

Abstract
Introduction:
Solid-phase synthesis and conjugation reactions of acids and amines using coupling reagents are common in organic synthesis, but rare in 18F radiochemistry. 4-[18F]Fluorobenzoic acid (FBA) is a useful building block, but is seldom used directly with coupling reagents for the preparation of 18F radiopharmaceuticals. To overcome the inconveniences associated with using [18F]FBA in conjugation reactions, we have developed a non-covalent solid-phase synthesis (SPS) strategy for the radiosynthesis of [18F]PARPi, a derivative of olaparib as a Poly (ADP-ribose) polymerase-1 (PARP-1) radioligand.
Methods:
Fluoro-, bromo- and iodo-benzoic derivatives of olaparib were synthesized, and their PARP-1 affinities were measured using a recently developed cell culture-based competitive assay. To produce [18F]PARPi, [18F]FBA was radiosynthesized and purified using a cation-exchange cartridge, and then trapped by an anion-exchange resin cartridge, on which the solid-phase radiosynthesis was carried out to produce the desired product. [18F]PARPi was evaluated in vivo in breast and prostate xenograft tumor models by microPET imaging, biodistribution and autoradiography.
Results:
The best derivatives of olaparib were identified as compound 4, 7 and 8. [18F]4 ([18F]PARPi) was radiosynthesized in high radiochemical yield, high molar activity and high radiochemical purity using this SPS strategy. The in vivo evaluation of [18F]PARPi demonstrates the PARP-1 specific uptake of [18F]PARPi in the animal models.
Conclusions:
This method is simple and efficient, having great potential for the synthesis of radiopharmaceuticals starting from [18F]FBA or other radiolabeled aromatic acids. Using [18F]PARPi prepared by this method, we demonstrated the promise of [18F]PARPi in the nuclear imaging of PARP-1 expression.
Keywords: Solid phase radiosynthesis, 4-[18F]fluorobenzoic acid, PARP-1, PET imaging, olaparib
Graphical Abstract
1. Introduction.
Solid-phase synthesis and conjugation reactions of acids and amines using coupling reagents are common in organic synthesis but very rare in 18F radiosynthesis [1–3]. 4-[18F]Fluorobenzoic acid ([18F]FBA) is a useful building block, commonly used as the active ester, N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB), which is generated from ([18F]FBA). The direct use of [18F]FBA is challenging because (a) basic conditions are required for the nucleophilic substitution reaction of [18F]fluoride; (b) strongly basic or acidic conditions are needed for the deprotection of the ester intermediates of [18F]FBA; and (c) side-products may interfere with the conjugation reactions. Recently, the direct use of [18F]FBA after labeling and deprotection was reported in the preparation of [18F]PARPi, a derivative of olaparib, for nuclear imaging of Poly (ADP-ribose) polymerase-1 (PARP-1) expression in tumors [4].
Poly (ADP-ribose) polymerase-1 (PARP-1) is an enzyme that plays a major role in repairing single-stranded DNA breaks and has been actively pursued as a therapeutic target for cancers [5] that are deficient in certain DNA repair mechanisms and thus depend on PARP-1 activity for survival [6] by inducing synthetic lethality. Olaparib, along with other PARP-1 inhibitors, has been approved by the US Food and Drug Administration (FDA) to treat certain cancers. Because of the important roles of PARP-1 in cancer treatment, several PARP-1 positron emission tomography (PET) imaging agents have been reported [7]. Among these, [18F]FluorThanatrace ([18F]FTT) [8] and [18F]PARPi [9] are undergoing clinical trials. The reported method for the radiosynthesis of [18F]PARPi uses a small amount of acid and base in a small volume of water, and then large quantities of the coupling reagent to overcome the challenges in the conjugation reaction in this aqueous medium. The result is satisfying but automation of the production is difficult. A one-pot labeling of [18F]PARPi is also reported, but with low radiochemical yields [10]. In this paper, we report the conjugation reaction of [18F]FBA on a solid-support for the rapid and convenient preparation of [18F]PARPi, and the evaluation of ([18F]PARPi for imaging PARP-1 expression in breast and prostate cancer xenograft models.
2. Method and General Information
2.1. General Information
All chemicals were obtained from standard commercial sources and used without further purification. All reactions were carried out by standard air-free and moisture-free techniques under an inert atmosphere with dry solvents unless otherwise stated. No-carrier added [18F]fluoride was produced by 18O(p, n)18F reaction through proton irradiation of 18O-enriched water (95%) using a RDS111 cyclotron. High performance liquid chromatography (HPLC) SpectraSYSTEM P4000 (Thermo Fisher Scientific, Waltham, MA, USA) was performed with an ultraviolet detector and a well-scintillation NaI (Tl) detector and associated electronics for radioactivity detection. Radio-TLC was accomplished using a Bioscan AR-2000 imaging scanner (Bioscan, Inc., Washington, DC, USA). Published methods were used for the synthesis of compound 1 [11], 3 and 4 and its derivatives 5, 6, 7, 8, and 9 [12]. All animal experiments were conducted under Washington University Animal Studies Committee IACUC-approved protocols in accordance with the recommendations of the National Research Council’s “Guide for the Care and Use of Laboratory Animals”.
2.2. Radiosynthesis of [18F]4 ([18F]PARPi)
[18F]Fluoride (1.85 GBq) in [18O]water was dried azeotropically in the presence of potassium carbonate (1 mg, 7.25 μmol) and kryptofix 222 (5.6 mg, 14.9 μmol) using typical drying protocol (3 × 1 mL acetonitrile/nitrogen/105 °C). Into the dried residue was added the labeling precursor 1 (3.5 mg, 9.8 μmol) in DMSO (300 μL) and heated at 83 °C for 6 min. For deprotection, 1N NaOH (100 μL) was added and the mixture was heated at 105 °C for 5 min. At room temperature, the mixture was neutralized with water (9 mL) and 1N HCl (125 μL), and then loaded onto a Waters MCX cartridge (6 cc), pretreated with methanol (10 mL) and water (10 mL). The cartridge was rinsed with water (20 mL), and then the radioactivity was eluted sequentially with methanol (4 mL) and water (20 mL). The eluted radioactivity was diluted with water to 40 mL, and loaded onto an MAX cartridge, which was prepared by repacking Waters Oasis MAX resin (200 mg from a 500 mg cartridge (cat# 186000865)) in a 3cc empty cartridge and treating the cartridge sequentially with methanol (9 mL), 0.5 N HCL (9 mL) and water (9 mL). The cartridge was rinsed with water (10 mL) and acetonitrile (3 mL) and dried by air (10 mL). A solution of the amine precursor 3 (2.5 mg, 6.8 μmol) and DIPEA (10 μL) in DMF (300 μL) was mixed with a solution of HATU (2.5 mg, 6.6 μmol) in DMF (100 μL) and loaded onto the MAX cartridge under gravity. After 10 min at room temperature, the product was eluted sequentially with acetonitrile (0.5 mL) and 0.2% TFA in water (4 mL). The eluted mixture was injected onto an HPLC column for purification (Column: Agilent SB-C18 250 × 9.4 mm 5 μm; Mobile phase: 31 % acetonitrile/69 % water/0.1 % TFA; Flow rate: 4 mL/min; UV: 254 nm). The radioactivity corresponding to [18F]4 was collected at 25–27 min and diluted with water to 40 mL. The solution was passed through a Waters HLB light cartridge. After the cartridge was rinsed with water (10 mL) to remove residual solvent, the radioactivity was eluted with ethanol in portions (0.2 mL per portion). The final dose was prepared in 10 % ethanol in saline. The molar activity and the radiochemical purity were both determined by analytical radio-HPLC (Column: Altima C18 250×4.6 mm 10 μm; Mobile phase: 40 % acetonitrile/60 % water/0.1 % TFA; Flow rate: 1.5 mL/min; UV: 254 nm), along with radio-TLC (silica gel/20 % methanol in dichloromethane) for free [18F]fluoride.
2.3. Radioactive competitive PAPR-1 affinity assay
PAPR-1 affinity assay was conducted using U251MG cell culture and using the selective PARP-1 radiotracer [3H]WC-DZ to obtain the dissociation constants (Ki values) and the pseudo-Hill coefficients (n’H values) of the ligands as we recently published [13].
2.4. Xenograft models
Mature athymic nu/nu mice from Charles River Laboratories (Wilmington, MA, USA) are allowed to acclimate in an AALAC accredited housing facility for at least 1 week prior to tumor implantation for these imaging studies. Female nu/nu mice (7 weeks old, n = 5) were implanted in the mammary fat pads (near the axillary lymph nodes) with 8 × 106 MDA-MB-231 breast cancer cells in ~100 μL Matrigel in PBS (v/v; 1:1). Imaging studies were conducted 20–21 days post implantation. Similarly, Male nu/nu mice (7 weeks old, n = 5) were implanted subcutaneously in the right flank with 3 × 106 PC-3 prostate cancer cells. The tumors were allowed to grow for 18–19 days before the imaging studies.
2.5. MicroPET
Tumor-bearing mice (baseline n =2; blocking n = 2) were placed in an induction chamber containing ~2% isoflurane/oxygen and then secured to a custom quadruple bed for placement of tail vein catheters; anesthesia was maintained via nose-cone at ~1% isoflurane/oxygen for the dynamic imaging procedure. The mice were injected with 7.4 MBq of [18F]4 and scanned for 60 min using Focus 220 and Inveon PET/CT scanners (Siemens, Knoxville, TN, USA). The standard uptake values (SUVs) were generated from manually drawn regions of interests for tumors and surrounding muscle and fat tissues. Visualization of specific uptake was determined by comparison of baseline scans with blocking scans where animals were pre-treated with olaparib (50 mg/kg, I.P., 5 mg/mL in 10% DMSO and 10% 2-hydroxy-propyl-β-cyclodextrin) 20 minutes before tracer administration.
2.6. Ex-vivo biodistribution and autoradiography
Biodistribution studies were also conducted to compare with microPET imaging data. At 60 min post tracer administration, the mice (baseline n = 3; blocking n = 2 for both PC-3 male and MDA-MB-231 female tumor-bearing mice) were sacrificed, and the blood, lungs, liver, spleen, kidneys, muscle, heart, brain, bone, pancreas, tumor, and fat (and prostate for male mice) were harvested, weighed, and counted in a Beckmann Gamma 8000 gamma counter (Beckman Coulter, Brea, CA, USA). The percentage of injected dose per gram of tissue (%ID/g) was determined by decay correction of the [18F]4 for each sample normalized to a standard of known weight, representing the injected dose. After radioactivity was counted, the tumors and muscles were then mounted on Superfrost Plus glass slides (Fisher Scientific, Pittsburgh, PA, USA). Finally, autoradiography-imaging studies were performed using a Packard InstantImager (Packard Instruments, Meriden, CT, USA).
3. Results and discussion
3.1. Characterization PARP-1 potential of olaparib derivatives
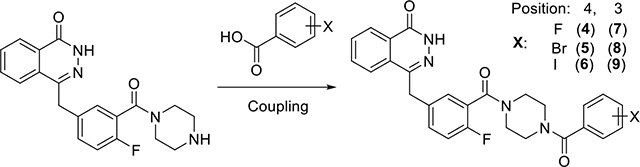
The fluoro-, bromo- and iodo-benzoic derivatives of olaparib, 4 (PARPi), 5, 6, 7, 8 and 9, which are suitable for solid-phase radiolabeling, were synthesized using the conjugation reaction as reported [12](Table 1). The PARP-1 affinity of these compounds were measured using a recently developed cell-based competitive assay using [3H]WC-DZ as the radioligand [13] (Figure S1). The Ki of fluoro-derivatives (4 and 7) are in the nM range, more potent than the bromo- and iodo-derivatives; The 3-substituted derivatives are more potent than the 4-substituted ones, which is consistent with the use of 3-substituted iodo-derivative (9) for PARP-1 targeted radiotherapy [14, 15].
Table 1.
Summary of PARP-1 affinities of olaparib derivatives
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Compound | 4 | 5 | 6 | 7 | 8 | 9 |
|
| ||||||
| Ki (nM)a | 5.74 ± 0.48 | 18.97 ± 4.21 | 39.18 ± 8.99 | 2.92 ± 0.22 | 5.30 ± 0.19 | 12.16 ± 0.89 |
|
| ||||||
| n′Hb | 0.74 ± 0.06 | 0.89 ± 0.05 | 1.00 ± 0.34 | 1.08 ± 0.09 | 1.03 ± 0.14 | 0.90 ± 0.11 |
Note:
Competitive binding for inhibition of the [3H]WC-DZ binding to PARP-1 in glioblastoma cells (U251MG)
Pseudo Hill coefficient (n′H values). Data are from three independent determinations, samples in triplicate. Mean ± SEM.
3.2. Solid-phase radiosynthesis
[18F]FBA is commonly used for the preparation of 18F radiopharmaceuticals using the active ester, [18F]SFB, and great effort has been taken in the preparation of [18F]SFB to simplify the procedure, shorten the synthesis time and increase the yields[16]. However, despite the fact that [18F]SFB needs to be obtained from [18F]FBA, the reactivity of [18F]SFB is also quite low. For example, due to the obvious time constraints associated with 18F radiochemistry, heating is required for the reaction of [18F]SFB with amine 3 to form [18F]4. Therefore, the direct conjugation of [18F]FBA with amines using coupling reagents would be ideal, but the harsh conditions needed for the labeling reaction (acid/base and water) are not compatible with the conjugation reaction, and amine side-products from the labeling reactions of quaternary ammonium precursors (e.g., 1) may interfere with the conjugation reaction. The reaction protocol reported for the preparation of [18F]PARPi ([18F]4) (nitro-precursor, small volume of reaction, and 10 mg HBTU) are consideration of these factors [4]. A summary of the reported synthesis of [18F]4 is presented in Table S1.
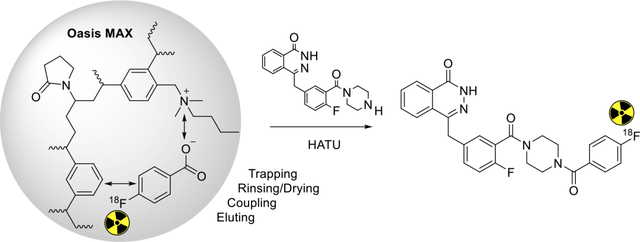
Reported here is a novel method of the radiosynthesis of [18F]4 on solid support, with the overall sequence shown in Scheme 1. The quaternary ammonium precursor 1 was used for the radiosynthesis of [18F]FBA 2, but before performing the coupling reaction using [18F]FBA on solid support or in solution, it proved critical to purify the [18F]FBA as obtained from the radiolabeling reaction mixture by removing any base or acid, salts, side-products and leftover precursor. As shown in Figure 1, absorption of [18F]FBA on a Waters Oasis MCX cartridge, followed by appropriate washing, can significantly remove the unwanted materials, resulting in very pure [18F]FBA (Figure S2) that can be eluted from the cartridge with methanol. Once diluted with water, [18F]FBA can then be extracted by passage of the water-methanol solution through a Waters Oasis MAX cartridge, which retains [18F]FBA using both lipophilic and ionic interactions. Once trapped, strong ionic interactions keeps [18F]FBA retained on the MAX cartridge, even during rinsing with acetonitrile, a convenient process for removal of water, which would interfere with the coupling reagents used for the conjugation reaction (Figure 1). The MAX resin was pre-treated with 0.5 N HCl to reduce the strong basicity of the resin (hydroxide as counter ion) to weak basicity (chloride as counter ion), a step that is essential for the success of the conjugation reaction on the MAX resin.
Scheme 1.

Radiosynthesis of [18F]4 by coupling of [18F]FBA 2 with 3 on an Oasis MAX cartridge (line 2) after radiofluorination of precursor 1 to produce 2 and purification of 2 on an Oasis MCX cartridge.
Figure 1.

Structures of Waters Oasis MCX and MAX resins and their interactions with substrates during the purification step and the coupling step, respectively. The anionic Oasis MCX retains the major by-product (4-carboxy-N,N,N-trimethylbenzenaminium salt) while allows the elution of [18F]FBA from the MCX resin by an organic solvent. The cationic Oasis MAX retains [18F]FBA on the MAX resin while allows the resin to be dried by rinsing with acetonitrile before the coupling reaction.
The void volume of the cartridge with the retained [18F]FBA is about 0.5 mL, so amine 3 and HATU in 0.5 mL DMF were loaded onto the cartridge, completely saturating the resin volume and immersing it fully in the reaction solution. After 10 min at room temperature, the reaction mixture was eluted with acetonitrile and water in sequence, resulting in a solution that can be directly injected for HPLC purification (Figure S3). About 80% of the radioactivity was eluted with the desired product [18F]4 as the major radioactive component. Starting from [18F]FBA, the decay corrected radiochemical conversion (RCC) of HPLC purified [18F]4 is 77.3 ± 1.5 % (n =3). The molar activity (Am) of [18F]4 at the end of synthesis is 49.0 ± 15.4 GBq/ μmol (1325 ± 416 mCi/μmol), which is in the range of Ams that we normally obtained. [18F]4 was confirmed by co-injection with authentic compound 4 on an analytical HPLC (Figure S4), and the radiochemical purity is >99%. The animal dose was formulated in 10% ethanol/saline.
Before concluding this section on radiosynthesis, it is worth noting that non-covalent solid-phase synthesis is a form of solid-phase synthesis whereby the organic substrate is retained on the solid phase by noncovalent interactions (such as lipophilic retention and ionic retention). In fact, the copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” radiosynthesis in an Oasis HLB cartridge was reported by us previously[17]. The conjugation reaction of [18F]FBA using a coupling reagent on the MAX cartridge also falls in the same category. This cartridge is stable in organic solvents. So, once the conjugation reaction is complete, the product becomes neutral and can be readily extracted from the resin with organic solvents, while prior to the coupling step, the anionic [18F]FBA remained firmly bound by ionic interactions with the resin material.
3.3. In vivo evaluation of [18F]4 for imaging PARP-1 expression in breast and prostate xenograft tumor models
Both in vitro and in vivo studies of [18F]4 by others have demonstrated its potential for PET imaging of PARP-1 expression in several cancers [4, 18], but not as yet in breast and prostate cancers. We previously reported on the use of the MDA-MB-231 tumor model [19] and PC-3 prostate tumor model [20] for the initial validation of [18F]FTT and [18F]WC-DZ-F, respectively, as PARP-1 PET tracers. Herein, we used both tumor models to validate [18F]4 that has been made by this novel solid-phase method. MDA-MB-231 human breast cancer has low PARP-1 expression. Nevertheless, its xenograft tumors in nu/nu female mice were clearly visualized by microPET using [18F]4 (Figure 2). These MDA-MB-231 tumors demonstrated increased uptake (SUV of 0.46 at 60 min) that was clearly distinguishable from background. Blocking studies with olaparib (50 mg/kg i.p., 20 min prior to tracer injection) resulted in decreased uptake of [18F]4 by the tumors down to background tissue activity levels.
Figure 2.

Representative microPET images of baseline (top panel) and blocking (bottom panel) studies (summed from 0 to 60 min) of [18F]4 in a female (left panel) and a male (right panle) athymic NCR Nu/Nu mouse subcutaneously implanted with MDA-MB-231 and PC-3 xenografts, respectively. The pseudo-color scale bar displays the relative SUV values and the tumors are circled and indicated by the yellow arrow. The MDA-MB-231 tumor uptake of [18F]4 is high and can be blocked by olaparib (50 mg/kg i.p., 20 min pre-treatment). The gut overwhelms PC-3 tumor uptake signals. The liver, stomach, intestine and colon had the highest radioactivity, SUV>4.
Previously We have demonstrated the PARP-1 specific uptake of [18F]4 and [18F]FTT in PC-3 cell uptake assay [20]. In the microPET imaging study of PC-3 xenograft tumors using [18F]4, it is difficult to distinguish the tumors from the gut because the tumors were implanted very close to the gut, which has high activity due to nearby fecal material (Figure 2). However, the post-PET biodistribution of [18F]4 in the PC-3 tumor-bearing mice and autoradiography of the tumors and muscles from these animals were carried out to evaluate the imaging potential of [18F]4 for prostate cancer.
The tracer biodistribution of [18F]4 in MDA-MB-231 and PC-3 bearing mice post imaging (60 min post-injection) is shown in Figure 3 and Table S2. [18F]4 showed high similarity in biodistribution in male and female mice. The highest uptake of [18F]4 was observed in liver and spleen, followed by pancreas. The uptake in liver was barely blocked by olaparib (50 mg/Kg, i.p.); however, uptake in spleen and pancreas were completely blocked by olaparib. Low uptake was observed in blood, muscle, heart, and fat in both groups, indicating no-PARP-1-mediated uptake and rapid wash-out. The fast clearance from non-target tissues/organs results in high tumor/muscle ratios, the favorable tracer property for PET imaging. The brain uptake of [18F]4 at 60 min post-injection is only 0.039 ± 0.001 %ID/g in female mice and 0.040 ± 0.004 %ID/g in male mice. In general, PARP-1 inhibitors are substrates of P-glycoprotein (PgP), which prevent them from crossing a functional blood-brain barrier (BBB) [21]. The significant uptake [18F]4 in the bone (about 1 %ID/g) should not be due to the defluorination of the tracer, but to the expression of PARP-1 in the bone since the uptake is blockable by olaparib. Actually, the same uptake and blockage of [18F]4 in the bone was previously reported in healthy nude mice [18]. The uptake in MDA-MB-231 tumors is 1.43 ± 0.19 %ID/g and was blocked by olaparib down to background tissue activity levels (0.32 ± 0.05 %ID/g). The uptake in PC-3 tumors is 1.10 ± 0.11 %ID/g with low blockage (0.55 ± 0.26 %ID/g), but the uptake in prostate is similar. However, since n = 2 in the blocking group, more studies are needed to see whether the difference is statistically significant between them. The uptake in MDA-MB-231 and PC-3 tumors and muscles is further showed in the autoradiograph of tumors and muscles as shown in Figure 4. Higher uptake in MDA-MB-231 than in PC-3 tumors was observed, and the uptake could be blocked down as far as background muscle level, which is low. The biodistribution data in both animal models are in agreement with that reported by others [4, 18], indicating the promise of [18F]4 as a PET tracer for imaging PARP-1 expression in breast and prostate cancers.
Figure 3.

Post-PET biodistribution of [18F]4 in (a) MDA-MB-231 in female mice and (b) PC-3 in male mice (n = 3 in control group and n = 2 in blocking group, blocked by olaparib (50 mg/Kg, i.p.)
Figure 4.

Autoradiography imaging of [18F]4 in MDA-MB-231 and PC-3 tumors collected after microPET imaging studies.
3.4. Potential application of solid phase radiosynthesis of other olaparib derivatives
Radiohalogen-labeled olaparib derivatives have great potential for PET imaging (18F, 76Br and 124I) and targeted radiotherapy (77Br, 123I,131I and221At). Other than [18F]4, several radiohalogen-labeled olaparib derivatives have been reported using different radiolabeling methods. For example, compound 5 and 6 have been labeled directly with 77Br and 125I respectively, using the copper-mediated nucleophilic radiohalogenation in very high yields and under mild conditions [22, 23]; Besides being labeled with 124I and 131I [24] using the N-hydroxysuccinimide (NHS) ester intermediates, compound 6 has been labeled with 123I using a solid state halogen exchange radioiodination methodology [12]; Compound 9 has been labeled with 123I and 131I for targeted radiotherapy [14, 15] using a multiple-step procedure (including radiosynthesis of an NHS ester, HPLC purification and conjugation). The solid phase radiosynthesis described above may provide a simple and feasible method to label olaparib derivatives, especially for 18F-labeled derivatives 3 and 4, which currently lack an efficient labeling method. However, this multiple-step solid-phase strategy is inferior to the aforementioned copper-mediated radiohalogenation for radiolabeling with other radiohalogens due to their longer half-lives and different radiation safety issues. The use of this method on other radiolabeled aromatic acids, especially for 18F-labeled ones appears to be promising and worth further investigation because this method can avoid a harsh radiolabeling condition (e.g. copper-mediated radiofluorination) for some labile moieties. Finally, because of the limitation of this method (non-homogenous reaction, 0.5 mL volume, 1 : 1 ratio of coupling agent vs amine), this method should have limited application in the radiolabeling of biomolecules.
4. Conclusion
Solid Phase radiosynthesis of [18F]PARPi ([18F]4) from [18F]FBA on anion-exchange resins is a simple and efficient method, which is compatible with automated synthesis modules. This method has great potential in the radiosynthesis of radiopharmaceuticals starting from [18F]FBA or other 18F and radiobromine/radioiodine labeled aromatic acids. The preclinical evaluation of [18F]PARPi ([18F]4), prepared by this method, demonstrated the promise of using [18F]PARPi ([18F]4) for PET imaging of PARP-1 expression in breast and prostate cancers.
Supplementary Material
Highlights.
Solid-phase radiosynthesis (SPRS) was carried out using ion exchange resins.
Several benzoic derivatives of olaparib show high potent for PARP-1.
[18F]PARPi was radiosynthesized efficiently via SPRS using [18F]FBA.
[18F]PARPi has promise for imaging PARP-1 expression in animal models.
Acknowledgement
This work was supported by the National Institutes of Health R01CA025836 (JAK), R01NS092865 (JX) and R01EB029752 (BER). We thank Robert Dennett and Brian Wingbermuehle for the production of [18F]fluoride and the staff of the Washington University Small Animal Imaging Facility for their technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Statement
The data supporting the findings of this article are available within the article and its supplementary materials.
References
- [1].Sutcliffe-Goulden JL, O’Doherty MJ, Marsden PK, Hart IR, Marshall JF, and Bansal SS. Rapid solid phase synthesis and biodistribution of 18F-labelled linear peptides. European Journal of Nuclear Medicine and Molecular Imaging 2002;29:754–9. [DOI] [PubMed] [Google Scholar]
- [2].Marik J, Hausner SH, Fix LA, Gagnon MKJ, and Sutcliffe JL. Solid-Phase Synthesis of 2-[18F]Fluoropropionyl Peptides. Bioconjugate Chemistry 2006;17:1017–21. [DOI] [PubMed] [Google Scholar]
- [3].Davis RA, Lau K, Hausner SH, and Sutcliffe JL. Solid-phase synthesis and fluorine-18 radiolabeling of cycloRGDyK. Org Biomol Chem 2016;14:8659–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Carney B, Carlucci G, Salinas B, Di Gialleonardo V, Kossatz S, Vansteene A, et al. Non-invasive PET Imaging of PARP1 Expression in Glioblastoma Models. Mol Imaging Biol 2016;18:386–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ferraris DV. Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. From concept to clinic. Journal of medicinal chemistry 2010;53:4561–84. [DOI] [PubMed] [Google Scholar]
- [6].Basu B, Sandhu SK, and de Bono JS. PARP inhibitors: mechanism of action and their potential role in the prevention and treatment of cancer. Drugs 2012;72:1579–90. [DOI] [PubMed] [Google Scholar]
- [7].Carney B, Kossatz S, and Reiner T. Molecular Imaging of PARP. Journal of nuclear medicine : official publication, Society of Nuclear Medicine 2017;58:1025–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Michel LS, Dyroff S, Brooks FJ, Spayd KJ, Lim S, Engle JT, et al. PET of Poly (ADP-Ribose) Polymerase Activity in Cancer: Preclinical Assessment and First In-Human Studies. Radiology 2017;282:453–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Schöder H, França PDDS, Nakajima R, Burnazi E, Roberts S, Brand C, et al. Safety and Feasibility of PARP1/2 Imaging with 18F-PARPi in Patients with Head and Neck Cancer. Clinical Cancer Research 2020;26:3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wilson TC, Pillarsetty N, and Reiner T. A one-pot radiosynthesis of [18F]PARPi. Journal of Labelled Compounds and Radiopharmaceuticals 2020;63:419–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Garg S, Kothari K, Thopate SR, Doke AK, and Garg PK. Design, Synthesis, and Preliminary in Vitro and in Vivo Evaluation of N-(2-diethylaminoethyl)-4-[18F]fluorobenzamide ([18F]-DAFBA): A Novel Potential PET Probe to Image Melanoma Tumors. Bioconjugate Chemistry 2009;20:583–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zmuda F, Malviya G, Blair A, Boyd M, Chalmers AJ, Sutherland A, et al. Synthesis and Evaluation of a Radioiodinated Tracer with Specificity for Poly(ADP-ribose) Polymerase-1 (PARP-1) in Vivo. Journal of Medicinal Chemistry 2015;58:8683–93. [DOI] [PubMed] [Google Scholar]
- [13].Zhou D, Chen H, Mpoy C, Afrin S, Rogers BE, Garbow JR, et al. Radiosynthesis and Evaluation of Talazoparib and Its Derivatives as PARP-1-Targeting Agents. Biomedicines 2021;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jannetti SA, Carlucci G, Carney B, Kossatz S, Shenker L, Carter LM, et al. PARP-1-Targeted Radiotherapy in Mouse Models of Glioblastoma. J Nucl Med 2018;59:1225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pirovano G, Jannetti SA, Carter LM, Sadique A, Kossatz S, Guru N, et al. Targeted Brain Tumor Radiotherapy Using an Auger Emitter. Clinical Cancer Research 2020;26:2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hou S, Phung DL, Lin W-Y, Wang M-w, Liu K, and Shen CK-F. Microwave-assisted one-pot synthesis of N-succinimidyl-4[18F]fluorobenzoate ([18F]SFB). Journal of visualized experiments : JoVE 2011:2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhou D, Chu W, Peng X, McConathy J, Mach RH, and Katzenellenbogen JA. Facile purification and click labeling with 2-[18F]fluoroethyl azide using solid phase extraction cartridges. Tetrahedron Lett 2015;56:952–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Carney B, Kossatz S, Lok BH, Schneeberger V, Gangangari KK, Pillarsetty NVK, et al. Target engagement imaging of PARP inhibitors in small-cell lung cancer. Nature Communications 2018;9:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhou D, Chu W, Xu J, Jones LA, Peng X, Li S, et al. Synthesis, [18F] radiolabeling, and evaluation of poly (ADP-ribose) polymerase-1 (PARP-1) inhibitors for in vivo imaging of PARP-1 using positron emission tomography. Bioorg Med Chem 2014;22:1700–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhou D, Xu J, Mpoy C, Chu W, Kim SH, Li H, et al. Preliminary evaluation of a novel 18F-labeled PARP-1 ligand for PET imaging of PARP-1 expression in prostate cancer. Nuclear Medicine and Biology 2018;66:26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Durmus S, Sparidans RW, van Esch A, Wagenaar E, Beijnen JH, and Schinkel AH. Breast Cancer Resistance Protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1) Restrict Oral Availability and Brain Accumulation of the PARP Inhibitor Rucaparib (AG-014699). Pharmaceutical Research 2015;32:37–46. [DOI] [PubMed] [Google Scholar]
- [22].Zhou D, Chu W, Voller T, and Katzenellenbogen JA. Copper-Mediated Nucleophilic Radiobromination of Aryl Boron Precursors: Convenient Preparation of a Radiobrominated PARP-1 Inhibitor. Tetrahedron letters 2018;59:1963–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Reilly SW, Makvandi M, Xu K, and Mach RH. Rapid Cu-Catalyzed [211At]Astatination and [125I]Iodination of Boronic Esters at Room Temperature. Organic letters 2018;20:1752–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Salinas B, Irwin CP, Kossatz S, Bolaender A, Chiosis G, Pillarsetty N, et al. Radioiodinated PARP1 tracers for glioblastoma imaging. EJNMMI Res 2015;5:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this article are available within the article and its supplementary materials.