

Abstract
Acute myeloid leukemia (AML) represents a set of heterogeneous myeloid malignancies hallmarked by mutations in epigenetic modifiers, transcription factors, and kinases1-5. It is unclear to what extent AML mutations drive chromatin 3D structure alteration and contribute to myeloid transformation. Here, we first performed Hi-C and whole-genome sequencing in 25 AML patient samples and seven healthy donor samples, and identified recurrent and subtype-specific alterations of A/B compartments, TADs, and chromatin loops. We then performed RNA-Seq, ATAC-Seq and CUT&Tag for CTCF, H3K27ac, and H3K27me3 in the same AML cohort, and identified extensive and recurrent AML-specific promoter-enhancer and promoter-silencer loops. We validated the role of repressive loops on their target genes by CRISPR deletion and interference experiments. Furthermore, we identified structural variation-induced enhancer-hijacking and silencer-hijacking events in AML samples. We demonstrated the role of hijacked enhancers in AML cell growth by CRISPR screening, and the downregulating role of hijacked silencers by CRISPRi de-repression. Finally, we performed whole-genome bisulfite sequencing in 20 AML and normal samples, and showed the delicate relationship between DNA methylation, CTCF binding and 3D genome structure. By treating the AML cells with the DNA hypomethylating agent and performing triple knockdown of DNMT1/3A/3B, we demonstrated the impact of manipulating DNA methylation on reverting 3D genome organization and gene expression. Overall, this study provides an invaluable resource for leukemia studies and highlighted the role of repressive-loops and hijacked cis-elements in human diseases.
Introduction
AML is a myeloid neoplasm characterized by differentiation blockade and clonal proliferation of abnormal myeloblasts in the bone marrow. The clinical course of AML is highly heterogeneous with variable molecular characteristics that are essential for risk stratification, prognostic, and therapeutic options1-4. Recent work has shown that different AML subtypes adopt unique landscape of chromatin accessibility, histone modifications, and binding of various transcription factors5-9.
The study of chromatin spatial organization in AML has been limited. Recent 3C-based work has suggested multiple layers of chromatin organization, including A/B compartment10, topologically associating domains (TADs)11,12, and chromatin loops. Interestingly, key chromatin structure proteins like STAG2, RAD21, SMC1/3, are recurrently mutated in AML13-15. Further, it has been shown that B and T acute lymphoblastic leukemia have altered chromatin conformation associated with lymphoid malignancies16-19. Therefore, it is imperative to unveil the altered 3D genome organization in AML and study their relationship with gene dysregulation and pathogenesis.
A major challenge to the study of 3D genome in AML and other cancers is the presence of frequent large structural variations (SVs), including inversions, deletions, duplications, and translocations. SVs have been shown to induce enhancer-hijacking in developmental diseases and different types of cancer20-22, and in particular, inv(3) can activate the EVI1 oncogene through such mechanism in AML21. However, genome-wide enhancer hijacking in AML have not been studied.
AML is also characterized by a global aberrant DNA methylation profile. The hypomethylating agent (HMA) 5-azacytidine (5-AZA) and its derivative decitabine are commonly used as therapeutic agents in AML. In this study, we also sought to delineate the relationship between DNA methylation and 3D genome structure, and whether HMA can restore the normal chromatin organization and gene regulation in AML cells.
Genomic and 3D genome data generation
We first performed in-situ Hi-C and RNA-seq in 32 primary samples, including 25 AML samples with more than 80% of myeloblast, four normal CD34+ hematopoietic stem progenitor cells (HSPC), and three normal peripheral blood mononuclear cells (PBMC) (Fig. 1a). The AML samples cover major driver genetic abnormalities, including mutations of NPM1, DNMT3A, TET2, KMT2B and RUNX1, FLT3-ITD, bi-allelic CEBPA, DEK-NUP214 t(6;9), RUNX1-RUNX1T1 t(8;21), BCR-ABL1 t(9;22), and CBFB-MYH11 inv(16). On average, we generated ~600 million paired-end reads in each Hi-C library. To identify AML and its subtype-specific enhancers and silencers, we performed ATAC-seq and CUT&Tag for histone 3 lysine 27 acetylation (H3K27ac) and histone 3 lysine27 trimethylation (H3K27me3). To identify the single nucleotide mutations (SNVs) and SVs, we performed PCR-free whole-genome sequencing (WGS) at ~40x coverage in 25 AML samples. To profile and study the impact of aberrant DNA methylation on chromatin structure in AML, we performed whole-genome bisulfite sequencing (WGBS) in two PBMC and 18 AML samples at ~30X coverage. In addition, we also performed CUT&Tag for CTCF in 10 AML samples. In total, we generated 199 genomic datasets from primary AML samples (Extended Data Fig. 1). An exemplary region containing all the epigenetic data in the same patient is shown in Fig. 1b.
Figure 1 ∣. Genome organization and compartment analysis in primary AML samples.

a, Summary of the AML samples and the genomic profiling assays performed in this study. b, Snapshot of an example region, showing Hi-C, ATAC-Seq, RNA-Seq, WGBS, and CUT&Tag for H3K27ac, H3K27me3 and CTCF data in the same patient (AML 027). The values for y-axis for ATAC-seq and CUT&Tag and the Hi-C data were normalized to sequencing depths. c, Left, unsupervised hierarchical clustering of AML and control samples based on the top 10% most variable first principal component (PC1) of the Hi-C matrices. Middle, mutation profiles of the known AML-relevant genes. Consensus mutations were not pre-selected but summarized from the clustering result. Right, an example region showing PC1 values and A/B compartment variations across samples. Squares on the heatmap demarcate samples with similar mutations. d, Number and proportion of A/B compartment switch in each AML sample, compared with HSPC. e, Gene expression alteration associated with change of A/B compartment. n=1,724 genes for A-to-B and n=867 for B-to-A. Genes are located inside recurrent compartment switch regions (in at least two AML samples). P value by two-sided Wilcoxon rank-sum test. Box plot: middle line denotes median, top/bottom of boxes denotes first/third quartiles and whiskers extend to 1.5 times the interquartile range of the first and third quartile. f, Clustering analysis of genomic regions (40kb bins) with differential Hi-C PC1 values, selected by one-way ANOVA analysis with P < 0.05. Samples were grouped by the gene mutation patterns. Representative COSMIC cancer census and AML-related genes in the corresponding regions are marked on the right.
Subtype-specific compartmentalization
To investigate whether AML samples possess unique chromatin compartmentalization, we performed unsupervised hierarchical clustering using the first principal component (PC1) of the Hi-C matrices. The samples were clustered into different groups, which accurately reflected the AML genetic subtypes (Fig. 1c), such as RUNX1 and CEBPA mutations. HSPC and PBMC samples were also grouped together, respectively. We noticed that a cluster of samples all contained mutations in the KMT2B gene, which has not been used as a dominant AML subtyping classifier before. On average, there was 3.2% A-to-B compartment switch and 2.9% B-to-A switch comparing AML samples with HPSC controls (Fig. 1d). Genes located in the A-to-B or B-to-A switching regions showed decreased or increased expression, respectively (Fig. 1e).
Moreover, the A/B compartment switching regions also grouped by their genetic mutation profile (Fig. 1f). For example, the WT1 gene showed B-to-A switch and expression activation exclusively in samples with TET2 and/or FLT3-ITD mutations, accompanied by the gain of ATAC-seq and H3K27ac peaks (Extended Data Fig. 2a-b). This is consistent with previous studies showing the association between WT1 activation and FLT3-ITD mutation23. Similarly, we observed other compartment switch regions unique to AML or AML subgroups, containing known AML gens such as POUA2F1, FGF13, and BCL11 (Extended Data Fig. 2c).
TAD alteration and gene expression
We predicted TADs at 40Kb resolution in all the samples using the DomainCaller pipeline11. Alterations in TAD boundaries were determined by comparing the span of each TAD in AML samples to HSPC. We defined three forms of TAD boundary alteration: expansion, shrink, and shift (Extended Data Fig. 3a). We identified 391 out of the 622 curated COSMIC cancer-related and AML-related genes located inside the altered TADs in at least one sample, and more than 100 are recurrent across multiple samples (Extended Data Fig. 3b)4,24,25. However, as shown in Extended Data Fig. 3c, the majority of TAD alterations did not affect the expression of genes, consistent with a recent study in multiple cancer types26.
AML-specific chromatin loops
We predicted chromatin loops at 10kb resolution using Peakachu27. On average, we identified ~200 AML-specific loops in each AML sample (Fig. 2a, Supplementary Table 1). In Fig. 2b, we showed the aggregate peak analysis (APA) for the 283 unique loops in AML sample 1021 but not in any of the HSPC samples. Only less than 4.8% of the AML-specific loops were in the CNV regions (Extended Data Fig. 4a-b), suggesting that they were not confounded by copy number variations (CNVs). The AML-specific loops also showed subtype-specific patterns and contained many known AML proto-oncogenes (such as MYCN, WT1, ERG, MEIS1, and RUNX1) in the loop anchors (Fig. 2c). Gene set enrichment analysis showed that genes on AML-specific loops were enriched in hematopoiesis and myeloid transformation pathways28,29 (Extended Data Fig. 4c). For example, MYCN formed a subtype-specific interaction with a cluster of co-occurring enhancers ~650Kb downstream, and this loop was only observed in AML samples with TET2/FLT3-ITD, CEBPA or STAG2/KRAS/NRAS mutations (Fig. 2d). Similarly, we observed sample-specific enhanced gene expression and loops for other oncogenes such as MEIS1 and ERG, linking their promoters with AML-specific enhancers (Extended Data Fig. 5).
Figure 2 ∣. AML and subtype-specific chromatin loops.

a, Number of AML-specific loops by comparing with four CD34+ HSPC, using the Gaussian mixture model of Peakachu (FDR < 5%). 22 AML samples that have more than 100 million uniquely mapped reads and minimally 20 million long-range reads (>20Kb) were included for this analysis. b, APA plot for the AML 1021-specific loops vs. four HSPC controls. c, Subtype-specific loop analysis for AML samples. Each row is a loop and the values are the loop probabilities from Peakachu. d, From top to bottom, genome browser tracks for RNA-seq, ATAC-seq, and CUT&Tag for H3K27ac, H3K27me3, and CTCF. The purple arc marks the loop anchors, which link the MYCN promoter to distal enhancers in samples with TET2 and CEBA mutations. e, Differential expression analysis for genes in recurrent AML-specific loops vs. HSPC. P value by two-tailed t-test. f, Heatmap of H3K27ac and H3K27me3 peaks in the anchors of AML-specific loops (AML 168 vs. HSPC). g, P-E, promoter-enhancer loops; P-P, promoter-promoter loops; P-S, promoter-silencer loops. Enhancer and silencer annotations were based on H3K27ac and H3K27me3 signals. When enhancers and silencers are present in the same 10Kb loop anchor, the annotation was determined by the ratio of H3K27ac vs. H3K27me3 signals. Details in the method section. h, For each gene in a P-E or P-S loop anchor, AML samples were grouped into two categories: with the P-E/P-S loop, and without either P-E or P-S loop for this gene. Then the average gene expression (TPM) within each category was calculated. P value calculated by two-sided Kruskal-Wallis H-test. Upper: n=4,948 genes. Lower: n=1,508 genes. Box plot: middle line denotes median, top/bottom of boxes denotes first/third quartiles and whiskers extend to 1.5 times the interquartile range of the first and third quartile.
Next, we studied the expression profiles of the genes located in the AML-specific loops. As shown in Fig. 2e, 220 genes were significantly upregulated in AML and 88 genes were downregulated. By comparing with H3K27ac and H3K27me3 peaks, we identified loop anchors containing enhancers or silencers (Fig. 2f). Furthermore, the majority of AML-specific loops co-occurred with the establishment of specific enhancer or silencer marks (Fig. 2f). Across all samples, 42.4% of AML-specific loops were between gene promoter and enhancers (P-E), and 11.2% are between promoter and silencers (P-S) (Fig. 2g, Supplementary Table 2). P-S loops account for 9.2% of all loops, with an average size of 169 kb (Extended Data Fig. 6a and 6b). The subtype-specific P-S and P-E loops were reported in Supplementary Table 3.
Characterizing promoter-silencer loops
70.45% of the P-S loops contains at least one CTCF binding sites at their anchors (30.38% for both anchors, and 40.07% for one anchor), suggesting that CTCF might play a role in P-S loop (Extended Data Fig. 6c). Next, we performed CUT&Tag for EZH2, a subunit of Polycomb repressive complex II, in THP-1 cells and observed that EZH2 binding sites mostly overlapped with H3K27me3 peaks in the P-S loop anchors (Extended Data Fig. 6d), consistent with the previously reported promoter-silencer loops30,31. Genes at AML-specific P-E loop anchors have much higher expression than those at P-S loop anchors (Extended Data Fig. 6e, P = 2.2E-16). For the same set of genes across different samples, they had higher expression when in P-E loops compared to samples where they were not in P-E nor P-S loops (Left panel in Fig. 2h, P=3.72E-21). Similar observation was made for genes in P-S loops (Right panel in Fig. 2h, P=5.24E-22).
To further confirm that the decrease in gene expression is due to P-S loops, we performed the following analysis: for each RefSeq gene that was expressed in at least one sample, we stratified the samples into 5 categories based on the distance between the TSS and the nearest non-looping H3K27me3 peaks (<10Kb, 10-50Kb, 50-100Kb, 100-200Kb, 200Kb-1Mb). As shown in Extended Data Fig. 6f, the local enrichment of H3K27me3 was correlated with decreased expression when they were within 100Kb to the gene promoters. However, for the non-looping H3K27me3 peaks more than 100Kb away from gene TSS, they did not show significant association with the decreased expression of the gene.
Validating promoter-silencer loops
To validate the P-S loops predicted by Hi-C data, we performed multiple high-resolution 4C and fluorescence in-situ hybridization (FISH) experiments. We first tested the P-S loops involving the IKZF2 gene, a gene frequently deleted in lymphoblastic leukemia32. There were two chromatin loops linking the IKZF2 promoter with two downstream silencers in multiple AML patients and Kasumi-1 cells (Fig. 3a). 4C-seq data in THP-1 confirmed the chromatin interactions between IKZF2 promoter and the silencers (orange tracks in Fig. 3a). Furthermore, FISH also confirmed that the 3D distance between the IKZF2 promoter and the downstream silencers was significantly shorter than the distance between the promoter and an upstream equidistant region in THP-1 and Kasumi-1 cell lines (Fig. 3b, Extended Data Fig. 6g). Across all samples, the IKZF2 expression was significantly lower in samples with the P-S loops (Fig. 3c). Analysis of the TCGA data showed that the lower expression of IKZF2 was associated with poorer prognosis in AML (Fig. 3d)33. We performed similar 4C-seq and DNA FISH experiments to validate another P-S loop involving the RTTN gene in both Kasumi-1 and THP-1 cells (Extended Data Fig. 7a-b). Again, the RTTN gene was expressed at lower level in samples with the P-S loop than in other samples (Extended Data Fig. 7c).
Figure 3 ∣. Identification and validation of repressive loops.

a, Hi-C and H3K27me3 CUT&Tag data in AML samples and cell line. IKZF2 P-S loops marked by black arrow. AML-798 did not have the P-S loops. Orange track: 4C for IKZF2 promoter. b, Left: DNA FISH image from a representative THP-1 cell. Labels: IKZF2 promoter (green, chr2:213,048,640-213,261,522), silencers (pink, chr2:212,609,118-212,811,039), and the control region (red, chr2:213,495,758-213,695,712). Right: Distance distribution (n=98 alleles). P value by two-sided Wilcoxon rank-sum test. c, IKZF2 expression in normal HSPC and PBMC (n=6), AML samples with the loop (n=8), and AML samples without the loop (n=17). P value by two-sided Student’s t-test. d, Kaplan-Meier plot for IKZF2 expression in TCGA AML GDC cohort (n=152). P value by log-rank test. e, H3K27me3 CUT&Tag data in Kasumi-1 cells in different conditions. Two clusters of silencers separately targeted CRISPR-dCas9-VP64. S4 and S5 were looped to IKZF2 promoter, while S1-S3 were not. Lower panel: linear distance. f, qPCR of IKZF2 expression (n=3 technical replicates in 2 biological replicates). P values by two-sided Student’s t-test. Data show mean ± s.e.m. g, Upper: defining two categories – genes looped to a silencer 200Kb-1Mb away (n=2,280) vs. genes with silencers at the same distance, but not looped to them (n=12,715). Lower: RNA expression of the two groups. P value by two-sided Wilcoxon rank-sum test. h, APA plot for AML-specific stripes and the same regions in four combined HSPCs and three combined PBMCs. i, A repressive stripe involving the KLF4 gene. j, KLF4 expression across normal samples (n=6), AML samples with P-S stripe on KLF4 (n=13), and AML samples w/o the stripe (n=12). P value by two-sided Student’s t-test. Box plot for b, c, g, j: middle line denotes median, top/bottom of boxes denotes first/third quartiles and whiskers extend to 1.5 times the interquartile range.
To examine the impact of silencers on their target genes, we performed multiple CRISPR deletion and CRISPR interference (CRISPRi) experiments. First, we deleted a ~50Kb silencer region (chr18:70,411,815-70,460,159) that looped to the RTTN promoter in Kasumi-1 cells (Extended Data Fig. 7d). We confirmed a heterozygous deletion in a single-cell derived clone by PCR and Sanger sequencing (Extended Data Fig. 7e and 7f). Deletion of this silencer increased the RTTN expression by 3 folds (Extended Data Fig. 7g, p=5E-5), dramatically slowed cell proliferation (Extended Data Fig. 7h), and decreased the sizes of the colonies by colony formation assay (Extended Data Fig. 7i-j).
Next, we performed more validation experiments for the P-S loops. There were two distal silencers linked to the IKZF2 gene (S4 and S5, Fig. 3e). We used CRISPR dCas9-VP64 to de-repress the silencers. In addition to non-specific sgRNAs, we also disrupted three nearby silencers not looped to the IKZF2 gene by CRISPRi (S1-S3, Fig. 3e) as additional control. Upon dCas9-VP64 expression with targeting S4 and R5, the IKZF2 RNA expression was significantly increased by over 35 fold (Fig. 3f, P=0.00017). CUT&Tag data confirmed that H3K27me3 signals at the targeted silencers were largely reduced (Fig. 3e). In contrast, disrupting the non-looping S1-S3 did not increase IKZF2 gene expression, indicating that the chromatin loop is critical for the distal silencers to impact their target genes.
Finally, we examined the genome-wide effect of P-S loops versus linear effect of heterochromatin compaction. To define high-resolution P-S loops, we performed HiChIP for H3K27me3 in Kasumi-1 cells. We compared the expression between genes with looped silencers vs. genes having silencers at the same range of distance (200kb to 1 Mb) but not looped to them. Genes with looped silencers showed significantly lower expression than the other group (Fig. 3g, P=6.87E-27). Taken together, these data suggest that the distal silencers can negatively impact their target gene expression through repressive loops.
Architectural stripes
Stripes were recently observed in Hi-C maps and proposed as evidence for the loop extrusion model 34. On average, we found 509 AML-specific stripes (length > 300kb, Supplementary Table 4). APA plot suggested that the stripe anchor interacted with a sliding zone in both directions (Fig. 3h). The stripes were enriched in super enhancers (Extended Data Fig. 8a-8b), consistent with previous findings 34. Surprisingly, we also identified stripes connecting promoters with silencers. For example, we observed a repressive stripe for the KLF4 gene in multiple AML samples (Fig. 3i). The whole sliding zone of this stripe was enriched in H3K27me3 signals in AML samples but not in controls (Fig. 3i, left panel), and this stripe was associated with decreased KLF4 expression (Fig. 3j). This is interesting because previous work suggested that KLF4 promotes myeloid cell differentiation and its downregulation contributes to AML leukemogenesis35. Overall, 10% of AML stripes were between gene promoters and predominantly repressive regions (Extended Data Fig. 8c). In each sample, genes located in the P-S stripe anchors had significantly lower expression than those in the P-E stripes (Extended Data Fig. 8d, P=8.74E-22, Kruskal-Wallis test). For the same genes across different samples, they were expressed at higher/lower levels when located in P-E/P-S stripes compared with when they were in neither types of stripes (Extended Data Fig. 8e, P=0.024, P=8.45E-13).
Detecting enhancer hijacking in AML
To detect “enhancer-hijacking” events22, we first identified SVs in 25 AML samples by WGS. In addition, as we previously demonstrated that certain aberrant Hi-C signals were indicative of SVs such as translocations and inversions (Extended Data Fig. 9a), we predicted SVs using Hi-C breakfinder20. SVs detected by WGS and Hi-C were then merged. On average, each AML sample has 1.7 large deletions (>1 Mb), 2.1 inversions (>1 Mb), and 13.2 inter-chromosomal translocations (Supplementary Table 5). Permutation analysis showed that translocations and deletions were enriched in proximity to AML or cancer-related genes (Extended Data Fig. 9b).
We predicted the SV-induced “neo-loops” using the NeoLoopFinder software36, which reconstructed the Hi-C maps surrounding the SV breakpoints according to SV types, loci and directions, and also normalizing the CNV effect. A representative example was shown in Extended Data Fig. 9c, where there was a fusion between chr7 and chr11 in AML 270, but not in HSPC. This neo-loop connected the CDK5 gene (located on chr7) to several enhancers on chr11. In contrast, there were no such inter-chromosomal Hi-C signals in HSPC. We performed the neo-loop analysis for all the AML samples, three AML cell lines (HL60, Kasumi-1, and THP-1), and a chronic myeloid leukemia cell line K56237. The number of neo-loops varies in different samples, depending on the number and the types of SVs (Supplementary Table 2).
To systematically identify recurrent enhancer-hijacking events, we defined the following three scenarios: 1) Same gene and hijacked enhancer pairs across different samples. Such events were usually formed across recurrent SVs, such as t(9;22) and inv(16). For example, we observed a recurrent enhancer-hijacking involved with the HSF4 gene in both sample 629 and 798 due to inv(16) (Fig. 4a); 2) Same gene linked with different enhancers in different samples. For example, the MYC gene (located on chr8) was looped with a cluster of enhancers on chr14 in sample 1360 due to t(8;14), but was looped with a different set of enhancers on chr11 in HL-60 cells due to t(8;11) (Fig. 4b). Similarly, the CBL gene was linked to enhancers on chr7 in sample 270, but in THP-1 cells, it was linked with enhancers on chr9 (Extended Data Fig. 9d); 3) Same enhancer linked to different genes. Fig. 4c shows such an example where the same enhancer was linked to different genes in different samples (ST7 and WNT2 genes on chr7 of sample 773, and POU2F3 gene on chr11 in sample 270). We summarized all recurrent enhancer hijacking events across samples and cell lines in Supplementary Table 6, and listed the subtype-specific enhancer-hijacking events in Supplementary Table 7. Furthermore, HSF4, MYC and CBL showed elevated expression in samples with enhancer hijacking(Fig. 4d, 4e, and Extended Data Fig. 9e) and genome-wide, genes with hijacked enhancers had significantly higher expression (Fig. 4f). To find what transcription factors (TFs) might be involved in enhancer hijacking, we performed motif search for the recurrently hijacked enhancers from scenario I and III, and identified TFs such as ERG, Fli1, Sox2, RUNX2, and CTCF (Fig. 4g). Motif analysis for all hijacked enhancers of each sample also confirmed the ETS (EHF, Elf4, GABPA, ERG, Fli1) family motifs (Extended Data Fig. 10a).
Figure 4 ∣. Identification, characterization, and screening of enhancer-hijacking in AML.

a-c, Different scenarios of recurrent enhancer-hijacking events induced by neo-loops (black circle). a, same gene (HSF4, orange bar) and enhancer (blue bar) pair in different samples. NeoLoopFinder was used to reconstruct the Hi-C map surrounding the SV. Virtual 4C was anchored at the gene promoter; b, same gene (MYC) with different enhancers in different samples; and c, same enhancer linked to different genes in different samples. d-e, Expression of HSF4 and MYC across all AML samples. f, Expression of all genes with hijacked enhancers (n=141 genes). P value by one-sided Wilcoxon rank-sum test. g, Homer motif analysis of recurrently hijacked enhancers across all samples. P value by binomial test. h, Design of the CRISPR screening experiment. This figure was created with BioRender.com. i, Fold change of each sgRNA abundance in the pre-screening vs. post-screening libraries. Each dot represents one sgRNA. Black dots: non-specific control sgRNAs. Red dots: the significantly depleted sgRNAs in the after-screening library. j, Luciferase/Renilla readout for 5 depleted enhancers from screening results (n=3 technical replicates in 2 biological replicates). Data show mean ± s.e.m. k, qPCR for NSMCE3 upon disruption of enhancer I separately by two sgRNAs in Kasumi-1 cells (n=3 technical replicates in 2 biological replicates). P value by two-sided Students’ t-test. Data show mean ± s.e.m. l, CERES gene dependency score by CRISPR-cas9 essentiality screens (DepMap 21Q2 Public). Grey dot: AML cell lines, n=26. Score<0: perturbation of a gene impairs cell growth. Box plot for f, l: middle line denotes median, top/bottom of boxes denotes first/third quartiles and whiskers extend to 1.5 times the interquartile range. m, CCK-8 assay for proliferation of Kasumi-1 targeting enhancer I. P value calculated by two-sided Student’s t-test (n=3 biological replicates). Data show mean ± s.e.m.
Function of hijacked enhancers
To investigate the impact of hijacked enhancers on AML cell survival and proliferation, we performed CRISPRi screening38 with the dCas9-KRAB-MeCP2 (CKM) system. There were 74 neo P-E loops involving 44 non-redundant enhancers in Kasumi-1 cells. We designed up to 6 sgRNAs for each enhancer and used 50 control sgRNAs in the screening library (Fig. 4h and Supplementary Table 8). 14 sgRNAs targeting 13 enhancers were significantly depleted in the post-screening library (Fig. 4i). We performed luciferase reporter assays to test the activity for five of them and all increased the luciferase expression (Fig. 4j).
To validate the CRISPRi screening result, we performed six individual CRISPRi experiments (Extended Data Fig. 10b). First, we tested enhancer I (Enh. I), which was predicted to regulate the NSMCE3 gene. When we disrupted Enh. I by CKM with two different sgRNAs, the NSMCE3 expression was reduced to 52% and 36%, respectively (Fig. 4k). The CUT&Tag data showed specific depletion of H3K37ac signals at the two targeted loci (Extended Data Fig. 10c), while there was no reduction of signals at NSMCE3 promoter. As additional control, we performed the CRISPRi experiment with the same sgRNAs in THP-1 cells, where there was no SV in this region and no enhancer hijacking. As expected, the CRISPRi experiment did not reduce the NSMCE3 expression (Extended Data Fig. 10d). Enh. II was predicted to upregulate both KBTBD7 and WBP4 gene expression, whereas Enh. III, IV, and V were predicted to upregulate the CYP20A1 gene. CRISPRi for each hijacked enhancer significantly reduced the expression of their target genes (Extended Data Fig. 10e).
To investigate the role of NSMCE3 and its hijacked enhancers in proliferation, we first examined the data from the Achilles project, which has systematically identified essential genes across hundreds of cancer cell lines. As shown in Fig. 4l, disruption of the NSMCE3 gene impaired cell growth in all of AML cell lines. We then performed CCK-8 assay in cells with sgRNAs targeting Enh 1. The results confirmed that both sgRNAs led to significant slower cell growth and proliferation (Fig. 4m), further suggesting that the hijacked enhancers might promote AML cell expansion through elevating their target gene expression.
Silencer hijacking
We noticed that SV-induced neo-loops could also link silencers with their target genes, and we define such events as silencer-hijacking, similar to the concept of enhancer-hijacking21,22. For example, we observed silencer-hijacking for two AML-related genes, JAK1 and KMT2C (Fig. 5a-b), both of which were associated with lower expression (Fig. 5c-d). Overall, 5.7% of all the neo-loops in this study lead to silencer hijacking, while 17.2% of the neo-loops introduce enhancer-hijacking (Fig. 5e). Genes linked with “hijacked” silencers showed decreased expression (Fig. 5f).
Figure 5 ∣. Identification and validation of silencer-hijacking in AML.

a-b, Two examples of silencer-hijacking. a is for the KMT2C gene in AML 270 and b is for the JAK1 gene in AML 168. Below the Hi-C maps are the H3K27me3 CUT&Tag data. c-d, KMT2C and JAK1 expression across all AML and normal samples. e, Pie chart showing percentages of different types of neo-loops. When enhancers and silencers are present in the same 10Kb loop anchor, annotation of P-E vs. P-S loops is determined by the ratio of H3K27ac vs. H3K27me3 signals (Details in the method section). f, Expression of genes with hijacked silencers (n=33 genes). P value by one-sided Wilcoxon rank-sum test. Box plot: line denotes the median, top/bottom of boxes denotes first/third quartiles and whiskers extend to 1.5 times the interquartile range. g, Design of the CRISPR interference experiment for the hijacked silencers. This figure was created with BioRender.com. h-i, Hijacked silencers that were targeted by dCas9-UTX in Kasumi-1 and THP-1 cells. Upper panel, reconstructed Hi-C maps for the regions surrounding the translocation breakpoints. Neo-loops are marked by blue circles. Promoters are marked by orange vertical bars and the green vertical bars highlight the hijacked silencers. Lower panel: CUT&Tag for H3K27me3 and dCas9 after CRISPRi treatment. The grey vertical bars highlight the regions with the most reduced H3K27me3 signals. j, qPCR results of EXD1 in Kasumi-1 cells (left) and ALG10 mRNA expression in THP-1 cells (right) when the hijacked silencers were de-repressed (n=3 technical replicates in 2 biological replicates). The control group underwent the same procedures with non-human genome targeting sgRNA. P value calculated by two-sided Student’s t-test. Data show mean ± s.e.m. k, List of the recurrent cancer-related genes whose promoters were located in the anchors of neo-loops.
We validated the function of hijacked silencers by the CRISPR dCas9-UTX (histone demethylase) system (Fig. 5g). First, we de-repressed a cluster of silencers that were predicted to regulate the EXD1 gene in Kasumi-1 cells (Fig. 5h). We also de-repressed another silencer, which was predicted to regulate the ALG10 gene in THP-1 cells (Fig. 5i). Upon the targeted de-repression of the hijacked silencers, the expressions of EXD1 and ALG10 were increased by 5.97 fold and 2.70 fold, respectively (Fig. 5j), confirming the repressive role of these hijacked silencers on their target genes.
Overall, we found 261 cancer-related genes whose promoters were located in the neo-loop anchors, 44 of which were recurrent across different samples. The detailed analysis were summarized in Fig. 5k, where we categorized them according to whether they were linked to hijacked enhancers or silencers and also by the types of SVs, including deletion, inversions and inter-chromosomal translocations.
Altered DNA methylation and 3D genome
DNA methylation alteration has been widely reported in cancer, and it has been shown to displace CTCF binding and induced pathogenic chromatin interactions in cancer39-43. To understand its relationship with unique chromatin organization in AML, we performed WGBS in 18 AML samples and two PBMC. We also downloaded WGBS and CTCF binding data in HSPC for analysis44,45. We observed variable methylation levels globally in AML samples (Extended Data Fig. 11a) and significantly higher methylation at CpG islands in AML compared with normal PBMC and HSPC (Fig. 6a), consistent with previous findings46. Sample 773 exhibited extremely high methylation at CpG islands, potentially because of the missense mutation (c.146A>G, p.Asp49Gly) in the SDHA gene, which has been associated with demethylation defects42. Therefore, we removed it from further downstream analysis. Across all samples, we observed subtype-specific DNA hypermethylation patterns (Fig. 6b).
Figure 6 ∣. Inhibition of DNA methylation restores chromatin structure and gene expression.

a, CG methylation levels for the top 10,000 most-variably methylated CpG islands across all samples. b, Hierarchical clustering of 12,409 differentially methylated regions (Z-score normalized). c, Experiment design for the DNMT TKD and 5-AZA treatment. d, WB results for DNMT3A/3B/1. MW: molecular weight. e, Stratum adjusted correlation coefficient analysis for Hi-C data by HiCRep. f, Venn diagrams comparing A/B compartment reversion between TKD and 5-AZA. A-to-B reversion: a 40-Kb bin in B compartment in normal HSPC, in A compartment under DMSO treatment, and in B compartment upon 5-AZA treatment or TKD. Vice versa for B-to-A reversion. g, Expressions of the genes in B-to-A reversion regions upon 5-AZA treatment (n=80 genes). P value by two-sided Wilcoxon rank-sum test. Box plot: line denotes median, top/bottom of boxes denotes first/third quartiles and whiskers extend to 1.5 times the interquartile range of the first and third quartile. h, Left, percentage of A/B compartment switch in AML cell line, compared with normal HSPCs; Upper right, percentage of switched compartments in cell line that are consistent with AML patient samples; Lower right, percentage of the stored compartments in AML cell line upon 5-AZA treatment compared with normal HSPCs. i, Compartmentalization saddle plots for DMSO and 5-AZA treatment. Left upper: B-B interaction; right lower: A-A interaction. Right upper and left lower: A-B interactions. The values are the average of each quarter square. j, Size distribution of chromatin loops. P value by two-sided Wilcoxon rank-sum test. k, APA plots for the different types of loops in different cells and conditions. AML-specific loops (n=1824); shared loops (n=8127). A significantly dissociated loop is determined when its fold change of PEAKACHU probability deviates from the Gaussian mixture model (mixture =2) with a P value < 0.05.
We observed that genes in the A compartment had lower methylation at transcription starting site (TSS) but higher methylation at gene bodies than genes in the B compartment (Extended Data Fig. 11b), consistent with the distinct roles of DNA methylations at different regions47. We observed that CTCF binding sites in AML samples were hypomethylated (Extended Data Fig. 11c). We also identified the loss of CTCF binding sites in AML samples that were potentially due to hypermethylation (Extended Data Fig. 11d). There are in average 76 such sites in each AML samples. Overall, 32.4% of hypermethylation-associated loss of CTCF binding overlap with TAD boundary switch in patient samples.
Current loop-extrusion model suggests that loop is mainly driven by Cohesin complex and stops at convergent CTCF sites48. Therefore, we investigated whether loss of CTCF binding is correlated with changes of local chromatin interactions (Extended Data Fig. 11e). As shown in Extended Data Fig. 11f, we collected the lost CTCF binding sites and plotted the aggregated differential Hi-C maps (AML 424 vs. HSPC 213), centered at the 142 lost CTCF sites associated with hypermethylation. We observed increased interactions across the lost CTCF binding sites, suggesting that disruption of CTCF site might lead to loss of insulation and change the local chromatin interaction profile. For example, we observed in four AML samples (424, 018, 546, and 472) a lost binding of CTCF, the motif of which was hypermethylated in those samples (Bar plot in Extended Data Fig. 11g). Correspondingly, we observed multiple gained chromatin loops appeared in these AML samples across the lost CTCF binding sites, including a loop linking WDR66 promoter with distal regions. These data suggest that DNA hypermethylation-induced loss of CTCF insulator can contribute to the gain of chromatin interactions in AML.
HMA/DNMT TKO reverses chromatin topology
To study whether the impact of DNA methylation on 3D genome structure is reversible, we first created an inducible DNMT3A/3B/1 triple-knockdown (TKD) in U937 AML cell line (Fig. 6c). The TKD cells were sequentially selected by puromycin (DNMT3A), sorted by GFP (DNMT3B) and RFP (DNMT1), and fully induced by doxycycline. We performed Western Blot (WB) in the TKD cells and confirmed that their protein expression levels were reduced (Fig. 6d).
In a parallel effort, we treated the U937 cells with DNA hypomethylating agent 5-AZA, at a low but effective dosage (0.5μM for 12 days), to mimic drug deliveries that are more physiologically tolerable and relevant to clinical dosing. The treatment slowed down the cell proliferation (Extended Data Fig. 12a). To examine whether such treatment will induce side effects, we performed a series of experiments and observed that the 0.5μM dosage did not induce a significant apoptosis (Extended Data Fig. 12b), DNA double strand break (Extended Data Fig. 12c), or cell cycle arrest (Extended Data Fig. 12d), while higher dosages (from 1μM to 8μM) indeed led to cell cycle arrest (from G2/M to G1/S phases) (Extended Data Fig. 12e).
Next, we performed WGBS in the 5-AZA treated cells and confirmed that global methylation was decreased from 74% to 39% (Extended Data Fig. 13a-b). We stratified the genome into CpG islands (CpGi), shores (<2Kb to CpGi), shelves (2Kb-4Kb to CpGi), and open sea (the rest of the genome). 5-AZA treatment dramatically decreased DNA methylation levels for all four regions, with the biggest changes in open sea (Extended Data Fig. 13c), as well as on CTCF binding motifs (Extended Data Fig. 13d).
Then, we performed Hi-C in both the DNMT TKD cells and the 5-AZA treated cells. Clustering analysis by Hi-CRep49 showed that 5-AZA treated and DNMT TKD cells were more similar to each other than the DMSO treated cells (Fig. 6e). We defined B-to-A compartment reversion as a region in B compartment under DMSO treatment but in A compartment in normal HSPC, and this region switched back to A compartment after 5-AZA treatment. We defined the A-to-B reversion in a similar way. Genome-wide, we found high consistency of the A/B compartment dynamics between the DNMT TKD and 5-AZA treatment. 865 out of the 905 (95.5%) TKD-induced A-to-B reversion regions overlapped with the A-to-B revision regions induced by 5-AZA treatment, and the overlap is 79% for the B-to-A reversion regions (Fig. 6f). As expected, genes in the B-to-A switching regions were significantly upregulated (Fig. 6g). More importantly, 84% of the A-to-B altered compartments in AML cell line were reminiscent of the same compartment switching in patient samples (Fig. 6h). Upon 5-AZA treatment, 55% of the A-to-B altered compartments were reversed to the A compartment. We also performed “saddle plot” analysis to examine the interactions between compartments, and the results showed that after 5-AZA treatment A/A and B/B interactions were decreased, while the interactions between A/B compartment were increased (Fig. 6i).
We investigated the relationship between compartment switches and CpG-density. The stable A compartments were slightly enriched for CpGi, while the stable B compartment regions were depleted of CpGi (Extended Data Fig. 13e). Interestingly, A-to-B compartment switching regions under 5-AZA treatment had lower density of CpGi. We further compared the methylation level changes in different A/B compartment switching regions for CpGi and open sea regions. We noticed that the CpGi originally in A compartment (A-to-A and A-to-B) were hypomethylated, while CpGi originally in B compartment (B-to-B and B-to-A) were hypermethylated. Upon 5-AZA treatment, methylation in all categories were reduced (Extended Data Fig. 13f). We observed the similar patterns for compartment switching regions in open seas.
Finally, we studied how 5-AZA or DNMT TKD affected chromatin loops. We noticed that there were more long-range interactions in the 5-AZA treated cells compared with cells treated with DMSO (Fig. 6j). We further identified 1,824 AML-specific loops and 8,127 shared loops by comparing with HSPC (Supplementary Table 9). Intriguingly, after the treatment, the AML-specific loops were much more weakened (31.6% significantly dissociated) compared with the shared loops (2.5% significantly dissociated) (Fig. 6k). Consistently, we observed similar loop dissociation pattern in the TKD cells (34.1% dissociated for AML specific loops, and 6.3% for shared loops). Compared with stable loops, the dissociated loop anchors had a higher percentage of CpGi at anchors (Extended Data Fig. 13g), and higher methylation level at CpGi in DMSO group, and a wider decrease of methylation after 5-AZA treatment (Extended Data Fig. 13h).
Discussion
In summary, through large-scale genomic study in primary AML samples, we identified subtype-specific distal enhancers and silencers, changes of 3D genome features such as compartment, TAD boundaries, and chromatin loops. In particular, we showed that repressive loops were widespread in the genome, and by CRISPR and CRISPRi experiments, we demonstrated long-range P-S loops as a novel mechanism of tumor suppressor repression in AML. An interesting future experiment would be to dissolve the chromatin interactions without changing the local chromatin status at the cis-regulatory elements.
By integrating WGS and Hi-C data, we identified hundreds of SV-induced neo-loops that linked the hijacked enhancers or silencers to their target genes. We showed that disruption of the hijacked enhancers could affect their target gene expression and impair cell proliferation and colony formation. It is important to further study the phenotypic effects in greater depth in the future, and animal models will be desirable to provide further biological and translational insights.
Finally, we showed that aberrant DNA methylation was associated with 3D genome alteration in AML. We demonstrated that the HMA exposure partially restored the chromatin structure, including reverting switched compartment and dissociating AML-specific loops. Therefore, it suggests that HMA treatment may potentially achieve therapeutic efficacy, at least in part, through restoration of normal chromatin architecture. Our finding is complementary with a recent study that showed altered DNA methylation in HCT116 colon cancer cells led to changes in H3K9me3 and impacted CTCF loop-extrusion barriers50. As we showed that DNA methylation could influence chromatin topology, combining HMA therapy with other agents that complement the restoration of normal genome architecture might increase therapeutic response and inform new mechanism-based therapeutic approaches to improve the treatment outcome in AML and other cancers.
Methods
I. Materials and Experiments
Primary sample collection
Human blood samples were obtained with consent from AML patients or healthy donors under the Penn State Hershey IRB-approved protocol STUDY00005272. Peripheral blood or bone marrow aspirates from AML patients and peripheral blood from healthy donors were collected and immediately subjected to selection for mononuclear cells using Ficoll-Paque PLUS density gradient media (GE Healthcare, 17-1440-02) following manufacturer’s instruction. Myeloblasts accounted for more than 90% of purified PBMC for most samples, and samples with blast cell concentrations less than 80% were further subjected to CD34+ selection as described in previous work5. Bone marrow HSPC was purchased from LONZA (Catalog #: 4M-105). Moreover, we utilized 8 datasets of HSPC from previous publication for analysis44,45,51,52.
Cell culture
Kasumi-1 cells (ATCC CRL-2724), HL60 cells (ATCC CCL-240), THP-1 cells (ATCC TIB-202) and U937 cells (CRL-1593) were purchased from ATCC. Cells were cultured following the manufacturer’s culture method. Kasumi cells were cultured with RPMI-1640 growth medium (Gibco, 11875093) containing 20% FBS. HL-60 cells were cultured with IMDM containing 20% FBS (Thermofisher 12440053). THP-1 cells were cultured with RPMI-1640 containing 10% FBS. U937 cells were cultured with RPMI-1640 containing 10% FBS.
In-situ Hi-C
One to two million cryopreserved primary samples or cell lines in a cell culture were spun down at 500g, resuspended in 1ml/million of RPMI 1640 medium and 10% fetal bovine serum, immediately crosslinked with 37% formaldehyde (MilliporeSigma 252549) to a final concentration of 2%, and incubated at room temperature for 10 minutes on a tube revolver at 16 rpm to mix. The resulting solution was quenched with a 2.5M glycine solution to a final concentration of 0.2M and incubated at room temperature for 5 minutes on a revolver. Cells were pelleted by centrifuging at 500g and 4°C for five minutes. The pellet was washed once with 1ml of cold 1X PBS by centrifuging at 500g and 4°C for five minutes and the supernatant was discarded. Cells were lysed with 250ul of lysis buffer to extract nuclei (10mM Tris-HCl pH8.0, 10mM NaCl, 0.2% Igepal CA630), mixed with 50ul of 50x protease inhibitor (Sigma, P8340), and incubated on ice for 15 minutes. Lysed cells were pelleted by centrifuging at 2500g and 4°C for five minutes and washed with 500ul of lysis buffer. Cell pellets were resuspended in 50ul of 0.5% sodium dodecyl sulfate (SDS), incubated at 62°C for 10 minutes, then quenched with 145ul of water and 25ul of 10% Triton X-100 (Sigma, 93443), and incubated at 37°C for 15 minutes. 25μl of 10X NEBuffer2 (NEB B7207) and 100 units of MboI restriction enzyme (NEB, R0147) were then added to the reaction for overnight DNA digestion at 37°C on the tube revolver. The digestion was then quenched by incubating at 62°C for 20 minutes. DNA was then end repaired and Biotin labeled with 50ul of fill-in master mix (37.5μl of 0.4mM biotin-14-dATP (Life Technologies, 19524-016), 1.5μl of 10mM dCTP, 1.5μl of 10mM dGTP, 1.5μl of 10mM dTTP, 8μl of 5U/μl DNA Polymerase I, Large (Klenow) Fragment (NEB, M0210)) and incubated at 37°C for 1.5 hours. DNA was then ligated with 900ul of ligation master mix (669ul of water, 120μl of 10X NEB T4 DNA ligase buffer (NEB, B0202), 100μl of 10% Triton X-100, 6μl of 20mg/ml Bovine Serum Albumin (MilliporeSigma B8667), 5μl of 400 U/ μl T4 DNA Ligase (NEB, M0202)) and incubated at room temperature for 4 hours with slow rotation. DNA was decrosslinked with 50ul of 20mg/ml proteinase K (QIAGEN 19133) and 120ul of 10% SDS and incubated at 55°C for 30 minutes. The reaction was quenched by adding 130μl of 5M sodium chloride and incubating at 68°C overnight. DNA was precipitated by adding 1.6X the volume of pure ethanol and 0.1X the volume of 3M pH 5.2 sodium acetate (MilliporeSigma S7899). The precipitate was incubated at −80°C for at least half an hour and centrifuged at max speed in 2°C for 15 minutes to discard supernatant. DNA was washed once with 700ul of 80% ethanol. The dried DNA pellet was dissolved in 130ul of 10 mM Tris-HCl, pH 8. The solution was sonicated to shear the DNA to an average size of 300-500bp using a Covaris sonicator with the following parameters set: PIP 140, duty factor 10, burst 200, and duration 58s-80s. 4ul of sheared DNA was run in 16ul of water on a 2% agarose gel to verify the size.
Biotin-labeled DNA was pulled down by washing 150μl of 10mg/ml Dynabeads MyOne Streptavidin T1 beads (Life technologies, 65602) with 400μl of 1X Tween Washing Buffer (TWB: 5mM Tris-HCl (pH 7.5); 0.5mM EDTA; 1M NaCl; 0.05% Tween 20) and the solution was discarded. The beads were resuspended in 300μl of 2X Binding Buffer (10mM Tris-HCl (pH 7.5); 1mM EDTA; 2M NaCl) and added to the sheared DNA. The DNA-bead mixtures were incubated at room temperature for 15 minutes with rotation. Beads were separated using a magnetic rack and the supernatant was discarded. Beads were washed with 600ul of TWB buffer twice. Sheared DNA was end repaired by resuspending the beads in 100ul of 1X NEB T4 DNA ligase buffer (NEB, B0202), separating the beads, resuspending in end repair master mix (88μl of 1X NEB T4 DNA ligase buffer with 10mM ATP (NEB B0202S), 2μl of 25mM dNTP mix, 5μl of 10U/μl NEB T4 PNK (NEB, M0201), 4μl of 3U/μl NEB T4 DNA polymerase I (NEB, M0203), 1μl of 5U/μl NEB DNA polymerase I, Large (Klenow) Fragment (NEB, M0210)), and incubating at room temperature for half an hour. Beads were washed twice with 500ul of TWB buffer, resuspended in 100μl of 1X Quick ligation reaction buffer (NEB, B6058), and recollected. To proceed with dATP attachment, beads were resuspended in 100μl of master mix (90μl of 1X NEBuffer 2, 5μl of 10mM dATP, 5μl of 5U/μl NEB Klenow exo minus (NEB, M0212)) and incubated at 37°C for 30 minutes. Beads were washed twice with 500ul of TWB buffer, resuspended in 100μl of 1X Quick ligation reaction buffer (NEB, B6058), and recollected. To proceed with adaptor ligation, beads were resuspended in 50μl of 1X NEB Quick ligation reaction buffer, 2μl of NEB DNA Quick ligase (NEB, M2200), 3ul of Illumina adaptor of choice, and incubated at room temperature for 15 minutes. Beads were washed with 600ul of TWB buffer and 100ul of 1X Tris buffer, resuspended in 50ul of 1X Tris buffer, and heated at 98°C for 10 minutes to elute the DNA off the beads. Beads were discarded. Size selection was performed to remove small DNA fragments by adding 0.8X-0.9X KAPA beads to the DNA elution, incubating at room temperature for 5 minutes, and collecting the beads while discarding the supernatant. Beads were washed twice with 500ul of 80% ethanol and eluted in 50ul of 1X Tris buffer. Library amplification was performed with 4-12 cycles of PCR with KAPA 2X library mix. Size selection was performed to remove small and large fragments using KAPA beads and maintain DNA fragments of 150bp-500bp. Libraries were sequenced as 150 bp paired-end reads with a raw sequencing depth between 300 million to 700 million read pairs per sample on platform Hiseq Xten or Novaseq.
CUT&Tag
The CUT & Tag experiments were performed exactly following the online protocol53: https://www.protocols.io/view/bench-top-cut-amp-tag-z6hf9b6?version_warning=no. For each targeted protein, 0.1 million cells were used. The following primary antibodies were used with 1:50 dilution: CTCF (Active motif 61311), H3K27ac (Active motif 39133), H3K27me3 (Cell signaling C36B11), dCas9 (Sigma-Aldrich #SAB4200701), Rabbit IgG (Cell signaling 2729). We use Guinea Pig anti-Rabbit IgG (H+L) secondary antibody (NBP1-72763) with 1:50 dilution. pA-Tn5 was bought from The earlier batch pA-Tn5 fusion protein was kindly gifted to us from Dr Steven Henikoff’s Lab. Final libraries were sequenced as 150 bp paired-end reads on platform Novaseq or Hiseq Xten with raw sequencing depths between 10 to 20 million read pairs.
ATAC-seq
ATAC-seq was performed following the published protocol with minimal modification54. Briefly, we centrifuged down 50,000 cells, washed the cells with PBS, and performed nuclei extraction with cold lysis buffer. We added an extra step of washing the nuclei with another 500ul of lysis buffer to further remove mitochondrial DNA. We then proceeded with the transposition reaction (Illumina Tagment DNA Enzyme and Buffer Large Kit 20034198) and purification steps (QIAGEN MinElute PCR Purification Kit Cat No./ID: 28004). We eluted the DNA in 20ul of elution buffer instead of the recommended 10ul of elution buffer to increase recovery rate. Then, we proceeded with the PCR amplification step, where 20ul of transposed DNA, 2.5ul of Nextera PCR primer 1, 2.5ul of Nexteral primer 2, and 25ul of KAPA HiFi HotStart Ready Mix master mix (KAPA KR0370) were used. We applied the PCR parameters indicated by the standard protocol with 11 cycles. We then performed size selection to remove small fragments using KAPA pure beads: 45ul of KAPA beads were added to 50ul of PCR solution, incubated for 15 minutes at room temperature, and a magnet was used to capture the beads and discard the supernatant. The beads were washed with 200ul of 80% ethanol twice and the residual ethanol was removed. We resuspend the beads in 20-50ul of pre-warmed 10mM Tris-HCl pH 8.0, incubated at 37°C for 10 minutes, and used a magnet to recollect the supernatant as the final library. We ran 1ul of the library on a 2% agarose gel to verify the footprint nucleosomes and confirm a successful assay. Libraries were sequenced as 150bp paired-end reads on platform Hiseq 4000 with 20 million raw read pairs per sample.
PCR-free whole genome sequencing
Genomic DNA was isolated by QIAGEN DNeasy Blood & Tissue Kits (69504) using 0.5 million cells. Concentration was detected by fluorometer or Microplate Reader (e.g. Qubit Fluorometer, Invitrogen). Sample integrity and purity were detected by Agarose Gel Electrophoresis. 1μg of genomic DNA was fragmented by Covaris. KAPA pure Magnetic beads (KK8000) were used to select DNA fragments with an average size of 300-400bp. DNA was quantified by Qubit fluorometer. The Fragments were subjected to end-repair and were then 3’ adenylated. Adaptors were ligated to the ends of these 3’ adenylated fragments. The double stranded products were heat denatured and circularized by the splint oligo sequence. The single stranded circle DNA was formatted as the final library. Library was qualified by the Agilent Technologies 2100 bioanalyzer. The library was amplified to make DNA nanoball (DNB) which have more than 300 copies of each molecule. The DNBs were loaded into the patterned nanoarray and sequenced as 150bp paired-end reads by combinatorial Probe-Anchor Synthesis (cPAS).
Whole genome bisulfite sequencing
DNA bisulfite treatment was performed using the EZ DNA Methylation-Gold Kit (catalog D5005, Zymo Research Corporation) according to the manufacturer’s instructions. The recovered bisulfite-converted single-stranded DNA was processed for library construction using the Accel-NGS@Methyl-seq DNA Library kit (catalog 30024, Swift BioSciences) as per manufacturer instructions. Briefly, using the Adaptase module, truncated adapter sequences were incorporated to the single-stranded DNA in a template-independent reaction through sequential steps. DNA was then enriched using 6 cycles of PCR with primers compatible with Illumina sequencing. The quantity and molecular size of the library was confirmed by Qubit HS DNA assay (ThermoFisher) and Tapestation 2200 system coupled with High Sensitivity D1000 ScreenTapes (Agilent). Illumina 8-nt dual-indices were used for multiplexing. Samples were pooled and sequenced on Illumina NovaSeq S4 sequencer for 150 bp read length in paired-end mode with an output of 580 million reads per sample.
RNA-seq
RNA was extracted using QIAGEN RNeasy Plus kit (74034). RNA quality was assessed by Agilent RNA ScreenTape on Agilent 2200 Tapestation and quantified by Qubit. The mRNA was enriched by poly-A selection and the second strand synthesis was performed with the NEBNext Ultra II Non-Directional RNA Second Strand Synthesis Module following manufacturer’s instructions (E6111S). Average final library size was between 380-400 bp. Illumina 8-nt dual-indices were used for multiplexing. Samples were pooled and sequenced on Illumina HiSeq X sequencer as 150 bp paired-end reads with an output of 40 million reads per sample.
CRISPR interference (de-repression) for Silencer Loops and Neo-Loops
CRISPR activation was used to validate the predicted silencer loops and repressive neo-loops. The single-guide RNA (sgRNA) sequences were designed using CRISPick from BROAD institute (https://portals.broadinstitute.org/gppx/crispick/public), targeting the silencer regions. The best sgRNAs were determined as the top-ranking sequences based on factors including on-target scores and off-target scores. To generate sgRNA expression vectors, each pair of the oligonucleotides for Spacers (Integrated DNA Technologies) were annealed into double-stranded DNA (Supplementary Table 10). The pool of the library for each were cloned into the sgLenti plasmid (Addgene 105996), which had been digested by AarI (Thermo Fisher Scientific) and gel-purified. The constructs were then transformed to Stbl3 competent cells for expansion (Invitrogen #C737303). Plasmids were extracted by Qiagen Midi-prep plus kit (Cat. 12941) and proper insertion was confirmed through Sanger Sequencing. The sgRNA were packed into lentivirus. Briefly, pMD2.G, psPAX2 and desired vectors were co-transfected into HEK293 T cells using Lipofectamine 3000 Transfection Kit (Thermo Fisher Scientific L3000015). The media was removed the next day and 10ml of fresh DMEM + 10% FBS media was added. Lentiviruses were harvested for three days and condensed using Amicon Ultra-15 Centrifugal Filter Units (Sigma UFC901024). The lentivirus was then transfected to Kasumi-1 or THP-1 cells, and the viable cells that are also RFP+ were selected by FACS sorting six days later.
pLV hUbC-VP64 dCas9 VP64-T2A-GFP (Addgene 59791) and dCas9-UTX (gift from Dr. Steven M. Offer, Mayo Clinic) were also separately packed into lentivirus by the procedures mentioned above. The lentivirus was transfected into Kasumi-1 or THP-1 cells that are stably expressing using sgRNA. Three days later, the expression of the dCas9-UTX fusion protein was tested by Western Blot (Sigma-Aldrich #SAB4200701) with expected shift to larger size (~130Kd to ~300Kd). Cells were then grown to sufficient numbers and the resulting changes in gene expression were quantified by qRT-PCR. The primers for qPCR are listed in the Supplementary Table 10.
CRISPR-deletion for Kasumi-1 RTTN Silencer Loop
sgRNAs for the CRISPR-Cas9 system were designed using a combination of benchling (https://www.benchling.com/), CHOPCHOP (https://chopchop.cbu.uib.no/), and CRISPick (https://portals.broadinstitute.org/gppx/crispick/public). Best sgRNAs were determined by selecting sequences that ranked highly in all three public databases (Supplementary Table 11). Factors such as on-target binding efficiency, off-target unspecific binding, GC content, and self-complementarity were considered. Sequences were given overhangs to facilitate sticky-end ligation with digested backbone. The left cut site used a BsmbI-V2 (NEB #R0580) digested LentiCRISPRv2GFP plasmid (Addgene 82416), and the right cut site used an AarI (Thermo Fisher Scientific) digested sgLenti plasmid (Addgene 105996). The sequences for sgRNA cloning were annealed, ligated, transformed, and positively selected using LB + 1% Ampicillin plates. Plasmids were extracted from positive clones and proper insertion was confirmed through Sanger Sequencing.
Lentiviruses were made using the Lipofectamine 3000 Transfection Kit (ThermoFisher L3000015). A mixture of Lipofectamine 3000 with Opti-Mem and a mixture of P3000 with pMD136 plasmid, pAX2 plasmid, and the desired virulent plasmid were combined. After a 15-minute incubation, the mixture was added dropwise to a 10cm plate of HEK293T cells with 5ml of DMEM + 10% FBS. The plates were incubated at 37°C for 4-6 hours before adding 3ml of media. The next day, the media was removed and 10ml of fresh DMEM + 10% FBS media was added. The media was collected for the next three days, storing the collection at 4°C and avoiding light exposure. After three collections, the virus-containing media was condensed using Amicon Ultra-15 Centrifugal Filter Units (Sigma UFC901024). Kasumi-1 cells were initially transfected by adding the condensed virus and polybrene (Millipore Sigma™ TR1003G), incubating for six hours, and then adding additional virus and polybrene to increase the transfection rate. The cells were checked for both GFP and RFP fluorescence before selecting for double-positive cells through flow-cytometry. The cells were single-cell sorted into 96-well plates as well as pooled into 15ml tubes and replated into 24-well plates. For the next week or two, cells were maintained and expanded as necessary. After sufficient proliferation, single-cell clones and pooled cells were lysed for genomic DNA, amplified by PCR, and analyzed via gel electrophoresis to determine successful cuts. PCR primers were designed such that the PCR product size was approximately 250bp for wildtype and 700bp when successfully deleted. The clones appeared with the 700bp band was further verified with Sanger sequencing. The clone with a successful deletion was expanded in culture. The qPCR was performed for RTTN RNA expression with the primer listed in Supplementary Table 11, using CRPSIR-processed Kasumi-1 cells without the deletion as the control.
CRISPR interference screening
CRISPR screening was carried out following the previously published protocol38,55. Briefly, the library for Spacer sequences (Supplementary Table 4) were designed using design_library.py and synthesized at IDT. Totally, 44 non-redundant enhancers in 74 pairs of P-E neo-loops from Kasumi-1 cells were targeted by 211 sgRNA, with each enhancer targeted by up to 6 sgRNAs. The library was PCR-amplified with 12 cycles using primer GTAACTTGAAAGTATTTCGA TTTCTTGGCTTTATATATCTTGT GGAAAGGACGAAACACC (forward), and ACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAG CTCTAAAAC (reverse), and was purified by QIAquick Gel Extraction Kit (Cat. 28706). Plasmid lentiGuide-Puro (Addgene, cat. 52963) was digested with BsmBI-V2 (NEB #R0580) at 50 °C for 15 minutes and 42 °C for 15 minutes, and purified with QIAquick Gel Extraction Kit. Gibson assembly (NEB #E2611S) was then performed to ligate the plasmid and amplified library, with the product purified by isopropanol precipitation. Pooled sgRNA library was electroporated into Endura ElectroCompetent cells (Lucigene Cat. 60242) with a final concentration of 25ng/ul, following the user manual. Transformed cells were transferred to large low-salt LB agar plates. Transformation efficiency was quantified by 1000 times dilution. 47,000 colonies were acquired, with each sgRNA covered by more than 200 colonies. Colonies were harvested from large plates by scraping and washing 5 times into ampicillin-containing LB and pelleting by centrifuge. The plasmid was extracted using QIAGEN Plasmid Plus Midi Kit (Cat. 12943).
The before-screening library was constructed by PCR using the pooled plasmid as template and NGS-Lib primers (NGS-Lib-Fwd-1: AATGATACGGCGACCACCGAGATCTA CACTCTTTCCCTACACGACGCTCTTCCGATCTTAAGTAGAGGCTTTATATATCTTGTGGAAAGGACGAAACACC; NGS-Lib-Fwd-2: AATGATACGGCGACCACCGAGATCTACACTC TTTCCCTACACGACGCTCTTCCGATCTACATGCTTAGCTTTATATATCTTGTGGAAAGGACGAAACACC; NGS-Lib-Rev-1: CAAGCAGAAGACGGCATACGAGATTCGCCTTGGTG ACTGGAGTTCAGACGTG TGCTCTTCCGATCTCCGACTCGGTGCCACTTTTTCAA; NGS-Lib-Rev-2: CAAGCAGAAGACGGCATACGAGATAT AGCGTCGTGACTGGAGTTCAGACGTG TGCTCTTCCGATCTCCGACTCGGTGCC ACTTTTTCAA), purified by Qiagen gel purification kit, and sent for sequencing on Illumina HiSeq Xten platform to assess the abundance and distribution of each sgRNA using python script count_spacers.py. The library passed QC by the following parameters: skew ratio = 2.53 (criterial <10); perfect match = 91.5 (>70%); undetected guides = 0 (<0.5%); Coverage = ~5,500 per sgRNA (>100). Lentivirus was then generated from the pooled plasmid as described in the method of CRISPR-deletion.
In parallel to the sgRNA library construction, lentiviruses were generated for Lenti_dCas9-KRAB-MeCP2 (Addgene #122205) as mentioned above. Viruses were transfected into Kasumi-1 cells as mentioned above. Cells underwent 15-day selection by 10ug/ml Blasticidine S hydrochloride (Millipore Sigma CAS Number: 3513-03-9) and one week recovery in Blasticidine-free medium, and the selected cells were tested for stable expression of dCas9-KRAB-MeCP2 by Western Blot. In two separate replicates of experiments, lentiviruses of sgRNA plasmids were then transfected into 2 million of the treated Kasumi-1 cells with an MOI<0.3. Cells were selected with 1.5ug/ml puromycin for 10 days and recovered in puromycin-free medium for one more week. Cells were then harvested for DNA extraction using Qiagen DNeasy Blood & Tissue Kit (Cat. 69504). The screening library was then constructed using the extracted DNA as a template and the NGS-Lib-Fwd and NGS-Lib-Rev as PCR primers. The PCR product from a bright band of 270-280bp was gel purified by Qiagen purification kit, and it was sequenced on an Illumina HiSeq Xten platform. The primary screening results were analyzed by RIGER38 and MAGECK56.
CRISPR interference screening validation
1). Enhancer reporter assay
We use reporter assay to validate the gene upregulation activity of five of the putative hijacked enhancers, for which the sgRNAs were significantly depleted during CRISPR screening. The enhancers were amplified using genomic DNA of Kasumi-1 cells as template, using the primers summarized in the Supplementary Table 12. PCR reaction was performed using Q5® High-Fidelity 2X Master Mix (Cat. M0492S), and the amplicon was cloned into pGL4.23[luc2/minimal promoter] purchased from Promega (cat. #E8411) by NEBuilder® HiFi DNA Assembly Master Mix (cat. E2621S). All insertions have been verified by Sanger sequencing. 0.5 million Kasumi-1 cells were transfected with 2ug of pGL4 constructs, 100ng Renilla for data normalization and 100ng GFP as transfection control using 6uL of TransIT®-2020 Transfection Reagent (cat. MIR5404), following manufacturer instructions. 48 hours post-transfection, luciferase activity was measured using Dual-Glo® Luciferase Assay System (cat. E2920) following manufacturer instructions, measurements were taken in triplicates. To control for cell number and transfection efficiency, firefly luciferase activity was normalized to renilla luciferase. Measurements were presented as a ratio relative to the activity of the pGL4.23-mini/P empty vector.
2). CRISPRi and qPCR of the predicted regulated gene
The five enhancer hijacking events were separately validated by first cloning their screening sgRNA into plasmid sgLenti (Supplementary Table 13), and the plasmid was packed for lentivirus generation. The original sgLenti plasmid with a stuffer sequence was also packaged for lentivirus generation as a control. Two million Kasumi-1 cells that stably express dCas9-KRAB-MeCP2, as mentioned above, were transfected with this sgRNA or the control lentivirus, respectively. Transfected cells were selected by puromycin (1.5ug/ml) for 6 days, and viable RFP+ cells were collected through fluorescent cell sorting. qPCR was performed to measure the impact on the predicted gene expression with primers listed in Supplementary Table 13. Cell phenotypic change associated with CRISPRi was tested by CCK-8 assay following the manufacture instruction (ab228554). Cell dependence to a gene was summarized by curating DepMap 21Q3 gene effect score of this gene in all AML cell lines (https://depmap.org/portal/).
3D DNA FISH
DNA FISH assays were performed as described with modifications57. THP1 and KASUMI-1 cells were fixed with 2% formaldehyde solution and nuclei extracted as described in the Hi-C method. The pellet was washed with PBS and stored at −80C. Crosslinked cells were thawed on ice and lysed in cold lysis buffer (10 mM Tris-HCl pH 8.0, 10 mM NaCl, 0.2% Triton-X with proteinase inhibitor) for 10 minutes, followed by washing with PBS. The pellet was resuspended in PBS and attached to glass plates (Superfrost Plus Microscope Slides, Fisher Scientific) by centrifugation at 300 g for 3 minutes. Cells were permeabilized in buffer containing 0.1% saponin and 0.1% Triton X-100 in PBS for 10 minutes at RT, incubated more than 20 minutes at RT with 20% glycerol in PBS, and freeze-thawed in liquid nitrogen three times. Then, cells were treated with 0.1 M HCl for 30 minutes at RT and permeabilized in 0.5% saponin and 0.5% Triton X-100 in PBS for 30 minutes at RT. For hybridization, nuclear DNA was denatured by incubating glass plates with buffer containing 2x SSC and 70% formamide for 2.5 minutes at 75C, followed by incubation with 2x SSC and 50% formamide for 30 seconds. All probes were made from BAC clones (BACPAC Genomics) CH17-330N5 (IKZF2 silencer, chr2:212,609,118-212,811,039), CH17-77N7 (IKZF2 promoter, chr2:213,048,640-213,261,522), CH17-9L22 (IKZF2 equal-distant control, chr2:213,495,758-213,695,712), CH17-338G21 (RTTN promoter, chr18:70,119,339-70,322,909), RP11-1145C7 (RTTN silencer, chr18:70,369,642-70,506,002), and CH17-472F14 (RTTN equal-distant control, chr18:69,842,716-70,042,911) by nick translation with chemical coupling using an Alexa Fluor succinimidyl ester (Alexa488 for CH17-77N7 and CH17-472F14; Alexa555 for CH17-330N5 and CH17-338G21; Alexa647 for CH17-9L22 and RP11-1145C7) (Thermo Fisher Scientific). The hybridization solution contained ~50 ng of each labeled probe, 6 mg of human Cot-1 DNA, and 10 mg of sheared salmon-sperm DNA in hybridization buffer (10% dextran sulfate, 50% formamide, 2x SSC). The probes were denatured at 75C for 5 minutes before use. Denatured cells and probes were sealed and incubated in a hybridization oven at 37C for overnight. On the next day, coverslips were removed and washed once in 2x SSC, 50% formamide solution for 15 minutes at 45C, three times in 2x SSC for 5 minutes at 45C, and once with 2x SSC for 5 minutes at RT with gentle agitation. Cells were washed once with PBS and stained with Hoechst dye. After washing with PBS, cells were mounted with Fluoromount-G mounting medium (Southern Biotech). Images were acquired on a confocal laser scanning microscope (Leica, Sp8 confocal) using a 63x objective lens (NA 1.40). A stack of ten to fifteen 0.3 μm thick optical sections was acquired for each field of view in the UV, green, red, and far-red channels. For the foci of interest, FISH signals were subjected to automated identification using custom macros in ImageJ. In the green, red and far-red channels, the centroid of each FISH signal was detected using auto local thresholding and the Euclidian distances to nearest alternate FISH probes were calculated.
DNMT3A/3B/1 Triple knockdown
For each gene, three shRNA were designed and oligos were synthesized at IDT (Sequence listed Supplementary Table 13). The DNMT3A shRNA were cloned into Tet-on-pLKO.1 (modified from Addgene #190853) using restriction enzymes AgeI (NEB #R3552) and EcoRI (NEB #R0101), DNMT1 shRNA were cloned to pLKO3G (Addgene #14748) using EcoRI and PacI (NEB #R0547), and DNMT3B were cloned into pLKO.1 mCherry (Addgene #128073) using AgeI and EcoRI. All plasmids were separately transformed into Stbl3 competent cells for expansion (Invitrogen #C737303). Each cloning was verified by Sanger sequencing. Each plasmid was extracted by endotoxin-free mini-prep (101Bio #W2106). The 3 plasmids for the same gene was pooled together for lentivirus generation and condensation following the protocol described above. The pLKO.1 vector (Addgene #10878) was used as control to produce lentivirus. 300 ul of each of the condensed DNMT3A/DNMT3B/DNMT1 shRNA lentivirus were pooled together to transfect 10 million U937 cells in the presence of 80 mg polybrene. Same process for the control lentivirus. Cells were selected in 1.5ug/ml puromycin for 10 days and dead cells were first removed by Ficoll (GE Healthcare, 17-1440-02) selection following the user manual, and cells was further selected for the top 4% of GFP and RFP double positive cells by FACS sorting. The selected cells were recovered in regular complete media for two days, and treated with 2.5 ug/ml Doxycycling for 12 days to induce long-term DNMT3A knockdown. The effect of triple knockdown was tested by Western blot with antibodies for DNMT3A (Santa Cruz sc-365769), DNMT3B (Cell signaling #D7070), DNMT1 (Cell signaling #D63A6) and GAPDH (Proteintech #60004-1-lg).
HiChIP experiment
HiChIP data was generated using the Arima-HiC+ Kit (P/N A101020), according to the manufacture’s protocol. Briefly, about 4 million cells was harvested and crosslinked with the 37% formaldehyde (MilliporeSigma 252549) in PBS to a final concentration of 2%, and incubated at room temperature for 10 minutes. The resulting solution was quenched with a Stop solution 1 and incubated at room temperature for 5 minutes. Crosslinked cells were treated following the Arima-HiC steps. Chromatin was sheared with Covaris S220 instrument and H3K27me3 (Cell signaling C36B11) antibody was used to bind the chromatin. HiChIP library was prepared using Swift Biosciences Accel-NGS 2S Plus DNA Library Kit following the manufacturer's instructions. The quantity and molecular size of the library was confirmed by Qubit HS DNA assay (ThermoFisher) and Tapestation 2200 system(Agilent). Library was sequenced on Illumina NovaSeq S4 (2x150bp) platform.
Assessment of apoptosis and DNA damage
U937 cells were cultured in either 0.5M 5-AZA or the same volume of DMSO for 12 days with alternate day drug media readministering. For apoptosis-positive control, cells were cultured in 100uM of Apoptosis Activator 2 drug (TOCRIS 2098) for six hours. For DNA damage positive control, U937 cells were irradiated with weakened UV light (13.4 watts from a germicidal bulb (G30T8) with minimized dosage penetrating cell culture dish lid, 15cm away, for three hours). The cells were then collected, washed with PBS, and lysed with RIPA Buffer (Sigma R0278) + Protease Inhibitor (Sigma 11873580001). SDS-PAGE was conducted on cell lysates, and the separated proteins were transferred to polyvinylidene difluoride membranes. The membranes were immunoblotted with antibodies for cleaved caspase-3 (Cell Signaling 9664, 1:1000 dilution), Phospho-Histone H2AX (Cell Signaling 9718, 1:1000 dilution), β-actin (abcam 8227, 1:2000 dilution), and GAPDH (Proteintech 60004, 1:50,000 dilution).
Cell Cycle Assay
The Propidium Iodide (PI) staining experiments were performed following a published protocol: https://www.nature.com/articles/nprot.2006.238. U937 cells were cultured in either 0.5M 5-AZA or the same volume of DMSO for 12 days with alternate day drug media readministering. For cell cycle arrest positive control, U937 cells were cultured in either 1uM, 2uM, 4uM, or 8uM of 5-AZA for 48 hours. The cells were collected and subjected to the above protocol’s “quick method” for thymocytes and non-adherent mononuclear cells. After staining, the cells were analyzed by flow cytometry: live cells were selected for, duplicate cells were removed, and peaks were gated to quantify cell cycle distribution.
5-AZA treatment
5-azacytidine (MilliporeSigma A2385-100MG) was dissolved in DMSO to make 100mM stock solutions which were aliquoted and stored in −80°C. Working solutions (0.5uM-10uM) were made by further diluting the stock solution using complete cell culture media. Media was changed every 24 hours with freshly made 5-AZA solution. Dead cells are removed by Ficoll-Paque PLUS density gradient media at the end of cell culture before cells are further processed for any profiling experiments.
Proliferation assay
Cell proliferation was independently performed with three technical replicates every 24 hours. Proliferation was measured by viable cell count with trypan-blue staining, MTT assay (abcam ab211091) and CCK-8 assay (ApexBio K1018) following the manufacturer’s instruction.
II. Informatics analysis
Data collection
Raw sequencing data generated in this study was downloaded with the BaseSpace Sequence Hub CLI tool (version 1.0.0). Publicly available data used in this study was downloaded with fastq-dump (version 2.9.6).
Point mutation and structural variants analysis
WGS reads were first aligned to human genome reference GRCh38 with BWA MEM (version 0.7.17-r1198). PCR duplicates were removed by Sambamba (version 0.7.0) 58. Uniquely mapped (MAPQ > 20) reads were retained for downstream variant detection. Point mutations including single nucleotide mutations and small indels were detected by Freebayes (version 1.2.0-17-ga78ffc0) with parameters “--min-alternate-count 2 --min-alternate-fraction 0.05 --min-repeat-entropy 1”, and minimal quality score of 20 was used to reduce false positive calls. To minimize the number of germline calls, point mutations that overlap with dbSNP150 mutations were removed. The functional effects of the filtered variants were annotated with SnpEff (version 4.3T) 59. Only variants annotated as high or moderate impact by SnpEff were used for downstream analysis. This final set of point mutations was further used to confirm the clinical molecular diagnosis of our AML samples, and both results were used for optimizing the subtyping of the AML samples.
Structural variants (SVs) were detected using WGS and Hi-C data as previously described20. Delly (version 0.7.7) 60 and Speedseq (version 0.1.2) 61 were used for detecting SVs in WGS data. Centromere, telomere and heterochromatin regions were excluded for SV detection. SV calls from Delly and Speedseq were merged and only SVs detected by both methods were kept to reduce false positives. Furthermore, the detected SVs were compared against the DGV database (version 2016-05-15) to reduce germline SV calls. For SV detection in Hi-C data, HiC Breakfinder 20 was used with default parameters. In this work, only large deletion (> 1Mb), inversion (> 1Mb) and inter-chromosomal translocations were considered for Neo-loop analysis.
Hi-C data analysis
Paired-end reads were first trimmed by Trim_Galore! (version 0.6.0) to remove adapters and low-quality bases with parameter “--paired”. Trimmed reads were then mapped to human genome GRCh38 by BWA MEM (version 0.7.17-r1198) with parameter “-SP5M”, and deduplicated with “pairtools dedup” (version 0.3.0). Hi-C pairs were generated with “pairtools parse” by removing unmapped/multimapped read pairs and rescuing single ligations in chimeric reads. Reads mapped to the same MboI restriction fragment are not informative to chromatin interactions and thus are removed for downstream analysis. Hi-C matrices and cooler files were generated with Cooler (version 0.8.6.post0) 62. Iterative correction and eigenvector decomposition (ICE) method was used for Hi-C normalization with “cooler balance” option. HiGlass, Juicebox, and 3D Genome Browser (http://3dgenome.org/) were used for visualization of Hi-C matrices63-65.
Hi-C compartments were identified at 40kb resolution using a “sliding window” strategy as previously described 66. First, the “exp” (expected) matrix was obtained by averaging Hi-C contacts at the same distance. Then the “obs/exp” (observed/expected) matrix was calculated by summing the observed Hi-C contacts within a window of 400kb centered at each bin divided by the sum of expected Hi-C contacts in the same window. A step size of 40kb was used to calculate the “obs/exp” value for all elements in the matrix. The “obs/exp” matrix was then converted to a Pearson Correlation matrix. The principal components were derived by calculating the covariance matrix of the Pearson Correlation matrix followed by eigenvector decomposition with the ‘eigen’ function in R. The first principal component (PC1) was used to assign the A and B compartment where regions with positive PC1 values correspond to A compartment and negative to B compartment based on their association with gene density.
Topologically associating domains (TADs) were identified at 40kb resolution by DomainCaller11. Briefly, a Directionality Index (DI) was calculated for each genomic bins at a window size of 2Mb. Then a Hidden Markov model was used to determine the up-or-downstream biased status for each genomic bin based on the DI scores. TADs were defined as continuous genomic regions starting from the first bin of a series of consecutive downstream biased bins to the last bin of the next series of consecutive upstream biased bins. TAD boundaries were defined as regions between two adjacent TADs that are less than 400kb. Insulation Scores were calculated at 10kb resolution using cooltools (version 0.2.0) with a sliding “diamond” window of 1 Mb. Insulation Score was derived as the average chromatin interactions within a “diamond” region along the diagonal of the ICE-normalized Hi-C matrix. Insulation boundaries were obtained by searching for all local minima of the log-transformed insulation scores.
Loop domains were identified at 10kb resolution using Peakachu, a machine-learning based method recently developed in our lab27. Peakachu reports a probability score associated with each loop which demarcates confidence of loop calling. The probability score is also positively associated with the loop intensity, making it convenient for differential loop analysis. Peakachu detects loops as CTCF or H3K27ac loops depending on a pre-trained CTCF model or H3K27ac model. In this work, loops were first detected using both models and only those detected by both models were reported to reduce the false positive loop calls.
Hi-C Stripes identification
To detect architectural stripes on the contact maps, we used an in-house implementation of the algorithm proposed by Vian L et al34. The implementation was based on Python and the cooler package62. Briefly, for each chromosome, we first extracted the upper-triangular contact matrix at 40K resolution, and then detected horizontal (3’) and vertical (5’) stripes separately by searching for consecutive pixels with contact signals significantly higher than the local backgrounds. To ensure specificity, we filter raw calls to retain stripes larger than 300kb (eight of 40Kb binds) to avoid calls that are less distinguishable from loop clusters or background noise.
The code for Stripe identification is available at https://github.com/XiaoTaoWang/StripeCaller.
In detail, to detect the horizontal stripes, the observed signal for each pixel was calculated by summing the signal of the pixel and the signal of 2 nearby pixels on the horizontal line:
where Mi,k is the raw contact frequency between bin i and k, and .
Accordingly, two local backgrounds (termed “top” and “bottom”) were defined to calculate the expected signal strength:
where Med indicates the median function, indicates the ICE (iterative correction and eigenvector decomposition) normalized matrix, , and . Using Poisson statistics, each pixel was tested whether the observed signal was significantly higher than the expected strength, and the candidate stripe pixels were defined by satisfying pvalue < 0.01 and fold change > 1.1. Then we detected stretches of 3 consecutive candidate pixels and further merged the stretches if they were separated by less than 2 pixels on the same row. Finally, because anchors of multiple stripes are usually located near each other, we proposed a local clustering algorithm to detect such clusters and represented each cluster as a single stripe.
Similar procedures were performed in detecting vertical stripes. Specifically, the observed signal was calculated on the vertical line of each pixel, and the expected signal strengths were calculated for two horizontal backgrounds (termed “left” and “right”):
where , , .
SV-induced neo-loop identification and simulation of 3D structure
Neo-loop induced by SVs were identified using NeoLoopFinder developed by our lab36. To infer the 3D structure of the reconstructed assembly, we adopted a probabilistic model, “pastis”, proposed by Varoquaux N et al67. Briefly, the chromatin is represented by the bead-on-a-string model, in which each bead corresponds to a 10Kb genomic region. By modeling the Hi-C contact signals fij as independent Poisson random variables, this method transforms the structure inference problem into a maximum likelihood problem and uses the L-BFGS-B algorithm to solve the optimization. The 3D coordinates were reported in .pdb format and visualized using PyMoL.
CUT & Tag data analysis
CUT & Tag sequencing reads were processed using ENCODE ChIP-seq pipeline (https://github.com/ENCODE-DCC/chip-seq-pipeline2). Specifically, reads were first trimmed by Trim_Galore! with “--paired” option, and then aligned to human genome reference GRCh38 with Bowtie2 (version 2.3.5.1)68. PCR duplicates were removed by Picard MarkDuplicates tool with “VALIDATION_STRINGENCY=LENIENT” option. MACS2 (version 2.2.4) was used for peak calling for histone marks and TFs with “-p 1e-2 --nomodel --shift 0 --keep-dup all -B --SPMR” options. Peaks in the ENCODE hg38 blacklist regions (http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/hg38-human/hg38.blacklist.bed.gz) were filtered out. Peaks with MACS2-reported q value < 10e-5 were retained for downstream analyses. Log transformed p values (−log10 p-value) were used for track visualization in University of California Santa Cruz (UCSC) genome browser and Integrative Genomics Viewer (IGV).
Whole genome bisulfite sequencing data analysis
WGBS reads were processed using the Bismark pipeline69. Reads were first trimmed by 10 base pairs on the 5’ end of both forward and reverse reads using Trim_Galore! with “--paired --clip_R1 10 --clip_R2 10” options. Trimmed reads were then mapped with “bismark” command and deduplicated using Bismark’s “deduplicate_bismark” tool with default parameters. Per-cytosine methylation level was obtained using methylpy (version 1.4.2)70 “call-methylation-state” tool with “--binom-test True --paired-end True” options. The average CpG methylation level for a genomic region was calculated as the accumulation of methylated reads over all CpG sites divided by the total reads over all CpG sites in the region.
ATAC-seq data analysis
ATAC-seq sequencing reads were processed using ENCODE ATAC-seq pipeline (https://github.com/ENCODE-DCC/atac-seq-pipeline). Specifically, reads were first trimmed by Cutadapt (version 2.4) with “-m 5 -e 0.2” options. Trimmed reads were then aligned to human genome reference GRCh38 with “bowtie2 -X2000 --mm” and deduplicated using Picard MarkDuplicates tool with “VALIDATION_STRINGENCY=LENIENT” option. Read alignments were shifted +4bp on “+” strand and −5bp on “-” strand to account for Tn5 insertion before peak calling. Peaks were called by MACS2 with “--shift −75 --extsize 150 --nomodel -B --SPMR --keep-dup all --call-summits” options, and filtered against the ENCODE hg38 blacklist (http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/hg38-human/hg38.blacklist.bed.gz). Peaks were then filtered by MACS2-reported p value (p < 10e-5). Peak summits were extended by 250bp on both sides to a final width of 500bp for all downstream analyses. Overlapped peaks were handled using an iterative-removal approach similarly as previously described71. The most significant peak was examined, and any peaks directly overlapping with it were removed. Then this process iterates to the next most significant peak until all peaks are not overlapped. We performed a “score per million” normalization of MACS2 peak scores (−log10 p-value) by dividing each individual peak score by the sum of all peak scores in the sample divided by 1 million.
RNA-seq data analysis
RNA-seq sequencing reads were analyzed following the ENCODE standard pipeline. Raw sequencing reads were first adapter-trimmed by Trimm_Galore! with “--paired” option, and aligned to human genome reference GRCh38 with STAR (version 2.5.3a_modified)72 with “--outSAMunmapped Within --outFilterType BySJout --outFilterMultimapNmax 20 --outFilterMismatchNmax 999 --outFilterMismatchNoverReadLmax 0.04 --alignIntronMin 20 --alignIntronMax 1000000 --alignMatesGapMax 1000000 --alignSJoverhangMin 8 --alignSJDBoverhangMin 1 --sjdbScore 1” options. RSEM (version 1.2.31)73 was used for transcript quantification with GENCODE v24 annotation and “--paired-end --estimate-rspd --calc-ci” options. TPM for all transcripts were quantile-normalized across all samples using the “normalize.quantiles” function in the “preprocessCore” library in R.
HiChIP data analysis
H3K27me3 HiChIP data for Kasumi-1 cells was analyzed and long-range chromatin interactions were identified with MAPS (v1.1.0)74. Interactions were detected at 25kb resolution and a maximum distance of 3Mb. Positive Poisson model was used for identifying significant interactions with an FDR of 2. MACS2 was used for H3K27me3 broad peaks calling with parameters “--broad --nolambda --broad-cutoff 0.3”. Repressive loops were defined as HiChIP interactions that anchored at gene TSS in one end and H3K27me3 peaks in the other end. We focused on long-range loops that have a distance between 200kb and 1Mb. These repressive loop-anchoring genes are termed “repressive mark target genes”. Genes that are within same distance to but not looped to the repressive marks are termed “repressive mark non-target genes”.
HiC correlation analysis
Clustering of DMSO-treated, 5-AZA-treated, and DNMT TKD cells was based on stratum-adjusted correlation coefficient (SCC) of Hi-C matrices49. SCC was calculated using the python implementation of HiCRep (https://github.com/cmdoret/hicreppy) with parameters “-v 5 -m 5000000” and 10kb resolution of Hi-C matrix.
Hi-C based clustering of AML samples
The first principal component (PC1) of the Hi-C matrix was used for unsupervised clustering of AML samples. PC1 was calculated at 40kb resolution from the ICE-normalized Hi-C data. Then coefficients of variation of PC1 across all samples and controls were calculated for each 40kb genomic bins. The top 10% of bins with the largest variation of PC1 were selected for deriving a correlation matrix of PC1 across all samples. Hierarchical clustering was performed on the correlation matrix using “complete” linkage and “euclidean” distance metrics.
Identification of AML and subtype-specific A/B compartments switch
We first included only genomic bins (n = 63472, bin size 40kb) that are all A compartments or B compartments four HSPC samples. We defined A-to-B switch when a region is in compartment in HSPC but in B compartment in AML samples, and vice versa for B-to-A switch.
For subtype-specific A/B compartment switch, we apply a one-way ANOVA analysis of the PC1 value of each genomic bin across AML and HSPC samples by treating samples within the same subtype as replicates using aov() in R. Genomic bins with significant differences (ANOVA test, P < 0.05) among subtype groups were retained. The resulting genomic bins were first filtered by requiring the maximum subtype mean of PC1 values greater than 0 and minimum smaller than 0 (namely, regions with A/B compartment switch among HSPC and AML subtypes), and further filtered by requiring the maximum subtype mean of absolute PC1 values at least two times higher than the minimum subtype mean of absolute PC1 values. The filtered genomic bins (n = 6700) were then hierarchically clustered using “complete” linkage and “euclidean” distance metrics.
Identification of TAD alteration in AML
To identify TAD alteration in AML, a set of TADs that are conserved (reciprocal overlap > 0.9) in all three HSPC was first compiled. Then this set of conserved TADs was compared against the TADs in each AML sample to generate a per-sample altered TADs list. TADs that are altered in more than one AML samples were defined as recurrent altered TADs. Expression of genes in samples with altered TADs was compared with the same genes in samples without altered TADs to identify up- and down-regulation of gene expression due to recurrent TAD alteration. The chromatin contacts between the promoter of representative deregulated genes and near regions were closely examined with virtual 4C plot.
Identification of AML-specific gain or loss of loops
AML gained or lost loops specific to HSPC were identified based on the loop probability from Peakachu with a Gaussian mixture model. First, Peakachu loop calls from pairs of each AML and each of the four HSPC samples, respectively, were merged and deduplicated. Then for each of the merged loops, fold change of Peakchu probability between AML and HSPC samples, and the reciprocal fold change were used as input for a Gaussian mixture model to determine significantly differential loops at an FDR of 0.05. The detailed algorithm can be found in: https://github.com/tariks/peakachu/tree/master/diffPeakachu. Each AML individual was compared to all four HSPC controls independently, and then the consensus from all four lists of differential loops were deemed as AML-specific gain or loss of loops. Differential loops specific to more than two AML individuals were defined as recurrent gain or loss of AML loops. Expression of genes in AML samples with recurrently differential loops were compared with the same genes in HSPC samples to identify dysregulation of gene expression due to differential loop formation. In virtual 4C plot, contacts were first averaged across AML and HSPC samples, and then normalized as a proportion to the contacts within the anchor itself.
Identification of AML subtype-specific loops
Differential loops between each AML sample and HSPCs were first identified as described above. Differential loops from each AML sample were then combined and de-duplicated, resulting in a set of 9,140 loops at 10kb bin size. For each loop, we obtained the Peakachu probability in all AML and HSPC samples. This forms a loop probability matrix with rows corresponding to loops and columns corresponding to all samples. We then calculated the average loop probability within the same subtype. We applied additional filters such that we required the maximum subtype-wise average of loop probability is at least 1.5 fold greater than the minimum subtype-wise average of loop probability. Then rows (loops) of the probability matrix were clustered by hierarchical clustering with “complete” linkage and “euclidean” distance.
Identification of most variable methylation CpG islands
CpG island (n = 31,144) coordinates for hg38 were downloaded from UCSC Table Browser. We estimated mCG levels for each CpG island in all samples with available WGBS data. We then calculated the standard variation of mCG levels for each individual CpG island across the all samples with available WGBS, and the top 10,000 CpG islands with highest variation among all sites were selected.
Subtype-associated differentially methylated regions
Differentially methylated regions (DMRs) were identified by methylpy DMRfind with FDR 0.1. Samples within the same subtype were treated as replicates for DMR identification. A CG methylation matrix of all DMRs across AML and HSPC samples was constructed and row-wise standardized to be centered at 0 by subtracting the mean of the methylation levels, followed by division of the standard deviation of the methylation levels in a row. The rows of the methylation matrix are then hierarchical clustered with “complete” linkage and “euclidean” distance for heatmap visualization.
Differential aggregated Hi-C analysis
The differentially methylated CpG sites between AML and HSPC 213 were identified if the methylation levels of CpG site were significantly different between these two samples (beta-binomial distribution p-value<0.01) and the methylation difference was greater than 0.3. We then merged the differentially methylated CpG sites within 500bp into DMRs. The DMRs with a length greater than 20bp (2 x length of consensus sequences of CTCF core motif) and higher methylation level in AML samples were defined as AML-hypermethylated regions. The CTCF peak sites in AML samples and HSPC which overlapped with the AML hypermethylated region were used as the center of the heatmap. The CTCF peak sites were stretched or shrunken to 100bp (5 bins) in the heatmap
To visualize the overall differential interaction patterns around hypermethylated AML-reduced CTCF binding site, we first normalized the 25-kb interaction matrix by Knight-Ruiz method and then divided each entry of the matrix by the expected interaction frequency at the corresponding genomic distance to get the obs/exp matrix. Then we generated a log2-transofrmed fold-change matrix between AML samples and HSPC-21375,76. The fold-change matrix was used to generate the differential aggregated matrices.
Loop classification based on histone marks
We annotated the AML-specific loops and neo-loops into promoter-promoter (P-P), promoter-enhancer (P-E), promoter-silencer (P-S) loops based on H3K27ac and H3K27me3 marks within the two anchors of the loops. P-P loops were defined as those with both anchors containing gene promoters but without any H3K27ac or H3K27me3 marks. P-E (R) loops have one anchor containing gene promoter while the other anchor H3K27ac (H3K27me) marks. For anchors with the presence of both H3K27ac and H3K27me3 peaks, we adapted a normalization method described in Corces et al. Science 2018[15], in which the MACS2 log P-value for each peak is normalized by the total sum of −log P-values of all peaks. We then defined P-E loops, if the largest −log P-value of all H3K27ac peaks are greater than two-fold of the largest −log P-value of all H3K27me3 peaks, and vice versa for P-S loops.
Gene Set Enrichment Analysis (GSEA) of genes associated with AML-specific loops
We first identified recurrent AML-specific loops as loops occurred in two or more AML samples but not in all control samples. We then identified a set of genes associated with the recurrent AML-specific loops. We performed GSEA analysis (version 4.0.2) on this set of genes that shows differential expression with a two-fold change of TPM compared with control averages. Gene sets database was set to “c2.all.v7.1.symbols.gmt” in GSEA. Gene sets “JAATINEN_HEMATOPOIETIC_STEM_CELL_UP” and “DIAZ_CHRONIC_MEYLOGENOUS_LEUKEMIA_UP” were chosen to illustrate the gene enriched for important hematopoietic pathways.
IKZF2 survival analysis with TCGA data
Survival analysis of IKZF2 was performed in the GDC TCGA LAML cohort. RNA-seq data and survival data for the cohort was downloaded from Xena Browser (https://xenabrowser.net/datapages/?cohort=GDC%20TCGA%20Acute%20Myeloid%20Leukemia%20(LAML)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443). Survival analysis was performed using survminer and survival R packages. Briefly, AML patients were grouped into high-expression (> 75% percentile) and low-expression (< 25% percentile) groups based on IKZF2 expression, and Kaplan-Meier curve is plotted between the two groups.
Saddle plot of Hi-C data with DMSO and 5-AZA treatment
We generated HiC saddle plots following guides from cooltools (https://github.com/mirnylab/cooltools). Briefly, the first eigenvector (E1) calculated at 25kb resolution was ranked ascendingly and then equally binned into 38 groups. Next, each genomic bin was assigned to these groups based on its E1 value. We then calculated the average of “obs/exp” contacts between bins in each group with bins in all other groups, resulting an obs/exp contact matrix whose rows and columns were ranked according to E1 values.
Statistics and reproducibility
The Western Blots for DNMT3A, DNMT3B, DNMT1, and GAPDH in the DNMT TKD experiment were repeated for three times with similar results (Fig. 7d). The Western Blots for yH2AX, Caspase-3, GAPDH, and β-actin under 5-AZA treatment were repeated for twice with similar results (Extended Data Fig. 12c-d). Two biological replications were applied to each of CRISPR interference, CRISPR deletion, and reporter assay with similar results. Each biological replicate contains three technical replicates. All other statistics and replications are specified in each method and figure legend.
Extended Data
Extended Data Fig. 1. List of genomic experiments performed in this study.
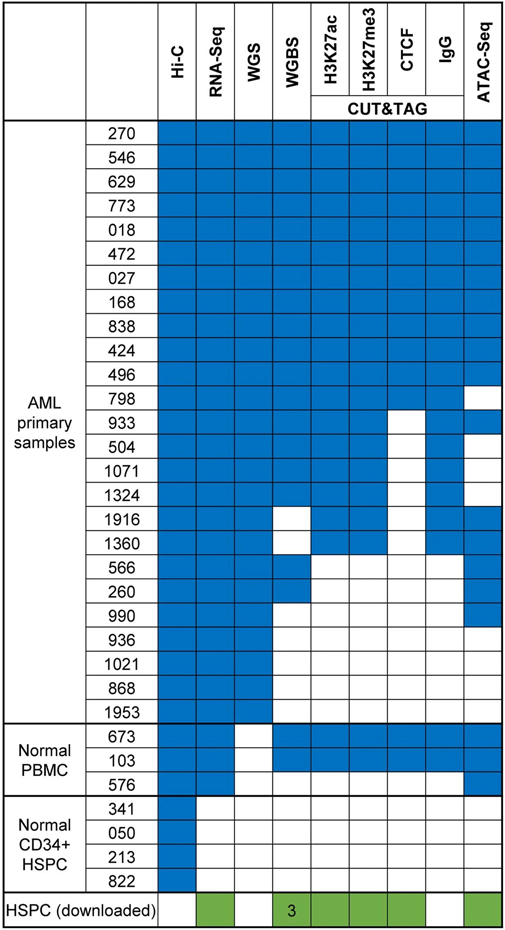
Extended Data Fig. 2. Examples of genes with A/B compartment switch.
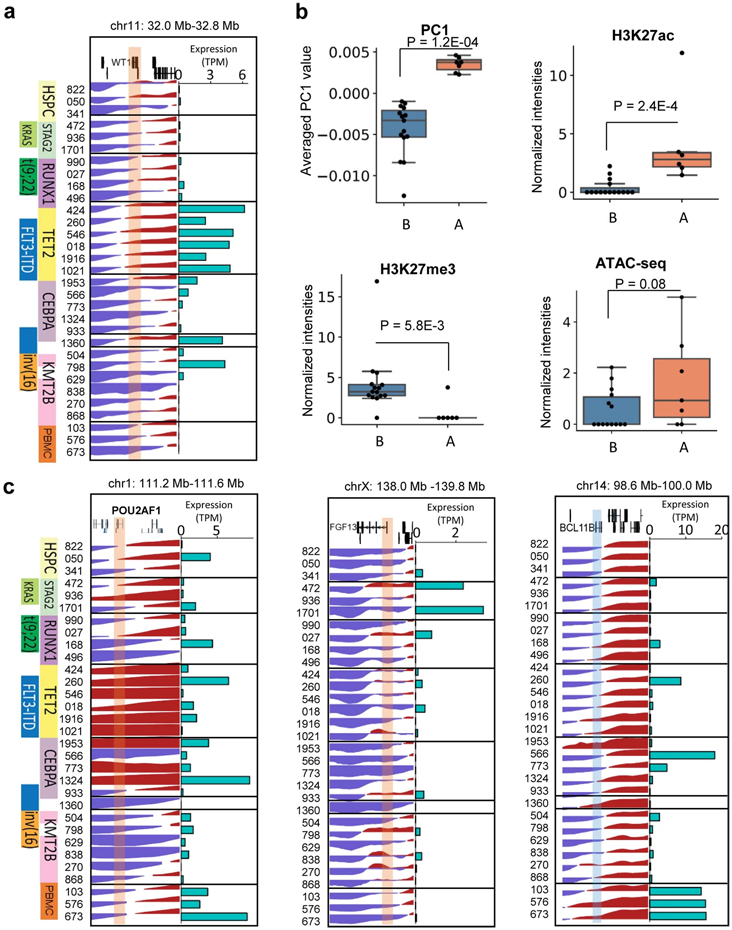
a and c, PC1 values and the expressions for WT1, POU2AF1, FGF13, and BCL11B genes. b, A compartment associated with higher H3K27ac and ATAC-seq signals and lower H3K27me3 intensities. Shown in figure are the ATAC-seq, CUT&Tag for H3K27ac and H3K27me3 signals at WT1 gene promoter, normalized to sequencing depths. Promoter is defined as within +/−1Kb of TSS. P value by two-sided Wilcoxon rank-sum test. PC1: A (n=15 samples) B (n=7 samples); H3K27ac: A (n=15) B (n=6); H3K27me3: A (n=15) B (n=6); ATAC-seq: A (n=14) B (n=7). Box plot: middle line denotes the median, top/bottom of boxes denotes first/third quartiles and whiskers extend to 1.5 times the interquartile range.
Extended Data Fig. 3. TAD boundary alteration and transcription.
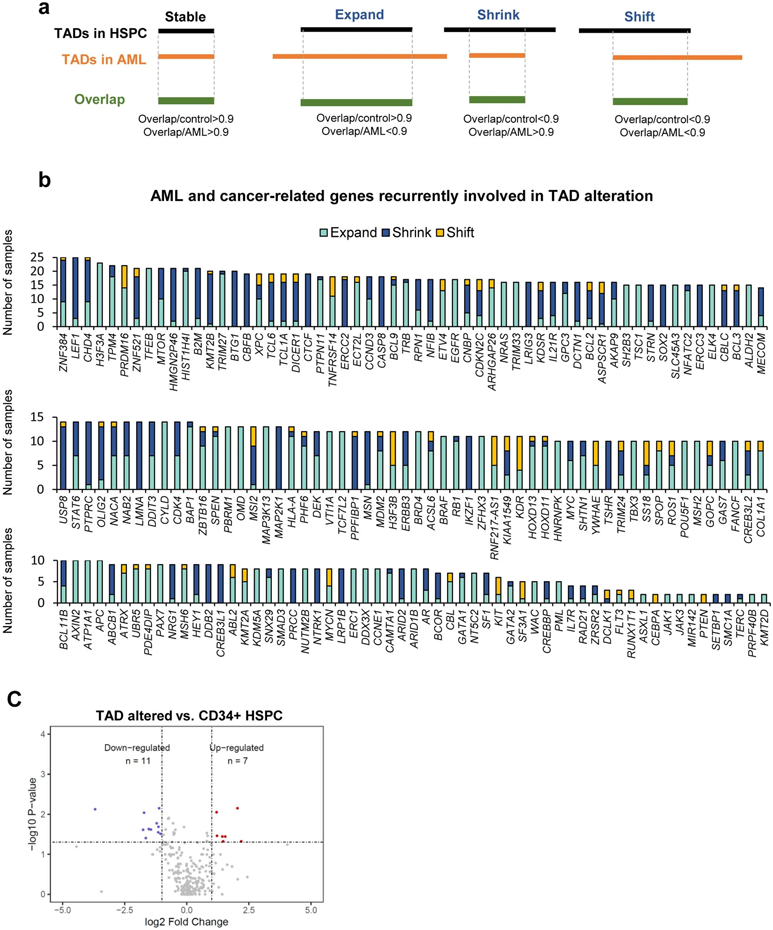
a, Illustration for how alteration of TAD boundary is defined in this study. b, COSMIC cancer-related genes that are located in TADs with recurrent change of boundary. Y axis is the number of incidences across different samples. c, Differential expression analysis of genes located inside recurrently altered TADs. P value by two-sided Student t-test.
Extended Data Fig. 4. AML-specific loop analysis.

a, Overlap of AML-specific loops with CNVs. Color scheme: neutral (blue), gain (orange), or loss (green) of copies. b, Genome-wide CNV profiles for all the 10kb bins. c, GSEA analysis for differentially expressed genes associated with AML-specific loops. P value is calculated by permutation test.
Extended Data Fig. 5. AML-specific loops associated with sample-specific open chromatins and gene expression.

a, Hi-C maps for the MEIS1 and ERG gene regions. Left lower halves are for AML samples and the right upper halves are for HSPC and PBMC. The loops are absent in all three PBMC samples (only one is plotted) and less frequent in the four HSPC samples (only one is plotted). b-c, RNA-Seq, CUT&Tag for H3K27ac and H3K27me3, and ATAC-Seq data for regions surrounding the three genes. Purple arcs indicate chromatin loops predicted in at least two AML samples. Shown in the figure are two representative AML samples.
Extended Data Fig. 6. Analysis of promoter-silencer (P-S) loops.
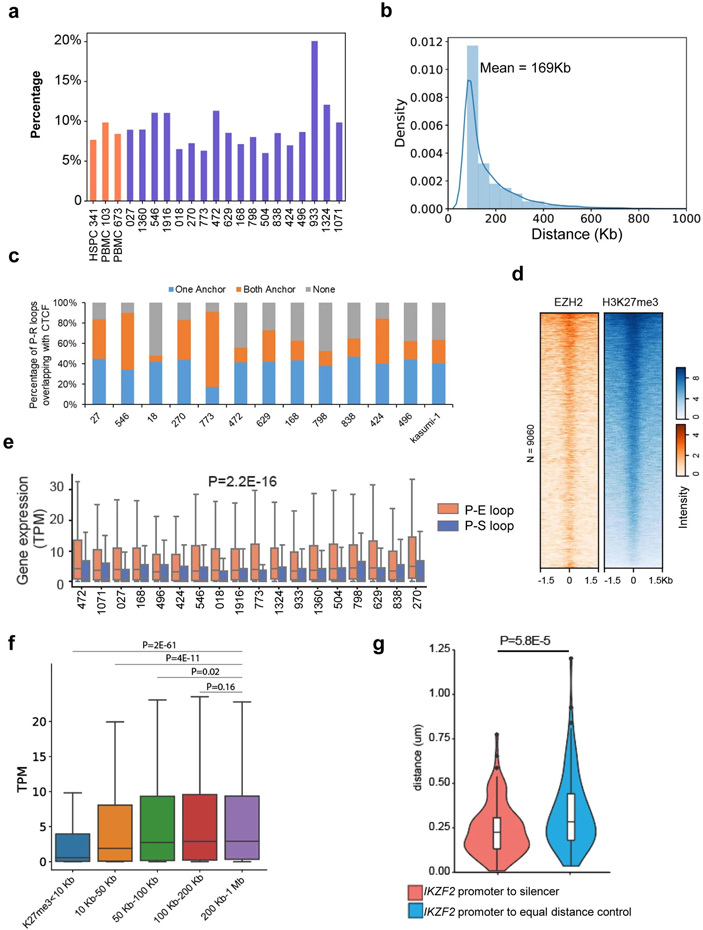
a, Percentage of P-S loops among all chromatin loops in all samples. b, Size distribution of P-S loops in all samples. c, Percentage of P-S loop anchors overlapping with CTCF binding peaks. d, Normalized EZH2 CUT&Tag signals at H3K27me3 peaks in the P-S loop anchors in THP-1 cells. e, Expression of genes that are located in the P-E vs. P-S loop anchors. P value was calculated by the two-sided Kruskal-Wallis test. P-E loops (n=31332), P-S loops (n=7912). Box plot: middle line denotes the median, top/bottom of boxes denotes first/third quartiles and whiskers extend to 1.5 times the interquartile range. f, Expression of genes in different patient samples with different distance to the nearest non-looping H3K27me3-marked silencer. P value calculated by one-sided Wilcoxon rank-sum test. N = 3809, 4403, 3865, 3217, 2490 genes (from left to right each category). Box plot: middle line denotes the median, top/bottom of boxes denotes first/third quartiles and whiskers extend to 1.5 times the interquartile range. g. Distance distribution between the IKZF2 promoter to control or its looped silencers in Kasumi-1 (n=112 alleles). P value by two-sided Wilcoxon rank-sum test. Box plot: middle line denotes the median, top/bottom of boxes denotes first/third quartiles and whiskers extend to 1.5 times the interquartile range.
Extended Data Fig. 7. Validation of a P-S loop for the RTTN gene.
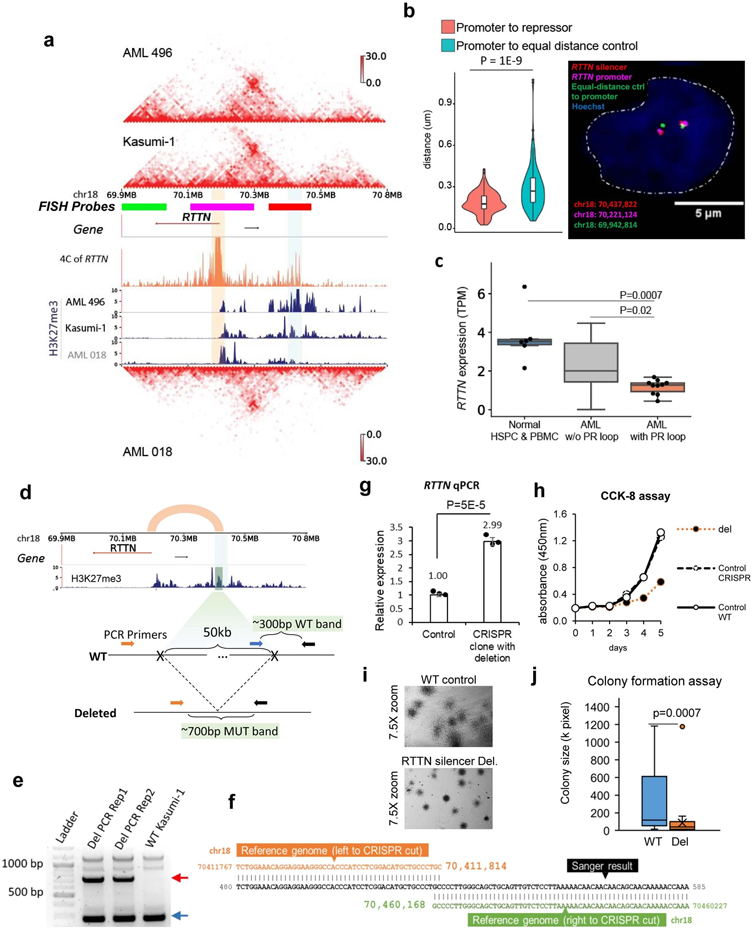
a, Hi-C and H3K27me3 CUT&Tag data in AML patient sample 496 and Kasumi-1 cells, which have the P-S loop for RTTN. AML sample 018 has the same loop but with less interacting signals and almost no H3K27me3 signals at the loop anchor. The orange tracks are the 4C data for RTTN in Kasumi-1 cells. b, (Right) DNA FISH imaging to measure the distance between RTTN promoters and the looped silencers in THP-1 cells. The control is the region on the other side of the RTTN promoter, with equal linear genomic distance. (Left) distance distribution between promoter and control or its looped silencer from 144 THP-1 cells (P value = 1E-9, two-sided Wilcoxon rank-sum test). Box plot: middle line denotes the median, top/bottom of boxes denotes first/third quartiles and whiskers extend to 1.5 times the interquartile range. c, RNA expression of RTTN across patient samples and normal controls. Normal HSPC&PBMC n=6 samples, AML w/o P-S loop n=13, AML with P-S loop n=10. P value was computed by the two-sided Student’s t-test. Box plot: middle line denotes the median, top/bottom of boxes denotes first/third quartiles and whiskers extend to 1.5 times the interquartile range. d, The design of PCR for detecting CRISPR deletion of the RTTN silencer (vertical bar in the H3K27me3 track). e, Two replicates of PCR results confirming the heterozygous deletion of the RTTN silencer. The red arrow points to the amplification of the CRISPR deletion junction (~700bp), and the blue arrow points to the wildtype band (~300bp). f. Sanger sequencing confirmed the CRISPR deletion of the silencer. g, qPCR results of RTTN expression in the clone with this heterozygous deletion vs. control group. The control group is other clones that underwent the same CRISPR system treatment without deletion (n=3 technical replicates in 2 biological replicates). P value by two-sided Student’s t-test. Data show mean ± s.e.m. h, CCK-8 assay results for proliferation of Kasumi-1 cells with the RTTN silencer deleted vs. both the wildtype and the CRISPR control cells in panel g (n=3 biological replicates). i, Stereomicroscope images of CFA cells on day 12 at 7.5X zoom. CFA was performed in n=3 replicates. j, Size (pixel) distribution of WT colonies (n=40) and the colonies with RTTN silencer deleted (n=35), measured by imageJ. P value calculated by two-sided Wilcoxon rank-sum test. Box plot: middle line denotes the median, top/bottom of boxes denotes first/third quartiles and whiskers extend to 1.5 times the interquartile range.
Extended Data Fig. 8. Analysis of chromatin stripes.
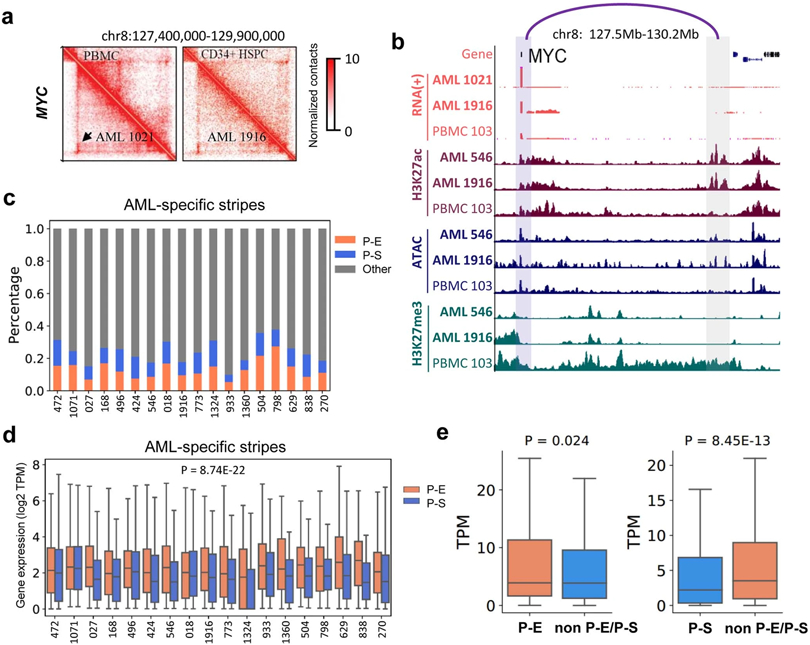
a, Hi-C maps for the MYC gene regions. Left lower panels are for AML samples and the right upper panels are for HSPC and PBMC. Black arrow marks a stripe. b, RNA-Seq, CUT&Tag for H3K27ac and H3K27me3, and ATAC-Seq data for regions surrounding the MYC genes. c, Classification of stripes based on whether the anchors contain gene promoters and the stripe zones contain enhancers (H3K27ac) or silencer marks (H3K27me3). d, In each patient samples, genes in enhancer stripes have higher expression than genes in silencer stripes. P-value by two-sided Kruskal–Wallis test. P-E stripes n=4310, P-S stripes n=2518. Box plot: middle line denotes the median, top/bottom of boxes denotes first/third quartiles and whiskers extend to 1.5 times the interquartile range. e. For each gene involved with a P-E stripe, samples were grouped into two categories: samples with P-E stripe, and samples with neither P-E nor P-S stripe for this gene. So was for P-S stripe analysis. Then the average gene expression (TPM) within each category was calculated. P value by two-sided Kruskal-Wallis test. Left: n=461 genes; Right: n=415 genes. Box plot: middle line denotes the median, top/bottom of boxes denotes first/third quartiles and whiskers extend to 1.5 times the interquartile range.
Extended Data Fig. 9. Detection and analysis of SV-induced neo-loops.
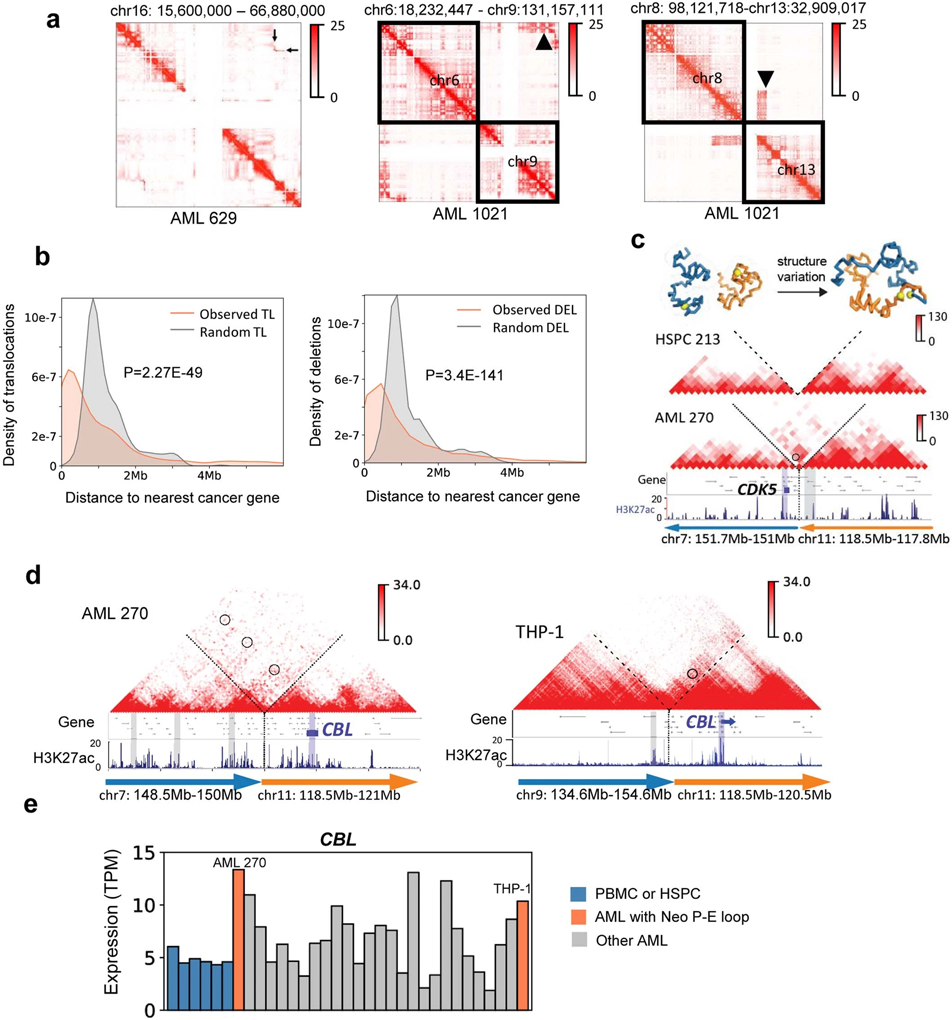
a, Detection of SVs in AML samples from Hi-C data, marked by the black arrow in Hi-C maps. Left: inv(16). Middle: t(6;9). Right: t(8;13). b, Distribution of genomic distances between translocations/deletions to the nearest cancer-related genes (COSMIC database). Expected value is calculated by random permutation of the SVs in the genome for 1000 times. P value is calculated by two sample Kolmogorov-Smirnov test. TL: translocation. DEL: deletion. c, An example of reconstructed Hi-C maps surrounding the SV breakpoint between chr7 and chr11 in the AML sample 270. We also showed the inter-chromosomal Hi-C map in the HSPC 213, where there is no visible inter-chromosomal interactions. The orientation of the SV is marked by the arrows, which always point from 5’ to 3’. The neo-loop for CDK5 is circled. Above the Hi-C: predicted 3D structure for the region in normal and AML sample visualized by PyMOL. d, Recurrent enhancer hijacking involving the CBL gene and enhancers. e, RNA expression of the CBL gene in all samples.
Extended Data Fig. 10. Enhancer hijacking analysis and validation.
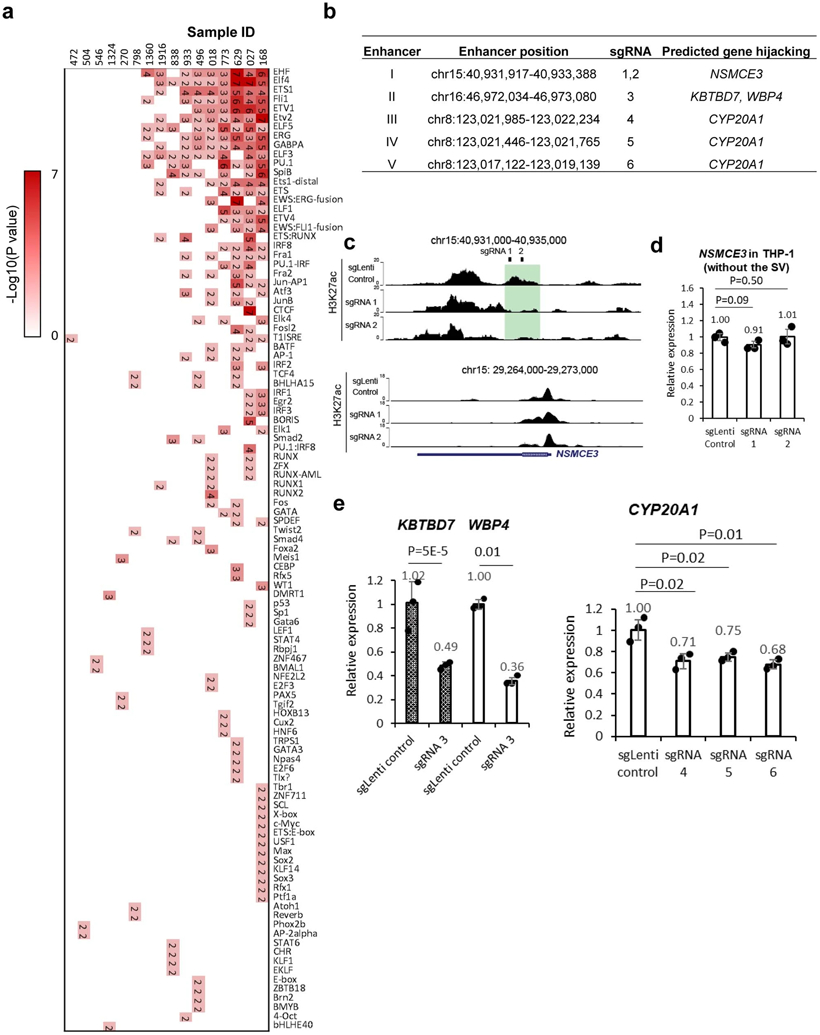
a, Motif enrichment analysis for all hijacked enhancers in AML. The number in each box is the P-value (−log10) calculated by binomial test. b, The list of hijacked enhancers being validated, the corresponding sgRNA, and their targeted genes over SV. c, H3K27ac at the sgRNA-targeted loci and the NSMCE3 gene promoter. d, qPCR relative gene expression in THP-1 cells expressing dCas9-KRAB-MeCP2 (n=3 technical replicates in 2 biological replicates). P value by two-sided Students’ t-test. Data show mean ± s.e.m. e, qPCR results of the targeted gene expression in Kasumi-1 cells with repressing the hijacked enhancers ((n=3 technical replicates in 2 biological replicates)). P value by two-sided Student’s t test. Data show mean ± s.e.m.
Extended Data Fig. 11. Aberrant DNA methylation in AML is associated with changed CTCF binding and chromatin structures.
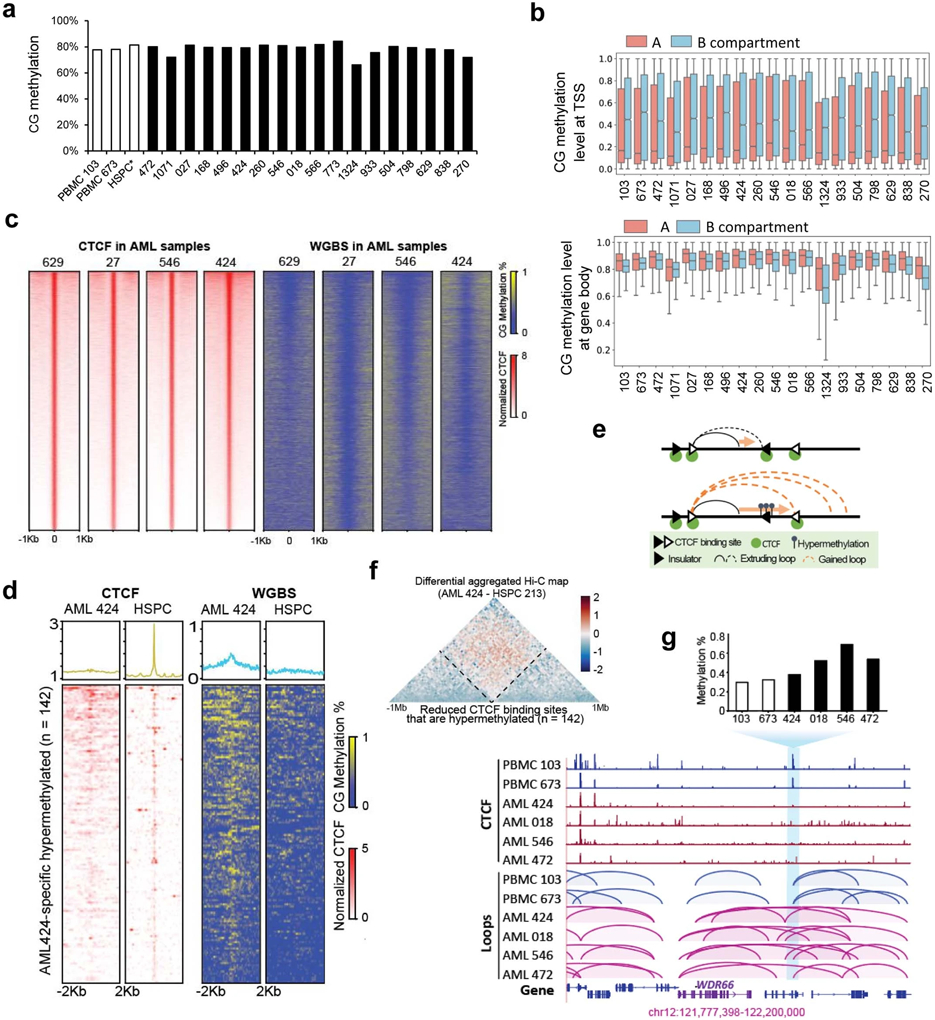
a, Genome-wide CG methylation level in HSPC, PBMC, and AML samples. Data for HSPC* was downloaded from Tovy et al., Cell Stem Cell, 2020. b, CG methylation levels at TSS regions (upper) and gene bodies (lower) in A and B compartment. (Upper) A (n= 531,112 TSS) B (n= 204,365 TSS), (Lower) A (n= 340,385 genes) B (156,852 genes). Box plot: middle line denotes median, top/bottom of boxes denotes first/third quartiles and whiskers extend to 1.5 times the interquartile range of the first and third quartile. c, (Left) Heatmaps of CTCF CUT&Tag signals in AML samples ranked by normalized peak intensity. (Right) Methylation levels surrounding the CTCF binding sites from the same samples. d, Heatmaps showing dynamic CTCF binding sites and methylation levels in AML 424 vs. HSPC. (Right) Hypermethylated sites in AML 424 vs. HSPC that also overlapped with CTCF binding sites in HSPC. Hypermethylation is defined as CpG methylation levels significantly different between two samples (beta-binomial distribution p-value<0.01) and at least 0.3 higher in AML sample. (Left) CTCF signals at the dynamic methylated regions. e, A model for gain of loops as a result of loop extrusion over loss of CTCF binding. f, For AML 424 and HSPC 213, we separately aggregated their Hi-C plots that are centered at the 142 reduced CTCF binding sites of AML 424 associated with hypermethylation. The two aggregated plots were then normalized by total contacts and distance, and the difference is calculated as log2 of fold change. The diamond-shaped region between the two dashed lines resided the interactions across the lost CTCF sites. g, Differential CTCF binding and chromatin loops. Blue vertical bar marks a differential CTCF binding site that was absent in four AML samples. The CTCF binding motifs were also hypermethylated in the AML samples. We observed additional loops across the lost CTCF binding sites in these AML samples.
Extended Data Fig. 12. Effect of 5-AZA treatment on DNA methylation and cell phenotypes.
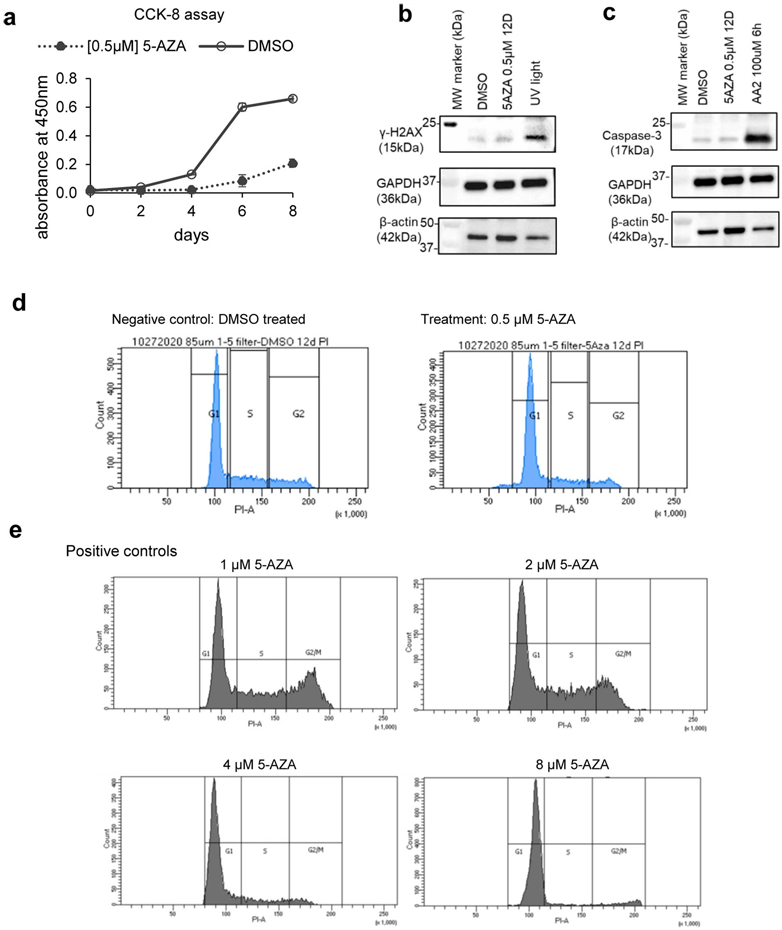
a, CCK-8 assay started with n=3 biological replicates of 1,000 cells per well in 96-well plate. The assay completed on day 8 as the DMSO group grow confluent. Data show mean ± s.e.m. MW: molecular weight. b, Western Blot results for Caspase-3. AA2 is Apoptosis Activator 2 (TOCRIS 2098). The experiments were independently repeated twice with similar results. c, Western Blot for yH2AX. UV light condition is detailed in method. The experiments were independently repeated twice with similar results. d, Flow cytometry result for cells treated with either DMSO or 5-AZA 0.5μM. e, Flow cytometry results for cells treated with 1μM, 2μM, 4μM, and 8μM 5-AZA for 2 days.
Extended Data Fig. 13. Impact of 5-AZA treatment on chromatin structure.
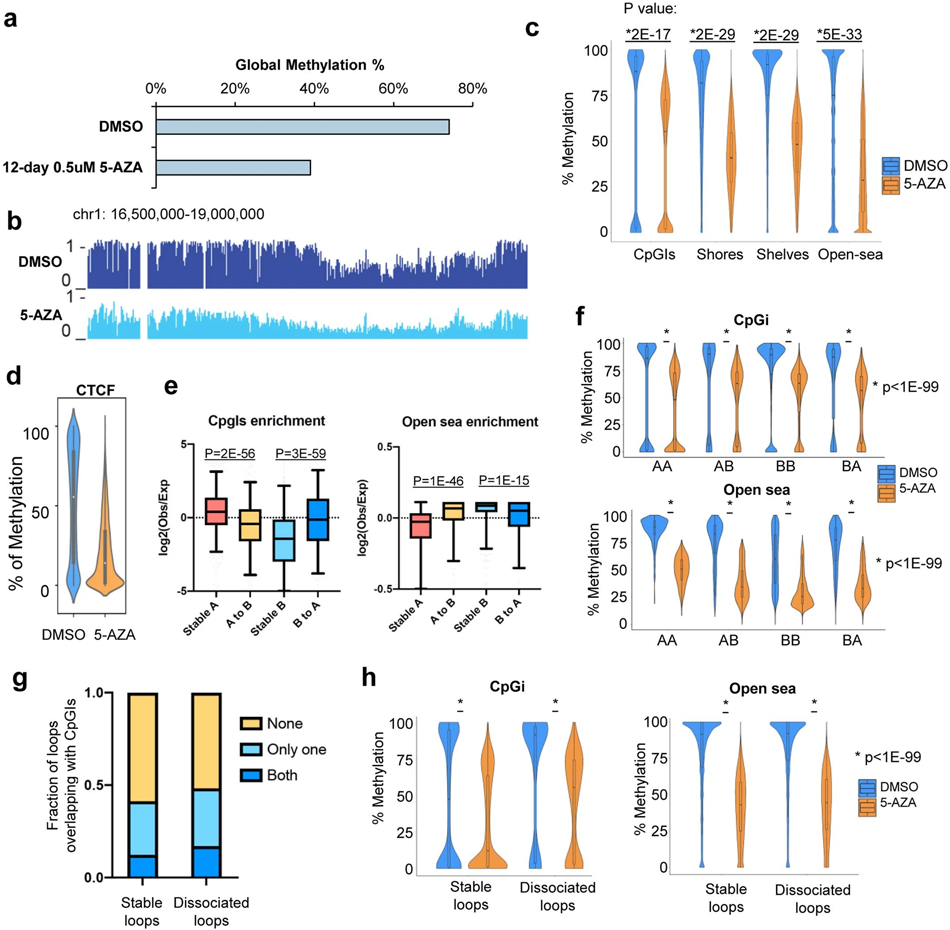
a, Global CG methylation levels of U937 under DMSO or 5-AZA treatment. b, A snapshot of DNA methylation levels across a genomic region. c-d, DNA methylation levels at CpGi, CpG shores (<2Kb), CpG shelves (2Kb-4Kb), open sea (rest of genome), and CTCF binding sites under 12-day DMSO or 0.5μM 5-AZA treatment. DMSO: n= 2,691,094 CpG types, 5-AZA n= 2,669,959 CpG types. CTCF binding sites were defined from HSPC cells. N=10,960 CTCF sites. P value by two-sided Wilcoxon rank-sum test. Violin plot: middle line denotes the median, top/bottom of boxes denotes first/third quartiles and boundary extend to 1.5 times the interquartile range. e, Enrichment of CpGi and open sea at each type of switching compartment. P value by two-sided Wilcoxon rank-sum test. From left to right for both CpGi and Open sea: n=808, n=372, n=636, n=338; Box plot: middle line denotes the median, top/bottom of boxes denotes first/third quartiles and whiskers extend to 1.5 times the interquartile range. f. Methylation levels for each type of switching compartment at CpGi (upper) and open sea (lower). From left to right: CpGi panel: n=20,566, n=450, n=4,651, n=910; Open sea panel: n=591, n=731, n=4,531, n=1,398. P value by two-sided Wilcoxon rank-sum test. AB: A in DMSO group and B in 5-AZA group. Violin plot: middle line denotes the median, top/bottom of boxes denotes first/third quartiles and boundary extend to 1.5 times the interquartile range. g, CpGi at one or both anchors of two types of loops under 5-Aza treatment. h, Methylation levels for two types of loops at CpGi and open sea. From left to right: CpGi panel: n=2,871, n=596; Open sea panel: n=117,177, n=20,499. P value by two-sided Wilcoxon rank-sum test. Violin plot: middle line denotes the median, top/bottom of boxes denotes first/third quartiles and boundary extend to 1.5 times the interquartile range.
Supplementary Material
Acknowledgements
F.Y. is supported by NIH grants R35GM124820, 1R01HG009906, R01HG011207, U01CA200060, and R24DK106766 (R.C.H. and F.Y.). We acknowledge the use of the Integrated Genomics Operation Core, funded by the Memorial Sloan Kettering Cancer Center Support Grant NIH P30 CA008748. This work was supported by a National Cancer Institute R35 CA197594-01A1 (R.L.L.), National Cancer Institute R01 CA216421 (R.L.L.), National Cancer Institute PS-OC U54 CA143869-05 (R.L.L), A.D.V. is supported by a National Cancer Institute career development grant K08 CA215317, the William Raveis Charitable Fund Fellowship of the Damon Runyon Cancer Research Foundation (DRG 117-15), and an Evans MDS Young Investigator grant from the Edward P. Evans Foundation. T.Y. is supported by U01DA053691. The CUT&Tag reagent pA-Tn5 was kindly provided as a gift from Steven Henikoff’s lab at Fred Hutchinson Cancer Research Center. The dCas9-UTX plasmid was kindly provided as a gift by Steven M. Offer’s lab at Mayo Clinic.
Footnotes
Competing interests
F.Y. is a cofounder of Sariant Therapeutics, Inc. Other authors declare no competing interests.
Data availability
The processed data from CUT&Tag, ATAC-seq, Hi-C, and WGBS of primary samples and cell lines and the raw sequencing data all cell lines are deposited at Gene Expression Omnibus (GEO) with the open session ID GSE152136. All processed data were generated by mapping to human reference genome GRCh38.
Code availability
The code for stripe identification is available in GitHub with the following link:
Reference
- 1.Dohner H et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129, 424–447, doi: 10.1182/blood-2016-08-733196 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dohner H, Weisdorf DJ & Bloomfield CD Acute Myeloid Leukemia. N Engl J Med 373, 1136–1152, doi: 10.1056/NEJMra1406184 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Arber DA et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127, 2391–2405, doi: 10.1182/blood-2016-03-643544 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Papaemmanuil E et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med 374, 2209–2221, doi: 10.1056/NEJMoa1516192 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Assi SA et al. Subtype-specific regulatory network rewiring in acute myeloid leukemia. Nat Genet 51, 151–162, doi: 10.1038/s41588-018-0270-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McKeown MR et al. Superenhancer Analysis Defines Novel Epigenomic Subtypes of Non-APL AML, Including an RARalpha Dependency Targetable by SY-1425, a Potent and Selective RARalpha Agonist. Cancer Discov 7, 1136–1153, doi: 10.1158/2159-8290.CD-17-0399 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harris WJ et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell 21, 473–487, doi: 10.1016/j.ccr.2012.03.014 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Luo H et al. CTCF boundary remodels chromatin domain and drives aberrant HOX gene transcription in acute myeloid leukemia. Blood 132, 837–848, doi: 10.1182/blood-2017-11-814319 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghasemi R, Struthers H, Wilson ER & Spencer DH Contribution of CTCF binding to transcriptional activity at the HOXA locus in NPM1-mutant AML cells. Leukemia, doi: 10.1038/s41375-020-0856-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lieberman-Aiden E et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dixon JR et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380, doi: 10.1038/nature11082 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nora EP et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385, doi: 10.1038/nature11049 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan J et al. Histone H3 lysine 4 monomethylation modulates long-range chromatin interactions at enhancers. Cell Res 28, 204–220 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rao SSP et al. Cohesin Loss Eliminates All Loop Domains. Cell 171, 305–320.e324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Viny AD et al. Cohesin Members Stag1 and Stag2 Display Distinct Roles in Chromatin Accessibility and Topological Control of HSC Self-Renewal and Differentiation. Cell Stem Cell 25, 682–696 e688, doi: 10.1016/j.stem.2019.08.003 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang M et al. Proteogenomics and Hi-C reveal transcriptional dysregulation in high hyperdiploid childhood acute lymphoblastic leukemia. Nat Commun 10, 1519, doi: 10.1038/s41467-019-09469-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diaz N et al. Chromatin conformation analysis of primary patient tissue using a low input Hi-C method. Nat Commun 9, 4938, doi: 10.1038/s41467-018-06961-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kloetgen A et al. Three-dimensional chromatin landscapes in T cell acute lymphoblastic leukemia. Nature genetics 52, 388–400, doi: 10.1038/s41588-020-0602-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang H et al. Non-coding germline GATA3 variants alter chromatin topology and contribute to pathogenesis of acute lymphoblastic leukemia. bioRxiv, 2020.2002.2023.961672, doi: 10.1101/2020.02.23.961672 (2020). [DOI] [Google Scholar]
- 20.Dixon JR et al. Integrative detection and analysis of structural variation in cancer genomes. Nat Genet 50, 1388–1398, doi: 10.1038/s41588-018-0195-8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Groschel S et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 157, 369–381, doi: 10.1016/j.cell.2014.02.019 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Northcott PA et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 511, 428–434, doi: 10.1038/nature13379 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spassov BV et al. Wilms' tumor protein and FLT3-internal tandem duplication expression in patients with de novo acute myeloid leukemia. Hematology 16, 37–42, doi: 10.1179/102453311X12902908411913 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Sondka Z et al. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat Rev Cancer 18, 696–705, doi: 10.1038/s41568-018-0060-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Metzeler KH et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood 128, 686–698, doi: 10.1182/blood-2016-01-693879 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Akdemir KC et al. Disruption of chromatin folding domains by somatic genomic rearrangements in human cancer. Nat Genet 52, 294–305, doi: 10.1038/s41588-019-0564-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salameh TJ et al. A supervised learning framework for chromatin loop detection in genome-wide contact maps. bioRxiv, 739698, doi: 10.1101/739698 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaatinen T et al. Global gene expression profile of human cord blood-derived CD133+ cells. Stem Cells 24, 631–641, doi: 10.1634/stemcells.2005-0185 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Diaz-Blanco E et al. Molecular signature of CD34(+) hematopoietic stem and progenitor cells of patients with CML in chronic phase. Leukemia 21, 494–504, doi: 10.1038/sj.leu.2404549 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Ngan CY et al. Chromatin interaction analyses elucidate the roles of PRC2-bound silencers in mouse development. Nat Genet 52, 264–272, doi: 10.1038/s41588-020-0581-x (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cai Y et al. H3K27me3-rich genomic regions can function as silencers to repress gene expression via chromatin interactions. bioRxiv, 684712, doi: 10.1101/684712 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kataoka K et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet 47, 1304–1315, doi: 10.1038/ng.3415 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Goldman MJ et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol 38, 675–678, doi: 10.1038/s41587-020-0546-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vian L et al. The Energetics and Physiological Impact of Cohesin Extrusion. Cell 173, 1165–1178 e1120, doi: 10.1016/j.cell.2018.03.072 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morris VA, Cummings CL, Korb B, Boaglio S & Oehler VG Deregulated KLF4 Expression in Myeloid Leukemias Alters Cell Proliferation and Differentiation through MicroRNA and Gene Targets. Mol Cell Biol 36, 559–573, doi: 10.1128/MCB.00712-15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X et al. Genome-wide detection of enhancer-hijacking events from chromatin interaction data in rearranged genomes. Nat Methods 18, 661–668, doi: 10.1038/s41592-021-01164-w (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phanstiel DH et al. Static and Dynamic DNA Loops form AP-1-Bound Activation Hubs during Macrophage Development. Mol Cell 67, 1037–1048 e1036, doi: 10.1016/j.molcel.2017.08.006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joung J et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc 12, 828–863, doi: 10.1038/nprot.2017.016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Achinger-Kawecka J et al. Epigenetic reprogramming at estrogen-receptor binding sites alters 3D chromatin landscape in endocrine-resistant breast cancer. Nat Commun 11, 320, doi: 10.1038/s41467-019-14098-x (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bell AC & Felsenfeld G Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405, 482–485, doi: 10.1038/35013100 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Figueroa ME et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 17, 13–27, doi: 10.1016/j.ccr.2009.11.020 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Flavahan WA et al. Altered chromosomal topology drives oncogenic programs in SDH-deficient GISTs. Nature 575, 229–233, doi: 10.1038/s41586-019-1668-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flavahan WA et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114, doi: 10.1038/nature16490 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tovy A et al. Tissue-Biased Expansion of DNMT3A-Mutant Clones in a Mosaic Individual Is Associated with Conserved Epigenetic Erosion. Cell Stem Cell 27, 326–335 e324, doi: 10.1016/j.stem.2020.06.018 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang X et al. Large DNA Methylation Nadirs Anchor Chromatin Loops Maintaining Hematopoietic Stem Cell Identity. Mol Cell 78, 506–521 e506, doi: 10.1016/j.molcel.2020.04.018 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Akalin A et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet 8, e1002781, doi: 10.1371/journal.pgen.1002781 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neri F et al. Intragenic DNA methylation prevents spurious transcription initiation. Nature 543, 72–77, doi: 10.1038/nature21373 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Sanborn AL et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci U S A 112, E6456–6465, doi: 10.1073/pnas.1518552112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang T et al. HiCRep: assessing the reproducibility of Hi-C data using a stratum-adjusted correlation coefficient. Genome Res 27, 1939–1949, doi: 10.1101/gr.220640.117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spracklin G et al. Heterochromatin diversity modulates genome compartmentalization and loop extrusion barriers. bioRxiv, 2021.2008.2005.455340, doi: 10.1101/2021.08.05.455340 (2021). [DOI] [Google Scholar]
Reference for Methods
- 51.Cesana D et al. Retrieval of vector integration sites from cell-free DNA. Nat Med 27, 1458–1470, doi: 10.1038/s41591-021-01389-4 (2021). [DOI] [PubMed] [Google Scholar]
- 52.Loke J et al. RUNX1-ETO and RUNX1-EVI1 Differentially Reprogram the Chromatin Landscape in t(8;21) and t(3;21) AML. Cell Rep 19, 1654–1668, doi: 10.1016/j.celrep.2017.05.005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaya-Okur HS et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 10, 1930, doi: 10.1038/s41467-019-09982-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buenrostro JD, Wu B, Chang HY & Greenleaf WJ ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 21–21 29 29, doi: 10.1002/0471142727.mb2129s109 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Diao Y et al. A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nat Methods 14, 629–635, doi: 10.1038/nmeth.4264 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li W et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554, doi: 10.1186/s13059-014-0554-4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bolland DJ, King MR, Reik W, Corcoran AE & Krueger C Robust 3D DNA FISH using directly labeled probes. J Vis Exp, doi: 10.3791/50587 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tarasov A, Vilella AJ, Cuppen E, Nijman IJ & Prins P Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034, doi: 10.1093/bioinformatics/btv098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cingolani P et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92, doi: 10.4161/fly.19695 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rausch T et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339, doi: 10.1093/bioinformatics/bts378 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chiang C et al. SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat Methods 12, 966–968, doi: 10.1038/nmeth.3505 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abdennur N & Mirny LA Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics 36, 311–316, doi: 10.1093/bioinformatics/btz540 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kerpedjiev P et al. HiGlass: web-based visual exploration and analysis of genome interaction maps. Genome Biol 19, 125, doi: 10.1186/s13059-018-1486-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Durand NC et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst 3, 99–101, doi: 10.1016/j.cels.2015.07.012 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Y et al. The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol 19, 151, doi: 10.1186/s13059-018-1519-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dixon JR et al. Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336, doi: 10.1038/nature14222 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Varoquaux N, Ay F, Noble WS & Vert JP A statistical approach for inferring the 3D structure of the genome. Bioinformatics 30, i26–33, doi: 10.1093/bioinformatics/btu268 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359, doi: 10.1038/nmeth.1923 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Krueger F & Andrews SR Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572, doi: 10.1093/bioinformatics/btr167 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schultz MD et al. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 523, 212–216, doi: 10.1038/nature14465 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Corces MR et al. The chromatin accessibility landscape of primary human cancers. Science 362, doi: 10.1126/science.aav1898 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, doi: 10.1093/bioinformatics/bts635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li B & Dewey CN RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323, doi: 10.1186/1471-2105-12-323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Juric I et al. MAPS: Model-based analysis of long-range chromatin interactions from PLAC-seq and HiChIP experiments. PLoS Comput Biol 15, e1006982, doi: 10.1371/journal.pcbi.1006982 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rao SS et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680, doi: 10.1016/j.cell.2014.11.021 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Knight PA & Ruiz D A fast algorithm for matrix balancing. IMA Journal of Numerical Analysis 33, 1029–1047, doi: 10.1093/imanum/drs019 (2012). [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The processed data from CUT&Tag, ATAC-seq, Hi-C, and WGBS of primary samples and cell lines and the raw sequencing data all cell lines are deposited at Gene Expression Omnibus (GEO) with the open session ID GSE152136. All processed data were generated by mapping to human reference genome GRCh38.
The code for stripe identification is available in GitHub with the following link: