

Abstract
Background: Very early-onset inflammatory bowel disease (VEOIBD) is defined as IBD in children under six years of age. We present outcome data of hematopoietic stem cell transplantation (HSCT) in the above children. Patients and methods: We performed a retrospective study in children under six years of age who underwent HSCT for VEOIBD with an identified monogenic disorder from December 2012 to December 2020. Results: Of the 25 children included, the underlying diagnosis was IL10R deficiency (n = 4), Wiskott-Aldrich syndrome (n = 4), Leukocyte adhesion defect (n = 4), Hyper IgM syndrome (n = 3), Chronic granulomatous disease (n = 2), and one each with XIAP deficiency, severe congenital neutropenia, Omenn syndrome, Hyper IgE syndrome, Griscelli syndrome, MHC Class II deficiency, LRBA deficiency, and IPEX syndrome. Donors included a matched family donor in 10(40%); a matched unrelated donor in 8 (32%), haploidentical in 7 (28%) (T depleted 16%, T replete with post-transplant cyclophosphamide12%). Conditioning was myeloablative in 84% ofHSCTs. We documented engraftment in 22 (88%) children, primary graft failure in 2 children (8%), mixed chimerism in 6 (24%) children with mortality in 4/6 children. Children with a sustained chimerism of > 95% did not have recurrence of any features of IBD. Overall survival was 64%, with a median follow-up of 55 months. Mixed chimerism was associated with a significantly increased risk of mortality (p-value = 0.001). Conclusions: VEOIBD caused by monogenic disorders can be offered HSCT. Early recognition, optimal supportive care, and complete chimerism are essential components to achieving survival.
Keywords: HSCT, VEOIBD, LRBA deficiency, IPEX syndrome, IL10R deficiency, Haploidentical
Introduction
Inflammatory bowel disease (IBD) results from dysregulated intestinal immune responses in genetically susceptible patients [1, 2]. In most patients, IBD displays polygenic inheritance. However, recent advances in next-generation sequencing have revealed many cases of childhood IBD that result from monogenic inheritance in the context of inborn errors of immunity (IEI) [3]. Children under six years of age who develop IBD are categorized as having very early-onset inflammatory bowel disease (VEOIBD). Disease development prior to age two is termed the infantile subtype while neonatal disease is diagnosed in patients less than 28 days [4].
The high rate of consanguinity in India leads to an increased rate of VEOIBD, which has a reported incidence of 19.2% [5]. The complex pathophysiology and ensuing immune dysregulation in VEOIBD often result in a challenging clinical course [2]. Here we discuss a series of children with underlying monogenic IEI presenting as VEOIBD who underwent HSCT at our centre.
Patients and Methods
We performed a retrospective study, where inclusion criteria were children aged less than six years with a diagnosis of VEOIBD and a genetically proven underlying monogenic IEI, and who underwent HSCT from December 2012 to December 2020.Published guidelines and criteria were used for diagnosing VEOIBD [6, 7], and all children fulfilling the criteria underwent whole-exome sequencing. Exclusion criteria included children more than six years of age with polygenic IBD or without an identified monogenic disorder. The hospital ethics committee approved the study, and all families signed a detailed informed consent.
High resolution HLA typing was performed for the child, siblings, and parents. In case of non-availability of a fully matched family donor (MFD) and based upon the mode of inheritance of the disorder, we performed unrelated searches in all registries. A haploidentical family donor was considered in case of unavailability of a matched unrelated donor (MUD). All haplo-matched donors were screened for donor-specific anti-HLA antibodies. Peripheral blood stem cells (PBSC) was the preferred stem cell source of stem cells in view of earlier engraftment, thereby reducing the duration of neutropenia and resultant risk of sepsis and decreased risk of graft rejection as compared to bone marrow stem cells. Bone marrow stem cells were harvested in case of donor weight less than 15 kg or in case of any contraindication to PBSC collection.
The chemotherapy used in conditioning was predominantly myeloablative, particularly in conditions with underlying immune dysregulation. Myeloablative conditioning (MAC) included Thiotepa/Treosulphan/Fludarabine or Fludarabine/Busulphan in the case of matched related donors. Anti-thymocyte globulin (ATG) was added in the case of MUD donors. Haploidentical HSCT with post-transplant cyclophosphamide (PTCy) at a dose of 50 milligram/kilogram (mg/kg) on Day + 3 and + 4, included conditioning with Fludarabine/Melphalan. Children with underlying severe combined immune deficiency underwent HSCT with reduced-intensity conditioning (RIC), including Fludarabine/Treosulphan. When TCR alpha beta depletion was employed, conditioning was myeloablative with Fludarabine/Treosulphan/Thiotepa/ATG. GVHD prophylaxis included tacrolimus, and methotrexate on day + 1, 3, 6 for bone marrow and day + 1, 3, 6, 11 for peripheral blood stem cells grafts. A short tandem repeat sequence by a polymerase chain reaction was used in case of same-sex donor for assessment of donor chimerism and Fluorescence in situ hybridization in case of sex mismatched HSCT. Donor chimerism was assessed on engraftment around D + 17, followed by D + 30, + 60, +90, 6 months and 1 year post HSCT. The National Institute of Health (NIH) Consensus guidelines were utilized for scoring GVHD [8].
Statistical Analysis
Data were analysed using the IBM SPSS version 25.0 (SPSS, Inc., Chicago, IL). Continuous variables were represented as mean ± S.D and the non-normal data as median + interquartile range. The categorical data was represented as percentages. The comparison of normally distributed and non-normally distributed continuous variables was done using the independent sample t-test and Mann – Whitney U test. Categorical variables were compared using the Chi-squared or Fisher’s exact test. The statistical analysis was carried out at a 95% confidence interval (CI). P value < 0.05 was considered statistically significant.
Results
Patient demographics
A total of 25 children with VEOIBD underwent HSCT at our centre. The age ranged from 1 to 93 months with a median of 17 months. The male: female ratio was 2:1 with a median follow-up of 55 months. The underlying diagnosis was IL10R deficiency in 4 (16%), Wiskott-Aldrich syndrome (WAS) in4 (16%), Leukocyte adhesion defect (LAD) in 4 (16%), Hyper IgM syndrome in 3 (12%), Chronic granulomatous disease (CGD) in 2 (8%), and one (4%) each with X-linked inhibitor of apoptosis protein (XIAP)deficiency, severe congenital neutropenia with ELANE mutation, Omenn syndrome, Hyper IgE syndrome with DOCK8 mutation, Griscelli syndrome, MHC Class II deficiency, lipopolysaccharide-responsive and beige-like anchor protein (LRBA) deficiency, and immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. In addition, VEOIBD was of the neonatal type in 4 (16%), childhood type in 3 (12%), and infantile type in 18 (72%) children.
Pre-HSCT events
All children had features suggestive of IBD at diagnosis, including recurrent episodes of loose stools, recurrent gastrointestinal bleeding, and diffuse abdominal pain. All children had failure to thrive with weight for age below the third centile. The child with LRBA deficiency received Abatacept till the start of conditioning. The child with IPEX syndrome was on multiple hormone replacements, including thyroid supplements and hydrocortisone which were continued. The co-morbidities documented before HSCT included pneumonia in 9 (36%), perianal ulcers in 8 (32%), meningitis with Klebsiella pneumoniae in 1 (4%) child, and multiple endocrinopathies in the child with IPEX syndrome.
Graft source and conditioning.
Types of donors were matched family donor in 10 (40%), MUD in 8 (32%), T depleted haploidentical HSCT with TCR alpha/beta depletion in 4 (16%), and T replete haploidentical HSCT with PTCy in 3 (12%) children. Peripheral blood stem cells were the predominant source of stem cells in 18 (72%) HSCTs, with bone marrow in 4 (16%) and umbilical cord blood unit in 2 (8%). Conditioning regimen was MAC in 21 (84%) and RIC in 4 (16%) HSCTs. The median CD34 dose for the matched family and matched unrelated donor HSCTs was 5 × 106/kg recipient body weight. In the T cell depleted haploidentical HSCTs, the median CD34 dose was 10 × 106/kg recipient body weight. In the T cell replete haploidentical HSCTs, CD34 dose was infused at a median of 7 × 106/kg, while maintaining a CD3 cell dose above 1.5 × 108/kg recipient body weight.
Engraftment, chimerism and graft failure
Engraftment by D + 14 to + 21 was documented in 22 (88%) children. One child with Omenn syndrome died before engraftment due to carbapenem-resistant gram-negative sepsis. We confirmed primary graft failure in 2 children (8%); one child with Wiskott-Aldrich syndrome undergoing a haplo-HSCT with PTCy and another child with IL10R deficiency who underwent a T cell depleted haplo-HSCT.
Mixed chimerism with a drop in donor chimerism after D + 45 was documented in 6 (24%) children. The underlying diagnosis in the above six children included IL10R deficiency in 2, and one each with WAS, MHC Class II deficiency, LAD and DOCK8 deficiency. Three children, each with LAD, DOCK8 deficiency, and IL10R deficiency, succumbed to sepsis following drop in chimerism. In the child with LAD, a drop in chimerism to 50% was documented with the septic event; while in the child with DOCK8 deficiency, recurrence of loose, frequent stools with streaks of blood correlated with a chimerism of 77%. He improved transiently with supportive care, however, succumbed to a septic event. In the child with IL10R deficiency, drop in chimerism to 60% correlated with rapid deterioration, with frequent blood-stained loose stools, metabolic acidosis and eventual decompensated septic shock.
One child with IL10R deficiency was stable with mixed chimerism following four whole blood donor lymphocyte infusions. Donor chimerism was detected to be 97% at 3 months post HSCT. The child was clinically stable, and hence was on follow up. Subsequently, chimerism dropped to 93.4% at 5 months post HSCT, and first DLI at a dose of 1 × 105/kg recipient body weight of donor T cells were infused. Further whole blood DLIs at graded doses were performed at chimerism levels of 75% (6 months), 68.5% (7 months) and 45% (9 months). Donor chimerism checked at 11 months post HSCT was 95% and child was clinically well.
One child with WAS underwent a haplo-HSCT following secondary graft failure of a MUD graft. The conditioning regimen during the first HSCT included fludarabine/busulphan/anti-thymocyte globulin. Conditioning regimen for the second HSCT included fludarabine/melphalan/PTCy. This patient did not engraft and family opted not to attempt a third transplant and continued best supportive care. The baby succumbed to the illness.
One child with MHC class II deficiency underwent a successful second matched sibling donor HSCT with a MAC regimen including fludarabine/treosulphan/thiotepa and the same donor. The first HSCT was conditioned with a reduced intensity regimen including fludarabine and treosulphan.
Supportive care
All children had documented hypokalemia and hypomagnesemia and were on regular corrections. All children were on parenteral nutrition with 50% dextrose, amino acids, and SMOF lipid infusions, and an elemental diet was allowed orally. In addition, we used continuous nasogastric feeds with semi-elemental feeds for all four children with IL10R deficiency. All children were on antifungal prophylaxis with micafungin or voriconazole based on the underlying condition and prior documented fungal infection, and antiviral prophylaxis with acyclovir.
Post HSCT complications
We documented neutropenic enterocolitis in 16 (64%) children, with Acinetobacterbaumanii and Klebsiellapneumoniae isolated in 2 (4%) children each. In addition, hematochezia during the neutropenic period was noted in the child with LAD type III. Seizures and posterior reversible encephalopathy syndrome (PRES) were documented in 2 (4%) children. All children recovered from the above complications except one with Omenn syndrome, who died of Acinetobacter sepsis before engraftment. Two children (4%) with Hyper IgM syndrome had documented Cryptosporidium parvum in stool and responded to nitazoxanide.
The cumulative incidence of acute GVHD at D + 100 was 4/25 (16%). Acute grade IV gut GVHD was recorded in two children with CGD. Both the children were treated with multiple lines of treatment including methylprednisolone, ruxolitinib and mycophenolate mofetil. Additionally, one child received etanercept. Both the children succumbed to grade IV gut GVHD. Grade I/II skin GVHD was documented in two children, which responded to steroids. In both these children, dose of calcineurin inhibitors were optimized and topical steroid ointment was also used.
Chronic mouth, skin, and eye (NIH score 2) were noted in 2 (4%) children. Calcineurin inhibitors were continued and both children responded to prednisolone continued for over 18 months at a dose of 1 mg/kg/day. They tolerated gradual steroid taper once clinical improvement was noted.
We documented cytomegalovirus reactivation in 6 (24%) children. Two children were diagnosed with Leukocyte adhesion defect, and one each with CGD, WAS, Hyper IgM syndrome and XIAP deficiency. Cytomegalovirus reactivation with a maximum viral copy number up to 5000 was detected after D + 30 post HSCT in all children and responded to 14 days of ganciclovir at a dose of 5 mg/kg/dose twice a day in all children. None of the above children had a drop in chimerism in relation to viral reactivation.
Survival and mortality
Overall survival in the cohort was 64%, with a median follow-up of 55 months (Fig. 1). Among the 9 (36%) children who died, the underlying diagnosis was CGD in 2, WAS in 2, IL10R deficiency in 2, and one each with LAD, DOCK8 deficiency, and Omenn syndrome. Causes of death included primary graft failure in 2 (4%), secondary graft failure in 4 (8%), grade IV gut GVHD in 2 (4%) and pre-engraftment gram-negative sepsis in 1 (2%). Mixed chimerism was associated with a significantly increased risk of mortality (p-value = 0.001).
Fig. 1.
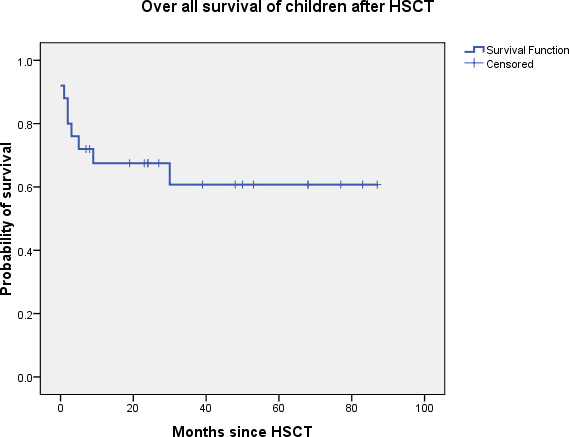
Kaplan Meier survival curve depicting overall survival of 64% with a median follow up of 55 months
Follow-up
Children with chimerism > 95%, sustained after 6 months post HSCT, had normal formed stools without any evidence of blood, and did not have recurrence of any clinical features based on validated IBD disease scoring systems.
The patient, disease, and HSCT details have been represented in Table 1.
Table 1.
Patients, disease and HSCT details of children included in the cohort n = 25
| Patient number | Diagnosis | Type of IBD | Age at the time of HSCT | Conditioning regimen | Source of stem cells | Donor type | Engraftment/ Chimerism | Follow up/ Months since HSCT |
|---|---|---|---|---|---|---|---|---|
| Patient 1 | LAD 1 | Infantile | 9 months | FLU, TREO | PBSC | MFD | Engrafted, MC, late graft rejection | Died/ 7 months |
| Patient 2 | CGD | Infantile | 2-year 1 month | FLU, BU, ATG | PBSC | MUD |
Engrafted, CC |
Died of grade IV GVHD/ 2 months |
| Patient 3 | LAD 3 | Neonatal | 2 year 5 months | FLU, TREO | PBSC | MFD |
Engrafted. CC |
Alive/ 41 months |
| Patient 4 | XIAP deficiency | Neonatal | 1 month | TT, FLU, TREO, ATG | PBSC | Haplo HSCT with TCR alpha/beta depletion |
Engrafted, CC |
Alive/ 30 months |
| Patient 5 | LAD 1 | Neonatal | 6 months | TT, FLU, TREO, ATG | PBSC | Haplo HSCT with TCR alpha/beta depletion |
Engrafted, CC |
Alive/ 29 months |
| Patient 6 | CGD | Infantile | 17 months | FLU, MEL | PBSC | Haplo HSCT with PTCy |
Engrafted, CC |
Died of grade IV GVHD/ 4months |
| Patient 7 | Severe congenital neutropenia with ELANE mutation | Childhood | 10years | TT, FLU, TREO | PBSC | MFD |
Engrafted, CC |
Alive/ 22 months |
| Patient 8 | LAD 2 | Neonatal | 4 months | TT, FLU, TREO, ATG | PBSC | MUD |
Engrafted, CC |
Alive/ 11 months |
| Patient 9 | Hyper IgM syndrome | Childhood | 5 years | FLU, BU | Bone marrow | MSD |
Engrafted, CC |
Alive/ 89 months |
| Patient 10 | Omenn syndrome | Infantile | 5 months | FLU, TREO | Bone marrow | MSD | Died before engraftment | Died of sepsis/ D + 8 |
| Patient 11 | WAS | Infantile | 16 months | FLU, MEL | PBSC | Haplo HSCT with PTCy |
Engrafted, CC |
Alive/ 72 months |
| Patient 12 | Hyper IgM syndrome | Infantile | 5 years | FLU, BU, ATG | PBSC | MUD |
Engrafted, CC |
Alive/ 86 months |
| Patient 13 | Hyper IgE syndrome with DOCK8 mutation | Infantile | 12 months | FLU, TREO | Bone marrow | MSD |
Engrafted, MC |
Died of sepsis/ 30 months |
| Patient 14 | WAS | Infantile | 2 years 9 months | FLU, BU, ATG | PBSC | MUD |
Engrafted, CC |
Alive/ 56 months |
| Patient 15 | Hyper IgM syndrome | Childhood | 5 years | TT, FLU, TREO | PBSC | MSD |
Engrafted, CC |
Alive/ 53 months |
| Patient 16 | WAS | Infantile | 2 years 1 month |
FLU, BU, ATG (1st HSCT) FLU, MEL (2nd HSCT) |
PBSC PBSC |
MUD Haplo HSCT with PTCy |
Engrafted, MC Primary graft failure |
Alive Died of sepsis/ D + 32 |
| Patient 17 | LRBA deficiency | Infantile | 21 months | TT, FLU, TREO | Bone marrow | MSD |
Engrafted, CC |
Alive/ 28 months |
| Patient 18 | MHC class II defect | Infantile | 2 years 8 months |
FLU, TREO (1st HSCT) TT, FLU, TREO (2nd HSCT) |
PBSC | MFD |
MC, Rejection Engrafted, CC |
Alive Alive/ 29 months |
| Patient 19 | WAS | Infantile | 2 years 8 months | FLU, BU | PBSC | Haplo HSCT with PTCy | Primary graft failure | Died of sepsis/ D + 35 |
| Patient 20 | IPEX syndrome | Infantile | 17months | FLU, BU, ATG | Umbilical cord blood unit | MUD |
Engrafted, CC |
Alive/ 92 months |
| Patient 21 | Griscelli syndrome | Infantile | 23 months | FLU, TREO, ATG | Umbilical cord blood unit | MUD |
Engrafted, CC |
Alive/ 88 months |
| Patient 22 | IL 10R deficiency | Infantile | 12 months | TT, FLU, TREO, ATG | PBSC | Haplo HSCT with TCR alpha/beta depletion |
Engrafted, CC |
Alive/ 53 months |
| Patient 23 | IL 10R deficiency | Infantile | 2 years | TT, FLU, TREO, ATG | PBSC | MUD |
Engrafted, MC |
Died of sepsis/ 4 months |
| Patient 24 | IL 10R deficiency | Infantile | 8 months | TT, FLU, TREO, ATG | PBSC | Haplo HSCT with TCR alpha/beta depletion | Primary graft failure | Died of sepsis/ D + 30 |
| Patient 25 | IL 10Rdeficiency | Infantile | 10 months | TT, FLU, TREO | PBSC | MFD |
Engrafted, MC |
Alive/ 11 months |
HSCT = hematopoietic stem cell transplantation, TT = thiotepa, TREO = treosulphan, FLU = fludarabine, BU = busulphan, ATG = anti-thymocyte globulin, CC = complete chimerism, MC = mixed chimerism, MSD = matched sibling donor, MFD = matched family donor, MUD = matched unrelated donor, Haplo HSCT = haploidentical HSCT, PBSC = peripheral blood stem cells, GVHD = graft versus host disease, LAD = leukocyte adhesion defect, CGD = chronic granulomatous disease, WAS = Wiskott-Aldrich syndrome, IPEX = immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome, XIAP = X-linked inhibitor of apoptosis protein, LRBA = lipopolysaccharide-responsive and beige-like anchor protein
Discussion
The study reports outcome data of HSCT in children with monogenic VEOIBD, with overall survival of 64%. Mixed chimerism was documented in 24% of children, which was significantly associated with a poor outcome (p value = 0.001). Complete resolution of IBD was observed in children with sustained complete donor chimerism.
Over 50 genes have been implicated in VEOIBD [9]. In addition, a recent publication has identified a novel exon deletion in the IL10RA gene, thus expanding the diagnostic pool [10]. Few groups have published data on HSCT in VEOIBD, including XIAP mutation and IL10R defects [11–13].
Myeloablative conditioning regimen was reported to be superior to RIC regimens [14, 15]. Worth et al. reported HSCT outcomes with a reduced toxicity conditioning with alemtuzumab, fludarabine, and treosulphan/melphalan in children with intractable enteropathy. In the above study, 26 children underwent 27 HSCTs, where 19 patients are alive at a median follow up of 51 months, and sustained improvement in enteropathy was noted in 17 of these children [16]. Rao et al. reported successful HSCT in IPEX syndrome with a resolution of symptoms in 4 patients with an alemtuzumab-based RIC regimen [17]. We used a MAC regimen in all patients in our cohort except for those with severe combined immune deficiency.
Early HSCT is crucial to alleviate IBD features and improve extraintestinal manifestations in children with monogenic VEOIBD. Patient selection is key to an optimal outcome. Lekbua et al. have identified colitis at an early age, failure to thrive, and organomegaly as risk factors for poor outcomes in children with VEOIBD and XIAP mutation and recommend early HSCT in this sub-group of patients [18]. Children with XIAP deficiency can continue to have persistent IBD and autoimmune features post a successful HSCT as well [19, 20].
Pre HSCT stabilization in conjunction with pediatric gastroenterologists is essential. Exclusive enteral nutrition and amino acid-based elemental formula have been reported as first-line treatments for VEOIBD [21, 22]. In addition, several targeted therapies have been studied as a bridge to allow HSCT in children with VEOIBD and inborn errors of immunity (IEI). Bakhtiar et al. have highlighted the need for targeted therapies prior to HSCT in patients with PIDD and immune dysregulation, in view of worse outcomes when patients underwent HSCT with advanced stages of poorly controlled infections and autoimmunity [23]. Abatacept is an effective CTLA4 agonist in children with CTLA4 and LRBA deficiencies [24]. Shouval et al. demonstrated that one of the pathogeneses of intestinal inflammation in IL10R defects includes loss of IL10 signalling through increased production of IL1 and the subsequent activation of CD4 + T cells [25]. Anti-IL1 therapy, including anakinra and canakinumab, has been used in sick children with IL10R defects. Perianal disease has been reported to be common in VEOIBD, particularly with IL10- and IL10- receptor signalling defects [26, 27]. Surgical procedures, particularly diverting colostomies, have been used in children with IBD [27, 28]. In our cohort, the child with LRBA deficiency was on Abatacept until HSCT, which has been stopped since. One child with an IL10R defect had undergone a colostomy before diagnosis in view of perianal disease, and was taken up for HSCT with the stoma.
Conclusion
VEOIBD caused by monogenic disorders can be offered HSCT. Children with complete chimerism of > 95% did not have any IBD related symptoms post-HSCT, and only those with mixed chimerism manifested gastrointestinal symptoms. Early recognition and optimal supportive care are essential components to achieving survival. Understanding the pathophysiology of these complex disorders would likely improve disease-free survival further.
Acknowledgements
We would like to acknowledge the immense contribution of the pediatric critical care team, stem cell pheresis, infectious disease specialists, and pediatric gastroenterologists in the management of these children.
Abbreviations
- VEOIBD
Very early onset inflammatory bowel disease
- HSCT
Hematopoietic stem cell transplantation
- GVHD
Graft versus host disease
- MUD
Matched unrelated donor
- MFD
Matched family donor
- MAC
Myeloablative conditioning
- RIC
Reduced intensity conditioning
- CMV
Cytomegalovirus
- EBV
Epstein Barr virus
- ATG
Anti thymocyte globulin
- IEI
Inborn errors of immunity
Authors’ contributions
SKM, RU, RR conceptualized the design of the study and wrote the manuscript; VVS, RC, HV helped with data collection and data entry; IJ helped with proof reading; BR did the statistical analysis.
Funding
None.
Declarations
Conflict of Interest
None.
Conflict of Interest
There are no conflicts of interest.
Footnotes
All co-authors have reviewed the manuscript and have contributed in a substantive and intellectual manner to the work described.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Snapper SB (2015 Aug) Very Early-Onset Inflammatory Bowel Disease. Gastroenterol Hepatol (N Y). 11:554–556 PMID: 27118953; PMCID: PMC4843045. 8 [PMC free article] [PubMed]
- 2.Kelsen JR, Russo P, Sullivan KE (2019 Feb) Early-Onset Inflammatory Bowel Disease. Immunol Allergy Clin North Am. 39:63–79. 10.1016/j.iac.2018.08.008. PMID: 30466773; PMCID: PMC6954002 1 [DOI] [PMC free article] [PubMed]
- 3.Almana Y, Mohammed R (2019 Mar) Current concepts in pediatric inflammatory bowel disease; IL10/IL10R colitis as a model disease. Int J Pediatr Adolesc Med 6(1):1–5. DOI: 10.1016/j.ijpam.2019.02.002 Epub 2019 Mar 12. PMID: 31304220; PMCID: PMC6603158 [DOI] [PMC free article] [PubMed]
- 4.Nameirakpam J, Rikhi R, Rawat SS et al (2019) Genetics on early onset inflammatory bowel disease: An update. Genes Dis. Oct 15;7(1):93–106. doi: 10.1016/j.gendis.2019.10.003. PMID: 32181280; PMCID: PMC7063406 [DOI] [PMC free article] [PubMed]
- 5.Srivastava A, Sathiyasekharan M, Jagadisan B et al (2020) Oct;32(10):1305–1311 Pediatric inflammatory bowel disease in India: a prospective multicentre study. Eur J Gastroenterol Hepatol. DOI: 10.1097/MEG.0000000000001859. PMID: 32796356 [DOI] [PubMed]
- 6.Kelsen JR, Sullivan KE, Rabizadeh S et al (2020) Mar;70(3):389–403 North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition Position Paper on the Evaluation and Management for Patients With Very Early-onset Inflammatory Bowel Disease. J Pediatr Gastroenterol Nutr. DOI: 10.1097/MPG.0000000000002567. PMID: 32079889 [DOI] [PMC free article] [PubMed]
- 7.Uhlig HH, Schwerd T, Koletzko S, COLORS in IBD Study Group and NEOPICS et al (2014 Nov) The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology 147(5):990–1007e3. DOI: 10.1053/j.gastro.2014.07.023 Epub 2014 Jul 21. PMID: 25058236; PMCID: PMC5376484 [DOI] [PMC free article] [PubMed]
- 8.Jagasia MH, Greinix HT, Arora M et al (2015 Mar) National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant 21(3):389–401e1 Epub 2014 Dec 18. PMID: 25529383; PMCID: PMC4329079 [DOI] [PMC free article] [PubMed]
- 9.Zheng HB, de la Morena MT, Suskind DL The Growing Need to Understand Very Early Onset Inflammatory Bowel Disease.Front Immunol. 2021 May26;12:675186. DOI: 10.3389/fimmu.2021.675186. PMID: 34122435; PMCID: PMC8187749. [DOI] [PMC free article] [PubMed]
- 10.Tang Z, Zhang P, Ji M et al (2021) Characterization of novel and large fragment deletions in exon 1 of the IL10RA gene in Chinese children with very early onset inflammatory bowel diseases. BMC Gastroenterol. Apr 13;21(1):167. DOI: 10.1186/s12876-021-01756-y. PMID: 33849446; PMCID: PMC8045347 [DOI] [PMC free article] [PubMed]
- 11.Cifaldi C, Chiriaco M, Di Matteo G et al Novel X-linked Inhibitor of Apoptosis Mutation in Very Early-Onset Inflammatory Bowel Disease Child Successfully Treated with HLA-Haploidentical Hematopoietic Stem Cells Transplant after Removal of αβ + T and B Cells.Front Immunol. 2017 Dec22;8:1893. DOI: 10.3389/fimmu.2017.01893. PMID: 29312354; PMCID: PMC5743702. [DOI] [PMC free article] [PubMed]
- 12.Murugan D, Albert MH, Langemeier J et al (2014 Apr) Very early onset inflammatory bowel disease associated with aberrant trafficking of IL-10R1 and cure by T cell replete haploidentical bone marrow transplantation. J Clin Immunol 34(3):331–339. doi: 10.1007/s10875-014-9992-8 Epub 2014 Feb 12. PMID: 24519095 [DOI] [PubMed]
- 13.Kotlarz D, Beier R, Murugan D et al (2012 Aug) Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology 143(2):347–355. doi: 10.1053/j.gastro.2012.04.045 Epub 2012 Apr 28. PMID: 22549091 [DOI] [PubMed]
- 14.Zhu L, Shi T, Zhong C et al IL-10 and IL-10 Receptor Mutations in Very Early Onset Inflammatory Bowel Disease. Gastroenterology Res. 2017 Apr;10(2):65–69. DOI: 10.14740/gr740w. Epub 2017 Apr 19. PMID: 28496525; PMCID: PMC5412537 [DOI] [PMC free article] [PubMed]
- 15.Karaca NE, Aksu G, Ulusoy E, et al. Early Diagnosis and Hematopoietic Stem Cell Transplantation for IL10R Deficiency Leading to Very Early-Onset Inflammatory Bowel Disease Are Essential in Familial Cases. Case Rep Immunol. 2016;2016:5459029. doi: 10.1155/2016/5459029.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Worth A, Nademi Z, Kammermeier J et al (2014) vol. 20 2 (pg. S89-S90)
- 17.Rao A, Kamani N, Filipovich A et al Successful bone marrow transplantation for IPEX syndrome after reduced-intensity conditioning. Blood. 2007 Jan 1;109(1):383-5. DOI: 10.1182/blood-2006-05-025072. Epub 2006 Sep 21. PMID: 16990602 [DOI] [PubMed]
- 18.Lekbua A, Ouahed J, O’Connell AE et al (2019 Jul) Risk-factors Associated With Poor Outcomes in VEO-IBD Secondary to XIAP Deficiency: A Case Report and Literature Review. J Pediatr Gastroenterol Nutr 69(1):e13–e18. DOI: 10.1097/MPG.0000000000002297 PMID: 31232887; PMCID: PMC6607918 [DOI] [PMC free article] [PubMed]
- 19.Marsh RA, Rao K, Satwant P et al (2013) Allogeneic hematopoietic cell transplantation for XIAP deficiency: an international survey reveals poor outcomes. Blood. Feb 7;121(6):877 – 83. DOI: 10.1182/blood-2012-06-432500. Epub 2012 Nov 6. PMID: 23131490; PMCID: PMC5162550 [DOI] [PMC free article] [PubMed]
- 20.Muller N, Fischer JC, Yabal M, et al. XIAP deficiency in hematopoietic recipient cells drives donor T-cell activation and GvHD in mice. Eur J Immunol. 2019;49:504–507. doi: 10.1002/eji.201847818. [DOI] [PubMed] [Google Scholar]
- 21.Ouched J, Spencer E, Kotlarz D et al (2020) Very Early Onset Inflammatory Bowel Disease: A Clinical Approach With a Focus on the Role of Genetics and Underlying Immune Deficiencies. Inflamm Bowel Dis. May 12;26(6):820–842. DOI: 10.1093/ibd/izz259. PMID: 31833544; PMCID: PMC7216773 [DOI] [PMC free article] [PubMed]
- 22.Arai K (2020 Sep) Very Early-Onset Inflammatory Bowel Disease: A Challenging Field for Pediatric Gastroenterologists. Pediatr Gastroenterol Hepatol Nutr. 23:411–422. 10.5223/pghn.2020.23.5.411. Epub 2020 Aug 27. PMID: 32953636; PMCID: PMC7481055 5 [DOI] [PMC free article] [PubMed]
- 23.Bakhtiar S, Fekadu J, Seidel MG et al Allogeneic Hematopoietic Stem Cell Transplantation for Congenital Immune Dysregulatory Disorders.Front Pediatr. 2019 Nov13;7:461. doi: 10.3389/fped.2019.00461. PMID: 31799221; PMCID: PMC6865355. [DOI] [PMC free article] [PubMed]
- 24.Lo B, Zhang K, Lu W et al (2015) AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science. Jul 24;349(6246):436 – 40. DOI: 10.1126/science.aaa1663. PMID: 26206937 [DOI] [PubMed]
- 25.Shouval DS, Biswas A, Kang YH et al (2016 Dec) Interleukin 1β Mediates Intestinal Inflammation in Mice and Patients With Interleukin 10 Receptor Deficiency. Gastroenterology 151(6):1100–1104. DOI: 10.10 [DOI] [PMC free article] [PubMed]
- 26.Begue B, Verdier J, Rieux-Laucat F et al (2011 Aug) Defective IL10 signaling defining a subgroup of patients with inflammatory bowel disease. Am J Gastroenterol 106(8):1544–1555. doi: 10.1038/ajg.2011.112 Epub 2011 Apr 26. PMID: 21519361 [DOI] [PubMed]
- 27.Kerur B, Benchimol EI, Fiedler K et al Natural History of Very Early Onset Inflammatory Bowel Disease in North America: A Retrospective Cohort Study. Inflamm Bowel Dis. 2021 Feb 16;27(3):295–302. DOI: 10.1093/ibd/izaa080. PMID: 32386060; PMCID: PMC8177809 [DOI] [PMC free article] [PubMed]
- 28.Maxwell EC, Dawany N, Baldassano RN et al (2017 Sep) Diverting Ileostomy for the Treatment of Severe, Refractory, Pediatric Inflammatory Bowel Disease. 65:299–305 DOI: 10.1097/MPG.0000000000001498. PMID: 28045769.53/j.gastro.2016.08.055. Epub 2016 Sep 28. PMID: 27693323; PMCID: PMC5124405. J Pediatr Gastroenterol Nutr3 [DOI] [PubMed]