

Abstract
Summary
As the amount of 3D chromosomal interaction data continues to increase, storing and accessing such data efficiently becomes paramount. We introduce Pairs, a block-compressed text file format for storing paired genomic coordinates from Hi-C data, and Pairix, an open-source C application to index and query Pairs files. Pairix (also available in Python and R) extends the functionalities of Tabix to paired coordinates data. We have also developed PairsQC, a collapsible HTML quality control report generator for Pairs files.
Availability and implementation
The format specification and source code are available at https://github.com/4dn-dcic/pairix, https://github.com/4dn-dcic/Rpairix and https://github.com/4dn-dcic/pairsqc.
1 Introduction
A Hi-C assay combines chromosome conformation capture with high-throughput sequencing to interrogate genome-wide chromosome organization at high resolution. Modern Hi-C and similar experiments (Krietenstein et al., 2020; Rao et al., 2014) can yield billions of paired genomic coordinates, representing a comprehensive set of 3D DNA contacts in a cell population. This paired coordinate information can be utilized to explore interactions between specific genomic regions. We sought a file format optimal for storing and querying this information.
One of the most widely used options for storing pairs of read-level coordinates is BAM (Li et al., 2009), a binary format that allows random access queries of genomic regions through Samtools. However, the BAM format does not support efficient querying of both coordinates in a pair, as it requires scanning the entire query result for the first coordinate of the pair, where the query result can span the whole genome. It also has an intrinsic redundancy of storing every pair of coordinates twice, so that the two coordinates are treated symmetrically. Moreover, BAM specifications are tightly defined and difficult to modify to circumvent these problems. A format used by Juicer (Durand et al., 2016) called merged_nodups.txt stores pairs of genomic coordinates non-redundantly, but it is intended to be an intermediate format and is not optimized for storage or querying. Tabix (Li, 2011) is a software tool that offers similar indexing and querying functionality to Samtools on block-compressed tab-delimited text files, thus allowing for flexibility in the file format. However, similar to the BAM format, indexing or querying a second coordinate in a pair is not supported by Tabix. BEDPE (Quinlan and Hall, 2010) is a format for storing paired genomic regions, but it is not economical for storing massive read-level Hi-C data due to its constraints of required and reserved fields.
To overcome these issues, we designed the Pairs file format, which avoids redundancy in storing paired data, and Pairix, an extension of Tabix to index and query over pairs of genomic coordinates. We named this format ‘Pairs’ given the colloquial language that refers to this type of files as ‘pairs’ files.
2 Results
2.1 Pairs format
The Pairs format consists of header lines and data lines. Each data line corresponds to a pair of genomic coordinates. There are four required fields (chr1, pos1, chr2 and pos2) corresponding to two chromosomes and positions. To make it Pairix-compatible, the content should be sorted by chr1, chr2, pos1, then pos2. There are three reserved fields (readID and strand1/2, but could be left blank) and two reserved field names that can be used for optional fields. To reduce redundancy, the Pairs format stores Hi-C data in an upper-triangular matrix instead of the whole matrix, ensuring that the second coordinate of the pair is larger than the first one. If the data matrix is asymmetric, e.g. MARGI for RNA–DNA interactions (Sridhar et al., 2017), the whole matrix could be stored.
A more detailed specification can be found at https://github.com/4dn-dcic/pairix/blob/master/pairs_format_specification.md. A Pairs file has the .pairs.gz extension after block-compression using bgzip (Li, 2011), and the extension of its Pairix index has .pairs.gz.px2 (px2 instead of tbi, a typical extension of a Tabix index). In terms of the size, a Hi-C BAM file containing 3.3 million reads is ∼870 MB, whereas the Pairs file containing 82% of the reads (2.7 million reads) is only ∼38 MB.
2.2 Queries
Figure 1 summarizes examples of three types of queries; intrachromosomal (e.g. getting all interactions between chrX:40 000 000–60 000 000 and chrX:80 000 000–100 000 000), interchromosomal (e.g. getting all interactions between chr1:100 000 000–300 000 000 and chrY:7 000 000–8 000 000) and 4C-like queries (e.g. getting all interactions between chr19:20 000 000–30 000 000 and any other region in the genome).
Fig. 1.
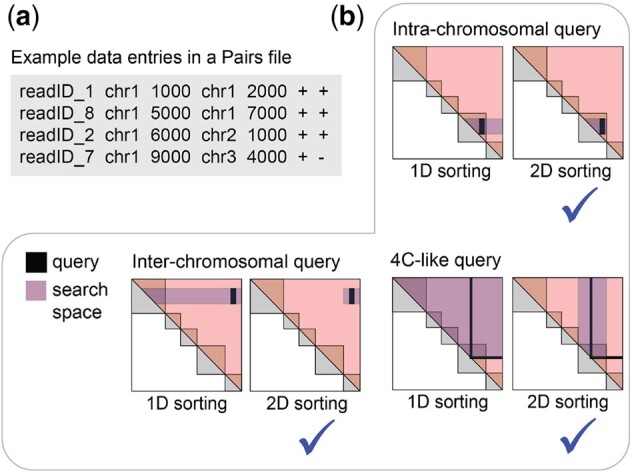
Pairs format and common types of queries for Pairs. (a) Pairs format, (b) comparison of search space between conventional 1D sorting and the 2D sorting adopted by Pairs and Pairix, for three types of queries over pairs of genomic loci. In all three scenarios, using a 2D sorted file involves a smaller search space
Figure 1 illustrates the search spaces for each example query with the conventional ‘1D sorting’ (sorting by chr1 then pos1) and with the ‘2D sorting’ that we adopted for Pairs and Pairix (sorting by chr1, chr2, pos1 then pos2). Clearly, the 2D sorting is more efficient.
2.3 Pairix
Pairix has four main additional features compared to Tabix. First, it can use pairs of chromosomes as hash keys, depending on the sorting mechanism. Second, it can parse both single- and paired-region queries. Third, it accepts Pairs as the default format. Fourth, the number of lines in a file is stored in the index and can be retrieved instantly. Most of the original Tabix functionalities are preserved. However, due to the modifications in the index and query structures, Tabix and Pairix are not mutually interchangeable, i.e. Tabix cannot be used with a Pairix index or vice versa. Pairix as well as Pypairix and Rpairix (Python and R libraries) are available at https://github.com/4dn-dcic/pairix and https://github.com/4dn-dcic/Rpairix.
2.4 Performance of Pairix
Creating an index file is a one-time task and its run time increases linearly with data size, taking 0.68 s per million lines of a Pairs file plus 9 s of overhead on an AMD EPYC 7571 CPU. Memory requirement saturates at about 200 Mb.
The performance improvement in queries using random access compared to not using random access is substantial, as expected. For a minimal Pairs file that contains over 300 million lines, retrieving an entry takes ∼1 s with Pairix, whereas scanning the whole file takes up to 30 min, depending on the genomic regions of interest.
Table 1 provides sample performance comparisons for 2D queries by Pairix and Tabix on a 2.4 GHz Quad-Core Intel Core i5 CPU. To perform 2D queries for pairs files with Tabix, the results of a 1D Tabix query were searched for the second region using awk (e.g. awk ‘$4==“chr13” && $5>=8 000 000 && $5 < 20 000 000’). The advantage of 2D indexing by Pairix is increasingly apparent for queries as the number of reads in the first region of the query increases. For some queries, Pairix was about 10 times as fast as Tabix. When we compared some 1D queries on SAM files, Tabix and Pairix performed similarly as well as Samtools on BAM files (Table 2).
Table 1.
Pairix performance for 2D querying of pairs files
| Query region | Reads in region 1 | Reads in regions 1 and 2 | Time (s) Pairix | Time (s) Tabix | Tabix/Pairix |
|---|---|---|---|---|---|
| chr1:10 Mb–20 Mb|chr1:8 Mb–20 Mb | 114 996 | 44 979 | 0.37 | 0.59 | 1.60 |
| 184 351 | 66 471 | 0.57 | 0.89 | 1.57 | |
| chr1:10 Mb–100 Mb|chr1:8 Mb–20 Mb | 1 052 486 | 44 979 | 0.60 | 4.34 | 7.22 |
| 1 284 965 | 66 471 | 0.76 | 5.42 | 7.13 | |
| chr1:|chr1:8 Mb–20 Mb | 2 133 783 | 44 979 | 0.90 | 8.92 | 9.91 |
| 3 609 461 | 82 884 | 1.20 | 15.39 | 12.86 | |
| chr1:|chr1:8 Mb–200 Mb | 2 133 783 | 1 307 808 | 2.41 | 10.02 | 4.16 |
| 3 609 461 | 1 019 677 | 2.26 | 16.39 | 7.25 | |
| chr1:|chr1: | 2 133 783 | 1 329 322 | 1.19 | 9.70 | 8.13 |
| 3 609 461 | 1 473 651 | 1.96 | 16.15 | 8.23 | |
| chr13:10 Mb–20 Mb|chr13:8 Mb–20 Mb | 114 707 | 53 103 | 0.37 | 0.56 | 1.51 |
| 138 405 | 57 006 | 0.52 | 0.71 | 1.46 |
Note: Pairix performs efficient 2D querying of pairs files. The relative advantage of Pairix seems to increase with the number of reads in the first chromosomal region of the 2D query, up to about 10 times for chromosome 1.
Table 2.
Pairix performance for 1D queries
| Query region | Reads in region | Time (s) Pairix | Time (s) Tabix | Time (s) Samtools |
|---|---|---|---|---|
| chr13:16 285 749–17 363 019 | 124 649 | 5.94 | 6.21 | 5.98 |
| chr13:16 285 749–17 363 019 | 17 094 | 0.82 | 0.85 | 0.71 |
2.5 PairsQC
PairsQC is an open-source quality control (QC) HTML report generator for Hi-C Pairs files based on Nozzle (Gehlenborg et al., 2013) and D3 (Bostock et al., 2011). The collapsible HTML report generated contains a summary table of various quality metrics and several QC plots for Hi-C. The source code and example reports can be found at https://github.com/4dn-dcic/pairsqc.
3 Discussion
Pairs, Pairix and PairsQC have been used to process and store filtered read-level Hi-C and Hi-C-like data for more than 600 experiment sets for the 4D Nucleome consortium (Dekker et al., 2017), with the results available at https://data.4dnucleome.org. The Pairs format is supported by Juicer (Durand et al., 2016), Cooler (Abdennur and Mirny, 2019) and Pairtools (https://github.com/mirnylab/pairtools).
Acknowledgements
We thank Nezar Abdenur, Anton Goloborodko and other 4D Nucleome members for the valuable discussion. We also thank Scott Kallgren for incorporating GenomicRanges (Lawrence et al., 2013) in Rpairix.
Funding
This work was supported by a grant from the National Institutes of Health Common Fund 4D Nucleome program [U01 CA200059] to P.J.P.
Conflict of Interest: none declared.
Contributor Information
Soohyun Lee, Department of Biomedical Informatics, Harvard Medical School, Boston, MA 02115, USA.
Clara R Bakker, Department of Biomedical Informatics, Harvard Medical School, Boston, MA 02115, USA.
Carl Vitzthum, Department of Biomedical Informatics, Harvard Medical School, Boston, MA 02115, USA.
Burak H Alver, Department of Biomedical Informatics, Harvard Medical School, Boston, MA 02115, USA.
Peter J Park, Department of Biomedical Informatics, Harvard Medical School, Boston, MA 02115, USA.
References
- Abdennur N., Mirny L.A. (2019) Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics, 36, 311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostock M. et al. (2011) D3 Data-Driven Documents. IEEE Trans. Vis. Comput. Graph., 17, 2301–2309. [DOI] [PubMed] [Google Scholar]
- Dekker J. et al. (2017) The 4D Nucleome project. Nature, 549, 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand N.C. et al. (2016) Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst., 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehlenborg N. et al. (2013) Nozzle: a report generation toolkit for data analysis pipelines. Bioinformatics, 29, 1089–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krietenstein N. et al. (2020) Ultrastructural details of mammalian chromosome architecture. Mol. Cell, 78, 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M. et al. (2013) Software for computing and annotating genomic ranges. PLoS Comput. Biol., 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2011) Tabix: fast retrieval of sequence features from generic TAB-delimited files. Bioinformatics, 27, 718–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S.S.P. et al. (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhar B. et al. (2017) Systematic mapping of RNA-chromatin interactions in vivo. Curr. Biol., 27, 602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]