

Abstract
Summary
echolocatoR integrates a diverse suite of statistical and functional fine-mapping tools to identify, test enrichment in, and visualize high-confidence causal consensus variants in any phenotype. It requires minimal input from users (a summary statistics file), can be run in a single R function, and provides extensive access to relevant datasets (e.g. reference linkage disequilibrium panels, quantitative trait loci, genome-wide annotations, cell-type-specific epigenomics), thereby enabling rapid, robust and scalable end-to-end fine-mapping investigations.
Availability and implementation
echolocatoR is an open-source R package available through GitHub under the GNU General Public License (Version 3) license: https://github.com/RajLabMSSM/echolocatoR.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Genome-wide association studies (GWAS) across a variety of phenotypes and quantitative trait loci (QTL) have identified many significant genetic associations. However, widespread non-independence between genomic variants due to linkage disequilibrium (LD) makes it difficult to distinguish causal variants from correlated non-causal variants (Pasaniuc and Price, 2017; Pritchard and Przeworski, 2001; Yang et al., 2011). Fine-mapping aims to identify the causal variant(s) and thus the mechanisms underlying a phenotype (Broekema et al., 2020; Hutchinson et al., 2020; Schaid et al., 2018; Spain and Barrett, 2015). This methodology has been especially important to the study of medical conditions such as diabetes (Gaulton et al., 2015; Mahajan et al., 2018), rheumatoid arthritis (Kichaev and Pasaniuc, 2015; Westra et al., 2018) and obesity (Zhang et al., 2018).
Many fine-mapping tools have been developed over the years (Broekema et al., 2020; Hutchinson et al., 2020; Schaid et al., 2018; Spain and Barrett, 2015), each of which can nominate partially overlapping sets of putative causal variants. It can therefore be useful to compare results from multiple fine-mapping methods with complementary strengths and weaknesses, such as the ability to model multiple causal variants or incorporate functional annotations. However, these powerful methods are underutilized in no small part due to technical reasons (e.g. lack of availability in the same programming language, idiosyncratic file inputs/outputs, gathering and formatting of datasets). We therefore developed echolocatoR, an open-source R package that conducts end-to-end statistical and functional fine-mapping, annotation, enrichment and plotting that only requires GWAS/QTL summary statistics as input (Fig. 1a). In addition, we have launched the echolocatoR Fine-mapping Portal (https://rajlab.shinyapps.io/Fine_Mapping_Shiny), an interactive database of standardized fine-mapping results across 11+ GWAS/QTL datasets ( Navarro et al., 2021; de Paiva Lopes et al., 2021; Schilder and Raj 2020). All of these fine-mapping results, plots and associated LD data are API-searchable and accessible, and can be imported directly into R via designated echolocatoR functions (see vignette: https://rajlabmssm.github.io/echolocatoR/articles/PD_loci_vignette.html).
Fig. 1.
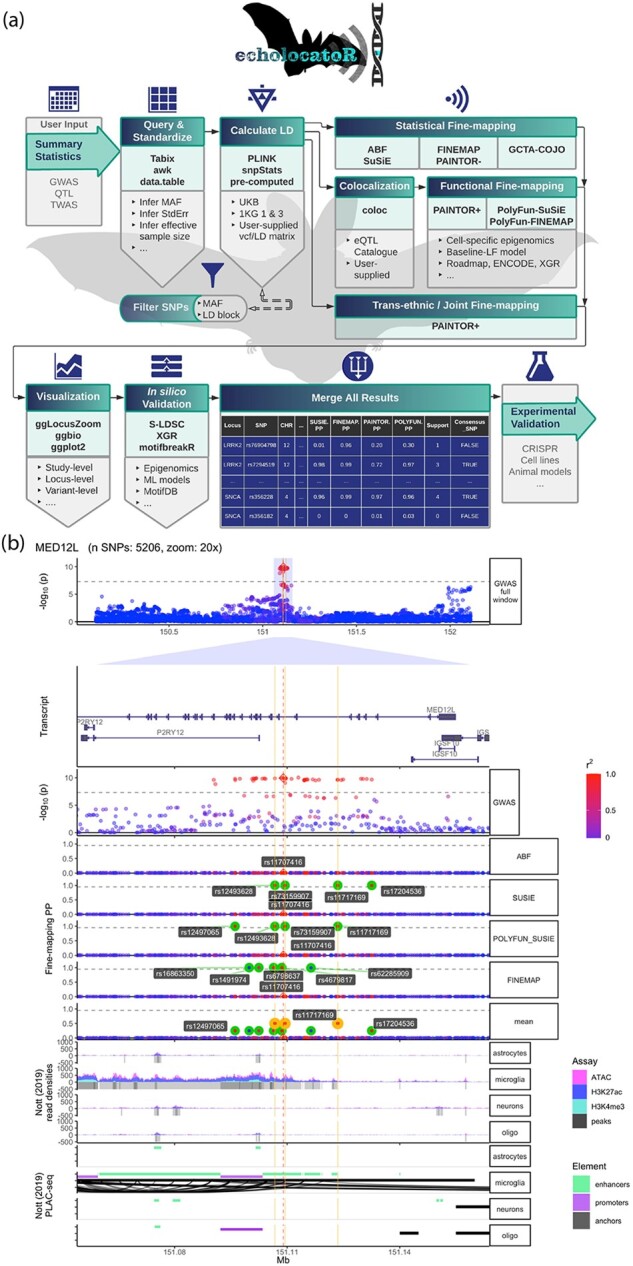
echolocatoR facilitates automated end-to-end fine-mapping. (a) Workflow of the echolocatoR pipeline: (i) user specifies the path to their full GWAS/QTL summary statistics, (ii) locus subsets are queried and saved in a standardized format, (iii) LD is extracted, computed from VCF or supplied by the user, (iv) statistical, functional and/or trans-ethnic/joint fine-mapping are performed, (v) results are visualized at study-, locus- and variant-level scales, (vi) in silico validation tests for differences in functional impact between SNP groups of interest, (vii) GWAS/QTL summary statistics, fine-mapping results and annotations are merged into a file with one SNP per row, (viii) narrowed SNPs lists can be targeted in validation experiments. (b) Example multi-track plot for the Parkinson’s Disease locus MED12L: (i) Manhattan plot of GWAS -log10(P-values) colored by the degree of correlation (r2) with the lead SNP, (ii) gene transcript models, (iii) GWAS -log10(P-values) zoomed in at 20×, (iv) per-SNP posterior probabilities (PP) from four different fine-mapping tools, (v) histogram and called peaks across multiple brain cell-type-specific epigenomic assays (Nott et al., 2019), (vi) cell-type-specific PLAC-seq interactions, PLAC-seq anchors, enhancers and promoters (Nott et al., 2019). The vertical red line indicates the location of the lead GWAS SNP, while the vertical gold lines indicate the location of Consensus SNPs
2 Implementation
The full echolocatoR fine-mapping pipeline can be run using just the finemap_loci() function, which ultimately produces an organized folder structure containing study- and locus-specific multi-tool fine-mapping results tables and annotated multi-track plots. If some stage of the pipeline has been run previously for a given locus, finemap_loci() will automatically detect and use the associated files, saving time for when testing different parameters. Most echolocatoR functions can run on a standard laptop (tested on a MacBook Pro with a 2.3 GHz Intel Core i5 processor and 8 GB 2133 MHz LPDDR3 memory), or take full advantage of its parallelizing capabilities on a high-performance computing (HPC) cluster.
2.1. Rapid, robust and scalable fine-mapping
By default, echolocatoR automatically indexes the user’s summary statistics file using Tabix (Li, 2011) for rapid on the fly querying. Locus-specific summary statistics are then extracted, standardized and filtered according to user-controllable parameters such as window size (±1 Mb surrounding the index SNP by default), minor allele frequency (MAF) threshold, LD block and many other features.
echolocatoR integrates a suite of existing fine-mapping tools, which currently includes: ABF (Benner et al., 2016; Wakefield, 2007; Wellcome Trust Case Control Consortium et al., 2012), GCTA-COJO (Yang et al., 2012), FINEMAP (Benner et al., 2016), SuSiE (Wang et al., 2020), PolyFun (Weissbrod et al., 2020) and PAINTOR (Kichaev et al., 2017), the latter of which can be run with (i.e. PAINTOR+) or without (PAINTOR-) functional annotations. Colocalization tests between pairs of GWAS and/or QTL can also be performed using coloc (Giambartolomei et al., 2014) to identify locus-specific phenotype-relevant tissues and cell types and prioritize GWAS/QTL datasets for joint functional fine-mapping.
Each fine-mapping tool produces its own 95% Credible Set (CS95%). The precise meaning of this term varies by tool but can be understood as the SNPs with 95% probability of being causal in the phenotype of interest. However, inter-tool comparisons have observed that there is substantial heterogeneity in their CS95% (Weissbrod et al., 2020), leading to questions about the validity of any single tool in all situations, which can be strongly influenced by the degree of LD complexity and the true number of causal SNPs (Pasaniuc and Price, 2017; Pritchard and Przeworski, 2001; Yang et al., 2011). We, therefore, define Consensus SNPs as those that were identified in the CS95% of two or more tools, representing high-confidence putative causal SNPs. Indeed, we have shown that these Consensus SNPs have significantly higher predicted regulatory impact than either lead GWAS SNPs or individual tool CS95% SNP sets in Parkinson’s Disease (PD) (Schilder and Raj, 2020). Within the results files, echolocatoR automatically adds columns for Support (the number of tools that a given SNP was in the CS95%), Consensus SNP status, as well as mean posterior probabilities (PP) across all fine-mapping tools used.
2.2. Extensive database access
A common barrier to performing accurate fine-mapping is access to the appropriate LD reference panels (Benner et al., 2017). Currently, API access is provided for 1000 Genomes Phases 1 & 3 (with selectable subpopulations) (The 1000 Genomes Project Consortium, 2015), UK Biobank (Bycroft et al., 2018; Sudlow et al., 2015; Weissbrod et al., 2020) or user-supplied VCF files or LD matrices. Unlike existing LD querying tools (Machiela and Chanock, 2015), echolocatoR does not restrict the size of LD matrices to allow comprehensive fine-mapping of all loci regardless of size or complexity.
2.3. Genome-wide annotations
Genome-wide annotations can be used to compute SNP-wise prior probabilities for functional fine-mapping (e.g. PolyFun, PAINTOR+). API access to a large compendium of genome-wide annotations and epigenomic data is provided, including tissue and/or cell type/line-specific chromatin marks from Roadmap (Bernstein et al., 2010; Satterlee et al., 2019), ENCODE (Jou et al., 2019), genic annotations through biomaRt (Durinck et al., 2009), HaploReg (Ward and Kellis, 2012; Zhbannikov et al., 2017), cell-type-specific epigenomic datasets (Nott et al., 2019; Corces et al., 2020) and hundreds of additional annotations through the R package XGR (Fang et al., 2016). catalogueR (https://github.com/RajLabMSSM/catalogueR), another R package developed by our group, provides rapid API access to full summary statistics from 112 uniformly reprocessed QTL datasets (across 21 studies) with parallelized Tabix queries. echolocatoR can utilize all genome-wide annotations and datasets to compare enrichment across different SNP groups (e.g. GWAS lead SNPs versus CS95% versus Consensus SNPs) using XGR (Fang et al., 2016), GoShifter (Trynka et al., 2015), S-LDSC (Bulik-Sullivan et al., 2015; Finucane et al., 2015; Gazal et al., 2017) and/or bootstrapping analyses.
2.4. In silico validation
We also built in API access to in silico validation datasets, including massively parallel reporter assays (MPRA) (Tewhey et al., 2018; van Arensbergen et al., 2019), S-LDSC heritability enrichment and predictions from multiple machine learning models trained on tissue- and cell-type-specific epigenomic annotations: Basenji (Kelley et al., 2018) and DeepSEA (Zhou and Troyanskaya, 2015) (provided by Dey et al. (2020)) as well as IMPACT (Amariuta et al., 2019). Finally, we integrated motifbreakR which uses a comprehensive set of algorithms and position weight matrices (n = 9933) to assess whether fine-mapped variants fall within sequence motifs and to what extent they disrupt binding to specific transcription factors (Coetzee et al., 2015).
2.5. Multi-track plotting
High-resolution multi-track plots are automatically generated for each locus (Fig. 1b) and can include any combination of the following tracks: Manhattan plots of GWAS/QTL P-values or tool-specific fine-mapping PP colored by LD with the lead SNP, mean PP, gene body models and all aforementioned genome-wide annotations. Plots can be further customized as returned patchwork or ggplot objects.
3 Conclusion
Overall, echolocatoR removes many of the primary barriers to perform a comprehensive fine-mapping investigation while improving the robustness of causal variant prediction through multi-tool consensus SNP identification and in silico validation using a large compendium of (epi)genome-wide annotations. Thus, we hope that echolocatoR will make fine-mapping a standard practice, thereby uncovering human disease etiology and accelerating the development of novel therapeutics.
Acknowledgements
The authors thank Elisa Navarro, Gloriia Novikova, Cecilia Lindgren and Teresa Ferreira for their valuable feedback and suggestions. They also thank Omer Weissbrod, Chris Glass and Alexi Nott for their guidance with data and/or tool integration. This work is dedicated to Robert Neil Cronin.
Funding
This work was supported by grants from the Michael J. Fox Foundation [Grant #14899 and #16743] and US National Institutes of Health [NIH NIA R01-AG054005]. B.M.S. was additionally supported by the UK Dementia Research Institute which receives its funding from UK DRI Ltd, funded by the UK Medical Research Council, Alzheimer's Society and Alzheimer's Research UK. This work was supported in part through the computational resources provided by Scientific Computing at the ISMMS.
Data availability
All code for echolocatoR is available on GitHub https://github.com/RajLabMSSM/echolocatoR. Fine-mapping results generated by echolocatoR can be found on the echolocatoR Fine-mapping Portal:https://rajlab.shinyapps.io/Fine_Mapping_Shiny.
Conflict of Interest: none declared.
Contributor Information
Brian M Schilder, Nash Family Department of Neuroscience & Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York 10029, NY, USA; Ronald M. Loeb Center for Alzheimer’s Disease, Icahn School of Medicine at Mount Sinai, New York 10029, NY, USA; Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York 10029, NY, USA; Icahn Institute for Data Science and Genomic Technology, Icahn School of Medicine at Mount Sinai, New York 10029, NY, USA; Estelle and Daniel Maggin Department of Neurology, Icahn School of Medicine at Mount Sinai, New York 10029, NY, USA.
Jack Humphrey, Nash Family Department of Neuroscience & Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York 10029, NY, USA; Ronald M. Loeb Center for Alzheimer’s Disease, Icahn School of Medicine at Mount Sinai, New York 10029, NY, USA; Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York 10029, NY, USA; Icahn Institute for Data Science and Genomic Technology, Icahn School of Medicine at Mount Sinai, New York 10029, NY, USA; Estelle and Daniel Maggin Department of Neurology, Icahn School of Medicine at Mount Sinai, New York 10029, NY, USA.
Towfique Raj, Nash Family Department of Neuroscience & Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York 10029, NY, USA; Ronald M. Loeb Center for Alzheimer’s Disease, Icahn School of Medicine at Mount Sinai, New York 10029, NY, USA; Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York 10029, NY, USA; Icahn Institute for Data Science and Genomic Technology, Icahn School of Medicine at Mount Sinai, New York 10029, NY, USA; Estelle and Daniel Maggin Department of Neurology, Icahn School of Medicine at Mount Sinai, New York 10029, NY, USA.
References
- Amariuta T. et al. ; RACI Consortium, GARNET Consortium. (2019) IMPACT: genomic annotation of cell-state-specific regulatory elements inferred from the epigenome of bound transcription factors. Am. J. Hum. Genet., 104, 879–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Arensbergen J. et al. (2019) High-throughput identification of human SNPs affecting regulatory element activity. Nat. Genet., 51, 1160–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benner C. et al. (2016) FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics, 32, 1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benner C. et al. (2017) Prospects of fine-mapping trait-associated genomic regions by using summary statistics from genome-wide association studies. Am. J. Hum. Genet., 101, 539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein B.E. et al. (2010) The NIH roadmap epigenomics mapping consortium. Nat. Biotechnol., 28, 1045–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broekema R.V. et al. (2020) A practical view of fine-mapping and gene prioritization in the post-genome-wide association era. Open Biol., 10, 190221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulik-Sullivan B.K. et al. ; Schizophrenia Working Group of the Psychiatric Genomics Consortium. (2015) LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet., 47, 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bycroft C. et al. (2018) The UK Biobank resource with deep phenotyping and genomic data. Nature, 562, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee S.G. et al. (2015) motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics, 31, 3847–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces M.R. et al. (2020) Single-cell epigenomic identification of inherited risk loci in Alzheimer’s and Parkinson’s disease. Nat. Genet., 52, 1158–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Paiva Lopes,K. et al. (2021) Atlas of genetic effects in human microglia transcriptome across brain regions, aging, and disease pathologies. Accepted at Nat. Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey K.K. et al. (2020) Evaluating the informativeness of deep learning annotations for human complex diseases. Nat. Commun., 11, 4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S. et al. (2009) Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc., 4, 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H. et al. (2016) XGR software for enhanced interpretation of genomic summary data, illustrated by application to immunological traits. Genome Med., 8, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane H.K., RACI Consortium. et al. (2015) Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet., 47, 1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton K.J. et al. ; DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium. (2015) Genetic fine mapping and genomic annotation defines causal mechanisms at type 2 diabetes susceptibility loci. Nat. Genet., 47, 1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazal S. et al. (2017) Linkage disequilibrium-dependent architecture of human complex traits shows action of negative selection. Nat. Genet., 49, 1421–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giambartolomei C. et al. (2014) Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet., 10, e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson A. et al. (2020) Fine-mapping genetic associations. Hum. Mol. Genet., 29, R81–R88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jou J. et al. (2019) The ENCODE Portal as an Epigenomics Resource. Curr. Protoc. Bioinf., 68, e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley D.R. et al. (2018) Sequential regulatory activity prediction across chromosomes with convolutional neural networks. Genome Res., 28, 739–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kichaev G. et al. (2017) Improved methods for multi-trait fine mapping of pleiotropic risk loci. Bioinformatics, 33, 248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kichaev G., Pasaniuc B. (2015) Leveraging functional-annotation data in trans-ethnic fine-mapping studies. Am. J. Hum. Genet., 97, 260–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2011) Tabix: fast retrieval of sequence features from generic TAB-delimited files. Bioinformatics, 27, 718–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machiela M.J., Chanock S.J. (2015) LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics, 31, 3555–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan A. et al. (2018) Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet., 50, 1505–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro,E. et al. (2021) Dysregulation of mitochondrial and proteo-lysosomal genes in Parkinson's disease myeloid cells. Accepted at Nat. Aging. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott A. et al. (2019) Brain cell type-specific enhancer-promoter interactome maps and disease risk association. Science, 366, 1134–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasaniuc B., Price A.L. (2017) Dissecting the genetics of complex traits using summary association statistics. Nat. Rev. Genet., 18, 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard J.K., Przeworski M. (2001) Linkage disequilibrium in humans: models and data. Am. J. Hum. Genet., 69, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterlee J.S. et al. (2019) The NIH common fund/roadmap epigenomics program: successes of a comprehensive consortium. Sci. Adv., 5, eaaw6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaid D.J. et al. (2018) From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet., 19, 491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilder B.M., Raj T. (2021) Fine-mapping of Parkinson’s disease susceptibility loci identifies putative causal variants. Accepted at Hum. Mol. Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spain S.L., Barrett J.C. (2015) Strategies for fine-mapping complex traits. Hum. Mol. Genet., 24, R111–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudlow C. et al. (2015) UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med., 12, e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewhey R. et al. (2018) Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell, 172, 1132–1134. [DOI] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trynka G. et al. (2015) Disentangling the effects of colocalizing genomic annotations to functionally prioritize non-coding variants within complex-trait loci. Am. J. Hum. Genet., 97, 139–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield J. (2007) A Bayesian measure of the probability of false discovery in genetic epidemiology studies. Am. J. Hum. Genet., 81, 208–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G. et al. (2020) A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Soc. Ser. B., 82, 1273–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward L.D., Kellis M. (2012) HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res., 40, D930–D934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissbrod,O. et al. (2020) Functionally informed fine-mapping and polygenic localization of complex trait heritability. Nature Genetics, 52, 1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium. et al. (2012) Bayesian refinement of association signals for 14 loci in 3 common diseases. Nat. Genet., 44, 1294–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra H.-J. et al. (2018) Fine-mapping and functional studies highlight potential causal variants for rheumatoid arthritis and type 1 diabetes. Nat. Genet., 50, 1366–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. et al. ; DIAbetes Genetics Replication and Meta-analysis (DIAGRAM) Consortium. (2012) Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet., 44, 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. et al. ; The GIANT Consortium. (2011) Genomic inflation factors under polygenic inheritance. Eur. J. Hum. Genet., 19, 807–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. et al. (2018) A fine-mapping study of central obesity loci incorporating functional annotation and imputation. Eur. J. Hum. Genet., 26, 1369–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhbannikov I.Y. et al. (2017) haploR: an R-package for querying web-based annotation tools. F1000Research, 6, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Troyanskaya O.G. (2015) Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods, 12, 931–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All code for echolocatoR is available on GitHub https://github.com/RajLabMSSM/echolocatoR. Fine-mapping results generated by echolocatoR can be found on the echolocatoR Fine-mapping Portal:https://rajlab.shinyapps.io/Fine_Mapping_Shiny.
Conflict of Interest: none declared.