

Abstract
Introduction
Stasis dermatitis (SD), also known as venous dermatitis, is a form of inflammatory dermatitis of the lower extremities that typically occurs in older individuals and represents a cutaneous manifestation of venous hypertension. Venous hypertension (also known as sustained ambulatory venous pressure) is most often due to retrograde blood flow, which occurs due to calf muscle pump failure. This failure is most commonly secondary to incompetent venous valves, valve destruction, or obstruction of the venous system. Many of the common symptoms associated with SD are caused by inflammatory processes.
Methods
This review summarizes the pathogenesis and key role of inflammation in SD by reviewing inflammatory biomarkers associated with SD. The literature was selected though a high-level PubMed search focusing on keywords relating to inflammation associated with SD.
Results
Venous reflux at the lower extremities causes venous hypertension, which leads to chronic venous insufficiency. High venous pressure due to venous hypertension promotes the local accumulation and extravasation of inflammatory cells across the vascular endothelium. Leukocyte trapping in the microcirculation and perivascular space is associated with trophic skin changes. Cell adhesion molecules are linked with the perpetuated influx of activated leukocytes into inflammatory sites. Here, inflammatory cells may influence the remodeling of the extracellular matrix by inducing the secretion of proteinases such as matrix metalloproteinases (MMPs). The increased expression of MMPs is associated with the formation of venous leg ulcers and lesions. Phosphodiesterase 4 activity has also been shown to be elevated in individuals with inflammatory dermatoses compared to healthy individuals.
Discussion
Because inflammation is a key driver of the signs and symptoms of SD, several of the highlighted biomarkers of inflammation represent potential opportunities to target and interrupt molecular pathways of cutaneous inflammation and, therefore, remediate the signs and symptoms of SD.
Conclusion
Understanding the pathogenesis of SD may help clinicians identify drivers of inflammation to use as potential targets for the development of new treatment options.
Keywords: Stasis dermatitis, Venous dermatitis, Inflammation, Venous hypertension, Pathogenesis
Plain Language Summary
Stasis dermatitis is a skin disease that affects the legs, most often of older people, with chronic venous insufficiency. Chronic venous insufficiency is when veins cannot return blood from the legs back to the heart. This leads to high blood pressure in veins and causes blood in those veins to flow backwards. If stasis dermatitis is left untreated, complications, including skin ulcers, can result. Other skin symptoms of stasis dermatitis include itchiness, scaling, and discoloration. Such skin symptoms can have a negative effect on a person’s quality of life. Inflammation that lasts a long time is likely the main link between the skin changes seen in people with stasis dermatitis and the increased pressure in leg veins. Several molecules are associated with the inflammation observed in stasis dermatitis, including white blood cells, matrix metalloproteinases, phosphodiesterase 4, and interleukin-31. Treatment for stasis dermatitis should focus both on the underlying chronic venous insufficiency and the associated skin issues. Identifying inflammatory markers and pathways could help treat the signs and symptoms associated with stasis dermatitis, including the skin symptoms.
Key Summary Points
| Stasis dermatitis is caused by venous hypertension due to venous reflux. |
| If undiagnosed or left untreated, stasis dermatitis may be a precursor to venous ulcers. |
| Chronic inflammation represents the most likely link between the venous hypertension and cutaneous changes observed in stasis dermatitis. |
| Interrupting the molecular pathways that sustain cutaneous inflammation may remediate the signs and symptoms of stasis dermatitis. |
Introduction
Venous reflux at the lower extremities causes venous hypertension (also known as sustained ambulatory venous pressure), which leads to chronic venous insufficiency. Stasis dermatitis (SD), also known as venous dermatitis, is an inflammatory dermatitis of the lower extremities that predominantly affects older individuals with underlying chronic venous insufficiency [1, 2]. SD typically presents as bilateral erythematous and eczematous patches and plaques of the lower extremities [3]. Estimates of the prevalence of chronic venous insufficiency vary widely depending on the characteristics of the populations examined. Approximately 2–6 million individuals in the USA have advanced forms of chronic venous insufficiency [2, 4]. In one study, SD was observed in 1.4% of individuals with varicose veins aged 15 years or more [5]. SD is more prevalent in older individuals and was found in 6.2% of individuals aged 65 years or more [6]. Results from similar studies indicated that SD was found in 5.9% of individuals with a mean age of 74 years (range 50–91 years) and in 6.9% of those with a mean age of 80 years (range 55–106 years) [2].
Early symptoms and signs of SD include pruritic, scaly, irritated, and discolored skin, especially in areas of the lower leg where varicose veins are present; additional symptoms and signs include aching and heaviness in one or both legs as well as swelling of the ankle and the lower leg [7]. The histologic epidermal changes linked to SD include hyperkeratosis, parakeratosis, acanthosis, and spongiosis. The histologic dermal changes associated with SD include extravasated erythrocytes, dermal fibrosis, perivascular lymphocytic infiltration, hemosiderin-laden macrophages, and proliferation of dilated small blood vessels with relatively thick walls in the papillary dermis [2]. SD is typically diagnosed on the basis of clinical findings and medical history [1, 8].
Several cutaneous disorders present similarly to SD, leading to misdiagnosis and mismanagement. More than 10% of cellulitis diagnoses are incorrect, with SD being the disease most frequently mistaken for cellulitis. Allergic forms of contact dermatitis are also often mistaken for SD. Moreover, allergic contact dermatitis may present simultaneously with SD, especially in patients with venous ulcers [1, 9]. SD can also resemble pigmented purpuric dermatoses, lymphedema, and sometimes neoplasms [1–3].
Several risk factors are associated with SD, including female sex, pregnancy, older age, obesity, prolonged immobility, family history of venous disease, chronic edema, heart failure, previous lower extremity trauma/surgery, injection drug use, calf muscle injury, and a history of deep venous thrombosis [1, 8, 10, 11]. SD is more common in patients who are obese and/or pregnant, especially those who are multigravida, because of the extra stress that is placed on the veins of their lower extremities [12]. Immobile patients may also develop SD due to the reduced tone and contractility of the musculature at their lower extremities rather than anatomical complications of the venous system. Failure to activate the calf muscle pump because of angle joint issues or muscle disease can also lead to SD, even in the absence of venous changes [10, 11, 13].
SD is an indication of underlying vascular pathology and, if undiagnosed or left untreated, is a precursor to venous ulcers [1, 2]. As was found in one study, 37–44% of patients with leg ulcers were diagnosed with SD [14]. Approximately 2.2 million individuals in Europe and more than 6 million individuals in the USA experience venous leg ulcers [15]. Venous ulcers can occur as either a single or multiple ulcers and are often located over the medial malleolus (Fig. 1). Untreated SD can present with oozing and red, eroded, and/or scaly patches and plaques. Other skin changes may coexist. Contact dermatitis and autoeczematization due to SD can also arise [1]. Patients may also develop acroangiodermatitis [2, 16]. Additional presentations of the legs in patients with SD include swelling, itching, tingling, leg aching, restless legs, cramps, and lipodermatosclerosis [2]. Lipodermatosclerosis is an indurated plaque in the medial malleolus that can be exquisitely painful in its acute form, with the chronic form presenting with skin induration and hyperpigmentation of the legs [17–19]. Secondary infections may also occur because of the disruption of the epidermal barrier. Allergic contact dermatitis in SD may be caused by allergen penetration through the deteriorated epidermal barrier as well as contact with potential allergens that are applied during treatment. Autoeczematization, also referred to as an “id reaction,” is the onset of a widespread, highly pruritic, erythematous, morbilliform or papulovesicular eruption in response to stimuli. The eruption often occurs at a distant location from the stimulus. Such stimuli could be due to a chronic inflammation from SD, contact dermatitis, or infections [1].
Fig. 1.
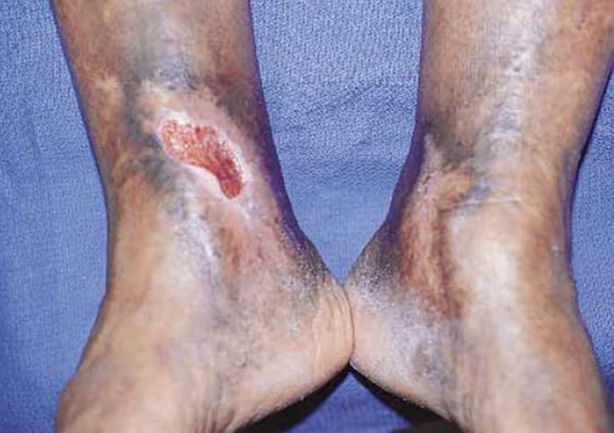
Venous ulcers located over the medial malleolus. Note the presence of hyperpigmentation. Reprinted with permission from Ref. [68], with permission from Isabel C. Valencia et al., 2001, Chronic venous insufficiency and venous leg ulceration
An improved understanding of the pathogenesis of SD could help in the development and utilization of more targeted and efficacious treatments. Accordingly, this review summarizes the pathogenesis and role of inflammation in SD. The PubMed literature search focused on keywords related to the inflammation associated with SD, including stasis dermatitis, varicose ulcers, venous stasis ulcers, stasis ulcers, venous ulcers, venous hypertension, varicose veins, venous eczema, venous stasis dermatitis, collagenases, dermatitis, inflammation, matrix metalloproteinases, and venous insufficiency. When indicated, venous intervention may remediate the underlying venous reflux associated with SD. When interventions are not feasible or fail, multiple long-term pharmaceutical and medical device options are available to treat the various dermatological symptoms associated with SD [1]. Non-invasive therapeutic approaches include leg elevation and walking [1]; however, such treatments were only shown to improve mild cases [2]. The initial treatment recommendation for SD is compression therapy in the form of compression bandages or stockings, which exert pressure to reduce ambulatory venous pressures, alleviating SD symptoms such as swelling and stasis skin changes [1, 2, 13, 20–22]. However, compression therapy often fails because of the gradual loss of elasticity of the device or as a result of patient nonadherence with a treatment plan [1, 23]. It has been reported that only 50–60% of patients with SD adhere to compression therapy [24]. Skincare treatments such as bland emollients are often used to alleviate the secondary skin changes associated with SD, including scaliness and pruritus [2]. High- or mid-potency topical corticosteroids can be intermittently used to reduce pruritus and inflammation in SD. The prolonged use of corticosteroids should be avoided because they may lead to cutaneous atrophy and systemic side effects, potentially increasing the risk of ulcer formation [1, 25–28]. Topical calcineurin inhibitors can also be used for the management of SD; however, their use has been associated with burning upon application and requires patient awareness of potential and theoretical long-term complications (e.g., increased risk of elevated serum creatinine, hyperlipidemia, malignancy, and diabetes mellitus) [25–27, 29–32]. Venous interventions are also available to treat the underlying venous reflux. Interventional radiology and surgical interventions may lead to the resolution of SD. However, the recurrence of SD-related complications remains a problem regardless of the treatment used [1, 33]. There is therefore a need for novel therapies that can elicit anti-inflammatory effects and target the cutaneous manifestations of SD with optimized risk–benefit profiles. However, research regarding novel treatment options for SD is lacking [2]. Nevertheless, because inflammation plays a vital role in SD pathogenesis, several novel therapies that target inflammatory pathways could be promising. Such therapies include Janus kinase (JAK) inhibitors, phosphodiesterase 4 (PDE4) inhibitors, and immunosuppressive therapies. A number of immunosuppressive therapies with anti-inflammatory properties, including azathioprine, cyclosporin, and mycophenolate mofetil, could also be potentially utilized in the treatment of SD [34–36]. Topical PDE4 inhibitors, such as crisaborole, could target the inflammation observed in SD while reducing unwanted side effects that are associated with systemic treatments [25]. Studies regarding the evaluation of JAK inhibitors targeting receptor-associated kinases, such as delgocitinib, are also anticipated for SD [34].
This article is based on previously conducted studies and scientific literature and does not contain any new studies with human participants or animals performed by any of the authors. No ethics approval was necessary for this review.
Prompt treatment and diagnosis of SD could ameliorate potential future complications, such as ulcers [2]. General practitioners may not recognize the severity of venous disorders such as SD and thereby may not refer their patients to the appropriate specialists. Overall, the misdiagnosis of SD is partially due to the lack of dermatology services in healthcare settings [37, 38]. However, dermatologic consultation is often not considered by inpatient providers when treating patients with lower extremity inflammation. The introduction of educational campaigns regarding SD for general practitioners could promote the development of “champions of care” in healthcare settings who are able to promptly initiate the appropriate diagnosis and treatment of the disease. One study found that the average patient with SD was seen by about 6.9 doctors throughout the course of their treatment. However, the high number of consulted physicians in SD was mainly due to the patients’ lack of adherence to treatment [38]. Accordingly, highlighting the importance of treatment adherence to patients could also improve disease outcome in SD.
Pathogenesis of Stasis Dermatitis
Venous Hypertension
SD is a cutaneous manifestation of venous hypertension that is the result of underlying chronic venous insufficiency [1, 2]. Chronic venous insufficiency of the lower extremities ranges from asymptomatic to severely symptomatic [39]. Chronic venous insufficiency has been clinically linked to sustained ambulatory venous pressures, whereby retrograde venous blood flow is induced by incompetent venous valves and venous flow obstruction as part of calf muscle pump failure at the lower extremities [2, 40]. Such venous reflux can occur at the superficial venous system, the deep venous system, or both [40]. When valve failure occurs at the superficial and perforating veins, venous pressure in the veins and venules of the skin and subcutaneous tissues remains high during ambulation, whereas it typically diminishes in normal legs. Increased venous pressure may lead to the reversal of blood flow. Sustained elevated venous pressure leads to the margination and extravasation of cells from the blood vessels and into the surrounding tissues. Such blood flow patterns lead to the restructuring of the superficial veins and venules, which are less supported from a mechanical perspective than deep veins. Associated alterations include valve cusp perforation, elongation, tearing, and disappearance [41]. In one study that included patients with venous disease, primary valvular incompetence was found in approximately 70–80% of cases; such valvular incompetence was due to trauma or deep vein thrombosis in 18–25% of cases [40].
Venous hypertension is associated with elevated and unstable venous pressure values; such venous pressure is mainly driven by two components: a hydrostatic and a hydrodynamic component. The hydrostatic component is determined by the weight of the blood in the venous system, whereas the hydrodynamic component is determined by the pressure generated by leg skeletal muscle contractions and the pressure in the capillary network. While a person is standing, the hydrostatic component and the capillary flow determine the venous pressure in the legs, reaching values of 80–90 mmHg. During ambulation, skeletal muscle contractions increase the pressure observed in the deep leg veins, causing venous blood to return to the heart under normal physiologic conditions. Accordingly, competent venous valves ensure the blood’s flow towards the heart, reducing venous pressure in the legs by emptying the superficial and deep venous systems and, hence, lowering the local venous pressure to less than 30 mmHg [2, 40, 41]. In the absence of competent valves, the reduction in the venous pressure at the lower extremities that is brought upon by leg movement is attenuated. If the perforator venous valves of the deep venous system are incompetent, increased venous pressure is observed during standing as well as during calf muscle contractions, which leads to the reflux of blood into the superficial venous system and into the microcirculation in the skin. This results in an overall and continuous high-pressure system. Accordingly, the vessel walls of the superficial and deep venous systems are distorted, leading to a chronic cycle of worsening valvular incompetence and venous hypertension [1, 40]. Chronic elevation of venous pressure is linked to inflammatory reactions in the venous valves, eventually leading to venous reflux and dysfunction as well as the elevation of upstream venous pressure [41]. Impaired venous valves limit the return of blood to the heart, which leads to backward venous flow, venous hypertension, and venous stasis [2]. In one study that included 360 patients with SD, it was demonstrated that increases in ambulatory venous pressure are associated with severe skin damage, including ulceration [42]. In another study of patients that had negative patch testing and negative deep venous insufficiency, venous hypertension was found to be the most probable cause of SD, thus demonstrating that venous hypertension alone can cause SD in the lower extremities [2, 43]. In the same study, successful classical vein surgery that targeted superficial venous reflux, including saphenectomy of incompetent superficial vein segments and ligation of proximal point of reflux, led to the complete resolution of SD symptoms in treated patients. Here, the lower leg dermatitis healed rapidly, completely, and without recurrence for an observation period of 1 year [43].
Stasis Dermatitis is an Inflammatory Disease
Leukocytes and Cell Adhesion Molecules
The initial theory that the cutaneous changes in SD were driven by fibrin cuffs encasing dermal capillaries, thereby impeding the diffusion of oxygen from the vasculature and into epidermal cells, has been replaced by the understanding that inflammation plays a key role in the observed cutaneous changes. Indeed, chronic inflammation represents the most likely link between venous hypertension and cutaneous changes, as suggested by the correlation between such hypertension and inflammation [2, 40]. A major driver in the progression of uncontrolled inflammation in chronic venous insufficiency is the extravasation of leukocytes across the vascular endothelium. Such extravasated leukocytes infiltrate nearby tissues, secrete high levels of inflammatory mediators, and recruit inflammatory cells, thereby perpetuating the inflammatory response. Moreover, high venous pressure due to venous hypertension promotes the local accumulation of inflammatory cells, such as macrophages and T cells [2, 44]. Thomas et al. [45] found that leukocytes repeatedly accumulate in the microcirculation and perivascular space of extremities with venous hypertension. Such leukocyte trapping is commonly seen during the earlier stages of chronic venous insufficiency, particularly in cases of SD [43]. A parallel study that examined the lower extremities of patients with chronic venous insufficiency found that leukocytes and membrane adhesion molecules were more common on proximal venous valve surfaces and venous walls than on distal venous regions. Chronic venous reflux is associated with increases in leukocyte–endothelial cell interactions as well as an increase in the number of activated leukocytes. Unsteady stress alterations on the vein walls as well as venous distensions may lead to the activation of polymorphonuclear cells, macrophages, and endothelial cells [41]. Indeed, increased numbers of macrophages, mast cells, and T lymphocytes were observed in the skin biopsies of lower extremities in individuals with chronic venous insufficiency [46]. As a result, leukocytes become attached to the endothelium of the vein walls and valve leaflets, leading to the necrosis and/or apoptosis of the endothelium, fibroblasts, smooth muscle cells, and the parenchymal cells of the venous wall. These changes may promote weakening and destruction of the venous wall structures and valve leaflets, resulting in the clinically observable venous changes [41]. The trophic skin changes seen in patients with venous hypertension may be due to the repeated accumulation of leukocytes in the microcirculation and the perivascular space [45]. Studies have shown that blood of the lower extremities in individuals with chronic venous disease is low in leukocytes, which suggests that leukocytes accumulate in regions of high venous pressure, leading to an inflammatory response [45–47]. Sustained high venous pressure also promotes the extravasation of erythrocytes at the affected regions [2, 44]. The extravasation of the erythrocytes is followed by the decomposition of hemoglobin, resulting in the excessive buildup of iron, which is stored at the affected tissue as hemosiderin and causes local hyperpigmentation. Further macrophage accumulation is induced by the buildup of the hemoglobin and hemosiderin; such macrophages alongside other cells promote inflammation (especially the accumulation of inflammatory macrophages) and induce the various histological features associated with SD, including vascular proliferation and epidermal spongiotic changes [44, 48]. The observed inflammatory response leads to local tissue damage [49].
The invasion of tissues by inflammatory cells is dependent on the transmigration of cells across the microvascular endothelium. This complex process of transmigration is mediated by cell adhesion molecules (CAMs). CAMs are expressed on the microvascular endothelial cells, whereas their receptors are found on the surface of leukocytes. The adhesion of leukocytes onto the endothelium is a multistep process [1, 2, 42]. The first step is the reversible binding of the endothelial cell selectin molecules, such as P-selectin and E-selectin, onto the sialyl Lewis X ligands on leukocytes. Leukocytes then firmly adhere onto the endothelial cells, which is required for the transmigration of leukocytes onto nearby tissues. This adhesion is mediated by intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1), found on endothelial cells, and their ligands lymphocyte function-associated antigen 1 (LFA-1) and very late antigen 4 (VLA-4), which are found on leukocytes. ICAM-1, VCAM-1, LFA-1, and VLA-4 are upregulated during the early stages of chronic venous insufficiency, including SD, and remain upregulated throughout latter stages (Fig. 2) [42, 43]. The highly expressed CAMs potentially promote skin tissue damage via the facilitation of an upregulated and perpetuated influx of activated leukocytes into the inflammatory sites [49]. Indeed, the aforementioned CAMs were found to be highly expressed on tissue-invading leukocytes and vascular endothelium at the edges of leg ulcers [50]. Such inflammatory changes lead to the release of several mediators, such as proteolytic enzymes, including collagenase and elastase, chemotactic factors, free radicals, and cytokines, which leads to an increase in vascular permeability [51].
Fig. 2.

ICAM-1, VCAM-1, LFA-1, and VLA-4 are upregulated in stasis dermatitis and remain upregulated in lipodermatosclerosis. Samples were obtained from healthy individuals (A–D), patients with stasis dermatitis (E–H), and patients with lipodermatosclerosis (I–L). Cryosections of all skin samples were immunohistochemically stained with monoclonal antibodies against ICAM-1, VCAM-1, LFA-1, and VLA-4. The bar indicates a length of 150 µm. Reprinted with permission from Ref. [34], with permission from Acta Dermato-Venereologica. ICAM-1 intercellular adhesion molecule 1, VCAM-1 vascular cell adhesion molecule 1, LFA-1 lymphocyte function-associated antigen 1, VLA-4 very late antigen 4
Matrix Metalloproteinases
Matrix metalloproteinases (MMPs) are proteolytic enzymes that are associated with vascular disease and cardiovascular remodeling, as well as the turnover of elastin, collagen, and other proteins of the extracellular matrix. The results of studies regarding ulcer fluid environments suggest a potential association between the activity of MMPs and the development of venous leg ulcers and skin lesions. Imbalances between the expression and activity of MMPs and endogenous tissue inhibitors of MMPs (TIMPs) may cause pathological alterations of the vein walls and valves, leading to the symptoms associated with chronic venous diseases. Increases in venous hydrostatic pressure may lead to endothelial cell injury and increased cellular permeability, resulting in the infiltration of leukocytes and vascular inflammation into the surrounding dermis. This then leads to loss of wall integrity, tissue fibrosis, valve degradation, and vein damage that is characteristic of the late stages of chronic venous insufficiency [52].
Activated endothelial and inflammatory cells can degrade components of the extracellular matrix, including collagen, elastin, laminin, and fibronectin, through the release of oxygen free radicals and MMPs [42, 53]. Several MMPs were linked to the degradation of structure-maintaining collagen macromolecules, with MMP-1, -2, and -13 degrading type I, IV, and V collagens as well as elastin. These MMPs are secreted by inflammatory cells such as neutrophils, eosinophils, and macrophages. MMP-1, -2, and -13 are associated with the pathogenesis of several skin disorders, including basal cell carcinoma, immune-bullous diseases, and granuloma annulare [2, 54]. Inflammatory cells influence the remodeling of the extracellular matrix by producing MMPs and by secreting cytokines that induce MMP gene expression, including tumor necrosis factor-α, interleukin (IL)-1, and transforming growth factor-β1 (TGFβ1) [54]. The increased expression of MMPs and other proteinases by the vessel wall breaks down the vascular extracellular matrix, leading to vascular permeability and edema [1]. Extravasated erythrocytes release ferritin and ferric ion, which may result in oxidative stress and the activation of additional MMPs; this process delays healing via promoting skin tissue damage and ulcer formation [2, 40, 55]. Hyperpigmentation of the skin, which is observed in SD, is due to hemosiderin deposition from extravasated erythrocytes, activation of MMPs, and inflammatory mediators [1, 2]. The activity of MMPs is tightly regulated by their inhibitors, TIMPs, with alterations in the balance between MMPs and TIMPs contributing to the pathological changes observed in venous diseases [52]. In one study, elevated levels of MMP-1, -2, and -13, as well as lower levels of TIMP-1 and -2, were present in the skin samples of patients with SD compared with healthy individuals. Such overexpression of MMP-1, -2, and -13 without the inhibitory effects of TIMP-1 and -2 may be due to cytokine-mediated induction. MMPs are associated with the degradation of extracellular matrix proteins, potentially leading to the remodeling of lesional skin and impaired healing in SD [54]. MMP-1, -2, and -13 may induce a number of histological features observed in SD, including spongiosis, alteration of papillary structures, and proliferation of small blood vessels in the papillary dermis [2]. Alterations in MMP-2 activity, in conjunction with events mediated via TGFβ1 (such as the differentiation of fibroblasts into myofibroblasts), were found to lead to the degradation of epidermal basement membrane collagen and the loss of epidermal integrity, creating an environment that is susceptible to the formation of venous ulcers (Figs. 3, 4) [53].
Fig. 3.

The possible roles of MMPs and TGFβ1 in the formation of venous ulcers.
Reproduced from Ref. [38], with permission from Elsevier. MMP matrix metalloproteinase, TGFβ1 transforming growth factor-β1, TIMP-1 tissue inhibitor of metalloproteinase-1
Fig. 4.

Pathophysiology of stasis dermatitis. MMP matrix metalloproteinase
Phosphodiesterase 4
PDE4 is a cyclic adenosine monophosphate (cAMP)-specific intracellular nonreceptor enzyme that regulates various fundamental functions, including inflammatory responses, as well as epithelial and endothelial barrier stability [25, 56, 57]. Patients with inflammatory diseases show elevated levels and activity of PDE4 compared with healthy individuals. As has been shown in studies, the overexpression and overactivity of PDE4 leads to increases in the production of inflammatory cytokines (Fig. 5), causing skin inflammation and disease exacerbation in patients with atopic dermatitis. The inhibition of PDE4 elevates intracellular cAMP levels, thereby modulating the inflammatory response and maintaining the immune balance. Targeting PDE4 has been shown to be an effective strategy for several inflammatory conditions, including psoriasis, atopic dermatitis, chronic obstructive pulmonary disease, asthma, inflammatory bowel diseases, rheumatoid arthritis, lupus, and neuroinflammation [56]. Accordingly, the inhibition of PDE4 may decrease the production of inflammatory cytokines, thereby limiting associated skin inflammation and disease exacerbation in SD [58, 59]. Emerging therapies for the treatment of secondary skin changes associated with SD such as skin inflammation could involve the application of nonsteroidal topical ointments, such as crisaborole. Crisaborole is a topical PDE4 inhibitor that has been approved for the treatment of mild-to-moderate AD in multiple countries and was recently investigated in a phase 2 study for SD treatment (NCT04091087) [60, 61].
Fig. 5.

The cAMP cascades involved in PDE4 inhibition.
Reproduced from Ref. [48], with permission from MDPI (Basel, Switzerland). 5′AMP 5-adenosine monophosphate, AC adenylyl cyclase, ATF-1 cAMP-dependent activating transcription factor 1, ATP adenosine triphosphate, Bcl-6 B cell lymphoma 6 protein, cAMP 3′,5′-cyclic adenosine monophosphate, CREB cAMP response element binding protein, Epac 1/2 exchange protein directly activated by cAMP 1 and 2, NF-κB nuclear factor kappa light chain enhancer of activated B cells, PDE4 phosphodiesterase 4, PDE4-I phosphodiesterase 4 inhibitor, PKA protein kinase A, Popeye Popeye domain family, Rac1 Ras-related C3 botulinum toxin substrate 1, Rap 1 Ras-related protein 1, RhoA Ras homolog family member A
Interleukin-31
Patients with SD often report having pruritus, which impairs quality of life and induces further cutaneous irritation and breakdown due to excessive scratching [1, 44]. Indeed, SD was the most frequently reported type of dermatitis to cause itch complaints in one study involving older adults [62]. Constant scratching aggravates wounds and increases the chances of developing skin infections; it may also result in skin thickening and lichenification [1, 44]. The pathogenesis of SD-linked pruritus is incompletely understood; however, it is likely nonhistaminergic because it is not remediated by use of antihistamines. Nonhistaminergic itch involves several itch mediators, such as cytokines/chemokines (e.g., IL-31), proteases and their receptors, amines, neuropeptides and their receptors (e.g., substance P and its receptor neurokinin-1 receptor), ion channels, and immune cells (e.g., mast cells, T cells, eosinophils, and basophils) [44]. IL-31 is a T-helper type (Th)2-related pruritogenic cytokine that has recently been implicated as a potential target for skin conditions involving itch. IL-31 is associated with exacerbating pruritus in various diseases, including psoriasis, atopic dermatitis, prurigo nodularis, and cutaneous T-cell lymphoma [63–65]. Increased IL-31 expression is found in the skin of patients with atopic dermatitis, prompting the release of inflammatory mediators and lowering the expression of molecules associated with maintaining the skin barrier [66]. The correlation between pruritus and IL-31 was highlighted in a study in which serum levels of IL-31 were found to be significantly higher in patients with chronic pruritus of unknown origin compared with those in healthy individuals [67].
A recent study has implicated dermal IL-31 from macrophages in inducing itch in SD [44]. The majority of IL-31-expressing cells in SD are CD68+ macrophages. The number of IL-31+CD68+ macrophages is significantly higher in patients with SD with severe pruritus compared with those with SD without severe pruritus. These CD68+ macrophages, which also coexpress CD163 (a macrophage-specific membrane marker), are classified as M2 macrophages (Fig. 6) [66]. The number of IL-31+CD68+ macrophages is positively correlated with the number of dermal C–C chemokine receptor type 4+ Th2 cells, basophils, and substance P+ cells, as well as dermal deposition of hemosiderin and periostin. Moreover, the number of IL-17+ cells is also higher in SD lesions, which is consistent with Th17 immunity predominating in SD. The number of IL-17+ cells is correlated with the number of IL-31+CD68+ cells but not with the presence of pruritus. IL-17 likely promotes M2 macrophage skewing, with M2 macrophages promoting Th17 cell expansion [44].
Fig. 6.

CD68+ macrophages express IL-31 and are correlated with pruritus in SD. Images of healthy skin and SD lesions with the quantification of staining are shown. a Epidermal expression of IL-31 was not higher in SD lesions. The number of CD68+ macrophages and IL-31+ cells was greater in SD lesions. b Most IL31+ cells expressed CD68. The number of CD68+/IL31+ cells was greater in SD lesions and was correlated with pruritus. c In SD with severe pruritus, CD68+ macrophages expressed CD163 (an M2 macrophage marker). The number of CD68+/CD163+ cells was greater in SD lesions and correlated with pruritus. *P < 0.05 (unpaired t test). Vertical bars indicate standard deviation. Dotted lines indicate the dermo-epidermal junction. The bar indicates 100 µm.
Reproduced from Ref. [29], with permission from Elsevier. AU arbitrary unit, CD cluster of differentiation, DAPI 4′,6-diamidino-2-phenylindole, IL-31 interleukin-31, NS not significant, SD stasis dermatitis, w/ with, w/o without
Conclusions
SD is a cutaneous manifestation of venous hypertension that is commonly seen in older patients with underlying venous insufficiency, wherein retrograde venous blood flow is induced by incompetent venous valves, venous flow obstruction, or muscle pump failure at the lower extremities. Treatments for SD focus primarily on identifying and eliminating primary causes of venous hypertension and insufficiency when feasible and, secondarily, on management of associated skin changes as well as preventing potential complications. Understanding the pathogenesis of SD may help clinicians effectively diagnose and treat SD. Several of the highlighted biomarkers of inflammation represent potential opportunities to individually or jointly interrupt molecular pathways which sustain cutaneous inflammation and, therefore, remediate the signs and symptoms of SD that diminish quality of life in patients with SD.
Acknowledgements
Funding
This review, as well as the journal’s Rapid Service Fee, was funded by Pfizer Inc., New York, NY, USA.
Medical Writing and Editorial Assistance
Editorial/medical writing support under the guidance of authors was provided by Mark Bloom, PhD, at ApotheCom, Yardley, PA, USA, and was funded by Pfizer Inc., New York, NY, USA, in accordance with Good Publication Practice (GPP 2022) guidelines (Ann Intern Med. 2022; 10.7326/M22-1460).
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
Writing—review and editing: Jonathan Silverberg, J. Mark Jackson, Robert Scott Kirsner, Roni Adiri, Gary Friedman, Xinghua Gao, Steven D. Billings, Urs Kerkmann.
Disclosures
Jonathan Silverberg served as an investigator for Celgene, Eli Lilly and Company, F. Hoffmann-LaRoche, Menlo Therapeutics, Realm Therapeutics, Regeneron, and Sanofi; as a consultant for Pfizer Inc., AbbVie, Anacor, AnaptysBio, Arena Pharmaceuticals, Dermavant, Dermira, Eli Lilly and Company, Galderma, GlaxoSmithKline, Glenmark, Incyte, Kiniksa Pharmaceuticals, LEO Pharma, Menlo Therapeutics, Novartis, Realm Therapeutics, Regeneron, and Sanofi; and as a speaker for Regeneron and Sanofi. J. Mark Jackson has received research, honoraria, consulting or speaking fees from AbbVie, Arcutis, Dermavant, Evommune, Janssen, Lilly, Novartis, Pfizer Inc., and UCB. Robert S. Kirsner served as a consultant for Pfizer Inc. Roni Adiri is an employee and shareholder of Pfizer Pharmaceuticals Israel Ltd. Gary Friedman is an employee and shareholder of Pfizer Inc. Urs Kerkmann is an employee and shareholder of Pfizer Pharma GmbH. Xing-Hua Gao reports the following: personal fees from Novartis, Pfizer, Astellas, Meda Pharma S.p.A., a Viatris company, Sanofi, Lilly, Bayer, LEO, GSK, Pierre Fabre, and Janssen, outside the submitted work. Steven D. Billings reports no relevant disclosures.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and scientific literature and does not contain any new studies with human participants or animals performed by any of the authors. No ethics approval was necessary for this review.
Data Availability
Data sharing is not applicable to this article as no data sets were generated or analyzed during the current study.
References
- 1.Rzepecki AK, Blasiak R. Stasis dermatitis: differentiation from other common causes of lower leg inflammation and management strategies. Curr Geriat Rep. 2018;7(4):222–227. doi: 10.1007/s13670-018-0257-x. [DOI] [Google Scholar]
- 2.Sundaresan S, Migden MR, Silapunt S. Stasis dermatitis: pathophysiology, evaluation, and management. Am J Clin Dermatol. 2017;18(3):383–390. doi: 10.1007/s40257-016-0250-0. [DOI] [PubMed] [Google Scholar]
- 3.Weaver J, Billings SD. Initial presentation of stasis dermatitis mimicking solitary lesions: a previously unrecognized clinical scenario. J Am Acad Dermatol. 2009;61(6):1028–1032. doi: 10.1016/j.jaad.2009.04.025. [DOI] [PubMed] [Google Scholar]
- 4.White JV, Ryjewski C. Chronic venous insufficiency. Persp Vasc Surg Endovasc Ther. 2005;17(4):319–327. doi: 10.1177/153100350501700406. [DOI] [PubMed] [Google Scholar]
- 5.Maffei FHA, Magaldi C, Pinho S, et al. Varicose veins and chronic venous insufficiency in Brazil: prevalence among 1755 inhabitants of a country town. Int J Epidem. 1986;15(2):210–217. doi: 10.1093/ije/15.2.210. [DOI] [PubMed] [Google Scholar]
- 6.Yalcin B, Tamer E, Toy GG, Oztas P, Hayran M, Alli N. The prevalence of skin diseases in the elderly: analysis of 4099 geriatric patients. Int J Dermatol. 2006;45(6):672–676. doi: 10.1111/j.1365-4632.2005.02607.x. [DOI] [PubMed] [Google Scholar]
- 7.American Academy of Dermatology. Eczema types: stasis dermatitis signs and symptoms. https://www.aad.org/public/diseases/eczema/types/stasis-dermatitis/symptoms. Accessed 1 July 2022.
- 8.Fiebig A, Krusche P, Wolf A, et al. Heritability of chronic venous disease. Human Gen. 2010;127(6):669–674. doi: 10.1007/s00439-010-0812-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silverberg JI, Hou A, Warshaw EM, et al. Prevalence and trend of allergen sensitization in patients with a diagnosis of stasis dermatitis referred for patch testing, North American contact dermatitis group data, 2001–2016. Arch Derm Res. 2022;314(9):857–67. [DOI] [PubMed]
- 10.Shiman MI, Pieper B, Templin TN, Birk TJ, Patel AR, Kirsner RS. Venous ulcers: a reappraisal analyzing the effects of neuropathy, muscle involvement, and range of motion upon gait and calf muscle function. Wound Repair Regen. 2009;17(2):147–152. doi: 10.1111/j.1524-475X.2009.00468.x. [DOI] [PubMed] [Google Scholar]
- 11.Pieper B, Kirsner RS, Templin TN, Birk TJ. Injection drug use: an understudied cause of venous disease. Arch Dermatol. 2007;143(10):1305–1309. doi: 10.1001/archderm.143.10.1305. [DOI] [PubMed] [Google Scholar]
- 12.Theodosat A. Skin diseases of the lower extremities in the elderly. Dermatol Clin. 2004;22(1):13–21. doi: 10.1016/S0733-8635(03)00113-X. [DOI] [PubMed] [Google Scholar]
- 13.Suehiro K, Morikage N, Murakami M, et al. A study of leg edema in immobile patients. Circ J. 2014;78(7):1733–1739. doi: 10.1253/circj.CJ-13-1599. [DOI] [PubMed] [Google Scholar]
- 14.Nazarko L. Diagnosis and treatment of venous eczema. Br J Community Nurs. 2009;14(5):188–194. doi: 10.12968/bjcn.2009.14.5.42076. [DOI] [PubMed] [Google Scholar]
- 15.Raffetto JD, Ligi D, Maniscalco R, Khalil RA, Mannello F. Why venous leg ulcers have difficulty healing: overview on pathophysiology, clinical consequences, and treatment. J Clin Med. 2020;10(1):1–34. doi: 10.3390/jcm10010029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palmer B, Xia Y, Cho S, Lewis FS. Acroangiodermatitis secondary to chronic venous insufficiency. Cutis. 2010;86(5):239–240. [PubMed] [Google Scholar]
- 17.Barron GS, Jacob SE, Kirsner RS. Dermatologic complications of chronic venous disease: medical management and beyond. Ann Vasc Surg. 2007;21(5):652–662. doi: 10.1016/j.avsg.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 18.Miteva M, Romanelli P, Kirsner RS. Lipodermatosclerosis. Dermatol Ther. 2010;23(4):375–388. doi: 10.1111/j.1529-8019.2010.01338.x. [DOI] [PubMed] [Google Scholar]
- 19.Kirsner RS, Pardes JB, Eaglstein WH, Falanga V. The clinical spectrum of lipodermatosclerosis. J Am Acad Dermatol. 1993;28(4):623–627. doi: 10.1016/0190-9622(93)70085-8. [DOI] [PubMed] [Google Scholar]
- 20.Cullum N, Nelson E, Fletcher A, Sheldon T. Compression for venous leg ulcers. Cochr Datab Syst Rev. 2001;2:CD000265. doi: 10.1002/14651858.CD000265. [DOI] [PubMed] [Google Scholar]
- 21.Al Shammeri O, AlHamdan N, Al-Hothaly B, Midhet F, Hussain M, Al-Mohaimeed A. Chronic venous insufficiency: prevalence and effect of compression stockings. Int J Health Sci. 2014;8(3):231–236. doi: 10.12816/0023975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suehiro K, Morikage N, Yamashita O, et al. Adherence to and efficacy of different compression methods for treating chronic venous insufficiency in the elderly. Phlebol. 2016;31(10):723–728. doi: 10.1177/0268355515608992. [DOI] [PubMed] [Google Scholar]
- 23.Raju S, Hollis K, Neglen P. Use of compression stockings in chronic venous disease: patient compliance and efficacy. Ann Vasc Surg. 2007;21(6):790–795. doi: 10.1016/j.avsg.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 24.Sippel K, Seifert B, Hafner J. Donning devices (foot slips and frames) enable elderly people with severe chronic venous insufficiency to put on compression stockings. Eur J Vasc Endovasc Surg. 2015;49(2):221–229. doi: 10.1016/j.ejvs.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 25.Paller AS, Tom WL, Lebwohl MG, et al. Efficacy and safety of crisaborole ointment, a novel, nonsteroidal phosphodiesterase 4 (PDE4) inhibitor for the topical treatment of atopic dermatitis (AD) in children and adults. J Am Acad Dermatol. 2016;75(3):494–503. doi: 10.1016/j.jaad.2016.05.046. [DOI] [PubMed] [Google Scholar]
- 26.Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis: section 2. Management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71(1):116–132. doi: 10.1016/j.jaad.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pariser D. Topical corticosteroids and topical calcineurin inhibitors in the treatment of atopic dermatitis: focus on percutaneous absorption. Am J Therap. 2009;16(3):264–273. doi: 10.1097/MJT.0b013e31818a975c. [DOI] [PubMed] [Google Scholar]
- 28.Hengge UR, Ruzicka T, Schwartz RA, Cork MJ. Adverse effects of topical glucocorticosteroids. J Am Acad Dermatol. 2006;54(1):1–5. doi: 10.1016/j.jaad.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 29.Dissemond J, Knab J, Lehnen M, Franckson T, Goos M. Successful treatment of stasis dermatitis with topical tacrolimus. Vasa. 2004;33(4):260–262. doi: 10.1024/0301-1526.33.4.260. [DOI] [PubMed] [Google Scholar]
- 30.Broeders JA, Ali UA, Fischer G. Systematic review and meta-analysis of randomized clinical trials (RCTs) comparing topical calcineurin inhibitors with topical corticosteroids for atopic dermatitis: a 15-year experience. J Am Acad Dermatol. 2016;75(2):410–419. doi: 10.1016/j.jaad.2016.02.1228. [DOI] [PubMed] [Google Scholar]
- 31.Abędź N, Pawliczak R. Efficacy and safety of topical calcineurin inhibitors for the treatment of atopic dermatitis: meta-analysis of randomized clinical trials. Postepy Dermatol Alergol. 2019;36(6):752–759. doi: 10.5114/ada.2019.91425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frankel HC, Qureshi AA. Comparative effectiveness of topical calcineurin inhibitors in adult patients with atopic dermatitis. Am J Clin Dermatol. 2012;13(2):113–123. doi: 10.2165/11597780-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 33.McDaniel HB, Marston WA, Farber MA. Recurrence of chronic venous ulcers on the basis of clinical, etiologic, anatomic, and pathophysiologic criteria and air plethysmography. J Vasc Surg. 2002;35(4):723–728. doi: 10.1067/mva.2002.121128. [DOI] [PubMed] [Google Scholar]
- 34.Klein B, Treudler R, Simon JC. JAK-inhibitors in dermatology - small molecules, big impact? Overview of the mechanism of action, previous study results and potential adverse effects. J Dtsch Dermatol Ges. 2022;20(1):19–24. doi: 10.1111/ddg.14668. [DOI] [PubMed] [Google Scholar]
- 35.Worm M, Bauer A, Elsner P, Mahler V, Molin S, Nielsen TS. Efficacy and safety of topical delgocitinib in patients with chronic hand eczema: data from a randomized, double-blind, vehicle-controlled phase IIa study. Br J Dermatol. 2020;182(5):1103–1110. doi: 10.1111/bjd.18469. [DOI] [PubMed] [Google Scholar]
- 36.Sawangjit R, Dilokthornsakul P, Lloyd-Lavery A, Lai NM, Dellavalle R, Chaiyakunapruk N. Systemic treatments for eczema: a network meta-analysis. Cochrane Datab Syst Rev. 2020;9(9):CD013206. [DOI] [PMC free article] [PubMed]
- 37.Sung CT, Taguines PR, Jacob SE. Stasis dermatitis. J Derm Nurs Assoc. 2019;11(3):134–136. [Google Scholar]
- 38.Nedorost S, White S, Rowland DY, et al. Development and implementation of an order set to improve value of care for patients with severe stasis dermatitis. J Am Acad Dermatol. 2019;80(3):815–817. doi: 10.1016/j.jaad.2018.10.034. [DOI] [PubMed] [Google Scholar]
- 39.Youn YJ, Lee J. Chronic venous insufficiency and varicose veins of the lower extremities. Kor J Intern Med. 2019;34(2):269–283. doi: 10.3904/kjim.2018.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bergan JJ, Schmid-Schönbein GW, Smith PDC, Nicolaides AN, Boisseau MR, Eklof B. Chronic venous disease. N Engl J Med. 2006;355(5):488–498. doi: 10.1056/NEJMra055289. [DOI] [PubMed] [Google Scholar]
- 41.Takase S, Pascarella L, Lerond L, Bergan JJ, Schmid-Schonbein GW. Venous hypertension, inflammation and valve remodeling. Eur J Vasc Endovasc Surg. 2004;28(5):484–493. doi: 10.1016/j.ejvs.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 42.Payne SP, London NJ, Newland CJ, Thrush AJ, Barrie WW, Bell PR. Ambulatory venous pressure: correlation with skin condition and role in identifying surgically correctible disease. Eur J Vasc Endovasc Surg. 1996;11(2):195–200. doi: 10.1016/S1078-5884(96)80051-7. [DOI] [PubMed] [Google Scholar]
- 43.Sippel K, Mayer D, Ballmer B, et al. Evidence that venous hypertension causes stasis dermatitis. Phlebol. 2011;26(8):361–365. doi: 10.1258/phleb.2010.010043. [DOI] [PubMed] [Google Scholar]
- 44.Hashimoto T, Kursewicz CD, Fayne RA, et al. Mechanisms of itch in stasis dermatitis: significant role of IL-31 from macrophages. J Invest Dermatol. 2020;140(4):850–859. doi: 10.1016/j.jid.2019.09.012. [DOI] [PubMed] [Google Scholar]
- 45.Thomas PR, Nash GB, Dormandy JA. White cell accumulation in dependent legs of patients with venous hypertension: a possible mechanism for trophic changes in the skin. Br Med J. 1988;296(6638):1693–1695. doi: 10.1136/bmj.296.6638.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilkinson L, Bunker C, Edwards J, Scurr J, Smith PC. Leukocytes: their role in the etiopathogenesis of skin damage in venous disease. J Vascul Surg. 1993;17(4):669–675. doi: 10.1016/0741-5214(93)90109-Y. [DOI] [PubMed] [Google Scholar]
- 47.Moyses C, Cederholm-Williams SA, Michel CC. Haemoconcentration and accumulation of white cells in the feet during venous stasis. Int J Microcirc Clin Exp. 1987;5(4):311–320. [PubMed] [Google Scholar]
- 48.Caggiati A, Rosi C, Casini A, et al. Skin iron deposition characterises lipodermatosclerosis and leg ulcer. Eur J Vasc Endovasc Surg. 2010;40(6):777–782. doi: 10.1016/j.ejvs.2010.08.015. [DOI] [PubMed] [Google Scholar]
- 49.Peschen M, Lahaye T, Hennig B, Weyl A, Simon JC, Vanscheidt W. Expression of the adhesion molecules ICAM-1, VCAM-1, LFA-1 and VLA-4 in the skin is modulated in progressing stages of chronic venous insufficiency. Acta Dermato-Venereol. 1999;79(1):27–32. doi: 10.1080/000155599750011651. [DOI] [PubMed] [Google Scholar]
- 50.Weyl A, Vanscheidt W, Weiss J, Peschen M, Schoepf E, Simon J. Expression of the adhesion molecules ICAM-1, VCAM-1, and E-selectin and their ligands VLA-4 and LFA-1 in chronic venous leg ulcers. J Am Acad Dermatol. 1996;34(3):418–423. doi: 10.1016/S0190-9622(96)90432-6. [DOI] [PubMed] [Google Scholar]
- 51.Kaur C, Sarkar R, Kanwar AJ, Attri AK, Dabra AK, Kochhar S. An open trial of calcium dobesilate in patients with venous ulcers and stasis dermatitis. Int J Dermatol. 2003;42(2):147–152. doi: 10.1046/j.1365-4362.2003.01709.x. [DOI] [PubMed] [Google Scholar]
- 52.Kucukguven A, Khalil AR. Matrix metalloproteinases as potential targets in the venous dilation associated with varicose veins. Curr Drug Targ. 2013;14(3):287–324. [PMC free article] [PubMed] [Google Scholar]
- 53.Saito S, Trovato MJ, You R, et al. Role of matrix metalloproteinases 1, 2, and 9 and tissue inhibitor of matrix metalloproteinase-1 in chronic venous insufficiency. J Vasc Surg. 2001;34(5):930–938. doi: 10.1067/mva.2001.119503. [DOI] [PubMed] [Google Scholar]
- 54.Herouy Y, Mellios P, Bandemir E, et al. Inflammation in stasis dermatitis upregulates MMP-1, MMP-2 and MMP-13 expression. J Dermatol Sci. 2001;25(3):198–205. doi: 10.1016/S0923-1811(00)00128-6. [DOI] [PubMed] [Google Scholar]
- 55.Wenk J, Foitzik A, Achterberg V, et al. Selective pick-up of increased iron by deferoxamine-coupled cellulose abrogates the iron-driven induction of matrix-degrading metalloproteinase 1 and lipid peroxidation in human dermal fibroblasts in vitro: a new dressing concept. J Invest Dermatol. 2001;116(6):833–839. doi: 10.1046/j.1523-1747.2001.01345.x. [DOI] [PubMed] [Google Scholar]
- 56.Li H, Zuo J, Tang W. Phosphodiesterase-4 inhibitors for the treatment of inflammatory diseases. Front Pharmacol. 2018;1048(9):1–21. doi: 10.3389/fphar.2018.01048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schick MA, Schlegel N. Clinical implication of phosphodiesterase-4-inhibition. Int J Mol Sci. 2022;23(3):1–19. doi: 10.3390/ijms23031209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rampersad SN, Wudwud A, Hubert F, Maurice DH. Adaptive phenotypic modulation of human arterial endothelial cells to fluid shear stress-encoded signals: modulation by phosphodiesterase 4D-VE-cadherin signalling. Cell Signal. 2016;28(7):741–748. doi: 10.1016/j.cellsig.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 59.Wan Q, Xu C, Zhu L, et al. Targeting PDE4B (phosphodiesterase-4 subtype B) for cardioprotection in acute myocardial infarction via neutrophils and microcirculation. Circul Res. 2022;131(5):442–455. doi: 10.1161/CIRCRESAHA.122.321365. [DOI] [PubMed] [Google Scholar]
- 60.Schlessinger J, Shepard JS, Gower R, et al. Safety, effectiveness, and pharmacokinetics of crisaborole in infants aged 3 to< 24 months with mild-to-moderate atopic dermatitis: a phase IV open-label study (CrisADe CARE 1) Am J Clin Dermatol. 2020;21(2):275–284. doi: 10.1007/s40257-020-00510-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.ClinicalTrials.gov. Study evaluating efficacy and safety of crisaborole in adults with stasis dermatitis. https://clinicaltrials.gov/ct2/show/NCT04091087. Accessed 26 Sep 2022.
- 62.Valdes-Rodriguez R, Mollanazar NK, González-Muro J, et al. Itch prevalence and characteristics in a Hispanic geriatric population: a comprehensive study using a standardized itch questionnaire. Acta Dermato-Venereol. 2015;95(4):417–421. doi: 10.2340/00015555-1968. [DOI] [PubMed] [Google Scholar]
- 63.Nattkemper LA, Martinez-Escala ME, Gelman AB, et al. Cutaneous T-cell lymphoma and pruritus: the expression of IL-31 and its receptors in the skin. Acta Derm Venereol. 2016;96(7):894–898. doi: 10.2340/00015555-2417. [DOI] [PubMed] [Google Scholar]
- 64.Nattkemper LA, Tey HL, Valdes-Rodriguez R, et al. The genetics of chronic itch: gene expression in the skin of patients with atopic dermatitis and psoriasis with severe itch. J Invest Dermatol. 2018;138(6):1311–1317. doi: 10.1016/j.jid.2017.12.029. [DOI] [PubMed] [Google Scholar]
- 65.Sonkoly E, Muller A, Lauerma AI, et al. IL-31: a new link between T cells and pruritus in atopic skin inflammation. J Allerg Clin Immunol. 2006;117(2):411–417. doi: 10.1016/j.jaci.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 66.Furue M, Furue M. Interleukin-31 and pruritic skin. J Clin Med. 2021;10(9):1906:1–1911. doi: 10.3390/jcm10091906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Salao K, Sawanyawisuth K, Winaikosol K, Choonhakarn C, Chaowattanapanit S. Interleukin-31 and chronic pruritus of unknown origin. Biomark Insight. 2020;15:1–4. doi: 10.1177/1177271920940712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Valencia IC, Falabella A, Kirsner RS, Eaglstein WH. Chronic venous insufficiency and venous leg ulceration. J Am Acad Dermatol. 2001;44(3):401–421. doi: 10.1067/mjd.2001.111633. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no data sets were generated or analyzed during the current study.