

Abstract
The human gut harbors native microbial communities, forming a highly complex ecosystem. Synthetic microbial communities (SynComs) of the human gut are an assembly of microorganisms isolated from human mucosa or fecal samples. In recent decades, the ever-expanding culturing capacity and affordable sequencing, together with advanced computational modeling, started a ‘‘golden age’’ for harnessing the beneficial potential of SynComs to fight gastrointestinal disorders, such as infections and chronic inflammatory bowel diseases. As simplified and completely defined microbiota, SynComs offer a promising reductionist approach to understanding the multispecies and multikingdom interactions in the microbe–host-immune axis. However, there are still many challenges to overcome before we can precisely construct SynComs of designed function and efficacy that allow the translation of scientific findings to patients’ treatments. Here, we discussed the strategies used to design, assemble, and test a SynCom, and address the significant challenges, which are of microbiological, engineering, and translational nature, that stand in the way of using SynComs as live bacterial therapeutics.
Keywords: human gut microbiome, synthetic microbial consortium, live bacterial therapeutic, rational design
Artificial human gut microbiota (synthetic microbial communities or SynComs) are assembled from microorganisms isolated from humans. SynComs offer a simplified model to study microbiome–host interactions. However, challenges remain in precisely constructing SynComs of designed functions and testing their functions. Here, strategies for designing, assembling, and testing SynComs are summarized, and the microbiological, engineering, and translational challenges are discussed.
Introduction
The human gut microbiota harbors microbial communities composed of fungi, bacteria, viruses, and protozoa, of which the microbe–microbe and microbe–host interactions are essential for human health. An imbalance of these interactions, reflected by shifted composition and function, has been associated with diseases. In recent decades, the human gut microbiota has caught substantial attention, progressing from descriptive observations and associations toward causality and mechanistic insights. However, the inherent complexity of the human gut microbiota makes it immensely challenging to establish causality and, subsequently, dissect mechanisms.
One emerging strategy to tackle this challenge is to use simplified, synthetic microbial communities (SynComs). SynComs are consortia of two or more known microbial species under (initially) defined conditions (Großkopf and Soyer 2014). SynComs maintain key features of natural microbial communities and, because of their reduced complexity and defined nature, have been increasingly used as a model system to study functional, ecological, and structural concepts of native microbiota. Construction and application of SynComs have been demonstrated in different contexts, such as crop resiliency (Knoth et al. 2014, Carrión et al. 2019), marine bacteria–plankton interaction (Fu et al. 2020), and inflammatory bowel diseases (van der Lelie et al. 2021).
Rodents, and specifically germ-free mice, are often used in studies where SynComs are used for preclinical testing and to study microbiome–host interactions in vivo. The similarities between mice and men in gut anatomy, histology, and physiology make it a preferred animal model (Hugenholtz and de Vos 2018). Gnotobiotic animal models, born germ-free and inoculated with (a panel of) microorganisms, have provided critical fundamental insights into host–microbiome interactions. However, there are several limitations of germ-free animals: the compromised development of the immune system; failed inoculation of single species, such as the important gut microbe Fecalibacterium prausnitzii(F. prausnitzii) (Miquel et al. 2015); lack of essential microbiota-specific metabolic functions, e.g. secondary bile acid production and short-chain fatty acid production. These limitations pose doubt about the relevance of the findings. In conventionally raised animals, the microbiota can vary among different housing conditions and labs (Laukens et al. 2016, Bär et al. 2020), challenging the reproducibility of the studies. To overcome these challenges regarding reproducibility, SynComs have been created to engraft better in their host (Eberl et al. 2019).
More fundamentally, SynComs can be used to study the function and interactions of microbes and their interaction with the host. The number of species in the human gut, combined with the individual variation, makes it challenging to decipher essential factors for a healthy microbiome (Rinninella et al. 2019, Eisenstein 2020). There is a clear indication that not the exact species but the function of the species (e.g. its metabolism) is essential (Krautkramer et al. 2021); hence, diversity can be understood in fewer (microbe-determined) functions. However, this needs experimental verification of the prospected role of a species in isolation and its function in a consortium with other species. SynComs are the most crucial tool for reproducibly studying the function in a relevant consortium.
Here, we systematically review SynComs of the human gut and the current strategies to construct and test their function. First, we introduce the functions of the gut microbiota that can be studied with SynComs. Next, we discuss the currently used strategies to design, assemble, and validate SynComs. Although all the bacterial species of SynComs discussed in this review are isolated from humans, the host used for testing these SynComs is mostly germ-free animals since; currently, there is no “germ-free” human available. In this regard, we present emerging tools, such as the germ-free miniature gut and in-silico models, that could help tackle the challenges in the design and testing of SynComs.
Functions of native microbial communities
Gut microbiota have many beneficial functions on the human host. Based on the involvement of the host and environment, this review classifies these functions into four categories: co-metabolism, fermentation, eco-resilience, and immune training (Fig. 1). First, co-metabolism implies the microbial metabolism of host-derived molecules. Second, fermentation is the direct production of compounds by microorganisms via the fermentation of dietary compounds. This function does not involve the host directly but does affect the host, especially the production of short-chain fatty acids, branched-chain fatty acids, drug metabolism, and the liberation of polyphenols from the host’s diet. Third, immune training occurs when the microbiota in the gut presents the host immune system with antigens like cell wall components, bacterial DNA, and RNA that prime the immune system. Last, the microbiota supplies eco-resilience to pathogens; this function works against the engraftment and overgrowth of the native microbial community by pathogens.
Figure 1.

The known beneficial effects of the gut microbiota on crucial functions in human health. Circles represent the main functional categories as identified in this review. Arrows with dashed lines indicate a relationship between functions of interest. Functions can have overlapping categories if they are defined in multiple categories.
Host-derived molecules involved in co-metabolism include primary bile acids (Long et al. 2017, Molinero et al. 2019), vitamins (Said and Mohammed 2006, LeBlanc et al. 2013), amino acids (Smith and Macfarlane 1997, Fernández and Zúñiga 2006, Lin et al. 2017), peptides, fatty acid amides and their derivatives, and neurotransmitters (Guo et al. 2017, Louis and Flint 2017, Koopman et al. 2021, Krautkramer et al. 2021). These metabolites can influence the host health directly by interacting with the host cells (Wong et al. 2006, Molinero et al. 2019, Müller et al. 2019, Parada Venegas et al. 2019, Silva et al. 2020) or indirectly by changing the structure and function of the native microbial community (Said and Mohammed 2006, Rossi et al. 2011).
The fermentation function of the microbiota, which is the anaerobic metabolism of substrates, includes the production of small molecules such as short-chain fatty acids (Wong et al. 2006, Bernalier-Donadille 2010). The substrates used for fermentation are exogenous chemicals, most notably components of the plant cell wall, which are comprised primarily of carbohydrate polymers that cannot be hydrolyzed by mammalian cells (Williams et al. 2017). While the host is not involved in the process of fermentation, the products of fermentation can be absorbed and subsequently used by the host to increase host fitness (Meijer et al. 2010, Parada Venegas et al. 2019, Silva, Bernardi and Frozza 2020). Moreover, fermentation products presented to the host have histological effects. It has been shown that fermentation products are critical in regulating cell growth, cell differentiation, and the barrier function of the gut epithelium (Hamer et al. 2008, Heitkemper et al. 2018). For example, compared to conventionally reared animals, the intestinal villi of germ-free mice are more expansive, longer, and show more microvasculature, while the crypts contain fewer cells (Stappenbeck et al. 2002, Hill and Artis 2010). The vascularization and short-chain fatty acid absorption stimulate salt and water uptake in the colon (Wong et al. 2006).
Additionally, gut microbiota can train the immune system (Wu and Wu 2012), which is demonstrated by the maldevelopment of the immune system of germ-free mice, typically showing insufficient maturation of B- and T-cells (Hapfelmeier et al. 2010), fewer lymphoid follicles as well as Peyer’s patches (Cebra et al. 1998, Bouskra et al. 2008, Jung et al. 2010). Mechanistically, there are quite a few ways for microbiota to influence the immune training of the host (reviewed in Hitch et al. 2022). One example of immune training can be attributed to short-chain fatty acids, especially butyrate. Butyrate has an immunomodulatory impact by regulating cell surface receptors and downstream signaling cascades that control inflammation (Meijer et al. 2010, Yang et al. 2018, Parada Venegas et al. 2019).
Lastly, the microbiota from a healthy subject represent a robust ecosystem that maintains the resistance to pathogen invasion and colonization, as illustrated, e.g. by a low risk of Clostridium difficile infections (CDI) (Ross et al. 2016). Factors that affect the stability of a community are cross-feeding, competition for nutrients, quorum sensing, and prey–predator relationships communities (Tomlin et al. 2005, Coyte et al. 2015, Thompson et al. 2016, Goyal et al. 2021). Predator–prey relationships are mediated by factors such as bacteriocins (Coyte and Rakoff-Nahoum 2019, Henriques et al. 2020, Heilbronner et al. 2021).
Design and applications of SynComs
Gut SynComs are designed to include one or more functions of human gut microbiota. To achieve this, bottom-up and top-down approaches have been developed (Fig. 2). Given that it is evidently and ethically impossible to study germ-free humans, germ-free animal models and in-vitro fermenters are mainly used to test these SynComs (Table 1).
Figure 2.

Overview on the strategies for designing, assembling, and testing SynComs. (A) The top-down approach starts at the inoculum and microbial composition profiling, followed by the selection and iteration cycle in which bacteria are added to an animal model, isolated, and subsequently sequenced for a further iteration cycle or ultimately the description of an adapted SynCom (Atarashi et al. 2013). (B) The bottom-up approach uses existing knowledge on the metagenome, abundances, and growth parameters of candidate microorganisms. Furthermore, when the SynCom is required to have a specific function, the candidate microorganisms can be adjusted accordingly (van der Lelie et al. 2021). The designed SynCom then enters iteration cycles in the in-vitro setting, being cultured in batches and sequenced to understand the composition. Lastly, the SynCom is tested in an in-vivo setting, being introduced to a murine model. The SynCom is then extracted from the model and sequenced after an incubation or experimental period. The functional profile is also checked to ensure the SynCom performed as expected. From there, feedback from the results can be used to start the iteration cycles again.
Table 1.
An overview of reported SynComs in chronological order.
| Yeara | Consortium name | Designb | Number of species | Number of phyla | Names of phyla presentc | Testing system | Reference |
|---|---|---|---|---|---|---|---|
| 2008 | - | TD | 7 | 4 | A, C, F, P | Murine | Martin et al. (2008) |
| 2009 | - | TD | 2 | 2 | B, F | Murine | Mahowald et al. (2009) |
| - | BU | 3 | 3 | A, F, P | Murine | Denou et al. (2009) | |
| - | TD | 2 | 2 | B, P | Murine | Goodman et al. (2009) | |
| 2011 | SIHUMI(x) | TD | 7/8 | 4 | A, B, F, P | Rat | Becker et al. (2011) |
| - | TD | 10 | 4 | A, B, F, P | Murine | Rezzonico et al. (2011) | |
| - | TD | 19 | 3 | A, B, F | Murine | McNulty et al. (2011) | |
| - | BU | 10 | 4 | A, B, F, T | Murine | Faith et al. (2011) | |
| 2013 | Shumix-9 | TD | 9 | 4 5 |
A, B, F, P A, B, F, FU, P |
Murine | Ganesh et al. (2013), Slezak et al. (2013, 2014) |
| Shumix-7 | TD | 7 | 4 | A, B, F, P | Rat | Woting et al. (2014) | |
| - | TD | 23 | 1 | F | Murine | Atarashi et al. (2013) | |
| - | TD | 2 | 2 | B, F | Rat | Wrzosek et al. (2013) | |
| - | TD | 2 | 2 | A, B | Murine | Marcobal et al. (2013) | |
| - | TD | 9 | 4 | A, B, F, P | Murine | Rey et al. (2013) | |
| - | TD | 14 | 2 | A, B, F | Murine | Reyes et al. (2013) | |
| RePOOPulate | BU | 33 | 4 | A,B,F,P | Human | Petrof et al. (2013) | |
| - | BU | 3 | 3 | B, E, F | Murine | (Shoaie et al. (2013) | |
| 2015 | - | TD | 4 | 3 | B, F | Murine | Buffie et al. (2015) |
| MET-2 | BU | 31 | 4 | A, B, F, P, V | Bioreactor | Yen et al. (2015) | |
| 2016 | - | TD | 14 | 5 | A, B, F, V | Murine | Desai et al. (2016) |
| MET-1 | BU | 33 | 5 | A, B, F, P, V | Murine, organoid | Martz et al. (2017), Munoz et al. (2016), Carlucci et al. (2019) | |
| 2017 | - | BU | 4 | 4 | B, F, P | Bioreactor | Pinto et al. (2017) |
| 2018 | - | BU | 12 | 4 | A, B, F, P | Microplate | Venturelli et al. (2018) |
| - | BU | 3 | 1 | F | Bioreactor | D'hoe et al. (2018) | |
| 2019 | SIHUMIx (8) | TD | 8 | 5 | A, B, F, P | Murine | Ring et al. (2019) |
| Simplified intestinal microbiota | TD | 10 | 4 | A, B, P, V | Murine | Kovatcheva-Datchary et al. (2019) | |
| 6-member clostridial consortium | BU | 6 | 1 | F | Murine | Abdel-Gadir et al. (2019) | |
| - | BU | 11 | 4 | B, F, FU | Murine | Tanoue et al. (2019) | |
| - | TD | 14 | 4 | A, B, F, P | Bioreactor | Gutiérrez and Garrido (2019) | |
| - | TD | 20 | 3 | A, B, F, P | Murine | Patnode et al. (2019) | |
| 2021 | - | BU | 25 | 4 | A, B, F, P | Microplate | Clark et al. (2021) |
| GUT-103, GUT-108 | BU | 17 11 |
4 5 |
B, F, V | Murine | van der Lelie et al. (2021) | |
| - | TD | 104 | 5 | A, B, F, P, V | Microplate | Cheng et al. (2021) | |
| MCC100 | TD | 100 | 5 | A, B, E, F, P | Bioreactor | Perez et al. (2021) |
Year of the earliest mention of the consortium. bThe design strategy of the SynCom: TD-top-down, BU-bottom-up. cAbbreviations of phyla: A-Actinobacteria, B-Bacteroidetes, E-Euryarchaeota (archaea), F-Firmicutes, FU-Fusobacteria, P-Proteobacteria, T-Thermodesulfobacteriota, and V-Verrucomicrobia.
Abundance-based assembly of SynComs (top-down)
The top-down design identifies the most abundant bacteria and assembles a subset that captures the diversity of the native gut microbiota (Vazquez-Castellanos et al. 2019). This strategy is based mainly on the relative abundance of bacterial species in human gut microbiota determined with advances in sequencing techniques from the fecal microbiota (Fig. 2A). There is a high variation of gut microbiota among individuals at the genera and species level, but the seven most common phyla are also known as Firmicutes, Bacteroides, Actinobacteria, Proteobacteria, Synergistetes, Verrucomicrobia, and Fusobacteria (Arumugam et al. 2011). Among these phyla, Firmicutes and Bacteroidetes represent ∼90% of the microbial sequences (Rinninella et al. 2019).
SynComs as a tool to generate reproducible humanized animal models
Defined microbiota in animal models increase the reproducibility of the experiments and further standardize the model systems. While an association of germ-free animal models with murine-derived defined communities, such as Altered Schaedler Flora, has been established since the 1960s (Wymore Brand et al. 2015), 50 years later, the “humanized” animal models have been generated with human gut isolate-derived consortia. For example, Becker et al. inoculated rats with a simplified human microbiota (SIHUMI) consortium, using dominant adult human intestinal microbiota members Anaerostipes caccae, Bacteroides thetaiotaomicron, Bifidobacterium longum, Blautia producta, C. ramosum, Escherichia coli, and Lactobacillus plantarum (Becker et al. 2011). The SIHUMI abundance-based consortium was able to stably colonize the caecum and colon of germ-free rats and was successfully transferred from mother to offspring. Notably, the concentration of individual species in SIHUMI-associated rats is similar to those in human feces. Additionally, the SIHUMI consortium is also stable in mice and bioreactors (Krause et al. 2020), leading to its wide use as a defined baseline microbiota consortium to study the effects of single species on host immunity (Ganesh et al. 2013, Lengfelder et al. 2019, Ring et al. 2019), metabolism (Woting et al. 2014, 2015), and development (Slezak et al. 2013, 2014). Despite the exceptional stability of SIHUMI consortia in rats and mice, short-chain fatty acid production is substantially lower than that in human fecal samples. The introduction of butyrate producers increased butyrate production by 56% (Becker et al. 2011) but did not reach the butyrate level observed in human feces. These results suggest that this consortium is not fully recapturing the functions of the native gut microbiota, calling for further development of consortia with higher complexity.
Iterative selection of minimal SynComs via decreasing species number
An excellent example of a top-down approach to select relevant species for protection against gastrointestinal disease is the study of Honda and colleagues. They studied the modulation of critical immune cells, CD4+ FOXP3+ Tregs, in antibiotic-treated mice (Atarashi et al. 2011). When antibiotics depleted gram-positive bacteria, which consist primarily of Clostridia, Treg frequencies decreased, which is associated with disease. Additionally, when the colonized germ-free mice with a fecal sample of 46 species, only the mice colonized with species from the Clostridium genus showed Treg accumulation. These results suggest that Clostridium species contribute to the immune response, but it remained unclear whether the results are translatable to indigenous human microbiota.
They tried to construct a SynCom with minimal members to find the relevant species while retaining the immunomodulatory function described above. First, they colonized germ-free mice with either healthy human fecal samples or feces treated with chloroform, as they previously reported that the chloroform-resistant fraction of mouse gut microbiota was enriched in Treg cell-inducing species (Atarashi et al. 2011). These adapted microbiotas were passaged through multiple generations. The adapted microbiota were sequenced, and the authors selected only the species with <99% 16S rRNA gene sequence identity to any other ones. This left the authors with 23 strains, which were inoculated into germ-free mice.
By iteratively constructing smaller SymComs from the 23 strains, it was found that only the 17 strains belonging to Clostridia clusters XIVa, IV, and XVIII colonized the mice and induced Treg cells (Atarashi et al. 2013). In both mice and rat models, adding the 17-strain SynCom affected Treg cell differentiation, accumulation, and function in the colon lamina propria (Atarashi et al. 2013). Interestingly, neither monoculture nor randomly selected subsets of the 17 strains have a comparable effect on Treg differentiation as the 17-strain SynCom, indicating this immunomodulatory effect is an emerging behavior of the 17-strain community. In a trinitrobenzene sulphonic acid-induced colitis specific-pathogen-free mouse model, the 17-strain SynCom attenuated colon shortening, reduced the histological disease features, and lowered the mortality rate of phosphate buffered saline-treated mice, likely attributed to the Foxp3-related pathway and inflammation regulation. Leveraging all this knowledge, the authors recently developed a smaller SynCom consisting of 11 members, different from the previously reported 17-mix. They revealed that it enhances both host resistance against Listeria monocytogenes and the therapeutic efficacy of immune checkpoint inhibitors in syngeneic tumor models (Tanoue et al. 2019).
Large SynComs as live bacterial therapeutics
One way to approach high, near-native complexity and hence promote emerging desired behavior is to increase the number of species in the consortia. A typical human fecal microbiota is estimated to contain a few hundred bacterial species, of which 66–69 species account for >99% of the detected sequence (Martínez et al. 2013, Schloissnig et al. 2013). These features pose several challenges when constructing a highly complex SynCom. First, it is laborious to simultaneously culture 66–69 bacterial strains and synchronously maintain their growth, given that they have different nutrient requirements and growth characteristics. Second, it is difficult to determine each strain’s abundance accurately and sensitively, especially for low-abundance species and closely related strains in the consortium. Third, it is unclear whether a stable community at such high complexity is achievable and reproducible over multiple passages. To tackle these challenges, Cheng et al. developed a new culture protocol to synchronize the growth of fast-growing and slow-growing bacteria (Cheng et al. 2021). To best synchronize the growth of 104 bacterial strains, Cheng et al. managed to use only two media, mega medium and chopped meat medium (Claros et al. 1995, Romano et al. 2015), and passage fast-growing strains more frequently than slow-growing strains. Furthermore, by adding species that are lowly abundant but highly prevalent, Cheng et al. managed to construct a 104-species SynCom, surpassing the 66–69 species mark, as mentioned before. This approach adds genetic redundancy by adding species that are phylogenetically close to each other. Additionally, to overcome the limitations of the available metagenomic read mapping algorithms, the authors developed a new algorithm, NinjaMap, which can quantify strain abundances across six orders of magnitude at high accuracy. This 104-species SynCom showed remarkable reproducibility, while the low-abundance species (relative abundance <10−4) were slightly more variable in their prevalence than the high-abundance species. These results suggest that it is feasible to recreate artificial microbiota to mimic the high complexity and diversity of native gut microbiota.
Highly complex SynComs open a new avenue to steer “dysbiotic” gut microbiota toward a more diverse and “healthy” state. Indeed, Perez et al. assembled a 100-species SynCom, MCC100, and supplemented MCC100 into “elderly” fecal microbiota in a fermentation bioreactor (Perez et al. 2021). They found that MCC100 increased the alpha diversity and the abundance of multiple species, such as Ba. fragilis, Bl. luti, Sutterella wadsworthensis, and F. prausnitzii, which are associated with healthy aging (Perez et al. 2021, Jeffery et al. 2016). Notably, the supplementation of MCC100 also increased opportunistic pathogens belonging to the Escherichia and Shigella genera, indicating that a thorough safety testing of SynComs is needed when they are considered as a candidate bacterial therapy.
Host-adapted selection of SynComs
Nevertheless, there are limitations to this abundance-based strategy from both a compositional and developmental point of view. The bacterial composition of fecal samples differs from the upper gastrointestinal tract and cannot recapitulate the difference between mucus-associated and gut luminal populations and therefore presents a biased view (Gorbach 1996, Stearns et al. 2011, Hillman et al. 2017). Furthermore, after inoculating the SynCom into germ-free animals, the microbial composition differs from the initially designed one owing to the host-specific microenvironment. To this end, other strategies are developed, such as the host-adaptation approach to assemble consortia. In the host-adaptation method, germ-free mice were inoculated with human fecal samples to humanize mice’s gut microbiota (Atarashi et al. 2013, Tanoue et al. 2019). Subsequently, the feces of the colonized mice were analyzed, and a consortium was built based on the bacterial composition identified in the “humanized” mice. These consortia take the assembly of microbial communities within the host into account but are more difficult to control due to biological variance. It remains a question of how translatable these host-adapted consortia are to humans, as microbial colonization was shown to be host-dependent (Nguyen et al. 2015).
Function-oriented assembly of SynComs (bottom-up)
Another primary strategy is to assemble SynComs to achieve specific functions, called bottom-up design (Fig. 2B). The bottom-up design focuses on assessing the function of individual species, followed by assembling species of designed functions to achieve specific emergent behavior, such as anti-inflammatory properties. Each species acts as a building block to achieve one or more designed functions, while some functions can only be achieved by the combination of multiple bacterial species. Because bottom-up design is function-oriented, the assembly of these SynComs is highly versatile in size and diversity of bacterial members. To date, most function-oriented SynComs are designed as candidate live bacterial therapeutics for aims such as combating infections or improving cancer therapy efficacy (Charbonneau et al. 2020). Specific aspects of SynComs as candidate therapeutics for gastrointestinal disorders are discussed in the next section.
Additionally, the rebirth of “culturomics” in human gut microbiota research rapidly and substantially expands our capacity to culture previously “unculturable” bacterial species in laboratories (Lagier et al. 2015, 2016, Poyet et al. 2019). Over 1000 microbial species in the human gut have been isolated and cultured, and the number is increasing (Rajilić-Stojanović and de Vos 2014, Sardelli et al. 2021). These resources enable researchers to assemble SynComs tailored to specific applications.
SynComs as live bacterial therapeutics for gastrointestinal disorders
Re-establishing gut microbial resilience to pathogen infection is a major driving force of rationally designed SynComs. For example, the SynCom MET-1, designed as a live bacterial biotherapeutic, was tested for treating CDI, one of the most frequent healthcare-associated, recurring, and antibiotic-resistant infections [Centers for Disease Control and Prevention (US) 2019, Balsells et al. 2019]. In CDI, C. difficile, an opportunistic pathogen, overgrows in the patient’s gut and produces toxins that ultimately disrupt the indigenous microbiota and induce severe local and systemic inflammation (Czepiel et al. 2019). Fecal microbiota transplant (FMT) offers a promising treatment for CDI (Kassam et al. 2013, Feuerstadt et al. 2022), but recently raises concerns about its safety due to its undefined nature and consequent rare severe incidence of bacteremia or fatal aspiration pneumonia after FMT (Baxter et al. 2015, DeFilipp et al. 2019). Moreover, because of the recipient’s diversity and need for compatibility with the donor’s fecal matter, differing immune reactions, transplantation efficacies, and consequently different treatment efficacies can be expected. These issues urge scientists to search for alternatives that are safe, effective, and wholly defined (Vázquez-Castellanos et al. 2019). In-vitro-created SynComs can provide this more efficient and safer way to treat dysbiosis in the human gut and intestinal inflammation. Especially for creating stable microbial populations in the human gut that are resilient toward environmental perturbations, SynComs could be an excellent alternative to FMT.
To test whether this alternative could accomplish a similar efficacy on CDI remission, Petrof et al. anaerobically recovered 62 bacterial isolates from a healthy female donor (Petrof et al. 2013). With these bacterial isolates, they assembled the SynCom RePOOPulate, which is ultimately composed of 33 multiantibiotic-sensitive isolates. Subsequently, RePOOPulate was deposited into the colon of two CDI patients during colonoscopy to investigate its efficacy in alleviating the symptoms and modulating the recipient’s fecal microbiota. The treatment with the RePOOPulate consortium induced the CDI symptoms’ remission, including restoring normal bowel movement and ceasing diarrhea. This remission lasted 24–26 weeks post-treatment, and the reduction of C. difficile toxin production was confirmed in one of the patients. Concurrently, a greater bacterial diversity was observed in both patients. Mechanistically, this is probably due to the decreased production of TcdA toxin and local and systemic inflammation reduction shown in a mouse model (Martz et al. 2017). In cells treated with purified TcdA toxin, RePOOPulate protected the barrier function by reducing the abundance of the TcdA toxin (Martz et al. 2017). The same group assembled a 58-strain SynCom, DEC58, and found similar effects as RePOOPulate on decreasing C. difficile growth and toxin secretion (Carlucci et al. 2019). While more extensive clinical trials are needed, these first-of-their-kind human trials demonstrated the promising potential of SynComs in rebuilding gastrointestinal resistance to microbial dysbiosis.
Furthermore, Martz et al. (2015) also performed experiments with a different pathogen than C. difficile, namely Salmonella typhimurium, with distinct mechanisms of pathogenesis compared to C. difficile; their MET-1 consortium consisted of the 33 RePOOPulate bacterial strains and as described above had been previously used to cure CDI patients. The authors showed that MET-1 also reduced systemic inflammation and helped to preserve the gut barrier function of mice infected by S. typhimurium (Martz et al. 2015). These results suggest that in some cases, one SynCom treatment fits multiple infections, possibly through identical mechanisms.
In multifaceted gastrointestinal disorders such as inflammatory bowel diseases, SynComs have been rationally designed to target specific functions. Chronic intestinal inflammation increases mucosal permeability, and intestinal microbial dysbiosis is a common pathophysiology of these gastrointestinal disorders. Using a bottom-up approach, van der Lelie et al. designed a 17-member consortium, GUT-103, to reduce inflammation and restore mucosal functions and intestinal microbiota (van der Lelie et al. 2021). Multiple functions are integrated purposely upon the design of GUT-103, including the production of short-chain fatty acids, tryptophan metabolites, synthesis of secondary bile acids, and release of antimicrobials and siderophores. The authors collected sequences based on published 16S rRNA studies and metadata from isolated strains. From there, they gathered isolated and metagenome-assembled genome sequences. The desired therapeutic functions were then deduced from the proteins encoded in the genome of the commensal human gut bacterial species. This approach yielded desirable genera for the authors to use in their SynCom. To select the specific species to use in their SynCom, the authors identified strains with complementary auxotrophies for essential metabolites like amino acids, vitamins, and co-factors to address the risk of competition and consecutive strain depletion. In a proof-of-concept test, the gavage of GUT-103 demonstrated its successful colonization in germ-free, wild-type, and IL10-deficient mice. Informed by the outcomes from GUT-103, the authors assembled another, more optimized consortium; GUT-108 (Ba. xylanisolvens, C. butyricum, C. scindens, Intestinimonas butyriciproducens, Eubacterium callanderi, Extibactersp.,Akkermansiasp.,C. symbiosum, Ba. uniformis, Bi. massiliensis, Barnesiellasp.). They achieved similar functions to those optimized in GUT-103 but used bacterial species that are quite different from those in GUT-103. The results demonstrated the successful application of a function-oriented strategy in designing SynComs and the feasibility of using various species to achieve similar designed functions.
Engraftment as a determinant for successful biotherapeutic use
SynComs, their design, and manufacturing opportunities have been gaining traction in recent years. However, the engraftment of the SynCom mostly comes as an afterthought, while it is an essential determinant for success. Engraftment success means that all the species in a SynCom transfer to a new test system or host organism. In the in-vitro setting, this can be covered relatively quickly, as experiments can be easily adjusted to fit the necessities to reach specific outcomes. However, in in-vivo and clinical settings, this is more difficult to achieve. In the field of FMTs, numerous studies have tried to understand the pillars of successful microbiome engraftment in the recipient (Schmidt et al. 2022). Adjusting the host environment seems essential for successful engraftment; recent findings from the FMT studies underline this: By analyzing the metagenomes from 316 FMTs pre and postintervention, Schmidt et al. found that the most critical determinants for the engraftment of microorganisms from donor to recipient are recipient factors and donor–recipient complementarity, both at the level of entire microbial communities and individual strains (Schmidt et al. 2022). In mice, the same principles hold in which the host factors seem to be the most important in the successful engraftment of microorganisms. While often germ-free rodents have been used, researchers have also used antibiotic-treated rodents to increase the engraftment success, similar to the antibiotic pretreatment given to patients that undergo FMT (Wos-Oxley et al. 2012, Hintze et al. 2014, Staley et al. 2017).
Emerging tools facilitate the design and testing of SynComs
While animal models, particularly germ-free animals, remain the cornerstone models in testing SynComs, other approaches such as computational modeling and microphysiological systems (also referred to as organ-on-chips) are emerging alternatives relevant to humans. With more rigorously verified nonanimal models available, it is foreseen that these alternative approaches will be employed in future fundamental and translational research on SynComs. This trend has been established since the introduction of regulation to replace, reduce, and refine the use of animals and will be facilitated by the recent passage of the so-called US “FDA Modernization Act of 2021,” which eliminates the animal testing mandate for new drugs, including live bacterial therapeutics.
Manufacturing a SynCom
Whilst improvements to culturing protocols are being proposed by researchers such as Cheng et al. (2021), as mentioned above, many SynComs are still being produced by individually growing all the members of the SynCom. Preferably, the microorganisms are used when they reach the exponential phase. However, the inherent differences between the microorganisms make it cumbersome to find a synchronous moment for the exponential phase. Precultures must be started at different times and combined when they all reach exponential growth. This experimental setup is beneficial for species that are difficult to grow together but is too laborious and costly for industry. One way to overcome this problem is to use co-cultivation techniques, where conditions are designed such that different bacterial strains can be precultured together (Kurt et al. 2021).
Computational approaches
Computational methods have innovative potential for two main applications: they could facilitate the selection of SynComs and help translate the results of SynComs to natural conditions. Selection of SynComs can be time-consuming because complicated interactions between its members cause difficulty in predicting what will happen when several species are combined. Members interact through metabolic interactions (e.g. mutualism, competition, commensalism, antibiosis, and predator–prey interactions) directly through toxins (e.g. bacteriocins) or indirectly by affecting the medium. The establishment of the communities can thus be facilitated by using computational techniques to find an optimal design based on predicted growth parameters (Stein et al. 2018, Clark et al. 2021, van den Berg et al. 2022).
For the first application, i.e. the selection of SynComs, computational models can parameterize the potential species and estimate which species combinations in which nutritional conditions could lead to stable SynComs or when high densities of beneficial species or compounds will be reached. These results would have to be verified with experiments, but the modeling effort can reduce the experimental conditions to be tested, which could considerably reduce time and investment. For the second application, to translate results to natural settings, models can be constructed from and validated with SynComs to be more reliable and simulate the conditions in more natural or larger communities. To aid in understanding dysbiosis, computational models can simulate stability or metabolite production in individuals with certain medical conditions that affect the microbiome. A different application where the computational model can be helpful is that many species are often changed when a person has dysbiosis. Well-adapted models can simulate alternative scenarios and thereby pinpoint which species or metabolites are likely causing dysbiosis and which are responding to changes in other consortium members.
Most efforts on computational approaches and SynComs at the moment are aimed at inferring community properties that cannot be measured, validating modeling techniques as well as testing how well models perform outside of the measured data points. Venturelli et al. (2018) used time-resolved density measurements to infer twelve species’ growth parameters and interaction coefficients. They found that though negative interactions dominate, positive interactions are also common. To predict the dynamics of the 12-species consortium, they fitted a generalized Lotka–Volterra model on the mono and bi-culture data, which showed good agreement with experimental validations. Growth in conditioned media showed that metabolic interactions are a likely key mechanism of the species interactions. Pinto et al. and D'hoe et al. characterized mono and bi-cultures in even more detail with time-resolved measurements of the extracellular metabolites (Pinto et al. 2017; D'hoe et al. 2018). Pinto et al. used these data to fit a monoculture growth model with inhibition terms for some metabolites and other species. They then predicted four species communities and could show a good fit, although one or two species mostly dominated the communities. D'hoe et al. fitted an ordinary differential equation model for both the species and the metabolites and evaluated the prediction of a three-species community. They concluded that the tri-culture dynamics could not be predicted based on only monoculture data. Including bi-culture data in the fit did significantly improve the tri-culture fit. However, they had to select a subset of experiments for the model fitting, showing that this model prediction remains challenging. In conclusion, the first efforts show that Lotka–Volterra-like growth models fit reasonably well. More specific growth models, including metabolite dynamics, can be generalized much better to new conditions but are more challenging to parameterize properly. Still, metabolites are not the only means of communication between bacteria. In fact, we still miss knowledge about species’ interactions mediated by cell-to-cell contact or quorum sensing compounds, explaining why predictions are not yet always matching experimental data satisfactorily.
SynComs and mathematical modeling are also used to predict the metabolic behavior of communities. Because there are plentiful metabolic interactions (e.g. cross-feeding) in the microbiota, the metabolic behavior of a community is not merely the sum of its parts, and a more holistic view is needed. The metabolic behavior of communities is of clinical relevance because many short-chain fatty acids and other metabolites produced by the microbiota have been identified as interacting with the host and associated with health and disease states. Gutiérrez and Garrido studied the effect of a 14-species community on the degradation of the prebiotic inulin using single-strain deletion experiments (Gutiérrez and Garrido 2019). Interestingly, the dropout of any single strain did not significantly influence the degradation of inulin and only had a slight impact on short-chain fatty acid production. They found that the butyrate producer changed the most in abundance as a response to other species, which was also observed by Venturelli et al. (2018). With the results, they propose a model of the interactions between species and metabolites. These findings indicate a certain degree of functional redundancy even in a simplified 14-species SynCom. The above-described models of Pinto et al. (2017) and D'hoe et al. (2018) predict metabolite profiles and species profiles. However, the difficulty of predicting all metabolites correctly also illuminates the challenges of predicting the metabolic behavior of communities.
Using computational methods to generalize the results from SynComs to the natural setting has not yet been attempted. The aim would be to model natural conditions and possible interventions and select or design possible dietary or prebiotic interventions. Although not all species in the natural setting can be grown in the lab, generalization might still be achieved by using the genomes of the noncultured species to understand their metabolism. Metabolism can be predicted from genome sequences to construct genome-scale metabolic models (GEMs), even with automated model generation tools (Magnúsdóttir et al. 2017, Machado et al. 2018), although the quality is sometimes low, and one has to be careful with which tools to use (Mendoza et al. 2019). GEMs can be combined in communities to resemble a natural setting and then simulated (Colarusso et al. 2021), e.g. with dynamic Flux Balance Analysis, where every timestep the species growth rates and others are calculated from external metabolites, and then the species densities and metabolite levels are updated (Mahadevan et al. 2002, Hjersted and Henson 2006, Meadows et al. 2010, Henson and Hanly 2014). Community models do not necessarily need to include all species; focusing on some key species with associated metabolic functions can already surface important dynamics [communities can be selected with, e.g. MiMiC (Kumar et al. 2021)]. An example of a (spatial) simulation of key species of lactose metabolism in infants and the dependency on oxygen is shown in the work of Versluis et al. (2022). The role of SynComs in this quickly expanding endeavor to model the microbiome with GEMs is to use them as a validation because that cannot be done in a natural setting (Weiss et al. 2022). Shoaie et al. created a system of three species and validated it in a mouse model (Shoaie et al. 2013). Before we can interpret how reliable predictions are, we need to know how well the models perform and their strengths and weaknesses. Therefore, combining wet lab, experimental SynCom analysis, and computational approaches are necessary to achieve new insights and applications.
Gut microphysiological systems
The advancement of tissue engineering, stem cell biology, mechanical engineering, and culturomics have brought advanced gut microphysiological systems (MPS), also known as gut-on-a-chip, forward as a promising approach in microbiome research. In 2012, Kim et al. (2012) reported a microfluidic device co-culturing human intestinal cell line Caco-2 with human isolate L. rhamnosus. The team demonstrated the feasibility of co-culturing host and bacterial cells under controlled continuous culturing conditions. A challenge that remained by then was the inclusion of primary human cells. This feature was soon made possible thanks to the isolation of Lgr5+ intestinal stem cells and sustainable 3D culture of them as organoids (Sato et al. 2009), which has been adopted to build primary human intestinal organoid-derived MPS.
In the context of SynComs, it is crucial to include primary human cells, which offer an option to test the personalized response of different hosts to a functional SynCom. As personalized responses have long been recognized in interventions of small molecule drugs, nutrition, and cell-based therapies, it is anticipated that a single SynCom will not function the same in all hosts. In addition, human fecal microbiota is highly individualized. To fully recapitulate the individualized host response to SynComs, individual microbiota background is required in the gut MPS.
One challenge to include microbiota in a gut MPS is that many gut microbes are susceptible to oxygen and require dissimilar nutrients. Recently, several groups have partly resolved this challenge by creating different compartments for human and microbial cells or operating the whole system in an anaerobic workstation (Ulluwishewa et al. 2015, Fofanova et al. 2019, Zhang et al. 2021). For example, Zhang et al. reported a mesofluidic GuMI system that enables the co-culture of primary human colon epithelium and the oxygen-intolerant F. prausnitzii and E. rectale at the oxic–anoxic interface (Zhang et al. 2021). Under continuous culture, F. prausnitzii reduced the transcription of genes in the NF–KB pathway in colon epithelium, mimicking the anti-inflammatory effects observed in human patients (Sokol et al. 2008). In another microfluidic device, Jalili-Firoozinezhad demonstrated the co-culturing of complex fecal microbiota and human intestinal epithelium (Jalili-Firoozinezhad et al. 2019). This device was used to test the protective effects of Enterococcus faecium against S. typhimurium infection in the gut MPS (Gazzaniga et al. 2021). These studies exemplified the systems that can be used to test SynComs in a robust and human-relevant manner.
Despite the promising potential of microbiome-competent gut-on-a-chip, challenges remain in disseminating these systems to facilitate the testing of SynComs by the end users. One barrier to microbiome researchers is the multidisciplinary nature needed to have such systems operational in the lab. To properly run a gut MPS in-house, it requires know-how of organoid culturing, anaerobic bacterial culture, and access to engineering expertise. This challenge calls for interdisciplinary collaborations among distinct fields and more support to build core facilities that will embrace the accessibility of state-of-the-art gut MPS technologies by end users.
Additionally, the current microbiome-competent gut MPS can be used to answer a limited number of questions, as they still lack certain vital features of a human in-vivo intestine. These features, such as 3D architectures (Wang et al. 2017), endothelial cells, immune cells (Shin and Kim 2018), and connection with other organs (Trapecar et al. 2020), are independently developed but have yet to be integrated into one single MPS.
Conclusions and future opportunities
We cannot identify one SynCom for all purposes, nor should we try to. Nonetheless, there are a few essential steps to keep in mind when creating a SynCom (Fig. 3). The first step is to identify the desired design rationale, being top-down or bottom-up. Both rationales have different sources of information that can be used to assemble a SynCom. The second step is to measure relevant parameters, e.g. the metabolites produced by the members of the SynCom, the metagenome, the pairwise interactions, and the host response to the individual members. From there, one can go immediately to the assembled SynCom, by combining the information gained in step two, or in-silico approaches can be deployed to further improve the composition and performance of the SynCom. Once the SynCom is assembled, it can be tested in the system of choice. The validation results can then be fed back to the in-silico models or used for downstream applications. SynComs have been a powerful tool for many applications, from studying microbe–microbe crosstalk to restoring microbiota function in humans. It is worth noting that a native-like complexity has been reached by assembling SynComs of 100 or more species, and clinical trials for biotherapeutics have been conducted.
Figure 3.
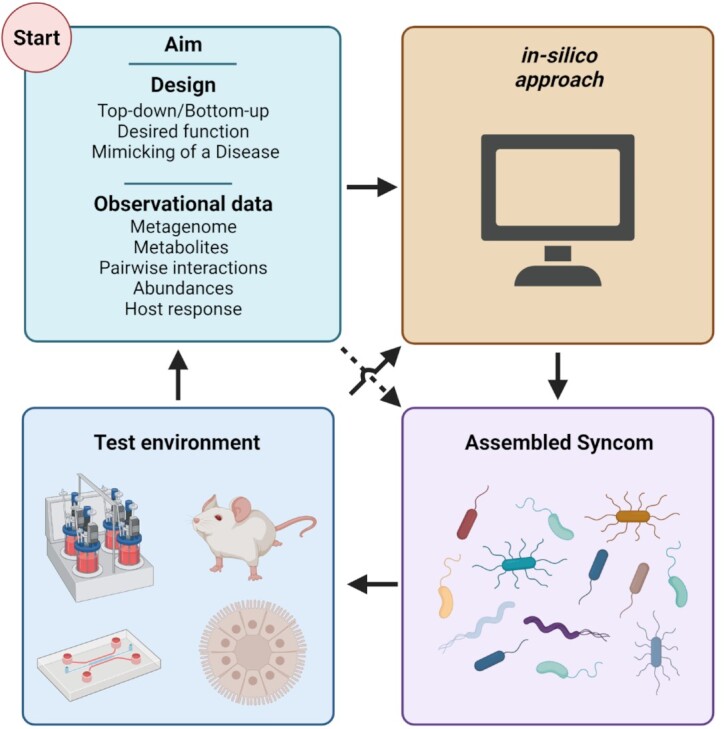
Design flowchart for the creation of SynComs. Blocks indicate steps to create a SynCom. Arrows indicate relationships, dashed arrow indicates a shortcut. Starting at the design phase, there are many factors that change the initial composition of a SynCom, most notably being the aim of the research that the SynCom will be used for, the design strategy used, and the observational data used. From there, an in-silico approach can help to predict the behavior of the SynCom and design the species and conditions, but could be skipped. Then, in-vitro testing commences, culturing the microorganisms and preparing them for the final step. In this final step, the SynCom is added to a test environment, which range from bioreactors to murine models and everything in between. The results from these experiments can next be fed back into the starting block, using prior knowledge for the initial design of a new SynCom.
These achievements in SynComs spark exciting opportunities to enhance their ability to better model host–microbe interactions and, ultimately, a better understanding of host health and diseases. The improvements can be categorized into four distinct categories that follow sequentially on each other. First, even though >1000 gut species have been isolated and cultivated, these species are mostly isolated from individuals living a western lifestyle. Bacterial species isolated from subjects with unindustrialized lifestyles, such as the Global Microbiome Conservancy storing fecal samples from traditional peoples around the world (https://microbiomeconservancy.org/), will diversify the biobank for designing SynComs. Even in individuals with industrialized lifestyles, there are still many uncultivable species that cannot be included in SynComs, calling for continuous effort on culturomics. Finally, there is a debate about how a healthy microbiome can be defined (Lloyd-Price et al. 2016, Eisenstein 2020). While certain microbiome features can be associated with good health, there is yet to be a consensus on the definition of a healthy microbiome (Bäckhed et al. 2012, Huttenhower et al. 2012, 2021, Shanahan et al. 2012, Ghosh et al. 2022). Until then, SynCom construction based on relative abundance will still be an adequate proxy to mimic the bacterial composition of healthy individuals and can assist in understanding what the makeup of a healthy microbiome is for individuals.
Second, the expansion of SynComs calls for a more streamlined and efficient assembly. For this, validated predictive models should be deployed to shorten the initial design phase of a SynCom. The studies that have used predictive models often did not include the metabolites in their models. With more studies and data becoming available, which include factors like metabolism, the models can make larger-scale predictions. Furthermore, GEMs are not yet used extensively in combination with SynComs but could link SynComs and microbiota models in vivo; hundreds of draft GEMs have been generated. By manually curating the GEMs using in-vitro verification experiments, these GEMs become more reliable. Advancements in methods for community simulation of growth with GEMs allow for the predictions of the metabolic potential of communities (Sen and Orešič 2019, Chowdhury and Fong 2020).
Third, the need to test more SynComs is anticipated to increase in the future, especially in translational biomedical research. Therefore, it requires continuous effort in developing, disseminating, and validating advanced in-vitro multicellular and multiorgan models to meet the research needs in SynComs. The discussion of using such in-vitro models will be facilitated by the recent change of mandatory animal testing US Food and Drug Administration. Nevertheless, the use of these new models resonates with the long-standing 3Rs concept (proposed in the 1960s) in animal testing replacement, reduction, and refinement.
Last, more clinical research is warranted, especially with the biotherapeutic goal in mind for SynComs. A potentially important application could be to increase microbiome resilience to pathogens, thereby reducing the global need for antibiotics, thus decreasing the speed at which antibiotic-resistant organisms spread.
While it is not discussed in this review, fungi, viruses, and archaea are essential components of native microbial communities in the human gut. Future research on constructing SynComs of fungi, bacteria, phages/viruses, or archaea will be challenging but can offer comprehensive model systems to study multikingdom ecosystem and their interaction with the host.
Taken together, SynComs offer many exciting research opportunities. The main challenges are the development of robust and reproducible synthetic consortia tailored to the research needs and scientific questions and the subsequent translation to the in-vivo setting.
Acknowledgements
We appreciate anonymous reviewers for their constructive comments and the editorial office for handling the submission.
Contributor Information
Pim T van Leeuwen, Molecular Biology and Microbial Food Safety, Swammerdam Institute for Life Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands.
Stanley Brul, Molecular Biology and Microbial Food Safety, Swammerdam Institute for Life Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands.
Jianbo Zhang, Molecular Biology and Microbial Food Safety, Swammerdam Institute for Life Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands.
Meike T Wortel, Molecular Biology and Microbial Food Safety, Swammerdam Institute for Life Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands.
Conflict of interest statement
The authors declare no conflict of interest.
Funding
P.T.v.L., M.T.W., J.Z., and S.B. are supported by the University of Amsterdam Research Priority Area Systems Biology Host–Microbiome Interactions. S.B. is supported by the University of Amsterdam Centre for Urban Mental Health. Finally, S.B., M.T.W., and J.Z. are supported by NWA-ORC project 1389.20.080.
References
- Abdel-Gadir A, Stephen-Victor E, Gerber GKet al. . Microbiota therapy acts via a regulatory T cell MyD88/RORγt pathway to suppress food allergy. Nat Med. 2019;25:1164–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam M, Raes J, Pelletier Eet al. . Enterotypes of the human gut microbiome. Nature. 2011;473:174–80. 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Oshima Ket al. . Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–36. 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Shima Tet al. . Induction of colonic regulatory T cells by indigenous clostridium species. Science. 2011;331:337–41. 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäckhed F, Fraser C, Ringel Yet al. . Defining a healthy human gut microbiome: current concepts, future directions, and clinical applications. Cell Host Microbe. 2012;12:611–22. 10.1016/j.chom.2012.10.012. [DOI] [PubMed] [Google Scholar]
- Balsells E, Shi T, Leese Cet al. . Global burden of Clostridium difficile infections: a systematic review and meta-analysis. J Glob Health. 2019;9:010407. 10.7189/jogh.09.010407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bär J, Leung J, Hansen Cet al. . Strong effects of lab-to-field environmental transitions on the bacterial intestinal microbiota of Mus musculus are modulated by Trichuris murisinfection. FEMS Microbiol Ecol. 2020;96:fiaa167. 10.1093/femsec/fiaa167. [DOI] [PubMed] [Google Scholar]
- Baxter M, Ahmad T, Colville Aet al. . Fatal aspiration pneumonia as a complication of fecal microbiota transplant. Clin Infect Dis. 2015;61:136–37. 10.1093/cid/civ247. [DOI] [PubMed] [Google Scholar]
- Becker N, Kunath J, Loh Get al. . Human intestinal microbiota: characterization of a simplified and stable gnotobiotic rat model. Gut Microbes. 2011;2:25–33. 10.4161/gmic.2.1.14651. [DOI] [PubMed] [Google Scholar]
- Berg N, Machado D, Santos Set al. . Ecological modelling approaches for predicting emergent properties in microbial communities. Nat Ecol Evol. 2022;6:855–65. 10.1038/s41559-022-01746-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernalier-Donadille A. Fermentative metabolism by the human gut microbiota. Gastroentérol Clin Biol. 2010;34:S16–22. 10.1016/S0399-8320(10)70016-6. [DOI] [PubMed] [Google Scholar]
- Bouskra D, Brézillon C, Bérard Met al. . Lymphoid tissue genesis induced by commensals through nod1 regulates intestinal homeostasis. Nature. 2008;456:507–10. 10.1038/nature07450. [DOI] [PubMed] [Google Scholar]
- Buffie CG, Bucci V, Stein RRet al. . Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015;517:205–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlucci C, Jones C, Oliphant Ket al. . Effects of defined gut microbial ecosystem components on virulence determinants of Clostridioides difficile. Sci Rep. 2019;9:885. 10.1038/s41598-018-37547-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrión V, Perez-Jaramillo J, Cordovez Vet al. . Pathogen-induced activation of disease-suppressive functions in the endophytic root microbiome. Science. 2019;366:606–12. 10.1126/science.aaw9285. [DOI] [PubMed] [Google Scholar]
- Cebra JJ, Periwal SB, Lee Get al. . Development and maintenance of the gut-associated lymphoid tissue (GALT): the roles of enteric bacteria and viruses. Dev Immunol. 1998;6:13–18. 10.1155/1998/68382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (U.S.) . 2019. Antibiotic Resistance Threats in the United States, 2019. Atlanta, GA: Centers for Disease Control and Prevention (U.S.). 10.15620/cdc:82532. [DOI] [Google Scholar]
- Charbonneau M, Isabella V, Li Net al. . Developing a new class of engineered live bacterial therapeutics to treat human diseases. Nat Commun. 2020;11:1738. 10.1038/s41467-020-15508-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Aranda-Díaz A, Jain Set al. . Systematic dissection of a complex gut bacterial community. bioRxiv 2021.06.15.448618. 2021. 10.1101/2021.06.15.448618. [DOI]
- Chowdhury S, Fong S. Computational modeling of the human microbiome. Microorganisms. 2020;8:197. 10.3390/microorganisms8020197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark R, Connors B, Stevenson Det al. . Design of synthetic human gut microbiome assembly and butyrate production. Nat Commun. 2021;12:3254. 10.1038/s41467-021-22938-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claros MC, Citron DM, Goldstein EJ. Survival of anaerobic bacteria in various thioglycolate and chopped meat broth formulations. J Clin Microbiol. 1995;33:2505–7. 10.1128/jcm.33.9.2505-2507.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colarusso A, Goodchild-Michelman I, Rayle Met al. . Computational modeling of metabolism in microbial communities on a genome-scale. Curr Opin Syst Biol. 2021;26:46–57. 10.1016/j.coisb.2021.04.001. [DOI] [Google Scholar]
- Coyte K, Rakoff-Nahoum S. Understanding competition and cooperation within the mammalian gut microbiome. Curr Biol. 2019;29:R538–44. 10.1016/j.cub.2019.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyte K, Schluter J, Foster K. The ecology of the microbiome: networks, competition, and stability. Science. 2015;350:663–66. 10.1126/science.aad2602. [DOI] [PubMed] [Google Scholar]
- Czepiel J, Dróżdż M, Pituch Het al. . Clostridium difficile infection: review. Eur J Clin Microbiol Infect Dis. 2019;38:1211–21. 10.1007/s10096-019-03539-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'hoe K, Vet S, Faust Ket al. . Integrated culturing, modeling and transcriptomics uncovers complex interactions and emergent behavior in a three-species synthetic gut community. Morgan X, Garrett WS (eds.). eLife. 2018;7:e37090. 10.7554/eLife.37090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFilipp Z, Bloom P, Soto Met al. . Drug-resistant E. coli bacteremia transmitted by fecal microbiota transplant. N Engl J Med. 2019;381:2043–50. 10.1056/NEJMoa1910437. [DOI] [PubMed] [Google Scholar]
- Denou E, Rezzonico E, Panoff J-Met al. . A Mesocosm of Lactobacillus johnsonii, Bifidobacterium longum, and Escherichia coli in the mouse gut. DNA Cell Biol. 2009;28:413–22. [DOI] [PubMed] [Google Scholar]
- Desai MS, Seekatz AM, Koropatkin NMet al. . A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell. 2016;167:1339–1353.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberl C, Ring D, Münch Pet al. . Reproducible colonization of germ-free mice with the oligo-mouse-microbiota in different animal facilities. Front Microbiol. 2019;10:2999. 10.3389/fmicb.2019.02999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenstein M. The hunt for a healthy microbiome. Nature. 2020;577:S6–8. 10.1038/d41586-020-00193-3. [DOI] [PubMed] [Google Scholar]
- Faith JJ, McNulty NP, Rey FEet al. . Predicting a Human Gut Microbiota’s Response to Diet in Gnotobiotic Mice. Science. 2011;333:101–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández M, Zúñiga M. Amino acid catabolic pathways of lactic acid bacteria. Crit Rev Microbiol. 2006;32:155–83. 10.1080/10408410600880643. [DOI] [PubMed] [Google Scholar]
- Feuerstadt P, Louie T, Lashner Bet al. . SER-109, an oral microbiome therapy for recurrent Clostridioides difficile infection. N Engl J Med. 2022;386:220–29. 10.1056/NEJMoa2106516. [DOI] [PubMed] [Google Scholar]
- Fofanova TY, Stewart CJ, Auchtung JMet al. . A novel human enteroid-anaerobe co-culture system to study microbial-host interaction under physiological hypoxia. bioRxiv 555755. 2019. 10.1101/555755. [DOI] [Google Scholar]
- Fu H, Uchimiya M, Gore Jet al. . Ecological drivers of bacterial community assembly in synthetic phycospheres. Proc Natl Acad Sci. 2020;117:3656–62. 10.1073/pnas.1917265117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesh B, Klopfleisch R, Loh Get al. . Commensal Akkermansia muciniphila exacerbates gut inflammation in SalmonellaTyphimurium-infected gnotobiotic mice. PLoS One. 2013;8:e74963. 10.1371/journal.pone.0074963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazzaniga F, Camacho D, Wu Met al. . Harnessing colon chip technology to identify commensal bacteria that promote host tolerance to infection. Front Cell Infect Microbiol. 2021;11:638014. https://www.frontiersin.org/articles/10.3389/fcimb.2021.638014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh T, Shanahan F, O'Toole P. Toward an improved definition of a healthy microbiome for healthy aging. Nat Aging. 2022;2:1054–69. 10.1038/s43587-022-00306-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AL, McNulty NP, Zhao Yet al. . Identifying Genetic Determinants Needed to Establish a Human Gut Symbiont in Its Habitat. Cell Host Microbe. 2009;6:279–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbach SL. Microbiology of the gastrointestinal tract. In: Baron S (ed.), Medical Microbiology, 4th edn. Galveston (TX): University of Texas Medical Branch at Galveston, 1996. http://www.ncbi.nlm.nih.gov/books/NBK7670/. [PubMed] [Google Scholar]
- Goyal A, Wang T, Dubinkina Vet al. . Ecology-guided prediction of cross-feeding interactions in the human gut microbiome. Nat Commun. 2021;12:1335. 10.1038/s41467-021-21586-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Großkopf T, Soyer O. Synthetic microbial communities. Curr Opin Microbiol. 2014;18:72–77. 10.1016/j.mib.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C-J, Chang F-Y, Wyche TPet al. . Discovery of reactive microbiota-derived metabolites that inhibit host proteases. Cell. 2017;168:517–26.e18. 10.1016/j.cell.2016.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez N, Garrido D. Species deletions from microbiome consortia reveal key metabolic interactions between gut microbes. mSystems. 2019;4:e00185–19. 10.1128/mSystems.00185-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamer HM, Jonkers D, Venema Ket al. . Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther. 2008;27:104–19. 10.1111/j.1365-2036.2007.03562.x. [DOI] [PubMed] [Google Scholar]
- Hapfelmeier S, Lawson MAE, Slack Eet al. . Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science. 2010;328:1705–9. 10.1126/science.1188454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilbronner S, Krismer B, Brötz-Oesterhelt Het al. . The microbiome-shaping roles of bacteriocins. Nat Rev Microbiol. 2021;19:726–39. 10.1038/s41579-021-00569-w. [DOI] [PubMed] [Google Scholar]
- Heitkemper MM, Cain KC, Shulman RJet al. . Stool and urine trefoil factor 3 levels: associations with symptoms, intestinal permeability, and microbial diversity in irritable bowel syndrome. Benef Microbes. 2018;9:345–55. 10.3920/BM2017.0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques S, Dhakan D, Serra Let al. . Metabolic cross-feeding in imbalanced diets allows gut microbes to improve reproduction and alter host behaviour. Nat Commun. 2020;11:4236. 10.1038/s41467-020-18049-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson M, Hanly T. Dynamic flux balance analysis for synthetic microbial communities. IET Syst Biol. 2014;8:214–29. 10.1049/iet-syb.2013.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill D, Artis D. Intestinal bacteria and the regulation of immune cell homeostasis. Annu Rev Immunol. 2010;28:623–67. 10.1146/annurev-immunol-030409-101330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillman E, Lu H, Yao Tet al. . Microbial ecology along the gastrointestinal tract. Microbes Environ. 2017;32:300–13. 10.1264/jsme2.ME17017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hintze KJ, Cox JE, Rompato Get al. . Broad scope method for creating humanized animal models for animal health and disease research through antibiotic treatment and human fecal transfer. Gut Microbes. 2014;5:183–91. 10.4161/gmic.28403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitch TCA, Hall LJ, Walsh SKet al. . Microbiome-based interventions to modulate gut ecology and the immune system. Mucosal Immunol. 2022;15:1095–113. 10.1038/s41385-022-00564-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjersted JL, Henson MA. Optimization of fed-batch Saccharomyces cerevisiae fermentation using dynamic flux balance models. Biotechnol Progr. 2006;22:1239–48. 10.1021/bp060059v. [DOI] [PubMed] [Google Scholar]
- Hugenholtz F, de Vos WM. Mouse models for human intestinal microbiota research: a critical evaluation. Cell Mol Life Sci. 2018;75:149–60. 10.1007/s00018-017-2693-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenhower C, Gevers D, Knight Ret al. . Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–14. 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalili-Firoozinezhad S, Gazzaniga FS, Calamari ELet al. . A complex human gut microbiome cultured in an anaerobic intestine-on-a-chip. Nat Biomed Eng. 2019;3:520–31. 10.1038/s41551-019-0397-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery IB, Lynch DB, O'Toole PW. Composition and temporal stability of the gut microbiota in older persons. ISME J. 2016;10:170–82. 10.1038/ismej.2015.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung C, Hugot J-P, Barreau F. Peyer’s patches: the immune sensors of the intestine. Int J Inflam. 2010;2010:823710. 10.4061/2010/823710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassam Z, Lee CH, Yuan Yet al. . Fecal microbiota transplantationForclostridium difficile infection: systematic review and meta-analysis. Am J Gastroenterol. 2013;108:500–8. 10.1038/ajg.2013.59. [DOI] [PubMed] [Google Scholar]
- Kim H, Huh D, Hamilton Get al. . Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip. 2012;12:2165–74. 10.1039/C2LC40074J. [DOI] [PubMed] [Google Scholar]
- Knoth J, Kim S-H, Ettl Get al. . Biological nitrogen fixation and biomass accumulation within poplar clones as a result of inoculations with diazotrophic endophyte consortia. New Phytol. 2014;201:599–609. 10.1111/nph.12536. [DOI] [PubMed] [Google Scholar]
- Koopman N, Katsavelis D, Hove ASet al. . The multifaceted role of serotonin in intestinal homeostasis. Int J Mol Sci. 2021;22:9487. 10.3390/ijms22179487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovatcheva-Datchary P, Shoaie S, Lee Set al. . Simplified Intestinal Microbiota to Study Microbe-Diet-Host Interactions in a Mouse Model. Cell Rep. 2019;26:3772–3783.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause JL, Schaepe SS, Fritz-Wallace Ket al. . Following the community development of SIHUMIx - a new intestinal in vitro model for bioreactor use. Gut Microbes. 2020;11:1116–29. 10.1080/19490976.2019.1702431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krautkramer KA, Fan J, Bäckhed F. Gut microbial metabolites as multi-kingdom intermediates. Nat Rev Microbiol. 2021;19:77–94. 10.1038/s41579-020-0438-4. [DOI] [PubMed] [Google Scholar]
- Kumar N, Hitch TCA, Haller Det al. . MiMiC: a bioinformatic approach for generation of synthetic communities from metagenomes. Microb Biotechnol. 2021;14:1757–70. 10.1111/1751-7915.13845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurt F, de Wouters T, Lacroix Cet al. . 2021. Method of manufacturing a consortium of bacterial strains. 20210355431, filed 15 October 2019, and issued 18 November 2021. https://patents.justia.com/patent/20210355431.
- Lagier J-C, Hugon P, Khelaifia Set al. . The rebirth of culture in microbiology through the example of culturomics to study human gut microbiota. Clin Microbiol Rev. 2015;28:237–64. 10.1128/CMR.00014-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier J-C, Khelaifia S, Alou MTet al. . Culture of previously uncultured members of the human gut microbiota by culturomics. Nat Microbiol. 2016;1:1–8. 10.1038/nmicrobiol.2016.203. [DOI] [PubMed] [Google Scholar]
- Laukens D, Brinkman BM, Raes Jet al. . Heterogeneity of the gut microbiome in mice: guidelines for optimizing experimental design. FEMS Microbiol Rev. 2016;40:117–32. 10.1093/femsre/fuv036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc JG, Milani C, Giori GSet al. . Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Current Opin Biotechnol. 2013;24:160–68. 10.1016/j.copbio.2012.08.005. [DOI] [PubMed] [Google Scholar]
- Lelie D, Oka A, Taghavi Set al. . Rationally designed bacterial consortia to treat chronic immune-mediated colitis and restore intestinal homeostasis. Nat Commun. 2021;12:3105. 10.1038/s41467-021-23460-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengfelder I, Sava IG, Hansen JJet al. . Complex bacterial consortia reprogram the colitogenic activity of Enterococcus faecalis in a gnotobiotic mouse model of chronic, immune-mediated colitis. Front Immunol. 2019;10:1420. 10.3389/fimmu.2019.01420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Liu W, Piao Met al. . A review of the relationship between the gut microbiota and amino acid metabolism. Amino Acids. 2017;49:2083–90. 10.1007/s00726-017-2493-3. [DOI] [PubMed] [Google Scholar]
- Lloyd-Price J, Abu-Ali G, Huttenhower C. The healthy human microbiome. Genome Med. 2016;8:51. 10.1186/s13073-016-0307-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long S, Gahan C, Joyce S. Interactions between gut bacteria and bile in health and disease. Mol Aspects Med. 2017;56:54–65. 10.1016/j.mam.2017.06.002. [DOI] [PubMed] [Google Scholar]
- Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol. 2017;19:29–41. 10.1111/1462-2920.13589. [DOI] [PubMed] [Google Scholar]
- Machado D, Andrejev S, Tramontano Met al. . Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 2018;46:7542–53. 10.1093/nar/gky537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnúsdóttir S, Heinken A, Kutt Let al. . Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat Biotechnol. 2017;35:81–89. 10.1038/nbt.3703. [DOI] [PubMed] [Google Scholar]
- Mahadevan R, Edwards JS, Doyle FJ. Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophys J. 2002;83:1331–40. 10.1016/S0006-3495(02)73903-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahowald MA, Rey FE, Seedorf Het al. . Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc Natl Acad Sci USA. 2009;106:5859–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcobal A, Kashyap PC, Nelson TAet al. . A metabolomic view of how the human gut microbiota impacts the host metabolome using humanized and gnotobiotic mice. ISME J. 2013;7:1933–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin F-PJ, Wang Y, Sprenger Net al. . Probiotic modulation of symbiotic gut microbial-host metabolic interactions in a humanized microbiome mouse model. Mol Syst Biol. 2008;8:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez I, Muller CE, Walter J. Long-term temporal analysis of the human fecal microbiota revealed a stable core of dominant bacterial species. PLoS One. 2013;8:e69621. 10.1371/journal.pone.0069621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martz SL, Guzman-Rodriguez M, He S-Met al. . A human gut ecosystem protects against C. difficile disease by targeting TcdA. J Gastroenterol. 2017;52:452–65. 10.1007/s00535-016-1232-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martz SL, McDonald JAK, Sun Jet al. . Administration of defined microbiota is protective in a murine Salmonella infection model. Sci Rep. 2015;5:16094. 10.1038/srep16094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNulty NP, Yatsunenko T, Hsiao Aet al. . The impact of a consortium of fermented milk strains on the gut microbiome of gnotobiotic mice and monozygotic twins. Sci Transl Med. 2011;3:106ra106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadows AL, Karnik R, Lam Het al. . Application of dynamic flux balance analysis to an industrial Escherichia colifermentation. Metab Eng. 2010; 12:150–60. 10.1016/j.ymben.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Meijer K, de Vos P, Priebe MG. Butyrate and other short-chain fatty acids as modulators of immunity: what relevance for health?. Curr Opin Clin Nutr Metab Care. 2010;13:715–21. 10.1097/MCO.0b013e32833eebe5. [DOI] [PubMed] [Google Scholar]
- Mendoza SN, Olivier BG, Molenaar Det al. . A systematic assessment of current genome-scale metabolic reconstruction tools. Genome Biol. 2019;20:158. 10.1186/s13059-019-1769-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miquel S, Leclerc M, Martin Ret al. . Identification of metabolic signatures linked to anti-inflammatory effects of Faecalibacterium prausnitzii. mBio. 2015;6:e00300–15. 10.1128/mBio.00300-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinero N, Ruiz L, Sánchez Bet al. . Intestinal bacteria interplay with bile and cholesterol metabolism: implications on host physiology. Front Physiol. 2019;10. https://www.frontiersin.org/article/10.3389/fphys.2019.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Hernández MAG, Goossens GHet al. . Circulating but not faecal short-chain fatty acids are related to insulin sensitivity, lipolysis and glp-1 concentrations in humans. Sci Rep. 2019;9:12515. 10.1038/s41598-019-48775-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz S, Guzman-Rodriguez M, Sun Jet al. . Rebooting the microbiome. Gut Microbes. 2016;7:353–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TA, Vieira-Silva S, Liston Aet al. . How informative is the mouse for human gut microbiota research?. Dis Model Mech. 2015;8:1–16. 10.1242/dmm.017400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parada Venegas D, Fuente MKD, Landskron Get al. . Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol. 2019;10:277. 10.3389/fimmu.2019.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patnode ML, Beller ZW, Han NDet al. . Interspecies Competition Impacts Targeted Manipulation of Human Gut Bacteria by Fiber-Derived Glycans. Cell. 2019;179:59–73.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez M, Ntemiri A, Tan Het al. . A synthetic consortium of 100 gut commensals modulates the composition and function in a colon model of the microbiome of elderly subjects. Gut Microbes. 2021;13:1–19. 10.1080/19490976.2021.1919464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrof EO, Gloor GB, Vanner SJet al. . Stool substitute transplant therapy for the eradication of Clostridium difficile infection: ‘RePOOPulating’ the gut. Microbiome. 2013;1:3. 10.1186/2049-2618-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto F, Medina DA, Pérez-Correa JRet al. . Modeling metabolic interactions in a consortium of the infant gut microbiome. Front Microbiol. 2017;8:2507. 10.3389/fmicb.2017.02507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyet M, Groussin M, Gibbons SMet al. . A library of human gut bacterial isolates paired with longitudinal multiomics data enables mechanistic microbiome research. Nat Med. 2019;25:1442–52. 10.1038/s41591-019-0559-3. [DOI] [PubMed] [Google Scholar]
- Rajilić-Stojanović M, Vos WM. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol Rev. 2014;38:996–1047. 10.1111/1574-6976.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey FE, Gonzalez MD, Cheng Jet al. . Metabolic niche of a prominent sulfate-reducing human gut bacterium. Proc Natl Acad Sci USA. 2013;110:13582–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A, Wu M, McNulty NPet al. . Gnotobiotic mouse model of phage-bacterial host dynamics in the human gut. Proc Natl Acad Sci USA. 2013;110:20236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezzonico E, Mestdagh R, Delley Met al. . Bacterial adaptation to the gut environment favors successful colonization. Gut Microbes. 2011;2:307–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring C, Klopfleisch R, Dahlke Ket al. . Akkermansia muciniphila strain ATCC BAA-835 does not promote short-term intestinal inflammation in gnotobiotic interleukin-10-deficient mice. Gut Microbes. 2019;10:188–203. 10.1080/19490976.2018.1511663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinninella E, Raoul P, Cintoni Met al. . What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms. 2019;7:14. 10.3390/microorganisms7010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano KA, Vivas EI, Amador-Noguez Det al. . Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-Oxide. mBio. 2015;6:e02481–14. 10.1128/mBio.02481-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CL, Spinler JK, Savidge TC. Structural and functional changes within the gut microbiota and susceptibility to Clostridium difficile infection. Anaerobe. 2016;41:37–43. 10.1016/j.anaerobe.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M, Amaretti A, Raimondi S. Folate production by probiotic bacteria. Nutrients. 2011;3:118–34. 10.3390/nu3010118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said HM, Mohammed ZM. Intestinal absorption of water-soluble vitamins: an update. Curr Opin Gastroenterol. 2006;22:140–46. 10.1097/01.mog.0000203870.22706.52. [DOI] [PubMed] [Google Scholar]
- Sardelli L, Perottoni S, Tunesi Met al. . Technological tools and strategies for culturing human gut microbiota in engineered in vitro models. Biotechnol Bioeng. 2021;118:2886–905. 10.1002/bit.27816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJet al. . Single lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–65. 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- Schloissnig S, Arumugam M, Sunagawa Set al. . Genomic variation landscape of the human gut microbiome. Nature. 2013;493:45–50. 10.1038/nature11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt TSB, Li SS, Maistrenko OMet al. . Drivers and determinants of strain dynamics following fecal microbiota transplantation. Nat Med. 2022;28:1902–12. 10.1038/s41591-022-01913-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen P, Orešič M. Metabolic Modeling of human gut microbiota on a genome scale: an overview. Metabolites. 2019;9:E22. 10.3390/metabo9020022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanahan F, Ghosh TS, O'Toole PW. The healthy microbiome—what is the definition of a healthy gut microbiome?. Gastroenterology. 2021;160:483–94. 10.1053/j.gastro.2020.09.057. [DOI] [PubMed] [Google Scholar]
- Shin W, Kim HJ. Intestinal barrier dysfunction orchestrates the onset of inflammatory host–microbiome cross-talk in a human gut inflammation-on-a-chip. Proc Natl Acad Sci. 2018;115:E10539–47. 10.1073/pnas.1810819115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoaie S, Karlsson F, Mardinoglu Aet al. . Understanding the interactions between bacteria in the human gut through metabolic modeling. Sci Rep. 2013;3:2532. 10.1038/srep02532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva YP, Bernardi A, Frozza RL. The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front Endocrinol. 2020;11: 25. https://www.frontiersin.org/article/10.3389/fendo.2020.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slezak K, Hanske L, Loh Get al. . Increased bacterial putrescine has no impact on gut morphology and physiology in gnotobiotic adolescent mice. Benef Microbes. 2013;4:253–66. 10.3920/BM2012.0047. [DOI] [PubMed] [Google Scholar]
- Slezak K, Krupova Z, Rabot Set al. . Association of germ-free mice with a simplified human intestinal microbiota results in a shortened intestine. Gut Microbes. 2014;5:176–82. 10.4161/gmic.28203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EA, Macfarlane GT. Dissimilatory amino acid metabolism in human colonic bacteria. Anaerobe. 1997;3:327–37. 10.1006/anae.1997.0121. [DOI] [PubMed] [Google Scholar]
- Sokol H, Pigneur B, Watterlot Let al. . Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of crohn disease patients. Proc Nat Acad Sci USA. 2008;105:16731–36. 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley C, Kaiser T, Beura LKet al. . Stable engraftment of human microbiota into mice with a single oral gavage following antibiotic conditioning. Microbiome. 2017;5:87. 10.1186/s40168-017-0306-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stappenbeck TS, Hooper LV, Gordon JI. Developmental regulation of intestinal angiogenesis by indigenous microbes via paneth cells. Proc Natl Acad Sci. 2002;99:15451–55. 10.1073/pnas.202604299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearns JC, Lynch MDJ, Senadheera DBet al. . Bacterial biogeography of the human digestive tract. Sci Rep. 2011;1:170. 10.1038/srep00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein RR, Tanoue T, Szabady RLet al. . Computer-guided design of optimal microbial consortia for immune system modulation. eLife. 2018;7:e30916. 10.7554/eLife.30916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanoue T, Morita S, Plichta DRet al. . A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature. 2019;565:600–5. 10.1038/s41586-019-0878-z. [DOI] [PubMed] [Google Scholar]
- Thompson JA, Oliveira RA, Xavier KB. Chemical conversations in the gut microbiota. Gut Microbes. 2016;7:163–70. 10.1080/19490976.2016.1145374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlin KL, Malott RJ, Ramage Get al. . Quorum-sensing mutations affect attachment and stability of Burkholderia cenocepaciabiofilms. Appl Environ Microbiol. 2005;71:5208–18. 10.1128/AEM.71.9.5208-5218.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapecar M, Communal C, Velazquez Jet al. . Gut-liver physiomimetics reveal paradoxical modulation of ibd-related inflammation by short-chain fatty acids. Cell Syst. 2020;10:223–39.e9. 10.1016/j.cels.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulluwishewa D, Anderson RC, Young Wet al. . Live Faecalibacterium prausnitzii in an apical anaerobic model of the intestinal epithelial barrier. Cell Microbiol. 2015;17:226–40. 10.1111/cmi.12360. [DOI] [PubMed] [Google Scholar]
- Vázquez-Castellanos JF, Biclot A, Vrancken Get al. . Design of synthetic microbial consortia for gut microbiota modulation. Curr Opin Pharmacol. 2019;49:52–59. 10.1016/j.coph.2019.07.005. [DOI] [PubMed] [Google Scholar]
- Venturelli OS, Carr AC, Fisher Get al. . Deciphering microbial interactions in synthetic human gut microbiome communities. Mol Syst Biol. 2018;14:e8157. 10.15252/msb.20178157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versluis DM, Schoemaker R, Looijesteijn Eet al. . A multiscale spatiotemporal model including a switch from aerobic to anaerobic metabolism reproduces succession in the early infant gut microbiota. MSystems. 2022;7:e00446–22. 10.1128/msystems.00446-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Gunasekara DB, Reed MIet al. . A microengineered collagen scaffold for generating a polarized crypt-villus architecture of human small intestinal epithelium. Biomaterials. 2017;128:44–55. 10.1016/j.biomaterials.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss AS, Burrichter AG, Raj ACDet al. . In vitro interaction network of a synthetic gut bacterial community. ISME J. 2022;16:1095–109. 10.1038/s41396-021-01153-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BA, Grant LJ, Gidley MJet al. . Gut fermentation of dietary fibres: physico-chemistry of plant cell walls and implications for health. Int J Mol Sci. 2017;18:2203. 10.3390/ijms18102203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JMW, Souza R, Kendall CWCet al. . Colonic health: fermentation and short chain fatty acids. J Clin Gastroenterol. 2006;40:235–43. 10.1097/00004836-200603000-00015. [DOI] [PubMed] [Google Scholar]
- Wos-Oxley ML, Bleich A, Oxley APAet al. . Comparative evaluation of establishing a human gut microbial community within rodent models. Gut Microbes. 2012;3:234–49. 10.4161/gmic.19934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woting A, Pfeiffer N, Hanske Let al. . Alleviation of high fat diet-induced obesity by oligofructose in gnotobiotic mice is independent of presence of bifidobacterium longum. Mol Nutr Food Res. 2015;59:2267–78. 10.1002/mnfr.201500249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woting A, Pfeiffer N, Loh Get al. . Clostridium ramosum promotes high-fat diet-induced obesity in gnotobiotic mouse models. mBio. 2014;5:e01530–01514. 10.1128/mBio.01530-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrzosek L, Miquel S, Noordine M-Let al. . Bacteroides thetaiotaomicron and Faecalibacterium prausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. BMC Biol. 2013;11:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H-J, Wu E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes. 2012;3:4–14. 10.4161/gmic.19320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wymore Brand M, Wannemuehler MJ, Phillips GJet al. . The altered Schaedler flora: continued applications of a defined murine microbial community. ILAR J. 2015;56:169–78. 10.1093/ilar/ilv012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Rodriguez V, Malphurs WLet al. . Butyrate regulates inflammatory cytokine expression without affecting oxidative respiration in primary astrocytes from spontaneously hypertensive rats. Physiol Rep. 2018;6:e13732. 10.14814/phy2.13732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen S, McDonald JAK, Schroeter Ket al. . Metabolomic analysis of human fecal microbiota: a comparison of feces-derived communities and defined mixed communities. J Proteome Res. 2015;14:1472–82. [DOI] [PubMed] [Google Scholar]
- Zhang J, Huang Y-J, Yoon JYet al. . Primary human colonic mucosal barrier crosstalk with super oxygen-sensitive Faecalibacterium prausnitzii in continuous culture. Med. 2021;2:74–98.e9. 10.1016/j.medj.2020.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]