HeLa digest (14). We quantified the number of consistent peptides per protein, where consistent peptides are defined as being present in 6/6 replicate runs. We chose this strict definition of ‘consistent’ as it is most clear for the discussion, although we recognize that algorithms may choose a different cutoff or have no cutoff at all. We found that of the 1420 proteins, 29% did not have any consistent peptides, 24% had one consistent peptide, 11% had two consistent peptides, 8% had three consistent peptides, and 28% had more than three consistent peptides (Fig. 4).
Fig. 4.

The number of consistent and inconsistent peptides in each protein. Proteins were categorized based on the number of consistent peptides (x-axis) and subdivided into the number of inconsistent peptides (colors within the stacked bar). For example, the first stacked bar counts how many proteins have no consistent peptides (∼400 proteins) and then subdivides it into the number of inconsistent peptides each protein contains. The majority have one inconsistent peptide (∼250 proteins in orange), meaning that proteins that do not contain any consistent peptides usually; only have one inconsistent peptide. Accurately quantifying such proteins would be very difficult.
We discuss the potential for volatility in protein quantification with three examples where there are fewer than three consistent peptides. In each example, the choice of summarization method (sum_all, topN, or median) has a dramatic impact on the reported protein quantitation value. Since there are not three consistent peptides in these examples, algorithms are forced to deal with inconsistent peptides which have missing data. We note that very few software tools describe how peptide (in)consistency is addressed, i.e., how are peptides filtered prior to summarization. They are similarly unclear about whether imputation is used on the peptide quantification matrix to partially ameliorate missing data prior to protein summarization. As we show in each of the demonstrative examples below, these decisions are critical. In examining the examples, assume that these are the final peptide quantitation values used as input to the protein summarization method. Any imputation, normalization, or filtering has already happened.
Figure 5A shows the metabolic protein Aconitase (ACO2), which is supported by two inconsistent peptides. Each peptide has similar amounts of missingness, but the missing values occur in different samples. In only one run (run5), both peptides are observed. Consider how sum_all and a median summarization method would perform differently on this protein. Sum_all would add the quantitative value of both peptides and report that the protein is most abundant in run5. We note again for emphasis that the sum_all method here does an implicit imputation of zero for the intensity of peptides with missing data. The median method would report nearly the opposite. With two values in run5, the median method would average the two peptide abundances, and therefore, rank run5 near the bottom compared to other runs in the experiment.
Fig. 5.

Proteins that contain inconsistent peptides. Each protein illustrates how quantitation methods are not designed to handle sparse data. The protein in panel A only contains inconsistent peptides and will be quantified differently depending on the summarization method used. The proteins in B and C illustrate that filtering out inconsistent peptides will drastically alter the results of quantitation.
Figure 5B shows ribosome related protein RRS1 which is supported by one consistent peptide and two inconsistent peptides. A topN or sum_all method would nominally consider all three peptides in creating a protein abundance. If all three peptides are used for quantitation, then the protein is highly abundant in run1, the second most abundant in the whole experiment. However, peptide VEEAIVAR is only present in 2/6 runs. If this peptide were to be filtered out prior to summarization due to inconsistent quantification, then run1 would rank near the bottom of the experiment. If this peptide were dropped, and then a median methodology was used, run1 would be at the very bottom. As mentioned above, most software tools do not describe this step of protein quantification in manuscripts or supplemental methods. Thus, it is not trivial to determine how scenarios such as this are resolved.
Figure 5C shows the peptide quantitation table for the metabolic protein phosphoglucomutase (PGM1). This protein has six identified peptides; two peptides are consistent and observed in every run within the experiment. Four peptides are inconsistent across the experiment. Consider how top3 would perform in this example. Several inconsistent peptides have the highest abundance, which would suggest their inclusion in the top3 calculation. Depending on how the three peptides are chosen (or perhaps fewer if three meritorious peptides are not found), the protein could be most abundant in run 1, 5, or 6. Similar volatility exists for this example protein using the sum_all or median method.
The thought experiment described above is provocative. Indeed, as standard proteomics software tools lack any clear documentation on how these situations are resolved, it is difficult to know how protein quantification is affected in reality by incomplete and sparse peptide quantification matrices. Fortunately, the scp library in R has implemented this step of protein rollup using a variety of methods (15). Using scp and the peptide matrices from Figure 5, we created the abundances for ACO2, RRS1, and PGM1 (Fig. 6) using the sum, top3, and median methods. As suggested by the thought experiment, we observe significant disagreement as to which run contains the most abundant protein. For example, in the quantification of PGM1, run6 could be the highest or lowest in abundance depending on the method that was used.
Fig. 6.

Various quantitation methods performed on proteins containing inconsistent peptides. The 'peptides' column shows the abundance of individual peptides for each example protein (ACO2, RRS1, and PGM1). The columns 'medians', 'sums', and 'top3′ show the quantitation result for each respective method, as calculated by the scp package. Note the difference between each method, particularly the median method and sum/top3.
One final note on protein quantification is the impact of imputation, which can be applied to either the peptide and/or protein abundance matrices. Computational methods for imputation are diverse and have been compared and reviewed elsewhere (94, 95, 96, 97, 98, 99), and the choice of method can affect the downstream interpretation of the experiment (100). Historically, many benchmarking comparisons and tool development efforts are pursued at the level of protein imputation (101, 102, 103). However, as seen in Figures 4 and 5, missing data is prevalent in peptide quantitation and has a significant impact on the resulting protein abundance calculation. Since missing data is more common in SCP than bulk proteomics, strategies should be evaluated in SCP for both peptide and protein-level imputation.
Possible Solutions
Protein quantitation methods for single-cell data must consider whether or not peptides making up a protein are consistent, since the more consistent a peptide is, the more reliable the quantitation will be. The proteins shown in Figures 5 and 6 demonstrate a volatility, depending on the type of protein summarization method employed. The confounding nature of these example proteins is quite common in SCP, with the vast majority of protein identifications having fewer than three consistent peptides. The importance of considering peptide consistency was demonstrated in a recent paper by the Vitek group, in which they developed an automated statistical approach that performs protein quantification using an informative subset of features (104). Feature is a generic term that refers to precursor ions in the case of DDA data, transitions in SRM data, and fragments in DIA data. Informative features are features that have few missing values, and for which intensity values are consistent with most of the protein intensity profiles across runs. Their method detects features that have inconsistent patterns, which can then be investigated and removed, facilitating more accurate peptide-level data. The suggested data presence criteria that comes from this method depends on the experimental design. Given that peptide quantitation matrices in SCP are much more sparse than in traditional bulk proteomics, we strongly suggest research into how peptide consistency affects protein quantitation. We also advocate for more transparency and better description of the summarization method in manuscripts and documentation of various software tools.
Conclusion
Single-cell proteomics has been making great strides in sample preparation, separation technologies, and measurement speed and sensitivity. However, the research to improve algorithms that interpret the resulting data is just beginning. This review considers the main types of algorithms used for peptide and protein identification and quantification, challenges of applying them to single-cell data, and possible solutions. Importantly, we note that every part of this identification/quantification workflow faces significant challenges, and thus all are targets for improvement. We note that none of the problems described in this review are exclusive to SCP. Rather, they were often ‘edge cases’ or rare occurrences that were not explicitly treated in bulk data. However, for SCP, the above challenges are major computational roadblocks that need to be addressed.
Peptide identification algorithms rely heavily on the number of peaks matched and the intensity of peptide ions being far above background noise. While these conditionals are met in bulk data, single-cell data has fewer ion peaks and the S/N ratio is substantially compressed. Some possible solutions are to 1) use a spectral library created from single-cell data, 2) use predicted MS2 spectra trained on single-cell data, 3) use semisupervised post hoc scoring tools, and 4) expand the feature space to include a diverse set of metrics. Peptide quantitation is also challenged as a result of fewer ions being detected, and the signal of peptide ions is much closer to the level of noise in comparison to bulk data. LFQ could struggle with multiple facets of feature finding and accurate quantitative calculation. TMT methods incorporate a carrier channel to improve peptide identification, however this impairs quantitative accuracy. The data generation methods have been exploring possible solutions, yet these have not often investigated the potential tradeoffs between identification and quantification, with several choices improving identification yet harming quantification. For example, shortening LC gradients improves ion signals but limits MS1 sampling which complicates quantitation. Moving forward, we need a greater integration between data generating and data analysis teams to explore viable solutions. Finally, protein quantitation algorithms rely on the presence of several consistent peptides. However, in single-cell data, we see a considerable amount of proteins with inconsistent peptides that the peptide quantitation algorithms are not robust enough to deal with them. There needs to be research into protein quantitation algorithms that can handle sparse data.
Data Availability
The data used in these analyses are six replicates of 0.2 ng. The data is uploaded to Massive (MSV000087524), and all code used to generate figures and analyze results can be found in the github repository, https://github.com/PayneLab/SCP_comp_review.
Conflict of interest
The authors declare no competing interests.
Acknowledgments
We thank members of the Payne lab for helpful discussion and critiques of the manuscript. We also thank a reviewer for suggesting Figure 6 and providing working code to create it. This work was supported by an NIGMS /NIH award (1R01GM147653) and a Sponsored Research Agreement from Biogen Inc.
Funding and additional information
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author contributions
H. B. and S. H. P. conceptualization; H. B. and S. H. P. formal analysis; H. B. and S. H. P. writing–original draft.
References
- 1.Xue V.W., Yang C., Wong S.C.C., Cho W.C.S. Proteomic profiling in extracellular vesicles for cancer detection and monitoring. Proteomics. 2021;21 doi: 10.1002/pmic.202000094. [DOI] [PubMed] [Google Scholar]
- 2.Khalilpour A., Kilic T., Khalilpour S., Álvarez M.M., Yazdi I.K. Proteomic-based biomarker discovery for development of next generation diagnostics. Appl. Microbiol. Biotechnol. 2017;101:475–491. doi: 10.1007/s00253-016-8029-z. [DOI] [PubMed] [Google Scholar]
- 3.Aebersold R., Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537:347–355. doi: 10.1038/nature19949. [DOI] [PubMed] [Google Scholar]
- 4.Lundberg E., Borner G.H.H. Spatial proteomics: a powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 2019;20:285–302. doi: 10.1038/s41580-018-0094-y. [DOI] [PubMed] [Google Scholar]
- 5.Huang Z., Ma L., Huang C., Li Q., Nice E.C. Proteomic profiling of human plasma for cancer biomarker discovery. Proteomics. 2017;17 doi: 10.1002/pmic.201600240. [DOI] [PubMed] [Google Scholar]
- 6.Specht H., Slavov N. Transformative opportunities for single-cell proteomics. J. Proteome Res. 2018;17:2565–2571. doi: 10.1021/acs.jproteome.8b00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramón Y Cajal S., Sesé M., Capdevila C., Aasen T., De Mattos-Arruda L., Diaz-Cano S.J., et al. Clinical implications of intratumor heterogeneity: challenges and opportunities. J. Mol. Med. Berl. Ger. 2020;98:161–177. doi: 10.1007/s00109-020-01874-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelly R.T. Single-cell proteomics: progress and prospects. Mol. Cell. Proteomics. 2020;19:1739–1748. doi: 10.1074/mcp.R120.002234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slavov N. Single-cell protein analysis by mass spectrometry. Curr. Opin. Chem. Biol. 2021;60:1–9. doi: 10.1016/j.cbpa.2020.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ctortecka C., Mechtler K. The rise of single-cell proteomics. Anal. Sci. Adv. 2021;2:84–94. doi: 10.1002/ansa.202000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vanderaa C., Gatto L. The current state of single-cell proteomics data analysis. arXiv. 2022 doi: 10.48550/ARXIV.2210.01020. [preprint] [DOI] [PubMed] [Google Scholar]
- 12.Marcotte E.M. How do shotgun proteomics algorithms identify proteins? Nat. Biotechnol. 2007;25:755–757. doi: 10.1038/nbt0707-755. [DOI] [PubMed] [Google Scholar]
- 13.Käll L., Vitek O. Computational mass spectrometry-based proteomics. PLoS Comput. Biol. 2011;7 doi: 10.1371/journal.pcbi.1002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boekweg H., Van Der Watt D., Truong T., Johnston S.M., Guise A.J., Plowey E.D., et al. Features of peptide fragmentation spectra in single-cell proteomics. J. Proteome Res. 2022;21:182–188. doi: 10.1021/acs.jproteome.1c00670. [DOI] [PubMed] [Google Scholar]
- 15.Vanderaa C., Gatto L. Replication of single-cell proteomics data reveals important computational challenges. Expert Rev. Proteomics. 2021;18:835–843. doi: 10.1080/14789450.2021.1988571. [DOI] [PubMed] [Google Scholar]
- 16.Schoof E.M., Furtwängler B., Üresin N., Rapin N., Savickas S., Gentil C., et al. Quantitative single-cell proteomics as a tool to characterize cellular hierarchies. Nat. Commun. 2021;12:3341. doi: 10.1038/s41467-021-23667-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li S., Tang H. Computational methods in mass spectrometry-based proteomics. Adv. Exp. Med. Biol. 2016;939:63–89. doi: 10.1007/978-981-10-1503-8_4. [DOI] [PubMed] [Google Scholar]
- 18.Noble W.S., MacCoss M.J. Computational and statistical analysis of protein mass spectrometry data. PLoS Comput. Biol. 2012;8 doi: 10.1371/journal.pcbi.1002296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Halder A., Verma A., Biswas D., Srivastava S. Recent advances in mass-spectrometry based proteomics software, tools and databases. Drug Discov. Today Technol. 2021;39:69–79. doi: 10.1016/j.ddtec.2021.06.007. [DOI] [PubMed] [Google Scholar]
- 20.Sinitcyn P., Rudolph J.D., Cox J. Computational methods for understanding mass spectrometry–based shotgun proteomics data. Annu. Rev. Biomed. Data Sci. 2018;1:207–234. [Google Scholar]
- 21.Ma B. Challenges in computational analysis of mass spectrometry data for proteomics. J. Comput. Sci. Technol. 2010;25:107–123. [Google Scholar]
- 22.Elias J.E., Gygi S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 23.Keller A., Nesvizhskii A.I., Kolker E., Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 24.Aebersold R., Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 25.Eng J.K., McCormack A.L., Yates J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 26.Mann M., Wilm M. Error-tolerant identification of peptides in sequence databases by peptide sequence tags. Anal. Chem. 1994;66:4390–4399. doi: 10.1021/ac00096a002. [DOI] [PubMed] [Google Scholar]
- 27.Tanner S., Shu H., Frank A., Wang L.-C., Zandi E., Mumby M., et al. Identification of posttranslationally modified peptides from tandem mass spectra. Anal. Chem. 2005;77:4626–4639. doi: 10.1021/ac050102d. [DOI] [PubMed] [Google Scholar]
- 28.Kong A.T., Leprevost F.V., Avtonomov D.M., Mellacheruvu D., Nesvizhskii A.I. MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods. 2017;14:513–520. doi: 10.1038/nmeth.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Craig R., Beavis R.C. Tandem: matching proteins with tandem mass spectra. Bioinforma. Oxf. Engl. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 30.Devabhaktuni A., Lin S., Zhang L., Swaminathan K., Gonzalez C.G., Olsson N., et al. TagGraph reveals vast protein modification landscapes from large tandem mass spectrometry datasets. Nat. Biotechnol. 2019;37:469–479. doi: 10.1038/s41587-019-0067-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Solntsev S.K., Shortreed M.R., Frey B.L., Smith L.M. Enhanced global post-translational modification discovery with MetaMorpheus. J. Proteome Res. 2018;17:1844–1851. doi: 10.1021/acs.jproteome.7b00873. [DOI] [PubMed] [Google Scholar]
- 32.Edwards N.J. PepArML: a meta-search peptide identification platform for tandem mass spectra. Curr. Protoc. Bioinforma. 2013;44:13.23.1–13.23.23. doi: 10.1002/0471250953.bi1323s44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sturm M., Bertsch A., Gröpl C., Hildebrandt A., Hussong R., Lange E., et al. OpenMS - an open-source software framework for mass spectrometry. BMC Bioinform. 2008;9:163. doi: 10.1186/1471-2105-9-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dorfer V., Pichler P., Stranzl T., Stadlmann J., Taus T., Winkler S., et al. MS Amanda, a universal identification algorithm optimized for high accuracy tandem mass spectra. J. Proteome Res. 2014;13:3679–3684. doi: 10.1021/pr500202e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim S., Pevzner P.A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 2014;5:5277. doi: 10.1038/ncomms6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 37.Nesvizhskii A.I. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J. Proteomics. 2010;73:2092–2123. doi: 10.1016/j.jprot.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Venable J.D., Dong M.-Q., Wohlschlegel J., Dillin A., Yates J.R. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat. Methods. 2004;1:39–45. doi: 10.1038/nmeth705. [DOI] [PubMed] [Google Scholar]
- 39.Bern M., Finney G., Hoopmann M.R., Merrihew G., Toth M.J., MacCoss M.J. Deconvolution of mixture spectra from ion-trap data-independent-acquisition tandem mass spectrometry. Anal. Chem. 2010;82:833–841. doi: 10.1021/ac901801b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsou C.-C., Avtonomov D., Larsen B., Tucholska M., Choi H., Gingras A.-C., et al. DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat. Methods. 2015;12:258–264. doi: 10.1038/nmeth.3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bruderer R., Bernhardt O.M., Gandhi T., Miladinović S.M., Cheng L.-Y., Messner S., et al. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol. Cell. Proteomics. 2015;14:1400–1410. doi: 10.1074/mcp.M114.044305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Navarro P., Kuharev J., Gillet L.C., Bernhardt O.M., MacLean B., Röst H.L., et al. A multicenter study benchmarks software tools for label-free proteome quantification. Nat. Biotechnol. 2016;34:1130–1136. doi: 10.1038/nbt.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuharev J., Navarro P., Distler U., Jahn O., Tenzer S. In-depth evaluation of software tools for data-independent acquisition based label-free quantification. Proteomics. 2015;15:3140–3151. doi: 10.1002/pmic.201400396. [DOI] [PubMed] [Google Scholar]
- 44.Searle B.C., Swearingen K.E., Barnes C.A., Schmidt T., Gessulat S., Küster B., et al. Generating high quality libraries for DIA MS with empirically corrected peptide predictions. Nat. Commun. 2020;11:1548. doi: 10.1038/s41467-020-15346-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Searle B.C., Pino L.K., Egertson J.D., Ting Y.S., Lawrence R.T., MacLean B.X., et al. Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nat. Commun. 2018;9:5128. doi: 10.1038/s41467-018-07454-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ge W., Liang X., Zhang F., Hu Y., Xu L., Xiang N., et al. Computational optimization of spectral library size improves DIA-MS proteome coverage and applications to 15 tumors. J. Proteome Res. 2021;20:5392–5401. doi: 10.1021/acs.jproteome.1c00640. [DOI] [PubMed] [Google Scholar]
- 47.Muntel J., Gandhi T., Verbeke L., Bernhardt O.M., Treiber T., Bruderer R., et al. Surpassing 10 000 identified and quantified proteins in a single run by optimizing current LC-MS instrumentation and data analysis strategy. Mol. Omics. 2019;15:348–360. doi: 10.1039/c9mo00082h. [DOI] [PubMed] [Google Scholar]
- 48.Siyal A.A., Chen E.S.-W., Chan H.-J., Kitata R.B., Yang J.-C., Tu H.-L., et al. Sample size-comparable spectral library enhances data-independent acquisition-based proteome coverage of low-input cells. Anal. Chem. 2021;93:17003–17011. doi: 10.1021/acs.analchem.1c03477. [DOI] [PubMed] [Google Scholar]
- 49.Wang Y., Lih T.-S.M., Chen L., Xu Y., Kuczler M.D., Cao L., et al. Optimized data-independent acquisition approach for proteomic analysis at single-cell level. Clin. Proteomics. 2022;19:24. doi: 10.1186/s12014-022-09359-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Demichev V., Messner C.B., Vernardis S.I., Lilley K.S., Ralser M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods. 2020;17:41–44. doi: 10.1038/s41592-019-0638-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Demichev V., Szyrwiel L., Yu F., Teo G.C., Rosenberger G., Niewienda A., et al. Dia-PASEF data analysis using FragPipe and DIA-NN for deep proteomics of low sample amounts. Nat. Commun. 2022;13:3944. doi: 10.1038/s41467-022-31492-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gessulat S., Schmidt T., Zolg D.P., Samaras P., Schnatbaum K., Zerweck J., et al. Prosit: Proteome-Wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods. 2019;16:509–518. doi: 10.1038/s41592-019-0426-7. [DOI] [PubMed] [Google Scholar]
- 53.Wang B., Wang Y., Chen Y., Gao M., Ren J., Guo Y., et al. DeepSCP: utilizing deep learning to boost single-cell proteome coverage. Brief. Bioinform. 2022;23:bbac214. doi: 10.1093/bib/bbac214. [DOI] [PubMed] [Google Scholar]
- 54.Fondrie W.E., Noble W.S. Machine learning strategy that leverages large data sets to boost statistical power in small-scale experiments. J. Proteome Res. 2020;19:1267–1274. doi: 10.1021/acs.jproteome.9b00780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Van Der Watt D., Boekweg H., Truong T., Guise A.J., Plowey E.D., Kelly R.T., et al. Benchmarking PSM identification tools for single cell proteomics. bioRxiv. 2021 doi: 10.1101/2021.08.17.456676. [preprint] [DOI] [Google Scholar]
- 56.Chen A.T., Franks A., Slavov N. DART-ID increases single-cell proteome coverage. PLoS Comput. Biol. 2019;15 doi: 10.1371/journal.pcbi.1007082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Declercq A., Bouwmeester R., Hirschler A., Carapito C., Degroeve S., Martens L., et al. MS2Rescore: data-Driven rescoring dramatically boosts immunopeptide identification rates. Mol. Cell. Proteomics. 2022;21:100266. doi: 10.1016/j.mcpro.2022.100266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Orsburn B.C. Time-of-Flight fragmentation spectra generated by the proteomic analysis of single human cells do not exhibit atypical fragmentation patterns. J. Proteome Res. 2023;22:1003–1008. doi: 10.1021/acs.jproteome.2c00715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu F., Haynes S.E., Teo G.C., Avtonomov D.M., Polasky D.A., Nesvizhskii A.I. Fast quantitative analysis of TimsTOF PASEF data with MSFragger and IonQuant. Mol. Cell. Proteomics. 2020;19:1575–1585. doi: 10.1074/mcp.TIR120.002048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pino L.K., Searle B.C., Bollinger J.G., Nunn B., MacLean B., MacCoss M.J. The skyline ecosystem: informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev. 2020;39:229–244. doi: 10.1002/mas.21540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Millikin R.J., Shortreed M.R., Scalf M., Smith L.M. Fast, free, and flexible peptide and protein quantification with FlashLFQ. Methods Mol. Biol. 2023;2426:303–313. doi: 10.1007/978-1-0716-1967-4_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pino L.K., Just S.C., MacCoss M.J., Searle B.C. Acquiring and analyzing data independent acquisition proteomics experiments without spectrum libraries. Mol. Cell. Proteomics. 2020;19:1088–1103. doi: 10.1074/mcp.P119.001913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Matthews D.E., Hayes J.M. Systematic errors in gas chromatography-mass spectrometry isotope ratio measurements. Anal. Chem. 1976;48:1375–1382. [Google Scholar]
- 64.Lipton M.S., Pasa-Tolic’ L., Anderson G.A., Anderson D.J., Auberry D.L., Battista J.R., et al. Global analysis of the deinococcus radiodurans proteome by using accurate mass tags. Proc. Natl. Acad. Sci. U. S. A. 2002;99:11049–11054. doi: 10.1073/pnas.172170199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu F., Haynes S.E., Nesvizhskii A.I. IonQuant enables accurate and sensitive label-free quantification with FDR-controlled match-between-runs. Mol. Cell. Proteomics. 2021;20:100077. doi: 10.1016/j.mcpro.2021.100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Woo J., Clair G.C., Williams S.M., Feng S., Tsai C.-F., Moore R.J., et al. Three-dimensional feature matching improves coverage for single-cell proteomics based on ion mobility filtering. Cell Syst. 2022;13:426–434.e4. doi: 10.1016/j.cels.2022.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kalxdorf M., Müller T., Stegle O., Krijgsveld J. IceR improves proteome coverage and data completeness in global and single-cell proteomics. Nat. Commun. 2021;12:4787. doi: 10.1038/s41467-021-25077-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thompson A., Schäfer J., Kuhn K., Kienle S., Schwarz J., Schmidt G., et al. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003;75:1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 69.Budnik B., Levy E., Harmange G., Slavov N. SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018;19:161. doi: 10.1186/s13059-018-1547-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cheung T.K., Lee C.-Y., Bayer F.P., McCoy A., Kuster B., Rose C.M. Defining the carrier proteome limit for single-cell proteomics. Nat. Methods. 2021;18:76–83. doi: 10.1038/s41592-020-01002-5. [DOI] [PubMed] [Google Scholar]
- 71.Ye Z., Batth T.S., Rüther P., Olsen J.V. A deeper look at carrier proteome effects for single-cell proteomics. Commun. Biol. 2022;5:150. doi: 10.1038/s42003-022-03095-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Woo J., Williams S.M., Markillie L.M., Feng S., Tsai C.-F., Aguilera-Vazquez V., et al. High-throughput and high-efficiency sample preparation for single-cell proteomics using a nested nanowell chip. Nat. Commun. 2021;12:6246. doi: 10.1038/s41467-021-26514-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Furtwängler B., Üresin N., Motamedchaboki K., Huguet R., Lopez-Ferrer D., Zabrouskov V., et al. Real-time search-assisted acquisition on a tribrid mass spectrometer improves coverage in multiplexed single-cell proteomics. Mol. Cell. Proteomics. 2022;21:100219. doi: 10.1016/j.mcpro.2022.100219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ting L., Rad R., Gygi S.P., Haas W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods. 2011;8:937–940. doi: 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Karp N.A., Huber W., Sadowski P.G., Charles P.D., Hester S.V., Lilley K.S. Addressing accuracy and precision issues in ITRAQ quantitation. Mol. Cell. Proteomics. 2010;9:1885–1897. doi: 10.1074/mcp.M900628-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Park J., Yu F., Fulcher J.M., Williams S.M., Engbrecht K., Moore R.J., et al. Evaluating linear ion trap for MS3-based multiplexed single-cell proteomics. Anal. Chem. 2023 doi: 10.1021/acs.analchem.2c03739. [DOI] [PubMed] [Google Scholar]
- 77.Domon B., Aebersold R. Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 2010;28:710–721. doi: 10.1038/nbt.1661. [DOI] [PubMed] [Google Scholar]
- 78.Whiteaker J.R., Lin C., Kennedy J., Hou L., Trute M., Sokal I., et al. A targeted proteomics-based pipeline for verification of biomarkers in plasma. Nat. Biotechnol. 2011;29:625–634. doi: 10.1038/nbt.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cappadona S., Baker P.R., Cutillas P.R., Heck A.J.R., van Breukelen B. Current challenges in software solutions for mass spectrometry-based quantitative proteomics. Amino Acids. 2012;43:1087–1108. doi: 10.1007/s00726-012-1289-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Blein-Nicolas M., Zivy M. Thousand and one ways to quantify and compare protein abundances in label-free bottom-up proteomics. Biochim. Biophys. Acta. 2016;1864:883–895. doi: 10.1016/j.bbapap.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 81.Dowell J.A., Wright L.J., Armstrong E.A., Denu J.M. Benchmarking quantitative performance in label-free proteomics. ACS Omega. 2021;6:2494–2504. doi: 10.1021/acsomega.0c04030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Al Shweiki M.R., Mönchgesang S., Majovsky P., Thieme D., Trutschel D., Hoehenwarter W. Assessment of label-free quantification in discovery proteomics and impact of technological factors and natural variability of protein abundance. J. Proteome Res. 2017;16:1410–1424. doi: 10.1021/acs.jproteome.6b00645. [DOI] [PubMed] [Google Scholar]
- 83.Pursiheimo A., Vehmas A.P., Afzal S., Suomi T., Chand T., Strauss L., et al. Optimization of statistical methods impact on quantitative proteomics data. J. Proteome Res. 2015;14:4118–4126. doi: 10.1021/acs.jproteome.5b00183. [DOI] [PubMed] [Google Scholar]
- 84.Nesvizhskii A.I., Aebersold R. Interpretation of shotgun proteomic data: the protein inference problem. Mol. Cell. Proteomics. 2005;4:1419–1440. doi: 10.1074/mcp.R500012-MCP200. [DOI] [PubMed] [Google Scholar]
- 85.Li Y.F., Radivojac P. Computational approaches to protein inference in shotgun proteomics. BMC Bioinform. 2012;13 Suppl 16(Suppl 16):S4. doi: 10.1186/1471-2105-13-S16-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang B., Chambers M.C., Tabb D.L. Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J. Proteome Res. 2007;6:3549–3557. doi: 10.1021/pr070230d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sticker A., Goeminne L., Martens L., Clement L. Robust summarization and inference in proteome-wide label-free quantification. Mol. Cell. Proteomics. 2020;19:1209–1219. doi: 10.1074/mcp.RA119.001624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Plubell D.L., Käll L., Webb-Robertson B.-J., Bramer L.M., Ives A., Kelleher N.L., et al. Putting humpty dumpty back together again: what does protein quantification mean in bottom-up proteomics? J. Proteome Res. 2022;21:891–898. doi: 10.1021/acs.jproteome.1c00894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang B., Pirmoradian M., Zubarev R., Käll L. Covariation of peptide abundances accurately reflects protein concentration differences. Mol. Cell. Proteomics. 2017;16:936–948. doi: 10.1074/mcp.O117.067728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Killinger B.J., Petyuk V.A., Wright A.T. Detecting differential protein abundance by combining peptide level P-values. Mol. Omics. 2020;16:554–562. doi: 10.1039/d0mo00045k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Suomi T., Corthals G.L., Nevalainen O.S., Elo L.L. Using peptide-level proteomics data for detecting differentially expressed proteins. J. Proteome Res. 2015;14:4564–4570. doi: 10.1021/acs.jproteome.5b00363. [DOI] [PubMed] [Google Scholar]
- 92.Ning Z., Zhang X., Mayne J., Figeys D. Peptide-centric approaches provide an alternative perspective to Re-examine quantitative proteomic data. Anal. Chem. 2016;88:1973–1978. doi: 10.1021/acs.analchem.5b04148. [DOI] [PubMed] [Google Scholar]
- 93.Matzke M.M., Brown J.N., Gritsenko M.A., Metz T.O., Pounds J.G., Rodland K.D., et al. A comparative analysis of computational approaches to relative protein quantification using peptide peak intensities in label-free LC-MS proteomics experiments. Proteomics. 2013;13:493–503. doi: 10.1002/pmic.201200269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Karpievitch Y.V., Dabney A.R., Smith R.D. Normalization and missing value imputation for label-free LC-MS analysis. BMC Bioinform. 2012;13 Suppl 16(Suppl 16):S5. doi: 10.1186/1471-2105-13-S16-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Webb-Robertson B.-J.M., Wiberg H.K., Matzke M.M., Brown J.N., Wang J., McDermott J.E., et al. Review, evaluation, and discussion of the challenges of missing value imputation for mass spectrometry-based label-free global proteomics. J. Proteome Res. 2015;14:1993–2001. doi: 10.1021/pr501138h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lazar C., Gatto L., Ferro M., Bruley C., Burger T. Accounting for the multiple natures of missing values in label-free quantitative proteomics data sets to compare imputation strategies. J. Proteome Res. 2016;15:1116–1125. doi: 10.1021/acs.jproteome.5b00981. [DOI] [PubMed] [Google Scholar]
- 97.Wang S., Li W., Hu L., Cheng J., Yang H., Liu Y. NAguideR: performing and prioritizing missing value imputations for consistent bottom-up proteomic analyses. Nucleic Acids Res. 2020;48:e83. doi: 10.1093/nar/gkaa498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shen M., Chang Y.-T., Wu C.-T., Parker S.J., Saylor G., Wang Y., et al. Comparative assessment and novel strategy on methods for imputing proteomics data. Sci. Rep. 2022;12:1067. doi: 10.1038/s41598-022-04938-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chion M., Carapito C., Bertrand F. Accounting for multiple imputation-induced variability for differential analysis in mass spectrometry-based label-free quantitative proteomics. PLoS Comput. Biol. 2022;18 doi: 10.1371/journal.pcbi.1010420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Taylor S.L., Ruhaak L.R., Kelly K., Weiss R.H., Kim K. Effects of imputation on correlation: implications for analysis of mass spectrometry data from multiple biological matrices. Brief. Bioinform. 2017;18:312–320. doi: 10.1093/bib/bbw010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Välikangas T., Suomi T., Elo L.L. A comprehensive evaluation of popular proteomics software workflows for label-free proteome quantification and imputation. Brief. Bioinform. 2018;19:1344–1355. doi: 10.1093/bib/bbx054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jin L., Bi Y., Hu C., Qu J., Shen S., Wang X., et al. A comparative study of evaluating missing value imputation methods in label-free proteomics. Sci. Rep. 2021;11:1760. doi: 10.1038/s41598-021-81279-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.O’Brien J.J., Gunawardena H.P., Paulo J.A., Chen X., Ibrahim J.G., Gygi S.P., et al. The effects of nonignorable missing data on label-free mass spectrometry proteomics experiments. Ann. Appl. Stat. 2018;12:2075–2095. doi: 10.1214/18-AOAS1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tsai T.-H., Choi M., Banfai B., Liu Y., MacLean B.X., Dunkley T., et al. Selection of features with consistent profiles improves relative protein quantification in mass spectrometry experiments. Mol. Cell. Proteomics. 2020;19:944–959. doi: 10.1074/mcp.RA119.001792. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used in these analyses are six replicates of 0.2 ng. The data is uploaded to Massive (MSV000087524), and all code used to generate figures and analyze results can be found in the github repository, https://github.com/PayneLab/SCP_comp_review.