

Summary
As modern biological sciences evolve from investigation of individual molecules and pathways to growing emphasis on global and systems-based processes, increasing efforts have focused on combining the study of genomics with that of the other omics technologies, including epigenomics, transcriptomics, quantitative proteomics, global analyses of post-translational modifications (PTMs) and metabolomics, to characterize specific biological or pathological processes. In addition, emerging genome-wide functional screening technologies further help researchers identify key regulators of immune functions. Derived from these multi-omics technologies, single cell sequencing analysis on multiple layers offers an overview of intra-tissue or intra-organ immune cell heterogeneity. In this review, we summarize advances in multi-omics tools to explore immune cell functions and applications of these multi-omics approaches in the analysis of clinical immune disorders, aiming to provide an outlook on the potential opportunities and challenges that these technologies pose in future investigation in the field of immunology.
Subject areas: Immunology, Cell biology, Bioinformatics
Graphical abstract
Immunology; Cell biology; Bioinformatics
Introduction
When attacked by pathogens, the immune system orchestrates comprehensive responses including activation of and communication between a diverse series of immune cells. As part of these processes, immune cells undergo active periods of transcription and translation which are strictly and precisely regulated to produce effector molecules including antibodies and cytokines. Therefore, the characterization of each process and their associated products (Figure 1) is indispensable for exploring the functions of immune cells and unraveling the underlying mechanisms in immune system responses.
Figure 1.

Summary of the key molecules and associated approaches involved in each regulatory layer
The process of information flowing from DNA to metabolites includes four parts and is placed in the Targeted molecules section. Typical approaches are divided into four levels according to different targeted molecules. Each group of molecules with similar chemical properties stands for one ‘omic’ layer, which constitutes multi-omics by connecting each other. scDNA-seq, single cell DNA sequencing; ATAC-seq, transposase-accessible chromatin sequencing; ChIP-seq, chromatin immunoprecipitation-sequencing; scRNA-seq, single cell RNA sequencing; snRNA-seq, single-nuclei RNA sequencing; SLAM-seq, thiol (SH)-linked alkylation for the metabolic sequencing of RNA; MS, massspectrometry; CyTOF, cytometry by time-of-flight; scTCR-seq, single cell T-cell receptor sequencing; scBCR-seq, single cell B-cell receptor sequencing; PTMs, post-translational modifications.
Although genomics opened the door to descriptions of genome-wide alterations, the ‘omics’ revolution allows for global and comprehensive descriptions of cellular states that extend beyond the linear DNA sequences. Omics is the analysis of a large quantity of data representing the whole set of molecules that make up the cells of an organism, including genes, proteins, lipids, and metabolites. Omics technologies that usually comprise genomics, epigenomics, transcriptomics, proteomics and metabolomics analyses permit interrogation of molecules from different regulatory levels and with both temporal and spatial resolution of biological activities in cells. The integration of these multi-parameter technologies, known as “multi-omics”, unveils not only the information of single regulatory layer but also the interwoven relationships between layers, enabling researchers to establish multi-dimensional molecular landscapes and elucidate the flow of information, from the original cause to the functional consequences or relevant interactions.1,2
Multi-omics tools have been increasingly used to reveal relevant signaling networks in immune cells. For instance, Tan et al.3 used two-dimensional liquid chromatography-tandem mass spectrometry (LC/LC-MS/MS) and multiplexed tandem-mass-tag (TMT) and quantified 8,431 proteins and 13,755 phospho-peptides in total. By integrating proteomics and phosphoproteomics data, they uncovered the signaling and bioenergetics pathway underlying T cell activation. Development of systems-based approaches to integrate data from large-scale omics also facilitates understanding of the immune cell functions. Bakker et al.4 developed a comprehensive systems biology approach and explored the influence of genetic and nongenetic host factors on the cytokine response by combining the data from large-scale genome, metagenome and metabolome studies with other baseline immunological parameters like the immune cell composition and hormone levels of each person involved.
Apart from the multi-omics approaches, the maturation of genome-wide functional screening technologies further provides researchers with potent tools to identify key regulators of immune functions. For example, genome-wide pooled CRISPR screens can be used to identify regulators of the T-cell receptor (TCR) responses.5 Complementing these multi-omics technologies, single cell sequencing analysis across multiple regulatory dimensions further offers an overview of the intra-tissue heterogeneity of those immune cells and is instrumental for identification of new subsets of immune cells that await functional characterization. Among the single cell sequencing analysis, single-cell mRNA sequencing has been used most widely in the field of immunology. Beyond identifying new subsets of immune cells, Schelker et al. created cell type-specific and indication-specific reference gene expression profiles by analyzing tumor-derived single-cell RNA sequencing data which could help with the estimation of immune cell content in a variety of tumors.6
Herein, our review is divided into three parts. First, we briefly summarize recent advances in multi-omics technologies and their use in the exploration of immune cell functions. Second, the latest applications of these multi-omics technologies in the investigation of clinical immune disorders are discussed. Finally, we provide an outlook on the potential opportunities and challenges that these technologies pose in future immunological research.
Multi-omics approaches to explore immune cell functions
Biological activities of immune cells can be classified into intracellular and intercellular ones. The hub in intracellular activity is the genes which control growth, development, and function of each immune cell. Different modifications at DNA, RNA, and protein levels as well as regulation at dynamic process of transcription and translation are mainly involved in regulating immune cell functions. On the basis of regulatory mechanisms, how to identify genetic variants and key genes, how to verify gene functions, and how to explore the regulation at each layer and coordinate the information from each layer to construct final pathways, are all challenging in investigating activities within immune cells. Intercellular activities which mainly contain interactions and communications among different immune cells, immune cells and microbiota, and immune cell and other type cells like cancer cells and cancer-related fibroblasts are also important questions in the quest for understanding these processes. In addition, as the products of cellular biological activities, metabolites act as executors of immune cell function which are versatile and warrant comprehensive profiling.
Besides, different immune cells are neighbored with other numerous types of cells, and relative distance between them may somewhat reflect the degree of interactions. But how to distinguish each type of cell and obtain the spatial information in situ requires special attention. Apart from known types of immune cells, there are still many unknown subtypes of immune cells, and thus identification and functional characterization of the novel immune cell subpopulations are needed. In this section, we discuss in detail viable approaches and examples in immunology field from a multi-omics perspective. Main technologies of each omics approach in the exploration of immune cell have been summarized in Table 1.
Table 1.
Main technologies of each omics approach in the exploration mechanistic investigations of immune cells
| Omics layer | Targeted molecule/problem | Main technologies |
|---|---|---|
| Genomics | DNA/Identification of genetic variants and key genes | Whole genome-seq: Whole genome sequencing (WGS) is a technique which aims to obtain all genes of a person including both coding and non-coding regions of a genome. |
| Whole exome-seq: Compared with WGS, whole exome sequencing (WES) focuses on sequencing of the coding regions of a genome, which is more efficient and cost-effective in exploring variants related to diseases. | ||
| Immunochip array: Immunochip is a genotyping chip technique especially used for identifying significant loci for variants in immune-related diseases. | ||
| scDNA-seq: Single-cell DNA sequencing (scDNA-seq) is a genomic approach used to decode variants, DNA modifications, and structural features of DNA at single cell level which may be ignored by using bulk DNA sequencing. | ||
| Epigenomics | DNA/Dissection of regulation at transcription level | ATAC-seq: Assay for Transposase-Accessible Chromatin with high-throughput sequencing (ATAC-seq) is a technique used for identifying chromatin accessibility through genome by taking advantage of hyperactive Tn5 transposase to insert the sequencing adapters into open chromatin and testing the replaced DNA fragment. |
| ChIP-seq: Chromatin immunoprecipitation followed by sequencing (ChIP–seq) is applied to profile the genome-wide features of DNA modifications including DNA methylation, histone modifications such as acetylation and methylation among others. | ||
| DNase-seq: DNase I hypersensitive sites sequencing (DNase-seq) is used to map the genome-wide active regulatory regions which are sensitive to DNase I. | ||
| 3D genome technologies: 3D genome technologies aim to study the three-dimensional (3D) structure of DNA and gene regulation, including chromosome conformation capture (3C), circularized chromosome conformation capture (4C), carbon copy chromosome conformation capture (5C), and Hi-C technology. | ||
| Genome-wide functional screening | DNA/Verification of gene function | CRISPR-based functional genomic screening technology: CRISPR-based functional genomic screening technology uses CRISPR tools to edit genome-wide DNAs, enabling perturbation of gene function and later measurement of a specific phenotype. |
| Transcriptomics | RNA/Identification of genetic variants and key genes | Bulk RNA-seq: Bulk RNA-sequencing, which is often referred as RNA-seq, is the most commonly used transcriptomic approach in which all RNAs from mixed samples including tissue, organ or a population of cells are extracted and sequenced through high-throughput technologies. The data from bulk RNA-seq represent an average expression of genes. |
| scRNA-seq: Single-cell RNA sequencing (scRNA-seq) is a transcriptomic approach in which all RNAs from single cells are extracted and sequenced through high-throughput technologies. In contrast with bulk RNA-seq, scRNA-seq focuses on a single cell level, which can be used for identifying new subtypes of cells and exploring the intracellular and extracellular interactions. | ||
| snRNA-seq: Single-nucleus RNA-sequencing (snRNA-seq) applies to individual nuclei capture and sequencing at single-cell level. | ||
| SLAM-seq: Thiol (SH)-linked alkylation of RNA for the metabolic labeling sequencing (SLAM-seq) is a transcriptomic approach for directly quantifying new synthesized mRNAs. | ||
| Translatomics | RNA/Dissection of regulation at translation level | Ribosome profiling: Ribosome profiling uses deep sequencing to globally monitor and measure the translation process in vivo. |
| Spatial transcriptomics | RNA/Presentation of immune cellular spatial distribution and interaction in tissue | Microdissection: Microdissection is a traditional technique used to separate tissue-specific cells with the help of inverted microscope and a fine glass needle. After these tissue-specific cells are isolated, RNA will be extracted and then sequenced. |
| In situ hybridization: In situ hybridization is a technique using a nucleotide probe labeled with radio material, fluorescence or other substance which can be shown visually to detect nucleotide sequences in cells, tissue section or bulk tissue. | ||
| In situ sequencing: In situ sequencing is a technique in which mRNA is sequenced directly in fixed tissue section or cells by using fluorescent probes. | ||
| In situ capturing: In situ capturing is a technique used to present the transcriptomics datain situ, which requires three steps to accomplish. Firstly, transcripts are captured and barcoded in the targeted tissue. Then, sequencing is completed outside the tissue. Finally, transcriptomics data is overlayed with the tissue image. | ||
| Proteomics and post-translational modification | Proteins/Dissection of regulation at post-translation level | Immunoassay: Immunoassay (IA) is a commonly used method basing on antibody-antigen combination, which is applied in identifying and measuring molecules. Immunoassay is divided into non-labeled immunoassay (including western blot and immunodiffusion) and labeled immunoassay (including radioimmunoassay (RIA), enzyme-linked immunosorbent assay (ELISA), immunohistochemical methods, and fluorescence-activated cell sorting. |
| MS: Mass spectrometry (MS) is an analytical detection technique used to ascertain specific chemical substances including oligonucleotides, proteins, carbohydrates and other metabolites of cells by sorting and testing gaseous ions in electric and magnetic fields according to the mass-to-charge ratios. | ||
| MS-based approaches: Developed from mass spectrometry, MS-based approaches, including GC-MS, LC-MS, LC-MS/MS and LC/LC-MS/MS, are used to profile specific chemical substances by different separation ways but with faster speed, higher efficiency and wider range of application. | ||
| CyTOF: Cytometry time of flight (CyTOF), also called mass cytometry, is developed from flow cytometry which uses antibodies conjugated with heavy metal ion instead of fluorochromes to detect intracellular or extracellular antigens of interest on single cells. | ||
| scTCR-seq: Single cell T cell receptor sequencing (scTCR-seq) is a method used to profile diversity and clonality of TCR repertoire and TCRαβ at single T cell level by multiplex PCR and high-throughput sequencing or Sanger sequencing. | ||
| scBCR-seq: Single cell B-cell receptor sequencing (scBCR-seq) is a method used to detect paired light and heavy BCR chains at single B cell level by amplification and high-throughput sequencing. | ||
| Metabolomics | Metabolites/Assistance in construction of metabolic pathways | MS: Mass spectrometry (MS) is an analytical detection technique used to ascertain specific chemical substances including oligonucleotides, proteins, carbohydrates and other metabolites of cells by sorting and testing gaseous ions in electric and magnetic fields according to the mass-to-charge ratios. |
| MS-based approaches: Developed from mass spectrometry, MS-based approaches, including GC-MS, LC-MS, LC-MS/MS and LC/LC-MS/MS, are used to profile specific chemical substances by different separation ways but with faster speed, higher efficiency and wider range of application. |
Identification of genetic variants and key genes
As the most mature discipline in the omics fields, genomics focuses on the linear DNA nucleotide sequences, encompassing both gene coding regions and non-coding regulatory and structural elements. When referring to genomics, there are several technologies including genotype arrays and next-generation sequencing (NGS) for both whole-genome and exome sequencing. Genomics technologies identify genes associated with the responsiveness of the immune system and immune diseases which have been described in two excellent reviews.7,8
Compared with genomics, transcriptomics examines the global transcriptional output of cells. In the field of transcriptomics, probe-based arrays and RNA-Seq are the two technologies most widely used. Gene expression analyses are capable of identifying key genes involved in immune responses under various conditions.9 Besides, key factors involved in the two modes of T cell activation have been uncovered by integration of transcriptomics with proteomics and metabolomics.10 In sum, based on array technology and deep sequencing, both known and unknown genetic variants can be identified in immune cells from DNA and RNA level. Significant and fine mapping of genetic loci can be achieved by combination of genomic or transcriptomic with other omics.
Verification of gene function, from genotype to phenotype
The advent of genome-wide functional screening technologies provides the opportunity for researchers to unbiasedly uncover key factors in certain biological processes and have been widely applied for seeking for novel immunotherapy targets in tumor cells.11,12 As a representative of genome-wide functional screening, CRISPR technology has evolved rapidly; it contains two components: the non-specific endonuclease termed Cas9 and a single stranded guide RNA (gRNA). The target of gRNA can be defined by users, and then Cas9 can modify any specific genomic locus under guidance of gRNA, which allows for specific and flexible knockout, repression, and activation of target genes, and enable researchers to achieve large-scale functional screening in a variety of scenarios like T cell activation13 and macrophage efferocytosis.14 Besides, improved CRISPR technology called SLICE have been used to uncover the key regulators of immune function in primary human T cells with higher efficiency of transduction.5 Overall, by engineering and selecting typical immune cells with pooled sgRNAs, CRISPR technology enables researchers to experimentally define gene function and explore potential mechanism of complex phenotypes in immune cells.
Dissection of regulatory process and revealing possible pathways in immune responses
Regulation on the transcription level
Covalent modifications of DNA and histones affect gene transcription and subsequent protein expression,15 with ramifications in disease pathogenesis, inspiring much interest in the investigation of the relationship between these modifications and diseases.16,17 Within the field of immunology, these modifications also play essential roles in immune cell fate determination, such as 5-hydroxymethylcytosine (5-hMC).18 Furthermore, epigenetic changes can lead to disorders of immune cells, contributing to the inflammatory changes accompanying diet-induced metabolic diseases.17
Epigenomics explores the genome-wide features of DNA modifications that do not affect the linear DNA sequence, including DNA methylation, histone modifications such as acetylation and methylation among others, as well as the three-dimensional conformation of genome.19 There are mainly four technologies in epigenomics: Assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq), chromatin immunoprecipitation followed by sequencing (ChIP–seq), DNase I hypersensitive sites sequencing (DNase-seq), and 3D genome technologies (Table 1). Among these technologies, ATAC-seq is most widely used. For instance, Liu et al. revealed that a transcription factor named NR4A1 served as an essential regulator in T cell dysfunction through ATAC-seq and RNA-seq.20 In addition, one study analyzed over 300 RNA-seq and ATAC-seq experiments from published reports of CD8+T cells in the context of cancer and infection, and identified a common differentiation trajectory in all types of dysfunctional T cells and related transcriptional drivers.21 To conclude, combination of ATAC-seq and RNA-seq assists in characterizing genome-wide epigenetic and gene expression features and identifying essential regulatory factors from transcription level in dysfunctional immune cells.
Besides, three-dimensional (3D) genome organization has been proven to act in various biological processes.22,23 As a potent tool, 3D genomics could not only show us the basic structures of DNA but also the underlying mechanism of DNA transcription.24 3D genomics mainly contains four types of technologies, which are chromosome conformation capture (3C), circularized chromosome conformation capture (4C), carbon copy chromosome conformation capture (5C), and Hi-C technology. 3C can only reveal two targeted chromatin interactions, whereas with development, 4C can reveal interactions of one with many other regions, and 5C is designed for many regions and many regions.25 On the basis of chromosome conformation capture, Hi-C takes advantage of next-generation sequencing and is able to reveal the interaction of all regions and all regions at genome-wide scale.26 Besides interactions between DNA regions, protein-mediated interactions between specific proteins and chromatins can be detected by ChIA-PET, which is a technique combining chromatin immunoprecipitation with 3C.27
Currently, Hi-C is the mostly used 3D genomics technology. One study used Hi-C to investigate how the lineage-specific 3D architecture was established and maintained during B cell differentiation, revealing that Pax5 participated in establishing and maintaining the lineage-specific genome organization of B cells from the beginning of differentiation to the plasmablast stage.28 The chromatin remodeler Brg1 was proved to function at different developmental stages of B cells by combined analysis of Hi-C, ATAC-seq, ChIP-seq and RNA-seq techniques,29which included regulation of contraction of locus encoding the immunoglobulin heavy chain (Igh) and promotion of the interaction between TFs and enhancers. Besides, by integration of multiple-enzyme Hi-C (3eHi-C), DNase-seq, ChIP-seq, and RNA-seq, many crucial factors were also identified in T cell differentiation.30 In conclusion, Hi-C is frequently coupled with epigenomic and transcriptomic technologies to realize interrogation of critical factors influencing 3D architecture at genome-wide scale during development of immune cells. Apart from investigation into development of T and B cells, 3D genomics has also been applied to present the variations of 3D genomic architecture during B-cell activation in germinal centers31 and T-cell activation.32
Regulation on the translation level
The cellular regulatory control of processes spanning translation of mRNAs to proteins is of great significance. By simultaneously measuring mRNA expression and protein levels, several investigations demonstrated that post-transcriptional gene regulation, including translation control, is actively involved in regulating the proteome landscape.33,34,35 Translatomics aims to probe into all components involved in the translation process including ribosomes, regulatory RNAs, translating mRNAs, and tRNAs.36 Exploring the translatomics of cells offers us the chance to gain a more holistic understanding of true proteins abundance.
As a potent method to monitor translation, ribosome profiling takes advantage of next-generation sequencing to identify ribosome-protected mRNA fragments which are shielded from nuclease digestion37 and has been applied for establishing mTOR translation signatures in mouse embryonic fibroblasts (MEFs) and human prostate cancer cells.38,39 In a recent research, ribosome profiling has been used for investigating the role of miR-223 in regulating inflammatory signaling and lipid metabolism in macrophages.40 A similar approach was also carried out to reveal the function of mTOR signals in resting CD4+T cells41 and ZFP36 RNA-binding proteins in active CD4+T cells.42 In addition to exploring functional mechanisms of key factors on the translation level, ribosome profiling was applied to characterize acute mRNA translation changes in naive CD4+T cells43 and CD8+ memory T cells44 following activation. All examples mentioned above performed ribosome profiling and RNA sequencing simultaneously to compare the transcriptional and the translation level alterations in immune cells under different conditions and then found that the regulatory mechanisms were at translation level instead of transcription level. Overall, ribosome profiling serves as a bridge between exploration of mRNA and protein, enabling us to gain insights into regulation of immune cell functions on the translation level.
Regulation on the PTMs level
Proteins play an indispensable role in immune responses and their functions can be regulated by PTMs.45 Proteomics aims to quantify the abundance of peptides, their various PTMs, and interactions between peptides. According to the sorting gaseous ions of sample molecules and principle of antibody-antigen combination, the proteomic technologies include mass spectrometry (MS), MS-based approaches, immunoassay, and cytometry time of flight (CyTOF). MS-based approaches, including GC-MS, LC-MS, LC-MS/MS and LC/LC-MS/MS, combine different physical separation ways with MS, for example, the GS-MS means separation by gas chromatography (GC) and detection by MS whereas LC-MS means separation by liquid chromatography (LC) and detection by MS.
As a powerful proteomics tool, MS offers great opportunity to gain biological insights into the system-wide characterization of information transmitted through downstream signal transduction networks. One study found that IL-2 could both positively and negatively regulate the phosphorylation of proteins in cytotoxic T lymphocytes (CTLs) by using stable isotope labeling by amino acids in cell culture (SILAC)-based quantitative high-resolution MS.46 Based on MS, many novel technologies have developed. Di-glycine remnant profiling is a kind of technology in which proteins with ubiquitination lysine side chains are digested into Lys-ϵ-Gly-Gly remnant that can be recognized by antibody and then endogenous ubiquitination sites are detected by MS. Dybas et al.47 used Di-glycine remnant profiling to uncover ubiquitylated proteins in activated CD4+T cells, uncovering that ubiquitination could serve as an important non-degradative mark with roles for ubiquitin chains in T cell signaling. In another example, Hubel et al.48 established an affinity enrichment with quantitative LC-MS strategy to identify the proteins associated with interferon-stimulated genes (ISGs) in innate immune cells.
Developed from LC-MS, the advent of liquid chromatography-tandem mass spectrometry (LC-MS/MS) enables both quantitative analyses of protein levels with greater capacity to distinguish between similar compounds. One study using ultra performance liquid chromatography/tandem mass spectrometry (UPLC-MS/MS) to characterize the molecular alterations of COVID-19 patients’ sera revealed dysfunction of complement system pathways and macrophages.49 In summary, though MS and MS-based approaches have different capacities of separation or detection, both of them enable us to widely define proteins and their modifications in immune cells. They help in selecting key factors on the PTM level and constructing complete biological pathways in immune cells, which will be discussed in the following section.
Analysis and construction of biological pathways in immune cell
In the beginning, pathway analysis is based on gene expression data and associated methods are mainly divided into three including the over-representation analysis (ORA), the Functional Class Scoring (FCS), and the topology-based (TB) methods.50 However, none of these methods can analyze or integrate data from other types, resulting in limited sample size, low reproducibility, and most importantly, constrained understanding of our immune system. Construction of a thorough and complete biological pathway requires multifaceted information from genes, proteins, metabolites, and the relationships among them, which could be realized by epigenomics, transcriptomics, proteomics, and metabolomics and subsequent integrative analysis across these data. Before formal introduction of how to conduct pathway analysis and construction by integrating multi-omics data, it is important to explain metabolomics of immune cell for readers to understand the following content. Immunometabolism is defined as the intrinsic metabolic pathways including glucolysis, glutaminolysis, fatty acid oxidation, oxidative phosphorylation, pentose phosphate, and Krebs cycle in modulating the fate and function of immune cells.51,52 As the effector functions and cellular fates of immune cells are inherently interwoven with cellular metabolism, many studies have explored immune cell function from the perspective of metabolome. Metabolomics aims to quantify diverse small molecules including amino acids, fatty acids, carbohydrates, nucleotides, and a diverse array of substances produced through cellular metabolism19 by using MS and MS-based approaches.
Back to pathway analysis and construction. After collecting multi-omics data from experiments or public database, integrative pathway analysis starts. First, proper integrative pathway analysis methods need to be carefully selected. Web-based tools and R packages are the most commonly used platform. Besides, 32 pathway analysis methods have been reviewed in detail50 and users can choose the optimal pathway analysis method according to their practical demands. The next step is to know how to use these methods. Integrative pathway analysis mainly incorporates two analysis stages: the pre-analysis and the pathway analysis.
In the pre-analysis stage, experimental data and pathway database are input with the right format (the omics expression matrices in.CSV or.txt format and pathways matrix in.GMT format). It is worth mentioning that most pathway analysis methods embed pathways information from public databases and users do not need to input pathway information without special demands. Quality control and data filtering are followed to guarantee the correctness of data and reduce the number of irrelevant features. After completing every check, identifier (ID) mapping is conducted aiming to combine multi-omics data with pathway information and make the gene/compound IDs consistent. When ID mapping finishes, there will be two optional choice including data matching (all expression data are matched) and pathway augmentation (validated multi-omics interaction are added) in certain methods.
In the pathway analysis stage, differential analysis is applied to omics data prepared by pre-analysis and differentially expressed genes are identified by adding a threshold. Next, based on the summary statistics from differential analysis, enrichment statistics for each pathway in each study or omics layers are able to be calculated and significant pathways can be further verified. In addition, constructing network by linking different omics layers is optional to complete before pathway analysis begins. Multi-omics data integration by classical methods (Fisher’s, minP, Stouffer’s, etc.) can be realized either in the pre-analysis stage or pathway analysis stage depending on the assumption and strategy of each method. Finally, the analysis results are visualized by charts (bar graphs, heatmap, etc.) and listed with their associated statistics (enrichment score, pvalue, etc.).
On the basis of multi-omics data integration, many biological pathways in immune cells have been uncovered. For instance, Tan et al.3 interrogated the proteome and phosphoproteome of naive T cells by using multiplexed TMT and LC/LC-MS/MS, quantifying 8,431 proteins and 13,755 phosphopeptides in total. By combining co-expression clustering and protein-protein interaction networks, they created a three-step computational pipeline for analysis and elucidated six mitochondrial pathways in activated T cells. This comprehensive pipeline could serve as a powerful tool to combine temporal multi-layer omics data (transcriptome, proteome and phosphoproteome) and various databases (PPI, TF-target and kinase-substrate) to reveal insights into T cell biology.
In addition to investigations of T cells, interaction of multiple cytokine signaling pathways in NK cells53 and definition of the diversities of regulatory networks in macrophages among different human populations54 are also achieved by combination of epigenomics and transcriptomics. Moreover, in combination with other technologies like RNA-Seq, ChIP-seq, and ATAC-seq, CRISPR-based functional screening tools provide researchers with a novel perspective to explore the relationship between different immune cellular states. One study developed a genome-wide retroviral CRISPR sgRNA library allowed for infection and utilization of CRISPR libraries in murine cells, which used to be a limitation for earlier lentiviral-based CRISPR approaches.55 Through this method, they discovered multiple genes involved in Th2 differentiation and interrogated these pathways through integration of RNA-Seq, ATAC-seq, and ChIP-seq datasets. Combined multi-omics analyses spanning distinct regulatory modalities revealed tight interconnection between Th2 activation and differentiation and exposed the key gene networks in regulating Th2 cell function.
Apart from normal physiologic functions, altered immunometabolism of infected CD4+T cells uncovered the influences of pathogens on immune cells. By analyzing transcriptome data, Guo et al. found that elevation of OXPHOS pathways in CD4+T cells infected with human immunodeficiency virus type-1 (HIV-1) is associated with poor prognosis. To further reveal more details concerning this pathway, integrative quantitative proteomics by (SILAC)-based quantitative high-resolution mass spectrometry and metabolic analysis showed that the mitochondrial innate immune receptor NLRX1 enhanced OXPHOS and glycolysis in CD4+T cells infected by HIV-1, which promoted viral replication.56 In conclusion, multi-omics data integration and analysis pave the way for building complex regulatory networks in immune cell, helping us get a better understanding of immune variation in various immune-related diseases.
Definition of immune cell types and their unique functional properties at single cell level
The complex architecture and associated higher order functions of the immune system rely on the highly specific immune cell types that coordinate with each other to prevent infections, eliminate pathogens, and generate immunologic memory. Thus, it is of great importance to define the cellular subsets of immune cells, to understand intra-tissue and inter-tissue heterogeneity, and to explore novel immune cell types and their unique functional properties (Figure 2). As a tool that has been used widely to identify the new cell types in many tissues, single-cell RNA sequencing and its associated technologies have also been applied to the immune system.57
Figure 2.

Combination of single cell sequencing, bulk sequencing and multi-omics analysis to dissect TME
Tumor tissue is mixed with various types of cells including fibroblasts, cancer cells and different immune cells. Bulk sequencing of tumor tissue provides average information of genomics, transcriptomics, proteomics, and metabolomics. By analyzing average information, tumor antigen and immune cell ratio can be gained for developing tumor vaccine, immune-related scores, and immune response biomarkers. Single cell analysis helps identify new types of immune cells and further exploration can be conducted in the same type of immune cells through multi-omics analysis and genome-wide functional screening to identify key regulatory genes, proteins and metabolites, which could potentially become new biomarkers and treatment targets. The tumor tissue can also be replaced by other clinical samples including blood, urine, feces, exhaled air and other tissues in the multi-omics analysis. PTMs, post-translational modifications.
One study combined cytometry by time-of-flight (CyTOF) and single-cell RNA sequencing to identify human blood dendritic cell precursors.58 Janela et al. used single-cell sequencing to determine that type I conventional dendritic cells (cDC1s) are key mediators of neutrophil activation and survival in the innate responses in cutaneous immunity.59 Through transcriptional profiling, they further identified two distinct cDC1 cell clusters through principal-component analysis. The less abundant cDC1 population expressed higher levels of secreted cytokines, including VEGFA, and enhanced neutrophil recruitment, revealing new roles for dendritic cell populations apart from antigen presentation. In addition to probing the potential functions of dendritic cells, Chakarov et al.also used single-cell RNA sequencing to describe the transcriptional profiles of individual interstitial macrophages (IM) isolated from lungs,60 identifying two distinct cell clusters. One cluster termed Lyve1hiMHCIIlo was consistently situated around blood vessels, whereas the other cluster termed Lyve1loMHCIIhi IMs surrounded nerves. Parallel IM populations were observed to exist in other tissues, including heart, adipose tissue and dermis.
Besides characterizing distinct cell subsets, single-cell RNA sequencing technology can also be used for both characterizing the heterogeneity of a population of immune cells and dissecting cell fate branch points by integrating data from other related technologies which have been described at length previously.61 One study focusing on rheumatoid arthritis (RA) coupled single-cell RNA sequencing with mass cytometry, unveiling the immune cell states in RA synovia and defining three distinct subsets of CD8+T cells.62 One strength of this study is the creation of a specific computational strategy, which was based on canonical correlation analysis to unify the multi-layer datasets from transcriptomic and proteomic profiles at the single-cell level. In addition, to profile the heterogeneity of the exhausted CD8+ T (Tex) cells in the context of HIV or lung cancer, Bengsch et al.developed a systems immunology approach63 by integration of epigenomic and transcriptomic data through ATAC-seq and single-cell RNA sequencing. They defined key exhaustion-specific genes and disease-induced changes of Tex cells in the chronic lymphocytic choriomeningitis virus (LCMV) system. Identification of these exhaustion genes informed development of a novel, focused single-cell proteomic profiling approach and identified nine distinct Tex cell clusters.
During the past two years, single-cell analyses have been increasingly utilized to explore the immune responses in COVID-19-infected patients. One study performed single-cell transcriptomics, surface proteomics, and T and B lymphocyte antigen receptor analyses of peripheral immune cells from COVID-19 patients with varying severities and found different kinds of T cells in severe and mild diseases.64 Similar single-cell analysis methods were also carried out to identify the components of peripheral blood mononuclear cells from COVID-19 patients with different free triiodothyronine (T3, thyroid hormone) serum values.9 Similarly, to develop immunologic disease severity biomarkers, another study assessed transcriptomes, surface protein markers, and T cell receptors sequence simultaneously in single peripheral immune cell from COVID-19 patients. Incorporated with the information of circulating proteins in patient blood, the authors characterized immune cell types and defined gene expression and metabolic states in each kind of immune cells.65
Apart from single-cell RNA sequencing, bulk RNA sequencing with deconvolution and data interpretation can also reveal the heterogeneity in tissue. For example, one Cell Linkage through Exploratory Matrices (ICLite) algorithm to deconvolve the result of bulk RNA sequencing of bronchoalveolar lavage cells, which was used to identify a special population of innate immune cells in which mitosis and IL-7 signaling are preserved.66
Presentation of the immune cellular spatial distribution and interaction in tissue
Cell-to-cell interaction
Cell-to-cell communication and interaction are ubiquitous in multicellular organism, which play indispensable roles in development, function, and dysfunction of immune system. For example, immunosuppressive TME depends on the crosstalk between immune cells and cancer cells67 as well as cancer-cell associated fibroblasts.68 Owing to the fact that various cytokines, growth factors, chemokines, exosomes and other effector molecules serve as the mediators in the intricate communication, how to dissect the communicational process and investigate the underlying mechanism are still challenging. Rieckmann et al. applied proteomics to explore the proteome state in 28 distinct human hematopoietic cell types.69 They identified over 10,000 different proteins in total and improved the limited amount of rare immune cells by using a single-run MS analysis approach. By combining the data from total and secreted proteomes, the authors unveiled fundamental intercellular crosstalk and novel connections between cell types, providing a new perspective for studying the immune system.
Apart from the intracellular proteins, extracellular vesicles (EVs) also serve as indispensable mediators in cell-to-cell communication. One study combined proteomics with other multi-omics (transcriptomics, metabolomics) tools to dissect the signaling properties of EVs from macrophages infected by Cryptococcus neoformans, revealing modulation of pathways such as extracellular matrix (ECM) receptors and phosphatidylcholine.70
According the relative distance between cells, intercellular communication is divided into three types encompassing paracrine, juxtacrine, and endocrine. Different relative distance may reflect different degrees of communication between cells. Exploring the spatial distribution of immune cells and other type cells in tissue can help us understand the communication ways and select proper cell populations (with closer distance) to study cell-cell communication. As the “Method of the Year 2020”, spatial transcriptomics (ST) serves as a potent tool for biologists to comprehend the complexity of multicellular organisms. Bulk RNA seq provides the average global information of transcripts in species and single-cell RNA seq profiles genes expression in single cells, however, none of them can present the spatial distribution of RNAs in situ. To solve this problem, ST was developed. ST encompasses a wide range of technologies which are mainly divided into four groups based on microdissection, in situ hybridization, in situ sequencing, and in situ capturing.71 Details of associated technologies are summarized in this review.72 Besides, Nitzan et al. took advantage of scRNA-seq data and developed a novel framework termed novoSpaRc to reconstruct spatial location of gene expression in several kinds of tissues without prior spatial information.73 Recently, spatial RNA sequencing (spRNA-seq) has emerged as a technology used for identifying RNAs in its native tissue location. By means of spRNA-seq, visualization and quantification of RNA in situ can be realized, which further helps researchers explore different types of cells and their interactions in tissues and organs.
When ST technologies are used in tissue sections, the entire information of gene expression in this section will be presented spatially, and one can select the areas of interest for further study. For example, one study used ST technologies to explore inflammatory signatures in rheumatoid arthritis (RA) biopsies and spondyloarthritis (SpA) biopsies. Based on the results from ST, researchers chose areas of mononuclear cells for deep analysis, identifying the interactions between T cells and B cells in RA, and tissue repair function in SpA.74 Besides selection of interested area, ST was also used in tumor microenvironment research to identify types of immune cell which were spatially close to cancer cells.75 It is more possible for immune cells in the vicinity of cancer cells to regulate the fate of tumor than these peripheral immune cells, and thus it is of great value to recognize the identity of immune cells surrounding cancer cells.
Although ST is able to provide spatial information of RNAs in tissue section, the non-single resolution limited its application. Therefore, combination of scRNA-seq and spRNA-seq is adopted to address this issue. Infiltrating immune cells in the tumor microenvironment vary in different kinds of tumors, which can be regarded as biomarkers and exert specific impacts on oncogenesis, tumor progression and metastasis. Cell-to-cell interaction plays a key role in understanding the underlying mechanisms, which including cancer cells to immune cells and the way back. One study integrated scRNA-seq and ST to uncover the communications between fibroblasts and macrophages in colorectal cancer (CRC).76 Researchers first profiled 54,103 cells from CRC tissue to identify the cellular composition and the enriched types of immune cells, finding the ratios of FAP+ fibroblasts and SPP1+ macrophages in samples were high. Then, the close position between the two kinds of immune cells was validated by ST and fluorescent in situ hybridization, indicating the possible cellular interactions. Deeper analysis of data from scRNA-seq and ST demonstrated that the interactions might be controlled by TGF-β, chemerin, and interleukin-1. In addition, clinical information showed that patients with FAP and SPP1 expression reacted poorly to anti-PD-L1 therapy, implicating the interaction of FAP+ fibroblasts and SPP1+ macrophages might be a potential target for CRC therapy. This study sets a good example for us to consider how to combine scRNA-seq and ST for the tumor microenvironmental investigation. The strategy includes four parts: (1) Identification of subpopulation of immune cells, (2) demonstration of the spatial information of these identified immune cells, (3) analysis of gene expression in immune cells with spatially close position, and (4) combination of clinical information to verify the result of the third step and offer potential target cues for therapy. In addition, integration methods of data from scRNA-seq and ST have also been benchmarked,77 offering the best choice of methods when it comes to map the spatial distribution of RNA transcripts or deconvolute the cell type of spots.
Microbe-immune interaction
The surface of the body, including the gastrointestinal, urogenital, and respiratory tracts as well as the skin, harbors enormous commensal microbiota. The most widely studied is gut microbiota considered to be advantageous to the host by facilitating digestion and eliminating pathogens.78 Gut microbiota has been proved to function in shaping both innate and adaptive immunity through influencing metabolites encompassing bile acids, short-chain fatty acids, and tryptophan metabolites.79 As a physical barrier between gut microbiota and immune cells, intestinal epithelial cells are also involved in the microbe-immune interactions by responding to both microbial and immune signals.80
Microbiomics aims to investigate all microbiota of a given community by meta-omics approaches involving metagenomics, metatranscriptomics, metaproteomics, metabolomics, and 16S RNA gene sequencing. Metagenomic technologies have been used globally to explore the compositional and functional alterations in microbiome between healthy controls and patients with cancer,81 autoimmune disease,82 and functional gastrointestinal disorder.83 In addition, microbial taxonomic shifts and prognosis related microbial taxa were also confirmed by metagenomics in patients with hepatobiliary cancers before and during anti-PD-1 therapy.84 However, metagenomics can only provide the information of composition and gene functions. Therefore, combining meta-omics of microbiota with multi-omics of host is pivotal in investigating the complicated interaction between host and microbiota. To probe into the effect of gut microbiota on immune response in humans who had taken vaccination, one study85 performed 16S rRNA gene sequencing to verify the changes in gut microbial composition in antibiotics-treated and control group. Then researchers profiled the innate and adaptive immune responses after subjects in the two groups vaccinated with influenza vaccine, finding antibiotics-driven difference of gut microbiota profoundly influenced the immune responses. Further metabolomic analysis revealed microbiota-related variation in bile acid metabolism which promoted inflammation.
Many immune-related diseases are the result of comprehensive pathogenic factors including microbial impact. Full-scale characterization of these complex diseases is a challenge for every researcher in this field but a combination of microbiomics and other omics can solve this tough problem. Take inflammatory bowel diseases (IBDs), for example. IBD including Crohn’ disease (CD) and ulcerative colitis (UC) are characterized by various degree of inflammation in gastrointestinal tract and colon.86 The pathogenesis of IBD is complex and heterogeneity of IBD patients can be reflected at genetic, molecular, microbial, and immunological levels. To get a comprehensive view of IBD, one study87 integrated metagenomics, metatranscriptomics, proteomics, and metabolomics, revealing alterations of microbial composition and transcription, metabolites pools, and antibodies in host serum during active period of IBD. Subsequent integrative analysis of multi-omics data demonstrated that microbial, biochemical, and host factors were all indispensable in IBD. As mentioned above, microbial bioactive compounds have been identified to be disturbed in IBD, but numerous compounds have not been well-characterized. Integration of metagenomics, metatranscriptomics and metaproteomics can help in deep profiling of unknown but functional microbial proteins and selection of prioritized ones which might play pivotal roles in host-microbiota interaction in IBD.88 In addition, metagenomics was applied to predict the IBD patients’ responses to anti-integrin therapy and anti-cytokine therapy (anti-TNF or anti-IL12/23) through definition of microbial characteristics. Moreover, by integrative analysis of paired metagenomics, metabolomics and metaproteomics data, potential mechanism of how gut microbiota influenced the therapy response was presented in this study.86
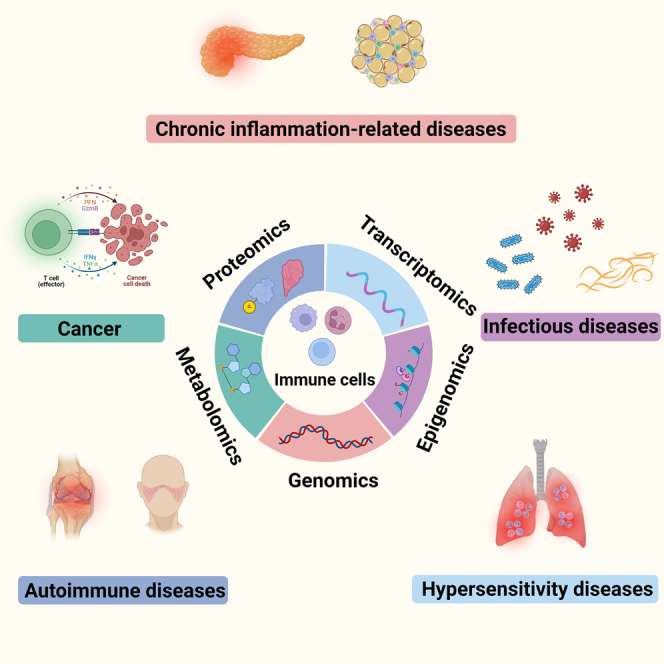
Clinical applications of immune cell-based multi-omics analysis
These multi-omics technologies serve as powerful tools applied not only in the laboratory to explore the functions and networks of immune cells using model organisms, but also in translational and clinical settings. Application of these techniques to clinical specimens such as primary human hematopoietic cells, primary human T cells and synovial tissues allows us to understand the pathophysiologic mechanisms underlying immune disorders to design potentially novel treatment strategies for related diseases. A considerable amount of work has been done to gain insights into human diseases from an immunological perspective, including allergic diseases, infection, cancer, autoimmune and chronic inflammation-related diseases89 (Figure 3).
Figure 3.

Summary of clinical applications of immune cell-based multi-omics analysis in the context of diseases
Immune cells are involved in wide spectrum of diseases, and specific diseases have their own distinct immunological hallmarks. Hypersensitivity diseases can occur in various tissues and organs including the nasal mucosa, trachea and skin, and functions of mast cells are significant in hypersensitivity disease. Infection can be caused by invasion of pathogens, and each pathogen is related to specific immune cells. Different types of tumors, even different subtypes, have featured composition of infiltrating immune cells and immune checkpoint molecules in TME. Dysfunction of immune cells is the key pathogenic factor in autoimmune disease and chronic inflammation-related disease. Thus, analyzing immune cells involved in these diseases through multi-omics approaches can help find new diagnosis biomarker, therapeutic targets and monitoring index in the clinical setting.
Hypersensitivity diseases
Hypersensitivity, also known as allergy, refers to increased immune reactivity to an antigen to which the body has been previously exposed. Hypersensitivity could be categorized into four types based on the time course of reaction and the underlying cellular and humoral mediators. The first three types are primarily immunoglobulin-mediated and consist of type I (immediate), type II (antibody-dependent), and type III (immune-complex mediated), whereas the fourth is predominantly lymphoid cell-mediated (delayed-type, type IV).90 Many locations in the human body can be involved in hypersensitivity diseases, including the nasal mucosa, trachea and skin.
Food allergy is a common type among allergic diseases, and contains several immune-mediated responses to food antigens, for instance, IgE-mediated food allergy. The diagnosis and risk management of food allergies remain challenging because of the diversity of clinical phenotypes and lack of effective biomarkers. Multi-omics approaches to identify and characterize biomarkers of peanut allergy (a relevant disease model) include development of Ig-E-signatures, basophil reactivity profiling, allergenomics, mass spectrometric resolution of peripheral allergen tracing, deep immune phenotyping, gene expression as well as gene methylation profiles, and these tools have been reviewed previously.91 Mast cells are immune cells of the myeloid lineage and normally exist in the connective tissue throughout the body to form a barrier to pathogen invasion, including parasites. Activation and degranulation of mast cells participate not only in the physiological conditions but also in the pathophysiology of many allergic diseases including asthma, anaphylaxis and gastrointestinal disorders.92 To understand the basic principles of mast cell functions, Plum et al. isolated primary human connective tissue mast cells93 and performed proteomics by quantitative mass spectrometry, defining these mast cells as a special cluster. Moreover, proteomics analyses identified 16 cell surface receptors which were highly expressed by these mast cells. This discovery indicating that mast cell depletion might serve as an effective treatment for patients who suffer from allergic diseases.
Infectious diseases
Invasion of pathogens including bacterial, fungal, viral, and parasitic organisms causes infectious diseases. The immune system protects against infectious diseases through prevention of pathogen entry, neutralization of pathogens, and development of immune memory responses; nevertheless, overexuberant and unchecked immune responses can contribute to ailments through the development of autoimmune processes and damage to normal tissues. In fact, such overactive immune responses have been implicated in the lethality of SARS-CoV2 infection. Thus, it is of great value to decipher the complicated processes by which immune responses are activated, maintained, and extinguished. A large number of studies have been carried out to dissect the immune response to SARS-CoV-2 using the potent multi-omics approaches since the outbreak of COVID-19. The composition and activation states of peripheral blood mononuclear cells in patients with COVID-19 will change as this disease develops. Using single-cell multi-omics, patients with severe infections displayed an expanded CD8+T cell repertoire and an increased ratio of CD8+ effector T cells to effector memory T cells, whereas circulating follicular helper T cells were more abundant in patients with mild disease.64 In addition, elevated apoptosis signatures in plasmacytoid dendritic cells and attenuated inflammation with increased fatty acid metabolism in NK cells was observed in severe COVID-19 infections.65 Sera and urine from patients with COVID-19 also have been used for detecting the proteins and metabolites related to inflammatory and immune responses through the combination of proteomic and metabolic analysis.94 These findings characterized the immune responses in COVID-19 patients, which were integrated with clinical data to provide information for the diagnosis, treatment and prognostic assessment of COVID-19 infections.
Apart from SARS-CoV-2, there are many other pathogens that can enter the human body and be pathogenic. HIV-infected patients with undetectable viral load in the absence of treatment with highly active antiretroviral therapy are known as “elite controllers”. Transcriptome sequencing of CD4+T cell samples from two elite controllers, two HIV positive infected patients, and two healthy controls allowed for construction of a co-expression networks based on the relationships among differentially expressed transcripts, offering clues into processes underlying efficient control of viral infection.95 In addition, Guo et al. adopted quantitative proteomics and metabolic analysis and demonstrated that NLRX1 could enhance OXPHOS and glycolysis in HIV-1-infected CD4+T cells, which fueled viral replication. Based on this mechanism, they utilized metformin to inhibit the mitochondrial respiratory chain complex-I and identified its utility in suppressing HIV-1 replication in the infected CD4+T cell.56 In addition to HIV, cytomegalovirus (CMV) is an influential factor in shaping host immune system. Specifically, memory T cells induced by CMV may have long-term impact on the immune system of host and can cross-react with some unrelated pathogens. Zhang et al. decoded T cell responses stimulated by CMV and showed that CMV induced CD4+T cells to experience distinct differentiation trajectories and to acquire cytotoxic phenotypes, with potential negative implications for transplant recipients.96
Extracellular vesicles (EVs), serve as communication mediators among cells and traffic various cargoes including proteins, lipids and RNAs. Deep profiling of EVs through multi-omics methods may assist in in-depth understanding of the mechanisms behind cell-to-cell communication, giving novel insights into potential targets for therapy. For example, EVs from C.neoformans-infected macrophages enhanced the inflammatory responses by inducing the differentiation of naive macrophages and activating immune-related pathways and potentiated stronger fungicidal effects. In addition, further multi-omics approaches identified unique proteins and lipids in EVs, which could be potential target for selecting or designing drugs for C.neoformans-related diseases.70
Cancer
With respect to cancer, the tumor microenvironment (TME) and tumor mutation burden (TMB) are important determinants of predicting clinical responses to immunotherapy. In recent years, the results from clinical studies exploring the composition of infiltrating immune cells and immune checkpoint molecules have shown that different types of tumors, even different subtypes of tumors, have their own special composition of infiltrating immune cells and immune checkpoint molecules in TME, which could serve as new therapy targets and immune signatures in the treatment of cancers. The benefits of PARP inhibition in patients with BRCA-associated triple-negative breast cancer (TNBC) indeed exist, but are transitory. By using high dimensional single cell profiling of human TNBC, Mehta et al. revealed that macrophages are predominant in the TNBC microenvironment. Through multi-omics profiling, they further uncovered that PARP inhibitors could drive not only anti-tumor macrophages but also pro-tumor macrophages. Combined PARP inhibitor and macrophage targeting therapy may prolong the effectiveness of PARP inhibition via maintaining a durable reprogramming of the TME.97
Similar studies investigating the infiltrating immune cells in TME also include ovarian cancer,98 lung cancer99 and colon cancer.100 Apart from single-cell based methods, a case from seventy-eight-year-old man with chronic lymphocytic leukemia showed the potential power of human T cell loss-of-function screens.101 Although CAR T cells are typically polyclonal with random integration of lentiviral constructs throughout the genome, in this case, 94% of CAR T cells were monoclonal at the peak of the patient’s responses, and derived from T cells with a lentiviral integration disrupting TET2 genes. Furthermore, this patient was later found to have a pre-existing hypomorphic mutation in the second allele of TET2. Therefore, by combining the two discoveries together, disruption of TET2 was proved to be an essential factor in the CAR T cell therapy for this patient. This case can also be a reminder for us to think about the previously described SLICE tool,5 which could potentially be applied for searching more systematically for genetic perturbations. CRISPR screening of CAR T cells is also applied for finding critical genes to enhance CAR therapeutic efficacy against glioblastoma.102
Autoimmune diseases
In normal conditions, the immune system functions to protect the body from invading pathogens, to maintain homeostasis, and to patrol for necrotic and neoplastic cells. However, immune dysfunction can occur because of a number of factors, leading to aberrant immune activation against normal host tissues and deleterious autoimmune effects. Examples of common autoimmune diseases include multiple sclerosis, systemic lupus erythematosus (SLE), rheumatoid arthritis (RA) and type1 diabetes.
Multi-omics approaches can be applied in investigating the pathogenesis, development and progression of autoimmune diseases, and can inform development of novel biomarkers and therapies.103 One excellent review summarized studies using multi-omics to explore the pathogenesis of systemic sclerosis and offered information for disease identification, therapeutic targets and potential biomarkers.104 In addition, multi-omics can be used for drug responses monitoring in autoimmune diseases.105 Dysfunction of immune cells plays an indispensable role in the pathophysiology of autoimmune diseases. Thus, decoding mechanisms underlying immune cell impairment and the relationships between impaired immune cells and autoimmune diseases will be highly informative. RA is associated with a highly polygenic genetic architecture including 120 identified RA susceptibility loci,106 with the main loci located in non-coding elements related to CD4+T cell pathways. To get a better understanding of the RA-specific signatures in CD4+T cells, Haet al. took advantage of multi-omics technology by profiling genome-wide variants, gene expression, and DNA methylation in CD4+T cells from 82 patients with RA and 40 healthy controls. They found that a great number of differentially expressed genes in CD4+T cells from patients with RA are affected by methylation changes which were driven by RA heritability-explaining variants.107 Another study revealed that the mechanism underlying the transcriptional abnormalities in CD4+T cells from children with active juvenile idiopathic arthritis (JIA) is associated with the alterations in CD4+T cells chromatin using ATAC-seq. Further combining known JIA genetic risk loci with data from multi-omics containing CTCF binding, ATAC-seq, and RNA-seq confirmed candidate target genes which had genetic influences on the transcriptional abnormalities.108 Multi-omics approaches have also been used to identify the target genes at JIA risk loci in neutrophils.109
Chronic inflammation-related diseases
Owing to the ubiquity and myriad functions of immune cells, an increasing number of pathophysiologic processes have been linked to immune dysregulation, with chronic inflammation acting as an important contributor to common diseases, such as obesity and type-2 diabetes.110 Immune cells residing in adipose tissue and in the gastrointestinal tract and liver can promote systemic inflammation and contribute to obesity, type-2 diabetes and other inflammation related diseases.111 Multi-omics approaches have demonstrated great potential in exploring the functions of immune cells in these diseases. White adipose tissue (WAT) consists of lipid-filled adipocytes and nonadipocyte cell populations, which plays an essential role in energy storage and endocrine regulation.112 One study113 employed single cell RNA-sequencing technology and analyzed human cells derived from healthy lean and obese patients, revealing 8 novel immune cell types. Distinct subsets of T cells, macrophages, dendritic cells and lymphoid cells were observed to increase in obese WAT. Besides, this study further analyzed single cell ligand-receptor pairs and upstream regulators and found obesity-related inflammatory interactomes and signalomes accumulated in WAT-resident immune cells. Apart from adipose tissues, single cell analysis was also applied to explore the immune components of pancreas in obesity and type-2 diabetes patients.114,115 To explore the relationship between type-2 diabetes and obesity, Eldakhakhny et al. analyzed immune signaling genes collected in public databases, identifying 10 immune-related pathways in type-2 diabetes and 4 immune-related pathways in obesity. There are two commonly altered pathway including hematopoietic cell lineage pathway and leukocyte transendothelial migration pathway in both diseases. Besides, five overexpressed genes were also found to be involved in TCR signaling including PTPN6, CD247, FOS, SPNS1 and PIK3R5.116
Conclusions and perspectives
The immune system is divided into immune organs, immune cells, and immune molecules. Immune cells are at the hub consisting of immune organ and secreting immune molecules. Therefore, investigations into normal and abnormal biological activities occurring in immune cells can help us better understand the functional and dysfunctional states of the immune system. Like other kinds of cells in our body, biological activities of immune cells are regulated by gene, the control center in every cell. Gene performs its function through a two-stage process including transcription and translation. In this continuous and dynamic process, each stage and its associated products are precisely and orderly controlled to maintain immune cell homeostasis and ensure proper functions. From DNA to RNA, and finally, the protein, these products are corresponded to genomics, transcriptomics, and proteomics whereas epigenomics, 3D genomics, translatomics and PTMs are involved in the regulation of these products. We used these multi-omics approaches to dissect this complicated process, trying to find enriched or disturbed genes, transcripts, proteins, and the connections between them (by combination of these multi-omics approaches), which can also be seen as pathways. All the pathways construct the cellular networks that essentially control growth, development, and function of immune cells. Immune cells in the body are classified into various types encompassing innate immunocytes (like neutrophils, basophils, eosinophils, mast cells, macrophages, dendritic cells and NK cells) and adaptive immunocytes (like T cells and B cells), which all live together with other types of cells or organisms like cancer cells and microbiota. Traditional bulk sequencing in each omics layer can only provide the average information of targeted tissue without detailed information of each type of cells. Single cell sequencing can solve this trouble by profiling heterogeneity, identifying rare or novel immune cell types, and defining immune cell states as well as other types of cells in targeted tissue. In this big family, communications and interplays between cell members and other organisms also have a profound impact on immune cell. On the basis of clear identification, combination of single-cell multi-omics data from different kinds of cells help to explore the intercellular interaction (Figure 4A) (including immune cells to immune cells and immune cells to other types of cells like cancer cells). Besides, the immunocyte-microbiota interplay can also be investigated by combination of multi-omics technologies and microbiomics.
Figure 4.

The general illustration of how omics technologies are integrated to tackle complex biological problems in immunology
(A). Complex biological activities of immune cells: various immune cells have their own states including active and inactive, functional and dysfunctional. Different states can be explained by different trajectories of transcription and translation. Regulative processes and products in intracellular activities can be measured by single cell multi-omics tools and associated data can be acquired in each immune cell. Combination of each single cell multi-omics data can reveal the interactions and communications in the same type immune cells with different states, different types of immune cells, and other types of cells like cancer cells and cancer-related fibroblasts. Microbe-immune interplay can also be decoded by integration of microbiomics and multi-omics data. In addition, degree of communication may depend on the relative distance between cells.
(B) Integration of multi-omics approaches and associated data to understand the complexities of the human immune landscape.
However, neither bulk sequencing nor single cell sequencing is able to provide the critical spatial information, which impedes further investigation into immune cell function and pathological changes. Spatial RNA sequencing is a state-of-the-art multi-omics technique which can offer information of not only the whole transcriptome data but also associated spatial distribution. Tumor tissue consists of diverse immune cell types frequently communicating with cancer cells in highly structured ways spatially and temporally. Using spatial RNA sequencing to dissect the tumor tissues can facilitate identification of specific type of immune cells whose spatial position is closer to cancer cells. It gives us an opportunity to select the target because closer spatial position represents more communications and interactions.
Using omics technologies to characterize molecules associated with different biological activities in immune cells sheds light on the complex mechanisms underlying our sophisticated immune system. Each omics technology offers us relevant data resources which can be iteratively reanalyzed and yield new insights long after being collected because of new conceptual advances or analytic methods. However, large-scale omics data generation and development of analytical methodology are virtually impossible for each research group to master individually, and thus coordinated integrative efforts will be vital for exploring the essence behind immune system and unveiling its many mysteries.117
Compared with omics technologies confined to a single modality, the combination of multi-omics approaches that integrate data sources from multiple modalities (Figure 4B) can give us deeper insights into the flow of information in immune cells. Apart from simply combining data from omics technologies, the development of analytic techniques to integrate multiple molecular datasets is a challenge for researchers aiming to uncover the truth of immunology by multi-omics technologies. Canonical correlation analysis (CCA) could serve as a powerful approach to integrate multi-dimensional biological data and has been successfully employed in several studies.62,118,119
For medical researchers, the ultimate goal is to serve patients more than just find these products and construct networks. Thus, how to apply multi-omics technology to tackle clinical problems is equally important. On the basis of strong experimental results from studies using multi-omics technologies in model organisms, the field would benefit from additional investigation using relevant human immune cells to determine the broader generalizability and translational capacity of these findings. The human peripheral blood mononuclear cells (PBMCs) are a crucial resource for investigation of immune responses, including the activation, proliferation, and differentiation of immune cells as well as metabolic reprogramming events that occur during this process.120 Apart from PBMCs, urine, feces, and exhaled air can also be tested by multi-omics approaches to gain metabolic information on immune cell population. Moreover, great technological progress has been in the omics field. One recently published study firstly found the existence of purinosomes which comprise nine enzymes and channel the pathway intermediates to synthesize purine nucleotides by using metabolomics and single-cell in situ three-dimensional chemical imaging. The appearance of RNA in situ conformation sequencing (RIC-seq) technology can help in globally profile the intra- and intermolecular RNA-RNA interactions,121 which can not only recapitulate known RNA secondary structures and tertiary interactions but also construct RNA-RNA three-dimensional interaction maps, opening a new avenue for exploration of immune cell functions. Besides, as an expansion of CyTOF, imaging mass cytometry (IMC) has been used for exploring the immune landscape in melanoma microenvironment.122,123 These emerging omics technologies, while inevitably endowing us with more potent analytical weapons to dissect immune functions, must be carefully selected based on the specific purpose of studies.
Acknowledgments
This work was supported by grants from National Natural Science Foundation of China (grant #82273255, #81822034, #81821002, and #81773119 to S.Z), National Key Research and Development Program of China (grant #2022YFA1106600, #2017YFA0106800 and #2018YFA0109200 to S.Z),Sichuan Science-Technology Project (grant ##22ZYZYTS0070 and #2019YFH0144 to S.Z), and Direct Scientific Research Grants from West China Second Hospital, Sichuan University (grant #KS021 and #K1907 to S.Z).
Author contributions
X.W., R.G., and S.Z. completed the conceptual design. X.W., D.F., and Y.Y. performed the literature review. X.W. and D.F. created the table and figures. All authors contributed to writing and final editing of the manuscript.
Declaration of interests
No conflict of interest is declared.
Contributor Information
Ryan C. Gimple, Email: ryangimple@gmail.com.
Shengtao Zhou, Email: shengtaozhou@scu.edu.cn.
References
- 1.Johnson Chavarria E.M. A primer of human genetics. Yale J. Biol. Med. 2016;89:603. [Google Scholar]
- 2.Civelek M., Lusis A.J. Systems genetics approaches to understand complex traits. Nat. Rev. Genet. 2014;15:34–48. doi: 10.1038/nrg3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tan H., Yang K., Li Y., Shaw T.I., Wang Y., Blanco D.B., Wang X., Cho J.H., Wang H., Rankin S., et al. Integrative proteomics and phosphoproteomics profiling reveals dynamic signaling networks and bioenergetics pathways underlying T cell activation. Immunity. 2017;46:488–503. doi: 10.1016/j.immuni.2017.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bakker O.B., Aguirre-Gamboa R., Sanna S., Oosting M., Smeekens S.P., Jaeger M., Zorro M., Võsa U., Withoff S., Netea-Maier R.T., et al. Integration of multi-omics data and deep phenotyping enables prediction of cytokine responses. Nat. Immunol. 2018;19:776–786. doi: 10.1038/s41590-018-0121-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shifrut E., Carnevale J., Tobin V., Roth T.L., Woo J.M., Bui C.T., Li P.J., Diolaiti M.E., Ashworth A., Marson A. Genome-wide CRISPR screens in primary human T cells reveal key regulators of immune function. Cell. 2018;175:1958–1971.e15. doi: 10.1016/j.cell.2018.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schelker M., Feau S., Du J., Ranu N., Klipp E., MacBeath G., Schoeberl B., Raue A. Estimation of immune cell content in tumour tissue using single-cell RNA-seq data. Nat. Commun. 2017;8:2032. doi: 10.1038/s41467-017-02289-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lenardo M., Lo B., Lucas C.L. Genomics of immune diseases and new therapies. Annu. Rev. Immunol. 2016;34:121–149. doi: 10.1146/annurev-immunol-041015-055620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pipkin M.E., Monticelli S. Genomics and the immune system. Immunology. 2008;124:23–32. doi: 10.1111/j.1365-2567.2008.02818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sciacchitano S., De Vitis C., D'Ascanio M., Giovagnoli S., De Dominicis C., Laghi A., Anibaldi P., Petrucca A., Salerno G., Santino I., et al. Gene signature and immune cell profiling by high-dimensional, single-cell analysis in COVID-19 patients, presenting Low T3 syndrome and coexistent hematological malignancies. J. Transl. Med. 2021;19:139. doi: 10.1186/s12967-021-02805-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schubert K., Karkossa I., Schor J., Engelmann B., Steinheuer L.M., Bruns T., Rolle-Kampczyk U., Hackermüller J., von Bergen M. A multi-omics analysis of mucosal-associated-invariant T cells reveals key drivers of distinct modes of activation. Front. Immunol. 2021;12:616967. doi: 10.3389/fimmu.2021.616967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pan D., Kobayashi A., Jiang P., Ferrari de Andrade L., Tay R.E., Luoma A.M., Tsoucas D., Qiu X., Lim K., Rao P., et al. A major chromatin regulator determines resistance of tumor cells to T cell–mediated killing. Science. 2018;359:770–775. doi: 10.1126/science.aao1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel S.J., Sanjana N.E., Kishton R.J., Eidizadeh A., Vodnala S.K., Cam M., Gartner J.J., Jia L., Steinberg S.M., Yamamoto T.N., et al. Identification of essential genes for cancer immunotherapy. Nature. 2017;548:537–542. doi: 10.1038/nature23477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shang W., Jiang Y., Boettcher M., Ding K., Mollenauer M., Liu Z., Wen X., Liu C., Hao P., Zhao S., et al. Genome-wide CRISPR screen identifies FAM49B as a key regulator of actin dynamics and T cell activation. Proc. Natl. Acad. Sci. USA. 2018;115 doi: 10.1073/pnas.1801340115. E4051–e4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi J., Wu X., Wang Z., Li F., Meng Y., Moore R.M., Cui J., Xue C., Croce K.R., Yurdagul A., Jr., et al. A genome-wide CRISPR screen identifies WDFY3 as a regulator of macrophage efferocytosis. Nat. Commun. 2022;13:7929. doi: 10.1038/s41467-022-35604-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piunti A., Shilatifard A. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science. 2016;352:aad9780. doi: 10.1126/science.aad9780. [DOI] [PubMed] [Google Scholar]
- 16.Multhaup M.L., Seldin M.M., Jaffe A.E., Lei X., Kirchner H., Mondal P., Li Y., Rodriguez V., Drong A., Hussain M., et al. Mouse-human experimental epigenetic analysis unmasks dietary targets and genetic liability for diabetic phenotypes. Cell Metab. 2015;21:138–149. doi: 10.1016/j.cmet.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raghuraman S., Donkin I., Versteyhe S., Barrès R., Simar D. The emerging role of epigenetics in inflammation and immunometabolism. Trends Endocrinol.Metab. 2016;27:782–795. doi: 10.1016/j.tem.2016.06.008. [DOI] [PubMed] [Google Scholar]
- 18.Nestor C.E., Lentini A., Hägg Nilsson C., Gawel D.R., Gustafsson M., Mattson L., Wang H., Rundquist O., Meehan R.R., Klocke B., et al. 5-Hydroxymethylcytosine remodeling precedes lineage specification during sifferentiation of human CD4(+) T Cells. Cell Rep. 2016;16:559–570. doi: 10.1016/j.celrep.2016.05.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hasin Y., Seldin M., Lusis A. Multi-omics approaches to disease. Genome Biol. 2017;18:83. doi: 10.1186/s13059-017-1215-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X., Wang Y., Lu H., Li J., Yan X., Xiao M., Hao J., Alekseev A., Khong H., Chen T., et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature. 2019;567:525–529. doi: 10.1038/s41586-019-0979-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pritykin Y., van der Veeken J., Pine A.R., Zhong Y., Sahin M., Mazutis L., Pe'er D., Rudensky A.Y., Leslie C.S. A unified atlas of CD8 T cell dysfunctional states in cancer and infection. Mol. Cell. 2021;81:2477–2493.e10. doi: 10.1016/j.molcel.2021.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beagrie R.A., Scialdone A., Schueler M., Kraemer D.C.A., Chotalia M., Xie S.Q., Barbieri M., de Santiago I., Lavitas L.M., Branco M.R., et al. Complex multi-enhancer contacts captured by genome architecture mapping. Nature. 2017;543:519–524. doi: 10.1038/nature21411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonev B., Cavalli G. Organization and function of the 3D genome. Nat. Rev. Genet. 2016;17:661–678. doi: 10.1038/nrg.2016.112. [DOI] [PubMed] [Google Scholar]
- 24.Li Y., Tao T., Du L., Zhu X. Three-dimensional genome: developmental technologies and applications in precision medicine. J. Hum. Genet. 2020;65:497–511. doi: 10.1038/s10038-020-0737-7. [DOI] [PubMed] [Google Scholar]
- 25.Johanson T.M., Chan W.F., Keenan C.R., Allan R.S. Genome organization in immune cells: unique challenges. Nat. Rev. Immunol. 2019;19:448–456. doi: 10.1038/s41577-019-0155-2. [DOI] [PubMed] [Google Scholar]
- 26.Belton J.M., McCord R.P., Gibcus J.H., Naumova N., Zhan Y., Dekker J. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods. 2012;58:268–276. doi: 10.1016/j.ymeth.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X., Luo O.J., Wang P., Zheng M., Wang D., Piecuch E., Zhu J.J., Tian S.Z., Tang Z., Li G., Ruan Y. Long-read ChIA-PET for base-pair-resolution mapping of haplotype-specific chromatin interactions. Nat. Protoc. 2017;12:899–915. doi: 10.1038/nprot.2017.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johanson T.M., Lun A.T.L., Coughlan H.D., Tan T., Smyth G.K., Nutt S.L., Allan R.S. Transcription-factor-mediated supervision of global genome architecture maintains B cell identity. Nat. Immunol. 2018;19:1257–1264. doi: 10.1038/s41590-018-0234-8. [DOI] [PubMed] [Google Scholar]
- 29.Bossen C., Murre C.S., Chang A.N., Mansson R., Rodewald H.R., Murre C. The chromatin remodeler Brg1 activates enhancer repertoires to establish B cell identity and modulate cell growth. Nat. Immunol. 2015;16:775–784. doi: 10.1038/ni.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu G., Cui K., Fang D., Hirose S., Wang X., Wangsa D., Jin W., Ried T., Liu P., Zhu J., et al. Transformation of accessible chromatin and 3D nucleome underlies lineage commitment of early T Cells. Immunity. 2018;48:227–242.e8. doi: 10.1016/j.immuni.2018.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bunting K.L., Soong T.D., Singh R., Jiang Y., Béguelin W., Poloway D.W., Swed B.L., Hatzi K., Reisacher W., Teater M., et al. Multi-tiered reorganization of the genome during B cell affinity maturation anchored by a germinal center-specific locus control region. Immunity. 2016;45:497–512. doi: 10.1016/j.immuni.2016.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robson M.I., de Las Heras J.I., Czapiewski R., Sivakumar A., Kerr A.R.W., Schirmer E.C. Constrained release of lamina-associated enhancers and genes from the nuclear envelope during T-cell activation facilitates their association in chromosome compartments. Genome Res. 2017;27:1126–1138. doi: 10.1101/gr.212308.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kristensen A.R., Gsponer J., Foster L.J. Protein synthesis rate is the predominant regulator of protein expression during differentiation. Mol. Syst. Biol. 2013;9:689. doi: 10.1038/msb.2013.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu R., Markowetz F., Unwin R.D., Leek J.T., Airoldi E.M., MacArthur B.D., Lachmann A., Rozov R., Ma'ayan A., Boyer L.A., et al. Systems-level dynamic analyses of fate change in murine embryonic stem cells. Nature. 2009;462:358–362. doi: 10.1038/nature08575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 36.Zhao J., Qin B., Nikolay R., Spahn C.M.T., Zhang G. Translatomics: the global view of translation. Int. J. Mol. Sci. 2019;20:212. doi: 10.3390/ijms20010212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ingolia N.T., Hussmann J.A., Weissman J.S. Ribosome profiling: global views of translation. Cold Spring Harb.Perspect.Biol. 2019;11:a032698. doi: 10.1101/cshperspect.a032698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thoreen C.C., Chantranupong L., Keys H.R., Wang T., Gray N.S., Sabatini D.M. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012;485:109–113. doi: 10.1038/nature11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hsieh A.C., Liu Y., Edlind M.P., Ingolia N.T., Janes M.R., Sher A., Shi E.Y., Stumpf C.R., Christensen C., Bonham M.J., et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nguyen M.A., Hoang H.D., Rasheed A., Duchez A.C., Wyatt H., Cottee M.L., Graber T.E., Susser L., Robichaud S., Berber İ., et al. miR-223 exerts translational control of proatherogenic genes in macrophages. Circ. Res. 2022;131:42–58. doi: 10.1161/circresaha.121.319120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Myers D.R., Norlin E., Vercoulen Y., Roose J.P. Active tonic mTORC1 signals shape baseline translation in naive T Cells. Cell Rep. 2019;27:1858–1874.e6. doi: 10.1016/j.celrep.2019.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moore M.J., Blachere N.E., Fak J.J., Park C.Y., Sawicka K., Parveen S., Zucker-Scharff I., Moltedo B., Rudensky A.Y., Darnell R.B. ZFP36 RNA-binding proteins restrain T cell activation and anti-viral immunity. Elife. 2018;7:e33057. doi: 10.7554/eLife.33057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manfrini N., Ricciardi S., Alfieri R., Ventura G., Calamita P., Favalli A., Biffo S. Ribosome profiling unveils translational regulation of metabolic enzymes in primary CD4(+) Th1 cells. Dev. Comp. Immunol. 2020;109:103697. doi: 10.1016/j.dci.2020.103697. [DOI] [PubMed] [Google Scholar]
- 44.Salloum D., Singh K., Davidson N.R., Cao L., Kuo D., Sanghvi V.R., Jiang M., Lafoz M.T., Viale A., Ratsch G., Wendel H.G. A rapid translational immune response program in CD8 memory T lymphocytes. J. Immunol. 2022;209:1189–1199. doi: 10.4049/jimmunol.2100537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beck H.C., Nielsen E.C., Matthiesen R., Jensen L.H., Sehested M., Finn P., Grauslund M., Hansen A.M., Jensen O.N. Quantitative proteomic analysis of post-translational modifications of human histones. Mol. Cell. Proteomics. 2006;5:1314–1325. doi: 10.1074/mcp.M600007-MCP200. [DOI] [PubMed] [Google Scholar]
- 46.Ross S.H., Rollings C., Anderson K.E., Hawkins P.T., Stephens L.R., Cantrell D.A. Phosphoproteomic analyses of interleukin 2 signaling reveal integrated JAK kinase-dependent and -independent networks in CD8(+) T Cells. Immunity. 2016;45:685–700. doi: 10.1016/j.immuni.2016.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dybas J.M., O'Leary C.E., Ding H., Spruce L.A., Seeholzer S.H., Oliver P.M. Integrative proteomics reveals an increase in non-degradative ubiquitylation in activated CD4(+) T cells. Nat. Immunol. 2019;20:747–755. doi: 10.1038/s41590-019-0381-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hubel P., Urban C., Bergant V., Schneider W.M., Knauer B., Stukalov A., Scaturro P., Mann A., Brunotte L., Hoffmann H.H., et al. A protein-interaction network of interferon-stimulated genes extends the innate immune system landscape. Nat. Immunol. 2019;20:493–502. doi: 10.1038/s41590-019-0323-3. [DOI] [PubMed] [Google Scholar]
- 49.Shen B., Yi X., Sun Y., Bi X., Du J., Zhang C., Quan S., Zhang F., Sun R., Qian L., et al. Proteomic and metabolomic characterization of COVID-19 patient Sera. Cell. 2020;182:59–72.e15. doi: 10.1016/j.cell.2020.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maghsoudi Z., Nguyen H., Tavakkoli A., Nguyen T. A comprehensive survey of the approaches for pathway analysis using multi-omics data integration. Brief. Bioinformatics. 2022;23:bbac435. doi: 10.1093/bib/bbac435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Makowski L., Chaib M., Rathmell J.C. Immunometabolism: from basic mechanisms to translation. Immunol. Rev. 2020;295:5–14. doi: 10.1111/imr.12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stathopoulou C., Nikoleri D., Bertsias G. Immunometabolism: an overview and therapeutic prospects in autoimmune diseases. Immunotherapy. 2019;11:813–829. doi: 10.2217/imt-2019-0002. [DOI] [PubMed] [Google Scholar]
- 53.Wiedemann G.M., Santosa E.K., Grassmann S., Sheppard S., Le Luduec J.B., Adams N.M., Dang C., Hsu K.C., Sun J.C., Lau C.M. Deconvoluting global cytokine signaling networks in natural killer cells. Nat. Immunol. 2021;22:627–638. doi: 10.1038/s41590-021-00909-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mishra B., Athar M., Mukhtar M.S. Transcriptional circuitry atlas of genetic diverse unstimulated murine and human macrophages define disparity in population-wide innate immunity. Sci. Rep. 2021;11:7373. doi: 10.1038/s41598-021-86742-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Henriksson J., Chen X., Gomes T., Ullah U., Meyer K.B., Miragaia R., Duddy G., Pramanik J., Yusa K., Lahesmaa R., Teichmann S.A. Genome-wide CRISPR Screens in T helper cells reveal pervasive crosstalk between activation and differentiation. Cell. 2019;176:882–896.e18. doi: 10.1016/j.cell.2018.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guo H., Wang Q., Ghneim K., Wang L., Rampanelli E., Holley-Guthrie E., Cheng L., Garrido C., Margolis D.M., Eller L.A., et al. Multi-omics analyses reveal that HIV-1 alters CD4(+) T cell immunometabolism to fuel virus replication. Nat. Immunol. 2021;22:423–433. doi: 10.1038/s41590-021-00898-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen H., Ye F., Guo G. Revolutionizing immunology with single-cell RNA sequencing. Cell. Mol. Immunol. 2019;16:242–249. doi: 10.1038/s41423-019-0214-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.See P., Dutertre C.-A., Chen J., Günther P., McGovern N., Irac S.E., Gunawan M., Beyer M., Händler K., Duan K., et al. Mapping the human DC lineage through the integration of high-dimensional techniques. Science. 2017;356:eaag3009. doi: 10.1126/science.aag3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Janela B., Patel A.A., Lau M.C., Goh C.C., Msallam R., Kong W.T., Fehlings M., Hubert S., Lum J., Simoni Y., et al. A subset of type I conventional dendritic cells controls cutaneous bacterial infections through VEGFalpha-mediated recruitment of neutrophils. Immunity. 2019;50:1069–1083.e8. doi: 10.1016/j.immuni.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 60.Chakarov S., Lim H.Y., Tan L., Lim S.Y., See P., Lum J., Zhang X.M., Foo S., Nakamizo S., Duan K., et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science. 2019;363:eaau0964. doi: 10.1126/science.aau0964. [DOI] [PubMed] [Google Scholar]
- 61.Papalexi E., Satija R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 2018;18:35–45. doi: 10.1038/nri.2017.76. [DOI] [PubMed] [Google Scholar]
- 62.Zhang F., Wei K., Slowikowski K., Fonseka C.Y., Rao D.A., Kelly S., Goodman S.M., Tabechian D., Hughes L.B., Salomon-Escoto K., et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat. Immunol. 2019;20:928–942. doi: 10.1038/s41590-019-0378-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bengsch B., Ohtani T., Khan O., Setty M., Manne S., O'Brien S., Gherardini P.F., Herati R.S., Huang A.C., Chang K.M., et al. Epigenomic-guided mass cytometry profiling reveals disease-specific features of exhausted CD8 T Cells. Immunity. 2018;48:1029–1045.e5. doi: 10.1016/j.immuni.2018.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stephenson E., Reynolds G., Botting R.A., Calero-Nieto F.J., Morgan M.D., Tuong Z.K., Bach K., Sungnak W., Worlock K.B., Yoshida M., et al. Single-cell multi-omics analysis of the immune response in COVID-19. Nat. Med. 2021;27:904–916. doi: 10.1038/s41591-021-01329-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu C., Martins A.J., Lau W.W., Rachmaninoff N., Chen J., Imberti L., Mostaghimi D., Fink D.L., Burbelo P.D., Dobbs K., et al. Time-resolved systems immunology reveals a late juncture linked to fatal COVID-19. Cell. 2021;184:1836–1857.e22. doi: 10.1016/j.cell.2021.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Camiolo M.J., Zhou X., Oriss T.B., Yan Q., Gorry M., Horne W., Trudeau J.B., Scholl K., Chen W., Kolls J.K., et al. High-dimensional profiling clusters asthma severity by lymphoid and non-lymphoid status. Cell Rep. 2021;35:108974. doi: 10.1016/j.celrep.2021.108974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marzagalli M., Ebelt N.D., Manuel E.R. Unraveling the crosstalk between melanoma and immune cells in the tumor microenvironment. Semin. Cancer Biol. 2019;59:236–250. doi: 10.1016/j.semcancer.2019.08.002. [DOI] [PubMed] [Google Scholar]
- 68.Mao X., Xu J., Wang W., Liang C., Hua J., Liu J., Zhang B., Meng Q., Yu X., Shi S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol. Cancer. 2021;20:131. doi: 10.1186/s12943-021-01428-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rieckmann J.C., Geiger R., Hornburg D., Wolf T., Kveler K., Jarrossay D., Sallusto F., Shen-Orr S.S., Lanzavecchia A., Mann M., Meissner F. Social network architecture of human immune cells unveiled by quantitative proteomics. Nat. Immunol. 2017;18:583–593. doi: 10.1038/ni.3693. [DOI] [PubMed] [Google Scholar]
- 70.Zhang L., Zhang K., Li H., Coelho C., de Souza Gonçalves D., Fu M.S., Li X., Nakayasu E.S., Kim Y.M., Liao W., et al. Cryptococcus neoformans-infected macrophages release proinflammatory extracellular vesicles: insight into their components by multi-omics. mBio. 2021;12:00279-21. doi: 10.1128/mBio.00279-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang L., Chen D., Song D., Liu X., Zhang Y., Xu X., Wang X. Clinical and translational values of spatial transcriptomics. Signal Transduct. Target.Ther. 2022;7:111. doi: 10.1038/s41392-022-00960-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Crosetto N., Bienko M., van Oudenaarden A. Spatially resolved transcriptomics and beyond. Nat. Rev. Genet. 2015;16:57–66. doi: 10.1038/nrg3832. [DOI] [PubMed] [Google Scholar]
- 73.Nitzan M., Karaiskos N., Friedman N., Rajewsky N. Gene expression cartography. Nature. 2019;576:132–137. doi: 10.1038/s41586-019-1773-3. [DOI] [PubMed] [Google Scholar]
- 74.Carlberg K., Korotkova M., Larsson L., Catrina A.I., Ståhl P.L., Malmström V. Exploring inflammatory signatures in arthritic joint biopsies with spatial transcriptomics. Sci. Rep. 2019;9:18975. doi: 10.1038/s41598-019-55441-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Van de Velde L.A., Allen E.K., Crawford J.C., Wilson T.L., Guy C.S., Russier M., Zeitler L., Bahrami A., Finkelstein D., Pelletier S., et al. Neuroblastoma formation requires unconventional CD4 T cells and arginase-1-dependent myeloid cells. Cancer Res. 2021;81:5047–5059. doi: 10.1158/0008-5472.Can-21-0691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Qi J., Sun H., Zhang Y., Wang Z., Xun Z., Li Z., Ding X., Bao R., Hong L., Jia W., et al. Single-cell and spatial analysis reveal interaction of FAP(+) fibroblasts and SPP1(+) macrophages in colorectal cancer. Nat. Commun. 2022;13:1742. doi: 10.1038/s41467-022-29366-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li B., Zhang W., Guo C., Xu H., Li L., Fang M., Hu Y., Zhang X., Yao X., Tang M., et al. Benchmarking spatial and single-cell transcriptomics integration methods for transcript distribution prediction and cell type deconvolution. Nat. Methods. 2022;19:662–670. doi: 10.1038/s41592-022-01480-9. [DOI] [PubMed] [Google Scholar]
- 78.Kayama H., Okumura R., Takeda K. Interaction between the microbiota, epithelia, and immune cells in the intestine. Annu. Rev. Immunol. 2020;38:23–48. doi: 10.1146/annurev-immunol-070119-115104. [DOI] [PubMed] [Google Scholar]
- 79.Ansaldo E., Farley T.K., Belkaid Y. Control of immunity by the microbiota. Annu. Rev. Immunol. 2021;39:449–479. doi: 10.1146/annurev-immunol-093019-112348. [DOI] [PubMed] [Google Scholar]
- 80.Soderholm A.T., Pedicord V.A. Intestinal epithelial cells: at the interface of the microbiota and mucosal immunity. Immunology. 2019;158:267–280. doi: 10.1111/imm.13117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Behary J., Amorim N., Jiang X.T., Raposo A., Gong L., McGovern E., Ibrahim R., Chu F., Stephens C., Jebeili H., et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat. Commun. 2021;12:187. doi: 10.1038/s41467-020-20422-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chu Y., Sun S., Huang Y., Gao Q., Xie X., Wang P., Li J., Liang L., He X., Jiang Y., et al. Metagenomic analysis revealed the potential role of gut microbiome in gout. NPJ Biofilms Microbiomes. 2021;7:66. doi: 10.1038/s41522-021-00235-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mars R.A.T., Yang Y., Ward T., Houtti M., Priya S., Lekatz H.R., Tang X., Sun Z., Kalari K.R., Korem T., et al. Longitudinal multi-omics reveals subset-specific mechanisms underlying irritable bowel syndrome. Cell. 2020;182:1460–1473.e17. doi: 10.1016/j.cell.2020.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mao J., Wang D., Long J., Yang X., Lin J., Song Y., Xie F., Xun Z., Wang Y., Wang Y., et al. Gut microbiome is associated with the clinical response to anti-PD-1 based immunotherapy in hepatobiliary cancers. J. Immunother. Cancer. 2021;9:e003334. doi: 10.1136/jitc-2021-003334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hagan T., Cortese M., Rouphael N., Boudreau C., Linde C., Maddur M.S., Das J., Wang H., Guthmiller J., Zheng N.Y., et al. Antibiotics-driven gut microbiome perturbation alters immunity to vaccines in humans. Cell. 2019;178:1313–1328.e13. doi: 10.1016/j.cell.2019.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee J.W.J., Plichta D., Hogstrom L., Borren N.Z., Lau H., Gregory S.M., Tan W., Khalili H., Clish C., Vlamakis H., et al. Multi-omics reveal microbial determinants impacting responses to biologic therapies in inflammatory bowel disease. Cell Host Microbe. 2021;29:1294–1304.e4. doi: 10.1016/j.chom.2021.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lloyd-Price J., Arze C., Ananthakrishnan A.N., Schirmer M., Avila-Pacheco J., Poon T.W., Andrews E., Ajami N.J., Bonham K.S., Brislawn C.J., et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569:655–662. doi: 10.1038/s41586-019-1237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang Y., Bhosle A., Bae S., McIver L.J., Pishchany G., Accorsi E.K., Thompson K.N., Arze C., Wang Y., Subramanian A., et al. Discovery of bioactive microbial gene products in inflammatory bowel disease. Nature. 2022;606:754–760. doi: 10.1038/s41586-022-04648-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nieman D.C., Lila M.A., Gillitt N.D. Immunometabolism: a multi-omics approach to Interpreting the Influence of exercise and diet on the immune system. Annu. Rev. Food Sci. Technol. 2019;10:341–363. doi: 10.1146/annurev-food-032818-121316. [DOI] [PubMed] [Google Scholar]
- 90.Momtazmanesh S., Rezaei N. In: Encyclopedia of Infection and Immunity. Rezaei N., editor. Elsevier; 2022. Hypersensitivity; pp. 243–258. [DOI] [Google Scholar]
- 91.Czolk R., Klueber J., Sørensen M., Wilmes P., Codreanu-Morel F., Skov P.S., Hilger C., Bindslev-Jensen C., Ollert M., Kuehn A. IgE-mediated peanut allergy: current and novel predictive biomarkers for clinical phenotypes uiing multi-omics approaches. Front. Immunol. 2020;11:594350. doi: 10.3389/fimmu.2020.594350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Krystel-Whittemore M., Dileepan K.N., Wood J.G. Mast cell: a multi-functional master cell. Front. Immunol. 2015;6:620. doi: 10.3389/fimmu.2015.00620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Plum T., Wang X., Rettel M., Krijgsveld J., Feyerabend T.B., Rodewald H.-R. Human mast cell proteome reveals unique lineage, putative functions, and structural basis for cell ablation. Immunity. 2020;52:404–416.e5. doi: 10.1016/j.immuni.2020.01.012. [DOI] [PubMed] [Google Scholar]
- 94.Tian W., Zhang N., Jin R., Feng Y., Wang S., Gao S., Gao R., Wu G., Tian D., Tan W., et al. Immune suppression in the early stage of COVID-19 disease. Nat. Commun. 2020;11:5859. doi: 10.1038/s41467-020-19706-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen C., Lu X., Wu N. RNA sequencing of CD4 T-cells reveals the relationships between lncRNA-mRNA co-expression in elite controller vs. HIV-positive infected patients. PeerJ. 2020;8:e8911. doi: 10.7717/peerj.8911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang W., Morris A.B., Peek E.V., Karadkhele G., Robertson J.M., Kissick H.T., Larsen C.P. CMV status drives distinct trajectories of CD4+ T cell differentiation. Front. Immunol. 2021;12:620386. doi: 10.3389/fimmu.2021.620386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mehta A.K., Cheney E.M., Hartl C.A., Pantelidou C., Oliwa M., Castrillon J.A., Lin J.R., Hurst K.E., de Oliveira Taveira M., Johnson N.T., et al. Targeting immunosuppressive macrophages overcomes PARP inhibitor resistance in BRCA1-associated triple-negative breast cancer. Nat. Cancer. 2021;2:66–82. doi: 10.1038/s43018-020-00148-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhu J., Zhou Q., Pan M., Zhou C. Multi-omics analysis of the prognosis and therapeutic significance of circadian clock in ovarian cancer. Gene. 2021;788:145644. doi: 10.1016/j.gene.2021.145644. [DOI] [PubMed] [Google Scholar]
- 99.Wu J., Xu C., Guan X., Ni D., Yang X., Yang Z., Wang M. Comprehensive analysis of tumor microenvironment and identification of an immune signature to predict the prognosis and immunotherapeutic response in lung squamous cell carcinoma. Ann. Transl. Med. 2021;9:569. doi: 10.21037/atm-21-463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yi T., Zhang Y., Ng D.M., Xi Y., Ye M., Cen L., Li J., Fan X., Li Y., Hu S., et al. Regulatory network analysis of mutated genes based on multi-omics data reveals the exclusive features in tumor immune microenvironment between left-sided and right-sided colon cancer. Front. Oncol. 2021;11:685515. doi: 10.3389/fonc.2021.685515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fraietta J.A., Nobles C.L., Sammons M.A., Lundh S., Carty S.A., Reich T.J., Cogdill A.P., Morrissette J.J.D., DeNizio J.E., Reddy S., et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. 2018;558:307–312. doi: 10.1038/s41586-018-0178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang D., Prager B.C., Gimple R.C., Aguilar B., Alizadeh D., Tang H., Lv D., Starr R., Brito A., Wu Q., et al. CRISPR screening of CAR T Cells and cancer stem cells reveals critical dependencies for cell-based therapies. Cancer Discov. 2021;11:1192–1211. doi: 10.1158/2159-8290.Cd-20-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Alcazar O., Hernandez L.F., Nakayasu E.S., Nicora C.D., Ansong C., Muehlbauer M.J., Bain J.R., Myer C.J., Bhattacharya S.K., Buchwald P., Abdulreda M.H. Parallel multi-omics in high-risk subjects for the identification of integrated biomarker signatures of type 1 diabetes. Biomolecules. 2021;11:383. doi: 10.3390/biom11030383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zuo X., Zhang L., Luo H., Li Y., Zhu H. Systematic approach to understanding the pathogenesis of systemic sclerosis. Clin.Genet. 2017;92:365–371. doi: 10.1111/cge.12946. [DOI] [PubMed] [Google Scholar]
- 105.Tasaki S., Suzuki K., Kassai Y., Takeshita M., Murota A., Kondo Y., Ando T., Nakayama Y., Okuzono Y., Takiguchi M., et al. Multi-omics monitoring of drug response in rheumatoid arthritis in pursuit of molecular remission. Nat. Commun. 2018;9:2755. doi: 10.1038/s41467-018-05044-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kwon Y.C., Lim J., Bang S.Y., Ha E., Hwang M.Y., Yoon K., Choe J.Y., Yoo D.H., Lee S.S., Lee J., et al. Genome-wide association study in a Korean population identifies six novel susceptibility loci for rheumatoid arthritis. Ann. Rheum. Dis. 2020;79:1438–1445. doi: 10.1136/annrheumdis-2020-217663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ha E., Bang S.Y., Lim J., Yun J.H., Kim J.M., Bae J.B., Lee H.S., Kim B.J., Kim K., Bae S.C. Genetic variants shape rheumatoid arthritis-specific transcriptomic features in CD4(+) T cells through differential DNA methylation, explaining a substantial proportion of heritability. Ann. Rheum. Dis. 2021;80:876–883. doi: 10.1136/annrheumdis-2020-219152. [DOI] [PubMed] [Google Scholar]
- 108.Tarbell E., Jiang K., Hennon T.R., Holmes L., Williams S., Fu Y., Gaffney P.M., Liu T., Jarvis J.N. CD4+ T cells from children with active juvenile idiopathic arthritis show altered chromatin features associated with transcriptional abnormalities. Sci. Rep. 2021;11:4011. doi: 10.1038/s41598-021-82989-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li J., Yuan X., March M.E., Yao X., Sun Y., Chang X., Hakonarson H., Xia Q., Meng X., Li J. Identification of target genes at juvenile idiopathic arthritis GWAS loci in human neutrophils. Front. Genet. 2019;10:181. doi: 10.3389/fgene.2019.00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lu J., Zhao J., Meng H., Zhang X. Adipose tissue-resident immune cells in obesity and type 2 diabetes. Front. Immunol. 2019;10:1173. doi: 10.3389/fimmu.2019.01173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Luck H., Khan S., Kim J.H., Copeland J.K., Revelo X.S., Tsai S., Chakraborty M., Cheng K., Tao Chan Y., Nøhr M.K., et al. Gut-associated IgA(+) immune cells regulate obesity-related insulin resistance. Nat. Commun. 2019;10:3650. doi: 10.1038/s41467-019-11370-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Church C.D., Berry R., Rodeheffer M.S. In: Methods in Enzymology. Macdougald O.A., editor. Academic Press; 2014. Chapter three - isolation and study of adipocyte precursors; pp. 31–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hildreth A.D., Ma F., Wong Y.Y., Sun R., Pellegrini M., O'Sullivan T.E. Single-cell sequencing of human white adipose tissue identifies new cell states in health and obesity. Nat. Immunol. 2021;22:639–653. doi: 10.1038/s41590-021-00922-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ying W., Lee Y.S., Dong Y., Seidman J.S., Yang M., Isaac R., Seo J.B., Yang B.H., Wollam J., Riopel M., et al. Expansion of islet-resident macrophages leads to inflammation affecting β Cell proliferation and function in obesity. Cell Metab. 2019;29:457–474.e5. doi: 10.1016/j.cmet.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wu M., Lee M.Y.Y., Bahl V., Traum D., Schug J., Kusmartseva I., Atkinson M.A., Fan G., HPAP Consortium. Kaestner K.H. Single-cell analysis of the human pancreas in type 2 diabetes using multi-spectral imaging mass cytometry. Cell Rep. 2021;37:109919. doi: 10.1016/j.celrep.2021.109919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Eldakhakhny B.M., Al Sadoun H., Choudhry H., Mobashir M. In-Silico study of immune system associated genes in case of type-2 diabetes with insulin action and resistance, and/or obesity. Front. Endocrinol. 2021;12:641888. doi: 10.3389/fendo.2021.641888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Subramanian I., Verma S., Kumar S., Jere A., Anamika K. Multi-omics data integration, interpretation, and its application. Bioinform.Biol. Insights. 2020;14 doi: 10.1177/1177932219899051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Witten D.M., Tibshirani R., Hastie T. A penalized matrix decomposition, with applications to sparse principal components and canonical correlation analysis. Biostatistics. 2009;10:515–534. doi: 10.1093/biostatistics/kxp008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Parkhomenko E., Tritchler D., Beyene J. Sparse canonical correlation analysis with application to genomic data integration. Stat. Appl. Genet. Mol. Biol. 2009;8 doi: 10.2202/1544-6115.1406. Article 1. [DOI] [PubMed] [Google Scholar]
- 120.Sen P., Kemppainen E., Orešič M. Perspectives on systems modeling of human peripheral blood mononuclear cells. Front. Mol. Biosci. 2017;4:96. doi: 10.3389/fmolb.2017.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cai Z., Cao C., Ji L., Ye R., Wang D., Xia C., Wang S., Du Z., Hu N., Yu X., et al. RIC-seq for global in situ profiling of RNA-RNA spatial interactions. Nature. 2020;582:432–437. doi: 10.1038/s41586-020-2249-1. [DOI] [PubMed] [Google Scholar]
- 122.Hoch T., Schulz D., Eling N., Gómez J.M., Levesque M.P., Bodenmiller B. Multiplexed imaging mass cytometry of the chemokine milieus in melanoma characterizes features of the response to immunotherapy. Sci. Immunol. 2022;7:eabk1692. doi: 10.1126/sciimmunol.abk1692. [DOI] [PubMed] [Google Scholar]
- 123.Moldoveanu D., Ramsay L., Lajoie M., Anderson-Trocme L., Lingrand M., Berry D., Perus L.J.M., Wei Y., Moraes C., Alkallas R., et al. Spatially mapping the immune landscape of melanoma using imaging mass cytometry. Sci. Immunol. 2022;7:eabi5072. doi: 10.1126/sciimmunol.abi5072. [DOI] [PubMed] [Google Scholar]