

ABSTRACT
Pseudorabies virus (PRV) infection activates inflammatory responses to release robust proinflammatory cytokines, which are critical for controlling viral infection and clearance of PRV. However, the innate sensors and inflammasomes involved in the production and secretion of proinflammatory cytokines during PRV infection remain poorly studied. In this study, we report that the transcription and expression levels of some proinflammatory cytokines, including interleukin 1β (IL-1β), IL-6, and tumor necrosis factor alpha (TNF-α), are upregulated in primary peritoneal macrophages and in mice during PRV infection. Mechanistically, Toll-like receptor 2 (TLR2), TLR3, TLR4, and TLR5 were induced by the PRV infection to enhance the transcription levels of pro-IL-1β, pro-IL-18, and gasdermin D (GSDMD). Additionally, we found that PRV infection and transfection of its genomic DNA triggered AIM2 inflammasome activation, apoptosis-related speckle-like protein (ASC) oligomerization, and caspase-1 activation to enhance the secretion of IL-1β and IL-18, which was mainly dependent on GSDMD, but not GSDME, in vitro and in vivo. Taken together, our findings reveal that the activation of the TLR2-TLR3-TRL4-TLR5-NF-κB axis and AIM2 inflammasome, as well as GSDMD, is required for proinflammatory cytokine release, which resists the PRV replication and plays a critical role in host defense against PRV infection. Our findings provide novel clues to prevent and control PRV infection.
IMPORTANCE PRV can infect several mammals, including pigs, other livestock, rodents, and wild animals, causing huge economic losses. As an emerging and reemerging infectious disease, the emergence of PRV virulent isolates and increasing human PRV infection cases indicate that PRV is still a high risk to public health. It has been reported that PRV infection leads to robust release of proinflammatory cytokines through activating inflammatory responses. However, the innate sensor that activates IL-1β expression and the inflammasome involved in the maturation and secretion of proinflammatory cytokines during PRV infection remain poorly studied. In this study, our findings reveal that, in mice, activation of the TLR2-TLR3-TRL4-TLR5-NF-κB axis and AIM2 inflammasome, as well as GSDMD, is required for proinflammatory cytokine release during PRV infection, and it resists PRV replication and plays a critical role in host defense against PRV infection. Our findings provide novel clues to prevent and control PRV infection.
KEYWORDS: pseudorabies virus, TLRs, AIM2 inflammasome, GSDMD, IL-1β
INTRODUCTION
Pseudorabies (PR; also named Aujeszky’s disease), caused by pseudorabies virus (PRV) (Aujeszky’s disease virus or Suid herpesvirus 1), has caused substantial economic losses to pig factories in some countries. PRV is a complex DNA virus that belongs to the family Herpesviridae and the subfamily Alphaherpesvirinae. Without specific host tropism, PRV can infect a wide variety of animal species, including ruminants, carnivores, rodents, and lagomorphs. Pigs are considered the only natural hosts for PRV. Piglets and finishing pigs infected with virulent PRV isolates caused acute death and severe clinical symptoms (1). In addition to pigs, cattle, sheep, cats, dogs, raccoons, minks, and skunks can all be infected by PRV, causing “mad itch,” neurological symptoms, or death (2). Recently, potential human cases of PRV infection have been reported sporadically with conjunctivitis, dermatitis, fatal encephalitis, and prominent central nervous system disorders (3, 4), suggesting that PRV still poses a significant threat to public health. Therefore, it is important to strengthen the investigation of the pathogenic mechanism of PRV infection.
Virus-induced inflammatory responses, composed of immune cells and inflammatory cytokines, execute a kind of complex protective mechanism to recognize and remove the invading virus and promote the repair of damaged tissues. Many sensors are involved in host antiviral inflammatory responses by recognizing viral infection and activating downstream signaling pathways to induce the production of antiviral cytokines and chemokines, including Toll-like receptors (TLRs), cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING), DNA damage, and gamma interferon (IFN-γ)-inducible protein 16 (IFI16), etc. (5). Previous studies showed that TLRs activated IL-1 receptor-associated kinase 1 (IRAK1) and tumor necrosis factor receptor-associated factor 6 (TRAF6) in a myeloid differentiation factor 88 (MyD88)-dependent manner, which then promoted nuclear factor NF-κB to enter the nucleus to induce the expression of pro-interleukin 1β (IL-1β) and pro-IL-18 (6, 7).
Inflammasomes are multiprotein complexes that consist of cytoplasmic inflammatory receptors, apoptosis-related speckle-like protein (ASC), and inflammation-related caspases. Upon viral infection, an active inflammasome leads to the maturation and secretion of proinflammatory cytokines to induce inflammatory responses, which play a critical role in host defense against viral infection (8). Among several major inflammasomes, the most intensely studied is the NLRP3 inflammasome, which can be activated upon exposure to various pathogens, as well as a number of structurally diverse pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs), and environmental irritants, including bacteria, viruses, reactive oxygen-free radicals (ORS), ATP, and so on. Both RNA viruses, such as Sendai virus (SeV), respiratory syncytial virus (RSV), vesicular stomatitis virus (VSV), and hepatitis C virus (HCV), and some DNA viruses, including adenovirus (ADV) and herpes simplex virus 1 (HSV-1), can activate the NLRP3 inflammasome (9, 10). Upon stimulation, NLRP3 recruits ASC and pro-caspase-1 to form the NLRP3 inflammasome complex. After autoactivation, caspase-1 directly leads to the maturation and secretion of IL-1β and IL-18 by cleaving pro-IL-1β and pro-IL-18 (8). Additionally, ligand requirements for AIM2 inflammasome activation are quite permissive, including cytosolic double-stranded DNA (dsDNA) from virus, bacteria, or the host itself-damaged genomic DNA and mitochondria DNA. Previous studies indicated that AIM2 was proposed to function in cytosolic surveillance for DNA viruses and might contribute to autoimmune responses against self-DNA in systemic lupus erythematosus (11). Subsequently, the released IL-1β binds its receptors to induce the release of other proinflammatory cytokines (such as tumor necrosis factor [TNF] and IL-6) and induce Th17 bias in cellular adaptive responses (12, 13), while IL-18 plays an essential role in the induction of IFN-γ and Th1 responses (14).
Gasdermins (GSDMs) are a characterized protein family encoded by six paralogous genes, Gsdma, Gsdmb, Gsdmc, Gsdmd, Gsdme (also known as Dfna5), and Dfnb59 (15). GSDMB and GSDME can be cleaved by the death-inducing proteases granzymes (Gzms) A and B in natural killer (NK) cells and cytotoxic T lymphocytes (CTLs), respectively, to activate pyroptosis during killer cell attack, which was previously thought to be noninflammatory (16, 17). GSDMC can be cleaved by caspase 8 in TNF-mediated death receptor signaling to trigger pyroptosis (18). However, it is not clear how GSDMA is activated. As a key executioner of inflammasome-induced pyroptosis and the release of inflammatory cytokines, GSDMD is the most important GSDM member and has been widely studied. GSDMD is a substrate of caspase-1 in the canonical inflammasome and caspase-4/5 (known as caspase-11 in mice) in the noncanonical inflammasome, which plays a central role in some inflammation-related diseases (19, 20). The proteolytic cleavage of GSDMD and GSDME liberates the N-terminal fragments (GSDMD-N, GSDME-N), which form pores in cell membranes to induce pyroptosis and IL-1β secretion (20, 21). Then, the released GSDMD, inflammatory cytokines, and chemokines are transported to amplify inflammation in the tissue, where immune cells are recruited to repair damaged tissue and clear viral infection.
Similar to HSV-1 and varicella-zoster virus (VZV), PRV infection also induces strong inflammatory responses, which cause inflammatory response-related diseases (22, 23). In this study, we systematically investigated the mechanism by which PRV infection induces inflammatory responses in vitro and in vivo in mice. We present biochemical and genetic evidence showing that, in mice, PRV infection enhances the production and secretion of IL-1β by targeting the TLR2-TLR3-TRL4-TLR5-NF-κB axis and AIM2 inflammasome, which is mainly dependent on the activation of GSDMD.
RESULTS
PRV infection induces IL-1β and IL-18 maturation and secretion in vitro and in vivo.
To investigate whether the levels of several proinflammatory cytokines, including pro-IL-1β, pro-IL-18, and TNF-α, are upregulated upon PRV infection, mouse peritoneal macrophages were isolated and infected with PRV at a multiplicity of infection (MOI) of 1, 5, or 10 for 12 h or infected with PRV at an MOI of 1 for 3, 6, or 12 h as indicated. The quantitative PCR (qPCR) results showed that the mRNA levels of pro-IL-1β, pro-IL-18, and TNF-α were significantly upregulated (Fig. 1A to F). Additionally, the protein levels of secreted IL-1β in the cell supernatants were also increased in a dose- and time-dependent manner after PRV infection (Fig. 1G to J). In line with these results, Western blot results illustrated that pro-caspase-1 was activated to produce active caspase-1 (p10) and that pro-IL-1β was cleaved to produce mature IL-1β (p17) in PRV-infected mouse peritoneal macrophages (Fig. 1K and L). As ASC oligomerization is a direct indicator of inflammasome activation (8), we further examined whether PRV infection induced ASC oligomerization and inflammasome complex formation in mouse peritoneal macrophages. As shown in Fig. 1M, the AIM2-ASC-caspase-1 complex, but not the NLRP3-ASC-caspase-1 complex, was detected in the PRV-infected group, suggesting that the AIM2 inflammasome was activated in PRV-infected mouse peritoneal macrophages. Additionally, ASC was diffusely distributed in the nucleus and cytoplasm in mock-infected macrophages, whereas ASC formed distinct small specks in PRV-infected macrophages, suggesting that PRV infection facilitated ASC oligomerization (Fig. 1N and O). Taken together, our findings demonstrate that PRV infection activates the AIM2 inflammasome, resulting in the maturation and secretion of IL-1β.
FIG 1.

PRV infection induces IL-1β maturation and secretion in vitro. Mouse peritoneal macrophages isolated from C57BL/6 mice were infected with PRV at an MOI of 1, 5, or 10 for 12 h or infected with PRV at MOI of 1 for 3, 6, or 12 h as indicated. Mouse peritoneal macrophages were pretreated with LPS (1 μg/mL) for 8 h followed by treatment with 5 mM ATP for another 2 h, which was used as a positive control. The mRNA levels of IL-1β (A, D), IL-18 (B, E), and TNF-α (C, F) were detected by qPCR. The levels of secreted IL-1β in cell supernatants, PRV DNA copy number, and the protein levels of pro-IL-1β, IL-1β (p17), pro-caspase-1, and caspase-1 (p10) in cell lysates were detected by ELISA (G, H), qPCR (I, J), and Western blotting (K, L), respectively. (M, N) Mouse peritoneal macrophages were either mock infected or infected with PRV at an MOI of 1 for 12 h. The cell lysates were coimmunoprecipitated with anti-ASC monoclonal antibody (mAb) or mouse IgG. (M) The immunoprecipitants and the whole-cell lysates were detected with anti-NLRP3, AIM2, ASC, caspase-1, and GAPDH antibodies, respectively. Alternatively, the oligomerization of ASC was examined under confocal microscopy. The scale bars represent 10 μm in panel N. (O) The statistical analysis of ASC dots. The results shown are representative of three independent experiments (one-way ANOVA) in panels A to J. *, P < 0.05; **, 0.001 < P < 0.01; ***, P < 0.001. All error bars show standard deviations (SDs).
To assess whether PRV infection induces inflammatory responses in vivo, wild-type (WT) mice were infected with PRV (2,000 PFU/mouse) by tail vein injection. As shown in Fig. 2A, 10 out of 10 WT mice (100%) died in the PRV-infected group at 5 days postinfection (dpi), whereas 3 out of 3 WT mice (0%) survived in the control group injected with phosphate-buffered saline (PBS) at 10 dpi. The protein levels of secreted IL-1β, IL-6, IL-18, and TNF-α in the serum from the mice infected with PRV for 24 h were significantly higher than those from the control mice injected with PBS (Fig. 2B to E). Correspondingly, the mRNA levels of Il-1β, Il-6, Il-18, and Tnf-α genes in the spleen, lung, and brain from the PRV-infected mice were also significantly upregulated (Fig. 2F to I). Additionally, in the tested tissues, the spleen showed the highest transcriptional levels of Il-1β, Il-6, Il-18, and Tnf-α genes. Consistent with these results, the PRV DNA copy numbers in the blood, spleen, lung, and brain were significantly increased in the PRV-infected mice after infection for 24 h (Fig. 2J and K). These results demonstrate that PRV infection activates inflammatory responses in vivo.
FIG 2.

PRV infection induces IL-1β and IL-18 expression and secretion in mice. (A) Survival rate of WT mice (10 mice in the PRV-infected group and 3 mice in the PBS-infected group). (B to E) Detection of proinflammation cytokines (IL-1β, IL-6, IL-18, and TNF-α) in serum by ELISA. (F to I) Detection of the mRNA levels of proinflammation cytokines (IL-1β, IL-6, IL-18, and TNF-α) in the spleen, lung, and brain by qPCR. (J, K) Detection of PRV DNA copy number in blood, spleen, lung, and brain. The results shown are representative of three independent experiments (n = 3 mice per group). *, P < 0.05; **, 0.001 < P < 0.01; ***, P < 0.001. All error bars show standard deviations (SDs).
PRV infection enhances IL-1β levels through the TLR2-TLR3-TRL4-TLR5-NF-κB pathway.
To elucidate the underlying molecular mechanisms by which PRV infection upregulates pro-IL-1β production, we first assessed the effect of TLRs on pro-IL-1β expression in PRV-infected mouse peritoneal macrophages. Mouse peritoneal macrophages were pretreated with different inhibitors of TLRs, MyD88, and NF-κB signaling and then infected with PRV for 24 h. As shown in Fig. 3A to D, AN-3485 (inhibitor of TLR2, TLR3, TLR4, and TLR5), T6167923 (inhibitor of MyD88), Hydroxychloroquine (inhibitor of TLR7 and TLR9), and JSH-23 (inhibitor of NF-κB) were harmless to mouse peritoneal macrophages. Notably, qPCR and Western blot results demonstrated that only the mRNA and protein levels of pro-IL-1β induced by PRV infection were significantly suppressed in mouse peritoneal macrophages pretreated with AN-3485, T6167923, and JSH-23 (Fig. 3E to L). In accordance with these results, the secreted IL-1β levels induced by PRV were significantly inhibited in AN-3485-, T6167923-, and JSH-23-pretreated mouse peritoneal macrophages but were not affected by Hydroxychl (Fig. 3M to P).
FIG 3.

PRV infection enhances IL-1β transcription, and expression was dependent on the TLR2-TLR3-TRL4-TLR5-NF-κB axis. (A to D) Mouse peritoneal macrophages were primed with TLR2, TLR3, TLR4, and TLR5 inhibitor AN-3485 (1 μM) (A), MyD88 inhibitor T6167923 (20 μM) (B), TLR7 and TLR9 inhibitor Hydroxychl (20 μM) (C), or NF-κB inhibitor JSH-23 (20 μM) (D), respectively, for 12, 24, or 36 h. Cell viability was analyzed by detecting LDH release. Positive indicates the total LDH level in the cell and supernatant. (E to P) Mouse peritoneal macrophages were pretreated with AN-3485 (0.1, 0.5, or 1 μM) (E, I), T6167923 (5, 10, or 20 μM) (F, J), Hydroxychl (5, 10, or 20 μM) (G, K), or JSH-23 (2, 10, 20, or 40 μM) (H, L), respectively, for 1 h and then mock infected or infected with PRV at MOI of 1 for another 24 h. The mRNA and protein levels of pro-IL-1β were detected with qPCR and Western blot, respectively. The secretion levels of IL-1β in the cell supernatant were detected by ELISA in panels M to P. The results shown are representative of three independent experiments (one-way ANOVA) (panels A to H and M to P). **, 0.001 < P < 0.01; ***, P < 0.001; ns, no significance. All error bars show standard deviations (SDs).
To further define the roles of TLR2, TLR3, TLR4, and TLR5 involved in enhancement of IL-1β production during PRV infection, HEK293T cells were transfected with an NF-κB reporter and a plasmid expressing mouse TLR2, TLR3, TLR4, or TLR5 for 24 h, respectively. Then, the cells were infected with PRV. As shown in Fig. 4A to D, we found that, without coexpressing TLRs, the activity of the NF-κB reporter with PRV infection was higher than that of control cells without PRV infection, suggesting that PRV infection itself can also trigger NF-κB activation (black columns 1 and 2), which is consistent with previous studies (24, 25). Additionally, our results indicated that overexpression of TLR2, TLR3, TLR4, and TLR5 enhanced NF-κB reporter’s activity during PRV infection (black columns 2 and 4). Consistent with these results, Western blot results also demonstrated that overexpression of TLR2, TLR3, TLR4, and TLR5 in HEK293T cells strengthened the production of pro-IL-1β during PRV infection (Fig. 4E to H). Taken together, these findings suggest that TLR2-TLR3-TRL4-TLR5-NF-κB may play an important role in the production of proinflammatory cytokines during PRV infection.
FIG 4.

Overexpressed TLR2, TLR3, TLR4, and TLR5 enhance IL-1β production during PRV infection. (A to D) HEK293T cells were transfected with the NF-κB reporter or a plasmid expressing TLR2, TLR3, TLR4, and TLR5 (200 ng/each). After transfection for 24 h, the cells were infected with PRV, and then luciferase assays were performed. The experiment shown is a representative experiment of three independent experiments with the mean ± SD of three technical replicates (one-way ANOVA) in panels A to D. **, 0.001 < P < 0.01; ***, P < 0.001. (E to H) HEK293T cells were transfected with plasmids expressing the indicated proteins. After transfection for 24 h, the cells were infected with PRV for 8 h and were assessed by Western blot analysis.
Shutting off the TLR2-TLR3-TRL4-TLR5-NF-κB pathway enhances PRV replication.
To further investigate the effect of the TLR2-TLR3-TRL4-TLR5-NF-κB pathway on IL-1β expression and virus replication in PRV-infected mouse peritoneal macrophages, the cells were transfected with small interfering RNA (siRNAs) targeting Tlr2, Tlr3, Tlr4, Tlr5, Tlr7, Tlr8, and Tlr9 and Myd88 genes to downregulate TLRs and Myd88 expression and then infected with PRV. The knockdown efficiency of Tlr and Myd88 genes was confirmed with qPCR and Western blot analysis, respectively (Fig. 5A and B). Indeed, knockdown of the expression of Tlr2, Tlr3, Tlr4, Tlr5, or Myd88 substantially reduced the mRNA and protein levels of IL-1β induced by PRV infection (Fig. 5C and D), whereas knockdown of the expression of Tlr7, Tlr8, and Tlr9 had no effect on the production of IL-1β induced by PRV infection (Fig. 5E and F). In addition, the confocal experimental results also showed that PRV infection promoted the transport of NF-κB (p65) from the cytoplasm to the nucleus (Fig. 5G and H). To confirm this result, the effect of PRV infection on the nuclear translocation of NF-κB was also analyzed by Western blotting. As shown in Fig. 5I, PRV infection augmented the amount of NF-κB in the nucleus in a dose-dependent manner, in line with the notion that NF-κB could be activated upon PRV infection (26). Notably, inhibiting the activities of TLR2, TLR3, TLR4, TLR5, Myd88, and NF-κB or knocking down the expression of Tlr2, Tlr3, Tlr4, Tlr5, or Myd88 genes increased the PRV genomic DNA and 50% tissue culture infective dose (TCID50) (Fig. 5J to M). Taken together, these results implicated that PRV infection strengthened IL-1β production through the TLR2-TLR3-TRL4-TLR5-NF-κB axis. Shutting off the TLR2-TLR3-TRL4-TLR5-NF-κB pathway can inhibit the IL-1β level and enhance PRV replication.
FIG 5.

Knockdown of TLR2, TLR3, TLR4, TLR5, and Myd88 enhances PRV replication. (A, B) Mouse peritoneal macrophages were transfected with control siRNA (siNC) and siRNAs targeting Tlr2, Tlr3, Tlr4, Tlr5, Tlr7, Tlr8, Tlr9, and Myd88 genes for 48 h or 72 h. The mRNA and protein levels of Tlr2, Tlr3, Tlr4, Tlr5, Tlr7, Tlr8, Tlr9, and Myd88 genes were detected with qPCR (A) and Western blotting (B). (C to F) Mouse peritoneal macrophages were transfected with control siRNA (siNC) and siRNAs targeting Tlr2, Tlr3, Tlr4, Tlr5, Tlr7, Tlr8, Tlr9, and Myd88 genes for 48 h and then mock infected or infected with PRV at MOI of 1 for another 24 h. The mRNA and protein levels of pro-IL-1β were detected with qPCR (C, E) and Western blotting (D, F). (G to I) Mouse peritoneal macrophages were infected with PRV for 24 h. (G) The subcellular locations of NF-κB (green) and the nuclear marker DAPI (blue) were visualized with confocal microscopy. (H) The percentage of the NF-κB localized in the cellular nucleus was calculated. The scale bars represent 5 μm. (I) The cell lysates were used for cytoplasmic and nuclear separation and analyzed by Western blotting. (J to M) Referring to the methods of panels C to F, PRV genomic DNA and the TCID50 were detected. Results shown are representative of three independent experiments (mean ± SD) or are representative of three independent experiments with similar results (one-way ANOVA) in panels C, E, and J to M. *, P < 0.05; **, 0.001 < P < 0.01; ***, P < 0.001; ns, no significance.
The AIM2 inflammasome is required for PRV-induced IL-1β secretion.
It has been reported that virus infection-induced inflammasome activation promotes caspase-1 activation and pro-IL-1β cleavage, resulting in IL-1β maturation and secretion (8). To test which inflammasome activation is required for activating caspase-1 and IL-1β secretion in mice during PRV infection, mouse peritoneal macrophages were pretreated with the NLRP3 inhibitor MCC950 or the caspase-1 inhibitor VX-765, respectively, and then mock infected or infected with PRV. As shown in Fig. 6A and B, these two inhibitors did not affect cell viability. Enzyme-linked immunosorbent assay (ELISA) results revealed that VX-765 treatment virtually abolished IL-1β secretion, whereas MCC950 treatment did not affect IL-1β secretion in PRV-infected mouse peritoneal macrophages (Fig. 6C and D), suggesting that NLRP3 inflammation may not be involved in PRV-induced inflammatory responses.
FIG 6.

PRV-induced IL-1β secretion is dependent on the AIM2 inflammasome. (A, B) Mouse peritoneal macrophages were pretreated with an inhibitor of NLRP3, MCC950 (2 μM) (A), or an inhibitor of caspase-1, VX-765 (2 μM) (B), for 12, 24, and 36 h. The cell viability was assessed by detecting LDH release. Positive indicates the total LDH level in the cell and supernatant. (C, D) Mouse peritoneal macrophages from WT mice were pretreated with MCC950 (C) or VX-765 (D) for 1 h, followed by PRV infection (MOI = 1, 5, or 10) for 12 h. The secreted IL-1β levels in the supernatants were measured by ELISA. (E, F) Mouse peritoneal macrophages from WT, Nlrp3−/−, Aim2−/−, ASC−/−, and caspase-1−/− mice were mock infected or infected with PRV (MOI = 1, 5, or 10) for 12 h. The pro-caspase-1 activation was analyzed (E), and the secreted IL-1β level in the supernatants was measured by ELISA (F). (G, H) HEK293T cells were transfected with plasmids expressing HA-Aim2, HA-ASC, and HA-pro-caspase-1, together with Flag-pro-IL-1β, to reconstruct the AIM2 inflammasome system and then infected with PRV at an MOI of 1 or 5 for 12 h. Then, the expression and secretion of IL-1β were detected by Western blotting (G) and ELISA (H), respectively. (I, J) Mouse peritoneal macrophages from WT, Nlrp3−/−, and Aim2−/− mice were mock infected or infected with PRV for 12 h. The ASC oligomerization was analyzed by fluorescence microscopy. The scale bars represent 10 μm. Data are representative of three independent experiments with three biological replicates (mean ± SD in panels A to F and H; one-way ANOVA in panels A, B, and H; two-way ANOVA in panels C to F). **, 0.001 < P < 0.01; ***, P < 0.001; ns, no significance.
To test whether AIM2 inflammasome activation is required for active caspase-1 and IL-1β secretion during PRV infection, we generated and identified Nlrp3−/−, Aim2−/−, ASC−/−, and caspase-1−/− mice (Fig. 7A to F). Subsequently, mouse peritoneal macrophages isolated from Nlrp3−/−, Aim2−/−, and WT mice were mock infected or infected with PRV at MOI of 1, 5, or 10 for 12 h. As shown in Fig. 6E and F, PRV infection still induced caspase-1 activation in mouse peritoneal macrophages isolated from Nlrp3−/− and WT mice but not in mouse peritoneal macrophages isolated from Aim2−/− mice. Consistent with these results, PRV infection also enhanced IL-1β secretion in mouse peritoneal macrophages isolated from Nlrp3−/− and WT mice but not in mouse peritoneal macrophages isolated from Aim2−/−, ASC−/−, and caspase-1−/− mice. These results indicate that the AIM2 inflammasome, but not the NLRP3 inflammasome, is involved in PRV-induced caspase-1 activation and IL-1β secretion. Notably, it was reported that in mice, active caspase-11 may also be involved in the noncanonical pyroptotic pathway and involved in IL-1β secretion (8). In our results, we found that the caspase-1 inhibitor VX-765 completely inhibited PRV-induced inflammatory responses, and macrophage isolated from caspase-1−/− mice could not release IL-1β after PRV infection (Fig. 6D and F), so we concluded that other caspases might not participate in this biological process.
FIG 7.

Construction and identification of knockout mice. (A to D, G to H) Construction strategy of Nlrp3−/−, Aim2−/−, ASC−/−, caspase-1−/−, Gsdmd−/−, and Gsdme−/−mice. (E, F, and I) Identification of knockout mice by Western blotting.
To further confirm these results, a recombinant AIM2 inflammasome system was reconstructed in vitro in which HEK293T cells were transfected with plasmids expressing AIM2, ASC, pro-caspase-1, and pro-IL-1β. The programmed cells were mock infected or infected with PRV at an MOI of 1 or 5 for 12 h. The result showed that the maturation and secretion of IL-1β (p17) were upregulated when the cells were infected with PRV (Fig. 6G and H). These results indicate that the AIM2 inflammasome plays an essential role in PRV-induced IL-1β secretion. Previous studies showed that AIM2 inflammasome activation required the adaptor protein ASC oligomerization to bridge AIM2 and pro-caspase-1 (8). Therefore, we assessed ASC oligomerization in PRV-infected mouse peritoneal macrophages from WT, Nlrp3−/−, and Aim2−/− mice. These results showed that ASC was diffusely distributed in the nucleus and cytoplasm in the mock-infected macrophages, while ASC formed distinct small specks in WT and Nlrp3−/− macrophages upon PRV infection. Notably, Aim2 deficiency significantly inhibited PRV-induced ASC oligomerization (Fig. 6I and J). Overall, these results demonstrate that the AIM2 inflammasome is essential for PRV-induced IL-1β secretion in vitro.
The AIM2 inflammasome is critical for host defense against PRV.
To explore the role of Aim2-mediated inflammatory responses during PRV infection, Nlrp3−/− and Aim2−/− mice and their WT littermates were challenged with PRV by tail vein injection. We noticed that, compared with WT and Nlrp3−/− mice, Aim2−/− mice were more susceptible to PRV infection, and all Aim2−/− mice died after 3 dpi (Fig. 8A). Consistent with these results, the secreted IL-1β levels in the serum from Aim2−/− mice were significantly lower than those from WT and Nlrp3−/− mice, while the IL-6 and TNF-α levels had no obvious change among these mice (Fig. 8B to D). Notably, the mRNA levels of IL-1β in the liver, lung, brain, and spleen from these mice were not different, suggesting that Aim2 knockout had no effect on IL-1β transcription (Fig. 8E). It is worth noting that the PRV DNA copy numbers in these tissues from Aim2−/− mice were higher than those of WT and Nlrp3−/− mice (Fig. 8F). Additionally, pathological examinations demonstrated that the lung tissues from WT and NLRP3−/− mice exhibited less necrosis of bronchioles and alveolar epithelial cells than those from Aim2−/− mice (Fig. 8G and H). Taken together, we proposed that AIM2-mediated inflammatory responses play critical roles in host defense against PRV infection.
FIG 8.

AIM2 inflammasome is critical for host defense against PRV infection in mice. (A) Survival rates of WT, Nlrp3−/−, and Aim2−/− mice infected with PRV at 20 PFU/each mouse by tail vein injection (n = 10 per group). Statistical significance was determined by the log-rank test. (B to H) The WT and Nlrp3−/−- and Aim2−/−-deficient mice were infected with PRV at 20 PFU/each mouse by tail vein injection for 24 h. The levels of IL-1β (B), IL-6 (C), and TNF-α (D) in serum were detected by ELISA. The mRNA levels of IL-1β (E) and the PRV DNA copy number (F) in the tissues were analyzed with qPCR. (G, H) Pathological lesions of mice (H&E staining) from the lung were shown. Data are representative of three independent experiments with three biological replicates (mean ± SD in panels B to F) (two-way ANOVA in panels B to F). *, P < 0.05; **, 0.001 < P < 0.01; ***, P < 0.001; ns, no significance.
PRV genomic DNA activates the AIM2 inflammasome to induce IL-1β secretion.
To examine whether the PRV replication process is required for IL-1β expression and secretion, mouse peritoneal macrophages were mock infected or infected with live PRV or UV-inactivated PRV. We noticed that the mRNA and protein levels of pro-IL-1β were increased in PRV-infected mouse peritoneal macrophages. However, UV-inactivated PRV only triggered a low inflammatory response (Fig. 9A and B). Consistent with these results, live PRV infection, but not UV-inactivated PRV, induced pro-caspase-1 activation and IL-1β secretion in the cell supernatant (Fig. 9C and D). Consistently, in vivo, PRV infection induced pro-IL-1β upregulation in the lung, spleen, and brain, as well as IL-1β secretion in the serum of mice, while the UV-inactivated PRV could not induce pro-IL-1β upregulation in the lung and brain and IL-1β secretion in the serum, except for a weaker upregulation of pro-IL-1β in the spleen of mice (Fig. 9E and F). These results suggest that the viral replication process is required for PRV-induced inflammatory responses and that the tegument proteins of PRV are not involved in this biological process.
FIG 9.

PRV genomic DNA activates AIM2 inflammasome to induce IL-1β secretion in vitro and in vivo. (A to D) Mouse peritoneal macrophages were mock infected or infected with PRV or infected with UV-inactivated PRV (UV inactivated for 3, 6, or 12 h, respectively) at MOI of 10 for 12 h. The mRNA levels of pro-IL-1β and the protein levels of pro-IL-1β, cleaved IL-1β, and activated IL-1β (p17) in the cell lysates were measured by qPCR (A) and Western blot analysis (B), respectively. The caspase-1 activity in cell lysates and the secreted IL-1β in the cell supernatants were analyzed by Caspase-Glo 1 inflammasome assay kits (C) and ELISA (D), respectively. (E, F) WT mice were infected with PRV or UV-inactivated PRV at 2,000 PFU/mouse by tail vein injection for 24 h. (E) The mRNA levels of IL-1β in the tissues were analyzed with qPCR. (F) The secretion level of IL-1β in serum was detected by ELISA. (G to J) PRV genomic DNA was extracted and transfected into mouse peritoneal macrophages at 1, 2, and 5 μg/mL for 24 h. The active caspase-1 in cell lysates was also analyzed (G). The mRNA, protein, and secreted levels of IL-1β were measured by qPCR (H), Western blotting (I), and ELISA (J), respectively. (K) HEK293T cells were transfected with pCAGGS-Flag-Nlrp3 or pCAGGS-HA-Aim2 for 24 h. Cell lysates were harvested and coincubated with biotinylated PRV genomic DNA (10 μg) at 4°C for 12 h. Streptavidin-Sepharose beads were then added and rotated at 4°C for 4 h. The beads were washed and analyzed by immunoblotting with the indicated antibodies. (L to N) PRV genomic DNA was transfected into mouse peritoneal macrophages at 2 μg/mL for 24 h. (L, M) The ASC oligomerization was examined with anti-ASC mAb under confocal microscopy, and the cell lysates were coimmunoprecipitated with anti-ASC mAb or mouse IgG. (N) The immunoprecipitants and whole-cell lysates were detected with anti-NLRP3, AIM2, ASC, caspase-1, and GAPDH antibodies. The scale bars represent 5 μm. Data are representative of three independent experiments with three biological replicates (mean ± SD in panels A, C to H, and J and one-way ANOVA in panels A, C to H, and J). **, 0.001 < P < 0.01; ***, P < 0.001.
Previous studies demonstrated that recognition of cytosolic DNA by AIM2 was essential for AIM2 inflammasome activation (8). Therefore, we next evaluated the ability of PRV DNA to trigger pro-caspase-1 activation, IL-1β expression, and secretion in vitro. As shown in Fig. 9G to J, we found that PRV DNA could induce caspase-1 activation, pro-IL-1β expression, and IL-1β secretion in a dose-dependent manner after transfection of PRV genomic DNA into mouse peritoneal macrophages for 24 h. Moreover, DNA pulldown results showed that AIM2 bound PRV genomic DNA but not NLRP3 (Fig. 9K). Furthermore, transfection of PRV DNA in mouse peritoneal macrophages facilitated ASC oligomerization and the formation of the AIM2-ASC-caspase-1 complex but not the NLRP3-ASC-caspase-1 complex (Fig. 9L to N). Overall, these findings indicate that PRV genomic DNA can induce the maturation and secretion of IL-1β through activating the AIM2 inflammasome.
Transfection with intact PRV DNA into cells may produce viral proteins and live viral particles, which may activate inflammatory responses. To further prove it, the 5′-untranslated region (5′-UTR) and 3′-UTR transcripts of the PRV genome were synthesized in vitro (Table 1) and transfected into mouse peritoneal macrophages. Most notably, both the 5′ UTR and 3′ UTR transcripts of PRV only induced moderate expression of pro-IL-1β and pro-IL-18 (Fig. 10A and B), as well as secretion of IL-1β (Fig. 10C). Interestingly, we found that the 5′-UTR and 3′-UTR transcripts could induce caspase-1 activation and ASC oligomerization (Fig. 10D to F). These data demonstrate that the 5′-UTR and 3′-UTR transcripts of PRV were sufficient for caspase-1 activation and ASC oligomerization but not pro-IL-1β and IL-18 transcription. Taken together, our results reveal that the PRV replication process is indispensable for PRV-induced inflammatory responses.
TABLE 1.
Primers sequences used for 5′-UTR and 3′-UTR transcript amplification
| Gene | Sequence (5′→3′) |
|---|---|
| 5′-UTR | F, CCCCCCAGCCCCTCCGCTCCCCCTTTCCCC |
| R, CCCACGGCGGCTGGCGGCGGACGGCGGTGC | |
| 3′-UTR | F, GCGGGGCGCCCCCTCGGCCCGGCTGGACTC |
| R, TAATCTGCATAACCCCTCCCCCTAATCTGC |
FIG 10.

The 5′ UTR and 3′ UTR of PRV genome DNA were sufficient for caspase-1 activation and ASC oligomerization but not pro-IL-1β transcription. (A to F) The 5′-UTR and 3′-UTR transcripts of PRV genomic DNA were amplified and transfected into mouse peritoneal macrophages isolated from C57BL/6 mice for 24 h. The mRNA levels of IL-1β (A) and IL-18 (B) were measured by qPCR, and the secretion levels of IL-1β were measured by ELISA (C). The protein levels of pro-caspase-1, cleaved caspase-1, and active caspase-1 (p10) in cell lysates were analyzed by Western blotting (D). (E, F) ASC oligomerization was examined with anti-ASC mAb under confocal microscopy. The scale bars represent 10 μm. Data are representative of three independent experiments with three biological replicates (mean ± SD in panels A to C).
GSDMD is mainly involved in PRV infection-induced IL-1β secretion in mice.
GSDMD and GSMDE are important substrates of inflammatory caspases involved in canonical and noncanonical inflammatory responses. To investigate whether GSDMD and GSDME are involved in the release of inflammatory cytokines induced by PRV infection in mice, mouse peritoneal macrophages derived from WT, Gsdmd−/−, and Gsdme−/− mice (Fig. 7G to I) were mock infected or infected with PRV at MOI of 1 for 24 h. As shown in Fig. 11A to C, knockout of the Gsdmd or Gsdme gene did not affect the transcription level of pro-IL-1β, whereas knockout of the Gsdmd gene obviously decreased the IL-1β levels in the cell supernatants, suggesting that GSDMD may be related to IL-1β secretion in the process of PRV infection in mice. In addition, we noticed that PRV infection positively regulated the expression and cleavage of GSDMD (Fig. 11D). Consistently, the in vivo results showed that, compared to those of WT and Gsdme−/− mice, IL-1β and IL-18 levels in serum from Gsdmd−/− mice were significantly decreased upon PRV infection for 24 and 48 h, indicating that the secretion of IL-1β and IL-18 induced by PRV was mainly dependent on the activation of GSDMD in mice (Fig. 11E and F). Interestingly, knockout of the Gsdmd gene affected the replication of PRV in the spleen, lung, and brain in mice (Fig. 11G). Taken together, our results reveal that during PRV infection, GSDMD acts as a main executor and is activated to promote the release of IL-1β and IL-18 in mice.
FIG 11.

GSDMD acts as an executor to promote the release of IL-1β and IL-18 induced by PRV in vitro and in vivo. (A to D) Mouse peritoneal macrophages were isolated from WT, Gsdmd−/−, and Gsdme−/− mice and infected with PRV at MOI of 1 for 24 h. The transcription level of pro-IL-1β and the expression and secretion level of IL-1β were measured with qPCR (A), Western blotting (B), and ELISA (C), respectively. (D) The expression and activation (GSDMD-N fragment) of GSDMD were detected by Western blotting. (E, F) WT, Gsdmd−/−, and Gsdme−/− mice were mock infected (n = 3) or infected with PRV (n = 3) at 2,000 PFU/mouse for 24 h and 48 h, and the secretion levels of IL-1β and IL-18 in the serum were detected by ELISA. (G) The PRV DNA copy number in the tissues was detected with qPCR. Data are representative of three independent experiments with three biological replicates (mean ± SD in panels A, C, E to G; two-way ANOVA in panels A, C, and E to G). *, P < 0.05; **, 0.001 < P < 0.01; ***, P < 0.001; ns, no significance.
DISCUSSION
PR is considered a reemerging infectious disease, consistently threatening the livestock industry in some countries. In 2010, epidemics of highly pathogenic PRV variants caused huge economic losses, which led to great difficulties in the prevention and control of PRV. Recently, some of these PRV variants were associated with rare cases of presumed PRV infection in humans and reportedly associated with severe respiratory symptoms, neurological symptoms, and even death (4, 27), suggesting that some of these PRV variants possibly pose a threat to public health. In this study, we found that PRV infection induced the release of inflammatory cytokines and severe inflammatory cell infiltration in the lung of infected mice. Mechanistically, in mice, we elucidated that PRV infection enhanced the expression of pro-IL-1β and pro-IL-18 by targeting the TLR2-TLR3-TRL4-TLR5-NF-κB axis. Using knockout mice as a model, we found that the AIM2 inflammasome was activated by PRV infection to drive the maturation and secretion of IL-1β and IL-18 in mice, which was mainly dependent on the activation of GSDMD (Fig. 12).
FIG 12.
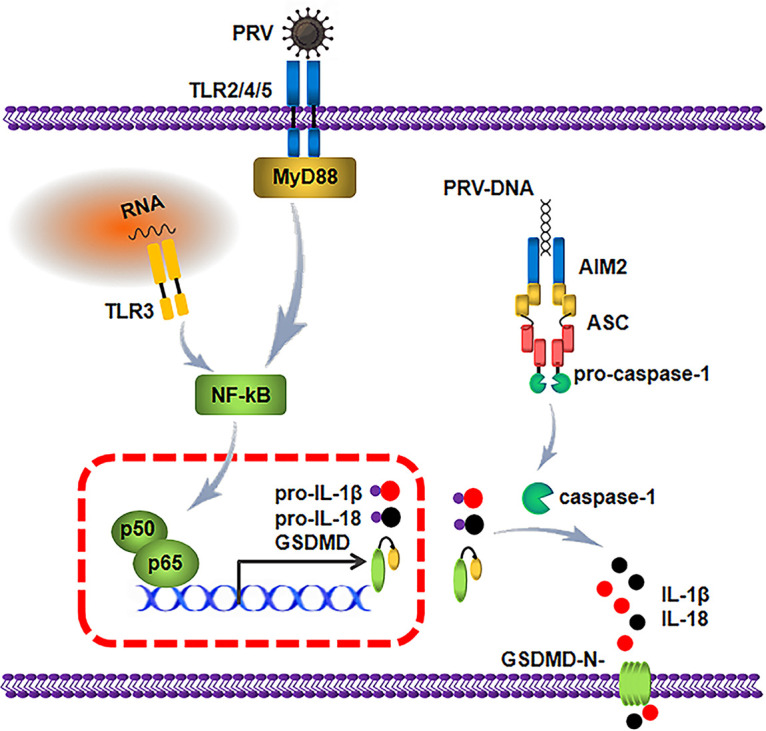
Schematic model showing the mechanism by which PRV infection induces the inflammatory response. During PRV infection and replication in mice, the TLR2-TLR3-TRL4-TLR5-NF-κB axis is activated to promote p65 translocation to the nucleus, which enhances the expression of pro-IL-1β, pro-IL-18 and GSDMD. Additionally, PRV genomic DNA is captured by AIM2, which results in the assembly of the AIM2 inflammasome by recruiting ASC and pro-caspase-1. The active caspase-1 cleaves GSDMD, pro-IL-1β, and IL-18 to secrete the biologically active IL-1β and IL-18.
IL-1 family members, most notably IL-1β and IL-18, play very important roles in host antiviral defense. IL-1β is largely responsible for acute phase responses, which include fever, anorexia, somnolence, and induction of Th17 bias in the cellular adaptive responses (12). IL-18 is essential for the induction of IFN-γ production and Th1 responses (14). Notably, these cytokines activate neutrophils and macrophages to phagocytose the invading pathogen and to release toxic oxygen, nitrogen radicals, and IFNs to eliminate the pathogen (28, 29). Previous studies have demonstrated that the TLR-NF-κB axis is involved in inducing the production of inflammatory cytokines upon viral infection by recognizing a variety of conserved microbial PAMPs to activate intracellular signaling pathways (30). Ye et al. reported that PRV infection upregulated the mRNA levels of pro-IL-1β and TNF-α through NF-κB signaling (26). Consistently, we found that PRV infection augmented the amount of NF-κB in the nucleus in a dose-dependent manner, in line with the notion that NF-κB could be activated upon PRV infection (24).
Each TLR has a specific function, and different TLRs may be involved in NF-κB activity and pro-IL-1β upregulation through different mechanisms (5, 31). For example, the innate resistance to alphaherpesvirus HSV-1 is mediated by MyD88 and activated by multiple TLRs (TLR2, TLR3, TLR7, and TLR9) (32). Cytomegalovirus (CMV), a double-stranded DNA virus with a capsule, can activate several TLRs (TLR2/CD14, TLR3, TLR9) (31). In addition to members of Herpesviridae, HCV was also reported to mediate the natural immune response process through TLR2, TLR3, TLR4, and TLR7 (31). The majority of TLRs sense pathogen components on the cell surface, whereas TLR3, TLR7, TLR8, and TLR9 play a critical role in the recognition of nucleic acids (33). TLR2 and TLR4 recognize many different microbial components of bacteria, fungi, and protozoa, as well as envelope proteins of viruses (such as human cytomegalovirus [HCMV], vaccinia virus [VACV], RSV, and HSV-1). TLR3 and TLR7/8 are usually present in endosomal compartments, where they sense double-stranded RNA (dsRNA) and single-stranded RNA (ssRNA) of some RNA viruses, respectively (34). TLR9 is essential for recognizing viral DNA and synthetic oligodeoxynucleotides containing unmethylated CpG dinucleotides (CpG DNA) (35). In our study, the use of specific inhibitors and siRNAs targeting Tlr2, Tlr3, Tlr4, Tlr5, and Myd88 genes significantly suppressed the expression and secretion of IL-1β in mouse peritoneal macrophages. In accordance with these results, overexpression of TLR2, TLR3, TLR4, and TLR5 enhanced the reporter’s activity of NF-κB and IL-1β expression during PRV infection. These results suggest that TLR2, TLR3, TLR4, TLR5, and MyD88 signaling play important roles in enhancing pro-IL-1β expression upon PRV infection. As a DNA virus, PRV infection can activate TLR3 in mice to induce the production of pro-IL-1β. Since TLR3 not only recognizes dsRNA from the genome of RNA viruses but also senses the viral RNA generated during the life cycle of DNA viruses, we propose that it is the viral RNA generated during PRV infection that activates TLR3, leading to the induction of inflammatory cytokines. Similar to our study, the infection of the TLR3-expressing human postmitotic neuron-derivative cell line NT2-N with HSV-1 triggers IL-6 and IFN regulatory factor 1 (IRF1) mRNA production (36). Furthermore, TLR5 could mediate the signal induced by flagellin (37). To date, few studies have reported the recognition of viral components by TLR5. Our study provides additional evidence that TLR5 is required for inducing the upregulated expression of IL-1β after PRV infection, which expands our understanding of TLR5 function. Additionally, we found that PRV infection triggered TLR2 and TLR4 activation to generate pro-IL-1β and enhance the inflammatory response, which may be dependent on the envelope protein of PRV. The detailed molecular mechanisms need to be further explored. It should be noted that the use of knockout mice deficient in TLR2, TLR3, TLR4, TLR5, and Myd88 could make our results more convincing. Unfortunately, we could not successfully obtain all knockout mice.
Viral infection induces inflammasome activation to release inflammatory cytokines and chemokines, which activate host antiviral immune responses. It was reported that many inflammasomes, including NLRP3, NLRC4, NLRP1b, and IFI16, were involved in inflammatory responses after viral infection. Increasing evidence has shown that AIM2 plays key roles in atherosclerosis (38), shaping neurodevelopment (11) and sensing viral DNA to switch on antiviral innate immunity (39). The NLRP3 inflammasome is activated upon exposure to whole pathogens as well as a number of structurally diverse PAMPs, DAMPs, and environmental irritants. It was reported that both RNA viruses, such as SeV, RSV, VSV, and HCV, and some DNA viruses, including ADV and HSV-1, could also activate the NLRP3 inflammasome (12, 13). Previous studies have shown that PRV infection produces many inflammatory cytokines, including IL-1β, IL-6, TNF-α, and MCP-1 (23). However, the mechanism by which PRV infection induces inflammatory cytokine secretion is not clear. Recently, (26) found that PRV infection could not directly induce inflammasome activation, whereas the NLRP3 inflammasome was activated for IL-1β maturation in mouse macrophages treated with PRV and ATP (26). In this study, to determine which inflammasome was mainly involved in PRV-induced inflammatory responses in mice, the ASC antibody was used to perform immunoprecipitation-mass spectrometry (IP-MS)-based experiments. We found that only AIM2 was involved in the PRV-induced inflammatory response in mice (data not shown). Furthermore, we demonstrated that the maturation and secretion of IL-1β and IL-18 were dependent on the activation of the AIM2 inflammasome in vitro and in vivo. Compared to WT and Nlrp3−/−, knockout of Aim2, Asc, and caspase-1 genes significantly decreased IL-1β production and increased PRV replication. Consistent with these results, we observed more inflammatory cell damage in the lung of Aim2−/− mice. Combining our results and previous relevant conclusions, we propose that the AIM2 inflammasome enhances the upregulation of the IL-1β secretion during PRV infection, which may be further enhanced by ATP.
Notably, humans, rodents, and horses are the only species that possess AIM2, whereas porcine cannot express AIM2 (40, 41). In this study, we want to figure out how PRV infection activates inflammatory responses in mice by using several knockout mice as models. These data of the current manuscript cannot be extrapolated to the pigs. In our previous study, we found that the mRNA levels of pro-IL-1β in the tissues of PRV-infected pigs were also significantly upregulated, but PRV infection failed to induce the secretion of IL-1β in pig serum (42). Notably, we found that the expression of NLRP3 was positively regulated in PRV-infected mouse peritoneal macrophages and RAW 264.7 cells (see Fig. S1 in the supplemental material), suggesting that the upregulation of NLRP3 induced by PRV infection may play a significant role, although it is not involved in IL-1β secretion. Further work will focus on this axis to explore the function of NLRP3 and its molecular mechanisms in the process of PRV infection in pigs and mice.
In this study, we noticed that UV-inactivated PRV could not induce the expression of pro-IL-1β or the secretion of IL-1β, suggesting that the PRV replication process was required for PRV infection-induced inflammatory responses, as PRV replication produces large amounts of viral DNA, which may be recognized by cytosolic DNA sensors. Therefore, we proposed that transfection of PRV genomic DNA could be recognized by AIM2 in mouse peritoneal macrophages, which induced AIM2 inflammasome activation and IL-1β maturation and secretion. As expected, we found that AIM2 could recognize PRV genomic DNA without sequence specificity and induced AIM2 inflammasome activation. Notably, both 5′-UTR and 3′-UTR transcripts of the PRV genome are sufficient to trigger AIM2 inflammasome activation but only induce a few transcriptions of pro-IL-1β and the secretion of IL-1β. In general, we propose that the PRV replication process is essential for the activation of the TLR2-TLR3-TRL4-TLR5-NF-κB axis and AIM2 inflammasome to induce the transcription, expression, and secretion of IL-1β.
Pyroptosis is characterized by the activation of inflammatory caspases. Gasdermin family members, such as GSDMD, a critical mediator of pyroptosis, can be cleaved within a linker between the N-terminal domain and the C-terminal domain by inflammatory-related caspases (19, 20). Subsequently, the N-terminal domain of GSDMD (GSDMD-N1–275) oligomerizes to be inserted into the plasma membrane, resulting in pore formation and the loss of osmotic homeostasis, leading to cell swelling and death. This process also participates in the release of proinflammatory cytokines, including IL-1β and IL-18 (43). Released IL-1β and IL-18 affect innate immunity and adaptive immunity, which contribute greatly to host antiviral responses and the development of autoimmune and inflammatory diseases (44). In this study, we found that GSDMD was upregulated and cleaved in PRV-infected mouse peritoneal macrophages, which was required for PRV infection-induced IL-1β secretion in vitro and in vivo. Thus, we propose that PRV infection-induced AIM2 inflammasome activation promotes caspase-1 activation, resulting in GSDMD cleavage and IL-1β secretion.
MATERIALS AND METHODS
Plasmids.
The NF-κB reporter and TK-Renilla reporter were obtained from Hong Tang. To construct plasmids expressing Flag-tagged NLRP3, IL-1β, GSDMD, GSDME, TLRs, and hemagglutinin (HA)-tagged AIM2 and caspase-1, the cDNAs corresponding to these proteins were amplified by standard reverse transcription-PCR (RT-PCR) using total RNA extracted from mouse peritoneal macrophages as the templates and then cloned into the pCAGGS-HA and pCAGGS-Flag vectors, respectively. All constructs were validated by DNA sequencing. The primers used in this study are available upon request (Table 2).
TABLE 2.
PCR primers used for constructing the plasmids
| Plasmid | Primer (5′–3′) |
|---|---|
| Flag-Nlrp3 | F, GATGACGACGATAAGGAATTCATGACGAGTGTCCGT |
| R, ATTAAGATCTGCTAGCTCGAGCTACCAGGAAATCTC | |
| Flag-IL-1β | F, GATGACGACGATAAGGAATTCATGGCAACTGTTCCT |
| R, ATTAAGATCTGCTAGCTCGAGTTAGGAAGACACGGA | |
| Flag-Gsdmd | F, GATGACGACGATAAGGAATTCATGCCATCGGCCTTTGAG |
| R, ATTAAGATCTGCTAGCTCGAGCTAACAAGGTTTCTGGCC | |
| Flag-Gsdme | F, GATGACGACGATAAGGAATTCATGTTTGCCAAAGCAACT |
| R, ATTAAGATCTGCTAGCTCGAGCTAGTCTTGACCTGTAGC | |
| Flag-Tlr2 | F, GATGACGACGATAAGGAATTCATGCTACGAGCTCTTTGG |
| R, ATTAAGATCTGCTAGCTCGAGCTAGGACTTTATTGCAGT | |
| Flag-Tlr3 | F, GATGACGACGATAAGGAATTCATGAAAGGGTGTTCCTCT |
| R, ATTAAGATCTGCTAGCTCGAGTTAATGTGCTGAATTCCG | |
| Flag-Tlr4 | F, GATGACGACGATAAGGAATTCATGATGCCTCCCTGGCTC |
| R, ATTAAGATCTGCTAGCTCGAGTCAGGTCCAAGTTGCCGT | |
| Flag-Tlr5 | F, GATGACGACGATAAGGAATTCATGGCATGTCAACTTGAC |
| R, ATTAAGATCTGCTAGCTCGAGCTAGGAAATGGTTGCTAT | |
| Flag-Tlr9 | F, GATGACGACGATAAGGAATTCATGGTTCTCCGTCGAAGG |
| R, ATTAAGATCTGCTAGCTCGAGTTCTGCTGTAGGTCCCCG | |
| HA-Aim2 | F, GTTCCAGATTACGCTGAATTCATGGAGAGTGAGTACCGG |
| R, ATTAAGATCTGCTAGCTCGAGTCACTCCACACTTTTCAT | |
| HA-Caspase-1 | F, GTTCCAGATTACGCTGAATTCATGGCTGACAAGATC |
| R, ATTAAGATCTGCTAGCTCGAGTTAATGTCCCGGGAA |
Cell lines and viruses.
Human HEK293T cells were purchased from American Type Culture Collection (Manassas, VA) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 mg/mL streptomycin at 37°C with 5% CO2. Mouse peritoneal macrophages were isolated from mice after injection of thioglycolate (Merck) and cultured in RPMI 1640 medium supplemented with 10% FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin at 37°C with 5% CO2. The PRV-TJ strain was kindly provided by Tongqing An (Harbin Veterinary Research Institute [HVRI], China).
Antibodies and chemical reagents.
Anti-NLRP3 (catalog no. ab263899), anti-AIM2 (catalog no. ab119791), anti-caspase-1 (procaspase +p10) (catalog no. ab179515), anti-IL-1β (catalog no. ab234437), anti-GSDMD (catalog no. ab209845), and anti-cleaved N-terminal GSDMD (catalog no. ab215203) antibodies were purchased from Abcam Biotechnology. The anti-Flag tag monoclonal antibody (catalog no. 66008-4-Ig), anti-HA tag monoclonal antibody (catalog no. 66006-2-Ig), anti-GAPDH (catalog no. 60004-1-Ig), anti-lamin B1 (catalog no. 12987-1-AP), anti-ASC (catalog no. 7494-1-Ig), and anti-phospho-NF-κB p65 (CST; catalog no. 3033S) were purchased from Proteintech and Cell Signaling Technology, respectively. DAPI (4′,6-diamidino-2-phenylindole; Sigma-Aldrich; catalog no. D9542), lipopolysaccharide (LPS; Beyotime; catalog no. S1732), ATP (MedChemExpress; catalog no. HY-B2176), AN-3485 (MedChemExpress; catalog no. HY-18325), T6167923 (MedChemExpress; catalog no. HY-19744), hydroxychloroquine sulfate (Selleckchem; catalog no. S4430), JSH-23 (ApexBio; catalog no. B1645), Belnacasan (VX-765) (Selleckchem; catalog no. S2228), Caspase-Glo 1 inflammasome assay (Promega; catalog no. G9951), and Alexa Fluor 488-labeled goat anti-rabbit IgG (Thermo Fisher Scientific; catalog no. A11008) were purchased from the corresponding technology companies.
Mice.
Specific-pathogen-free (SPF) C57BL/6 mice were purchased from Liaoning Changsheng Biotechnology Co., Ltd. (Liaoning, China). Nlrp3−/−, Asc−/−, and Caspase-1−/− mice (C57BL/6), generated by homologous recombination technology, were purchased from Saiye Biotechnology Co., Ltd. (Guangzhou, China). Aim2−/− mice were provided by Yongjun Yang. Gsdmd−/− and Gsdme−/− mice were provided by Feng Shao. The genotypes of these mice were confirmed by PCR using the primers listed in Table 3 and Western blot analysis. The knockout mice and their wild-type littermates (6 to 8 weeks old) were used throughout the experiments. All mice were generated and housed in SPF barrier facilities at the HVRI of the Chinese Academy of Agricultural Sciences (CAAS) (Harbin, China). All animal experiments were performed according to animal protocols approved by the Subcommittee on Research Animal Care at the HVRI.
TABLE 3.
PCR primers used for identifying the genotypes of mice
| Genotype | Primer (5′–3′) |
|---|---|
| Nlrp3−/− | F, GTTTTCATTCCTGCACTGCCAGTG |
| R, CAAAAACCCTTCTGTTTACTCACTC | |
| Asc−/− | F, GCTGACTTCCTGGTCTTG |
| R, CCACGGTGCTAAGTTCAA | |
| Caspase-1−/− | F, GCTGTATGTGAAAGGGACATTTTGC |
| R, CTGCCAGGTAGCAGTCTTCATTAC | |
| Aim2−/− | F, GTTTGGCTCAGAAATGTCCAG |
| R1, CCCACATAACCTGGGATTAGTT | |
| R2, AAGGGTCTTTGAGCACCAGA | |
| Gsdmd−/− | F, TGGCAGTGGTGGAAGACTCTAC |
| R, GATATCTGGTAGCAGGCAGAAGCAAG | |
| Gsdme−/− | F, CCATTACTGTGGCTAAAGAGGGG |
| R1, TCCTAAACTCCTGCGGAAGACA | |
| R2, GCCTAGCTTTGAAGTCTAATGTTGTCCAG |
Western blot analysis.
The cells were harvested in lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 1% Triton X-100, and 10% glycerol) supplemented with protease and phosphatase inhibitor phenylmethylsulfonyl fluoride (PMSF). Protein samples were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore), which were blocked with 5% nonfat milk (Biosharp) in Tris-buffered saline with Tween 20 (TBST) for 2 h at room temperature. Incubation with primary antibody (1:1,000) was performed at 4°C overnight and then incubated with goat anti-mouse IRDye 800CW or goat anti-rabbit IRDye 800CW (Odyssey) for 1 h at room temperature. Immunoblot results were visualized with an Odyssey 2-color infrared fluorescence imaging system (Odyssey CL, USA).
ELISA.
The concentrations of these inflammatory cytokines, including IL-1β, IL-6, IL-18, and TNF-α, in cell culture supernatant and the serum of mice were measured using the ELISA kits according to the manufacturer’s instructions. ELISA kits for IL-1β, IL-6, and IL-18 were purchased from Abclonal Technology, and ELISA kits for TNF-α were purchased from Thermo Fisher Scientific.
Cell viability assay.
Lactate dehydrogenase (LDH) assays were performed to determine cell viability. Mouse peritoneal macrophages, isolated from WT or knockout mice, were treated with different inhibitors of TLRs, Myd88, or inflammasomes. The cell culture supernatant was harvested and cleared by centrifugation at 2,000 × g for 5 min, and 50 μL was used to perform the LDH assay according to the manufacturer’s instructions (absorption at 490 and 680 nm for correction).
Confocal microscopy analysis.
Mouse peritoneal macrophages were fixed for 30 min in 4% paraformaldehyde in 1× phosphate-buffered saline (PBS; pH 7.4) after being infected with PRV for 24 h. Then, the fixed cells were permeabilized for 15 min with 0.3% Triton X-100 in 1× PBS and then blocked in 1× PBS with 10% bovine serum for 60 min. The cells were incubated with ASC or NF-κB antibodies and then stained with Alexa Fluor 488-labeled goat anti-rabbit IgG. ASC oligomerization or the subcellular localization of NF-κB was visualized using a Zeiss LSM 980 laser scanning fluorescence microscope (Carl Zeiss AG, Oberkochen, Germany) under a 63× oil objective. Zeiss processing system software was used to determine the positive rate (%) of NF-κB localized in the cellular nucleus.
DNA pulldown assay.
293T cells were transfected with pCAGGS-Flag-Nlrp3 or pCAGGS-HA-Aim2 and harvested 24 h posttransfection (hpt). The cells were lysed in lysis buffer containing PMSF. Ten micrograms of PRV genomic DNA were prepared and biotinylated according to the manufacturer’s instructions (Novagen) and then incubated with cell lysates at 4°C for 12 h. Streptavidin-Sepharose beads (Novagen) were then added and rotated at 4°C for 4 h. Afterward, the beads were washed extensively with PBS and analyzed by immunoblotting with the indicated antibodies.
RNA interference.
Mouse peritoneal macrophages were transfected with the indicated siRNA (Gene Pharma) for 48 h at a final concentration of 120 pM. Transfections were performed with GP-transfect-Mate reagent (Gene Pharma) according to the manufacturer’s instructions in Opti-MEM medium. The Opti-MEM medium was replaced with RPMI medium (Sigma) containing 10% FBS after 12 hpt. The knockdown efficiency was assessed by qPCR after 48 hpt. The siRNA sequences of Tlr genes and Myd88 are shown in Table 4. Mouse peritoneal macrophages were transfected with negative-control siRNA (siNC) or the indicated siRNAs for 48 h and then infected with PRV (MOI = 1). After infection for another 12 h, the total RNA or cell lysates were harvested and assessed.
TABLE 4.
siRNA sequences of Tlrs and Myd88 genes
| Gene | siRNA sequence |
|---|---|
| Tlr2 | GCCUUGACCUGUCUUUCAATT |
| UUGAAAGACAGGUCAAGGCTT | |
| Tlr3 | CACUCCACAUCAUUAUUAUTT |
| AUAAUAAUGAUGUGGAGUGTT | |
| Tlr4 | GCUAUAGCUUCUCCAAUUUTT |
| AAAUUGGAGAAGCUAUAGCTT | |
| Tlr5 | GACCAAACAUUCAGAUUAUTT |
| AUAAUCUGAAUGUUUGGUCTT | |
| Tlr7 | GCCGGUGAUAACAGAUACUTT |
| AGUAUCUGUUAUCACCGGCTT | |
| Tlr8 | CUGGAAGACAACCAGUUAUTT |
| AUAACUGGUUGUCUUCCAGTT | |
| Tlr9 | CCGCAAGAAGGUAUCCUUUTT |
| AAAGGAUACCUUCUUGCGGTT | |
| Myd88 | GCCUAUCGCUGUUCUUGAATT |
| UUCAAGAACAGCGAUAGGCTT |
RNA extraction and qPCR.
For detection of the mRNA levels of Il-1β, Il-6, Il-18, Tnf-α, Tlrs, and Myd88 genes, total RNA was extracted using TRIzol reagent (Invitrogen), and the reverse transcription products were amplified with a PrimeScript RT reagent kit (TaKaRa). Reverse transcription products were amplified with TB Green premix Ex Taq II (TaKaRa) according to the manufacturer’s instructions. The mRNA levels of these genes were normalized to Gapdh (glyceraldehyde-3-phosphate dehydrogenase). The final mRNA levels of genes in this study were calculated using the comparative cycle threshold method. The qPCR primers are listed in Table 5.
TABLE 5.
Primers used for qPCR amplification of the mouse genes
| Gene | Sequence (5′→3′) |
|---|---|
| m-Gapdh | F, TGGCCTTCCGTGTTCCTAC |
| R, GAGTTGCTGTTGAAGTCGCA | |
| m-Il-1β | F, GAAATGCCACCTTTTGACAGTG |
| R, TGGATGCTCTCATCAGGACAG | |
| m-Il-6 | F, ACAAAGCCAGAGTCCTTCAGA |
| R, TCCTTAGCCACTCCTTCTGT | |
| m-Il-18 | F, GACAGCCTGTGTTCGAGGATATG |
| R, TGTTCTTACAGGAGAGGGTAGAC | |
| m-Tnf-α | F, ACTGAACTTCGGGGTGATCG |
| R, TCTTTGAGATCCATGCCGTTG | |
| m-Tlr2 | F, ACAGCAAGGTCTTCCTGGTTCC |
| R, GCTCCCTTACAGGCTGAGTTCT | |
| m-Tlr3 | F, GTCTTCTGCACGAACCTGACAG |
| R, TGGAGGTTCTCCAGTTGGACCC | |
| m-Tlr4 | F, AGCTTCTCCAATTTTTCAGAACTTC |
| R, TGAGAGGTGGTGTAAGCCATGC | |
| m-Tlr5 | F, TCCTGACCAGAGCACATTTGCC |
| R, CCTTCAGTGTCCCAAACAGTCG | |
| m-Tlr7 | F, GTGATGCTGTGTGGTTTGTCTGG |
| R, CCTTTGTGTGCTCCTGGACCTA | |
| m-Tlr8 | F, AAGTGCTGGACCTGAGCCACAA |
| R, CCTCTGTGAGGGTGTAAATGCC | |
| m-Tlr9 | F, GCTGTCAATGGCTCTCAGTTCC |
| R, CCTGCAACTGTGGTAGCTCACT | |
| m-Myd88 | F, ACCTGTGTCTGGTCCATTGCCA |
| R, GCTGAGTGCAAACTTGGTCTGG |
For PRV genome copies, glycoprotein B (gB)-specific absolute quantification PCR was performed. PRV genomic DNA was extracted from the cells, tissues, or EDTA-treated whole peripheral blood by using the universal genomic DNA kit (Axygen) and applied for gB-specific absolute quantification PCR to precisely detect PRV genome copies. The primers and TaqMan probe sequences are listed in Table 6. The final PRV genome copies were calculated based on the standard curve established with the recombinant pCAGGS-Flag-gB plasmid as the template and presented as the mean ± standard deviation (SD) from three replicated wells.
TABLE 6.
Primers and TaqMan probe sequences used in this study
| Primer name | Sequence (5′→3′) |
|---|---|
| gB-probe-F | ACGGCACGGGCGTGATC |
| gB-probe-R | ACTCGCGGTCCTCGAGCA |
| gB-TaqMan-probe | FAM-CTCGCGCGACCTCATCGAGCCCTGCAC-MGB |
Viral infection.
For mouse infection, 8- to 10-week-old and sex-matched Nlrp3−/−, Aim2−/−, Gsdmd−/−, and Gsdme−/− mice and their WT littermates were intraperitoneally injected with PRV (2,000 PFU/mouse). For survival experiments, the survival of mice was monitored every day after PRV infection. Serum from PRV-infected mice was collected for ELISA analysis 24 h postinfection (hpi), and the tissues (spleen, lung, and brain) were collected for qPCR, PRV genome copies, or histological analysis.
Histological analysis.
The tissues of mice infected with PRV were fixed in a 10% formalin neutral buffer solution overnight. Histological analysis of tissue damage was assessed by standard hematoxylin and eosin (H&E) staining. The results were analyzed by light microscopy. Representative views of the tissue sections are shown.
Statistical analysis.
All data were analyzed by Prism software (GraphPad; version 8.0). Statistical analysis was performed by unpaired two-tailed Student’s t test for two-group comparisons, log-rank test for survival experiments, or one-way analysis (ANOVA) of Dunnett’s multiple-comparison test for comparisons of more than two groups or two-way ANOVA for comparisons of more than two groups with two or more time points. Statistical significance was determined with P values of <0.05 were considered to be statistically significant, where *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ns, no significance.
Ethics statement.
All animal experiments were performed according to animal protocols approved by the Subcommittee on Research Animal Care at the HVRI and carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the Ministry of Science and Technology of the People’s Republic of China.
ACKNOWLEDGMENTS
We thank Feng Shao from Peking University for generating the Gsdmd−/− and Gsdme−/− mice. We also thank Yongjun Yang from Jilin University for generating the Aim2−/− mice.
This study was supported by the State Key Laboratory of the Veterinary Biotechnology Program (SKLVBP202101) and the Natural Science Foundation of Heilongjiang Province of China (grant no. C2016061).
We declare no conflict of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Li Huang, Email: highlight0315@163.com.
Changjiang Weng, Email: wengchangjiang@caas.cn.
Jae U. Jung, Lerner Research Institute, Cleveland Clinic
REFERENCES
- 1.Sehl J, Teifke JP. 2020. Comparative pathology of pseudorabies in different naturally and experimentally infected species—a review. Pathogens 9:633. 10.3390/pathogens9080633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanson RP. 1954. The history of pseudorabies in the United States. J Am Vet Med Assoc 124:259–261. [PubMed] [Google Scholar]
- 3.Ai JW, Weng S, Cheng Q, Cui P, Li YJ, Wu HL, Zhu YM, Xu B, Zhang WH. 2018. Human endophthalmitis caused by pseudorabies virus infection, China, 2017. Emerg Infect Dis 24:1087–1090. 10.3201/eid2406.171612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Q, Wang X, Xie C, Ding S, Yang H, Guo S, Li J, Qin L, Ban F, Wang D, Wang C, Feng L, Ma H, Wu B, Zhang L, Dong C, Xing L, Zhang J, Chen H, Yan R, Wang X, Li W. 2021. A novel human acute encephalitis caused by pseudorabies virus variant strain. Clin Infect Dis 73:e3690–e3700. 10.1093/cid/ciaa987. [DOI] [PubMed] [Google Scholar]
- 5.Barton G, Medzhitov R. 2003. Toll-like receptor signaling pathways. Science 300:1524–1525. 10.1126/science.1085536. [DOI] [PubMed] [Google Scholar]
- 6.Medzhitov R, Preston-Hurlburt PP, Janeway CAJ. 1997. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388:394–397. 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 7.Matsumoto F, Saitoh SI, Fukui R, Kobayashi T, Miyake K. 2003. Mammalian Toll-like receptors. Curr Opin Immunol 15:5–11. [DOI] [PubMed] [Google Scholar]
- 8.Schroder K, Tschopp J. 2010. The inflammasomes. Cell 140:821–832. 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 9.Wang W, Hu D, Wu C, Feng Y, Li A, Liu W, Wang Y, Chen K, Tian M, Xiao F, Zhang Q, Shereen MA, Chen W, Pan P, Wan P, Wu K, Wu J. 2020. STING promotes NLRP3 localization in ER and facilitates NLRP3 deubiquitination to activate the inflammasome upon HSV-1 infection. PLoS Pathog 16:e1008335. 10.1371/journal.ppat.1008335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choudhury SM, Ma XS, Abdullah SW, Zheng HX. 2021. Activation and inhibition of the NLRP3 inflammasome by RNA viruses. J Inflamm Res 14:1145–1163. 10.2147/JIR.S295706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lammert CR, Frost EL, Bellinger CE, Bolte AC, McKee CA, Hurt ME, Paysour MJ, Ennerfelt HE, Lukens JR. 2020. AIM2 inflammasome surveillance of DNA damage shapes neurodevelopment. Nature 580:647–652. 10.1038/s41586-020-2174-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C. 2009. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 30:576–587. 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dinarello CA. 1996. Biologic basis for interleukin-1 in disease. Blood 87:2095–2147. 10.1182/blood.V87.6.2095.bloodjournal8762095. [DOI] [PubMed] [Google Scholar]
- 14.Dinarello C. 1999. IL-18: a TH1-inducing, proinflammatory cytokine and new member of the IL-1 family. J Allergy Clin Immunol 103:11–24. 10.1016/s0091-6749(99)70518-x. [DOI] [PubMed] [Google Scholar]
- 15.Tamura M, Tanaka S, Fujii T, Aoki A, Komiyama H, Ezawa K, Sumiyama K, Sagai T, Shiroishi T. 2007. Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics 89:618–629. 10.1016/j.ygeno.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, Junqueira C, Meza-Sosa KF, Mok TMY, Ansara J, Sengupta S, Yao Y, Wu H, Lieberman J. 2020. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579:415–420. 10.1038/s41586-020-2071-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, Wang Y, Li D, Liu W, Zhang Y, Shen L, Han W, Shen L, Ding J, Shao F. 2020. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 368:eaaz7548. 10.1126/science.aaz7548. [DOI] [PubMed] [Google Scholar]
- 18.Hou J, Zhao R, Xia W, Chang C-W, You Y, Hsu J-M, Nie L, Chen Y, Wang Y-C, Liu C, Wang W-J, Wu Y, Ke B, Hsu JL, Huang K, Ye Z, Yang Y, Xia X, Li Y, Li C-W, Shao B, Tainer JA, Hung M-C. 2020. Author correction: PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol 22:1396. 10.1038/s41556-020-00599-1. [DOI] [PubMed] [Google Scholar]
- 19.Li S, Wu Y, Yang D, Wu C, Ma C, Liu X, Moynagh PN, Wang B, Hu G, Yang S. 2019. Gasdermin D in peripheral myeloid cells drives neuroinflammation in experimental autoimmune encephalomyelitis. J Exp Med 216:2562–2581. 10.1084/jem.20190377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao J, Wang C, Yao J-C, Alippe Y, Xu C, Kress D, Civitelli R, Abu-Amer Y, Kanneganti T-D, Link DC, Mbalaviele G. 2018. Gasdermin D mediates the pathogenesis of neonatal-onset multisystem inflammatory disease in mice. PLoS Biol 16:e3000047. 10.1371/journal.pbio.3000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. 2017. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun 8:14128. 10.1038/ncomms14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun W, Liu S, Huang X, Yuan R, Yu J. 2021. Cytokine storms and pyroptosis are primarily responsible for the rapid death of mice infected with pseudorabies virus. R Soc Open Sci 8:210296. 10.1098/rsos.210296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ren CZ, Hu WY, Zhang JW, Wei YY, Yu ML, Hu TJ. 2021. Establishment of inflammatory model induced by Pseudorabies virus infection in mice. J Vet Sci 22:e20. 10.4142/jvs.2021.22.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Romero N, Waesberghe CV, Favoreel HW. 2020. Pseudorabies virus infection of epithelial cells leads to persistent but aberrant activation of the NF-κB pathway, inhibiting hallmark NF-κB-induced proinflammatory gene expression. J Virol 94:e00196-20. 10.1128/JVI.00196-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Romero N, Favoreel HW. 2021. Pseudorabies virus infection triggers NF-κB activation via the DNA damage response but actively inhibits NF-κB-dependent gene expression. J Virol 95:e01666-21. 10.1128/JVI.01666-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chao Y, Qh A, Jj C, Gang LA, Dx A, Zheng ZC, Lp A, Yp A, Rfa B. 2021. ATP-dependent activation of NLRP3 inflammasome in primary murine macrophages infected by pseudorabies virus. Vet Microbiol 259:109130. 10.1016/j.vetmic.2021.109130. [DOI] [PubMed] [Google Scholar]
- 27.Müller T, Hahn EC, Tottewitz F, Kramer M, Klupp BG, Mettenleiter TC, Freuling C. 2011. Pseudorabies virus in wild swine: a global perspective. Arch Virol 156:1691–1705. 10.1007/s00705-011-1080-2. [DOI] [PubMed] [Google Scholar]
- 28.Taylor PR, Martinez-Pomares L, Stacey M, Lin H-H, Brown GD, Gordon S. 2005. Macrophage receptors and immune recognition. Annu Rev Immunol 23:901–944. 10.1146/annurev.immunol.23.021704.115816. [DOI] [PubMed] [Google Scholar]
- 29.Orange JS, Ballas ZK. 2006. Natural killer cells in human health and disease. Clin Immunol 118:1–10. 10.1016/j.clim.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 30.Kawai T, Akira S. 2007. TLR signaling. Semin Immunol 19:24–32. 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 31.Xagorari A, Chlichlia K. 2008. Toll-like receptors and viruses: induction of innate antiviral immune responses. Open Microbiol J 2:49–59. 10.2174/1874285800802010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RW. 2004. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci USA 101:1315–1320. 10.1073/pnas.0308057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barton GM. 2007. Viral recognition by Toll-like receptors. Semin Immunol 19:33–40. 10.1016/j.smim.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 34.Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford C, Wagner H, Bauer S. 2004. Species-specific recognition of single-stranded RNA via Toll-like receptor 7 and 8. Science 303:1526–1529. 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 35.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. 2000. A Toll-like receptor recognizes bacterial DNA. Nature 408:740–745. 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 36.Prehaud C, Megret F, Lafage M, Lafon M. 2005. Virus infection switches TLR-3-positive human neurons to become strong producers of beta interferon. J Virol 79:12893–12904. 10.1128/JVI.79.20.12893-12904.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A. 2001. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410:1099–1103. 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 38.Fidler TP, Xue C, Yalcinkaya M, Hardaway B, Abramowicz S, Xiao T, Liu W, Thomas DG, Hajebrahimi MA, Pircher J, Silvestre-Roig C, Kotini AG, Luchsinger LL, Wei Y, Westerterp M, Snoeck H-W, Papapetrou EP, Schulz C, Massberg S, Soehnlein O, Ebert B, Levine RL, Reilly MP, Libby P, Wang N, Tall AR. 2021. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature 592:296–301. 10.1038/s41586-021-03341-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, Hardy LL, Garceau V, Sweet MJ, Ross IL, Hume DA, Stacey KJ. 2009. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323:1057–1060. 10.1126/science.1169841. [DOI] [PubMed] [Google Scholar]
- 40.Cridland JA, Curley EZ, Wykes MN, Schroder K, Sweet MJ, Roberts TL, Ragan MA, Kassahn KS, Stacey KJ. 2012. The mammalian PYHIN gene family: phylogeny, evolution and expression. BMC Evol Biol 12:1471–2148. 10.1186/1471-2148-12-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dawson HD, Smith AD, Chen C, Urban JF. 2017. An in-depth comparison of the porcine, murine and human inflammasomes; lessons from the porcine genome and transcriptome. Vet Microbiol 202:2–15. 10.1016/j.vetmic.2016.05.013. [DOI] [PubMed] [Google Scholar]
- 42.Zhou Q, Zhang L, Liu H, Ye G, Huang L, Weng C. 2022. Isolation and characterization of two pseudorabies virus and evaluation of their effects on host natural immune responses and pathogenicity. Viruses 14:712. 10.3390/v14040712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Felderhoff-Mueser U, Sifringer M, Polley O, Dzietko M, Leineweber B, Mahler L, Baier M, Bittigau P, Obladen M, Ikonomidou C, Bührer C. 2005. Caspase-1–processed interleukins in hyperoxia-induced cell death in the developing brain. Ann Neurol 57:50–59. 10.1002/ana.20322. [DOI] [PubMed] [Google Scholar]
- 44.Netea MG, Simon A, van de Veerdonk F, Kullberg B-J, Van der Meer JWM, Joosten LAB. 2010. IL-1β processing in host defense: beyond the inflammasomes. PLoS Pathog 6:e1000661. 10.1371/journal.ppat.1000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Download jvi.00003-23-s0001.docx, DOCX file, 0.1 MB (96.8KB, docx)