

Abstract
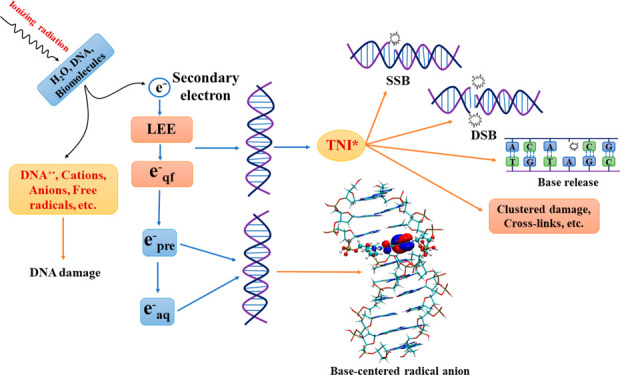
Reactions of radiation-produced secondary electrons (SEs) with biomacromolecules (e.g., DNA) are considered one of the primary causes of radiation-induced cell death. In this Review, we summarize the latest developments in the modeling of SE attachment-induced radiation damage. The initial attachment of electrons to genetic materials has traditionally been attributed to the temporary bound or resonance states. Recent studies have, however, indicated an alternative possibility with two steps. First, the dipole-bound states act as a doorway for electron capture. Subsequently, the electron gets transferred to the valence-bound state, in which the electron is localized on the nucleobase. The transfer from the dipole-bound to valence-bound state happens through a mixing of electronic and nuclear degrees of freedom. In the presence of aqueous media, the water-bound states act as the doorway state, which is similar to that of the presolvated electron. Electron transfer from the initial doorway state to the nucleobase-bound state in the presence of bulk aqueous media happens on an ultrafast time scale, and it can account for the decrease in DNA strand breaks in aqueous environments. Analyses of the theoretically obtained results along with experimental data have also been discussed.
Introduction
The information required for cellular function and structure is stored in genetic material. Most organisms, including humans, have deoxyribonucleic acid (DNA) as the genetic material. However, ribonucleic acid (RNA) is the genetic material in many organisms, such as retroviruses.1 DNA is considered the pillar of evolution since it plays a vital role in the propagation of parental traits to offspring and in the development of new traits which help living beings adapt better to the external environment. The double helix structure of DNA2 is known to be extremely stable, which is aided by the highly efficient DNA-repair mechanism.3 However, the efficiency of the repair mechanism is pushed to its limits by DNA-damaging agents, such as ionizing radiation.4−9 The unrepaired lesions, as well as the lesions that are not repaired correctly (i.e., nonfidelity repair), in the genetic material can lead to accumulation of lesions that lead to genomic instability and ultimately to mutation, cell death, and neoplastic transformation.4 Damage to DNA consists of base damage, release of unaltered bases, single-strand breaks (SSBs), double-strand breaks (DSBs), tandem lesions (e.g., [5′-8]-cyclopurine nucleosides), inter- and intrastrand cross-links, DNA–protein cross-links, etc.10−17 The lesions involving the sugar–phosphate backbone such as SSB and DSB, tandem lesions including cross-links when formed in proximity along with other lesions (clustered lesions), often lead to radiation-induced cell death.4,5,8,9,18−21 In this Review, we attempt to summarize recent theoretical and experimental advances in the literature on an important pathway, viz., dissociative electron attachment (DEA), that is involved in the reductive pathway of radiation damage to DNA (vide infra). Although cells contain both nuclear and mitochondrial DNA, we focus the discussion on the former, even though the latter has also attracted a lot of attention recently.22,23
There are multiple simultaneously occurring pathways that are involved in the interaction between ionizing radiation and DNA. These are direct, quasidirect, and indirect pathways.11,14,24,25 The direct pathway involves direct radiation–DNA interaction (Reactions 1 and 2, Scheme 1), thereby leading to ionization, excitation, and reduction of the target molecule (i.e., DNA); the ionizations result in the formation of DNA cation radicals [holes or unpaired spins (DNA•+)] and in the ejection of electrons (excess electrons).10,11,14,24−33 Owing to the random nature of the interaction of radiation with DNA, the ionization events leading to hole and excess electron formation along with the excitation events are also random in nature (i.e., can occur anywhere on the DNA) and occur in approximate proportions to the number of electrons present at that site.13,14,24,26,34−37
Scheme 1. Schematic Representation of Direct, Quasidirect, and Indirect Effects Caused by Ionizing Radiation on DNA.

The high-energy electrons ejected via the initial ionization events cause further ionizations and excitations. These secondary ionization events result in a cascade of medium- and low-energy electrons (LEEs) that cause further ionizations and numerous excitation events.13,34−36 Similar to holes, excess radiation-produced electrons add randomly to DNA and produce DNA anion radicals (DNA•–) that undergo various reactions (see Schemes 1, 3, and 4, and ref (25)).
Scheme 3. Hydrogen Atom Adduct of a Nucleobase Is Identical to a Two-Step Reaction Where an Electron and Proton Are Subsequently Added to the Nucleobase.

Here, dR is 2′-deoxyribose. Nature (oxidizing and reducing) of the radicals are mentioned.10,11 The 5-yl radical is of β-oxoalkyl radical type which are oxidizing in nature.10
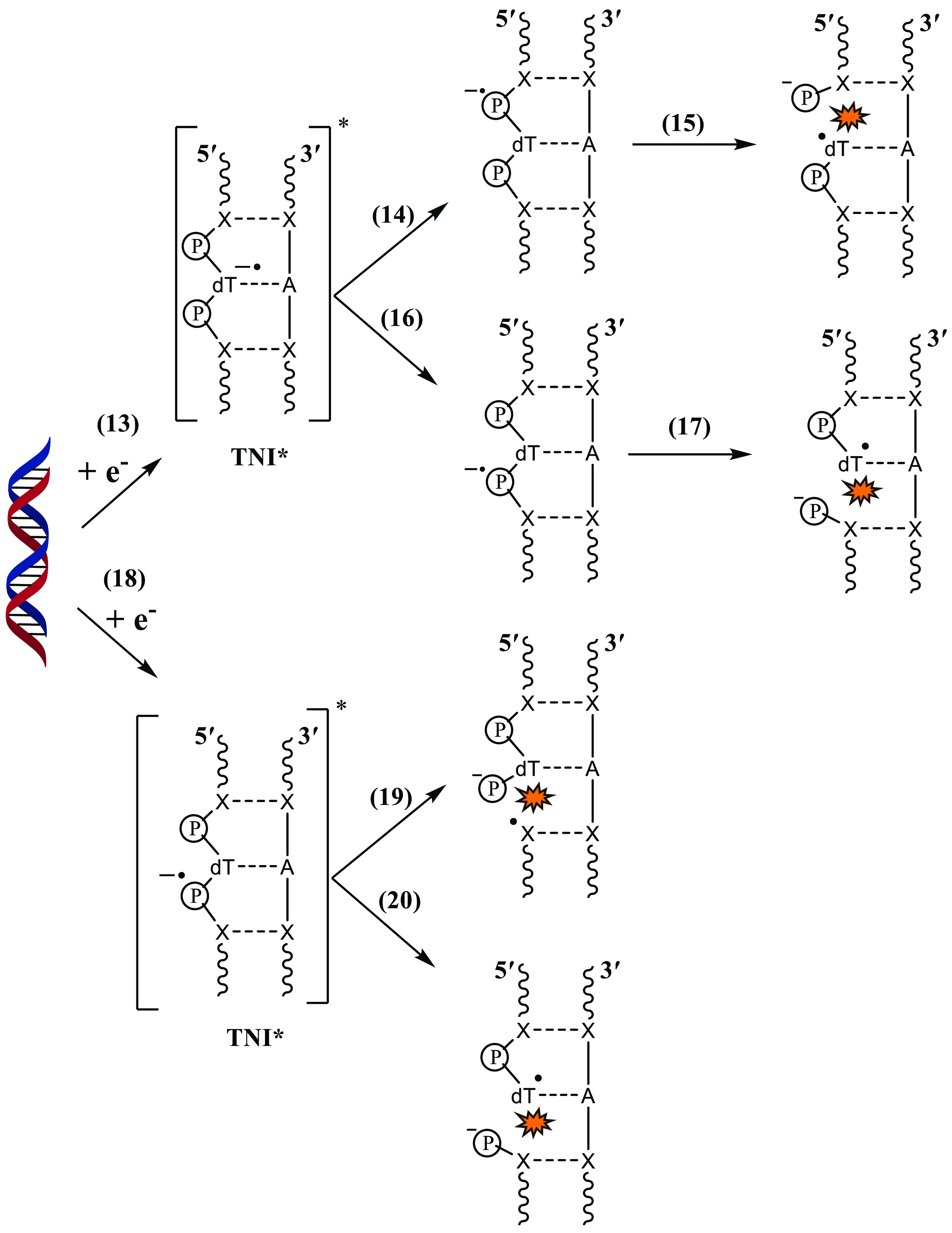
Scheme 4. Various SSB Pathways through the Formation of a Phosphate-Centered TNI*.
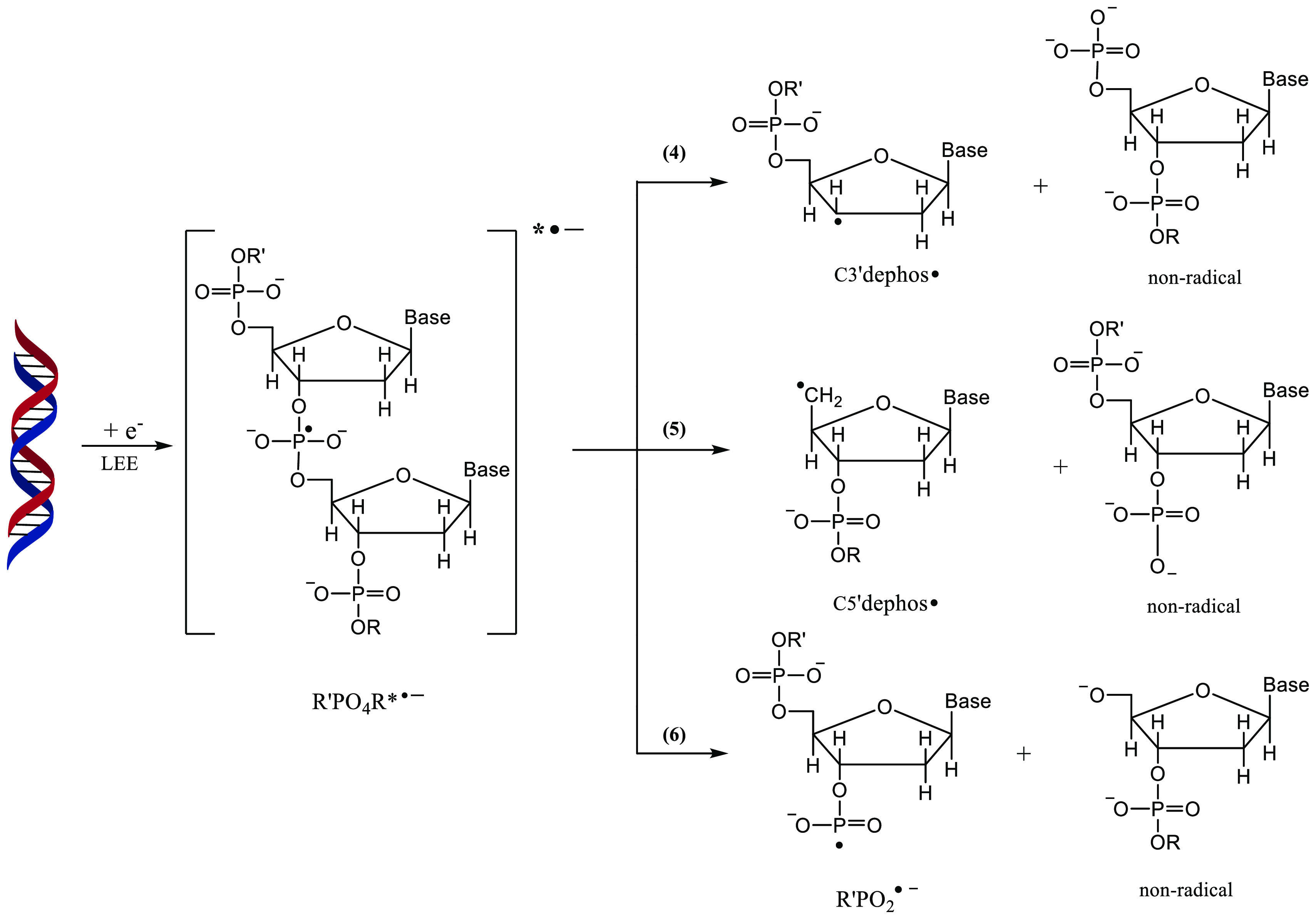
Processes 4 and 5, which result in the formation of C3′dephos• and C5′dephos•, account for nearly a majority of the products.11,14,25 R′PO2•– and the nonradical anion moiety formed in process 6 are due to the P–O bond cleavage which occurs only to a minor extent.11,14,25
DNA is also closely associated with water molecules, histone proteins, etc., whose concentration in the vicinity of DNA largely depends on its organizational state.10,38 Ionization of these species bound to DNA results in the formation of the corresponding cation radicals (for e.g., H2O•+) and of excess electrons. H2O•+ could transfer the holes to DNA (Reactions 4 and 5), resulting in the formation of additional DNA•+, and the addition of these excess electrons to DNA results in the formation of additional DNA•–.11,14,16,39−41 Damage to DNA that occurs due to these DNA-radicals constitutes the quasidirect pathway. Direct and quasidirect effects are combined together and called direct-type effects.25 In the indirect pathway of radiation action, the high-energy photons interact with the aqueous medium of the cell nucleus surrounding the DNA, resulting in the ionization of water molecules present in the bulk environment. Ionizing radiation could either ionize water molecules to generate H2O•+ and secondary electrons or lead to the formation of the water molecule in an excited state (H2O*).16,17 Both H2O•+ and H2O* are highly reactive and can dissociate as shown in Reactions 9 and 10.16,24 This results in the formation of an array of secondary products such as hydroxyl radicals (•OH), hydrogen atoms (H•), and fully solvated or aqueous electrons (eaq–), which in turn attack the DNA.10,42,43
Because of the simultaneous and competitive reactions, free radical chemistry is highly complex. Pulse radiolysis experiments using DNA-model compounds (nucleosides, nucleotides, polynucleotides, and highly polymeric DNA) established that the free radical reactions that are involved in radiation damage to DNA via the indirect pathway are nearly diffusion-controlled.4,10,24,25 •OH adds to the double bonds on the nucleobase, e.g., at C4/C5 and at C8 in the purine and at the C5=C6 double bond in the pyrimidines; it can also abstract an H atom from the sugar moiety.4,10,24,25 A combination of pulse radiolysis in aqueous solutions at ambient temperature, electron spin resonance (ESR) studies in frozen aqueous solutions at low temperature, and product analyses established that, in the case of nucleosides/nucleotides, the extent of •OH attack to the nucleobase is ca. 80% via addition as compared to the deoxyribose-sugar (via H atom abstraction), where the latter amounts to only about 20%.4,10,44,45 When experiments on short double-stranded DNA (dsDNA) and oligonucleotides are considered, the extent of hydrogen abstraction by •OH from the sugar moiety is ≤10%, and addition to the nucleobase amounts to ≥90%.10 Nevertheless, radical attack to DNA generates sugar or base-centered free radicals that subsequently lead to damage that is difficult to repair (e.g., strand breaks, tandem lesions, etc.).10,44 As mentioned above, due to the high concentration of the macromolecules in cells and because of the compact organizational status (e.g., chromosomes) of biomacromolecules, DNA damage via the direct-type effect becomes significant, and the role played by secondary electrons (see above) becomes important.13−15,46−48
The class of secondary electrons that plays a major role in DNA damage is the LEEs that possess energy in the range 0–20 eV.42,46,49−52 They are abundantly generated when high-energy radiation interacts with the cellular environment.53 Since LEEs typically possess energies lower than the ionization threshold (7.5–10 eV)54,55 of biomolecules such as DNA, LEE–biomolecule interaction results in the formation of excited transient negative ions (TNI*) with lifetimes in the range 10–12–10–15 s.56 Once the TNI* is formed, either it can undergo autodetachment to yield the neutral molecule and the additional electron, or it could relax back to the radical anion in the ground state releasing the additional energy to the environment.56 Several reviews14,24 on electron attachment to DNA refer to the ground-state radical anion as a ground-state transient negative ion (TNI), although it is a misnomer, as the ground state of many of the DNA-subunit anion radicals is actually a dipole-bound state.57,58
The formation of the TNI* could also lead to the rupture of the molecule and is known as DEA (Scheme 2).56,59−62 Interaction of LEE with DNA results in the formation of SSBs, DSBs, base release, cross-links, multiple damage sites, etc., via DEA.62−76 Theoretical modeling of DEA is highly challenging because it involves the coupling between nuclear and electronic degrees of freedom of a molecule.56 At the same time, the TNIs are difficult to characterize experimentally due to their short lifetimes.
Scheme 2. Schematic Representation of DEA to Molecule AB, Which Results in the Dissociation of AB to A and B–

(AB–)* is the TNI* formed upon electron attachment which can either undergo autodetachment to form the neutral molecule and the extra electron or undergo dissociation to produce the fragments A and B–.56,59−62
Once the secondary electrons are produced in the aqueous medium, they rapidly undergo inelastic collisions with the surrounding water molecules and lose energy to form LEEs (see Figure 1). In the absence of electron-scavengers, LEE in the aqueous solution eventually forms solvated electrons (eaq–). The lifetime of eaq is in the range of several hundred microseconds and varies widely with respect to experimental conditions.42,49,79 Since eaq– are trapped in a deep potential well with a VDE (vertical detachment energy) of ∼3.4 eV,80−82 the possibility of eaq causing DEA is rather low.83−85 However, several pulse radiolysis experiments and theoretical studies have shown that eaq– can react with nucleobases14,50,86 to form stable anions. This reaction occurs at near diffusion-controlled rates and leads to scavenging of electrons by the nucleobase.10,50,87,88 Contrary to these findings, Abel and co-workers proposed that the nucleobase should not scavenge eaq since VDE is higher than the electron affinity of nucleobases (∼1.7–2.2 eV).81 However, one should consider the adiabatic electron affinity (AEA) of eaq– instead of VDE while drawing conclusions regarding the binding of eaq with biomolecules.88 The AEA of eaq– (∼1.6 eV)78 is lower than that of nucleobases89,90 which clearly shows that eaq can bind to them. If eaq– adds to the biomolecules, the corresponding radical anion would be formed which would have a strong Lewis base character.10,11 This species can abstract a proton from the surrounding environment and form a neutral radical. This neutral radical is identical to a hydrogen atom adduct (Scheme 3).
Figure 1.
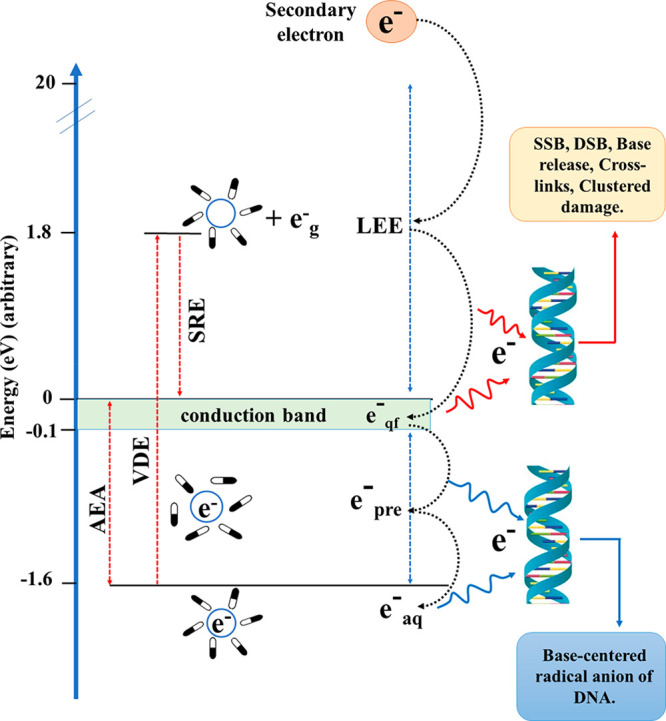
Schematic representation of the formation of eaq– from secondary electrons in an aqueous cellular environment. Vertical detachment energy (VDE) of eaq = 3.4 eV, and AEA is the adiabatic electron affinity (1.6 eV).77,78 SRE is the solvent reorganization energy which is equal to the difference between VDE and AEA.
In the process of the formation of eaq– from LEE, several intermediate states are formed, such as quasifree electrons (eqf) and presolvated electrons (epre–) (Figure 1).53,91,92 These states of the electron in the aqueous environment are classified based on their energy and lifetime (Figure 1).53,91,92 eqf generally lies near the water conduction band and has energy ∼0 eV,14 whereas secondary electrons with energy between −0.1 and −1.6 eV are considered as epre–. epre are one of the most abundant products of radiation–water interaction and have a lifetime <500 fs.91 However, recent experimental studies indicate that epre– do not take part in DEA.14,93 Instead, epre rapidly become solvated to form eaq–.91 Recent pulse radiolysis studies by Ma et al. have shown that epre and eaq– do not cause DEA to aqueous nucleosides and nucleotides.93 Their picosecond pulse-radiolysis results contradict the study reported by Lu and co-workers94 who employed femtosecond laser pump–probe spectroscopy to suggest the formation of TNI* of purine monophosphate nucleotides in solution, which may subsequently lead to SSB formation via DEA. However, the more reliable picosecond pulse-radiolysis measurements showed that nucleotides of guanine and adenine are poor scavengers of epre at lower concentrations (≤50 mM).93 Therefore, interaction of epre– and eaq with DNA does not lead to SSBs and DSBs in aqueous solutions. Instead, it is the eqf–, which are known as the LEE in solutions, that can cause damage to DNA.24
LEE attachment-induced DNA damage is unique because it is associated with direct strand break along with the predominant formation of a neutral C-central radical species at the C3′-site (C3′dephos•) and at the C5′-site (C5′dephos•) (Scheme 4).10,11,13−16,25 The carbon-centered radical with the phosphate group attached (C3′•, C5′•) or without (C1′•) are formed in the oxidative pathway of DNA damage involving H-abstraction from the deoxyribose sugar moiety by •OH (indirect effect).4,10,12,14,95 These same radicals are also formed by ionization–deprotonation and hole transfer from the excited base cation radical to the sugar moiety followed by deprotonation (direct-type effect).11,13−16 C3′•, C4′•, and C5′• subsequently lead to sugar–phosphate bond breaking by β-phosphate elimination.4,10,95−98 Adhikary and co-workers have detected the ESR spectral signatures of C5′• (produced via oxidative pathway of radiation damage) and C3′dephos• (produced by reductive pathway through DEA) in both ion-beam and γ-ray irradiated hydrated DNA (degree of hydration = 12–14 water molecules/nucleotide), where both anion radical and cation radical scavengers were used to isolate the ESR spectrum in irradiated DNA due to only neutral backbone radical (sugar radical) species.35 The ESR studies by the Adhikary–Sevilla laboratory and the collaborative work between Adhikary and Mostafavi groups have established that formation of the neutral sugar radical in the backbone via the direct-type effect involves various pathways: (a) ionization–deprotonation, (b) hole transfer from excited base cation radicals to the sugar moiety by deprotonation, (c) phosphate-to-sugar hole transfer, and (d) base-to-sugar hole transfer.11,13−16,99−101
Mechanism of TNI-Induced DNA Damage
In the past two decades, a large number of experimental studies have been reported in the literature which have investigated the interaction of LEEs with DNA.62−76 One of the first experimental studies on electron attachment-induced DNA strand breaks was reported by Sanche and co-workers,63 where dry DNA, having only the structural water molecules intact, was exposed to electrons having kinetic energies as low as 3.5 eV. Gel electrophoresis of the DNA sample was performed to quantify the damaged (SSBs and DSBs) and undamaged DNA. It was observed that the yield of SSBs depends upon the kinetic energy of the incident electron and is a maximum at specific values of the kinetic energy, which was inferred due to the involvement of resonance or the TNI-based pathway in the electron attachment process. These TNIs are formed when the incoming electron populates the unoccupied molecular orbital of the parent molecule. Depending upon the energy of incoming electrons, shape and core-excited resonances are formed in the DNA subunits.102 The shape resonances are generally formed when an electron with energy in the range 0–4 eV is captured by the σ* or π* molecular orbital of closed shell neutral species (see Figure 2). These are known as shape resonances due to the fact that it is the shape of the interaction potential by virtue of which the incoming electron is temporarily trapped. Such a potential barrier is generated by the attractive polarization of the neutral molecule and the repulsive centrifugal interaction of the incoming electron. Since the extra electron occupies the unoccupied molecular orbital, the parent molecular electronic configuration is not perturbed. Therefore, its decay via the DEA channel is allowed by Koopmans’ theorem. It is a one-electron process commonly known as one-particle resonance, while the interaction of electrons with energy above 4 eV with the DNA subunits results in the formation of core-excited resonances (Figure 2).102
Figure 2.
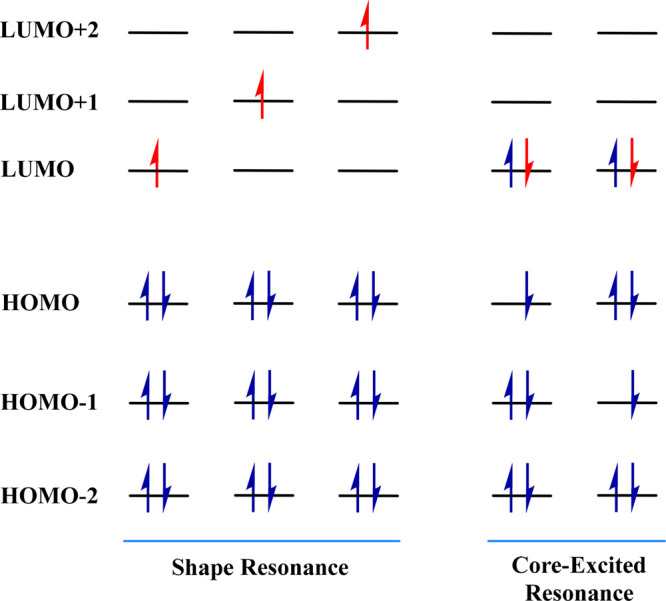
Schematic representation of the orbital level diagram of shape and core-excited resonance.
In core-excited resonances, the energy of the incoming electron excites another electron from the inner core-orbital to an unoccupied molecular orbital resulting in the formation of one hole in the inner core and two particles in vacant molecular orbitals. These are also known as one-hole two-particle resonances. Since the electronic configuration of the inner orbitals is perturbed, decay of such transient anions via electron detachment is dominated by the electron correlation and is Koopmans' forbidden. Two types of core-excited resonances, core-excited Feshbach and core-excited shape resonances, are distinguished based on the energy of the resulting transient negative ion. If the resulting anion has energy above the neutral excited state, such resonances are known as core-excited shape resonance. Here also, the electron is trapped due to the shape of the interaction potential. At the same time, the ones that lie below the neutral excited state are known as core-excited Feshbach resonances.
Sanche and co-workers suggested that the core-excited resonance states formed by the attachment of electrons to the DNA bases’ π* orbitals initiate the bond breaking which lead to SSBs in DNA.63,66 This conclusion was suggested based on the maxima of strand break-yield vs electron energy plots.63 These core-excited resonance states can be represented as e– + π2π*0 → π1π*2 where the incoming electron initially attaches to the π* orbital of nucleobase and simultaneously excites another electron from the π to π* orbital. However, Burrow and co-workers103 have shown that the attachment of LEE <3 eV can lead to the formation of shape resonance states where the additional electron gets attached to the low-lying π* orbitals of DNA bases.
The first theoretical study emphasizing the role of π* shape resonance states in LEE-induced DNA strand breaks was performed by Simons and co-workers,104 soon after the first experiment from Sanche’s group.63 Although Sanche and co-workers63 had used electron beams with energy from 3 to 20 eV in their experiment, Simons and co-workers104 have analyzed if electrons with energy <3 eV could induce strand breaks in DNA, similar to that suggested by Burrow and co-workers.103 With cytosine mononucleotide as a DNA model system, Simons and co-workers have proposed a mechanism for the DEA involving the shape resonance state formed due to the electron initially captured into the π* orbital of the base. The potential energy surface (PES) generated by stretching of the C–O bond shows a small barrier of 13 kcal/mol connecting the initial π* anionic state to the σ* dissociative state. The initially formed π* shape resonance state goes through this barrier to cause C–O bond rupture leading to single strand breaks in DNA. This finding was later supported by the experimental investigation of Martin et al., which showed that shape resonances are involved in the DNA strand breaks caused by the electron having energy in the range of 0–4 eV.105 Based on their experiment with short DNA strands deposited on metal surfaces, they have also concluded that the sugar–phosphate C–O bond cleavage of the DNA backbone is the result of electron transfer from the nucleobase π* resonance state of DNA to the σ* antibonding molecular orbital of the C–O sugar–phosphate bond. The theoretically calculated rate of cleavage suggests that such a transition can happen within the lifetime of the resonance state, making the bond cleavage feasible via this process.106 Since the resonance cross section is inversely related to the energy, and the lifetime also increases with a decrease in energy, the low-lying shape resonances have been claimed to possess high efficiency in causing DNA strand breaks.106,107
However, the sugar–phosphate C–O bond cleavage leading to DNA strand breaks can also occur due to the initial formation of phosphate-centered shape resonances by direct attachment of LEE to the phosphate group.65,69,108,109 Li et al.108 have performed DFT calculations to understand the behavior of the anion radical formed by the attachment of near zero eV electrons directly to the DNA backbone. They have chosen a sugar–phosphate–sugar model system where the nucleobase was replaced by an amino group. From the PESs generated along the 3′C–O and 5′C–O bond stretching, the activation barrier for both 3′C–O and 5′C–O bond dissociation was reported to be around 10 kcal/mol.108 The reported activation energy values suggest that the C–O bond cleavage leading to strand breaks is thermodynamically favorable. However, these TNIs are metastable states, which cannot be treated by standard bound state quantum chemical methods and require special techniques.110−114 Recent coupled cluster113 and SAC-CI115 studies have shown that the first two resonance states of all five nucleobases have a lifetime of several femtoseconds. From their time-dependent wave packet studies on nucleotide model systems, Sarma and co-workers have shown that electron attachment to the nucleotide can lead to the formation of both nucleobase109,116,117 and phosphate-centered shape resonances.118 According to their work, the phosphate-centered shape resonances have a higher lifetime and lead to preferential cleavage of the 5′C–O bond over 3′C–O bond dissociation.118
Zheng et al.119 investigated the collision of electrons with energy in the range 4–15 eV on an oligonucleotide tetramer (GCAT) in the presence and absence of guanine or adenine. They showed that the extent of LEE-induced C–O bond cleavage is significantly reduced when one or more abasic sites are present in the tetramer. This indicates that the electron transfer from the nucleobase to the phosphate group plays an essential role in the cleavage of the sugar–phosphate bond, and restricting this electron transfer can reduce the strand breaks in DNA. The incident 6 eV electron electronically excites a base before transferring to the C–O-centered σ* orbital. However, from her experiments on gas-phase 2′-deoxycytidine-5′-monophosphate (5′-dCMP), Kopyra69 showed that the majority of the strand breaks (∼85%) in DNA is caused by direct electron attachment to the phosphate group. The recent LC-MS/MS experiment on the TpT DNA model system by Wagner and co-workers72 has shown that even very low-energy electrons (∼1.8 eV) can lead to strand breaks in DNA through the formation of a shape resonance anion, which subsequently decays via DEA mechanism. The extent of C–O bond cleavage at the C3′ position has been found to be 3-fold greater than that observed at the C5′ position. Experimentally120 observed resonance features in the regions 4–6 and 9–11 eV are attributed to core-excited resonances. However, these resonances are challenging to model theoretically due to the inherent high complexity of such resonances, and consequently, very few theoretical calculations are available in the literature for the core-excited resonances of DNA. Recently, Fennimore and Matsika121 suggested that there exists a high degree of mixing between three π* shape resonance and π1 (π*)2 core-excited resonance in uracil, which can affect the lifetime of both states.
Once the base-centered TNI is formed, the attached electron can also transfer to the dissociative σ* orbital of the N-glycosidic bond, leading to the base release.64,67 In addition to the sugar–phosphate and N-glycosidic bond cleavage, the DEA can lead to the rupture of the other bonds in DNA. Illenberger and co-workers have highlighted the possibility of the formation of nucleobase-centered resonances with the subsequent release of the hydrogen atoms in all of the DNA bases.122−125 Mark and co-workers have reported the loss of H atoms (both N1 and N3) from cytosine and thymine at incident electron energies near 1.1–1.5 eV.124,126 They have suggested the involvement of π* shape resonance centered on the nucleobase as the energy of π* orbitals lie in this range. Burrow et al.,127 on the other hand, attributed the release of hydrogen atoms to vibrational Feshbach resonances (VFRs). The DNA nucleobases possess high dipole moments and can support the formation of a dipole-bound (DB) state.58,128 In such cases, the initial electron attachment is governed by the long-range charge–dipole interaction, where the incoming electron is trapped in the highly diffuse orbitals. Experimentally, the DB states are reported to lie 62–86 meV (±8 meV) below the neutral ground state.129 Here, the excess electron attached due to the dipolar attraction potential gets enough time to stay close to the molecule and can lead to the excitation of nearby vibrational levels of the neutral molecule resulting in VFRs. Burrow et al.127 have observed sharp peaks in the DEA spectra of uracil and thymine nucleobases for incoming electrons below 3 eV of energy. The sharp structure in the DEA cross sections of uracil at 1 eV has been assigned as VFRs occurring due to N1–H bond vibrations. Here, the extra electron occupies the dissociative σ* orbital, lying between the N1 and H bond, which leads to H atom loss at the N1 site. In comparison, the resonance associated with dissociation of the N3–H bond lies somewhat higher in energy at 1.8 eV.
Role of the Dipole-Bound (DB) Anionic State in DEA
Most studies on DEA in the literature attribute resonance states of DNA as the reason for the generation of various types of DNA-lesions, e.g., SSBs, DSBs, base damages (BDs), and clustered damage (multiple damage sites).104,106,107,130,131 Although base-centered and phosphate-centered core-excited and shape resonances may play an essential role in electron attachment-induced DNA strand breaks, one needs to consider the other competing pathways as well. It is even more important because DNA has been proven to be an extremely stable molecule.3 Regardless of the large number (4 × 104 per MeV)14,24,46,47 of LEEs that are generated in the cellular medium,53 the rate of strand breaks in DNA has continued to be minimal, ensuring the survival of life. When DEA experiments on short single-stranded DNA-oligomers of defined sequences in the gas phase, microsolvated, and deposited on metal surfaces are considered, there are essentially three types of bound anionic states that are important. The first one is the DB radical anion state, which is formed due to a charge–dipole interaction between the additional electron and DNA. The formation of DB anion is aided by the strong dipole moment of DNA. Even the nucleobases, the simplest model systems of DNA, have dipole moments with magnitudes >2.5 D (except adenine) which ensures the formation of DB anions. The additional electron density is located away from the nuclear framework in the DB anion (see Figure 3a) of nucleobases which results in minimal distortion in geometry as compared to the neutral molecule.128 On the contrary, in the second kind of bound-anionic state, which is called the π*-type valence-bound (VB) state, the extra electron occupies the vacant valence π*-orbital of the system (see Figure 3b).128 Therefore, the additional electron density is localized on the molecule which causes significant geometric distortions as compared to the neutral molecule. The third bound-anionic state, perhaps the most important in terms of DNA strand breaks, is the σ*-type VB radical anionic state (see Figure 3c), which lies along either the sugar–phosphate (C–O) or sugar–nucleobase bonds (C–N). Base release or strand breaks occur once the electron gets transferred to this state which is assisted by the vibrational degree of freedom along the respective bonds.
Figure 3.

EA-EOM-DLPNO-CCSD natural orbital representing (a) DB, (b) π*-type valence-bound, and (c) σ*-type valence-bound radical anionic states of 2′-deoxycytidine-3′-monophosphate. Reproduced with permission from ref (132). Copyright 2021 American Chemical Society.
It is plausible that more than one kind of anionic state can be present in DNA.133 Therefore, before we delve further into the description of these anionic states, it is necessary to establish the level of theory that can treat all three of these different anionic states on equal footing. The various theoretical methods available in the literature to calculate electron affinities can be broadly classified into two classes. The first one is the Δ-based method, where separate calculations need to be done for both neutral and anion to obtain the electron affinity. The second one is the direct energy difference-based methods which include EOM-CC,134−138 Green’s function-based methods,139 linear response methods, and methods based on polarizable propagators.139−141 Most of the previous computational studies on electron attachment-induced DNA damage in the literature use the Δ-based technique along with density functional theory (DFT) to determine the electron affinity of model systems of DNA. Ref (86) comprehensively reviews the DFT-based studies on electron attachment to DNA subunits. However, the standard DFT methods are plagued with the issue of self-interaction in the case of anions.142 The Δ-based wave function methods, although free from self-interaction, are still prone to variational collapse when large basis sets are used.143 The EOM-CC method is free from the problem of variational collapse and allows one to access the ground and excited states of the anion in the same calculation. However, the high computational cost and large storage requirements limit its application beyond small molecules. Therefore, one needs to use approximations to reduce the computational cost of the EOM-CCSD method. Dutta and co-workers have reported a domain-based local pair natural orbital (DLPNO)-based144 implementation of the EOM-CCSD method (EOM-DLPNO-CCSD) for the EA145 and IP146 of large molecules. Tripathi et al. have recently shown that pair natural orbital-based approximation provides the best balance between accuracy and computational cost for studying radiation-induced damage to genetic material and can treat different anionic states of the DNA subunits on an equal footing.57
Now, all of the DNA bases (except adenine), the guanine–cytosine (GC) base pair, and some of the nucleotides show a stable DB state (see Table 1). The simulation of these DB states is often tricky and requires specially constructed basis sets.58 The DB anionic state can act as a gateway for electron capture. The trapped electron can subsequently get transferred to the σ*-type or π*-type VB anionic state. The transfer of the additional electron from the initial DB state to the σ*-type VB state can lead to DNA single-strand breaks (SSB). On the other hand, SSB could be avoided if a stable π*-type VB anion is formed from the DB state. This is because our study has already shown that once a base-centered anion is formed, the subsequent transfer of an electron to the dissociative σ*-type state has a large kinetic barrier.132 The π*-type VB anion of DNA nucleobases are not adiabatically bound.58,128 However, stable π*-type VB anionic states are observed in Watson–Crick GC and adenine–thymine (AT) base pairs,128 and in microsolvated analogues148−150 of nucleobases. Also, the transitions from the DB to the VB state in these systems are very feebly allowed optically. However, molecular vibrations can also cause a transition from the DB state to the π*-type or σ*-type VB states.
Table 1. Vertical Electron Affinities (meV) in the PNO-Based EOM-CCSD Method and Dipole Moments in Debye Units Corresponding to the DB States of the DNA Subunit.
Modeling the vibrational transition in DNA subunits using multidimensional PES would be tedious and computationally expensive. One can take a one-dimensional cut of the PES or draw a potential energy curve along a single unified coordinate. The initial electron attachment in the WC GC base pair leads to the formation of a doorway-DB state, from which the electron gets transferred to the π*-type VB state.128Figure 4 presents the potential energy curve (PEC) along a linear transit between the DB and VB geometries.128 The intermediate geometries in the PEC were obtained by the following expression:
| 3 |
where R represents a particular geometric parameter such as bond length, bond angle, and dihedral angle for the intermediate geometry. RDB and RVB are the corresponding geometric parameters for the DB and VB anionic states, respectively. The λ is a dimensionless parameter that is varied from 0 to 1 to obtain the intermediate geometries. When λ = 0, it corresponds to the DB anion geometry, and if λ = 1, the VB anion geometry is obtained. The PEC shows avoided crossing between the ground and the first excited state of the anion. This leads to the breakdown of the Born–Oppenheimer approximation, and one can no longer apply the adiabatic picture.151 Sommerfeld152,153 have previously used a simple diabatization scheme introduced by Köppel154,155 and co-workers to solve a similar problem. In his 2002 work, he used the DB and VB states of nitromethane as the basis for constructing diabatic PEC.152 Diabatic PESs, unlike adiabatic surfaces, can cross each other, and the electronic coupling between them was calculated by fitting a simple avoided crossing model potential to the diabatic basis.
| 4 |
Here, V1 are V2 are defined using harmonic potential at the coordinate λ. The value of W that Sommerfeld calculated was lower by one order in magnitude than the vertical excitation energy of both anionic states in their equilibrium geometry. Therefore, the nitromethane anion (W = 30 meV) comes in the weak coupling regime. Based on first-order perturbation theory, the rate of transition between the diabatic states is proportional to W2. Hence, the obtained value of W indicates that the VB state will be populated quickly from the initially formed DB state, which acts as the doorway for electron capture. He also obtained comparable results for uracil and 5-chloro-uracil.153
Figure 4.

Adiabatic and diabatic PES corresponding to the linear transit from the DB to the VB state of GC. Reproduced with permission from ref (128). Copyright 2019 American Chemical Society.
Our recent works have demonstrated the existence of doorway mechanisms for the formation of a stable VB state of various model systems of DNA in the gas phase and under microsolvation.128,132,148−150,156 For the GC base pair, we have used the same diabatization scheme employed by Sommerfeld to obtain the coupling between DB and VB states. The W was found to be 12 meV which is 1 order of magnitude less than the vertical excitation energies at the equilibrium geometries of the diabatic states, which means that the anionic states are in the threshold of the weak coupling limit. Under this scenario, the rate constant for the transition from the initial electronic DB state to the final π*-type VB state can be calculated approximately using the Marcus law.157
| 5 |
Here, ΔG0 is the free energy difference between the DB and VB anionic states without considering the entropy contribution, and λRis the reorganization energy (see Figure 5). The additional electron in the π*-type VB states in GC is localized on the cytosine. The calculated rate constant for the transition of the electron from DB to π*-type VB states of the GC base pair was found to be 4.1 × 1011 s–1, significantly higher than the one obtained for isolated cytosine nucleobase (1.6 × 105 s–1). However, one should also note that the coupling constant for cytosine was more than double that of the GC base pair,128 and the results contradict the simple W2 dependency proposed earlier.152,153 This trend in the rate can be attributed to the difference between the ΔG0 value of the two systems. The GC base pair forms a stable VB anionic state when compared to its neutral ground state. However, the VB anion of cytosine is unbound, and as a result, the rate of transfer of the electron from DB to π*-type VB states is 6 orders of magnitude lower.
Figure 5.
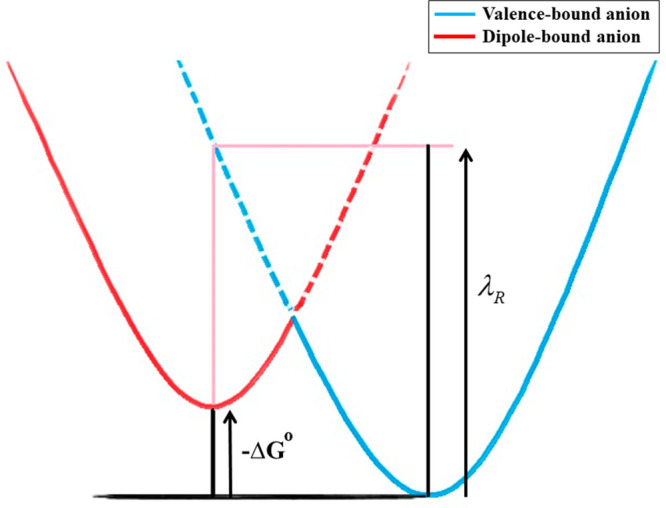
Schematic representation of the electron transfer between DB and VB states. The solid line indicates the ground-state adiabatic surface of the anion, and the dashed line indicates the diabatic surface corresponding to the first excited state of the anion. Reproduced with permission from ref (128). Copyright 2019 American Chemical Society.
The transfer of an electron from the DB state to the σ*-type VB states using the 2′-deoxycytidine-3′-monophosphate (3′-dCMPH) model system was also studied.132 From the PEC for 3′-dCMPH C–O bond elongation (Figure 6), one can see that the DB state of the nucleotide is formed initially. As the C–O bond length increases, a σ*-type VB state is formed which subsequently becomes the ground state of the anion on further elongation of the bond. The PEC shows an avoided crossing between the ground and excited state in the adiabatic PEC of the 3′-dCMPH anion. It indicated that the transition of the electron from the initial DB state to σ*-type VB states happens due to the mixing of electronic and nuclear degrees of freedom. The population of dissociative σ*-type VB states will lead to the cleavage of the C–O bond. However, the rate of C–O bond cleavage in the 3′-dCMPH model system starting from a stable DB state has been found to be extremely slow as compared to the formation of the π*-type nucleobase-centered VB states. This shows that the doorway mechanism cannot lead to strand break. However, it can lead to the formation of a stable anionic state, which will be competitive to the resonance-induced strand break. It should be noted that the doorway states mentioned above are bound anionic states where the extra electron is stabilized by charge–dipole interactions. However, the large dipole moments in DNA subunits can lead to dipole-bound resonance states.127 Recently, Kim and co-workers observed C–I bond cleavage in the iodophenoxide anion due to the transfer of an additional electron from the DB Feshbach resonance state to the σ*-type dissociative VB state.158,159 A DB state-based doorway mechanism was also suggested for DNA damage observed when terpyridine-platinum intercalated plasmid DNA was irradiated with LEEs.160 Similar mechanisms may also prevail in electron attachment-induced DNA strand break.
Figure 6.
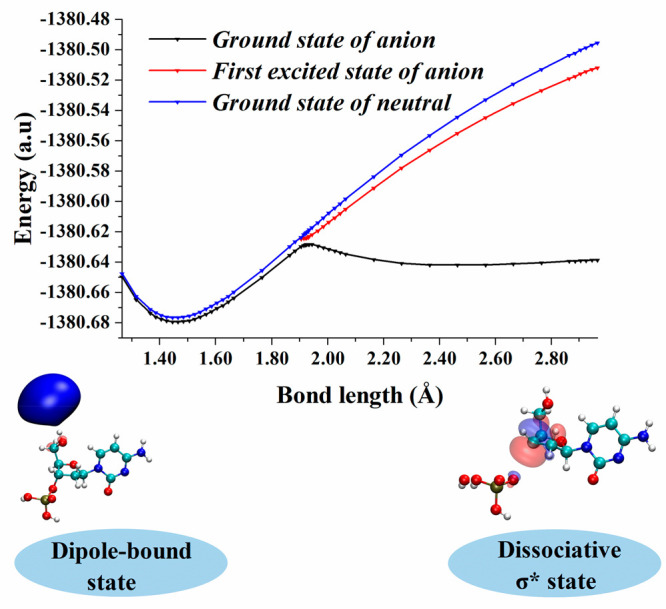
Adiabatic (right) PEC for C–O bond dissociation from the DB state of 2′-deoxycytidine-3′-monophosphate. Reproduced with permission from ref (132). Copyright 2021 American Chemical Society.
Effect of the Environment
The cellular environment is drastically different from the dilute solutions used in experimental studies. Obviously, one cannot expect the experimental and computational studies done on gas-phase and sparsely solvated model systems of DNA to simulate the actual process of electron attachment to DNA. However, they could still shed insights into the electron attachment-induced radiation damage to DNA. Water molecules constitute a major part of the cellular environment. Fedor and co-worker161−163,203 have shown that the presence of water molecules suppresses the DEA-induced fragmentation. Their mass spectrometric studies on 5′-dCMP showed that the extent of sugar–nucleobase and phosphate P–O bond breaks decreased with the increase in the number of water molecules bound to the nucleotide.163 Furthermore, when microsolvated pyrimidine nucleobases were bombarded with LEE, the formation of the corresponding hydrated anions was preferred over fragmentation pathways.161,203 Mostafavi and co-workers52 have demonstrated that the nucleosides can scavenge the epre– within 400 fs. Therefore, to properly understand the mechanism of DNA strand break in the condensed environment, one needs to include the effect of aqueous solvation in the simulations. The simplest way to include the solvation effect in quantum chemical calculations is to use the implicit solvation models. They add very little computational overhead on gas-phase calculations. The Barone–Tomasi polarizable continuum model (PCM)164,165 is one of the most popular implicit solvation models widely used for simulating electron attachment to genetic material in an aqueous environment.86,166,167 Simons and co-workers have found that the presence of PCM solvation preferably stabilized the radical anionic state of 3′-dCMPH compared to the neutral.104 The nucleotide anion radical was unbound at the Hartree–Fock level of theory used by them, and electron correlation is known to increase the stability of the anion. Sanche and co-workers have shown that the use of DFT methods leads to bound anions in DNA subunits, and the AEA of their sugar–phosphate–sugar model increased from 0.03 to 0.88 eV at the B3LYP/6-31+G-(d) level, when the effect of aqueous solvent was considered.108 The calculated electron affinity values are extremely sensitive to the used theoretical method and the basis set. A long spread of theoretically predicted AEA values for purine and pyrimidine nucleobases is available in the literature.86 For example, the gas-phase AEA of guanine varies from 0.07 eV168 at the B3LYP/TZ2P++ level to −0.7 eV169 calculated at the MP2/6-31+G(D) level of theory. However, the AEA for the guanine in aqueous solution, calculated using the PCM model, is generally found to be positive.170,171 One can observe a similar trend for the other nucleobases86 as well, when the gas-phase result is compared to that in the aqueous solution. Kumar et al. attempted to combine the effect of explicit water–DNA interaction and that of the bulk solvent environment using the microhydrated GC base pair in PCM with the B3LYP functional and 6-31+G** basis set.172 The AEA values for the hexahydrated and the decahydrated GC base pair were 0.74 and 0.95 eV, respectively. Interestingly, upon PCM’s introduction, the AEA for both GC–water clusters increased to 1.77 eV. The PCM-based studies have also been reported using other model systems such as nucleosides, nucleotides, dinucleotides, etc.104,106,130,173−176 These studies indicated that electron attachment to nucleotides in both the gas phase and solvent favored the formation of base-centered radical anions.173−175 The presence of a negatively charged phosphate group in the nucleotide makes it different from the other model systems such as nucleobase and nucleobase pairs. In the biological environment, the PO4 negative charge is largely neutralized by counterions such as K+ and Na+. In several theoretical studies86 where mono- and diphosphate nucleotides are used as model systems, the phosphate oxygen is protonated or methylated to neutralize it. However, Gu et al. obtained similar values of AEA, vertical electron affinity (VEA), and VDE for 2′-deoxythymidine-5′-monophosphate (5 ′-dTMP–) and its protonated analogue (5′-dTMPH) using the B3LYP/DZP++ level of theory, along with PCM solvation.173 This indicates that the presence of counterions in an aqueous solution may not influence the electron affinity of nucleotides. Sevilla and Kumar have tried to simulate the bulk solvation effect on 5′-dTMP by solvating it with 11 water molecules and treating the whole system in PCM.177 In addition to the increased ability to take up extra electrons, the aqueous 5′-dTMP cluster also exhibited a higher barrier height for 5′C–O bond cleavage compared to that calculated in the gas phase at the B3LYP/6-31++G** level of theory. Other attempts to determine the barrier height of 3′C–O and 5′C–O bond break in the radical anions of 2′-deoxypyrimidine monosphates,174,178 2′-deoxypurine monophosphates,179 2′-deoxypyrimidine-3′,5′-diphosphate,180 and 2′-deoxypurine-3′,5′-diphosphate181,182 have also resulted in the increase in barrier height in aqueous solutions.
The PCM model is successful in giving a qualitative picture of electron attachment in the aqueous environment, where the additional electron density is localized on the nuclear framework. Continuum-based solvation models consider the molecule in a predefined cavity, and the dielectric continuum exists outside the cavity. For an accurate description of the system, it is essential that the electron density exists within the cavity. However, the electron density can spread well beyond the cavity for diffuse states such as the DB state and surface-bound states183,184 (where an electron exists at the air–water interface). This makes PCM-based solvation models unsuitable for describing the DB anionic state in the presence of a solvent.185 Apart from this issue, PCM provides a crude approximation to the solute–solvent interactions such as the hydrogen-bonding and chemical processes such as proton transfer from solvent to solute that significantly contribute to the stabilization of the radical anions of DNA in the aqueous media. A combination of microsolvation and PCM methods, previously used by Kumar et al.,172 might be one of the possible ways of taking care of the explicit interactions between DNA and surrounding water molecules. However, Simons and co-workers185 have recently shown that such an approach would still fail to describe DB and surface-bound states.
Explicit solvation of DNA model systems with molecular water in theoretical simulations would address the shortcoming of PCM-like solvation models. However, considering all water molecules in a bulk solvated-DNA system, even at a semiempirical level, is not practically feasible due to the high computational cost. Kohanoff and co-workers suggested that the computational bottleneck can be avoided by considering a smaller number of water molecules (∼30) along with periodic boundary conditions (PBC) to simulate a fully solvated DNA subunit.186 Their first principle dynamics on solvated nucleobases showed that the excess electron, which is initially delocalized over nucleobase and water molecules, becomes localized on the nucleobase under ∼15 fs.186 They also performed a constrained ab initio molecular dynamics (MD) simulation to study the 3′C–O bond cleavage in 3′-monophosphate nucleotides.187 Their simulations have shown that barrier heights for the 3′C–O bond cleavage in the aqueous medium are higher than those in the gas phase for all of the nucleotides.187 McAllister et al. further analyzed the effect of water to nucleobase proton transfer on the 3′C–O and N-glycosidic (C–N) bond rupture.188 For the sugar–phosphate bond cleavage, protonation of the nucleobase resulted in an increase in the free energy barrier of all 3′-monophosphate nucleotides except the 3′-dAMPH.188 Moreover, the 3′C–O bond of purine nucleotides is more vulnerable to rupture upon electron attachment than pyrimidine nucleotide in the solution. They have also found that the barrier heights for 3′C–O and C–N bond break reactions in water were comparable in pyrimidine nucleotides. Although the sugar–phosphate and the N-glycosidic bond breaks contribute to the majority of DEA products, one should not ignore the significance of base damages due to the cleavage of the nucleobase nitrogen–hydrogen (N–H) bond. When the additional electron gets transferred from the nucleobase π* resonance to the N–H σ* orbital due to the coupling of the electronic degrees of freedom with the N–H vibrational mode, it can lead to bond rupture. Kohanoff and co-workers have modeled this process in solution using first-principle MD simulations.189 They found that, in the gas phase, the N–H bond breaks with 1.67 eV of kinetic energy supplied to the bond vibrational mode.189 Their study shows that, in the presence of explicit water molecules, the hydrogen atom release the required energy >5 eV. This clearly shows the protective role water molecules play through the formation of extensive hydrogen bonding with the genetic material.
The first principle MD simulation at the DFT level has played a great role to enhance our understanding of the electron attachment to the components of DNA and the subsequent chemical processes.85 However, due to the high computational cost, if one has to achieve the required time scales of those reactions through MD simulation, the number of water molecules in the simulation will be restricted to <100, even with enhanced sampling techniques. Such a small number of water molecules are not sufficient to model the effect of the bulk solvent environment in the process of interest. The QM/MM approach is a potential solution to this issue. Hugosson and co-workers190 have used DFT-based QM/MM simulations to determine the free energy barriers of the sugar–phosphate bond break in 3′- and 5′-dCMP radical dianions. The calculated free energy barriers for both 3′C–O (∼35 kcal/mol) and 5′C–O (∼42 kcal/mol) show that sugar–phosphate bond ruptures are unlikely to occur in an aqueous environment.
The DFT-based QM/MM studies, as described above, mostly considered the ground state of the anions. However, the experiments by Mostafavi and co-workers52,93 have shown that the excited state of the anion plays a critical role in the electron attachment process. The electron attachment to DNA subunits in the bulk water may differ considerably from that observed in the microsolvated models. Nevertheless, the microsolvated systems can help to understand the effect of explicit solvent–solute interaction on the electron attachment process. Those results could bridge the studies conducted in gas-phase and bulk-solvated model systems of DNA. The microsolvation studies have shown that the surrounding water molecules can stabilize π* shape resonances in nucleobases,191,192 which has recently been verified experimentally by Verlet and co-workers.90 A similar stabilizing effect is observed for the stable π* VB state. The existence148 of the doorway mechanism for electron attachment has been observed for microsolvated uracil, where the DB state acts as a doorway for electron capture, similar to that observed for the gas-phase GC base pair (see Figure 7). The initial electron attachment leads to the formation of a DB state which acts as a doorway for electron capture. The electron subsequently gets transferred to the π* type VB state by a mixing of electronic and nuclear degrees of freedom. The rate constant for the DB to VB state transition for microhydrated uracil is three orders greater than that observed in gas-phase uracil. This shows that even the presence of a single solvent molecule can enhance the rate of formation of the stable VB anion. The hydrogen bonding interaction between the water molecule and uracil stabilizes the anion and increases the rate of transitions from the DB to the VB state. Although the DB anion is unlikely to exist in the bulk water medium, the recent works from our group have shown that a doorway mechanism still exists for electron attachment to nucleobases in the bulk aqueous environment.148,150 In the case of the electron attachment to nucleobase in bulk water, the solvent-bound anionic state acts as the doorway state for electron attachment.148 EA-EOM-DLPNO-CCSD/aug-cc-pVDZ(+5s5p4d)-based QM/MM studies have shown that the incoming electron initially becomes bound to the water molecules. The water-bound electronic state is similar to the presolvated electrons, and their nature is not influenced by the presence of uracil in the system.148 The electron subsequently gets transferred to the π*-type VB state by mixing of electronic and nuclear degrees of freedom.
Figure 7.

Uracil monohydrate anion (a) natural orbital corresponding to the DB state, (b) natural orbital corresponding to the VB state, (c) adiabatic surface corresponding to the ground and first excited state, and (d) diabatic dipole and VB states. Reproduced with permission from ref (148). Copyright 2020 American Chemical Society.
Only one bound state was observed at the DB geometry in the monosolvated uracil. In the bulk solvated uracil, at least six bound anionic states existed even at the initial snapshot from the QM/MM trajectory. All six bound anionic states were reported to be solvent-bound initially. Bound states where the additional electron density is localized on uracil appeared as an excited state at 3.0 fs (see Figure 8). This state eventually becomes the ground state, and the solvent-bound state becomes the first excited state at 5.5 fs. However, the complete transfer of electrons and subsequent reorganization of the solvent molecules take around 500 fs. This process results in an effective electron scavenging mechanism by the nucleobase, where the presolvated electron reacts with solvated uracil to form an anionic complex where the extra electron is localized on water. The electron is then transferred from the water molecules to the uracil. The electron transfer rate is found to be very rapid at 2.1 × 1012 M s–1. The plot of the time evolution of the detachment energies of the bulk solvated uracil anion shows (see Figure 9) multiple avoided crossing similar to that observed in the microsolvated uracil. The water molecules stabilize the nucleobase-bound anions by extensive hydrogen bonds, and the rapid formation of the stable nucleobase-bound anion will be competitive to the DEA-induced sugar–phosphate bond cleavage, which can explain the suppression of DNA-strand break in the presence of an aqueous environment.
Figure 8.
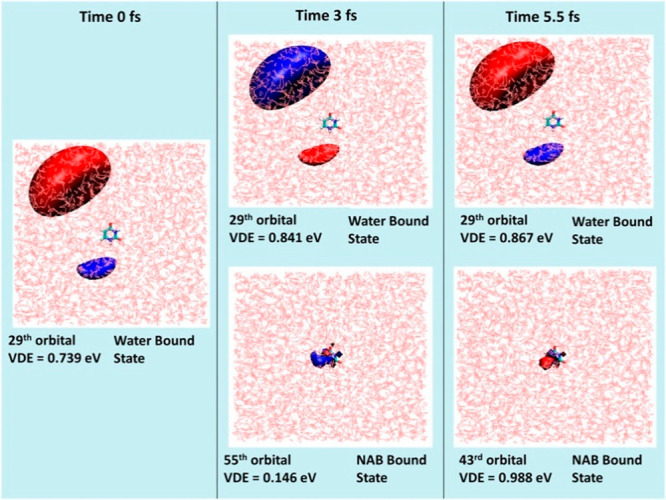
Molecular orbitals corresponding to the most dominant transition in the EA-EOM-DLPNO-CCSD method depicting time evolution of the anionic state formed by attachment of the bulk-solvated electron to uracil. Reproduced with permission from ref (148). Copyright 2020 American Chemical Society.
Figure 9.

Time evolution of detachment energy of the (left) bound anionic states and (right) diabatic water-bound and uracil bound states for the attachment of electron to uracil in bulk water environment. Reproduced with permission from ref (148). Copyright 2020 American Chemical Society.
The water-bound state-based doorway mechanism has also been observed for aqueous cytosine150 and GC base pairs.149 It has been observed that the stability of the bulk solvation is extremely sensitive to the level of theory used for the calculations. One needs to use a big water box, a large basis set with a sufficient number of diffuse functions, including polarization effect in the MM region and extensive sampling, to get a reasonable agreement with the experimental results.193 Matsika and co-workers have reported similar conclusions for electron attachment to solvated uracil.194,195
In addition to the aqueous media, the other molecular components present in the biological environment can also participate in electron attachment-induced radiation damage to DNA. Especially the histone proteins present in the close vicinity of DNA can play a crucial role in modulating the electron attachment process. Solomun et al. are among the first to show that the protein can effectively inhibit electron attachment-induced DNA strand break.196 Sanche and co-workers have also demonstrated that a high concentration of amino acid leads to protective action against LEE.197 The gas phase studied by Bowen and co-workers198,199 has shown that the electron attachment to nucleobase-amino acid complex leads to a barrier-free proton transfer between anionic nucleobase and amino acids, which stabilizes the excess negative charge. The QM/MM simulation by Kohanoff and co-workers200 has also shown that the glycine environment can act as a protective shield against the electron attachment to thymine.
We have recently demonstrated that electron attachment to nucleobases in the presence of an amino acid environment happens through a doorway mechanism,201 where the amino acid bound states act as a doorway for electron capture. Figure 10 presents the sequence of events observed in the electron attachment to cytosine in glycine solution. Initially, the electron is localized on the bulk glycine molecules. The extra electron density subsequently gets transferred to the nucleobase at around 100 fs and is followed by glycine to cytosine proton transfer. The presence of the amino acid protects the DNA subunits in two ways. First, the initial electron density is localized on the amino acid and away from the nucleobase. Therefore, the presence of amino acid leads to a physical shielding of the nucleobase and reduces the possibility of the formation of base-centered resonance states161,202,203 responsible for DNA strand cleavage. Second, the glycine can stabilize the nucleobase-bound anion by hydrogen bonding and proton transfer, increasing its electron scavenging effect and reducing the availability of secondary electrons leading to resonance-induced strand break. This kind of protection can be referred to as a chemical mechanism of protection.
Figure 10.

Evolution of the lowest anionic state of cytosine in the presence of a bulk glycine environment.
Summary of DNA Strand-Break Mechanisms
The mechanism shown in Scheme 5 involves the formation of a ground-state anion where the additional electron is localized on the nucleobase.174,175,178,180,181 The transfer of the electron from the nucleobase to the sugar–phosphate moiety has to occur to cause the sugar–phosphate bond to break. As mentioned earlier, the proton transfer from the complementary base (Scheme 5, process 7) would compete with the nucleobase to phosphate electron transfer (Scheme 5, processes 9 and 11). DNA bases are known to scavenge eaq– from aqueous solution to form base-centered radical anions.4,10 The base-centered radical anion of DNA is formed either by attachment of eaq or by the decay of base-centered resonances to their bound anionic ground state. Since eaq– bound-anionic states do not cause strand breaks in DNA, process 7 should be the dominant pathway compared to the processes shown in Reactions 9 and 11. This assumption is also supported by the kinetically favored inter-base-pair proton transfer in the DNA-ion radical.86,204−207 The proton transfer in the GC base pair anion radical takes place in picosecond (∼10 ps) time scales, as theoretically shown by Shiga and co-workers.204 One could attempt to obtain a qualitative picture of the kinetics of proton transfer from the barrier height of the process. A low barrier height would indicate that proton transfer is a kinetically favored pathway. Various theoretical studies86,204−207 have predicted the GC proton transfer to be a low-barrier process. Hence, DNA strand breaks may not occur through the mechanism shown in Scheme 5.
Scheme 5. Possible Pathways after the Formation of a Base-Centered Radical Anion.
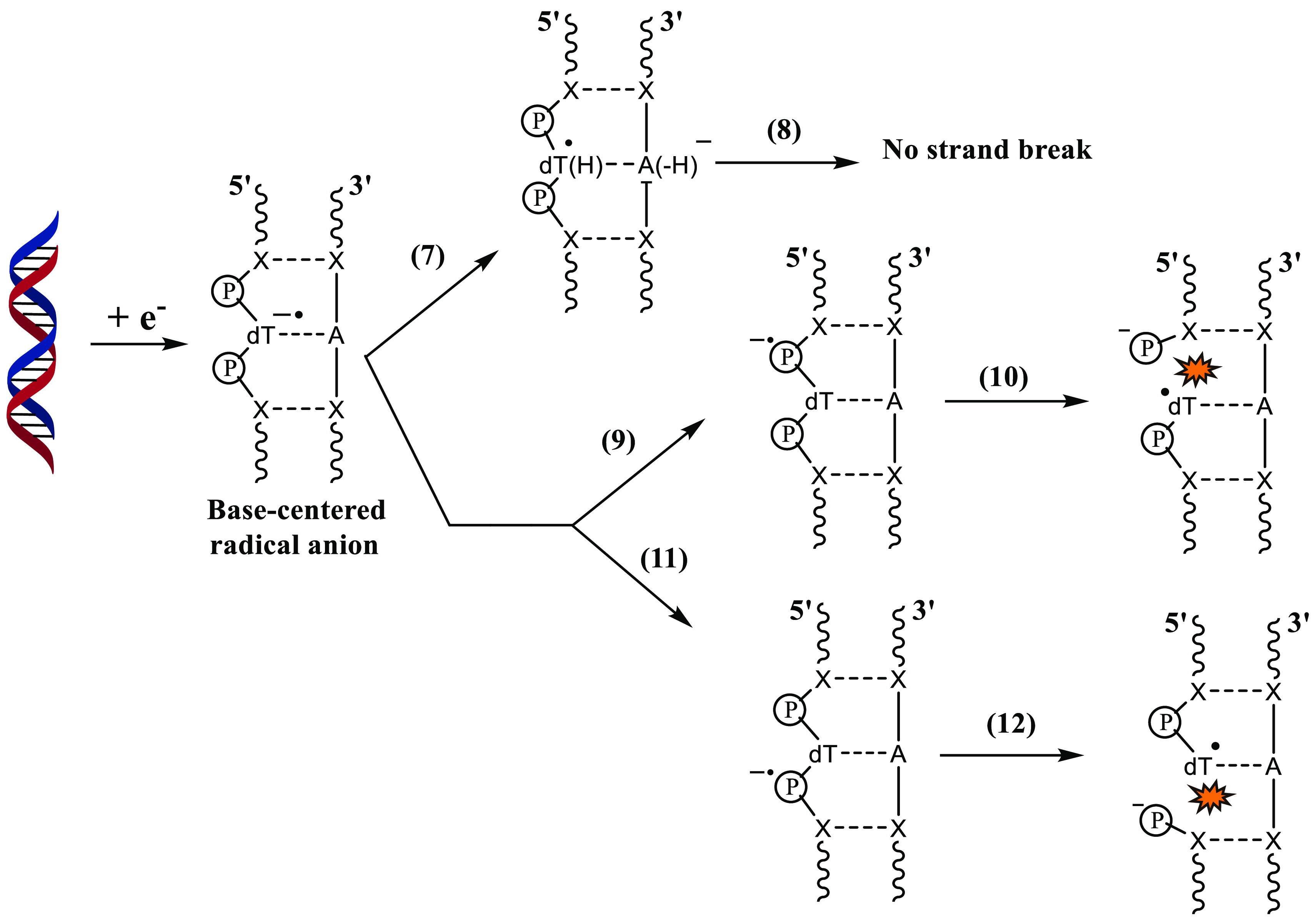
The second pathway (Scheme 6, process 13) involves the base-centered or phosphate-centered resonances. Once the phosphate-centered TNI* is formed in process 14, it may lead to either 3′C–O or 5′C–O bond rupture as shown in Scheme 4. However, we have only shown the latter in the scheme (process 15) to avoid it from becoming more complex, although the actual process it represents is very complex. The role of base-centered shape resonance was first proposed by Simons and co-workers.104,106,107,130 Shape resonance in DNA is formed in the energy range from 0 to ∼4 eV.105 The TNI*s detected at energies >5 eV and less than the ionization threshold of DNA are core-excited resonances.102,120 Sanche’s group suggested that core-excited resonances are responsible for DSBs,102,120,208 and shape resonances are responsible for SSBs105 in DEA to DNA. Once the base-centered excited TNI is formed, the electron may get transferred to the phosphate group which would eventually lead to 3′C–O or 5′C–O bond cleavage. Several theoretical studies108,131,209 had initially suggested that direct electron attachment to the DNA backbone could also occur, which was later verified experimentally by Kopyra.69 Once the phosphate-centered TNI* is formed, either by direct LEE attachment or by transfer of the additional electron from base-centered TNI*, the sugar–phosphate bond rupture could occur at 3′C–O or 5′C–O sites. Among the sugar–phosphate and sugar–nucleobase bonds, which one is more prone to DEA is still under debate. Wagner and co-workers employed ∼1.8 eV electrons to study the DEA pathway in thymidylyl-(3′-5′)-thymidine.72 Their results showed that the majority of DEA products were due to sugar–phosphate bond break. LC-MS/MS analysis of the products further showed that only a minor quantity of the products was formed due to base release. However, when the model DNA systems were changed to a 16-mer oligonucleotide, unaltered nucleobase formed due to sugar–nucleobase rupture was the major DEA product.76 They attribute this observation to the delocalization of the additional electron within the long DNA strand.
Scheme 6. Possible Strand Break Pathways Caused by the Formation of Base-Centered and Phosphate-Centered TNI* in DNA.
The last pathway of sugar–phosphate bond break involves initial electron attachment to form a dipole-bound anion (see Scheme 7).132 Our recent work on 3′-dCMPH showed that the valence-bound anion formation is favored over sugar–phosphate and sugar–nucleobase bond cleavage.132 Therefore, the dipole-bound state may not lead to DNA strand break or base release.
Scheme 7. Three Possible Pathways after the Formation of the Dipole-Bound Anion of the NPN System in the Gas Phase: Formation of the π*-type Valence-Bound Anion (a), C3′–O Bond Break (b), and C5′–O Bond Rupture (c).

Detection of LEE-Mediated DNA-Radicals in Irradiated DNA
The radical species formed in irradiated DNA due to LEE-mediated reaction (e.g., C3′dephos•, Scheme 4) have been identified by ESR spectroscopy in ion-beam irradiated hydrated DNA (degree of hydration = 12–14 water molecules/nucleotide) and in gamma-irradiated hydrated DNA (degree of hydration = 12–14 water molecules/nucleotide).11,13,14,35,210−212 However, ROPO2•– (Scheme 4) was detected in ion-beam irradiated hydrated DNA only.11,13,14,35,210−212 These results have been reviewed.11,13,14
Relevance to the Clinic (The Role of Radiation-Produced Electrons Involved in the Radiosensitization, i.e., Augmentation of Radiation Damage in Cells)
Experimental techniques including ESR studies of irradiated DNA-model compounds and of irradiated hydrated DNA, picosecond pulse radiolysis of DNA-model compounds, and theoretical studies have unequivocally established that pyrimidine bases are more electron affinic than the purine bases.24,52,57,77,93 Therefore, the C5-modified pyrimidine (especially thymine) nucleosides are being extensively explored for use as radiosensitizers. New base and nucleoside derivatives are synthesized that have similar or better DEA yields in the 0–3 eV region, and these modified nucleosides would not lose the C5-modification of the thymine base by thymidylate synthetase.213−221 These electron-mediated base-radicals would augment formation of the radical species that are precursor to the DNA-lesions, and in addition, the incorporated modified nucleosides could affect the repair processes to augment the radiation-induced cell death. These results show how radiation-produced electrons could be used to improve the chemoradiotherapeutic efficacy.213−220 Several platinum-based radiosensitizers are also being investigated.160
Future Directions
As described above, a majority of the experimental14,68,69,72,74 and theoretical studies85,86 on secondary electron attachment-induced radiation damage are performed on small DNA subunits, oligonucleotides, and short double-stranded DNA. However, experimental studies on the whole DNA are also available in the literature.20,70,73 A small model system allows one to gain insight into the mechanism of electron attachment. However, in real life, the symmetry (or lack of it), length of the DNA chain, organization of DNA, and its microenvironment play an important role in determining the site of electron attachment and formation of the subsequent lesions. Thus, one needs to perform experiments and theoretical studies with larger and more realistic models. This will require developing new experimental techniques and formulating faster and more accurate theoretical models.
The use of artificial intelligence and quantum computing will play a major role in the theoretical simulations of larger DNA model systems in the coming days. A lot of work is being done and needs to be done in that direction.
Acknowledgments
A.K.D. acknowledges the support from IIT Bombay, DST-SERB CRG (Project no. CRG/2022/005672), and Matrix (Project no. MTR/2021/000420) projects, DST-Inspire Faculty Fellowship (Project no. DST/INSPIRE/04/2017/001730), CSIR-India (Project no. 01(3035)/21/EMR-II), ISRO (Project no. RD/0122-ISROC00-004), Prime Minister’s Research Fellowship for financial support, IIT Bombay super computational facility, and C-DAC supercomputing resources (PARAM Smriti, PARAM Brahma) for computational time. A.A. acknowledges the support from National Cancer Institute of the National Institutes of Health (Grant RO1CA045424) and the National Science Foundation under Grant No. CHE-1920110.
Author Contributions
All authors contributed to this work equally, and the final version has been approved by all authors.
The authors declare no competing financial interest.
References
- Coffin J. M., Hughes S. H., Varmus H. E., Eds. Retroviruses; Cold Spring Harbor: NY, 1997. [PubMed] [Google Scholar]
- Watson J. D.; Crick F. H. C. Genetical Implications of the Structure of Deoxyribonucleic Acid. Nature 1953, 171 (4361), 964–967. 10.1038/171964b0. [DOI] [PubMed] [Google Scholar]
- Modrich P. Mechanisms in E. Coli and Human Mismatch Repair (Nobel Lecture). Angew. Chem., Int. Ed. 2016, 55 (30), 8490–8501. 10.1002/anie.201601412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill P.; Jonah C. D.; Rao B. S. M. Radiation-Induced Damage in DNA. Stud. Phys. Theor. Chem. 2001, 87, 585–622. 10.1016/S0167-6881(01)80023-9. [DOI] [Google Scholar]
- Cadet J.; Bellon S.; Douki T.; Frelon S.; Gasparutto D.; Muller E.; Pouget J.-P.; Ravanat J.-L.; Romieu A.; Sauvaigo S. Radiation-Induced DNA Damage: Formation, Measurement, and Biochemical Features. J. Environ. Pathol. Toxicol. Oncol. 2004, 23, 33–44. 10.1615/JEnvPathToxOncol.v23.i1.30. [DOI] [PubMed] [Google Scholar]
- Berthel E.; Ferlazzo M. L.; Devic C.; Bourguignon M.; Foray N. What Does the History of Research on the Repair of DNA Double-Strand Breaks Tell Us?—A Comprehensive Review of Human Radiosensitivity. Int. J. Mol. Sci. 2019, 20 (21), 5339. 10.3390/ijms20215339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obodovskiy I.Radiation Therapy. In Radiation; Elsevier, 2019; pp 387–396. [Google Scholar]
- Dizdaroglu M., Lloyd R. S., Dizdaroglu M., LLoyd R. S., Eds. DNA Damage, DNA Repair and Disease; The Royal Society of Chemistry, 2020; Vol. 1. [Google Scholar]
- Dizdaroglu M., Lloyd R. S., Dizdaroglu M., LLoyd R. S., Eds. DNA Damage, DNA Repair and Disease; The Royal Society of Chemistry, 2020; Vol. 2. [Google Scholar]
- von Sonntag C.Free-Radical-Induced DNA Damage and Its Repair: A Chemical Perspective, 1st ed.; Springer: Berlin, Heidelberg, 2006. [Google Scholar]
- Adhikary A.; Becker D.; Sevilla M. D.. Electron Spin Resonance of Radicals in Irradiated DNA. In Applications of EPR in Radiat. Res.; Lund A., Shiotani M., Eds.; Springer International Publishing: Cham, 2014; pp 299–352. [Google Scholar]
- Dizdaroglu M. Oxidatively Induced DNA Damage and Its Repair in Cancer. Mutation Research/Reviews in Mutation Research 2015, 763, 212–245. 10.1016/j.mrrev.2014.11.002. [DOI] [PubMed] [Google Scholar]
- Sevilla M. D.; Becker D.; Kumar A.; Adhikary A. Gamma and Ion-Beam Irradiation of DNA: Free Radical Mechanisms, Electron Effects, and Radiation Chemical Track Structure. Radiat. Phys. Chem. 2016, 128, 60–74. 10.1016/j.radphyschem.2016.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Becker D.; Adhikary A.; Sevilla M. D. Reaction of Electrons with DNA: Radiation Damage to Radiosensitization. Int. J. Mol. Sci. 2019, 20 (16), 3998. 10.3390/ijms20163998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kant M.; Jaruga P.; Coskun E.; Ward S.; Stark A. D.; Baumann T.; Becker D.; Adhikary A.; Sevilla M. D.; Dizdaroglu M. Ne-22 Ion-Beam Radiation Damage to DNA: From Initial Free Radical Formation to Resulting DNA-Base Damage. ACS Omega 2021, 6 (25), 16600–16611. 10.1021/acsomega.1c01954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Wang F.; Mostafavi M. Ultrafast Chemistry of Water Radical Cation, H2O•+, in Aqueous Solutions. Molecules 2018, 23 (2), 244–244. 10.3390/molecules23020244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauduel Y.; Pommeret S.; Migus A.; Antonetti A. Some Evidence of Ultrafast H2O+-Water Molecule Reaction in Femtosecond Photoionization of Pure Liquid Water: Influence on Geminate Pair Recombination Dynamics. Chem. Phys. 1990, 149 (1–2), 1–10. 10.1016/0301-0104(90)80126-I. [DOI] [Google Scholar]
- Kanaar R.; Hoeijmakers J. H. J.; van Gent D. C. Molecular Mechanisms of DNA Double-Strand Break Repair. Trends Cell Biol. 1998, 8 (12), 483–489. 10.1016/S0962-8924(98)01383-X. [DOI] [PubMed] [Google Scholar]
- Ward J. F. The Complexity of DNA Damage: Relevance to Biological Consequences. Int. J. Radiat. Biol. 1994, 66 (5), 427–432. 10.1080/09553009414551401. [DOI] [PubMed] [Google Scholar]
- Sahbani S. K.; Girouard S.; Cloutier P.; Sanche L.; Hunting D. J. The Relative Contributions of DNA Strand Breaks, Base Damage and Clustered Lesions to the Loss of DNA Functionality Induced by Ionizing Radiation. Radiat. Res. 2014, 181 (1), 99–110. 10.1667/RR13450.1. [DOI] [PubMed] [Google Scholar]
- Georgakilas A. G.; O'Neill P.; Stewart R. D. Induction and Repair of Clustered DNA Lesions: What Do We Know So Far?. Radiat. Res. 2013, 180 (1), 100–109. 10.1667/RR3041.1. [DOI] [PubMed] [Google Scholar]
- Costa R. A. P.; Romagna C. D.; Pereira J. L.; Souza-Pinto N. C. The Role of Mitochondrial DNA Damage in the Citotoxicity of Reactive Oxygen Species. Journal of Bioenergetics and Biomembranes 2011, 43 (1), 25–29. 10.1007/s10863-011-9329-8. [DOI] [PubMed] [Google Scholar]
- Greaves L. C.; Reeve A. K.; Taylor R. W.; Turnbull D. M. Mitochondrial DNA and Disease. J. Pathol. 2012, 226 (2), 274–286. 10.1002/path.3028. [DOI] [PubMed] [Google Scholar]
- Ma J.; Denisov S. A.; Adhikary A.; Mostafavi M. Ultrafast Processes Occurring in Radiolysis of Highly Concentrated Solutions of Nucleosides/Tides. Int. J. Mol. Sci. 2019, 20 (19), 4963. 10.3390/ijms20194963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker D.; Kumar A.; Adhikary A.; Sevilla M. D.. Gamma and Ion-beam DNA Radiation Damage: Theory and Experiment. DNA Damage, DNA Repair and Disease; Dizdaroglu M., Lloyd R. S., Eds.; Royal Society of Chemistry: London, 2020; Vol. 2, pp 426–457. [Google Scholar]
- Becker D.; Adhikary A.; Sevilla M. D.. The Role of Charge and Spin Migration in DNA Radiation Damage. In Charge Migration in DNA: Perspectives from Physics, Chemistry, and Biology; Chakraborty T., Ed.; Springer: Berlin, Heidelberg, 2007; pp 139–175. [Google Scholar]
- de la Lande A.; Denisov S.; Mostafavi M. The Mystery of Sub-Picosecond Charge Transfer Following Irradiation of Hydrated Uridine Monophosphate. Phys. Chem. Chem. Phys. 2021, 23 (37), 21148–21162. 10.1039/D0CP06482C. [DOI] [PubMed] [Google Scholar]
- Bernhard W. A.Radical Reaction Pathways Initiated by Direct Energy Deposition in DNA by Ionizing Radiation. In Radical and Radical Ion Reactivity in Nucleic Acid Chemistry; John Wiley & Sons, 2009; pp 41–68. [Google Scholar]
- Close D. M.From the Primary Radiation Induced Radicals in DNAConstituents to Strand Breaks: Low Temperature EPR/ENDOR Studies. In Radiation Induced Molecular Phenomena in Nucleic Acids: A Comprehensive Theoretical and Experimental Analysis; Shukla M. K., Leszczynski J., Eds.; Springer Netherlands: Dordrecht, 2008; pp 493–529. [Google Scholar]
- Genereux J. C.; Barton J. K. Mechanisms for DNA Charge Transport. Chem. Rev. 2010, 110 (3), 1642–1662. 10.1021/cr900228f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanvah S.; Joseph J.; Schuster G. B.; Barnett R. N.; Cleveland C. L.; Landman U. Oxidation of DNA: Damage to Nucleobases. Acc. Chem. Res. 2010, 43 (2), 280–287. 10.1021/ar900175a. [DOI] [PubMed] [Google Scholar]
- Wagenknecht H.-A.Principles and Mechanisms of Photoinduced Charge Injection, Transport, and Trapping in DNA. In Charge Transfer in DNA; Wiley-VCH Verlag GmbH & Co. KGaA, 2005; pp 1–26.
- Peluso A.; Caruso T.; Landi A.; Capobianco A. The Dynamics of Hole Transfer in DNA. Molecules 2019, 24 (22), 4044. 10.3390/molecules24224044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker D.; Adhikary A.; Sevilla M. D.. Mechanisms of Radiation-Induced DNA Damage: Direct Effects. In Recent Trends in Radiation Chemistry; World Scientific, 2010; pp 509–542. [Google Scholar]
- Adhikary A.; Becker D.; Palmer B. J.; Heizer A. N.; Sevilla M. D. Direct Formation of the C5′-Radical in the Sugar–Phosphate Backbone of DNA by High-Energy Radiation. J. Phys. Chem. B 2012, 116 (20), 5900–5906. 10.1021/jp3023919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary A.; Kumar A.; Palmer B. J.; Todd A. D.; Sevilla M. D. Formation of S–Cl Phosphorothioate Adduct Radicals in DsDNA S-Oligomers: Hole Transfer to Guanine vs Disulfide Anion Radical Formation. J. Am. Chem. Soc. 2013, 135 (34), 12827–12838. 10.1021/ja406121x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Marignier J.-L.; Pernot P.; Houée-Levin C.; Kumar A.; Sevilla M. D.; Adhikary A.; Mostafavi M. Direct Observation of the Oxidation of DNA Bases by Phosphate Radicals Formed under Radiation: A Model of the Backbone-to-Base Hole Transfer. Phys. Chem. Chem. Phys. 2018, 20 (21), 14927–14937. 10.1039/C8CP00352A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterworth P. J. Lehninger: Principles of Biochemistry (4th Edn) Nelson, D. L.; and Cox, M. C., W. H. Freeman & Co., New York, 1119 pp (plus 17 pp glossary), ISBN 0-7167-4339-6 (2004). Cell Biochemistry and Function 2005, 23 (4), 293–294. 10.1002/cbf.1216. [DOI] [Google Scholar]
- Swarts S. G.; Sevilla M. D.; Becker D.; Tokar C. J.; Wheeler K. T. Radiation-Induced DNA Damage as a Function of Hydration: I. Release of Unaltered Bases. Radiat. Res. 1992, 129 (3), 333–344. 10.2307/3578034. [DOI] [PubMed] [Google Scholar]
- Becker D.; La Vere T.; Sevilla M. D. ESR Detection at 77 K of the Hydroxyl Radical in the Hydration Layer of Gamma-Irradiated DNA. Radiat. Res. 1994, 140 (1), 123–129. 10.2307/3578577. [DOI] [PubMed] [Google Scholar]
- La Vere T.; Becker D.; Sevilla M. D. Yields of OH in Gamma-Irradiated DNA as a Function of DNA Hydration: Hole Transfer in Competition with OH Formation. Radiat. Res. 1996, 145 (6), 673–680. 10.2307/3579357. [DOI] [PubMed] [Google Scholar]
- Herbert J. M.; Coons M. P. The Hydrated Electron. Annu. Rev. Phys. Chem. 2017, 68 (1), 447–472. 10.1146/annurev-physchem-052516-050816. [DOI] [PubMed] [Google Scholar]
- von Sonntag C.Radiation-Induced DNA Damage: Indirect Effects. In Recent Trends in Radiation Chemistry; World Scientific, 2010; pp 543–562. [Google Scholar]
- Dizdaroglu M.; Jaruga P. Mechanisms of Free Radical-Induced Damage to DNA. Free Radic. Res. 2012, 46 (4), 382–419. 10.3109/10715762.2011.653969. [DOI] [PubMed] [Google Scholar]
- Schulte-Fröhlinde D.; Bothe E. Identification of a Major Pathway of Strand Break Formation in Poly U Induced by OH Radicals in Presence of Oxygen. Z. Naturforsch. 1984, 39c (3–4), 315–319. 10.1515/znc-1984-3-423. [DOI] [PubMed] [Google Scholar]
- Alizadeh E.; Sanche L. Precursors of Solvated Electrons in Radiobiological Physics and Chemistry. Chem. Rev. 2012, 112 (11), 5578–5602. 10.1021/cr300063r. [DOI] [PubMed] [Google Scholar]
- Baccarelli I.; Bald I.; Gianturco F. A.; Illenberger E.; Kopyra J. Electron-Induced Damage of DNA and Its Components: Experiments and Theoretical Models. Phys. Rep. 2011, 508 (1), 1–44. 10.1016/j.physrep.2011.06.004. [DOI] [Google Scholar]
- Rak J.; Chomicz L.; Wiczk J.; Westphal K.; Zdrowowicz M.; Wityk P.; Żyndul M.; Makurat S.; Golon Ł. Mechanisms of Damage to DNA Labeled with Electrophilic Nucleobases Induced by Ionizing or UV Radiation. J. Phys. Chem. B 2015, 119 (26), 8227–8238. 10.1021/acs.jpcb.5b03948. [DOI] [PubMed] [Google Scholar]
- Walker D. C. The Hydrated Electron. Quart. Rev. 1967, 21 (1), 79–108. 10.1039/qr9672100079. [DOI] [Google Scholar]
- Steenken S. Purine Bases, Nucleosides, and Nucleotides: Aqueous Solution Redox Chemistry and Transformation Reactions of Their Radical Cations and E∼ and OH Adducts. Chem. Rev. 1989, 89 (3), 503–520. 10.1021/cr00093a003. [DOI] [Google Scholar]
- Kumar A.; Sevilla M. D.. Low-Energy Electron (LEE)-Induced DNA Damage: Theoretical Approaches to Modeling Experiment. In Handbook of Computational Chemistry; Leszczynski J., Ed.; Springer: Dordrecht, The Netherlands, 2015, 1–63. [Google Scholar]
- Ma J.; Kumar A.; Muroya Y.; Yamashita S.; Sakurai T.; Denisov S. A.; Sevilla M. D.; Adhikary A.; Seki S.; Mostafavi M. Observation of Dissociative Quasi-Free Electron Attachment to Nucleoside via Excited Anion Radical in Solution. Nat. Commun. 2019, 10 (1), 102–102. 10.1038/s41467-018-08005-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimblott S. M.; LaVerne J. A. On the Radiation Chemical Kinetics of the Precursor to the Hydrated Electron. J. Phys. Chem. A 1998, 102 (17), 2967–2975. 10.1021/jp980496v. [DOI] [Google Scholar]
- Pluhařová E.; Schroeder C.; Seidel R.; Bradforth S. E.; Winter B.; Faubel M.; Slavíček P.; Jungwirth P. Unexpectedly Small Effect of the DNA Environment on Vertical Ionization Energies of Aqueous Nucleobases. J. Phys. Chem. Lett. 2013, 4 (21), 3766–3769. 10.1021/jz402106h. [DOI] [Google Scholar]
- Pluhařová E.; Slavíček P.; Jungwirth P. Modeling Photoionization of Aqueous DNA and Its Components. Acc. Chem. Res. 2015, 48 (5), 1209–1217. 10.1021/ar500366z. [DOI] [PubMed] [Google Scholar]
- Fabrikant I. I.; Eden S.; Mason N. J.; Fedor J.. Recent Progress in Dissociative Electron Attachment: From Diatomics to Biomolecules, 1st ed.; Elsevier Inc., 2017; Vol. 66, p 657. [Google Scholar]
- Tripathi D.; Dutta A. K. The Performance of Approximate EOM-CCSDfor Ionization Potential and Electron Affinity of Genetic Material Subunits: A Benchmark Investigation. Int. J. Quantum Chem. 2022, 122 (15), e26918. 10.1002/qua.26918. [DOI] [Google Scholar]
- Tripathi D.; Dutta A. K. Bound Anionic States of DNA and RNA Nucleobases: An EOM-CCSD Investigation. Int. J. Quantum Chem. 2019, 119 (9), e25875. 10.1002/qua.25875. [DOI] [Google Scholar]
- Wang X.-D.; Xuan C.-J.; Feng W.-L.; Tian S. X. Dissociative Electron Attachments to Ethanol and Acetaldehyde: A Combined Experimental and Simulation Study. J. Chem. Phys. 2015, 142 (6), 064316 10.1063/1.4907940. [DOI] [PubMed] [Google Scholar]
- Ptasinska S. A Missing Puzzle in Dissociative Electron Attachment to Biomolecules: The Detection of Radicals. Atoms 2021, 9 (4), 77. 10.3390/atoms9040077. [DOI] [Google Scholar]
- Bald I.; Čurík R.; Kopyra J.; Tarana M.. Dissociative Electron Attachment to Biomolecules. In Nanoscale Insights into Ion-Beam Cancer Therapy; Solov’yov A. V., Ed.; Springer International Publishing: Cham, 2017; pp 159–207. [Google Scholar]
- Pan X.; Cloutier P.; Hunting D.; Sanche L. Dissociative Electron Attachment to DNA. Phys. Rev. Lett. 2003, 90 (20), 208102. 10.1103/PhysRevLett.90.208102. [DOI] [PubMed] [Google Scholar]
- Boudaïffa B.; Cloutier P.; Hunting D.; Huels M. A.; Sanche L. Resonant Formation of DNA Strand Breaks by Low-Energy (3 to 20 EV) Electrons. Science 2000, 287 (5458), 1658–1660. 10.1126/science.287.5458.1658. [DOI] [PubMed] [Google Scholar]
- Abdoul-Carime H.; Gohlke S.; Fischbach E.; Scheike J.; Illenberger E. Thymine Excision from DNA by Subexcitation Electrons. Chem. Phys. Lett. 2004, 387 (4–6), 267–270. 10.1016/j.cplett.2004.02.022. [DOI] [Google Scholar]
- Zheng Y.; Cloutier P.; Hunting D. J.; Sanche L.; Wagner J. R. Chemical Basis of DNA Sugar-Phosphate Cleavage by Low-Energy Electrons. J. Am. Chem. Soc. 2005, 127 (47), 16592–16598. 10.1021/ja054129q. [DOI] [PubMed] [Google Scholar]
- Sanche L. Low Energy Electron-Driven Damage in Biomolecules. Eur. Phys. J. D 2005, 35 (2), 367–390. 10.1140/epjd/e2005-00206-6. [DOI] [Google Scholar]
- Zheng Y.; Cloutier P.; Hunting D. J.; Wagner J. R.; Sanche L. Phosphodiester and N-Glycosidic Bond Cleavage in DNA Induced by 4–15 EV Electrons. J. Chem. Phys. 2006, 124 (6), 064710. 10.1063/1.2166364. [DOI] [PubMed] [Google Scholar]
- Li Z.; Cloutier P.; Sanche L.; Wagner J. R. Low-Energy Electron-Induced DNA Damage: Effect of Base Sequence in Oligonucleotide Trimers. J. Am. Chem. Soc. 2010, 132 (15), 5422–5427. 10.1021/ja9099505. [DOI] [PubMed] [Google Scholar]
- Kopyra J. Low Energy Electron Attachment to the Nucleotide Deoxycytidine Monophosphate: Direct Evidence for the Molecular Mechanisms of Electron-Induced DNA Strand Breaks. Phys. Chem. Chem. Phys. 2012, 14 (23), 8287–8289. 10.1039/c2cp40847c. [DOI] [PubMed] [Google Scholar]
- Sahbani S. K.; Sanche L.; Cloutier P.; Bass A. D.; Hunting D. J. Loss of Cellular Transformation Efficiency Induced by DNA Irradiation with Low-Energy (10 EV) Electrons. J. Phys. Chem. B 2014, 118 (46), 13123–13131. 10.1021/jp508170c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y.; Dong Y.; Hunting D.; Zheng Y.; Sanche L. Unified Mechanism for the Generation of Isolated and Clustered DNA Damages by a Single Low Energy (5–10 EV) Electron. J. Phys. Chem. C 2017, 121 (4), 2466–2472. 10.1021/acs.jpcc.6b12110. [DOI] [Google Scholar]
- Khorsandgolchin G.; Sanche L.; Cloutier P.; Wagner J. R. Strand Breaks Induced by Very Low Energy Electrons: Product Analysis and Mechanistic Insight into the Reaction with TpT. J. Am. Chem. Soc. 2019, 141 (26), 10315–10323. 10.1021/jacs.9b03295. [DOI] [PubMed] [Google Scholar]
- Dong Y.; Gao Y.; Liu W.; Gao T.; Zheng Y.; Sanche L. Clustered DNA Damage Induced by 2–20 EV Electrons and Transient Anions: General Mechanism and Correlation to Cell Death. J. Phys. Chem. Lett. 2019, 10 (11), 2985–2990. 10.1021/acs.jpclett.9b01063. [DOI] [PubMed] [Google Scholar]
- Dong Y.; Liao H.; Gao Y.; Cloutier P.; Zheng Y.; Sanche L. Early Events in Radiobiology: Isolated and Cluster DNA Damage Induced by Initial Cations and Nonionizing Secondary Electrons. J. Phys. Chem. Lett. 2021, 12 (1), 717–723. 10.1021/acs.jpclett.0c03341. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Zheng Y.; Sanche L. Low-Energy Electron Damage to Condensed-Phase DNA and Its Constituents. Int. J. Mol. Sci. 2021, 22 (15), 7879. 10.3390/ijms22157879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari B.; Huwaidi A.; Robert G.; Cloutier P.; Bass A. D.; Sanche L.; Wagner J. R. Shape Resonances in DNA: Nucleobase Release, Reduction, and Dideoxynucleoside Products Induced by 1.3 to 2.3 EV Electrons. J. Phys. Chem. B 2022, 126 (28), 5175–5184. 10.1021/acs.jpcb.2c01851. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Walker J. A.; Bartels D. M.; Sevilla M. D. A Simple Ab Initio Model for the Hydrated Electron That Matches Experiment. J. Phys. Chem. A 2015, 119 (34), 9148–9159. 10.1021/acs.jpca.5b04721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz H. A. Enthalpy and Entropy of Formation of the Hydrated Electron. J. Phys. Chem. 1991, 95 (17), 6697–6701. 10.1021/j100170a058. [DOI] [Google Scholar]
- Lapointe F.; Wolf M.; Campen R. K.; Tong Y. Probing the Birth and Ultrafast Dynamics of Hydrated Electrons at the Gold/Liquid Water Interface via an Optoelectronic Approach. J. Am. Chem. Soc. 2020, 142 (43), 18619–18627. 10.1021/jacs.0c08289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y.; Shen H.; Sekiguchi K.; Kurahashi N.; Mizuno T.; Suzuki Y. I.; Suzuki T. Direct Measurement of Vertical Binding Energy of a Hydrated Electron. Phys. Chem. Chem. Phys. 2010, 12 (15), 3653–3655. 10.1039/b925741a. [DOI] [PubMed] [Google Scholar]
- Siefermann K. R.; Liu Y.; Lugovoy E.; Link O.; Faubel M.; Buck U.; Winter B.; Abel B. Binding Energies, Lifetimes and Implications of Bulk and Interface Solvated Electrons in Water. Nat. Chem. 2010, 2 (4), 274–279. 10.1038/nchem.580. [DOI] [PubMed] [Google Scholar]
- Shreve A. T.; Yen T. A.; Neumark D. M. Photoelectron Spectroscopy of Hydrated Electrons. Chem. Phys. Lett. 2010, 493 (4–6), 216–219. 10.1016/j.cplett.2010.05.059. [DOI] [Google Scholar]
- Nabben F. J.; Karman J. P.; Loman H. Inactivation of Biologically Active DNA by Hydrated Electrons.. Int. J. Rad. Biol. and Related Studies in Physics, Chemistry and Medicine 1982, 42 (1), 23–30. 10.1080/09553008214550881. [DOI] [PubMed] [Google Scholar]
- Kuipers G. K.; Lafleur M. V. M. Characterization of DNA Damage Induced by Gamma-Radiationderived Water Radicals, Using DNA Repair Enzymes. Int. J. Radiat. Biol. 1998, 74 (4), 511–519. 10.1080/095530098141384. [DOI] [PubMed] [Google Scholar]
- Kohanoff J.; McAllister M.; Tribello G. A.; Gu B. Interactions between Low Energy Electrons and DNA: A Perspective from First-Principles Simulations. J. Phys.: Condens. Matter 2017, 29 (38), 383001. 10.1088/1361-648X/aa79e3. [DOI] [PubMed] [Google Scholar]
- Gu J.; Leszczynski J.; Schaefer H. F. Interactions of Electrons with Bare and Hydrated Biomolecules: From Nucleic Acid Bases to DNA Segments. Chem. Rev. 2012, 112 (11), 5603–5640. 10.1021/cr3000219. [DOI] [PubMed] [Google Scholar]
- Buxton G. V.; Greenstock C. L.; Helman W. P.; Ross A. B. Critical Review of Rate Constants for Reactions of Hydrated Electrons, Hydrogen Atoms and Hydroxyl Radicals (·OH/·O– in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17 (2), 513–886. 10.1063/1.555805. [DOI] [Google Scholar]
- Kumar A.; Adhikary A.; Shamoun L.; Sevilla M. D. Do Solvated Electrons (Eaq-) Reduce DNA Bases? A Gaussian 4 and Density Functional Theory- Molecular Dynamics Study. J. Phys. Chem. B 2016, 120 (9), 2115–2123. 10.1021/acs.jpcb.5b11269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A. K.; Sengupta T.; Vaval N.; Pal S. Electron Attachment to DNA and RNA Nucleobases: An EOMCC Investigation. Int. J. Quantum Chem. 2015, 115 (12), 753–764. 10.1002/qua.24892. [DOI] [Google Scholar]
- Cooper G. A.; Clarke C. J.; Verlet J. R. R. Low-Energy Shape Resonances of a Nucleobase in Water. J. Am. Chem. Soc. 2023, 145, 1319. 10.1021/jacs.2c11440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik D. H.; Lee I.-R.; Yang D.-S.; Baskin J. S.; Zewail A. H. Electrons in Finite-Sized Water Cavities: Hydration Dynamics Observed in Real Time. Science 2004, 306 (5696), 672–675. 10.1126/science.1102827. [DOI] [PubMed] [Google Scholar]
- Savolainen J.; Uhlig F.; Ahmed S.; Hamm P.; Jungwirth P. Direct Observation of the Collapse of the Delocalized Excess Electron in Water. Nat. Chem. 2014, 6 (8), 697–701. 10.1038/nchem.1995. [DOI] [PubMed] [Google Scholar]
- Ma J.; Wang F.; Denisov S. A.; Adhikary A.; Mostafavi M. Reactivity of Prehydrated Electrons toward Nucleobases and Nucleotides in Aqueous Solution. Sci. Adv. 2017, 3 (12), 1701669. 10.1126/sciadv.1701669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. R.; Nguyen J.; Lu Q. B. Bond Breaks of Nucleotides by Dissociative Electron Transfer of Nonequilibrium Prehydrated Electrons: A New Molecular Mechanism for Reductive DNA Damage. J. Am. Chem. Soc. 2009, 131 (32), 11320–11322. 10.1021/ja902675g. [DOI] [PubMed] [Google Scholar]
- Dedon P. C. The Chemical Toxicology of 2-Deoxyribose Oxidation in DNA. Chem. Res. Toxicol. 2008, 21 (1), 206–219. 10.1021/tx700283c. [DOI] [PubMed] [Google Scholar]
- Dizdaroglu M.; Von Sonntag C.; Schulte-Fröhlinde D. Strand Breaks and Sugar Release by.Gamma.-Irradiation of DNA in Aqueous Solution. J. Am. Chem. Soc. 1975, 97 (8), 2277–2278. 10.1021/ja00841a051. [DOI] [PubMed] [Google Scholar]
- Stelter L.; von Sonntag C.; Schulte-Fröhlinde D. Radiation Chemistry of DNA-Model Compounds, VIII Dephosphorylation Products from Reactions of OH Radicals with Ribose-5-Phosphate in Aqueous Solution. The Effect of Oxygen. Zeitzchrift fuer Naturforschung B 1975, 30 (7-8), 609–615. 10.1515/znb-1975-7-827. [DOI] [Google Scholar]
- Halliwell B.; Adhikary A.; Dingfelder M.; Dizdaroglu M. Hydroxyl Radical Is a Significant Player in Oxidative DNA Damage in Vivo. Chem. Soc. Rev. 2021, 50 (15), 8355–8360. 10.1039/D1CS00044F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Denisov S. A.; Marignier J.-L.; Pernot P.; Adhikary A.; Seki S.; Mostafavi M. Ultrafast Electron Attachment and Hole Transfer Following Ionizing Radiation of Aqueous Uridine Monophosphate. J. Phys. Chem. Lett. 2018, 9 (17), 5105–5109. 10.1021/acs.jpclett.8b02170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarek R.; Ward S.; Debnath D.; Jacobs T.; Stark A. D.; Korczyński D.; Kumar A.; Sevilla M. D.; Denisov S. A.; Shcherbakov V.; Pernot P.; Mostafavi M.; Dembinski R.; Adhikary A. One Way Traffic: Base-to-Backbone Hole Transfer in Nucleoside Phosphorodithioate. Chemistry 2020, 26 (43), 9495–9505. 10.1002/chem.202000247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisov S. A.; Ward S.; Shcherbakov V.; Stark A. D.; Kaczmarek R.; Radzikowska-Cieciura E.; Debnath D.; Jacobs T.; Kumar A.; Sevilla M. D.; Pernot P.; Dembinski R.; Mostafavi M.; Adhikary A. Modulation of the Directionality of Hole Transfer between the Base and the Sugar-Phosphate Backbone in DNA with the Number of Sulfur Atoms in the Phosphate Group. J. Phys. Chem. B 2022, 126 (2), 430–442. 10.1021/acs.jpcb.1c09068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alizadeh E.; Orlando T. M.; Sanche L. Biomolecular Damage Induced by Ionizing Radiation: The Direct and Indirect Effects of Low-Energy Electrons on DNA. Annu. Rev. Phys. Chem. 2015, 66, 379–398. 10.1146/annurev-physchem-040513-103605. [DOI] [PubMed] [Google Scholar]
- Aflatooni K.; Gallup G. A.; Burrow P. D. Electron Attachment Energies of the DNA Bases. J. Phys. Chem. A 1998, 102 (31), 6205–6207. 10.1021/jp980865n. [DOI] [Google Scholar]
- Barrios R.; Skurski P.; Simons J. Mechanism for Damage to DNA by Low-Energy Electrons. J. Phys. Chem. B 2002, 106 (33), 7991–7994. 10.1021/jp013861i. [DOI] [Google Scholar]
- Martin F.; Burrow P. D.; Cai Z.; Cloutier P.; Hunting D.; Sanche L. DNA Strand Breaks Induced by 0–4 EV Electrons: The Role of Shape Resonances. Phys. Rev. Lett. 2004, 93 (6), 6–9. 10.1103/PhysRevLett.93.068101. [DOI] [PubMed] [Google Scholar]
- Berdys J.; Anusiewicz I.; Skurski P.; Simons J. Damage to Model DNA Fragments from Very Low-Energy (<1 EV) Electrons. J. Am. Chem. Soc. 2004, 126 (20), 6441–6447. 10.1021/ja049876m. [DOI] [PubMed] [Google Scholar]
- Simons J. How Do Low-Energy (0.1–2 EV) Electrons Cause DNA-Strand Breaks?. Acc. Chem. Res. 2006, 39 (10), 772–779. 10.1021/ar0680769. [DOI] [PubMed] [Google Scholar]
- Li X.; Sevilla M. D.; Sanche L. Density Functional Theory Studies of Electron Interaction with DNA: Can Zero EV Electrons Induce Strand Breaks?. J. Am. Chem. Soc. 2003, 125 (45), 13668–13669. 10.1021/ja036509m. [DOI] [PubMed] [Google Scholar]
- Bhaskaran R.; Sarma M. The Role of the Shape Resonance State in Low Energy Electron Induced Single Strand Break in 2′-Deoxycytidine-5′-Monophosphate. Phys. Chem. Chem. Phys. 2015, 17 (23), 15250–15257. 10.1039/C5CP00126A. [DOI] [PubMed] [Google Scholar]
- Moiseyev N.Non-Hermitian Quantum Mechanics; Cambridge University Press, 2011. [Google Scholar]
- Sajeev Y.; Ghosh A.; Vaval N.; Pal S. Coupled Cluster Methods for Autoionisation Resonances. Int. Rev. Phys. Chem. 2014, 33 (3), 397–425. 10.1080/0144235X.2014.935585. [DOI] [Google Scholar]
- Jagau T. C.; Bravaya K. B.; Krylov A. I. Extending Quantum Chemistry of Bound States to Electronic Resonances. Annu. Rev. Phys. Chem. 2017, 68, 525–553. 10.1146/annurev-physchem-052516-050622. [DOI] [PubMed] [Google Scholar]
- Fennimore M. A.; Matsika S. Electronic Resonances of Nucleobases Using Stabilization Methods. J. Phys. Chem. A 2018, 122 (16), 4048–4057. 10.1021/acs.jpca.8b01523. [DOI] [PubMed] [Google Scholar]
- Thodika M.; Fennimore M.; Karsili T. N. V.; Matsika S. Comparative Study of Methodologies for Calculating Metastable States of Small to Medium-Sized Molecules. J. Chem. Phys. 2019, 151 (24), 244104–244104. 10.1063/1.5134700. [DOI] [PubMed] [Google Scholar]
- Kanazawa Y.; Ehara M.; Sommerfeld T. Low-Lying Π* Resonances of Standard and Rare DNA and RNA Bases Studied by the Projected CAP/SAC-CI Method. J. Phys. Chem. A 2016, 120 (9), 1545–1553. 10.1021/acs.jpca.5b12190. [DOI] [PubMed] [Google Scholar]
- Bhaskaran R.; Sarma M. Effect of Quantum Tunneling on Single Strand Breaks in a Modeled Gas Phase Cytidine Nucleotide Induced by Low Energy Electron: A Theoretical Approach. J. Chem. Phys. 2013, 139 (4), 045103–045103. 10.1063/1.4815975. [DOI] [PubMed] [Google Scholar]
- Bhaskaran R.; Sarma M. Low Energy Electron Induced Cytosine Base Release in 2′-Deoxycytidine-3′-Monophosphate via Glycosidic Bond Cleavage: A Time-Dependent Wavepacket Study. J. Chem. Phys. 2014, 141 (10), 104309–104309. 10.1063/1.4894744. [DOI] [PubMed] [Google Scholar]
- Bhaskaran R.; Sarma M. Low-Energy Electron Interaction with the Phosphate Group in DNA Molecule and the Characteristics of Single-Strand Break Pathways. J. Phys. Chem. A 2015, 119 (40), 10130–10136. 10.1021/acs.jpca.5b08000. [DOI] [PubMed] [Google Scholar]
- Zheng Y.; Wagner J. R.; Sanche L. DNA Damage Induced by Low-Energy Electrons: Electron Transfer and Diffraction. Phys. Rev. Lett. 2006, 96 (20), 208101–208101. 10.1103/PhysRevLett.96.208101. [DOI] [PubMed] [Google Scholar]
- Luo X.; Zheng Y.; Sanche L. DNA Strand Breaks and Crosslinks Induced by Transient Anions in the Range 2–20 EV. J. Chem. Phys. 2014, 140 (15), 155101–155101. 10.1063/1.4870519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fennimore M. A.; Matsika S. Core-Excited and Shape Resonances of Uracil. Phys. Chem. Chem. Phys. 2016, 18 (44), 30536–30545. 10.1039/C6CP05342D. [DOI] [PubMed] [Google Scholar]
- Hanel G.; Gstir B.; Denifl S.; Scheier P.; Probst M.; Farizon B.; Farizon M.; Illenberger E.; Märk T. D. Electron Attachment to Uracil: Effective Destruction at Subexcitation Energies. Phys. Rev. Lett. 2003, 90 (18), 188104. 10.1103/PhysRevLett.90.188104. [DOI] [PubMed] [Google Scholar]
- Abdoul-Carime H.; Gohlke S.; Illenberger E. Site-Specific Dissociation of DNA Bases by Slow Electrons at Early Stages of Irradiation. Phys. Rev. Lett. 2004, 92 (16), 168103. 10.1103/PhysRevLett.92.168103. [DOI] [PubMed] [Google Scholar]
- Ptasińska S.; Denifl S.; Grill V.; Märk T. D.; Scheier P.; Gohlke S.; Huels M. A.; Illenberger E. Bond-Selective H– Ion Abstraction from Thymine.. Angew. Chem., Int. Ed. 2005, 44 (11), 1647–1650. 10.1002/anie.200461739. [DOI] [PubMed] [Google Scholar]
- Abdoul-Carime H.; Langer J.; Huels M. A.; Illenberger E. Decomposition of Purine Nucleobases by Very Low Energy Electrons.. Eur. Phys. J. D - Atomic, Molecular, Optical and Plasma Physics 2005, 35 (2), 399–404. 10.1140/epjd/e2005-00194-5. [DOI] [Google Scholar]
- Denifl S.; Ptasińska S.; Probst M.; Hrušák J.; Scheier P.; Märk T. D. Electron Attachment to the Gas-Phase DNA Bases Cytosine and Thymine. J. Phys. Chem. A 2004, 108 (31), 6562–6569. 10.1021/jp049394x. [DOI] [Google Scholar]
- Burrow P. D.; Gallup G. A.; Scheer A. M.; Denifl S.; Ptasinska S.; Märk T.; Scheier P. Vibrational Feshbach Resonances in Uracil and Thymine. J. Chem. Phys. 2006, 124 (12), 124310–124310. 10.1063/1.2181570. [DOI] [PubMed] [Google Scholar]
- Tripathi D.; Dutta A. K. Electron Attachment to DNA Base Pairs: An Interplay of Dipole- and Valence-Bound States. J. Phys. Chem. A 2019, 123 (46), 10131–10138. 10.1021/acs.jpca.9b08974. [DOI] [PubMed] [Google Scholar]
- Schiedt J.; Weinkauf R.; Neumark D. M.; Schlag E. W. Anion Spectroscopy of Uracil, Thymine and the Amino-Oxo and Amino-Hydroxy Tautomers of Cytosine and Their Water Clusters. Chem. Phys. 1998, 239 (1–3), 511–524. 10.1016/S0301-0104(98)00361-9. [DOI] [Google Scholar]
- Berdys J.; Anusiewicz I.; Skurski P.; Simons J. Theoretical Study of Damage to DNA by 0.2–1.5 EV Electrons Attached to Cytosine. J. Phys. Chem. A 2004, 108 (15), 2999–3005. 10.1021/jp035957d. [DOI] [Google Scholar]
- Kumar A.; Sevilla M. D. The Role of Πσ* Excited States in Electron-Induced DNA Strand Break Formation: A Time-Dependent Density Functional Theory Study. J. Am. Chem. Soc. 2008, 130 (7), 2130–2131. 10.1021/ja077331x. [DOI] [PubMed] [Google Scholar]
- Narayanan S J J.; Tripathi D.; Dutta A. K. Doorway Mechanism for Electron Attachment Induced DNA Strand Breaks. J. Phys. Chem. Lett. 2021, 12 (42), 10380–10387. 10.1021/acs.jpclett.1c02735. [DOI] [PubMed] [Google Scholar]
- Hendricks J. H.; Lyapustina S. A.; de Clercq H. L.; Bowen K. H. The Dipole Bound-to-Covalent Anion Transformation in Uracil. J. Chem. Phys. 1998, 108 (1), 8–11. 10.1063/1.475360. [DOI] [Google Scholar]
- Sekino H.; Bartlett R. J. A Linear Response, Coupled-Cluster Theory for Excitation Energy. Int. J. Quantum Chem. 1984, 26 (S18), 255–265. 10.1002/qua.560260826. [DOI] [Google Scholar]
- Nooijen M.; Bartlett R. J. Equation of Motion Coupled Cluster Method for Electron Attachment. J. Chem. Phys. 1995, 102 (9), 3629–3647. 10.1063/1.468592. [DOI] [Google Scholar]
- Kowalski K.; Piecuch P. The Active-Space Equation-of-Motion Coupled-Cluster Methods for Excited Electronic States: Full EOMCCSDt. J. Chem. Phys. 2001, 115 (2), 643. 10.1063/1.1378323. [DOI] [Google Scholar]
- Musial M.; Bartlett R. J. Equation-of-Motion Coupled Cluster Method with Full Inclusion of Connected Triple Excitations for Electron-Attached States: EA-EOM-CCSDT. J. Chem. Phys. 2003, 119 (4), 1901–1901. 10.1063/1.1584657. [DOI] [Google Scholar]
- Krylov A. I. Equation-of-Motion Coupled-Cluster Methods for Open-Shell and Electronically Excited Species: The Hitchhiker’s Guide to Fock Space. Annu. Rev. Phys. Chem. 2008, 59, 433–462. 10.1146/annurev.physchem.59.032607.093602. [DOI] [PubMed] [Google Scholar]
- Cederbaum L. S.; Domcke W. Theoretical Aspects of Ionization Potentials and Photoelectron Spectroscopy: A Green’s Function Approach. Advances in chemical physics 2007, 36, 205–344. 10.1002/9780470142554.ch4. [DOI] [Google Scholar]
- Trofimov A. B.; Schirmer J. An Efficient Polarization Propagator Approach to Valence Electron Excitation Spectra.. J. Phys. B: At., Mol. Opt. Phys. 1995, 28 (12), 2299–2299. 10.1088/0953-4075/28/12/003. [DOI] [Google Scholar]
- Dreuw A.; Wormit M. The Algebraic Diagrammatic Construction Scheme for the Polarization Propagator for the Calculation of Excited States.. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2015, 5 (1), 82–95. 10.1002/wcms.1206. [DOI] [Google Scholar]
- Kim M.-C.; Sim E.; Burke K. Communication: Avoiding Unbound Anions in Density Functional Calculations. J. Chem. Phys. 2011, 134 (17), 171103. 10.1063/1.3590364. [DOI] [PubMed] [Google Scholar]
- Voora V. K.; Kairalapova A.; Sommerfeld T.; Jordan K. D. Theoretical Approaches for Treating Non-Valence Correlation-Bound Anions. J. Chem. Phys. 2017, 147 (21), 214114. 10.1063/1.4991497. [DOI] [PubMed] [Google Scholar]
- Riplinger C.; Neese F. An Efficient and near Linear Scaling Pair Natural Orbital Based Local Coupled Cluster Method. J. Chem. Phys. 2013, 138 (3), 034106 10.1063/1.4773581. [DOI] [PubMed] [Google Scholar]
- Dutta A. K.; Saitow M.; Demoulin B.; Neese F.; Izsák R. A Domain-Based Local Pair Natural Orbital Implementation of the Equation of Motion Coupled Cluster Method for Electron Attached States. J. Chem. Phys. 2019, 150 (16), 164123. 10.1063/1.5089637. [DOI] [PubMed] [Google Scholar]
- Dutta A. K.; Saitow M.; Riplinger C.; Neese F.; Izsák R. A Near-Linear Scaling Equation of Motion Coupled Cluster Method for Ionized States. J. Chem. Phys. 2018, 148 (24), 244101. 10.1063/1.5029470. [DOI] [PubMed] [Google Scholar]
- Desfrançois C.; Abdoul-Carime H.; Schermann J. P. Electron Attachment to Isolated Nucleic Acid Bases. J. Chem. Phys. 1996, 104 (19), 7792–7792. 10.1063/1.471484. [DOI] [Google Scholar]
- Mukherjee M.; Tripathi D.; Dutta A. K. Water Mediated Electron Attachment to Nucleobases: Surface-Bound vs Bulk Solvated Electrons. J. Chem. Phys. 2020, 153 (4), 044305 10.1063/5.0010509. [DOI] [PubMed] [Google Scholar]
- Ranga S.; Mukherjee M.; Dutta A. K. Interactions of Solvated Electrons with Nucleobases: The Effect of Base Pairing. ChemPhysChem 2020, 21 (10), 1019–1027. 10.1002/cphc.202000133. [DOI] [PubMed] [Google Scholar]
- Verma P.; Ghosh D.; Dutta A. K. Electron Attachment to Cytosine: The Role of Water. J. Phys. Chem. A 2021, 125 (22), 4683–4694. 10.1021/acs.jpca.0c10199. [DOI] [PubMed] [Google Scholar]
- Köuppel H.; Domcke W.; Cederbaum L. S.. Multimode Molecular Dynamics Beyond the Born-Oppenheimer Approximation. In Advances in Chemical Physics; John Wiley & Sons, Ltd, 1984; pp 59–246. [Google Scholar]
- Sommerfeld T. Coupling between Dipole-Bound and Valence States: The Nitromethane Anion. Phys. Chem. Chem. Phys. 2002, 4 (12), 2511–2516. 10.1039/b202143a. [DOI] [Google Scholar]
- Sommerfeld T. Intramolecular Electron Transfer from Dipole-Bound to Valence Orbitals: Uracil and 5-Chlorouracil. J. Phys. Chem. A 2004, 108 (42), 9150–9154. 10.1021/jp049082u. [DOI] [Google Scholar]
- Pacher T.; Cederbaum L. S.; Köppel H.. Adiabatic and Quasidiabatic States in a Gauge Theoretical Framework. In Advances in Chemical Physics; Advances in Chemical Physics; 1993; pp 293–391. [Google Scholar]
- Thiel A.; Köppel H. Proposal and Numerical Test of a Simple Diabatization Scheme. J. Chem. Phys. 1999, 110 (19), 9371–9383. 10.1063/1.478902. [DOI] [Google Scholar]
- Narayanan S J J.; Bachhar A.; Tripathi D.; Dutta A. K. Electron Attachment to Wobble Base Pairs. J. Phys. Chem. A 2023, 127, 457. 10.1021/acs.jpca.2c07469. [DOI] [PubMed] [Google Scholar]
- Subotnik J. E.; Vura-Weis J.; Sodt A. J.; Ratner M. A. Predicting Accurate Electronic Excitation Transfer Rates via Marcus Theory with Boys or Edmiston-Ruedenberg Localized Diabatization. J. Phys. Chem. A 2010, 114 (33), 8665–8675. 10.1021/jp101235a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D. H.; Kim J.; Eun H. J.; Kim S. K. Experimental Observation of the Resonant Doorways to Anion Chemistry: Dynamic Role of Dipole-Bound Feshbach Resonances in Dissociative Electron Attachment. J. Am. Chem. Soc. 2022, 144 (35), 16077–16085. 10.1021/jacs.2c06334. [DOI] [PubMed] [Google Scholar]
- Kang D. H.; Kim J.; Eun H. J.; Kim S. K. State-Specific Chemical Dynamics of the Nonvalence Bound State of the Molecular Anions. Acc. Chem. Res. 2022, 55 (20), 3032–3042. 10.1021/acs.accounts.2c00512. [DOI] [PubMed] [Google Scholar]
- Ouyang L.; Lin H.; Zhuang P.; Shao Y.; Khosravifarsani M.; Guérin B.; Zheng Y.; Sanche L. DNA Radiosensitization by Terpyridine-Platinum: Damage Induced by 5 and 10 EV Transient Anions. Nanoscale 2023, 15, 3230. 10.1039/D2NR05403E. [DOI] [PubMed] [Google Scholar]
- Kočišek J.; Pysanenko A.; Fárník M.; Fedor J. Microhydration Prevents Fragmentation of Uracil and Thymine by Low-Energy Electrons. J. Phys. Chem. Lett. 2016, 7 (17), 3401–3405. 10.1021/acs.jpclett.6b01601. [DOI] [PubMed] [Google Scholar]
- Kočišek J.; Grygoryeva K.; Lengyel J.; Fárník M.; Fedor J. Effect of Cluster Environment on the Electron Attachment to 2-Nitrophenol. Eur. Phys. J. D 2016, 70 (4), 98. 10.1140/epjd/e2016-70074-0. [DOI] [Google Scholar]
- Kočišek J.; Sedmidubská B.; Indrajith S.; Fárník M.; Fedor J. Electron Attachment to Microhydrated Deoxycytidine Monophosphate. J. Phys. Chem. B 2018, 122 (20), 5212–5217. 10.1021/acs.jpcb.8b03033. [DOI] [PubMed] [Google Scholar]
- Cossi M.; Barone V.; Cammi R.; Tomasi J. Ab Initio Study of Solvated Molecules: A New Implementation of the Polarizable Continuum Model. Chem. Phys. Lett. 1996, 255 (4–6), 327–335. 10.1016/0009-2614(96)00349-1. [DOI] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105 (8), 2999–3094. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- Gu J.; Xie Y.; Schaefer H. F. Electron Attachment to DNA Single Strands: Gas Phase and Aqueous Solution. Nucleic Acids Res. 2007, 35 (15), 5165–5172. 10.1093/nar/gkm135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J.; Xie Y.; Schaefer H. F. Guanine Nucleotides: Base-Centered and Phosphate-Centered Valence-Bound Radical Anions in Aqueous Solution. J. Phys. Chem. B 2010, 114 (2), 1221–1224. 10.1021/jp911103f. [DOI] [PubMed] [Google Scholar]
- Wesolowski S. S.; Leininger M. L.; Pentchev P. N.; Schaefer H. F. Electron Affinities of the DNA and RNA Bases. J. Am. Chem. Soc. 2001, 123 (17), 4023–4028. 10.1021/ja003814o. [DOI] [PubMed] [Google Scholar]
- Sevilla M. D.; Besler B.; Colson A. O. Ab Initio Molecular Orbital Calculations of DNA Radical Ions. 5. Scaling of Calculated Electron Affinities and Ionization Potentials to Experimental Values. J. Phys. Chem. 1995, 99 (3), 1060–1063. 10.1021/j100003a032. [DOI] [Google Scholar]
- Harańczyk M.; Gutowski M. Finding Adiabatically Bound Anions of Guanine through a Combinatorial Computational Approach.. Angew. Chem., Int. Ed. 2005, 44 (40), 6585–6588. 10.1002/anie.200501671. [DOI] [PubMed] [Google Scholar]
- Haranczyk M.; Gutowski M.; Li X.; Bowen K. H. Adiabatically Bound Valence Anions of Guanine. J. Phys. Chem. B 2007, 111 (51), 14073–14076. 10.1021/jp077439z. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Sevilla M. D.; Suhai S. Microhydration of the Guanine-Cytosine (GC) Base Pair in the Neutral and Anionic Radical States: A Density Functional Study. J. Phys. Chem. B 2008, 112 (16), 5189–5198. 10.1021/jp710957p. [DOI] [PubMed] [Google Scholar]
- Gu J.; Xie Y.; Schaefer H. F. Electron Attachment to Nucleotides in Aqueous Solution. ChemPhysChem 2006, 7 (9), 1885–1887. 10.1002/cphc.200600294. [DOI] [PubMed] [Google Scholar]
- Bao X.; Wang J.; Gu J.; Leszczynski J. DNA Strand Breaks Induced by Near-Zero-Electronvolt Electron Attachment to Pyrimidine Nucleotides. Proc. Natl. Acad. Sci. U.S.A. 2006, 103 (15), 5658–5663. 10.1073/pnas.0510406103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J.; Xie Y.; Schaefer H. F. Near 0 EV Electrons Attach to Nucleotides. J. Am. Chem. Soc. 2006, 128 (4), 1250–1252. 10.1021/ja055615g. [DOI] [PubMed] [Google Scholar]
- Li X.; Sanche L.; Sevilla M. D. Base Release in Nucleosides Induced by Low-Energy Electrons: A DFT Study. Radiat. Res. 2006, 165 (6), 721–729. 10.1667/RR3568.1. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Sevilla M. D. Low-Energy Electron Attachment to 5′-Thymidine Monophosphate: Modeling Single Strand Breaks through Dissociative Electron Attachment. J. Phys. Chem. B 2007, 111 (19), 5464–5474. 10.1021/jp070800x. [DOI] [PubMed] [Google Scholar]
- Gu J.; Wang J.; Leszczynski J. Electron Attachment-Induced DNA Single Strand Breaks: C3′- O3′ σ-Bond Breaking of Pyrimidine Nucleotides Predominates. J. Am. Chem. Soc. 2006, 128 (29), 9322–9323. 10.1021/ja063309c. [DOI] [PubMed] [Google Scholar]
- Schyman P.; Laaksonen A. On the Effect of Low-Energy Electron Induced DNA Strand Break in Aqueous Solution: A Theoretical Study Indicating Guanine as a Weak Link in DNA. J. Am. Chem. Soc. 2008, 130 (37), 12254–12255. 10.1021/ja805000g. [DOI] [PubMed] [Google Scholar]
- Gu J.; Wang J.; Leszczynski J. Electron Attachment-Induced DNA Single-Strand Breaks at the Pyrimidine Sites. Nucleic Acids Res. 2010, 38 (16), 5280–5290. 10.1093/nar/gkq304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J.; Wang J.; Leszczynski J. Comprehensive Analysis of DNA Strand Breaks at the Guanosine Site Induced by Low-Energy Electron Attachment. ChemPhysChem 2010, 11 (1), 175–181. 10.1002/cphc.200900656. [DOI] [PubMed] [Google Scholar]
- Gu J.; Wang J.; Leszczynski J. Low Energy Electron Attachment to the Adenosine Site of DNA. J. Phys. Chem. B 2011, 115 (49), 14831–14837. 10.1021/jp207801e. [DOI] [PubMed] [Google Scholar]
- Sagar D. M.; Bain C. D.; Verlet J. R. R. Hydrated Electrons at the Water/Air Interface. J. Am. Chem. Soc. 2010, 132 (20), 6917–6919. 10.1021/ja101176r. [DOI] [PubMed] [Google Scholar]
- Matsuzaki K.; Kusaka R.; Nihonyanagi S.; Yamaguchi S.; Nagata T.; Tahara T. Partially Hydrated Electrons at the Air/Water Interface Observed by UV-Excited Time-Resolved Heterodyne-Detected Vibrational Sum Frequency Generation Spectroscopy. J. Am. Chem. Soc. 2016, 138 (24), 7551–7557. 10.1021/jacs.6b02171. [DOI] [PubMed] [Google Scholar]
- Anusiewicz I.; Skurski P.; Simons J. Fate of Dipole-Bound Anion States When Hydrated. J. Phys. Chem. A 2020, 124 (10), 2064–2076. 10.1021/acs.jpca.0c00360. [DOI] [PubMed] [Google Scholar]
- Smyth M.; Kohanoff J. Excess Electron Localization in Solvated DNA Bases. Phys. Rev. Lett. 2011, 106 (23), 238108–238108. 10.1103/PhysRevLett.106.238108. [DOI] [PubMed] [Google Scholar]
- Smyth M.; Kohanoff J. Excess Electron Interactions with Solvated DNA Nucleotides: Strand Breaks Possible at Room Temperature. J. Am. Chem. Soc. 2012, 134 (22), 9122–9125. 10.1021/ja303776r. [DOI] [PubMed] [Google Scholar]
- McAllister M.; Smyth M.; Gu B.; Tribello G. A.; Kohanoff J. Understanding the Interaction between Low-Energy Electrons and DNA Nucleotides in Aqueous Solution. J. Phys. Chem. Lett. 2015, 6 (15), 3091–3097. 10.1021/acs.jpclett.5b01011. [DOI] [PubMed] [Google Scholar]
- McAllister M.; Kazemigazestane N.; Henry L. T.; Gu B.; Fabrikant I.; Tribello G. A.; Kohanoff J. Solvation Effects on Dissociative Electron Attachment to Thymine. J. Phys. Chem. B 2019, 123 (7), 1537–1544. 10.1021/acs.jpcb.8b11621. [DOI] [PubMed] [Google Scholar]
- Schyman P.; Laaksonen A.; Hugosson H. W. Phosphodiester Bond Rupture in 5′ and 3′ Cytosine Monophosphate in Aqueous Environment and the Effect of Low-Energy Electron Attachment: A Car–Parrinello QM/MM Molecular Dynamics Study. Chem. Phys. Lett. 2008, 462 (4–6), 289–294. 10.1016/j.cplett.2008.07.106. [DOI] [Google Scholar]
- Sieradzka A.; Gorfinkiel J. D. Theoretical Study of Resonance Formation in Microhydrated Molecules. II. Thymine-(H2O)n, n = 1,2,3,5. J. Chem. Phys. 2017, 147 (3), 034303–034303. 10.1063/1.4993946. [DOI] [PubMed] [Google Scholar]
- Cornetta L. M.; Coutinho K.; Varella M. T. D. N. Solvent Effects on the Π* Shape Resonances of Uracil. J. Chem. Phys. 2020, 152 (8), 084301–084301. 10.1063/1.5139459. [DOI] [PubMed] [Google Scholar]
- Mukherjee M.; Tripathi D.; Brehm M.; Riplinger C.; Dutta A. K. Efficient EOM-CC-Based Protocol for the Calculation of Electron Affinity of Solvated Nucleobases: Uracil as a Case Study. J. Chem. Theory Comput. 2021, 17 (1), 105–116. 10.1021/acs.jctc.0c00655. [DOI] [PubMed] [Google Scholar]
- Anstöter C. S.; Matsika S. Understanding the Interplay between the Nonvalence and Valence States of the Uracil Anion upon Monohydration. J. Phys. Chem. A 2020, 124 (44), 9237–9243. 10.1021/acs.jpca.0c07407. [DOI] [PubMed] [Google Scholar]
- Anstöter C. S.; Dellostritto M.; Klein M. L.; Matsika S. Modeling the Ultrafast Electron Attachment Dynamics of Solvated Uracil. J. Phys. Chem. A 2021, 125 (32), 6995–7003. 10.1021/acs.jpca.1c05288. [DOI] [PubMed] [Google Scholar]
- Solomun T.; Skalický T. The Interaction of a Protein-DNA Surface Complex with Low-Energy Electrons. Chem. Phys. Lett. 2008, 453 (1–3), 101–104. 10.1016/j.cplett.2007.12.078. [DOI] [Google Scholar]
- Ptasińska S.; Li Z.; Mason N. J.; Sanche L. Damage to Amino Acid–Nucleotide Pairs Induced by 1 EV Electrons. Phys. Chem. Chem. Phys. 2010, 12 (32), 9367–9372. 10.1039/b926267a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutowski M.; Dabkowska I.; Rak J.; Xu S.; Nilles J.M.; Radisic D.; Bowen Jr K.H. Barrier-Free Intermolecular Proton Transfer in the Uracil-Glycine Complex Induced by Excess Electron Attachment. Eur. Phys. J. D - Atomic, Molecular, Optical and Plasma Physics 2002, 20 (3), 431–439. 10.1140/epjd/e2002-00168-1. [DOI] [Google Scholar]
- Da̧bkowska I.; Rak J.; Gutowski M.; Nilles J. M.; Stokes S. T.; Bowen K. H. Barrier-Free Intermolecular Proton Transfer Induced by Excess Electron Attachment to the Complex of Alanine with Uracil. J. Chem. Phys. 2004, 120 (13), 6064–6071. 10.1063/1.1666042. [DOI] [PubMed] [Google Scholar]
- Gu B.; Smyth M.; Kohanoff J. Protection of DNA against Low-Energy Electrons by Amino Acids: A First-Principles Molecular Dynamics Study. Phys. Chem. Chem. Phys. 2014, 16 (44), 24350–24358. 10.1039/C4CP03906H. [DOI] [PubMed] [Google Scholar]
- Verma P.; Narayanan S J J.; Dutta A. K. Electron Attachment to DNA: The Protective Role of Amino Acids. J. Phys. Chem. A 2023, 10.1021/acs.jpca.2c06624. [DOI] [PubMed] [Google Scholar]
- Neustetter M.; Ulia Aysina J.; Ferreira Da Silva F.; Denifl S. The Effect of Solvation on Electron Attachment to Pure and Hydrated Pyrimidine Clusters. Angew. Chem., Int. Ed 2015, 54 (31), 9124–9126. 10.1002/anie.201503733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poštulka J.; Slavíček P.; Fedor J.; Fárník M.; Kočišek J. Energy Transfer in Microhydrated Uracil, 5-Fluorouracil, and 5-Bromouracil. J. Phys. Chem. B 2017, 121 (38), 8965–8974. 10.1021/acs.jpcb.7b07390. [DOI] [PubMed] [Google Scholar]
- Honda T.; Minoshima Y.; Yokoi Y.; Takayanagi T.; Shiga M. Semiclassical Dynamics of Electron Attachment to Guanine–Cytosine Base Pair. Chem. Phys. Lett. 2015, 625, 174–178. 10.1016/j.cplett.2015.03.006. [DOI] [Google Scholar]
- Colson A. O.; Besler B.; Sevilla M. D. Ab Initio Molecular Orbital Calculations on DNA Base Pair Radical Ions: Effect of Base Pairing on Proton-Transfer Energies, Electron Affinities, and Ionization Potentials. J. Phys. Chem. 1992, 96 (24), 9787–9794. 10.1021/j100203a039. [DOI] [Google Scholar]
- Szyperska A.; Rak J.; Leszczynski J.; Li X.; Ko Y. J.; Wang H.; Bowen K. H. Valence Anions of 9-Methylguanine–1-Methylcytosine Complexes. Computational and Photoelectron Spectroscopy Studies. J. Am. Chem. Soc. 2009, 131 (7), 2663–2669. 10.1021/ja808313e. [DOI] [PubMed] [Google Scholar]
- Szyperska A.; Rak J.; Leszczynski J.; Li X.; Ko Y. J.; Wang H.; Bowen K. H. Low-Energy-Barrier Proton Transfer Induced by Electron Attachment to the Guanine···Cytosine Base Pair. ChemPhysChem 2010, 11 (4), 880–888. 10.1002/cphc.200900810. [DOI] [PubMed] [Google Scholar]
- Zheng Y.; Sanche L. Influence of Organic Ions on DNA Damage Induced by 1 EV to 60 KeV Electrons. J. Chem. Phys. 2010, 133 (15), 155102. 10.1063/1.3505046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Sevilla M. D. Role of Excited States in Low-Energy Electron (LEE) Induced Strand Breaks in DNA Model Systems: Influence of Aqueous Environment. ChemPhysChem 2009, 10 (9–10), 1426–1430. 10.1002/cphc.200900025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker D.; Bryant-Friedrich A.; Trzasko C.; Sevilla M. D. Electron Spin Resonance Study of DNA Irradiated with an Argon-Ion Beam: Evidence for Formation of Sugar Phosphate Backbone Radicals. Radiat. Res. 2003, 160 (2), 174–185. 10.1667/RR3037. [DOI] [PubMed] [Google Scholar]
- Shukla L. I.; Pazdro R.; Becker D.; Sevilla M. D. Sugar Radicals in DNA: Isolation of Neutral Radicals in Gamma-Irradiated DNA by Hole and Electron Scavenging. Radiat. Res. 2005, 163 (5), 591–602. 10.1667/RR3347. [DOI] [PubMed] [Google Scholar]
- Becker D.; Adhikary A.; Tetteh S. T.; Bull A. W.; Sevilla M. D. Kr-86 Ion-Beam Irradiation of Hydrated DNA: Free Radical and Unaltered Base Yields. Radiat. Res. 2012, 178 (6), 524–537. 10.1667/RR3066.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdrowowicz M.; Chomicz L.; Żyndul M.; Wityk P.; Rak J.; Wiegand T. J.; Hanson C. G.; Adhikary A.; Sevilla M. D. 5-Thiocyanato-2′-Deoxyuridine as a Possible Radiosensitizer: Electron-Induced Formation of Uracil-C5-Thiyl Radical and Its Dimerization. Phys. Chem. Chem. Phys. 2015, 17 (26), 16907–16916. 10.1039/C5CP02081F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosnowska M.; Makurat S.; Zdrowowicz M.; Rak J. 5-Selenocyanatouracil: A Potential Hypoxic Radiosensitizer. Electron Attachment Induced Formation of Selenium Centered Radical. J. Phys. Chem. B 2017, 121 (25), 6139–6147. 10.1021/acs.jpcb.7b03633. [DOI] [PubMed] [Google Scholar]
- Makurat S.; Zdrowowicz M.; Chomicz-Mańka L.; Kozak W.; Serdiuk I. E.; Wityk P.; Kawecka A.; Sosnowska M.; Rak J. 5-Selenocyanato and 5-Trifluoromethanesulfonyl Derivatives of 2′-Deoxyuridine: Synthesis, Radiation and Computational Chemistry as Well as Cytotoxicity. RSC Adv. 2018, 8 (38), 21378–21388. 10.1039/C8RA03172J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameixa J.; Arthur-Baidoo E.; Meißner R.; Makurat S.; Kozak W.; Butowska K.; Ferreira da Silva F.; Rak J.; Denifl S. Low-Energy Electron-Induced Decomposition of 5-Trifluoromethanesulfonyl-Uracil: A Potential Radiosensitizer. J. Chem. Phys. 2018, 149 (16), 164307. 10.1063/1.5050594. [DOI] [PubMed] [Google Scholar]
- Wen Z.; Peng J.; Tuttle P. R.; Ren Y.; Garcia C.; Debnath D.; Rishi S.; Hanson C.; Ward S.; Kumar A.; Liu Y.; Zhao W.; Glazer P. M.; Liu Y.; Sevilla M. D.; Adhikary A.; Wnuk S. F. Electron-Mediated Aminyl and Iminyl Radicals from C5 Azido-Modified Pyrimidine Nucleosides Augment Radiation Damage to Cancer Cells. Org. Lett. 2018, 20 (23), 7400–7404. 10.1021/acs.orglett.8b03035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meißner R.; Makurat S.; Kozak W.; Limão-Vieira P.; Rak J.; Denifl S. Electron-Induced Dissociation of the Potential Radiosensitizer 5-Selenocyanato-2′-Deoxyuridine. J. Phys. Chem. B 2019, 123 (6), 1274–1282. 10.1021/acs.jpcb.8b11523. [DOI] [PubMed] [Google Scholar]
- Mudgal M.; Dang T. P.; Sobczak A. J.; Lumpuy D. A.; Dutta P.; Ward S.; Ward K.; Alahmadi M.; Kumar A.; Sevilla M. D.; Wnuk S. F.; Adhikary A. Site of Azido Substitution in the Sugar Moiety of Azidopyrimidine Nucleosides Influences the Reactivity of Aminyl Radicals Formed by Dissociative Electron Attachment. J. Phys. Chem. B 2020, 124 (50), 11357–11370. 10.1021/acs.jpcb.0c08201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Bahry T.; Denisov S. A.; Adhikary A.; Mostafavi M. Quasi-Free Electron-Mediated Radiation Sensitization by C5-Halopyrimidines. J. Phys. Chem. A 2021, 125 (36), 7967–7975. 10.1021/acs.jpca.1c05974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adjei D.; Reyes Y.; Kumar A.; Ward S.; Denisov S. A.; Alahmadi M.; Sevilla M. D.; Wnuk S. F.; Mostafavi M.; Adhikary A. Pathways of Dissociative Electron Attachment Observed in 5- and 6-Azidomethyluracil Nucleosides: Nitrogen (N2) Elimination vs. Azide anion (N3®) Elimination. J. Phys. Chem. B 2023, 127, 1563. 10.1021/acs.jpcb.2c08257. [DOI] [PMC free article] [PubMed] [Google Scholar]