

Abstract
Vaccines have been a hugely successful public health intervention, virtually eliminating many once common diseases of childhood. However, they have had less success in controlling endemic pathogens including Mycobacterium tuberculosis, herpesviruses and HIV. A focus on vaccine-mediated generation of neutralizing antibodies, which has been a successful approach for some pathogens, has been complicated by the emergence of escape variants, which has been seen for pathogens such as influenza viruses and SARS-CoV-2, as well as for HIV-1. We discuss how vaccination strategies aimed at generating a broad and robust T cell response may offer superior protection against pathogens, particularly those that have been observed to mutate rapidly. In particular, we consider here how a focus on generating resident memory T cells may be uniquely effective for providing immunity to pathogens that typically infect (or become reactivated in) the skin, respiratory mucosa or other barrier tissues.
Subject terms: Vaccines, T cells
In this Review, Rotrosen and Kupper focus on the generation of tissue resident memory T cells (TRM cells) by vaccines. They discuss our current understanding of TRM cell generation by different vaccine platforms and explain why focusing on this population of cells is important for future vaccination strategies.
Introduction
Vaccines are one of the most effective and inexpensive public health interventions after clean water and hand hygiene1,2. They have been instrumental in the elimination of polio in the United States and in the eradication of smallpox worldwide3. Vaccine immunogenicity has historically been judged by antibody titres, which are often relied upon as a surrogate marker of protection4. This is based on their proven clinical association with protective immunity and the ease, speed and reproducibility of such assessments across laboratories. Antibody assays require small volumes of blood, remain stable over time with banked sera and are readily commercialized. Efforts to understand the role of T cell immunity elicited by vaccines have generally focused on T cells circulating in peripheral blood, and these studies are technically much more challenging and involve many more variables5,6. In the present Review, we focus on whether a recently described subset of memory T cells — namely tissue resident memory T cells (TRM cells) — may also have a key role in vaccine-induced protective immunity.
Although sampling blood provides a useful approximation of systemic humoral immunity, the recent appreciation that most memory T cells reside in peripheral tissues highlights the need for better characterization of immune cells within tissues and organs7. There is a greater awareness of the subsets of memory T cells that do not recirculate, namely TRM cells, and a growing appreciation of their role in immune homeostasis and protection7–11. TRM cells and other T cells in tissues greatly outnumber circulating T cells; for example, each square centimetre of human skin is home to one million T cells, and phenotypical analyses suggest that more than 50% of these are TRM cells, making skin TRM cells twice as abundant as circulating memory T cells in the peripheral blood10. TRM cells and other tissue T cells also provide rapid and potent recall responses in the skin and mucosal tissues12. However, obtaining useable numbers of T cells from tissue in patients is not straightforward and most methods are not available outside a handful of academic laboratories, posing challenges for studies in humans. Much of what we know about TRM cells has come from mouse models, which for many reasons are only partially translatable to human biology8,10,13,14. In this Review, we first highlight some key features in the biology of TRM cells. We then discuss how to enhance their role in vaccine-induced immune protection, and propose that a focus on tissue T cells, including TRM cells, should be more routinely integrated into early-stage vaccine research and development.
T cells in tissue and TRM cell biology
TRM cells were initially described, somewhat provocatively, in the setting of systemic viral infection15,16. Although it was already known that effector memory T cells (TEM cells) are found in peripheral tissues, TRM cells are a distinct subset of memory T cells characterized by long-term residency in peripheral non-lymphoid tissues. TRM cell populations from different tissue sites have been shown to share a core gene expression profile, as well as to have tissue-specific differences in gene expression17–21. Other memory T cells that are found in the extravascular space of tissue are heterogeneous; in human skin, T cells with relatively shorter and longer ‘dwell’ times in skin have been identified22. These non-TRM cell populations eventually exit the skin and enter peripheral blood. This is in contrast to almost all TRM cells, which remain as long-term residents within the tissue23. Both CD4+ and CD8+ TRM cell populations have been described in skin, with CD8+ TRM cells associated with antiviral immunity and CD4+ TRM cells more closely linked with immunity to bacteria and fungi24–26. A comparison of the transcriptional profiles of resident and circulating T cells from multiple tissues demonstrated that TRM cells from sites across the body (for example, lungs, skin and gut) have a core conserved transcriptional signature that distinguishes them from both TEM cells and central memory T cells (TCM cells), although the markers originally used to discriminate these cells from each other nearly 20 years ago have limitations17,27,28. The transcriptomes of TEM cells and TCM cells reveal that these cell types more closely resemble each other than they do TRM cells17,20,29. Their anatomic location allows TRM cells to act as immunological sentries, functioning as ‘alarm’ cells that are programmed to persist in tissues and elicit a rapid recall immune response upon antigen encounter30,31 (Fig. 1). Fundamentally, they have been shown to provide enhanced immunity against re-infection and to accelerate pathogen clearance32,33.
Fig. 1. CD8+ TRM cell reactivation upon secondary pathogen encounter.

Tissue resident memory T cells (TRM cells) are uniquely positioned within tissues to respond rapidly to pathogen re-encounter, and this response is multifaceted. Upon recognition of cognate antigen, which is presented to them by epithelial cells or antigen-presenting cells (APCs), CD8+ TRM cells rapidly secrete inflammatory cytokines (for example, IFNγ and TNF). The downstream effects of these cytokines include upregulation of adhesion molecules on endothelial cells (such as VCAM1, ICAM1 and E-selectin) and expression of chemokines, which in turn facilitates recruitment of circulating lymphocytes (B cells and T cells), dendritic cell maturation, and the recruitment and activation of natural killer cells. In addition, some TRM cells express high levels of granzyme B at baseline, leading to direct lysis of infected host cells. Reactivation leads to TRM cell proliferation within tissues as well as recruitment of peripheral lymphocytes that become TRM cells. TCM cell, central memory T cell; TEM cell, effector memory T cell.
Identification of TRM cells by surface phenotype has been debated for some time, and certain markers that are closely associated with TRM cell biology (for example, CD69 and CD103) are neither universally nor continuously expressed by TRM cells34. One controversy regarding TRM cells as originally defined is the absence of recirculation once they have taken up tissue residence8,11,35,36. Although this can be measured in mouse models by parabiosis or treatment with trafficking inhibitors, it is considerably more difficult to assess in humans. More recently, this property of indefinite residence has been challenged; there are reports of (formerly) TRM cells leaving tissue and entering the circulation37,38. Whether TRM cells represent a terminally differentiated population that cannot leave tissue or whether there is built in plasticity to reverse some elements of the TRM cell ‘programme’ is increasingly debated39. It could be argued that the original requirement that TRM cells can ‘never’ migrate out of tissue does not allow for the potential for plasticity of biological systems. A better question is under what conditions certain TRM cells can leave tissue, and what adaptive immune advantage is conferred. We will attempt to address this question below.
Development of TRM cells
TRM cell development continues to be studied extensively. Two general models of TRM cell development from naive T cell precursors have emerged, which for simplicity we will term the ‘local divergence’ and the ‘systemic divergence’ models (Fig. 2). The local divergence model proposes that pluripotent effector T cells enter tissues and are influenced by local signals to differentiate into TRM cells and establish long-term tissue residence22,27,29,40. In contrast, the systemic divergence model proposes that there is a subpopulation of circulating effector T cells in blood that are already poised to enter the tissue and differentiate into TRM cells. In this model, a subset of T cells are preconditioned to have greater capacity to migrate into inflamed tissue and to respond to the environmental signals within the tissue that drive TRM cell differentiation27,39,41–44. Upon reflection, these two models need not be mutually exclusive. Evidence supporting the notion that precursors of TRM cells undergo their maturation after antigen encounter and, in addition, differentiate further in peripheral tissues includes reports on the common clonal origin of TCM cells and TRM cells (bearing the same CDR3 sequence as assessed by high throughput sequencing) following skin immunization42. It remains unknown exactly which factors prime some incompletely differentiated effector T cells to acquire a TRM cell identify when exposed to tissue microenvironments, although transforming growth factor-β (TGFβ) appears to be a dominant signal19. It is also not clear why other effector T cells do not respond to these tissue factors and, instead, develop into mature circulating TEM cells and TCM cells or else undergo apoptosis in the tissue. It is unlikely that this differentiation process is simply stochastic. A recent study using single-cell RNA sequencing analysis of gut TRM cells over time was unable to identify circulating precursors of TRM cells before the effector T cells entered the tissue45, supporting the concept of local divergence. In favour of the systemic divergence model, a subset of phenotypically distinct circulating mature T cells have been identified; these are presumed to be dedicated TRM precursor cells as they share more than 90% of the transcriptional profile of authentic TRM cells in tissue39,46. This subset is made up of T cells that differ from each other transcriptionally in accordance with the tissue to which they home27,39,46,47. It is proposed that these TRM cell precursors in peripheral blood have been imprinted with tissue-specific homing molecules shortly after antigen encounter in the lymph node48. A model that is a compromise between these two extremes proposes that in skin (for example), activated dendritic cells of the classical DC1 subset that express TGFβ migrate to draining lymph nodes and cross-present antigens to activate naive T cells47,48; these activated T cells divide asymmetrically, with some progeny being directed towards a TRM cell programme19 whereas others retain plasticity and become TCM cells42, including some TCM cells expressing skin-homing markers49. Once resident in tissue, TRM cells are poised to participate in host defence. It is increasingly appreciated, however, that some TRM cells may develop the capacity to respond to antigenically related autoantigens, contributing to autoimmune disease in joints and skin, either spontaneously or after therapy with immune checkpoint inhibitors8–11,50–52.
Fig. 2. Two models of TRM cell divergence.

a, Recent studies propose there is a population of ‘circulating TRM cell precursors’ that are phenotypically distinct from central memory T cells (TCM cells), effector memory T cells (TEM cells) and tissue resident memory T cells (TRM cells). The transcriptional profiles of these putative circulating TRM cell precursors resemble those of TRM cells, and the cells are thought to be destined to become TRM cells. This is in line with the model of systemic divergence. b, Other data support the theory of local divergence of TRM cells, in which TRM cells differentiate within tissues from multipotent or pluripotent effector T cells in the early stages of the immune response. These two theories should be viewed as potentially complementary, and that there may, in fact, be significant overlap between these theories in TRM cell development in vivo.
What defines a TRM cell?
Retention of TRM cells within tissues is a precondition for residency and is mediated by a combination of variables. Some of the first changes are downregulation of the transcription factor Kruppel-like factor 2 (KLF2) and upregulation of CD69 on T cells within the destination tissue20. Both KLF2 and CD69 mediate tissue retention of developing TRM cells via their actions on the sphingosine-1-phosphate receptor 1 (S1PR1), which under normal conditions promotes cell egress from tissues via efferent lymph8,53. More recently, a related receptor S1PR5 has also been implicated in enforcing tissue residence54. In addition to CD69, many TRM cells are positive for CD103 (also known as αE integrin)27. In skin, most CD8+ TRM cells are CD103+; CD103− TRM cells are fewer in number and more motile9,27,36. However, as noted earlier, TRM cell surface markers are not uniformly expressed in all tissues. CD103 expression may be limited to certain tissues and cell subsets as CD103− TRM cells have been found in tissues such as the brain55,56. In addition, co-expression of CD69 and CD103 does not guarantee tissue residency, highlighting the importance of parabiosis and intravascular labelling and other functional techniques in determining the true nature of residency26,57. A recent study examining TRM cell development over time with single-cell RNA sequencing found that genes associated with T cell receptor (TCR) activation (for example, Nr4a2) and AP1 dimerization partners (for example, Junb, Fosl) were associated with development of gut TRM cells in mice after infection, even at very late time points, but it is unknown whether this is generalizable to other tissues45.
The respective tissue microenvironment almost certainly plays a significant role in TRM cell differentiation and maintenance, and transcriptional analyses indicate heterogeneity in gene expression signatures across TRM cells in different tissues8. It is tempting to speculate that different tissue microenvironments act via distinct signalling pathways to drive TRM cell differentiation and maintenance, thus leading to the generation of different TRM cell populations throughout the body18,19,58,59. It remains unclear whether these populations contribute to pathogen control in different ways, or whether different pathogens in the same tissue generate different TRM cell populations. The tissue architecture of the microenvironment may also influence the location of TRM cells within a tissue. In the skin, for example, CD4+ TRM cells reside predominantly in the dermis, whereas CD8+ TRM cells localize to the epidermis. CD103 staining is far more intense in the CD8+ TRM cell population8,60. In the epidermis, CD8+ herpes simplex virus (HSV)-reactive CD103+ TRM cells display a crawling dendritic cell-like migration pattern, probing keratinocyte junctions in a manner that suggests active surveillance for infected keratinocytes61. In the lungs, the relative abundance of extracellular matrix components may influence TRM cell localization. It has been proposed that CD4+ TRM cells and CD8+ TRM cells, respectively, gravitate towards areas rich in distinct collagens8,9. Thus, TRM cell subtypes may have evolved mechanisms to localize within mucosal and epithelial tissues in ways that enhance the likelihood of a rapid coordinated response to local infection40,62,63.
What maintains TRM cells in tissue?
If vaccines are to be designed to generate TRM cells, then understanding the survival dynamics of TRM cells becomes important. A recent human study showed TRM cell clones surviving in the skin for up to 10 years following bone marrow transplantation34. One variable mediating TRM cell survival in this study was a stem cell-like profile adopted by TRM cells, which promoted superior survival of these cells when exposed to myeloablative radio-chemotherapy. Upon stimulation with cognate antigen, these TRM cells became active, changing their metabolism and undergoing clonal expansion. Other important mediators thought to influence TRM cell survival include IL-15 and ICOS1 (refs. 27,64). Fatty acid internalization and oxidative metabolism appear to be an additional mechanism for enhancing TRM cell survival, and there is evidence that there is variation on this theme in different tissues. For example, whereas fatty acid-binding protein 4 (FABP4) and FABP5 appear to be important for fatty acid metabolism in skin TRM cells, FABP1 seems to be critical for liver TRM cell metabolism and persistence29,65. However, our overall understanding of TRM cell maintenance remains limited and is a key area for future research.
Impact of infection and inflammation on TRM cell biology
In humans, TRM cells are thought to accumulate in tissues over time in response to repeated infections and to provide protective immunity against previously encountered pathogens33,66. TRM cells express anti-apoptotic factors, including BCL2, which may support their maintenance in tissues8,27. However, the duration of TRM cell survival differs between tissues. For example, TRM cells in the lung do not persist for as long as those in the skin and other tissues; the reasons for this are still unclear but are important when considering vaccines to pulmonary pathogens. A recent review discussed that lung TRM cells may, in fact, migrate into the mediastinal lymph nodes after some time of residence within the lungs, rather than undergoing cell death24,67. It was recently shown that pools of TRM cells in the skin can be replenished by both the replication of existing TRM cells and the recruitment of circulating precursors50. Upon re-infection or antigen encounter following vaccination, there is local proliferation of TRM cells as well as egress of some TRM cells to lymphoid tissue, where they may again take up residence38,39,68. TRM cells that egress from tissues have been shown in one report to re-enter the circulation, where they have a high propensity to home back to their tissue of origin37. In a skin xenograft model, a subset of CD4+ TRM cells were identified that had the ability to leave the skin to join the circulating pool of T cells as CD103+ T cells and to later re-enter the skin at distant tissue sites to form skin TRM cells after local infection39,69. This general observation is consistent with multifocal skin diseases known to involve TRM cells, such as psoriasis and mycosis fungoides36,70. The development of inflammatory patches and plaques at new sites in the skin remote from the original inflamed sites is likely to involve this mechanism; however, whether this is truly dedifferentiation of TRM cells or migration and maturation of what have been termed ‘migratory memory’ T cells is unknown10. The study of epigenetic changes that occur with TRM cell differentiation is in its infancy, and whether these changes are reversed in ‘former’ TRM cells will be important to document. Much remains undiscovered regarding TRM cell longevity, replenishment and regeneration. Many of the factors that may drive egress of TRM cells from tissue into the circulation also remain obscure at this time.
TRM cell generation following tissue infection has been studied in both humans and mice, and several factors have been identified that contribute to the induction of TRM cells. In the lungs, it is now generally accepted that tissue microenvironmental niches receptive for TRM cells are generated during and after clearance of viral infection, and TRM cells are spatially compartmentalized and maintained near sites of pathogen entry and accumulation71,72. It may be that longer lived TRM cells specific for respiratory pathogens reside in the oropharyngeal mucosa, which may be an alternative site to target for TRM cells. In another study, TRM cells accumulated at sites of tissue injury and regeneration73. Both observations suggest local tissue regulation of TRM cell maintenance. Antigen processing also contributes to the generation of all T cell memory, including TRM cells. In the case of CD8+ TRM cell generation, classical DC1s have the ability to cross-present antigen to naive CD8+ T cells in lymph nodes, which appears to be critical for antiviral CD8+ TRM cell development46,47,58. Much of what is known about gut TRM cells derives from studies of lymphocytic choriomeningitis (LCMV) infection, which generates effector T cells from splenic naive T cells after intravenous challenge. However, LCMV delivered intraperitoneally enters the mediastinal node74 and leads to the seeding of all lymphoid and non-lymphoid tissues with effector T cells that mature into TRM cells. Do such cells educated in the spleen versus the lymph node differ in their behaviour or homing properties? And do LCMV-specific effector T cells that enter intestinal tissue encounter virally infected cells and perform effector functions, or simply passively differentiate in the gut microenvironment? There may be fundamental differences between the TRM cells that arise in the setting of acute tissue inflammation, TCR activation and immune protection (for example, during infections with influenza A virus (IAV) in the lungs, or with vaccinia virus (VACV) or HSV in the skin) and those that are seeded into non-inflamed tissues by systemic infection (for example, in infection with a non-cytopathic virus such as LCMV in multiple tissues), although there are some clear commonalities.
Vaccines: can they efficiently generate TRM cells?
Above, we have provided an overview of the current understanding of the factors that influence TRM cell generation and their maintenance. In this section we will more specifically concentrate on relevant research related to vaccines and TRM cells, primarily highlighting findings in animal models, as well as the substantial gaps in our knowledge. Naturally acquired infection in peripheral tissues generally elicits a long-lived T cell memory response, including the induction of TRM cells. However, relatively little is known about the generation of TRM cells by vaccines, and the contribution of other tissue-dwelling T cells, including TRM cells, to vaccine-induced protection33,75. Newly available research tools and approaches could be more widely applied to parse out the roles of TRM cells, circulating T cells and antibodies in vaccine-mediated protection against infection. Overall awareness of and interest in TRM cells in vaccine-elicited immunity should be an element of early-stage vaccine research and development efforts. Although there are practical limitations to such research in humans, much can be learned at a fundamental level in small animal and non-human primate models. Finally, we comment below on the factors that may prove important in generating TRM cells, including, first, the route of antigen administration; second, the mode of antigen delivery; and last, the role of vaccine adjuvants.
With most vaccines, generation of neutralizing antibody is associated with protection against a specific pathogen and remains a goal of vaccine development. Less attention has been paid to the generation of memory T cells, which is also associated with protection but is less likely to be measured in vaccine studies. It is assumed that for the generation of TRM cells, the immune challenge (whether through natural infection or immunization) ought to occur in a peripheral tissue site that is destined to recruit these cells. Current intramuscular vaccines do not elicit TRM cells, at least whenever this has been studied76,77. In animal models, parsing out whether protection from infectious challenge is mediated predominantly by T cells or B cells is now achievable75. Examples include the use of B cell-depleting or antibody-depleting strategies prior to infection, which helps focus attention on the role of T cells75. In addition, T cell-depleting antibodies (which typically deplete T cells from the blood but not from tissues) or agents that limit T cell egress from lymph nodes or from the circulation can more directly discriminate between the roles of circulating T cells and TRM cells77,78. For example, after inoculation with a vaccine that generates a TRM cell, followed by challenge infection, pathogen clearance is unaffected by blocking the circulating T cell response (via treatment with FTY720, an S1P1R agonist), pointing to the central role of TRM cells in these immune responses14,75,79,80. Parabiosis models can also pinpoint the relative contributions of circulating T cells and TRM cells57. New technology promises to answer many open questions; for example, TRM cells may now be quantified and the spatial and functional relationships of TRM cells in the overall tissue architecture may be characterized through in situ high-resolution multiplex imaging as well as through deep immune profiling and repertoire analysis (for instance, using CODEX, TCR sequencing, single-cell RNA sequencing and related technologies).
Vaccine administration: impact of delivery route on TRM cells
Intramuscular injection into the deltoid or gluteus maximus muscle is the most common route of vaccine administration. It is generally well tolerated in children and adults and is convenient and accessible. The musculature is well vascularized and drained by lymphatics, enabling recruitment of immune cells from blood and rapid delivery of vaccine antigens to the draining lymph nodes, which is clearly sufficient for the induction of humoral immunity. Despite these practical clinical advantages, there are substantial disadvantages of this approach for T cell immunity77. Unlike skeletal muscle, skin and other epithelial tissues are consistently exposed to the external environment and have been evolutionarily shaped accordingly11. In other studies, intratracheal, intranasal or intravenous81 delivery of experimental vaccines for Mycobacterium tuberculosis and respiratory syncytial virus (RSV) induced a TRM cell response that was more robust and more protective against subsequent challenge as compared with intramuscular or intraperitoneal delivery51,82,83. It could be argued that skeletal muscle has not faced any evolutionary pressure from infectious pathogens (Fig. 3). As such, muscle has relatively few dendritic cells, and intramuscular immunization generates weak CD8+ T cell and TRM cell responses in mice as compared with mucosal and epidermal immunization75. Adjuvant-mediated recruitment of dendritic cells to muscle presumably involves their transit from blood.
Fig. 3. Host immune responses to different routes of vaccine administration.
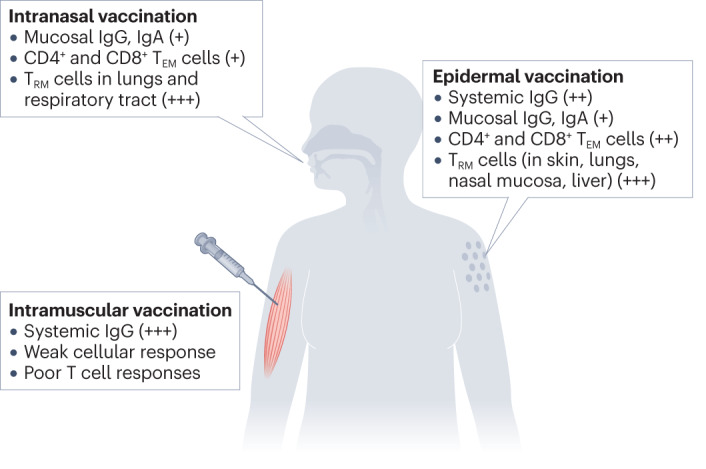
Both vaccine formulation and route of administration shape the immune response generated by the host immune system. Intranasal vaccination leads to the production of mucosal IgA and the generation of CD4+ and CD8+ effector memory T cells (TEM cells), and a tissue resident memory T cell (TRM cell) response in the respiratory tract. Intramuscular injection leads to a humoral immune response that produces systemic IgG, but this route generates relatively weak effector and memory T cell responses, most likely because there are few dendritic cells in muscle. Epidermal vaccine administration results in strong humoral and cell-mediated immune responses. This route generates systemic production of IgG, as well as mucosal IgG and IgA. It also elicits a strong cellular response with the generation and proliferation of both TEM cells and TRM cells in skin and also in distant tissues, including the lungs, nasal mucosa and liver. Adding adjuvants to vaccine formulations can potentially enhance and shift the immune response. For instance, adjuvanted intramuscular vaccinations have greater immunogenicity and may, in some cases, generate mucosal antibodies in addition to a systemic antibody response.
Vaccination against IAV has historically involved immunization with whole virus, either inactivated or attenuated, with the primary intent of generating neutralizing antibodies. Intramuscular injection of inactivated IAV has been shown to elicit a particularly poor CD8+ T cell response84. In a comparison of a live attenuated and an inactivated influenza vaccine, intranasal delivery of the live attenuated vaccine generated both neutralizing antibodies and lung TRM cells80. Intraperitoneal or subcutaneous delivery of the inactivated vaccine generated neutralizing antibodies, but not lung TRM cells. Neither systemic (intraperitoneal) administration of the live attenuated vaccine nor intranasal delivery of the inactivated IAV vaccine elicited a measurable TRM cell response, suggesting a requirement for both the route of vaccine administration and a live attenuated formulation to generate a robust TRM cell response. Mucosal vaccines have been recently reviewed (see refs. 85,86). Briefly, emerging data suggest that mucosal vaccination against respiratory pathogens can elicit tissue immunity and prevent or eliminate infection at the site of entry. This is in comparison with current intramuscular SARS-CoV-2 vaccines, which prevent severe illness and mortality but lack substantial efficacy in preventing upper respiratory infection and viral transmission.
In mouse and hamster studies, a CpG-adjuvanted, recombinant SARS-CoV-2 vaccine targeting the Omicron Spike protein was administered via the intramuscular and intranasal routes87. Intranasal vaccination was superior in producing cross-neutralizing antibodies and promoted more rapid clearance of virus following Omicron challenge. The intranasal route elicited a T helper 1 cell (TH1 cell)-biased Spike-specific CD4+ and CD8+ splenic T cell response that was cross-reactive (to pre-Omicron variants) and likely contributed to vaccine-mediated protection.
Humoral and cellular responses were assessed in a mouse model comparing intramuscular and intranasal administration of a chimpanzee adenoviral vector vaccine targeting the SARS-CoV-2 Spike protein88. Although both routes of administration resulted in comparable serum-neutralizing antibody titres, a single intranasal dose generated superior immune protection against SARS-CoV-2 challenge compared with a two-dose regimen given intramuscularly. Furthermore, a single intranasal immunization prevented both upper and lower respiratory tract infection by SARS-CoV-2. Following intranasal, but not intramuscular, immunization, CD103+CD69+ T cells were present in the lung, likely representing TRM cells. The authors suggested that the superior protection conferred by the intranasal route was due to the mucosal immune response generated following intranasal vaccination and pointed to the potential value of strategies to discern which elements of the immune response contributed to protection (for example, B cell and T cell depletion, passive transfer or parabiosis experiments).
A series of elegant studies dissected mechanisms of vaccine-elicited B cell and T cell protection and directly compared intranasal and parenteral (subcutaneous or intramuscular) routes of administration. The authors developed IAV and SARS-CoV-2 protein subunit vaccines formulated in a carbomer-based nanoemulsion adjuvant system (Adjuplex) including either the Toll-like receptor 9 (TLR9) agonist CpG or the TLR4 agonist glucopyranosyl lipid A (GLA). The Adjuplex adjuvant system is a strong promoter of dendritic cell antigen cross-presentation and a potent inducer of CD8+ T cell responses to protein subunit vaccines. Immunization by either the intranasal or subcutaneous route led to effective control of SARS-CoV-2 Beta variant infection in mice with intact antibody responses, but only the intranasal route induced protection against the virus in the absence of antibody-mediated neutralization89–91. Protection in response to intranasal vaccination was associated with durable T cell-mediated immunity and with T cells in the lung parenchyma that expressed CD103, CD69 and CD49a. In contrast, subcutaneous administration of the SARS-CoV-2 vaccine was associated with abundant splenic and lung CD8+ T cells, but these were localized predominantly to the vasculature, and lacked cell surface markers of TRM cells. Thus, a growing body of work points to the possibility of fine-tuning mucosal immunization with a series of adjuvants to generate tissue-specific immunity mediated by TRM cells.
One immunization strategy to elicit a tissue-targeted TRM cell response is the ‘prime and pull’ approach that has been used in animal models92,93. The ‘prime’ step involves conventional parenteral immunization, followed by the ‘pull’ step which relies on topical application of an innate immune inflammatory stimulus (for instance, chemokine, adjuvant or another inflammatory mediator) to recruit activated T cells into particular tissues21,94,95. This method successfully recruited and retained effector T cells in local tissue environments and generated TRM cells for at least 1 year in some models21. This suggests that uncommitted effector T cells generated by parenteral inflammation can be altered by recruitment into inflamed peripheral tissues, suggesting that for these cells, TRM cell programming occurs after tissue entry. However, it is not clear how this method can be applied to an entire tissue, as opposed to a specific site within a tissue. Additionally, it is unclear which T cells are best at forming TRM cells when ‘pulled’ into a tissue, and whether current or recent TCR activation is required. At the very least, these experiments clearly demonstrate that persistent antigen stimulation is not required for the establishment of tissue residence. TRM cell precursors ‘pulled’ into tissue by inflammation clearly can establish residence, but whether they differ in fundamental ways from TRM cells that experience TCR activation in their destination tissue requires further study.
The route of administration as well as the TRM cell response elicited by VACV may have contributed to the success of the smallpox vaccine; subsequent human studies were able to confirm this, and show that the appearance of a skin ‘pox’ lesion conferred better immunity, even when the mode of intended vaccination was subcutaneous or intramuscular96,97. The cellular requirements for the evolution of a pox lesion are undefined, but it appears to involve epidermal infection by live VACV. Studies of recombinant VACV in mice demonstrated that epidermal disruption (skin scarification) was best at eliciting a T cell response (including TRM cells and TCM cells), which itself is sufficient to provide protection against subsequent cutaneous and lethal respiratory challenges; this protection did not require neutralizing antibodies75. Modified vaccinia virus Ankara (MVA) is a highly attenuated VACV licensed as a third-generation smallpox and monkeypox vaccine and has been widely used as an investigational vaccine vector98. Because it is replication-incompetent and lacks approximately 10% of the VACV genome, immunization via epidermal disruption with MVA avoids many of the undesirable side effects of VACV99–101, including the formation of a florid pox lesion that heals with a noticeable scar. MVA is licensed for intramuscular or subcutaneous administration, whereas only VACV is licensed for human administration by skin scarification/epidermal disruption. Epidermal disruption (in distinction from intradermal immunization) may be important in recruiting and activating dendritic cells at the immunization site47, through the release of cytokines or by the generation of ‘danger’ signals. Immunogenicity of an ovalbumin (OVA) peptide-expressing MVA construct (MVAOVA) was recently assessed in mice. MVAOVA was administered via epidermal disruption or via intradermal, intratracheal, subcutaneous or intramuscular routes. Epidermal disruption generated a more robust T cell response that was transcriptionally unique from the other routes tested77. Others have suggested that vaccines designed to recruit TRM cells to the respiratory tract should be administered intranasally102; however, recent experiments of poxvirus vector vaccines have generated a measurable TRM cell response via epidermal disruption77,103. Poxvirus vector vaccines are uniquely suited for skin, and in this study, skin scarification elicited a superior lung TRM cell response compared with all but the intratracheal route and was superior in protecting mice against lethal VACVOVA challenge. Importantly, these data showed a dose-sparing effect on TRM cell generation by vaccination via epidermal disruption compared with intramuscular, subcutaneous and intraperitoneal routes, with intradermal being dose sparing but at a lower level. Previous studies have also shown a dose-sparing effect of MVA vaccine in eliciting neutralizing antibodies when delivered intradermally versus subcutaneously104. Current concerns surrounding limited vaccine stockpiles raise interesting prospects for future research105. Specifically, the Jynneos vaccine against smallpox and monkeypox was recently granted emergency use authorization by the US Food and Drug Administration (FDA) and a clinical trial is currently recruiting patients to evaluate vaccine immunogenicity when administered intradermally at 20% of the conventional dose (NCT05512949). Further studies comparing this or other MVA vaccines by various routes of administration, including epidermal disruption, could be informative. Combined, such studies could highlight the potential value of further dose de-escalation via intradermal administration and development and testing of monkeypox vaccine products suitable for administration by skin scarification or similarly needle-free techniques.
Of note, the effector T cells elicited by MVAOVA skin scarification or by the intratracheal route had overlapping transcriptional profiles77. A fuller appreciation of the immune mechanisms by which skin scarification elicits lung TRM cells (in addition to cutaneous TRM cells) and allows for dose-sparing vaccination will require further exploration. Similarly, it may be instructive to compare various modes of cutaneous immunization, including epidermal disruption and epidermal injection with a particular focus on the generation of TRM cells.
Vaccine platforms
Few vaccine platforms have been rationally designed or rigorously evaluated to assess their role in the generation of TRM cells and other tissue-dwelling T cells. One group designed a peptide vaccine targeting wild-type and in silico optimized HLA-A*0201-restricted CD8+ T cell epitopes derived from 11 structural, non-structural and accessory proteins of SARS-CoV-2. Following a single subcutaneous injection, mice generated abundant draining lymph node and splenic CD8+ T cells with surface markers and a phenotype suggestive of the potential for maturation into lung and mucosal barrier TRM cells, although this was not validated by sampling those sites106. Another approach uses a pH-dependent antigen-delivery system allowing for release of the vaccine’s ‘cargo’ antigen at a favourable pH, and enhancing antigen processing by the MHC class I pathway. This strategy prolonged antigen presentation leading to a more robust CD8+ T cell response40. Thus, the platform harnessed two factors known to promote CD8+ TRM cells in natural infection — namely, enhancing antigen processing by the MHC class I pathway and extending the duration of antigen presentation.
Adenoviral vector vaccines, such as VACV vectors, mimic viral infection to elicit an immune response, although the normal target tissue of the parent virus is different107. Recently developed mRNA-based vaccines employ a unique mechanism that relies on host cell machinery to synthesize and present antigen to the immune system. In this way, they simulate certain features of natural infection, eliciting a combined cellular and humoral response. Several leading SARS-CoV-2 vaccines rely on adenoviral vectors (AstraZeneca, Janssen/JNJ and Sputnik-5) or on mRNA platforms (Pfizer/BioNTech and Moderna)108–111. Early results suggest that the adenoviral-vectored and mRNA SARS-CoV-2 vaccines generally elicit robust CD4+ T cell and, to a lesser extent, CD8+ T cell responses, although one study of an adenoviral-vectored SARS-CoV-2 vaccine demonstrated comparable CD4+ and CD8+ T cell responses to immunization112. The published clinical results describe circulating T cells, so any extension to TRM cells is hypothetical. Preclinical studies of intranasally delivered adenoviral-vectored vaccines have demonstrated a strong, focused immune response against the receptor binding domain of SARS-CoV-2 Spike protein through induction of mucosal IgA in addition to serum-neutralizing antibodies, CD4+ and CD8+ T cells and a population of CD8+ TRM cells103,113. In a humanized mouse model utilizing a heat-shock protein chaperone that promotes vaccine antigen cross-presentation, a SARS-CoV-2 Spike protein vaccine elicited TRM cells in the lungs and airways114. A comprehensive analysis by systems vaccinology of the Pfizer/BioNTech COVID vaccine suggests that the mRNA platform does not elicit lung TRM cells when administered via the intramuscular route in mice, despite measurable CD8+ T cell and natural killer cell responses115. A recent mouse model study of an mRNA SARS-CoV-2 vaccine compared different strategies of vaccination, including contralateral versus ipsilateral prime–boost doses, as well as intravenous and intranasal administration116. Recent studies of mRNA SARS-CoV-2 and influenza vaccines in humans have tested the intradermal route of administration. These vaccines have been shown to be safe and effective via this route when compared with intramuscular administration117,118. In one study, the intradermal route led to a less robust cellular immune response when compared with the intramuscular route, although it was delivered intradermally at only 20% of the dose used intramuscularly117. This study showed the greatest generation of a circulating T cell and lung TRM cell response occurred when combining intramuscular injection with a subsequent intranasal booster dose.
Current concerns about vaccine-induced antibody waning and the emergence of SARS-CoV-2 antibody-escape variants underscore the potential importance of CD8+ TRM cells in long-term protection against COVID-19. Antibodies to either the Pfizer or Moderna vaccine (targeting the original Spike protein) cross-react poorly, if at all, with the Spike from the Omicron BA.1, BA.2 or BA.5 viral variants119, suggesting that the correlation between the number of vaccine doses and the reduction in serious illness and/or hospitalizations is likely, at least in part, to be T cell-mediated120. Indeed, mRNA vaccine-elicited T cell responses have been implicated in protection against recent variants of concern121,122. Newer mRNA constructs may address this protective antibody deficiency, at least temporarily. However, it is likely that the selective pressure on the virus induced by the newer vaccines will drive the emergence of additional escape variants.
As noted above, pulmonary TRM cells generally do not persist long term and the immunity they confer may be relatively transient. SARS-CoV-2 breakthrough infections in fully vaccinated individuals are characterized by asymptomatic or mildly symptomatic upper airway involvement with transient but high levels of viral replication in nasopharyngeal mucosa123. Circulating SARS-CoV-2-specific CD4+ and CD8+ T cells have been found in a high proportion of convalescent individuals with COVID-19. These T cells target a wide range of SARS-CoV-2 proteins in addition to the immunodominant Spike. SARS-CoV-2-reactive CD4+ T cells were also found in ~50% of unexposed individuals, consistent with such cells being generated in response to prior exposure to common cold coronaviruses, highlighting a potential role for these cells in cross-protective immunity124,125. The presence of SARS-CoV-2-specific T cells in bronchoalveolar fluid and biopsies from patients with COVID-19 and the presence of TRM cells in lung biopsies taken from patients up to 10 months following infection with SARS-CoV-2 point to their potential role in immune protection126,127. In addition, such TRM cells could be isolated from cadaveric tissues of patients who had survived infection with COVID-19 and died of other causes. Again, these findings point to the importance of tissue-specific immunity and studies to explore these issues following SARS-CoV-2 immunization are practical in small and large animal models. Furthermore, rational, pan-coronavirus vaccine design should incorporate conserved CD4+ and CD8+ T cell epitopes along with novel vaccination strategies to promote long-term tissue residence126.
Vaccine adjuvants
As summarized above, TRM cell generation following vaccination is influenced by both the route of administration (favoured by mucosal or epidermal administration) as well as the vaccine formulation (with live attenuated vaccines being most effective). Adjuvants offer the possibility of either enhancing the TRM cell response generated by these methods or, possibly, circumventing these requirements14. The focus of adjuvant research has long been on enhancement of the antibody response. This follows in that alum, the only adjuvant in FDA-licensed vaccines for many decades, is primarily a driver of TH2-type humoral responses and not a strong promotor of cell-mediated immunity. Newer adjuvants or combination adjuvants, especially those that promote TH1 cell and TH17 cell immunity, also appear to be good promotors of CD4+ and CD8+ T cells14. Deploying rationally designed adjuvants may ultimately prove helpful in enabling parenteral vaccines to overcome an apparent requirement for mucosal or epidermal delivery to optimally elicit TRM cells. A good example of this is the development of the carbomer-based nanoemulsion adjuvant system (Adjuplex) incorporating either GLA or CpG, as discussed previously.
Shingles presents an interesting opportunity to consider TRM cell-targeted vaccines over antibody-targeted vaccines. This is because varicella zoster virus (VZV) and other herpesviruses are reactivated in the skin, where VZV-specific and HSV-specific TRM cells have been shown to reside. Therefore, vaccination strategies aimed at boosting skin TRM cells may elicit faster and stronger immune responses upon primary pathogen encounter and viral reactivation. One group found that Zostavax, a live attenuated, subcutaneous, endogenously adjuvanted VZV vaccine, had no effect on the proportion of VZV-specific TRM cells within the skin128. In clinical trials, Zostavax had 61.1% efficacy in adults aged 60 years and older129. By contrast, Shingrix is a recombinant, AS01B-adjuvanted vaccine that is highly protective against VZV, exhibiting 97.2% overall efficacy in adults aged 50 years and older130. The Shingrix vaccine relies on the glycoprotein E antigen — the most abundant glycoprotein expressed on VZV-infected cells and a target of both neutralizing antibodies and T cells. Unadjuvanted glycoprotein E does not elicit a strong immune response. However, when glycoprotein E is combined with the AS01B adjuvant system it generates robust glycoprotein E-specific antibody and CD4+ and CD8+ T cell responses131. Thus, Shingrix relies on glycoprotein E to direct the immune response and AS01B to shape and enhance the response. No tissue samples were collected as part of this study by Heineman et al.131; however, an ongoing clinical trial plans to identify and characterize cutaneous TRM cells following Shingrix vaccination132.
In general, vaccines that generate T cell responses have important potential advantages over vaccines geared primarily towards antibody production. Certain vaccines that induce a robust T cell (and TRM cell) response target highly conserved, mostly internal and/or non-structural, viral proteins133. Antibody-mediated neutralization, on the other hand, relies primarily on recognition of surface conformational epitopes, which are known to be less well conserved across viral strains and subject to evasive mutation. By engineering vaccines to elicit a significant T cell response, we can circumvent evasion of the humoral immune response through viral recombination and antigenic drift. T cell vaccine responses are therefore capable of broad heterosubtypic protection across viral strains80, and T cell vaccines are thus good candidates for universal influenza or pan-coronavirus vaccines.
Challenges and future directions
There remain many challenges to achieving a more complete understanding of the roles of circulating T cells and tissue resident T cells in vaccine-mediated protective immunity. Defining the subsets of circulating T cells that are destined to become TRM cells49, what prompts their egress from blood to repopulate local TRM cell pools and what stimulates their regeneration within tissues are promising areas for study. Emerging and future insights into these issues would identify questions that could be addressed in vaccine clinical trials. The mRNA vaccine platform proved very successful with SARS-CoV-2 vaccines in the COVID-19 pandemic and may become a leading strategy for future vaccine development. Next to nothing is known regarding the effects of mRNA vaccines on the generation of TRM cells, although a recent study showed that the levels of TRM cells in nasopharyngeal samples increased after each of the two doses of the Pfizer BioNTech mRNA COVID-19 vaccine134. However, longitudinal sampling could be informative in light of the previously noted decline in nasopharyngeal immunity seen in SARS-CoV-2 breakthrough infections. Gaining a better understanding of the T cell and TRM cell responses to mRNA vaccines and devising strategies to enhance those responses in rodent and non-human primate models could be of immediate value.
The major barrier to studying T cell-targeted vaccines is obtaining human tissue samples required to study tissue resident T cells7. The development and use of tissue banks to address the dearth of human tissue samples available for research has been invaluable in other research settings. The Network for Pancreatic Organ Donors with Diabetes (nPOD) is one such tissue bank that collects and distributes cadaveric pancreatic and other tissue samples from individuals with recent onset of type 1 diabetes or those who are at increased risk of developing type 1 diabetes. A similar effort to facilitate access to relevant tissues and enable studies of T cell vaccine would be valuable in furthering our understanding of these vaccines. Dr Donna Farber has been uniquely successful in studying human TRM cell biology in multiple tissues. The success of the Farber laboratory in leveraging the organ donor process in New York can serve as a model for the study of human tissue T cells, including TRM cells. Lungs that are not suitable for transplantation and nasopharyngeal, gastrointestinal or urogenital tissues could be banked and distributed to study vaccine-elicited protection at these mucosal sites. Indeed, the expansion of such tissue banks for use in vaccine studies has recently been proposed7.
One promising area of research may be the identification of better cutaneous immunization platforms, in addition to epidermal disruption or skin scarification, that generate skin and lung TRM cells. Whether classical epidermal disruption can be refined with other approaches remains unexplored. A challenge to skin-directed vaccines is the potential for reactogenicity and local inflammatory responses, thus demonstrating safety and tolerability of such vaccines will be important. Newer, more potent and well-tolerated adjuvants may alleviate such concerns. More data are needed to elucidate and deconvolute the respective roles of antibodies, circulating T cells and TRM cells in immune responses, defining how they differ and where they overlap.
Acknowledgements
T.S.K. is supported by National Institutes of Health (NIH) grants R01 AI127654 and R01 AR065807.
Glossary
- Central memory T cells
(TCM cells). A subset of long-lived memory T cells that express CCR7 and CD62L, allowing them to home to and patrol secondary lymphoid organs for known pathogens to respond more quickly to a subsequent infection.
- Effector memory T cells
(TEM cells). A subset of memory T cells that express integrins and chemokine receptors, allowing them to localize to inflamed tissues and mediate a rapid and potent immune response following repeat encounter with a known antigen.
- Tissue resident memory T cells
(TRM cells). A subset of memory T cells that are distinct in that they take up long-term residence within a peripheral tissue. Here, they can quickly mount a robust immune response upon pathogen encounter in non-lymphoid tissue.
Author contributions
The authors contributed equally to all aspects of the article.
Peer review
Peer review information
Nature Reviews Immunology thanks P. Scott and the other, anonymous, reviewers for their contribution to the peer review of this work.
Competing interests
T.S.K. has financial interest in Pellis Therapeutics, a start-up biotech company based in Boston. E.R. declares no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ehreth J. The global value of vaccination. Vaccine. 2003;21:596–600. doi: 10.1016/S0264-410X(02)00623-0. [DOI] [PubMed] [Google Scholar]
- 2.Rémy V, Zöllner Y, Heckmann U. Vaccination: the cornerstone of an efficient healthcare system. J. Mark. Access. Health Policy. 2015;3:27041. doi: 10.3402/jmahp.v3.27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greenwood B. The contribution of vaccination to global health: past, present and future. Philos. Trans. R. Soc. B: Biol. Sci. 2014;369:20130433. doi: 10.1098/rstb.2013.0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pollard AJ, Bijker EM. A guide to vaccinology: from basic principles to new developments. Nat. Rev. Immunol. 2021;21:83–100. doi: 10.1038/s41577-020-00479-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gilbert SC. T-cell-inducing vaccines — what’s the future. Immunology. 2012;135:19–26. doi: 10.1111/j.1365-2567.2011.03517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Todryk SM. T cell memory to vaccination. Vaccines. 2018;6:84. doi: 10.3390/vaccines6040084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farber DL. Tissues, not blood, are where immune cells function. Nature. 2021;593:506–509. doi: 10.1038/d41586-021-01396-y. [DOI] [PubMed] [Google Scholar]
- 8.Mueller SN, Mackay LK. Tissue-resident memory T cells: local specialists in immune defence. Nat. Rev. Immunol. 2016;16:79–89. doi: 10.1038/nri.2015.3. [DOI] [PubMed] [Google Scholar]
- 9.Topham, D. J. & Reilly, E. C. Tissue-resident memory CD8+ T cells: from phenotype to function. Front. Immunol. 9, 10.3389/fimmu.2018.00515 (2018). [DOI] [PMC free article] [PubMed]
- 10.Watanabe R, et al. Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci. Transl. Med. 2015;7:279ra239. doi: 10.1126/scitranslmed.3010302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park CO, Kupper TS. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat. Med. 2015;21:688–697. doi: 10.1038/nm.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark RA, et al. The vast majority of CLA+ T cells are resident in normal skin. J. Immunol. 2006;176:4431–4439. doi: 10.4049/jimmunol.176.7.4431. [DOI] [PubMed] [Google Scholar]
- 13.Ariotti S, Haanen JB, Schumacher TN. Behavior and function of tissue-resident memory T cells. Adv. Immunol. 2012;114:203–216. doi: 10.1016/B978-0-12-396548-6.00008-1. [DOI] [PubMed] [Google Scholar]
- 14.Wilk MM, Mills KHG. CD4 TRM cells following infection and immunization: implications for more effective vaccine design. Front. Immunol. 2018;9:1860. doi: 10.3389/fimmu.2018.01860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hogan RJ, et al. Activated antigen-specific CD8+ T cells persist in the lungs following recovery from respiratory virus infections. J. Immunol. 2001;166:1813–1822. doi: 10.4049/jimmunol.166.3.1813. [DOI] [PubMed] [Google Scholar]
- 16.Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001;291:2413–2417. doi: 10.1126/science.1058867. [DOI] [PubMed] [Google Scholar]
- 17.Milner JJ, Goldrath AW. Transcriptional programming of tissue-resident memory CD8+ T cells. Curr. Opin. Immunol. 2018;51:162–169. doi: 10.1016/j.coi.2018.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Milner JJ, et al. Heterogenous populations of tissue-resident CD8+ T cells are generated in response to infection and malignancy. Immunity. 2020;52:808–824.e7. doi: 10.1016/j.immuni.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Christo SN, et al. Discrete tissue microenvironments instruct diversity in resident memory T cell function and plasticity. Nat. Immunol. 2021;22:1140–1151. doi: 10.1038/s41590-021-01004-1. [DOI] [PubMed] [Google Scholar]
- 20.Mackay LK, Kallies A. Transcriptional regulation of tissue-resident lymphocytes. Trends Immunol. 2017;38:94–103. doi: 10.1016/j.it.2016.11.004. [DOI] [PubMed] [Google Scholar]
- 21.Mackay LK, et al. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc. Natl Acad. Sci. USA. 2012;109:7037–7042. doi: 10.1073/pnas.1202288109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schenkel JM, Masopust D. Tissue-resident memory T cells. Immunity. 2014;41:886–897. doi: 10.1016/j.immuni.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masopust D, Soerens AG. Tissue-resident T cells and other resident leukocytes. Annu. Rev. Immunol. 2019;37:521–546. doi: 10.1146/annurev-immunol-042617-053214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van de Wall, S., Badovinac, V. P. & Harty, J. T. Influenza-specific lung-resident memory CD8+ T cells. Cold Spring Harb. Perspect Biol. 13, 10.1101/cshperspect.a037978 (2021). [DOI] [PMC free article] [PubMed]
- 25.Lange J, Rivera-Ballesteros O, Buggert M. Human mucosal tissue-resident memory T cells in health and disease. Mucosal Immunol. 2022;15:389–397. doi: 10.1038/s41385-021-00467-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng MZM, Wakim LM. Tissue resident memory T cells in the respiratory tract. Mucosal Immunol. 2021 doi: 10.1038/s41385-021-00461-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mackay LK, et al. The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013;14:1294–1301. doi: 10.1038/ni.2744. [DOI] [PubMed] [Google Scholar]
- 28.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 29.Pan Y, et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature. 2017;543:252–256. doi: 10.1038/nature21379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ariotti S, et al. T cell memory. Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science. 2014;346:101–105. doi: 10.1126/science.1254803. [DOI] [PubMed] [Google Scholar]
- 31.Schenkel JM, et al. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science. 2014;346:98–101. doi: 10.1126/science.1254536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gebhardt T, et al. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 2009;10:524–530. doi: 10.1038/ni.1718. [DOI] [PubMed] [Google Scholar]
- 33.Jiang X, et al. Skin infection generates non-migratory memory CD8+ TRM cells providing global skin immunity. Nature. 2012;483:227–231. doi: 10.1038/nature10851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strobl, J. et al. Long-term skin-resident memory T cells proliferate in situ and are involved in human graft-versus-host disease. Sci. Transl. Med. 12, 10.1126/scitranslmed.abb7028 (2020). [DOI] [PMC free article] [PubMed]
- 35.Masopust D, Schenkel JM. The integration of T cell migration, differentiation and function. Nat. Rev. Immunol. 2013;13:309–320. doi: 10.1038/nri3442. [DOI] [PubMed] [Google Scholar]
- 36.Clark RA. Resident memory T cells in human health and disease. Sci. Transl. Med. 2015;7:269rv261. doi: 10.1126/scitranslmed.3010641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fonseca R, et al. Developmental plasticity allows outside-in immune responses by resident memory T cells. Nat. Immunol. 2020;21:412–421. doi: 10.1038/s41590-020-0607-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beura LK, et al. T cells in nonlymphoid tissues give rise to lymph-node-resident memory T cells. Immunity. 2018;48:327–338.e5. doi: 10.1016/j.immuni.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kok L, Masopust D, Schumacher TN. The precursors of CD8+ tissue resident memory T cells: from lymphoid organs to infected tissues. Nat. Rev. Immunol. 2021 doi: 10.1038/s41577-021-00590-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knight FC, et al. Mucosal immunization with a pH-responsive nanoparticle vaccine induces protective CD8+ lung-resident memory T cells. ACS Nano. 2019;13:10939–10960. doi: 10.1021/acsnano.9b00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herndler-Brandstetter D, et al. KLRG1+ effector CD8+ T cells lose KLRG1, differentiate into all memory T cell lineages, and convey enhanced protective immunity. Immunity. 2018;48:716–729.e8. doi: 10.1016/j.immuni.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gaide O, et al. Common clonal origin of central and resident memory T cells following skin immunization. Nat. Med. 2015;21:647–653. doi: 10.1038/nm.3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chun, et al. Human CD1c+ dendritic cells drive the differentiation of CD103+CD8+ mucosal effector T cells via the cytokine TGF-β. Immunity. 2013;38:818–830. doi: 10.1016/j.immuni.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bourdely P, et al. Transcriptional and functional analysis of CD1c+ human dendritic cells identifies a CD163+ subset priming CD8+CD103+ T cells. Immunity. 2020;53:335–352.e8. doi: 10.1016/j.immuni.2020.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kurd, N. S. et al. Early precursors and molecular determinants of tissue-resident memory CD8+ T lymphocytes revealed by single-cell RNA sequencing. Sci. Immunol. 5, 10.1126/sciimmunol.aaz6894 (2020). [DOI] [PMC free article] [PubMed]
- 46.Kok, L. et al. A committed tissue-resident memory T cell precursor within the circulating CD8+ effector T cell pool. J. Exp. Med. 217, 10.1084/jem.20191711 (2020). [DOI] [PMC free article] [PubMed]
- 47.Iborra S, et al. Optimal generation of tissue-resident but not circulating memory T cells during viral infection requires crosspriming by DNGR-1+ dendritic cells. Immunity. 2016;45:847–860. doi: 10.1016/j.immuni.2016.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mani, V. et al. Migratory DCs activate TGF-β to precondition naive CD8+ T cells for tissue-resident memory fate. Science366, 10.1126/science.aav5728 (2019). [DOI] [PMC free article] [PubMed]
- 49.Matos TR, et al. Central memory T cells are the most effective precursors of resident memory T cells in human skin. Sci. Immunol. 2022;7:eabn1889. doi: 10.1126/sciimmunol.abn1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park SL, et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat. Immunol. 2018;19:183–191. doi: 10.1038/s41590-017-0027-5. [DOI] [PubMed] [Google Scholar]
- 51.Li N, et al. RSV recombinant candidate vaccine G1F/M2 with CpG as an adjuvant prevents vaccine-associated lung inflammation, which may be associated with the appropriate types of immune memory in spleens and lungs. Hum. Vaccin. Immunother. 2019;15:2684–2694. doi: 10.1080/21645515.2019.1596710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Samat, A. A. K., van der Geest, J., Vastert, S. J., van Loosdregt, J. & van Wijk, F. Tissue-resident memory T cells in chronic inflammation — local cells with systemic effects? Cells10, 10.3390/cells10020409 (2021). [DOI] [PMC free article] [PubMed]
- 53.Skon CN, et al. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 2013;14:1285–1293. doi: 10.1038/ni.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Evrard, M. et al. Sphingosine 1-phosphate receptor 5 (S1PR5) regulates the peripheral retention of tissue-resident lymphocytes. J. Exp. Med. 219, 10.1084/jem.20210116 (2022). [DOI] [PMC free article] [PubMed]
- 55.Vincenti I, et al. Tissue-resident memory CD8+ T cells cooperate with CD4+ T cells to drive compartmentalized immunopathology in the CNS. Sci. Transl. Med. 2022;14:eabl6058. doi: 10.1126/scitranslmed.abl6058. [DOI] [PubMed] [Google Scholar]
- 56.Wakim LM, et al. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J. Immunol. 2012;189:3462–3471. doi: 10.4049/jimmunol.1201305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang H, Gavil NV, Koewler N, Masopust D, Jameson SC. Parabiosis in mice to study tissue residency of immune cells. Curr. Protoc. 2022;2:e446. doi: 10.1002/cpz1.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Retamal-Diaz, A. et al. Contribution of resident memory CD8+ T cells to protective immunity against respiratory syncytial virus and their impact on vaccine design. Pathogens8, 10.3390/pathogens8030147 (2019). [DOI] [PMC free article] [PubMed]
- 59.Masopust D, Vezys V, Wherry EJ, Barber DL, Ahmed R. Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J. Immunol. 2006;176:2079–2083. doi: 10.4049/jimmunol.176.4.2079. [DOI] [PubMed] [Google Scholar]
- 60.Gebhardt T, et al. Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature. 2011;477:216–219. doi: 10.1038/nature10339. [DOI] [PubMed] [Google Scholar]
- 61.Ariotti S, et al. Tissue-resident memory CD8+ T cells continuously patrol skin epithelia to quickly recognize local antigen. Proc. Natl Acad. Sci. USA. 2012;109:19739–19744. doi: 10.1073/pnas.1208927109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Puksuriwong S, et al. Modified vaccinia Ankara-vectored vaccine expressing nucleoprotein and matrix protein 1 (M1) activates mucosal M1-specific T-cell immunity and tissue-resident memory T cells in human nasopharynx-associated lymphoid tissue. J. Infect. Dis. 2020;222:807–819. doi: 10.1093/infdis/jiz593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rosato PC, Beura LK, Masopust D. Tissue resident memory T cells and viral immunity. Curr. Opin. Virol. 2017;22:44–50. doi: 10.1016/j.coviro.2016.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peng C, et al. Engagement of the costimulatory molecule ICOS in tissues promotes establishment of CD8+ tissue-resident memory T cells. Immunity. 2022;55:98–114.e5. doi: 10.1016/j.immuni.2021.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Frizzell, H. et al. Organ-specific isoform selection of fatty acid-binding proteins in tissue-resident lymphocytes. Sci. Immunol. 5, 10.1126/sciimmunol.aay9283 (2020). [DOI] [PubMed]
- 66.Davies B, et al. Cutting edge: tissue-resident memory T cells generated by multiple immunizations or localized deposition provide enhanced immunity. J. Immunol. 2017;198:2233–2237. doi: 10.4049/jimmunol.1601367. [DOI] [PubMed] [Google Scholar]
- 67.Carbone, F. R. Unique properties of tissue-resident memory T cells in the lungs: implications for COVID-19 and other respiratory diseases. Nat. Rev. Immunol. 1–7, 10.1038/s41577-022-00815-z (2022). This review highlights the short lifespan of TRMcells within the lung parenchyma, suggesting these cells may egress from the lung to the mediastinal nodes as well as undergoing cell death as previously believed. [DOI] [PMC free article] [PubMed]
- 68.Stolley, J. M. et al. Retrograde migration supplies resident memory T cells to lung-draining LN after influenza infection. J. Exp. Med. 217, 10.1084/jem.20192197 (2020). [DOI] [PMC free article] [PubMed]
- 69.Klicznik MM, et al. Human CD4+CD103+ cutaneous resident memory T cells are found in the circulation of healthy individuals. Sci. Immunol. 2019;4:eaav8995. doi: 10.1126/sciimmunol.aav8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Campbell JJ, Clark RA, Watanabe R, Kupper TS. Sezary syndrome and mycosis fungoides arise from distinct T-cell subsets: a biologic rationale for their distinct clinical behaviors. Blood. 2010;116:767–771. doi: 10.1182/blood-2009-11-251926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim TS, Hufford MM, Sun J, Fu Y-X, Braciale TJ. Antigen persistence and the control of local T cell memory by migrant respiratory dendritic cells after acute virus infection. J. Exp. Med. 2010;207:1161–1172. doi: 10.1084/jem.20092017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Turner DL, et al. Lung niches for the generation and maintenance of tissue-resident memory T cells. Mucosal Immunol. 2014;7:501–510. doi: 10.1038/mi.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takamura S, et al. Specific niches for lung-resident memory CD8+ T cells at the site of tissue regeneration enable CD69-independent maintenance. J. Exp. Med. 2016;213:3057–3073. doi: 10.1084/jem.20160938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Olson MR, McDermott DS, Varga SM. The initial draining lymph node primes the bulk of the CD8 T cell response and influences memory T cell trafficking after a systemic viral infection. PLoS Pathog. 2012;8:e1003054. doi: 10.1371/journal.ppat.1003054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu L, et al. Epidermal injury and infection during poxvirus immunization is crucial for the generation of highly protective T cell-mediated immunity. Nat. Med. 2010;16:224–227. doi: 10.1038/nm.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Maes T, et al. Below the surface: twenty-five years of seafloor litter monitoring in coastal seas of North West Europe (1992–2017) Sci. Total. Env. 2018;630:790–798. doi: 10.1016/j.scitotenv.2018.02.245. [DOI] [PubMed] [Google Scholar]
- 77.Pan, Y. et al. Epicutaneous immunization with modified vaccinia Ankara viral vectors generates superior T cell immunity against a respiratory viral challenge. NPJ Vaccines6, 10.1038/s41541-020-00265-5 (2021). [DOI] [PMC free article] [PubMed]
- 78.Liu L, Fuhlbrigge RC, Karibian K, Tian T, Kupper TS. Dynamic programming of CD8+ T cell trafficking after live viral immunization. Immunity. 2006;25:511–520. doi: 10.1016/j.immuni.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 79.Lapuente D, et al. IL-1β as mucosal vaccine adjuvant: the specific induction of tissue-resident memory T cells improves the heterosubtypic immunity against influenza A viruses. Mucosal Immunol. 2018;11:1265–1278. doi: 10.1038/s41385-018-0017-4. [DOI] [PubMed] [Google Scholar]
- 80.Zens, K. D., Chen, J. K. & Farber, D. L. Vaccine-generated lung tissue-resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight1, 10.1172/jci.insight.85832 (2016). [DOI] [PMC free article] [PubMed]
- 81.Layton ED, et al. T cells specific for a mycobacterial glycolipid expand after intravenous bacillus Calmette–Guerin vaccination. J. Immunol. 2021;206:1240–1250. doi: 10.4049/jimmunol.2001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Counoupas, C. et al. Mucosal delivery of a multistage subunit vaccine promotes development of lung-resident memory T cells and affords interleukin-17-dependent protection against pulmonary tuberculosis. NPJ Vaccines5, 10.1038/s41541-020-00255-7 (2020). [DOI] [PMC free article] [PubMed]
- 83.Schwarz B, et al. Viruslike particles encapsidating respiratory syncytial virus M and M2 proteins induce robust T cell responses. ACS Biomater. Sci. Eng. 2016;2:2324–2332. doi: 10.1021/acsbiomaterials.6b00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Koutsakos, M. et al. Circulating TFH cells, serological memory, and tissue compartmentalization shape human influenza-specific B cell immunity. Sci. Transl. Med. 10, 10.1126/scitranslmed.aan8405 (2018). [DOI] [PubMed]
- 85.Lavelle EC, Ward RW. Mucosal vaccines — fortifying the frontiers. Nat. Rev. Immunol. 2022;22:236–250. doi: 10.1038/s41577-021-00583-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lavelle EC, Ward RW. Publisher Correction: Mucosal vaccines — fortifying the frontiers. Nat. Rev. Immunol. 2022;22:266. doi: 10.1038/s41577-021-00599-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lam JH, et al. Artificial cell membrane polymersome-based intranasal Beta spike formulation as a second generation COVID-19 vaccine. ACS Nano. 2022;16:16757–16775. doi: 10.1021/acsnano.2c06350. [DOI] [PubMed] [Google Scholar]
- 88.Hassan AO, et al. A single-dose intranasal ChAd vaccine protects upper and lower respiratory tracts against SARS-CoV-2. Cell. 2020;183:169–184.e13. doi: 10.1016/j.cell.2020.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kingstad-Bakke B, et al. Vaccine-induced systemic and mucosal T cell immunity to SARS-CoV-2 viral variants. Proc. Natl Acad. Sci. USA. 2022;119:e2118312119. doi: 10.1073/pnas.2118312119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marinaik CB, et al. Programming multifaceted pulmonary T cell immunity by combination adjuvants. Cell Rep. Med. 2020;1:100095. doi: 10.1016/j.xcrm.2020.100095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee W, et al. Carbomer-based adjuvant elicits CD8 T-cell immunity by inducing a distinct metabolic state in cross-presenting dendritic cells. PLoS Pathog. 2021;17:e1009168. doi: 10.1371/journal.ppat.1009168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shin H, Iwasaki A. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature. 2012;491:463–467. doi: 10.1038/nature11522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shin H, Iwasaki A. Tissue-resident memory T cells. Immunol. Rev. 2013;255:165–181. doi: 10.1111/imr.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gopinath S, Lu P, Iwasaki A. Cutting edge: the use of topical aminoglycosides as an effective pull in “prime and pull” vaccine strategy. J. Immunol. 2020;204:1703–1707. doi: 10.4049/jimmunol.1900462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fowell DJ, Kim M. The spatio-temporal control of effector T cell migration. Nat. Rev. Immunol. 2021;21:582–596. doi: 10.1038/s41577-021-00507-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Belongia EA, Naleway AL. Smallpox vaccine: the good, the bad, and the ugly. Clin. Med. Res. 2003;1:87–92. doi: 10.3121/cmr.1.2.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McClain DJ, et al. Immunologic responses to vaccinia vaccines administered by different parenteral routes. J. Infect. Dis. 1997;175:756–763. doi: 10.1086/513968. [DOI] [PubMed] [Google Scholar]
- 98.Pittman PR, et al. Phase 3 efficacy trial of modified vaccinia Ankara as a vaccine against smallpox. N. Engl. J. Med. 2019;381:1897–1908. doi: 10.1056/NEJMoa1817307. [DOI] [PubMed] [Google Scholar]
- 99.Hruby DE. Vaccinia virus vectors: new strategies for producing recombinant vaccines. Clin. Microbiol. Rev. 1990;3:153–170. doi: 10.1128/CMR.3.2.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hessel A, et al. MVA vectors expressing conserved influenza proteins protect mice against lethal challenge with H5N1, H9N2 and H7N1 viruses. PLoS One. 2014;9:e88340. doi: 10.1371/journal.pone.0088340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Antoine G, Scheiflinger F, Dorner F, Falkner FG. The complete genomic sequence of the modified vaccinia Ankara strain: comparison with other orthopoxviruses. Virology. 1998;244:365–396. doi: 10.1006/viro.1998.9123. [DOI] [PubMed] [Google Scholar]
- 102.Ahn, J. H. et al. Nasal ciliated cells are primary targets for SARS-CoV-2 replication in the early stage of COVID-19. J. Clin. Invest. 131, 10.1172/JCI148517 (2021). [DOI] [PMC free article] [PubMed]
- 103.Lund FE, Randall TD. Scent of a vaccine. Science. 2021;373:397–399. doi: 10.1126/science.abg9857. [DOI] [PubMed] [Google Scholar]
- 104.Frey SE, et al. Comparison of lyophilized versus liquid modified vaccinia Ankara (MVA) formulations and subcutaneous versus intradermal routes of administration in healthy vaccinia-naive subjects. Vaccine. 2015;33:5225–5234. doi: 10.1016/j.vaccine.2015.06.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zia, S. FDA to stretch monkeypox vaccine supply via intradermal injection. The Scientisthttps://www.the-scientist.com/news-opinion/fda-to-stretch-monkeypox-vaccine-supply-via-intradermal-injection-70362 (2022).
- 106.Gauttier V, et al. Tissue-resident memory CD8 T-cell responses elicited by a single injection of a multi-target COVID-19 vaccine. BioRxiv. 2020 doi: 10.1101/2020.08.14.240093. [DOI] [Google Scholar]
- 107.Lapuente D, Ruzsics Z, Thirion C, Tenbusch M. Evaluation of adenovirus 19a as a novel vector for mucosal vaccination against influenza A viruses. Vaccine. 2018;36:2712–2720. doi: 10.1016/j.vaccine.2018.02.075. [DOI] [PubMed] [Google Scholar]
- 108.Anderson EJ, et al. Safety and immunogenicity of SARS-CoV-2 mRNA-1273 vaccine in older adults. N. Engl. J. Med. 2020;383:2427–2438. doi: 10.1056/NEJMoa2028436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sahin U, et al. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature. 2020;586:594–599. doi: 10.1038/s41586-020-2814-7. [DOI] [PubMed] [Google Scholar]
- 110.Ewer KJ, et al. T cell and antibody responses induced by a single dose of ChAdOx1 nCoV-19 (AZD1222) vaccine in a phase 1/2 clinical trial. Nat. Med. 2021;27:270–278. doi: 10.1038/s41591-020-01194-5. [DOI] [PubMed] [Google Scholar]
- 111.Sadoff J, et al. Interim results of a phase 1–2a trial of Ad26.COV2.S COVID-19 vaccine. N. Engl. J. Med. 2021;384:1824–1835. doi: 10.1056/NEJMoa2034201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Alter G, et al. Immunogenicity of Ad26.COV2.S vaccine against SARS-CoV-2 variants in humans. Nature. 2021;596:268–272. doi: 10.1038/s41586-021-03681-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.King, R. G. et al. Single-dose intranasal administration of AdCOVID elicits systemic and mucosal immunity against SARS-CoV-2 in mice. Preprint at bioRxiv 2020.2010.2010.331348 (2020).
- 114.Fisher E, et al. Induction of SARS-CoV-2 protein S-specific CD8+ T cells in the lungs of gp96-Ig-S vaccinated mice. Front. Immunol. 2020;11:602254. doi: 10.3389/fimmu.2020.602254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li C, et al. Mechanisms of innate and adaptive immunity to the Pfizer-BioNTech BNT162b2 vaccine. Nat. Immunol. 2022 doi: 10.1038/s41590-022-01163-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Künzli M, et al. Route of self-amplifying mRNA vaccination modulates the establishment of pulmonary resident memory CD8 and CD4 T cells. bioRxiv. 2022 doi: 10.1101/2022.06.02.494574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Intapiboon, P. et al. Immunogenicity and safety of an intradermal BNT162b2 mRNA vaccine booster after two doses of inactivated SARS-CoV-2 vaccine in healthy population. Vaccines (Basel)9, 10.3390/vaccines9121375 (2021). [DOI] [PMC free article] [PubMed]
- 118.Feldman RA, et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine. 2019;37:3326–3334. doi: 10.1016/j.vaccine.2019.04.074. [DOI] [PubMed] [Google Scholar]
- 119.Abu-Raddad LJ, et al. Effect of mRNA vaccine boosters against SARS-CoV-2 Omicron infection in Qatar. N. Engl. J. Med. 2022;386:1804–1816. doi: 10.1056/NEJMoa2200797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hurme A, et al. Long-lasting T cell responses in BNT162b2 COVID-19 mRNA vaccinees and COVID-19 convalescent patients. Front. Immunol. 2022;13:869990. doi: 10.3389/fimmu.2022.869990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Noh JY, Jeong HW, Kim JH, Shin E-C. T cell-oriented strategies for controlling the COVID-19 pandemic. Nat. Rev. Immunol. 2021;21:687–688. doi: 10.1038/s41577-021-00625-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Goel RR, et al. mRNA vaccines induce durable immune memory to SARS-CoV-2 and variants of concern. Science. 2021;374:abm0829. doi: 10.1126/science.abm0829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mostafa HH, et al. SARS-CoV-2 infections in mRNA vaccinated individuals are biased for viruses encoding spike E484K and associated with reduced infectious virus loads that correlate with respiratory antiviral IgG levels. J. Clin. Virol. 2022;150–151:105151. doi: 10.1016/j.jcv.2022.105151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Grifoni A, et al. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell. 2020;181:1489–1501.e15. doi: 10.1016/j.cell.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sette A, Crotty S. Pre-existing immunity to SARS-CoV-2: the knowns and unknowns. Nat. Rev. Immunol. 2020;20:457–458. doi: 10.1038/s41577-020-0389-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Poon MML, et al. SARS-CoV-2 infection generates tissue-localized immunological memory in humans. Sci. Immunol. 2021;6:eabl9105. doi: 10.1126/sciimmunol.abl9105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Grau-Exposito J, et al. Peripheral and lung resident memory T cell responses against SARS-CoV-2. Nat. Commun. 2021;12:3010. doi: 10.1038/s41467-021-23333-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Patel NP, et al. Impact of Zostavax vaccination on T-cell accumulation and cutaneous gene expression in the skin of older humans after varicella zoster virus antigen-specific challenge. J. Infect. Dis. 2018;218:S88–S98. doi: 10.1093/infdis/jiy420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Oxman MN, et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N. Engl. J. Med. 2005;352:2271–2284. doi: 10.1056/NEJMoa051016. [DOI] [PubMed] [Google Scholar]
- 130.Lal H, et al. Efficacy of an adjuvanted herpes zoster subunit vaccine in older adults. N. Engl. J. Med. 2015;372:2087–2096. doi: 10.1056/NEJMoa1501184. [DOI] [PubMed] [Google Scholar]
- 131.Heineman TC, Cunningham A, Levin M. Understanding the immunology of Shingrix, a recombinant glycoprotein E adjuvanted herpes zoster vaccine. Curr. Opin. Immunol. 2019;59:42–48. doi: 10.1016/j.coi.2019.02.009. [DOI] [PubMed] [Google Scholar]
- 132.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/study/NCT04403139 (2022).
- 133.Sridhar, S. Heterosubtypic T-cell immunity to influenza in humans: challenges for universal T-cell influenza vaccines. Front. Immunol. 7, 10.3389/fimmu.2016.00195 (2016). [DOI] [PMC free article] [PubMed]
- 134.Ssemaganda A, et al. Expansion of cytotoxic tissue-resident CD8+ T cells and CCR6+CD161+CD4+ T cells in the nasal mucosa following mRNA COVID-19 vaccination. Nat. Commun. 2022;13:3357. doi: 10.1038/s41467-022-30913-4. [DOI] [PMC free article] [PubMed] [Google Scholar]