

Abstract
Motivation
Conservation is broadly used to identify biologically important (epi)genomic regions. In the case of tumor growth, preferential conservation of DNA methylation can be used to identify areas of particular functional importance to the tumor. However, reliable assessment of methylation conservation based on multiple tissue samples per patient requires the decomposition of methylation variation at multiple levels.
Results
We developed a Bayesian hierarchical model that allows for variance decomposition of methylation on three levels: between-patient normal tissue variation, between-patient tumor-effect variation and within-patient tumor variation. We then defined a model-based conservation score to identify loci of reduced within-tumor methylation variation relative to between-patient variation. We fit the model to multi-sample methylation array data from 21 colorectal cancer (CRC) patients using a Monte Carlo Markov Chain algorithm (Stan). Sets of genes implicated in CRC tumorigenesis exhibited preferential conservation, demonstrating the model’s ability to identify functionally relevant genes based on methylation conservation. A pathway analysis of preferentially conserved genes implicated several CRC relevant pathways and pathways related to neoantigen presentation and immune evasion. Our findings suggest that preferential methylation conservation may be used to identify novel gene targets that are not consistently mutated in CRC. The flexible structure makes the model amenable to the analysis of more complex multi-sample data structures.
Availability and implementation
The data underlying this article are available in the NCBI GEO Database, under accession code GSE166212. The R analysis code is available at https://github.com/kevin-murgas/DNAmethylation-hierarchicalmodel.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
DNA methylation provides cells with a mechanism to regulate gene transcription, whereby methylation at CpG dinucleotides generally induces gene repression (Dawson and Kouzarides, 2012; Feinberg and Tycko, 2004). Genome-wide patterns of DNA methylation have been demonstrated to reliably distinguish cancerous from normal tissue, indicating a significant role of methylation in tumorigenesis (Irizarry et al., 2009; Lam et al., 2016; Lewin et al., 2007; Mitchell et al., 2014). However, the exact mechanisms by which these epigenetic alterations drive neoplastic transformation remain poorly characterized (Dawson and Kouzarides, 2012).
The recent development of methylation microarrays that assess genome-wide profiles of CpG methylation have enabled epigenomic studies in the context of a broad range of genetic disease including cancer (Bibikova et al., 2011; Mansell et al., 2019; Pidsley et al., 2016). To date, the utility of these assays has primarily been established through differential methylation studies that seek to identify CpG sites with consistent changes in methylation between normal and diseased tissues (Frigola et al., 2005; Irizarry et al., 2009; Mitchell et al., 2014). With this primary focus on differential methylation, little attention has so far been paid to the concept of epigenomic conservation. Indeed, analogously to DNA sequence conservation, methylation conservation can identify genomic regions of particular importance during cancer initiation (Hansen et al., 2011; Hvitfeldt et al., 2020; Naccarati et al., 2007; Ryser et al., 2018; Sottoriva et al., 2015; Teschendorff and Widschwendter, 2012; Yates et al., 2017).
The basic concept of epigenomic conservation implies that as genomes replicate, functionally unimportant sites exhibit larger drift due to random replication errors compared with important functional regions, where changes in methylation status have a detrimental impact on fitness leading to reduced proliferation and negative or purifying selection (Sottoriva et al., 2015). Therefore, during tumor growth, more variation would be expected to accumulate in non-functional regions relative to functional regions. Experimentally, comparison of genome-wide methylation patterns between multiple samples of the same tumor provides an opportunity to identify regions with lower drift, that is, preferential conservation.
Recent studies have analyzed methylation conservation in normal and cancerous tissues by quantifying variation supported by a multiple-sampling approach (Hvitfeldt et al., 2020; Ryser et al., 2018). Using pairwise distance (PWD) metrics to compare methylation patterns within and between patients, these studies found preferential conservation in promoters and expressed genes. However, the use of PWD is limited because it cannot capture the natural hierarchy of tissues between and within patients, which in turn renders a robust analysis of multiple tissue types from multiple patients difficult. For example, direct PWD comparison of tumor samples between patients does not account for the underlying between-patient variation of normal tissue methylation, and thus cannot directly identify loci that are universally conserved (in both tumor and normal tissue) and those that are preferentially conserved within tumors (Ryser et al., 2018).
To address this methodological gap, we propose here a Bayesian hierarchical modeling approach that enables variance decomposition of methylation across the between-patient and within-patient levels. Based on the different variation components, the model enables identification of preferential conservation at individual CpG sites, individual genes and gene pathways. We illustrate the approach on a cohort of 21 colorectal cancer patients.
2 Materials and methods
2.1 Tumor samples
Tissue samples were collected from 21 patients diagnosed with colorectal tumors, including 5 adenomas and 16 carcinomas (Table 1). We had previously analyzed data from 16 of the 21 tumors in another study (Ryser et al., 2018). From each tumor, two bulk samples (∼0.5 cm3) were obtained from opposite sides of the lesions (n = 42) (Fig. 1A). From 6 of the patients, an additional bulk sample was obtained from adjacent normal colon tissue (n = 6). The samples were obtained at the USC Keck School of Medicine as excess tissues taken in the course of routine clinical care with Institutional Review Board approval.
Table 1.
Tumor sample metadata
| Tumor | Type (stage) | Size (cm) | Matched normal? |
|---|---|---|---|
| A | Adenoma | 5.6 | No |
| K | Adenoma | 6 | No |
| P | Adenoma | 3.5 | No |
| S | Adenoma | 6 | No |
| X | Adenoma | 2.5 | No |
| C | Cancer (3) | 6.4 | Yes |
| D | Cancer (1) | 2 | No |
| E | Cancer (1)a | 6.1 | Yes |
| F | Cancer (1) | 1.8 | No |
| G | Cancer (3) | 3.5 | No |
| H | Cancer (4) | 4 | Yes |
| I | Cancer (4) | 8 | No |
| J | Cancer (3) | 5 | Yes |
| K* | Cancer (1) | 3.5 | Yes |
| M | Cancer (2) | 3 | No |
| N | Cancer (1) | 2.3 | No |
| O | Cancer (3) | 9.5 | No |
| T | Cancer (3) | 5.7 | No |
| U | Cancer (2) | 3.9 | No |
| W | Cancer (1)b | 3.4 | Yes |
| Z | Cancer (3) | 2.7 | No |
Note: Supporting metadata for each colorectal tumor patient, with tumor type (adenoma or carcinoma), stage (carcinoma only), size, and if a matched normal sample was taken.
POLE mutated CRC.
MSI + CRC.
Fig. 1.

DNA methylation model. (A) Schematic of multi-sampling approach used to inform the hierarchical model. Two tumor samples were collected from opposite sides of each colorectal tumor, along with a normal colon tissue sample in some patients. (B) Overview of modeling approach including DNA methylation array data and TCGA-based priors, which were incorporated into the hierarchical model by Stan’s Bayesian MCMC sampling algorithm. Model fits were used to evaluate methylation conservation successively on the CpG-site, gene and pathway levels. (C) Depiction of hierarchical model on a hypothetical set of samples from normal (yellow) and tumor (red, blue) tissues. Fixed effects at the normal (μ) and tumor (μ + ν) levels are estimated along with random effects at the hierarchical levels: normal tissue methylation (σP), normal-tumor differential methylation (σPT), within-tumor methylation drift (σT).
2.2 Methylation assay
DNA methylation in the bulk samples was measured with the Infinium MethylationEPIC 850K BeadChip Microarray (Illumina) on 866 091 CpG sites across the genome. In short, the EPIC array utilizes bisulfite conversion to convert unmethylated cytosines to uracil, where methylation blocks this process. The resulting bisulfite-converted product is hybridized to paired fluorescent red/green probes specific for the DNA sequence around a single CpG site, with the color corresponding to the unmethylated or methylated state. Fluorescence intensities are directly measured, and the ratio of methylation signal to total signal can be used to calculate a methylation reading (beta value) for each CpG site in the array.
2.3 Pre-processing and quality control
EPIC array data were pre-processed in R using the minfi Bioconductor package using minfi’s ‘noob’ pre-processing method and subsequently converted to beta values (Aryee et al., 2014). Density plots were examined to ensure bimodal density as a first inspection of quality control. Calculating percent of probes covered and minfi’s getQC command provided additional quality metrics. Two samples were discarded due to poor data quality (>1% of sites with missing values), and sequencing was repeated for these samples. Beta values were subsequently transformed to M-values via the logit function (Du et al., 2010). To avoid sex-specific methylation bias, CpG sites mapping to sex-chromosomes (19 632 out of 866 091 sites, or 2.2%) were removed from analysis. In addition, sites with likely single-nucleotide polymorphisms (SNPs) were identified via the MethylToSNP package (LaBarre et al., 2019). Using a reliability score cutoff of 0.5, MethylToSNP identified 377 CpG sites suspicious for SNPs that were subsequently removed from analysis.
2.4 Hierarchical model
Considering each CpG site in the array independently, the M-value yij of sample j in patient i was modeled as
| (1) |
where j = 0 for normal tissue samples, j = 1,2 for tumor samples and δij is the Kronecker delta (δij = 1 if i = j, δij = 0 otherwise); that is, the third term is non-zero only for tumor samples. In (1), the M-value of normal tissue samples is determined by a random intercept of μ + αi where αi∼N(0,σP2). The (unmeasured) tumor mean is derived from the patient’s normal tissue by adding a random slope ν + βi where βi∼N(0,σPT2). Finally, the tumor bulk samples are derived from the tumor mean by adding a random effect γij∼N(0,σT2). Residual variation for all samples is captured by an error term εij∼N(0,σE2). In summary, the above mixed-effects model implements a hierarchical variance decomposition of DNA methylation at the following levels: between-patient normal tissue variation (σP), between-patient tumor-effect variation (σPT) and within-patient tumor variation (σT).
2.5 Model fitting
For each CpG site, the hierarchical model (1) was fit to the M-values of patient samples using an adaptive Bayesian Monte Carlo Markov Chain (MCMC) algorithm as implemented in Stan (R package rstan) (Carpenter et al., 2017; Team, 2020). In short, Stan uses the adaptive Hamiltonian Monte Carlo No-U-Turn Sampler (NUTS) to approximate the posterior distributions of the model parameters (Carpenter et al., 2017). For each CpG site, we ran 4 independent chains of length 2000 (including 200 warm-up iterations) with adapt_delta = 0.999. No chain thinning was applied. For each model parameter and the log-posterior-likelihood variable, the following posterior summary statistics were stored: mean, standard error of mean, median, effective sample size and Gelman-Rubin convergence diagnostic R^. After fitting all CpG sites, sites with R^ >1.1 in the log-posterior-likelihood (8740 out of 866 091 sites, or 1.0%) were considered to have insufficient convergence and were removed from analysis.
Model fitting was performed on a high-performance compute cluster, using batch tasks to analyze 10 000 CpG sites per batch (that is, 87 batch tasks to cover all 866 091 sites) and parallel processing on 24–32 CPUs per task to perform the MCMC fitting procedure. Total run time was less than 24 hours.
2.6 Prior distributions
Because of limited sample size, non-informative uniform priors resulted in identifiability issues and insufficient sampling convergence. Therefore, informative prior parameter distributions were constructed based on empirical estimates from an independent dataset from The Cancer Genome Atlas (TCGA) (Supplementary Table S1, Supplementary Fig. S1A). Specifically, we used a bimodal Gaussian mixture prior for μ, a Cauchy prior for ν and gamma priors for σP2, σPT2, σT2 and σE2. Prior distributions were fit to the TCGA data for each parameter and then variance-relaxed by a factor of 3, while maintaining the same mode. The effect of the choice of priors on the modeling results was examined through a series of sensitivity analyses, which demonstrated that the variance-relaxed priors did not lead to over-constrained posterior distributions (Supplementary Fig. S1B).
2.7 CpG conservation score
A key quantity of interest for conservation is the within-patient tumor methylation variance σT2 relative to the between-patient normal methylation variance σP2. Indeed, genome regions that are essential for survival and growth of tumor cells are expected to be more conserved compared to genome regions that are non-coding or of little importance to tumor cell survival. To delineate between loci where conservation is important for both normal and malignant cells and loci where conservation is uniquely important to malignant cells, we introduced a log2-transformed conservation score of the between-patient normal variance normalized by the within-patient tumor variance
| (2) |
For c > 0, tumor variation is lower than normal variation indicating relative gain of conservation of methylation at that site. Conversely for c < 0, tumor variation is greater than normal variation indicating relative loss of conservation at that site. To obtain a single score per site, we used the posterior median of c as a summary statistic.
2.8 Functional region analysis
To assess differences in methylation conservation by genomic regions, each CpG site was assigned to a single CpG-island region (island, shore, shelf, sea) and one or more gene regulatory regions (TSS1500, TSS200, 5′-UTR, 1stExon, Body, ExonBnd, 3′-UTR), based on UCSC annotations in the EPIC array manifest (Illumina). Conservation scores in each CpG-island region were compared using a non-parametric one-way Kruskal–Wallis test, with post hoc Tukey test for comparisons between groups. Because individual sites could belong to multiple regulatory regions, a linear regression model with indicators for each regulatory region was used to model the CpG conservation scores, by which each region was assessed for the statistical significance of its respective slope in the regression model.
2.9 CpG genomic distance analysis
For gene-associated CpG sites, we further assessed how conservation varied with the CpG-site genomic distance (in base pairs, or bp) from the gene start site (Hvitfeldt et al., 2020). Genomic distance was calculated using the CpG genomic position defined in the EPIC array manifest and gene start position defined by Ensembl (Yates et al., 2020). We built on a previous version of this CpG-distance analysis which only used the positive distance from the left-most CpG in each gene (Hvitfeldt et al., 2020). We instead calculated CpG distance based on the Ensembl gene start position accounting for the direction of the gene such that reverse-stranded genes were calculated as having CpG distance increasing in the negative direction. This gene-direction correction avoided CpG sites at the far end of reverse-stranded genes, which would have lower genomic position due to the gene being oriented in the reverse direction, from being considered as close to the gene start site. CpG distance was then binned by 100 bp and CpG conservation scores were averaged within each bin.
2.10 Gene conservation score
To obtain conservation scores at the gene level, the conservation scores of CpG sites belonging to a given gene were averaged. Of note, individual sites may belong to multiple genes. To establish statistical significance of conservation at the gene level, we used an adjusted bootstrapping technique (Hvitfeldt et al., 2020). More precisely, to account for the correlated spatial structure of adjacent CpG sites within genes, the null distribution for bootstrapping was constructed as follows: for a gene containing n CpG sites in the EPIC array, we performed 1000 random draws of consecutive CpG sites (ordered by genomic position) belonging to single genes of size n or larger. Genes with less than 5 array CpG sites and the top 5% of genes with the greatest number of array CpG sites were excluded. Out of 27 373 total genes, 4027 (14.7%) were removed from the final analysis based on these criteria.
2.11 Pathway conservation analysis
All genes above the 95th percentile of the bootstrapped gene conservation score were designated as significantly conserved. These genes were fed into a pathway enrichment analysis using the Reactome pathway analysis online tool (Jassal et al., 2020). In short, Reactome statistically tests for pathway enrichment using an over-representation analysis, producing an enrichment probability based on the hypergeometric distribution which is then corrected for false discovery rate (FDR) via Benjamini–Hochberg procedure (Benjamini and Hochberg, 1995). Pathways with FDR < 0.05 were considered to be significantly conserved.
2.12 Code availability
All data processing, model fitting and analysis code, along with a tutorial for how to run the analysis, is available on GitHub: https://github.com/kevin-murgas/DNAmethylation-hierarchicalmodel.
3 Results
3.1 Overview
We developed a hierarchical Bayesian model to capture DNA methylation variation across different tissue types of multiple patients (Fig. 1C). More precisely, the model estimates the overall mean normal tissue methylation state (μ), the change between normal and tumor methylation (ν) and random effects at three hierarchical levels: between-patient normal tissue variation (σP), between-patient tumor-effect variation (σPT) and within-patient tumor variation (σT). We applied the approach to two bulk samples each from tumors of 21 colorectal cancer patients, along with normal colon tissue samples from 6 of the patients. Samples were profiled using Infinium MethylationEPIC microarray to measure the fraction of methylation at each of 866 091 CpG sites in the genome. Modeling the CpG sites in the EPIC array as independent processes, the hierarchical model was fit to the data using a Bayesian MCMC algorithm via the software Stan under prior distributions informed by an independent dataset from The Cancer Genome Atlas (TCGA).
3.2 Model inference
A model fit example for a select CpG site is shown in Figure 2A, with corresponding posterior distributions shown in Figure 2B. In total, we fit the model to 837 534 CpG sites (Fig. 2C). The posterior medians of the fixed effect intercept (μ) were bimodally distributed across CpG sites, indicating populations of primarily demethylated or methylated sites, respectively. Overall, 313 077 sites (37.4%) were demethylated as indicated by a negative posterior median of μ, while 524 457 sites (62.6%) were methylated as indicated by a positive posterior median of μ. As reflected by the symmetric prior distribution of the fixed effect slope (ν), we did not make a priori assumptions about the direction of methylation change from normal to tumor (i.e. hypomethylation or hypermethylation). Examining the posterior medians of ν, a decrease in methylation (hypomethylation in tumors) was observed in 580 890 (69.4%) of sites as indicated by a negative posterior median of ν, while an increase in methylation (hypermethylation in tumors) was observed in 256 644 (30.6%) of sites as indicated by a positive posterior median of ν. The posterior medians of the hierarchical random effect variation (σ) parameters were unimodally distributed across all CpG sites. On average, these posterior medians were highest at the between-patient tumor-effect level (σPT, mean 0.509) compared to the between-patient normal tissue level (σP, mean 0.250) and within-patient tumor level (σT, mean 0.249).
Fig. 2.
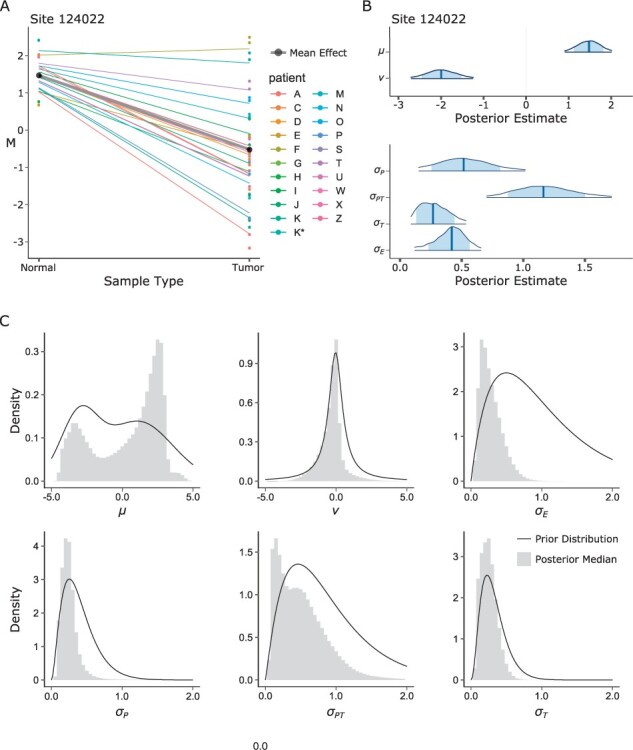
Bayesian hierarchical model fits. (A) Example fit at CpG site 124022. Thick black line is normal mean and tumor slope (fixed effects intercept μ and slope ν), colored lines are model estimates for each patient. (B) Posterior distributions for each of the six main parameters of the model for CpG site 124022. (C) Histograms of posterior median estimates for each parameter; continuous lines are the corresponding prior distributions.
3.3 CpG-site conservation
We sought to determine which methylation sites were fundamentally conserved during tumor growth. To this end, we used the relative conservation score c to compare within-patient methylation variation of tumor tissue samples (σT) relative to the between-patient variation of normal tissue samples (σP). The genome-wide distributions of conservation scores for gene-associated and non-gene-associated CpG sites are show in Figure 3A. On average, the gene-associated CpG sites were more conserved (mean 0.140) compared to non-gene-associated sites (mean −0.182; two-sample t-test: P < 0.001).
Fig. 3.
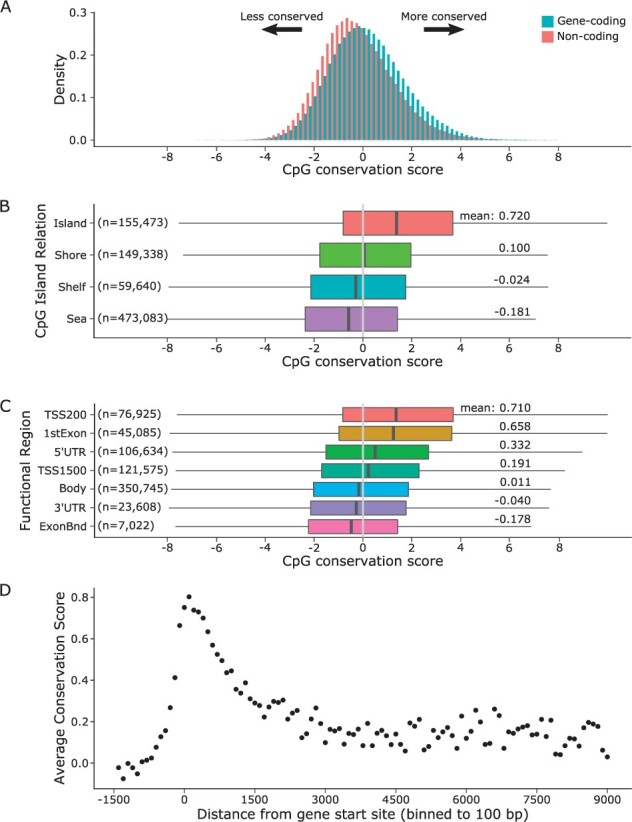
Gene conservation score. (A) CpG conservation score distributions are shown with color designating gene-associated sites. (B) Violin plots of CpG conservation score are shown for each CpG-island region: island, shore, shelf and sea. Below each violin are listed the mean conservation scores. (C) Violin plots of CpG conservation score are shown for each of four functional regions: transcription start site 200 bases (TSS200), first exon (1stExon), 5-prime untranslated region (5′-UTR), transcription start site 1500 bases (TSS1500), gene body (Body), 3-prime untranslated region (3′-UTR), exon boundary (ExonBnd). Below each violin are listed the mean conservation scores. (D) Average CpG conservation score as a function of CpG distance from gene start site (in bp).
3.4 Regional conservation effects
To examine the degree of methylation conservation with respect to genomic regions, we performed three distinct analyses. First, we calculated the average conservation score within distinct genomic regions based on relationship to CpG islands: island, shore, shelf and sea (Fig. 3B). Average conservation scores were significantly different between CpG-island regions (one-way Kruskal–Wallis test: P < 0.001, with post hoc Tukey tests: P < 0.001 for all comparisons). The highest conservation was found on islands and lowest in the sea. Next, we examined parameter averages within each type of gene regulatory region (Fig. 3C), defined by RefGene: transcription start site (TSS) ±200 bp (TSS200), TSS ±1500 bp (TSS1500), 5-prime untranslated region (5′-UTR), first exon (1stExon), gene body (Body), exon boundary (ExonBnd), 3-prime untranslated region (3′-UTR). We found significant differences in methylation conservation for all regulatory regions (multiple linear regression: all regions P < 0.001), with the highest conservation in TSS200 and lowest conservation in ExonBnd. Finally, we examined CpG conservation as a function of the genomic distance of the site from its corresponding gene start position (Fig. 3D), defined by Ensembl gene database (Yates et al., 2020). This distance analysis revealed a peak of conservation in the window of 500 to 2000 bp with respect to the gene start site. The maximum conservation was reached in the 0-100 bp bin with an average conservation score of 0.802, suggesting that this region may be critical for epigenetic regulation.
3.5 Gene conservation
To illustrate the utility of methylation conservation at the gene level, we highlight here three genes with presumed biological relevance in normal colon tissue and colorectal cancer: TUBA1A, TTN and HLA-A (Fig. 4A). TUBA1A is a structural gene important for both normal and cancer cell function (Lewis and Cowan, 1990). Accordingly, we found that the conservation scores of CpG sites in TUBA1A were distributed around 0, indicating that the gene is equally relevant to both normal and cancer tissues (mean score: −0.292; one-sample Wilcoxon rank-sum test: P = 0.2861). TTN is a cardiomyocyte protein, and therefore of no known relevance to either normal or neoplastic colon cells. In agreement with this, there was no evidence of methylation conservation based on the scores for the CpG sites in TTN. In fact, we found statistically significant loss of conservation (mean score: −0.900; P < 0.001). Finally, HLA-A is an antigen-presentation gene with established functional significance in CRC (Menon et al., 2002). Accordingly, we found that the CpG scores of HLA-A fell to the right of 0 (mean score: 3.33; P < 0.001), consistent with preferential conservation in the tumor.
Fig. 4.
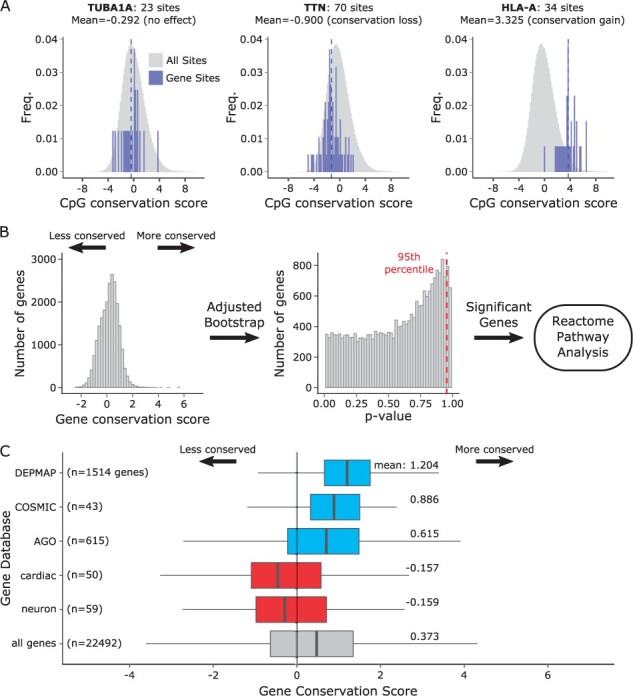
Gene conservation score. (A) Three example genes with known biological significance. Gene CpG conservation scores are shown as blue histograms over the gray distribution of all CpG sites. Blue dashed lines indicate the gene conservation score as the mean of all CpG sites in the gene. TUBA1A: alpha tubulin subunit 1A, expected to be conserved equally in normal and tumor. TTN: titin, expected to not be conserved in tumors. HLA-A: human leukocyte antigen A, expected to be conserved in tumors. (B) Gene conservation scores and adjusted bootstrap P-values. Significant genes were subsequently fed into Reactome pathway analysis. (C) Distributions of gene conservation scores of various gene sets. A positive shift indicating conservation is observed in essential cancer genes from DEPMAP and two colorectal cancer gene databases, COSMIC and Atlas of Genetics in Oncology, shown in blue. A negative shift indicating loss of conservation is observed in two sets of genes irrelevant to normal and cancerous colon tissue, cardiac progenitor differentiation genes (cardiac) and neuron marker genes (neuron), shown in red. Distribution of all genes is shown in gray.
To enable ranking of genes by degree of methylation conservation, we defined a gene conservation score as the average conservation score of all CpG sites in each gene (Fig. 4B). Based on this gene-level score, we examined two established lists of colorectal cancer-associated genes as identified by the COSMIC Cancer Census (43 genes) and the Atlas of Genetics and Cytogenetics in Oncology (AGO; 615 genes), along with a list of essential fitness genes in cancer cell lines (including CRC cell line DLD1) determined by CRISPR knockout as part of the Cancer Dependency Map project (DEPMAP; 1514 genes) (Hart et al., 2015; Huret et al., 2012; Tate et al., 2019). Compared to the mean conservation score of 0.373 across all genes, we found increased conservation in each of the gene sets, COSMIC (mean: 0.886; two-sample t-test: P < 0.001), AGO (mean: 0.614; P < 0.001) and DEPMAP (mean: 1.204; P < 0.001) (Fig. 4C).
As a negative control, we examined genes with no known function in colon which should show no evidence of conservation. We utilized two curated gene sets from Molecular Signatures Database (MSigDB) as examples: cardiac progenitor differentiation genes (cardiac; C2: WikiPathways Cardiac Progenitor Differentiation, 59 genes) and neuron marker genes (neuron; C2: Lein Neuron Markers, 50 genes) (Lein et al., 2007; Liberzon et al., 2011; Martens et al., 2021). In contrast to the CRC-related gene sets above, we found decreased conservation in both cardiac (mean: −0.157; two-sample t-test: P < 0.01) and neuron (mean: −0.159; P < 0.005) gene sets. These findings demonstrate the ability of the conservation score to delineate functionally relevant from functionally irrelevant gene sets.
3.6 Reactome pathway analysis
An adjusted bootstrap technique allowed us to determine statistical significance of gene conservation score (Hvitfeldt et al., 2020). Out of 22 493 genes, 1794 were found to be significantly preferentially conserved within tumors above the 95th percentile of their respective null distribution (Fig. 4B). Taking these significantly conserved genes, we performed a pathway analysis using Reactome, which yielded 33 significant pathways with FDR < 0.05 (Table 2) (Jassal et al., 2020). Additional data including gene hits in each pathway are listed in Supplementary Table S2. In agreement with a previous analysis on a subset of the same cancers, the enriched pathways include 13 immune-related pathways and the previously reported SLIT-ROBO signaling pathway (Beggs et al., 2013; Ryser et al., 2018). Other pathways were generally related to RNA metabolism and protein translation.
Table 2.
Pathway analysis of significantly conserved genes
| Pathway name | Category | FDR |
|---|---|---|
| Antigen presentation: folding, assembly and peptide loading of class I MHC | Immune presentation | 2.79E-14 |
| ER-phagosome pathway | Immune presentation | 2.79E-14 |
| Endosomal/vacuolar pathway | Immune presentation | 2.79E-14 |
| Antigen processing-cross presentation | Immune presentation | 2.79E-14 |
| Interferon signaling | Immune signaling | 2.79E-14 |
| Interferon gamma signaling | Immune signaling | 2.79E-14 |
| Interferon alpha/beta signaling | Immune signaling | 2.79E-14 |
| Class I MHC mediated antigen processing and presentation | Immune presentation | 4.65E-09 |
| Non-sense-mediated decay (NMD) | RNA metabolism | 1.46E-04 |
| NMD enhanced by the exon junction complex (EJC) | RNA metabolism | 1.46E-04 |
| Immunoregulatory interactions between a lymphoid and a non-lymphoid cell | Immune signaling | 2.02E-04 |
| Cytokine signaling in immune system | Immune signaling | 2.37E-04 |
| NMD independent of the EJC | RNA metabolism | 5.45E-04 |
| Eukaryotic translation elongation | Protein translation | 6.21E-04 |
| Metabolism of RNA | RNA metabolism | 9.77E-04 |
| Peptide chain elongation | Protein translation | 0.001 |
| Response of EIF2AK4 (GCN2) to amino acid deficiency | Metabolism; signaling | 0.001 |
| GTP hydrolysis and joining of the 60S ribosomal subunit | Protein translation | 0.001 |
| L13a-mediated translational silencing of Ceruloplasmin expression | Protein translation | 0.001 |
| Eukaryotic translation initiation | Protein translation | 0.001 |
| Cap-dependent translation initiation | Protein translation | 0.001 |
| Regulation of expression of SLITs and ROBOs | Transcription; signaling | 0.001 |
| Eukaryotic translation termination | Protein translation | 0.004 |
| Cellular response to starvation | Signaling | 0.006 |
| Formation of a pool of free 40S subunits | Protein translation | 0.009 |
| Translocation of ZAP-70 to immunological synapse | Immune presentation | 0.014 |
| SRP-dependent cotranslational protein targeting to membrane | Protein translation | 0.015 |
| Phosphorylation of CD3 and TCR zeta chains | Immune presentation | 0.026 |
| PD-1 signaling | Immune signaling | 0.026 |
| Selenocysteine synthesis | Amino acid metabolism | 0.040 |
| Folding of actin by CCT/TriC | Protein folding | 0.042 |
| Signaling by ROBO receptors | Signaling | 0.049 |
| Viral mRNA translation | Translation; disease | 0.049 |
Note: Pathway analysis was performed by taking the list of genes which were significantly conserved at a level of the 95th percentile in the adjusted bootstrap, and feeding this list into Reactome’s online pathway enrichment tool. Resulting pathways were selected based on FDR at a significance level alpha = 0.05, here reporting pathway names, category and FDR values. Detailed results including significant gene names and gene hits in each pathway are included in Supplementary Table S2.
4 Discussion
In this study, we developed a hierarchical mixed-effect model to quantify the conservation of DNA methylation during tumor growth. We successfully applied the model to a dataset comprising multiple colorectal cancer (CRC) patients. Using a Bayesian MCMC method with TCGA-informed priors, we fit the model at each of 866 091 CpG sites across the genome with successful model convergence in over 99% of sites. Using a novel conservation score that compares within-tumor variation of methylation to between-patient normal variation, we identified individual CpG sites, genes and functional gene pathways that were significantly conserved during colorectal tumor growth.
Regional analyses of methylation conservation revealed several trends of increased relative conservation in gene-coding and functional regions. Gene-associated CpG sites exhibited higher methylation conservation, suggesting reduced methylation drift in functional compared to non-functional areas of the genome. Similar to previous findings in normal colon tissue, methylation conservation was higher in CpG-island-associated sites and sites near or upstream of the transcription start site (TSS), indicating increased epigenetic regulation in these critical regulatory regions (Hvitfeldt et al., 2020). Within genes, conservation was most pronounced in a neighborhood of 2000 bp around the gene start site, consistent with previous findings of conserved CpG regions near the start site (Hvitfeldt et al., 2020; Irizarry et al., 2009). The mechanistic role of methylation in these intra-gene regions, however, is complex and not fully understood, and could range from gene silencing to alternative transcription (Irizarry et al., 2009). These results further support our motivation for exploring relative conservation as an indicator of functional importance during tumor growth.
Gene analyses of methylation conservation matched biological expectations in select example genes of known biological relevance for normal colon tissue and its neoplastic transformation. Of particular interest are genes that are conserved within tumors but not between normal colon tissue of different patients, that is genes with a high conservation score. Indeed, such genes can identify biological pathways that are uniquely important during tumorigenesis, but less so in normal colon tissue. When analyzing two established colorectal cancer gene sets (AGO, COSMIC), and a cancer essential fitness gene set (DEPMAP), we found a moderate but statistically significant preferential conservation in all three sets. Conversely, many genes that had a high conservation score in our analyses were not included in the cancer gene sets. Because these sets are based on DNA mutation patterns, our findings suggest that DNA mutation analyses alone may overlook genes that play important functional roles in tumorigenesis. Indeed, some genes may not exhibit a consistent mutational burden (mutations may even be deleterious to their function) but instead contribute to tumorigenesis through epigenetic dysregulation.
Pathway analyses of the most preferentially tumor-conserved genes revealed enrichment of 33 pathways, including 13 immune-related pathways and other pathways that were previously found to be enriched in CRC, such as the SLIT-ROBO signaling pathway (Beggs et al., 2013; Ryser et al., 2018). The strong enrichment of immune pathways, including antigen-presentation pathways, may indicate an important role of epigenetic regulation in facilitating evasion of immune surveillance against tumor neoantigens during colorectal tumorigenesis. Conservation of major histocompatibility complex genes such as HLA-A may indeed be necessary for successful evasion of immune surveillance during tumor growth, thus providing a potential explanation for preferential conservation in this gene (DuPage et al., 2012; Menon et al., 2002). Interestingly, most mismatch-repair-proficient CRCs do not respond to checkpoint blockade immunotherapy, indicating that their neoantigens are not usually recognized by the immune system (Le et al., 2015; Overman et al., 2018).
Previous studies explored conservation in DNA methylation by non-parametric analyses of PWD between the methylation states of multiple samples (Hvitfeldt et al., 2020; Ryser et al., 2018). The PWD approach has several limitations; for instance, it is not suitable for variance decomposition across different between- and within-patient levels, and it relies on (often arbitrary) cutoffs for the definition of relative conservation during tumor growth. The model-based approach introduced here offers several advantages. First, thanks to integration of multiple samples from different patients and tissue types, it enables a natural means for hierarchical variance decomposition. Second, the introduced relative conservation score enables straightforward identification of individual CpG sites, genes and pathways that are preferentially conserved during tumor growth, without invoking pre-specified cut-offs. Third, the Bayesian inference approach facilitates robust quantification of posterior uncertainty in the parameter estimates, including the different variation components.
A limitation of our study is the relatively small sample size, in particular for normal colon samples, which led to practical parameter identifiability issues in the absence of informative prior distributions. We addressed this issue by construction of prior distributions based on an independent dataset from the TCGA database and by performing sensitivity analyses around the informative priors. Furthermore, all studies utilizing methylation arrays are subject to biases from genetic variation (somatic DNA mutations and germline SNPs) proximal to methylation probes (Daca-Roszak et al., 2015; LaBarre et al., 2019). To mitigate the risk of confounding due to such variation, we excluded 377 CpG sites suspicious for SNPs, and we restricted gene-level analyses to genes with 5 CpG sites or more. With a median of 21 CpG sites among the remaining included genes, the impact of residual genetic variation is expected to have a limited effect on conclusions at both the gene and pathway levels. Finally, we note that by modeling CpG sites as independent processes, our approach currently does not account for spatial dependencies between sites.
Our findings warrant further investigation to determine the clinical significance of the genes found to be strongly conserved during CRC growth in this cohort of patients. For instance, accompanying gene expression data (RNA-sequencing, microarray or quantitative-PCR) would reveal if gene expression levels also reflect the same conservation seen in methylation. From the modeling perspective, potential future work could focus on incorporating additional hierarchical levels. For example, tissue sampling at a glandular or cellular level could be used to add additional levels of within-tissue hierarchy. Additional hierarchical levels could also be constructed by grouping cancers by molecular sub-types or including data from a broader range of cancers. Therefore, our model should not be considered definite but rather a starting point for more complex variation hierarchies. Finally, we note that although we focused on methylation conservation for the purposes of this study, the model can also be used to analyze patterns of differential methylation.
In summary, we have developed a statistical framework that enables identification of regions of the genome where methylation status is preferentially conserved during tumor growth, and we illustrated the potential clinical implications of these analyses on a cohort of colorectal cancer patients. Preferentially conserved genes and pathways as identified in our study provide an opportunity for discovery of targeted therapeutic strategies that hone in on genes where epigenetic regulation is subject to evolutionary pressure.
Supplementary Material
Acknowledgement
The authors thank Duke Data+ program, hosted by the Rhodes Information Initiative at Duke University, during which preliminary analyses for the project were conducted.
Funding
This work was supported by the National Science Foundation [DMS 1614838 to M.D.R.].
Conflict of Interest: none declared.
Data availability
The DNA methylation array data have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO) database under the accession code GSE166212. All other data supporting the findings of this study are available within the article and its Supplementary Information.
Contributor Information
Kevin A Murgas, Department of Biomedical Informatics, Stony Brook University School of Medicine, Stony Brook, NY 11794, USA.
Yanlin Ma, Center for Public Health Genomics, University of Virginia, Charlottesville, VA 22903, USA.
Lidea K Shahidi, Department of Electrical and Computer Engineering, Duke University, Durham, NC 27708, USA.
Sayan Mukherjee, Department of Statistical Science, Duke University, Durham, NC 27708, USA; Department of Computer Science, Duke University, Durham, NC 27708, USA; Department of Mathematics, Duke University, Durham, NC 27708, USA; Department of Bioinformatics and Biostatistics, Duke University, Durham, NC 27710, USA.
Andrew S Allen, Department of Bioinformatics and Biostatistics, Duke University, Durham, NC 27710, USA; Duke Center for Statistical Genetics and Genomics, Duke University, Durham, NC 27710, USA.
Darryl Shibata, Department of Pathology, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA.
Marc D Ryser, Department of Mathematics, Duke University, Durham, NC 27708, USA; Department of Population Health Sciences, Duke University Medical Center, Durham, NC 27701, USA.
References
- Aryee M.J. et al. (2014) Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics, 30, 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs A.D. et al. (2013) Whole-genome methylation analysis of benign and malignant colorectal tumours. J. Pathol., 229, 697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B, 57, 289–300. [Google Scholar]
- Bibikova M. et al. (2011) High density DNA methylation array with single CpG site resolution. Genomics, 98, 288–295. [DOI] [PubMed] [Google Scholar]
- Carpenter B. et al. (2017) Stan: a probabilistic programming language. J. Stat. Softw., 76, 1-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daca-Roszak P. et al. (2015) Impact of SNPs on methylation readouts by Illumina Infinium HumanMethylation450 BeadChip Array: implications for comparative population studies. BMC Genomics, 16, 1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson M.A., Kouzarides T. (2012) Cancer epigenetics: from mechanism to therapy. Cell, 150, 12–27. [DOI] [PubMed] [Google Scholar]
- Du P. et al. (2010) Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics, 11, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M. et al. (2012) Expression of tumour-specific antigens underlies cancer immunoediting. Nature, 482, 405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg A.P., Tycko B. (2004) The history of cancer epigenetics. Nat. Rev. Cancer, 4, 143–153. [DOI] [PubMed] [Google Scholar]
- Frigola J. et al. (2005) Differential DNA hypermethylation and hypomethylation signatures in colorectal cancer. Hum. Mol. Genet., 14, 319–326. [DOI] [PubMed] [Google Scholar]
- Hansen K.D. et al. (2011) Increased methylation variation in epigenetic domains across cancer types. Nat. Genet., 43, 768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T. et al. (2015) High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell, 163, 1515–1526. [DOI] [PubMed] [Google Scholar]
- Huret J.-L. et al. (2012) Atlas of genetics and cytogenetics in oncology and haematology in 2013. Nucleic Acids Res., 41, D920–D924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hvitfeldt E. et al. (2020) Epigenetic conservation is a beacon of function: an analysis using Methcon5 Software for studying gene methylation. JCO Clin. Cancer Inf., 4, 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry R.A. et al. (2009) The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet., 41, 178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jassal B. et al. (2020) The reactome pathway knowledgebase. Nucleic Acids Res., 48, D498–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBarre B.A. et al. (2019) MethylToSNP: identifying SNPs in Illumina DNA methylation array data. Epigenet. Chromatin, 12, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam K. et al. (2016) DNA methylation based biomarked in colorectal cancer: a systematic review. Biochim. Biophys. Acta Rev. Cancer, 1866, 106–120. [DOI] [PubMed] [Google Scholar]
- Le D.T. et al. (2015) PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Eng. J. Med., 372, 2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein E.S. et al. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature, 445, 168–176. [DOI] [PubMed] [Google Scholar]
- Lewin J. et al. (2007) Comparative DNA methylation analysis in normal and tumour tissues and in cancer cell lines using differential methylation hybridization. Int. J. Biochem. Cell Biol., 39, 1539–1550. [DOI] [PubMed] [Google Scholar]
- Lewis S.A., Cowan N.J. (1990) Tubulin genes: structure, expression, and regulation. In: Avila J. (ed.) Microtubule Proteins, 1st edn., CRC Press, Boca Raton, pp. 37–66. [Google Scholar]
- Liberzon A. et al. (2011) Molecular signatures database (MSigDB) 3.0. Bioinformatics, 27, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansell G. et al. (2019) Guidance for DNA methylation studies: statistical insights from the Illumina EPIC array. BMC Genomics, 20, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens M. et al. (2021) WikiPathways: connecting communities. Nucleic Acids Res., 49, D613–D621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon A.G. et al. (2002) Down-regulation of HLA-A expression correlates with a better prognosis in colorectal cancer patients. Lab. Invest., 82, 1725–1733. [DOI] [PubMed] [Google Scholar]
- Mitchell S.M. et al. (2014) A panel of genes methylated with high frequency in colorectal cancer. BMC Cancer, 14, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naccarati A. et al. (2007) Sporadic colorectal cancer and individual susceptibility: a review of the association studies investigating the role of DNA repair genetic polymorphisms. Mutat. Res. Rev. Mutat., 635, 118–145. [DOI] [PubMed] [Google Scholar]
- Overman M.J. et al. (2018) Where we stand with immunotherapy in colorectal cancer: deficient mismatch repair, proficient mismatch repair, and toxicity management. Am. Soc. Clin. Oncol. Educ. Book, 38, 239–247. [DOI] [PubMed] [Google Scholar]
- Pidsley R. et al. (2016) Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol., 17, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryser M.D. et al. (2018) Epigenetic heterogeneity in human colorectal tumors reveals preferential conservation and evidence of immune surveillance. Sci. Rep., 8, 17292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottoriva A. et al. (2015) A Big Bang model of human colorectal tumor growth. Nat. Genet., 47, 209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate J.G. et al. (2019) COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res., 47, D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team S.D. (2020) RStan: the R interface to Stan. R package version, 2.19.3. http://mc-stan.org.
- Teschendorff A.E., Widschwendter M. (2012) Differential variability improves the identification of cancer risk markers in DNA methylation studies profiling precursor cancer lesions. Bioinformatics, 28, 1487–1494. [DOI] [PubMed] [Google Scholar]
- Yates A.D. et al. (2020) Ensembl 2020. Nucleic Acids Res., 48, D682–D688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates L.R. et al. (2017) Genomic evolution of breast cancer metastasis and relapse. Cancer Cell, 32, 169–184.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The DNA methylation array data have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO) database under the accession code GSE166212. All other data supporting the findings of this study are available within the article and its Supplementary Information.