

Abstract
Aims
Vascular stiffness increases with age and independently predicts cardiovascular disease risk. Epigenetic changes, including histone modifications, accumulate with age but the global pattern has not been elucidated nor are the regulators known. Smooth muscle cell-mineralocorticoid receptor (SMC-MR) contributes to vascular stiffness in ageing mice. Thus, we investigated the regulatory role of SMC-MR in vascular epigenetics and stiffness.
Methods and results
Mass spectrometry-based proteomic profiling of all histone modifications completely distinguished 3 from 12-month-old mouse aortas. Histone-H3 lysine-27 (H3K27) methylation (me) significantly decreased in ageing vessels and this was attenuated in SMC-MR-KO littermates. Immunoblotting revealed less H3K27-specific methyltransferase EZH2 with age in MR-intact but not SMC-MR-KO vessels. These ageing changes were examined in primary human aortic (HA)SMC from adult vs. aged donors. MR, H3K27 acetylation (ac), and stiffness gene (connective tissue growth factor, integrin-α5) expression significantly increased, while H3K27me and EZH2 decreased, with age. MR inhibition reversed these ageing changes in HASMC and the decline in stiffness genes was prevented by EZH2 blockade. Atomic force microscopy revealed that MR antagonism decreased intrinsic stiffness and the probability of fibronectin adhesion of aged HASMC. Conversely, ageing induction in young HASMC with H2O2; increased MR, decreased EZH2, enriched H3K27ac and MR at stiffness gene promoters by chromatin immunoprecipitation, and increased stiffness gene expression. In 12-month-old mice, MR antagonism increased aortic EZH2 and H3K27 methylation, increased EZH2 recruitment and decreased H3K27ac at stiffness genes promoters, and prevented ageing-induced vascular stiffness and fibrosis. Finally, in human aortic tissue, age positively correlated with MR and stiffness gene expression and negatively correlated with H3K27me3 while MR and EZH2 are negatively correlated.
Conclusion
These data support a novel vascular ageing model with rising MR in human SMC suppressing EZH2 expression thereby decreasing H3K27me, promoting MR recruitment and H3K27ac at stiffness gene promoters to induce vascular stiffness and suggests new targets for ameliorating ageing-associated vascular disease.
Keywords: Mineralocorticoid receptor, Vascular ageing, Vascular stiffness, Epigenetics, EZH2
Graphical Abstract
1. Introduction
Ageing is a universal physiological process and potent risk factor for cardiovascular disease (CVD). The population over 65 years of age is expected to increase to 22% by 2040 and over 40% of people over 65 years are expected to die of CVD.1 Vascular stiffness is one of the principal consequences of cardiovascular ageing and is associated with increased risk of coronary heart disease, stroke, and cardiovascular death, independent of blood pressure.1 Vascular stiffness is measured non-invasively by pulse wave velocity (PWV) and increases with age in males and females2,3 but the mechanism is unknown. The haemodynamic consequence of aortic stiffening is increased pulse pressure, which damages the microvasculature in downstream organs thereby contributing to ageing-related dysfunction of the kidney, heart, and brain. Thus, enhanced understanding of the process of vascular stiffness during cardiovascular ageing is necessary to identify rational new anti-ageing targets to prevent the adverse impact of ageing on cardiovascular health.
Vessel stiffness is a composite of the intrinsic stiffness of vascular cells, the composition of the extracellular matrix (ECM), and the attachments between the cells and matrix.4 Pathologically, vascular ageing is characterized by increased fibrosis, involving accumulation of collagen in the medial and adventitial layers of the remodelled artery,3 and decreased elasticity of the vessel wall due to an increment in matrix metalloproteinase (MMP) activity that reduces elastin integrity.3,5,6 Vascular smooth muscle cells (SMCs) contribute to all aspects of vascular stiffness as they evolve towards a senescence phenotype with advancing age. This includes production of ECM components and MMPs that contribute to vascular fibrosis and elastin degradation, cytoskeletal changes that affect intrinsic SMC stiffness, and modulation of surface proteins that interact with the ECM.6 Specifically, SMC produce connective tissue growth factor (CTGF), which promotes collagen synthesis. In addition, the SMC actin cytoskeleton attaches to the ECM through integrin proteins located at focal adhesions. The upstream regulators driving ageing-related changes in expression of SMC genes are not clear, but greater understanding could identify novel anti-ageing targets.
Ageing is associated with epigenetic changes that modulate gene expression independent of the DNA sequence.7,8 One such epigenetic mechanism is post-translational modifications of histone proteins around which DNA is packaged to form nucleosomes. Such chromatin modifications modulate nucleosome structure to regulate the accessibility of DNA regulatory sequences to transcription factors thereby regulating gene and protein expression.9 The activity of histone-modifying enzymes (methyltransferases, acetylases, deacetylases) in cardiovascular tissues have been associated with hypertension and cardiac hypertrophy in animal models8 and epigenetic mechanisms have been implicated in SMC phenotype modulation that contributes to vascular injury and atherosclerosis.10,11 However, global ageing-induced changes in histone modifications in the vasculature have not been interrogated nor is it clear how this may be regulated to contribute to vascular stiffness in the ageing vasculature.
The aldosterone-binding mineralocorticoid receptor (MR) plays a critical role in blood pressure regulation12,13 and has been implicated in vascular stiffness.14,15 In humans, vascular stiffness and fibrosis are increased in patients with primary hyperaldosteronism16 and MR antagonist treatment decreased vascular stiffness.17 The MR is a hormone-activated transcription factor that regulates vascular gene transcription by binding to DNA regulatory elements.18 Previous studies of our group reveal that MR expression increases with age in mouse vessels and that old mice treated with mineralocorticoid receptor antagonist or with the MR genetically knocked out from SMC (SMC-MR-KO) are protected from ageing-induced vascular fibrosis and stiffness14,15,19,20 but this has never been tested on human samples. Vascular gene expression profiling revealed that in aged mice, SMC-MR-KO promotes a global shift to decreased expression of a fibrosis-associated gene network that includes CTGF, integrin-α5, and matrix metalloprotease-2 (MMP2), genes that promote vascular stiffness. However, the mechanism by which the MR in SMC drives vascular ageing and whether contributes similarly to ageing in human SMC has never been explored.
Here we investigate the mechanism driving vascular stiffness gene expression with ageing by characterizing global changes in histone modifications in the ageing mouse vasculature comparing MR-intact to SMC-MR-KO mice by mass spectrometry. We identify a mechanism by which MR down-regulates the histone-modifying enzyme EZH2 to alter histone-H3 lysine-27 (H3K27) modifications and allowing MR recruitment to stiffness target genes to drive vascular stiffness in ageing mice and in primary low passage human SMC and human aortic tissue from young vs. old people.
2. Methods
2.1 Human tissue
De-identified human aortic tissue was obtained post-mortem from the NIH-supported National Disease Research Interchange and hence the medical ethics committee of participating centre (Tufts Medical Center) deemed this research to be exempt from human subjects research requirements.19
2.2 Human cell culture and treatment
Human aortic smooth muscle cell (HASMC) were treated with the MR antagonist spironolactone (1 μM, Research Plus, Inc, Barnegat, NJ), EZH2 inhibitor GSK126 (15 µM, MedChemExpress, Monmouth Junction, NJ), or H2O2 (1 μM, Sigma Aldrich, Saint Louis, MO) compared to their respective vehicle (DMSO or water), for 24, 48, and 72 h and cells harvested for mRNA (see Supplementary material online, Table S1), protein, and/or chromatin immunoprecipitation (ChIP) studies (see Supplementary material online, Table S2).
2.3 Animal studies
All mice were treated in accordance with US National Institutes of Health standards and all procedures were approved by the Tufts University Institutional Animal Care and Use Committee and conformed to the NIH guidelines for the care and use of laboratory animals. Twelve-month-old WT (C57BL/6J background) male mice were implanted in the subscapular region with pellets containing either placebo or spironolactone (two consecutive 60 day release pellets at 20 mg kg−1 day−1, Innovative Research of America) for total 120 days. Mice were anaesthetized using an induction chamber with 4% isoflurane mixed with 0.5 L/min of 100% O2. During the surgery, 1.5% isoflurane with 0.5 L/min O2 was maintained for analgesia. At the indicated time points, animals were euthanized by terminal blood and tissue harvest under isoflurane inhalation. Mice with tamoxifen-inducible SMC-specific deletion of MR gene (Nr3c2) were generated by crossing floxed MR (MRf/f) mice with SMA-Cre-ERT2 mice as previously described.14,19
2.4 Statistical analysis
Statistical analysis for the proteomic data was performed as described.21 For all other measurements, values are presented as mean ± SEM. Differences between two groups were analysed using Student’s t-test. Differences between multiple groups (mice ± spironolactone or ± SMC-MR at different ages) were analysed by two-way ANOVA followed by Tukey’s post hoc test. Pearson correlation coefficients were calculated to determine correlations in human tissue. Statistical analyses were performed using GraphPad Software Inc. version 8. Statistical significance was set at P < 0.05.
3. Results
3.1 MR expression increases with donor age in primary human SMCs
Vascular MR mRNA expression increases with age in rodents,22,23 but this has never been tested in humans, nor has protein been tested due to lack of a mouse MR antibody. Thus, MR protein was first quantified in primary low passage HASMCs from adult (average age 44 years) compared to aged (average age 78 years) donors. MR protein expression was 2.5 times higher in SMC from aged compared with adult cells (P < 0.05, Figure 1A). SMC from aged donors also demonstrated increased senescence as measured by senescence-associated β-galactosidase (SAβ-gal) staining, confirming maintenance of the ageing phenotype when cells are cultured in vitro (Figure 1B). We further measured protein expression of stiffness genes previously shown to be influenced by the presence of SMC-MR in ageing mouse vessels. CTGF and integrin-α5 were increased significantly (P < 0.05) in aged compared to adult HASMC (Figure 1C), while MMP2 expression did not significantly change with age in HASMC.
Figure 1.

Mineralocorticoid receptor (MR) and stiffness gene expression increase with age in primary human aortic smooth muscle cells (HASMC). Primary HASMC were compared from adult (average age 40s) vs. aged (average age late 70s) donors. (A) Representative immunoblots and quantification of MR protein expression by immunoblotting. n = 9 for each age group. (B) Representative HASMC images of senescence-associated (SA) β-galactosidase staining and quantification of SAβ-galactosidase staining. n = 4 of each age group. (C) Representative immunoblots and quantification of protein expression of select stiffness genes: connective tissue growth factor (CTGF), matrix metalloproteinase 2 (MMP2), and integrin-α5. n = 7 for each age group. Dot plots show the individual data points and bars indicate the mean with error bars indicating the SEM for relative protein expression normalized to GAPDH. Results were analysed using Student’s t-test. *P < 0.05, **P < 0.01.
3.2 Global changes in histone post-translational modifications distinguish young from aged mouse aortas
To explore the impact of ageing on epigenetic histone modifications in the vasculature, chromatin was isolated from six aortas each from young (3 month) and older (12 month) mice with MR present vs. SMC-MR-KO littermates at each age; 12 months of age was chosen as a time-point in which aortic fibrosis and stiffness are already evident in ageing mice and stiffness gene expression is attenuated when aged mice lack SMC-MR.14 Chromatin was digested to peptides and the levels of 68 histone post-translational modification states were quantified by mass spectrometry, as described.21 Principal component analysis of the mass spectrometry data revealed that the histone modification state readily and completely discriminates between young and old vessels (Figure 2A, principal Component 2 axis). Among the young aortas, the histone modification profile does not distinguish between the MR-intact vs. SMC-MR-KO vessels suggesting a limited role for SMC-MR in the histone modification state of young vessels. However, among the old aortas, principal Component 1 separated the vessels by genotype, with most of the aged MR intact mice falling on the left, further from the young vessels, and all of the old SMC-MR-KO vessels falling on the right (Figure 2A).
Figure 2.

Proteomic profiling of all aortic histone modifications from young and old mouse aortas distinguishes old from young vessels. Mass spectrometry-based profiling of histone modifications was performed on mouse aortic chromatin from 3 month and 12 month old mineralocorticoid receptor (MR)-intact and smooth muscle cells (SMC)-MR knock out littermates. (A) Principal component analysis (PCA) comparing the histone proteomic data and plotting by co-ordinates for principal Components 1 and 2, colour-coded by the age and genotype of each vessel. Principal Component 2 separates all vessels by age and principal Component 1 further separates most of the old MR-intact from old SMC-MR-KO vessels. (B) Hierarchical cluster analysis of global histone modification patterns in MR-intact and SMC-MR-KO mice young and old. Red indicates an increase and blue a decrease in the amount of each histone modification relative to the mean for all vessels. (C) Marker selection analysis identifies specific histone modifications that significantly distinguish young from old vessels. Histone modifications with a FDR < 0.05 are listed. ac, acetyl; me, methyl.
Hierarchical clustering of the histone marks similarly revealed a substantial difference in the global chromatin modification profile of old vs. young vessels that is further modified by the presence of SMC-MR only in the aged vessels (Figure 2B). All of the young mice cluster together on the left, without segregation by genotype. There is only one old aorta that clusters with the young vessels and that vessel came from a SMC-MR-KO mouse. Among the old vessels, there is further clustering with most of the MR-intact old vessels clustering together on the right and furthest away from the young vessels.
Marker selection analysis was next used to identify specific histone modifications that significantly distinguish between old and young vessels (Figure 2C).24 H2AK9ac increased and H2B acetylation state changed with age. Moreover, of the 10 histone marks that differ with age with a False discovery rate (FDR) < 0.05, 7 were changes in modification of H3K27 with an overall decrease in methylation and an increase in acetylation with advancing age (Figure 2C). Specifically, H3K27 mono (H3K27me1) and di-methylation (H3K27me2) marks are decreased in vessels from old mice (regardless of the methylation state of the H3K36 on the same peptide), and the suppression of H3K27 methylation appears attenuated in the absence of SMC-MR.
3.3 Ageing-associated changes in histone-modifying enzymes in mouse aorta and in primary human SMC
To begin to explore the mechanism for ageing-associated changes in the H3K27 histone modification pattern, we examined expression of enzymes implicated in histone methylation, including the H3K27 methyltransferase EZH2, the H3K27 demethylase UTX, and the histone methyltransferase G9a.25 We also quantified Cyclic adenosine monophosphate Response Element Binding (CREB)-binding protein (CBP) and PCAF/p300, MR transcriptional co-activators with histone acetyltransferase activity.26 Protein was quantified first in aortic lysates from young vs. old MR-intact and SMC-MR-KO mice (Figure 3A). Expression of PCAF/p300, G9a, and UTX all increased significantly with ageing in mouse aorta but this was independent of the presence of SMC-MR. Expression of the H3K27 methyltransferase, EZH2 was significantly reduced in old compared with young MR-intact mice. The decline in EZH2 with age was not evident in SMC-MR-KO aortas, resulting in significantly higher EZH2 expression in aortas from old SMC-MR-KO vs. MR-intact littermates. Similarly, CBP expression was significantly increased in old compared with young MR-intact mice and was significantly decreased in vessels from aged SMC-MR-KO mice compared to aged MR-intact littermates.
Figure 3.

Ageing-associated changes in histone modifications and histone-modifying enzymes in mouse aortas and primary human aortic SMC (HASMC). (A) Representative immunoblots and quantification of protein expression of enzymes involve in the methylation (me; EZH2, G9a, UTX) and acetylation (ac; CBP, PCAF/P300) of histones in aortas from mice with MR-intact vs. SMC-MR-KO. n = 6–7 of each age group. (B) Representative immunoblots and protein expression of mono (me1), di (me2), and tri (me3)-methylation and acetylation of histone 3 (H3) lysine 27 (H3K27) in primary HASMC from adult and aged donors. n = 6–12 of each age group. (C) Representative immunoblots and quantification of protein expression of enzymes involve in the methylation (EZH2, G9a, UTX) and acetylation (CBP, PCAF) of histone H3 in HASMC from adult and aged donors. Dot plots show the individual data points and bars indicate the mean with error bars indicating the SEM of protein expression normalized to β-tubulin or histone modifications normalized to total histone H3. n = 6–8 of each age group *P < 0.05; **P < 0.01. Mouse aorta results were analysed using two-way ANOVA with Tukey’s post hoc test. Human cell results were analysed using Student’s t-test.
We next examined whether these ageing-associated changes in H3K27 post-translational modifications and histone-modifying enzymes identified in mouse vessels are also present in adult vs. aged primary human cells (Figure 3B and C). In HASMC, H3K27 methylation was lower and acetylation was higher in primary cells from aged vs. adult donors. Specifically, there was a significant decrease in H3K27me2 and H3K27me3 and increase in H3K27ac in aged vs. adult human SMC (Figure 3B). Also consistent with the mouse vessels, expression of the methyltransferase, EZH2, was significantly reduced and the acetyltransferase, CBP, was increased in aged SMCs compared to adult cells (Figure 3C). The levels of the other histone-modifying enzymes (Ga9, UTX, and PCAF) did not differ with age in human SMCs. The decline with age in EZH2 is consistent with decreased H3K27 methylation and the rise in CBP could mediate acetylation of MR target promoters. Moreover, these two protein changes were consistent between mouse vessels and human cells. Thus, further studies focused on EZH and CBP.
3.4 MR inhibition in aged HASMC reverses the ageing EZH2/H3K27/stiffness phenotype
To test whether MR is driving the ageing phenotype in HASMC, cells from aged donors were treated with the MR antagonist spironolactone and the impact on histone-modifying enzyme expression, H3K27 modifications, stiffness gene expression, and SMC stiffness and adhesion properties were quantified. As expected, pharmacological MR inhibition did not significantly impact MR protein or mRNA expression (see Supplementary material online, Figure S1A and B). MR inhibition significantly increased EZH2 mRNA (see Supplementary material online, Figure S1C) and protein (Figure 4A) after 48 h, supporting that the lower levels of EZH2 in aged HASMC is at least in part driven by the MR. CBP mRNA was unchanged by MR inhibition (see Supplementary material online, Figure S1C) although protein expression increased suggesting that high CBP in aged HASMC is not transcriptionally regulated by the MR and that spironolactone might affect CBP at a post-transcriptional level. Consistent with the rise in EZH2, spironolactone treatment significantly increased H3K27me1 and H3K27me3 in association with decreased H3K27ac (Figure 4B). MR inhibition also significantly reduced the mRNA and protein level of the stiffness genes CTGF, MMP2, and integrin-α5 after 48 h (see Supplementary material online, Figure S1D–E). To test whether EZH2 histone methyltransferase activity is necessary for suppression of fibrosis genes with MR inhibition, aged HASMCs were treated for 48 h with spironolactone alone or co-treated with the EZH2 inhibitor GSK126. EZH2 inhibition for 48 h in HASMCs significantly decreased H3K27 methylation, confirming the efficacy of GSK126 inhibition of EZH2 function (see Supplementary material online, Figure S2). EZH2 inhibition with GSK126 prevented the spironolactone-induced reduction of CTGF and integrin-α5 (Figure 4C), further supporting that EZH2 contributes to the gene regulatory changes mediated by MR inhibition.
Figure 4.

MR inhibition in aged human cells reverses the impact of ageing on histone modifications, stiffness gene expression, and SMC stiffness. HASMCs from aged human donors were treated with vehicle or the MR antagonist spironolactone for 24, 48, and 72 h. Protein was quantified by immunoblotting relative to vehicle treatment at each time and representative immunoblots are shown for: (A) histone-modifying enzymes (EZH2 and CBP). n = 17 of each age group; (B) histone modifications [mono- (me1), di- (me2), tri (me3)-methylation, and acetylation (ac)] of H3K27. n = 10–17 of each age group. (C) HASMCs from aged human donors were treated with vehicle, MR antagonist spironolactone ± EZH2 inhibitor GSK128 for 48 h. Representative immunoblots and protein was quantification for stiffness genes (CTGF, MMP2, Integrin-α5) n = 6. (D) Schematic of a fibronectin (FN) functionalized AFM tip and probe interacting with the surface of a single SMC. Representative force curve as the probe is lowered (red) to indent the cell surface and induce upward deflection of the probe (indentation force) and is then retracted (blue) to rupture adhesions formed between fibronectin on the AFM tip and integrins on the SMC. AFM was performed on HASMC from aged donors treated with vehicle or spironolactone for 48 h. (E) SMC stiffness was calculated from the rising portion of the approach curve, n = 9 of each age group and (F) SMC adhesion to fibronectin was quantified by determining the probability of adhesion events (# of force curves with at least one adhesion event/total # of force curves collected) in HASMC. Dot plots show the individual data points and bars indicate the mean with error bars indicating the SEM in relative protein expression normalized to GAPDH (A and C) or histone modifications normalized to total histone H3 (B), n = 9 of each age group. Results were analysed using Student’s t-test (A, B, E, and F) or two-way ANOVA (C) with Tukey’s post hoc test. (A and B) *P < 0.05. vs. respective vehicle at each time (C–F) *P < 0.05, ** P < 0.01, *** P < 0.001.
Atomic force microscopy was next used to explore whether these molecular changes driven by SMC-MR impact intrinsic HASMCs stiffness (Figure 4D–F). MR inhibition significantly decreased the intrinsic stiffness of aged SMC (Figure 4E). Next, using an Atomic Force Microscopy (AFM) probe coated with fibronectin, the probability of adhesion of SMC integrins to fibronectin was quantified by measuring the frequency of an adhesion-rupture event between the coated probe and the SMC membrane.27 Treatment with MR antagonist significantly reduced the probability of adhesion of aged SMC to fibronectin (Figure 4F) consistent with the decrease in the integrin-α5 (fibronectin receptor) expression (Figure 4C).
3.5 Induction of an ageing phenotype in young HASMC induces MR and the epigenetic shift to decreased H3K27 methylation
This ageing mechanism was next tested in primary HASMC from young donors treated with H2O2, a previously identified inducer of an ageing phenotype in cultured cells.28 We first confirmed that H2O2 treatment for 48 h significantly increased SAβ-gal staining consistent with promoting cell senescence (Figure 5A). H2O2 treatment also significantly increased MR mRNA (see Supplementary material online, Figure S3A) and protein expression (Figure 5B) after 24 and 48 h. There was a concomitant decrease of EZH2 and an increase of CBP expression (Figure 5C, Supplementary material online, Figure S3B) consistent with the phenotype seen in aged HASMC and mouse vessels (Figure 3). The changes in histone-modifying enzymes further associated with a significant decrease in H3K27me1 at 24 h, H3K27me3 at 48 h, and increased H3K27ac at both times (Figure 5D).
Figure 5.

Induction of an ageing phenotype in young HASMC drives changes in MR, histone modifications, and stiffness gene expression. Primary human aortic SMC (HASMC) from adult donors (age 40s) were treated with hydrogen peroxide (H2O2) for the indicated time (24–72 h) to induce an ageing phenotype. (A) Representative HASMC images of SAβ-galactosidase staining and quantification of SAβ-galactosidase staining in HASMCs treated with vehicle or H2O2 for 24 h to confirm induction of cell senescence, n = 4. Protein was quantified by immunoblotting and representative immunoblots are shown for: (B) MR, n = 16; (C) histone-modifying enzymes EZH2 and CBP, n = 16; (D) histone modifications [mono- (me1), di- (me2), tri (me3)-methylation and acetylation (ac)] of H3K27, n = 12–19. (E) and stiffness genes (CTGF, MMP2, Integrin-α5), n = 12–16. (F and G) Chromatin immunoprecipitation (ChIP)-qPCR was performed in chromatin isolated from HASMC treated with vehicle or H2O2 for 24 h followed by PCR with primers specific to the CTGF, MMP2, and Integrin-α5 promoter gene loci. ChIP was performed in cells treated with vehicle (white bar) or H2O2 (grey bar) compared to control IgG with antibodies specific for: (F) H3K27 acetyl to indicate open chromatin, n = 4 experiments; or (G) MR to indicate the degree of MR enrichment at the promoter, n = 4 experiments. Dot plots show the individual data points and bars indicate the mean with error bars indicating the SEM in relative protein expression normalized to GAPDH (B, C, and E) or to total histone H3 (D). Protein results were analysed using two-way ANOVA (treatment, time) with Tukey’s post hoc test. ChIP results were analysed using Student’s t-test. *P < 0.05 vs. vehicle.
3.6 Induction of ageing in HASMC increases enrichment of MR and acetylated H3K27 at stiffness gene promoters and promotes stiffness gene expression
The epigenetic shift induced by H2O2 treatment of young HASMC also associated with a significant increase in mRNA expression (see Supplementary material online, Figure S3C) and protein level of the three representative stiffness genes, CTGF, MMP2, and integrin-α5 (Figure 5E). To test whether the H2O2-induced changes in global histone modifications altered locus-specific histone modifications at the stiffness gene promoters, ChIP-qPCR was performed using H3K27ac antibody. H2O2 treatment for 24 h significantly increased enrichment of H3K27ac over IgG control antibody at the promoters of the CTGF and integrin-α5 genes with no significant change in H3K27ac enrichment at the MMP2 promoter (Figure 5F). Since these three stiffness genes are putative SMC-MR target genes, we tested whether MR is recruited to these sites in HASMC by ChIP-qPCR with anti-MR antibody. H2O2 treatment significantly increased enrichment of MR over IgG at the promoters of all three stiffness gene promoters (Figure 5G). Primers targeting exon 2 of each gene, an atypical site for MR binding or gene regulation, were simultaneously tested as a negative control revealing no enrichment of H3K27ac or MR at those sequences (see Supplementary material online, Figure S3D and E).
3.7 MR inhibition in old mice reverses the epigenetic mechanism and attenuates vascular stiffening
To test if this MR-driven epigenetic vascular ageing mechanism can be modulated in vivo, 12-month-old mice were treated with the MR antagonist spironolactone vs. placebo by subcutaneous pellet for 4 months at a dose that does not significantly alter blood pressure in mice (see Supplementary material online, Figure S4). Spironolactone treatment in vivo significantly increased aortic EZH2 expression without affecting CBP (Figure 6A), further supporting that MR regulates EZH2 (but not CBP) expression in the ageing vasculature. This was associated with a significant increase in global mono and tri-methylation and decreased acetylation of H3K27 on histones isolated from mouse aortas from spironolactone-treated mice (Figure 6B). To examine if EZH2 is recruited to the stiffness gene promoters to induce locus-specific changes in histone modifications, ChIP-qPCR was performed using EZH2 or H3K27ac antibody (but not MR as there is no mouse MR-specific antibody). Spironolactone treatment significantly increased enrichment of EZH2 over IgG at the promoter region of the three stiffness genes (Figure 6C) and this was associated with decreased enrichment of H3K27ac at the same sites (Figure 6D). Protein expression of the stiffness genes, CTGF and integrin-α5, was significantly reduced in old mice treated with spironolactone compared to placebo, with no significant impact on MMP2 expression (Figure 6E). To assess the impact of these gene regulatory changes on the vasculature, PWV was measured to quantify vascular stiffness during spironolactone treatment and vascular fibrosis was assessed by histology at the end of the 4 months of treatment. In placebo-treated mice, vascular stiffness increased progressively from 12 to 14 and then 16 months and this was prevented by spironolactone, resulting in significantly decreased vascular stiffness in spironolactone vs. placebo-treated mice at 14 and 16 months of age (Figure 6F). Vascular fibrosis was also significantly reduced in mice treated with spironolactone (Figure 6G).
Figure 6.

Mineralocorticoid receptor antagonism attenuates epigenetic and gene expression changes with decreased vascular fibrosis and stiffness in aged mice. Twelve-month-old mice were treated with placebo (black) or MR antagonist (spironolactone, grey) for 4 months and aortas were isolated for protein quantification and ChIP (A–E). Representative immunoblots and protein quantification are shown for: (A) histone-modifying enzymes (EZH2 and CBP), n = 8–9; (B) histone modifications [mono- (me1), di- (me2), tri (me3)-methylation and acetylation (ac)] of H3K27, n = 8–9; and (E) and stiffness genes, CTGF, MMP2, Integrin-α5, n = 8–9. Relative protein expression is normalized to tubulin (A and E) or to total histone H3 (B). ChIP-qPCR data quantifies enrichment of (C) EZH2, n = 5 or (D) H3K27ac, vs. control IgG at the CTGF, MMP2, and Integrin-α5 promoter gene loci, n = 5. (F) Aortic stiffness measured by abdominal aortic pulse wave velocity (PWV) at randomization (12 months) and again after 2 (14 months) and 4 (16 months) months of treatment with placebo or spironolactone (n = 15–19 mice per group). (G) Vascular fibrosis quantified in aorta sections stained with Masson’s trichrome after the 4 months treatment with spironolactone or placebo. Representative images are displayed and scale bar = 10 μm, n = 9. Dot plots show the individual data points and bars indicate the mean with error bars indicating the SEM. (A–E and G) Results were analysed using Student’s t-test. (F) PWV results were analysed using two-way ANOVA with Tukey’s post hoc test, n = 4–8 of each age group. *P < 0.05, **P < 0.01.
3.8 Correlations of MR, histone modifications, and stiffness gene protein levels in human aortic tissue of with advancing age
Finally, we measured key proteins involved in this ageing pathway in human aortic tissue lysates and tested correlations with increasing age. Protein was isolated from aortic tissue from 11 individuals ranging in age from 29 to 72 years and immunoblotting was used to quantify MR, EZH2, H3K27 modifications, and stiffness genes. MR expression significantly positively correlated with age as did the stiffness genes CTGF, MMP2, and integrin-α5 (Figure 7A). H3K27 tri-methylation showed a significant negative correlation with age, while H3K27 acetylation trended in the opposite direction but was not statistically significant (Figure 7B). Although the correlation of EZH2 expression with age was not statistically significant, when EZH2 expression was compared to MR protein levels, there was a significant negative correlation in human vessels (P = 0.0176, Figure 7C), suggesting that EZH2 is linked more closely to the level of MR than to age itself in human tissue.
Figure 7.

Correlations between age and protein levels of mineralocorticoid receptor (MR), histone-modifying enzymes, histone modifications, and stiffness marker genes in human aortic tissue. Immunoblotting was performed in aortic tissue from adult donors from a range of ages. (A) Correlation between increasing age and MR, CTGF, MMP2, and Integrin-α5 protein in human aortic tissue. (B) Correlations between age and H3K27 modifications normalized to total H3 in human aortic tissue. (C) Correlation of aortic EZH2 protein level with age of the donor or with MR expression in human aortic tissue, n = 11. Pearson correlation coefficients and the degree of significance are indicated in each graph.
4. Discussion
These data support a new vascular ageing mechanism in which MR controls global histone post-translational modifications to regulate expression of genes that contribute to SMC and aortic stiffness. Specifically, we demonstrate for the first time that: (i) MR expression increases with age in primary, low passage, human aortic SMC, and correlates with age in whole aortic tissue from ageing humans; (ii) the global proteomic profile of histone modifications in mouse vessels changes profoundly with ageing with a significant overall decrease in H3K27 methylation; (iii) expression of the H3K27 methyltransferase EZH2 decreases with age in mouse vessels and in human SMCs in a MR-dependent manner and negatively correlates with MR expression in whole human aortic tissue; (iv) the ageing-induced decline in EZH2 associates with reduced H3K27 methylation and increased H3K27 acetylation in vitro and in vivo; (v) these epigenetic changes in ageing human SMC and mouse vessels correspond with increased expression of the vascular stiffness genes, CTGF and integrin-α5, previously identified vascular MR target genes; (vi) induction of an ageing phenotype in human SMC associates with increased MR enrichment and H3K27 acetylation at these stiffness gene promoters; and (vii) inhibition of MR in aged mice and aged HASMC reverses the entire process; increasing EZH2 and H3K27 methylation, increasing locus-specific EZH2 enrichment and decreasing H3K27 acetylation at stiffness gene promoters, decreasing vascular expression of CTGF and integrin-α5, and decreasing the stiffness and adhesiveness of aged human SMC in vitro by AFM and mouse aortic stiffness and fibrosis in vivo. Overall, these results are consistent with the model in graphical abstract in which rising MR in ageing vascular SMCs down-regulates EZH2 to globally shift to a more open chromatin thereby allowing MR to be recruited to promoters to transcriptionally up-regulate target genes involved in vascular stiffness. This mechanism provides multiple potential targets to prevent vascular stiffness in ageing humans, an independent risk factor for adverse cardiovascular outcomes and a mediator of organ damage with ageing.
Enhanced MR activity, particularly in the vasculature, has been implicated in many cardiovascular disorders including hypertension, atherosclerosis, and heart failure,29 all of which increase in incidence with ageing. In this study, we demonstrated for the first time that MR expression increases with age in primary human SMC and in human aortic tissue, as had previously been shown in mice.14,15 The positive correlation between age and MR expression in whole human aorta supports that the culture conditions model MR expression in the intact vessel. Previous studies show that MR expression increases in the setting of other cardiovascular risk factors like obesity or hypertension30 as well as in kidney fibrosis.31 MR is a hormone-activated transcription factor that is activated by aldosterone (and in some tissues glucocorticoids) to contribute to vascular dysfunction, fibrosis, and stiffness.14,15,19 The enzyme 11β-hydroxysteroid dehydrogenase Type 2 locally converts glucocorticoids to metabolites with poor affinity for the MR. Previous studies of our lab demonstrated that 11β-HSD is expressed and functions inactivates cortisol in HASMCs32 supporting that aldosterone is likely the main agonist in these cells. However, one conundrum in clinical studies is that MR antagonists are often found to be beneficial even when hormone levels are not elevated.32 The finding that MR itself increases in the ageing vasculature may explain its role in driving ageing vascular pathology even in the absence of elevated hormone levels. Particularly since MR has been shown to be activated in a hormone-independent manner by angiotensin II signalling and oxidative stress-dependent Rac1 signalling, both of which also increase with age.18,33
Epigenetic changes have been shown to accumulate in ageing tissues resulting in global changes in gene transcription that drives the ageing phenotype.7 Epigenetic changes include altered patterns of histone post-translational modifications, DNA methylation, and expression of non-coding RNAs. DNA is wrapped around nucleosomes composed of two copies each of H2A, H2B, H3, and H4. Changes in histone modifications have been implicated in SMC phenotype switching that contributes to CVD,9 but have not been interrogated systematically in vascular ageing. Here global histone modification proteomic profiling was performed for the first time in single mouse aortas revealing profound changes in bulk vascular histone modification that distinguished young from old vessels with 100% discrimination by PCA. Ageing vessels also had significant changes in the pattern of H2B(1–29) acetylation and increased H2AK9ac, modifications which deserve further investigation. Here we focused on the shift from H3K27 methylation to acetylation, as this modification integrated most of the top epigenetic changes, was reproduced in human cells, and was reversed by MR antagonism in vitro and in vivo. Also, decreased H3K27me2 and increased H3K27ac have been previously found in SMC from human carotid plaques compared to healthy arteries.34 In those studies, the healthy arteries were also from younger donors, further supporting a potential role for this epigenetic mechanism in vascular disease in ageing humans.34,35 H3K27 methylation silences genes by compacting chromatin. Once the methyl groups are removed, DNA-binding transcription factors, such as the MR, can bind to DNA regulatory elements to which they recruit histone acetyltransferases, including CBP and P300, which acetylate H3K27 to promote gene expression (Graphical abstract).9 Indeed, we confirmed that in aged HASMC, MR, and H3K27ac are enriched at stiffness gene promoters resulting in increased expression of CTGF, a key driver of collagen expression and fibrosis, and integrin-α5, the fibronectin receptor and component of focal adhesions that link the SMC cytoskeleton to the ECM. Moreover, this mechanism is reversed by MR antagonism in aged human SMC in vitro, resulting decreased SMC stiffness and adhesion to fibronectin by AFM, and in vivo in aged mice, with a decrease in vascular stiffness by PWV. This is also consistent with published findings that SMC-MR-KO mice are protected from vascular stiffness with ageing.14,19
Graphical Abstract.
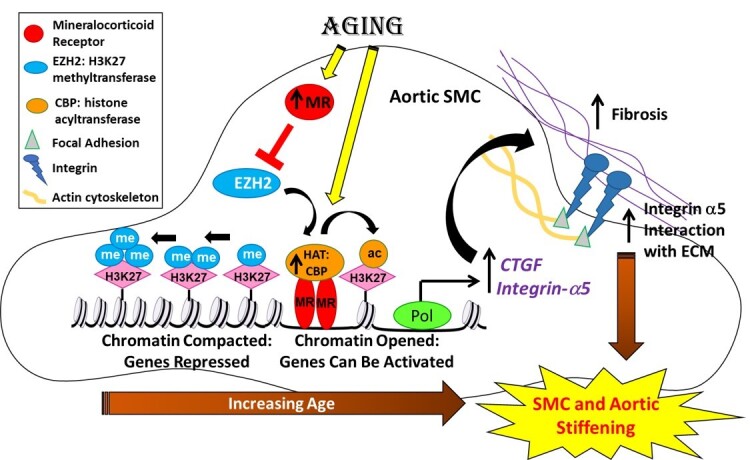
Investigation of histone-modifying enzymes yielded ample data supporting that EZH2 is negatively regulated by MR in the ageing vasculature. Indeed, EZH2 declined with age in mouse vessels and this was reversed by MR antagonism or deletion from SMC. This was consistent with decreased EZH2 in aged human SMC, which increases with MR blockade. EZH2 is a methyltransferase, which mono-, di-, and tri-methylates H3K27.36 EZH2 has been extensively studied in cancers where it is activated to drive tumour growth, cancer cell proliferation, and metastasis.33 While epigenetic mechanisms have been studied in the pathogenesis of heart failure,37 a role for EZH2 in ageing and specifically in vascular stiffness, has not previously been explored. Some studies suggest a role of EZH2 in the vascular function.38,39 EZH2 regulates cell adhesion and angiogenesis,40 maintains vasculature integrity via MMP9,38 and induces the proliferation of pulmonary SMCs.41 Here we show that MR induces a decline in EZH2 with age, which may contribute to vascular stiffness by de-methylation and activation of stiffness genes. This is supported by the observation that in aged mice, MR inhibition increased EZH2 expression and enrichment at the promoter stiffness genes and in SMC from aged humans, MR inhibition decreases vascular stiffness gene expression in an EZH2-dependent manner. Interestingly, in cells from patients with Hutchinson–Gilford Progeria syndrome, a rare condition of rapid early ageing, EZH2 expression and H3K27me3 was found to be decreased.42 Our data support overlap between the epigenetic mechanism driving universal vascular ageing as well as the rare premature ageing in progeria and suggests common therapies (MR antagonists) may be applied. Conversely, this study raises the possibility that EZH2 inhibitors under development for cancer therapy43 may accelerate vascular stiffness and promote premature vascular ageing.
Once H3K27 is de-methylated, it can be acetylated by histone acetyltransferases, of which there are many. We focused on two specific histone acetyltransferase proteins, PCAF/P300 and CBP, because they were previously reported to function as a MR co-activators to induce expression of MR target genes.26 In our study, CBP was consistently increased with age in mouse vessels and HASMCs and it was induced by H2O2 in cultured SMCs. However, whether CBP is directly regulated by the MR is less clear. While the ageing-induced rise in aortic CBP expression in MR-intact mice was prevented in SMC-MR-KO (but not PCAF/P300), MR inhibition did not decrease CBP expression in vitro or in vivo. CBP and P300 have been shown to be increased in abdominal aortic tissue from aneurysm patients vs. controls44 and to increase with age in the heart45 where they contribute to cardiac dysfunction. Thus, our findings are consistent with other studies implicating CBP as a driver of cardiovascular ageing pathology and suggests that the mechanism could involve co-activation of MR target genes; however, further studies are needed to clarify the detailed mechanisms.
One of the principal drivers of vascular stiffness is vascular fibrosis.5,14 We previously found that MR antagonism or SMC-MR-KO attenuates vascular stiffness and fibrosis with aging14 in association with down-regulation of stiffness genes including CTGF, MMP2, and integrin-α5.14 In this study, CTGF and integrin-α5 appear to be true SMC-MR vascular ageing target genes that are regulated through this epigenetic mechanism. Both are increased in aged vs. young mouse vessels and human cells and decrease with MR inhibition or SMC-MR deletion. H2O2-induced ageing of SMC increases H3K27ac and MR enrichment by ChIP at the CTGF and integrin-α5 promoters as well as expression of the mRNA and protein and this is reversed by MR inhibition in vivo. CTGF is an established MR target gene in multiple tissues including heart, kidney, and vessels where it promotes the production and extracellular deposition of collagen thereby contributing to fibrosis in many pathological conditions.29,46 Integrin-α5 and integrin-β1 together form the fibronectin receptor, which links the SMC actin cytoskeleton with the ECM and has been implicated in vascular stiffness in ageing.47 We focused on integrin-α5 because expression profiling showed it is specifically increased with age in mouse aorta, is decreased in aorta from aged SMC-MR-KO mice compared to MR-intact littermates,14 and was found by others to be increased in vessels exposed to hypertension in a SMC-MR-dependent manner.48 We propose that through this epigenetic mechanism, rising MR in ageing SMC drives transcription of CTGF, to promote extracellular collagen deposition, and integrin-α5, which mediates focal adhesions between SMC and the ECM to contribute to vascular stiffness (Graphical abstract).
The results for MMP2 are nuanced and suggest regulation by MR as well as other factors. While MMP2 expression is not increased in aged vs. young HASMC, when aged SMC are treated with MR antagonist, MMP2 goes down and when young HASMCs are treated with H2O2, MMP2 rises and MR localizes to MMP2 promoter. In vivo, spironolactone treatment did not significantly decrease MMP2 protein but it did decrease H3K27ac at the MMP2 promoter. These data suggest that the level of MMP2 expression in the vasculature is a composite of the impact of ageing with other stressors, which may include oxidative stress, and that MMP2 may be regulated by MR and other competing mechanisms depending on the pathologic milieu. This is important because MMP2 is implicated in the pathogenesis of abdominal aortic aneurysm (AAA), a vascular disease that is tightly linked with age, but also exacerbated by other factors including smoking and hypertension. In mouse models of AAA, MR drives aneurisms and MR antagonism is beneficial.49 Interestingly, a panel of histone H3 post-translational modifications were examined in different models of AAA with distinct changes in H3K27me depending on the model and timing of disease progression.50 Thus, future studies are needed to understand MR regulation of MMP2 in the ageing vasculature with clinical implications for AAA progression and treatment.
This study has several limitations that we have attempted to mitigate. First, mouse ageing is not completely analogous to human ageing. To address this, we integrate data from mouse models with studies in human primary cells and whole human tissue thereby providing greater confidence in the veracity of mechanisms when they are reproduced in multiple models. Similarly, cell culture studies are limited by changes in cellular functions when grown on plastic and without surrounding cells. We address this by using only low passage primary cells and by comparing key findings to the results in whole human tissues. Our human tissue sample size is also small and spans a large age range. While this limits the conclusions that can be drawn from the results, we mitigate the limitation by combining the tissue results with the data from cells and mice. Finally, the MR antagonist spironolactone is a very potent MR inhibitor but is not totally specific. This limitation is mitigated by using both MR antagonism and SMC-specific MR deletion and focusing on findings that implicate the MR consistently across multiple models.
Despite these limitations, this study provides several novel insights. We show for the first time that MR rises with age in human SMC and vessels where it drives vascular pathology. We also characterize the global histone post-translational modification profile of ageing vessels and show profound changes that completely distinguish young from old vessels. The combined data support a model (Graphical abstract) in which rising SMC-MR in ageing vessels decreases expression of the H3K27 methyltransferase EZH2, decreasing global and locus-specific aortic H3K27 methylation and acetylation such that MR is recruited to promoters of stiffness genes, including CTGF and integrin-α5, where histone acetyltransferases such as CBP (also increased with ageing) may be recruited to induce H3K27ac and increase stiffness gene expression. Vascular stiffness and MR activation are both associated with adverse cardiovascular outcomes that increase with ageing, including hypertension, heart failure, aneurism formation, and cardiovascular death. Elucidation of the role of MR as a driver of epigenetic changes in the ageing vasculature is important for the development of anti-ageing treatments (such as MR antagonists) and to more safely develop anti-cancer therapies (including EZH2 inhibitors).
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Supplementary Material
Contributor Information
Jaime Ibarrola, Molecular Cardiology Research Institute, Tufts Medical Center, 800 Washington Street, Box 80, Boston, MA 02111, USA.
Seung Kyum Kim, Molecular Cardiology Research Institute, Tufts Medical Center, 800 Washington Street, Box 80, Boston, MA 02111, USA; Department of Sports Science, Seoul National University of Science and Technology, 232 Gongneung-ro, Nowon-gu, 01811 Republic of Korea, Seoul, South Korea.
Qing Lu, Molecular Cardiology Research Institute, Tufts Medical Center, 800 Washington Street, Box 80, Boston, MA 02111, USA.
Jennifer J DuPont, Molecular Cardiology Research Institute, Tufts Medical Center, 800 Washington Street, Box 80, Boston, MA 02111, USA.
Amanda Creech, Broad Institute, Proteomics Platform, Cambridge, MA 02142, USA.
Zhe Sun, Department of Medical Pharmacology and Physiology, Dalton Cardiovascular Research Center, University of Missouri, Columbia, MO 65203, USA.
Michael A Hill, Department of Medical Pharmacology and Physiology, Dalton Cardiovascular Research Center, University of Missouri, Columbia, MO 65203, USA.
Jacob D Jaffe, Broad Institute, Proteomics Platform, Cambridge, MA 02142, USA.
Iris Z Jaffe, Molecular Cardiology Research Institute, Tufts Medical Center, 800 Washington Street, Box 80, Boston, MA 02111, USA.
Funding
This work was supported by a grant from the National Institutes of Health: HL119290 to I.Z.J and M.A.H.
Data availability
The data underlying this article will be shared on reasonable request to the corresponding author. See detailed methods in Online Methods Supplement.
Translational perspective.
These findings provide a new epigenetic mechanism whereby rising MR in ageing human SMC promotes vascular stiffness. Vascular stiffness contributes to common disorders of ageing including hypertension, heart and kidney failure, and stroke, yet no therapies successfully target vascular stiffness. Drugs that inhibit MR are already approved and used in the elderly. In addition, drugs targeting histone-modifying enzymes, including EZH2, are being developed to treat cancer. Thus, these results provide preclinical support for drugs that could be immediately tested to treat ageing-associated vascular stiffness and raise the potential for some cancer therapies to promote vascular stiffness.
References
- 1. North BJ, Sinclair DA. The intersection between aging and cardiovascular disease. Circ Res 2012;110:1097–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mitchell GF, Parise H, Benjamin EJ, Larson MG, Keyes MJ, Vita JA, Vasan RS, Levy D. Changes in arterial stiffness and wave reflection with advancing age in healthy men and women: the Framingham Heart Study. Hypertension 2004;43:1239–1245. [DOI] [PubMed] [Google Scholar]
- 3. Lakatta EG, Wang M, Najjar SS. Arterial aging and subclinical arterial disease are fundamentally intertwined at macroscopic and molecular levels. Med Clin North Am 2009;93:583–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cecelja M, Shanahan CM. Targeting cell stiffness: a paradigm shift in the treatment of aortic stiffness. Circ Res 2021;128:769–771. [DOI] [PubMed] [Google Scholar]
- 5. Harvey A, Montezano AC, Lopes RA, Rios F, Touyz RM. Vascular fibrosis in aging and hypertension: molecular mechanisms and clinical implications. Can J Cardiol 2016;32:659–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ponticos M, Smith BD. Extracellular matrix synthesis in vascular disease: hypertension, and atherosclerosis. J Biomed Res 2014;28:25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pal S, Tyler JK. Epigenetics and aging. Sci Adv 2016;2:e1600584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Illi B, Ciarapica R, Capogrossi MC. Chromatin methylation and cardiovascular aging. J Mol Cell Cardiol 2015;83:21–31. [DOI] [PubMed] [Google Scholar]
- 9. Handy DE, Castro R, Loscalzo J. Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation 2011;123:2145–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu R, Leslie KL, Martin KA. Epigenetic regulation of smooth muscle cell plasticity. Biochim Biophys Acta 2015;1849:448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol 2012;74:13–40. [DOI] [PubMed] [Google Scholar]
- 12. Rogerson FM, Fuller PJ. Mineralocorticoid action. Steroids 2000;65:61–73. [DOI] [PubMed] [Google Scholar]
- 13. Rossier BC, Staub O, Hummler E. Genetic dissection of sodium and potassium transport along the aldosterone-sensitive distal nephron: importance in the control of blood pressure and hypertension. FEBS Lett 2013;587:1929–1941. [DOI] [PubMed] [Google Scholar]
- 14. Kim SK, McCurley AT, DuPont JJ, Aronovitz M, Moss ME, Stillman IE, Karumanchi SA, Christou DD, Jaffe IZ. Smooth muscle cell-mineralocorticoid receptor as a mediator of cardiovascular stiffness with aging. Hypertension 2018;71:609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. DuPont JJ, Kim SK, Kenney RM, Jaffe IZ. Sex differences in the time course and mechanisms of vascular and cardiac aging in mice: role of the smooth muscle cell mineralocorticoid receptor. Am J Physiol Heart Circ Physiol 2021;320:H169–H180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bernini G, Galetta F, Franzoni F, Bardini M, Taurino C, Bernardini M, Ghiadoni L, Bernini M, Santoro G, Salvetti A. Arterial stiffness, intima-media thickness and carotid artery fibrosis in patients with primary aldosteronism. J Hypertens 2008;26:2399–2405. [DOI] [PubMed] [Google Scholar]
- 17. Savoia C, Touyz RM, Amiri F, Schiffrin EL. Selective mineralocorticoid receptor blocker eplerenone reduces resistance artery stiffness in hypertensive patients. Hypertension 2008;51:432–439. [DOI] [PubMed] [Google Scholar]
- 18. Jaffe IZ, Mendelsohn ME. Angiotensin II and aldosterone regulate gene transcription via functional mineralocortocoid receptors in human coronary artery smooth muscle cells. Circ Res 2005;96:643–650. [DOI] [PubMed] [Google Scholar]
- 19. McCurley A, Pires PW, Bender SB, Aronovitz M, Zhao MJ, Metzger D, Chambon P, Hill MA, Dorrance AM, Mendelsohn ME, Jaffe IZ. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat Med 2012;18:1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. DuPont JJ, Kenney RM, Patel AR, Jaffe IZ. Sex differences in mechanisms of arterial stiffness. Br J Pharmacol 2019;176:4208–4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Creech AL, Taylor JE, Maier VK, Wu X, Feeney CM, Udeshi ND, Peach SE, Boehm JS, Lee JT, Carr SA, Jaffe JD. Building the connectivity map of epigenetics: chromatin profiling by quantitative targeted mass spectrometry. Methods 2015;72:57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. DuPont JJ, McCurley A, Davel AP, McCarthy J, Bender SB, Hong K, Yang Y, Yoo JK, Aronovitz M, Baur WE, Christou DD, Hill MA, Jaffe IZ. Vascular mineralocorticoid receptor regulates microRNA-155 to promote vasoconstriction and rising blood pressure with aging. JCI Insight 2016;1:e88942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krug AW, Allenhöfer L, Monticone R, Spinetti G, Gekle M, Wang M, Lakatta EG. Elevated mineralocorticoid receptor activity in aged rat vascular smooth muscle cells promotes a proinflammatory phenotype via extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase and epidermal growth factor receptor-dependent pathways. Hypertension 2010;55:1476–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gopal S, Lu Q, Man JJ, Baur W, Rao SP, Litichevskiy L, Papanastasiou M, Creech AL, DeRuff KC, Mullahoo J, Officer A, Egri SB, Davison D, Jaffe JD, Jaffe IZ. A phosphoproteomic signature in endothelial cells predicts vascular toxicity of tyrosine kinase inhibitors used in CML. Blood Adv 2018;2:1680–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pan MR, Hsu MC, Chen LT, Hung WC. Orchestration of H3K27 methylation: mechanisms and therapeutic implication. Cell Mol Life Sci 2018;75:209–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fuse H, Kitagawa H, Kato S. Characterization of transactivational property and coactivator mediation of rat mineralocorticoid receptor activation function-1 (AF-1). Mol Endocrinol 2000;14:889–899. [DOI] [PubMed] [Google Scholar]
- 27. Sun Z, Martinez-Lemus LA, Trache A, Trzeciakowski JP, Davis GE, Pohl U, Meininger GA. Mechanical properties of the interaction between fibronectin and alpha5beta1-integrin on vascular smooth muscle cells studied using atomic force microscopy. Am J Physiol Heart Circ Physiol 2005;289:H2526–H2535. [DOI] [PubMed] [Google Scholar]
- 28. Caldini R, Chevanne M, Mocali A, Tombaccini D, Paoletti F. Premature induction of aging in sublethally H2O2-treated young MRC5 fibroblasts correlates with increased glutathione peroxidase levels and resistance to DNA breakage. Mech Ageing Dev 1998;105:137–150. [DOI] [PubMed] [Google Scholar]
- 29. Buonafine M, Bonnard B, Jaisser F. Mineralocorticoid receptor and cardiovascular disease. Am J Hypertens 2018;31:1165–1174. [DOI] [PubMed] [Google Scholar]
- 30. Epstein M, Calhoun DA. Aldosterone blockers (mineralocorticoid receptor antagonism) and potassium-sparing diuretics. J Clin Hypertens 2011;13:644–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ayuzawa N, Nagase M, Ueda K, Nishimoto M, Kawarazaki W, Marumo T, Aiba A, Sakurai T, Shindo T, Fujita T. Rac1-mediated activation of mineralocorticoid receptor in pressure overload-induced cardiac injury. Hypertension 2016;67:99–106. [DOI] [PubMed] [Google Scholar]
- 32. Nishiyama A. Pathophysiological mechanisms of mineralocorticoid receptor-dependent cardiovascular and chronic kidney disease. Hypertens Res 2019;42:293–300. [DOI] [PubMed] [Google Scholar]
- 33. Nagase M, Fujita T. Role of Rac1-mineralocorticoid-receptor signalling in renal and cardiac disease. Nat Rev Nephrol 2013;9:86–98. [DOI] [PubMed] [Google Scholar]
- 34. Greißel A, Culmes M, Burgkart R, Zimmermann A, Eckstein HH, Zernecke A, Pelisek J. Histone acetylation and methylation significantly change with severity of atherosclerosis in human carotid plaques. Cardiovasc Pathol 2016;25:79–86. [DOI] [PubMed] [Google Scholar]
- 35. Greißel A, Culmes M, Napieralski R, Wagner E, Gebhard H, Schmitt M, Zimmermann A, Eckstein H-H, Zernecke A, Pelisek J. Alternation of histone and DNA methylation in human atherosclerotic carotid plaques. Thromb Haemost 2015;114:390–402. [DOI] [PubMed] [Google Scholar]
- 36. Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, Dynlacht BD, Reinberg D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell 2008;32:503–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu CF, Tang WHW. Epigenetics in cardiac hypertrophy and heart failure. JACC Basic Transl Sci 2019;4:976–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Delgado-Olguín P, Dang LT, He D, Thomas S, Chi L, Sukonnik T, Khyzha N, Dobenecker MW, Fish JE, Bruneau BG. Ezh2-mediated repression of a transcriptional pathway upstream of Mmp9 maintains integrity of the developing vasculature. Development 2014;141:4610–4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mitić T, Caporali A, Floris I, Meloni M, Marchetti M, Urrutia R, Angelini GD, Emanueli C. EZH2 modulates angiogenesis in vitro and in a mouse model of limb ischemia. Mol Ther 2015;23:32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dreger H, Ludwig A, Weller A, Stangl V, Baumann G, Meiners S, Stangl K. Epigenetic regulation of cell adhesion and communication by enhancer of zeste homolog 2 in human endothelial cells. Hypertension 2012;60:1176–1183. [DOI] [PubMed] [Google Scholar]
- 41. Aljubran SA, Cox R, Tamarapu Parthasarathy P, Kollongod Ramanathan G, Rajanbabu V, Bao H, Mohapatra SS, Lockey R, Kolliputi N. Enhancer of zeste homolog 2 induces pulmonary artery smooth muscle cell proliferation. PLoS One 2012;7:e37712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS, Jenuwein T, Goldman RD. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci USA 2006;103:8703–8708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Duan R, Du W, Guo W. EZH2: a novel target for cancer treatment. J Hematol Oncol 2020;13:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Han Y, Tanios F, Reeps C, Zhang J, Schwamborn K, Eckstein HH, Zernecke A, Pelisek J. Histone acetylation and histone acetyltransferases show significant alterations in human abdominal aortic aneurysm. Clin Epigenetics 2016;8:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ghosh AK. p300 in cardiac development and accelerated cardiac aging. Aging Dis 2020;11:916–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Newfell BG, Iyer LK, Mohammad NN, McGraw AP, Ehsan A, Rosano G, Huang PL, Mendelsohn ME, Jaffe IZ. Aldosterone regulates vascular gene transcription via oxidative stress-dependent and -independent pathways. Arterioscler Thromb Vasc Biol 2011;31:1871–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lakatta EG, Levy D. Arterial cardiac aging: major shareholders in cardiovascular disease enterprises: part II: the aging heart in health: links to heart disease. Circulation 2003;107:346–354. [DOI] [PubMed] [Google Scholar]
- 48. Galmiche G, Pizard A, Gueret A, El Moghrabi S, Ouvrard-Pascaud A, Berger S, Challande P, Jaffe IZ, Labat C, Lacolley P, Jaisser F. Smooth muscle cell mineralocorticoid receptors are mandatory for aldosterone-salt to induce vascular stiffness. Hypertension 2014;63:520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu S, Xie Z, Daugherty A, Cassis LA, Pearson KJ, Gong MC, Guo Z. Mineralocorticoid receptor agonists induce mouse aortic aneurysm formation and rupture in the presence of high salt. Arterioscler Thromb Vasc Biol 2013;33:1568–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Greenway J, Gilreath N, Patel S, Horimatsu T, Moses M, Kim D, Reid L, Ogbi M, Shi Y, Lu XY, Shukla M, Lee R, Huo Y, Young L, Kim HW, Weintraub NL. Profiling of histone modifications reveals epigenomic dynamics during abdominal aortic aneurysm formation in mouse models. Front Cardiovasc Med 2020;7:595011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article will be shared on reasonable request to the corresponding author. See detailed methods in Online Methods Supplement.
Translational perspective.
These findings provide a new epigenetic mechanism whereby rising MR in ageing human SMC promotes vascular stiffness. Vascular stiffness contributes to common disorders of ageing including hypertension, heart and kidney failure, and stroke, yet no therapies successfully target vascular stiffness. Drugs that inhibit MR are already approved and used in the elderly. In addition, drugs targeting histone-modifying enzymes, including EZH2, are being developed to treat cancer. Thus, these results provide preclinical support for drugs that could be immediately tested to treat ageing-associated vascular stiffness and raise the potential for some cancer therapies to promote vascular stiffness.