

Abstract
Core histones including H2A, H2B, H3, and H4 are key modulators of cellular repair, transcription, and replication within eukaryotic cells, playing vital roles in the pathogenesis of disease and cellular responses to environmental stimuli. Traditional mass spectrometry (MS)-based bottom-up and top-down proteomics allows for the comprehensive identification of proteins and of post-translational modification (PTM) harboring proteoforms. However, these methodologies have difficulties preserving near-cellular spatial distributions because they typically require laser capture microdissection (LCM) and advanced sample preparation techniques. Herein, we coupled a matrix-assisted laser desorption/ionization (MALDI) source with a Thermo Scientific Q Exactive HF Orbitrap MS upgraded with ultrahigh mass range (UHMR) boards for the first demonstration of complementary high-resolution accurate mass (HR/AM) measurements of proteoforms up to 16.5 kDa directly from tissues using this benchtop mass spectrometer. The platform achieved isotopic resolution throughout the detected mass range, providing confident assignments of proteoforms with low ppm mass error and a considerable increase in duty cycle over other Fourier transform mass analyzers. Proteoform mapping of core histones was demonstrated on sections of human kidney at near-cellular spatial resolution, with several key distributions of histone and other proteoforms noted within both healthy biopsy and a section from a renal cell carcinoma (RCC) containing nephrectomy. The use of MALDI-MS imaging (MSI) for proteoform mapping demonstrates several steps toward high-throughput accurate identification of proteoforms and provides a new tool for mapping biomolecule distributions throughout tissue sections in extended mass ranges.
Graphical Abstract
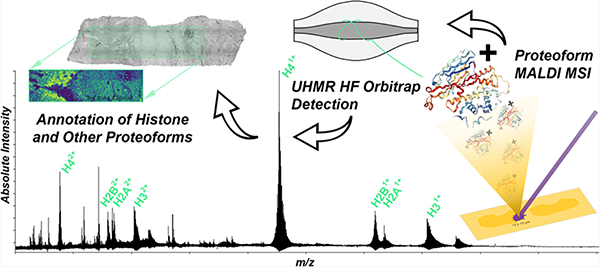
Within eukaryotic cells, regulation of DNA has key modulatory mechanisms, which promote repair, transcription, and replication. Conformational changes within the conserved structure of chromatin stem from the nucleosome, which is comprised of four highly dynamic core histones (H2A, H2B, H3, and H4).1,2 The backbones of these core histones, and the N-terminal tail, which directly interacts with DNA, have been found to harbor multiple post-translational modifications (PTMs) resulting in a multitude of unique proteoforms.3,4 Each proteoform potentially serves unique biological functions based upon the nucleosomes’ conformational change via histone PTMs, ultimately resulting in a highly complex histone code,5 and epigenetic interactions.6,7 These chromatin-related proteins are prime examples of dynamic modulators of pathogenesis and cellular repair within eukaryotic lifeforms and have been extensively studied through traditional proteomic methodologies 8
While functional studies of histones have been accomplished at the peptide level using bottom-up (BU) or middle-down (MD) mass spectrometry (MS), proteoform level identification solely from peptide level datasets remains a highly challenging aspect reliant on correlation-based bioinformatics tools 9 The vast majority of proteoform level characterizations are completed by top-down (TD) analyses of intact proteins.10,11 Recent advances combining laser capture microdissection (LCM) with TD-MS enabled identification of greater than 400 proteoforms from roughly 250 cells.12,13 However, such analyses require isolation of cells or tissue sections for protein extraction and subsequent liquid chromatography mass spectrometry (LC/MS), which have high specificity but relatively low throughput. This limits the feasibility and resolution of whole-tissue LCM-based LC/MS by microPOTS14 or nanoPOTS,15 and with the loss of cellular context, challenges arise for interrogation of the native heterogeneity in a spatially resolved manner. To this end, mass spectrometry imaging (MSI) has emerged as a powerful high-throughput supplement capable of nontargeted, spatially resolved measurements of biomolecules directly from tissuees.16
Various modalities have the capability of creating complementary ion images localizing not only xenobiotics, metabolites, lipids, and peptides but also intact proteoforms.17 Despite the widespread proteomic applications within MSI, the path forward is a challenging multidisciplinary effort reliant on instrument and method development.18 Several ambient imaging approaches have recently gained more traction for protein applications, allowing for the detection and TD dissociation of multiply charged proteoforms and even native protein complexes.19–24 Despite these advances, matrix-assisted laser desorption/ ionization (MALDI) remains the most commonly used tool for MSI-based proteomic applications.17,25 MALDI has been capable of cellular resolution MSI for intact protein analyses for nearly a decade,26,27 with several reports documenting distributions of proteins with masses over 100 kDa to recent upper bounds of 200 kDa.28,29 However, several distinct challenges arise, both from the sample preparation requirements and limited proteome coverage afforded by MSI.30
MSI of intact proteoforms has a unique set of prerequisites for ionization and instrumentation. However, a trade-off exists within MSI where throughput, spatial resolution, and mass resolution are interrelated variables and often one or more of these variables is compromised.31 Histone proteoforms have been directly analyzed from samples of the mouse brain via MALDI-MSI.32 Several reports have confidently annotated proteoforms directly from tissues using high-sensitivity and mass-resolution MS. For example, Fourier transform ion cyclotron resonance (FTICR) MS has been utilized for direct proteoform annotation by high-resolution accurate mass (HR/AM) alone. However, exhaustive standalone FTICR-MSI studies are usually not completed due to time constraints for imaging, despite exhibiting performance not attainable using faster and more commonly implemented linear time-of-flight (TOF) mass analyzers.33,34
Herein, we demonstrate the addition of a novel sample pretreatment through direct acidification of the tissue for the enhanced MALDI-MSI of core histones (H2A, H2B, H3, and H4), all of which can be detected through the first coupling of MALDI to an ultrahigh mass range (UHMR) Thermo Scientific Q Exactive high-field (HF) Orbitrap MS. While several reports have coupled various MALDI sources to Orbitrap MS, this is the first HR/AM demonstration of isotopically resolved proteoforms with high mass accuracy (i.e., low ppm mass error). This platform also demonstrates the same high-resolution performance as FTICR MS but with a significant reduction in requisite detection time per pixel in extended mass ranges. Herein, two distinct samples of the human kidney were studied at a near-cellular resolution to benchmark the new MSI platform. Key advantages of this instrument are exhibited for proteoform mapping directly from human kidney tissues, utilizing optimized protocols for histone proteoforms. Several differential and characteristic distributions of histone proteoforms within the sections were observed, alongside small protein signatures, which can be utilized as indicators of underlying processes and/or can serve as potential biomarkers of disease and trauma.
EXPERIMENTAL SECTION
Materials and Chemicals.
High-performance liquid chromatography (HPLC)-grade acetonitrile, isopropanol, and water were purchased from Fisher Scientific (Fair Lawn, NJ).Glacial acetic acid (AA, 99.99%) and trifluoroacetic acid (TFA, 99.5%) were Optima LC/MS Grade from Fisher Chemical (Fair Lawn, NJ). 200 proof ethanol was acquired from Decon Laboratories (King of Prussia, PA). Chloroform, 2′,5′-dihydroxyacetophenone (2,5-DHA), and cesium iodide were from Sigma-Aldrich (St. Louis, MO).
Samples.
Sectioning was performed at the University of Texas Health Science Center (UTHSC) at San Antonio. The UTHSC institutional review board has approved all of the experimental procedures (IRB No. HSC2017035H) and written consent was attained from all patients. Tissue specimens were obtained by surgical resection in the Department of Pathology, UTHSC. Tissue blocks were fresh-frozen in liquid nitrogen (without PBS pre-wetting) and stored at −80 °C until analysis. The tissue blocks were not embedded and were cryosectioned at 10 μm onto indium-tinoxide (ITO)-coated slides. Tissue blocks were stored for 3 months for the cancerous segment, prior to sectioning. After cryosectioning, tissue segments were shipped on dry ice and immediately placed within a −80 °C freezer. Prior to analysis, the cancerous tissue section was stored at −80 °C for 2 months.
Lipid Depletion Washes.
Pretreatment steps were evaluated for optimizing the global histone proteoform signal on human kidney and the optimized procedure was as follows. Samples were desiccated under 18 inHg of vacuum for 30 min and then washed in fresh solutions of 70% ethanol for 30 s, 100% ethanol for 30 s, Carnoy’s solution (6:3:1 v/v ethanol/chloroform/glacial acetic acid) for 2 min, 100% ethanol for 30 s, water with 0.2% TFA for 15 s, and immediately washed in 100% ethanol for 30 s. Samples were then dried by a stream of nitrogen gas prior to tissue acidification.
Tissue Acidification Pretreatment.
After washing, each slide was directly acidified with a solution of 5% acetic acid (v/v) in 50% ethanol using an HTX Technologies M5 Sprayer (Chapel Hill, NC) with a flow rate of 150 μL/min, a nozzle temperature of 30.0 °C, and the velocity set to 1250 mm/min with 10 PSI of nitrogen gas. A 5 s drying period was added between each of the passes with a 3 mm track spacing and a “CC” pattern. No delocalization was found to occur due to this pretreatment step.
MALDI Matrix Deposition.
Directly following the pre-extraction of proteins from tissues by acidification, the same sprayer was used to deposit 15 mg/mL 2,5-DHA in 90% acetonitrile with 0.2% TFA. The supernatant of the matrix was collected after sonication and centrifugation. The flow rate of the matrix was 150 μL/min with a nozzle temperature of 30.0 °C, and the velocity was set to 1300 mm/min with 10 PSI of nitrogen gas. The matrix was applied with a 2 mm track spacing and a “CC” pattern, and the matrix coverage was calculated to be 277 μg/cm2. The matrix was then recrystallized with 5% acetic acid solution in water at 38.5 °C and dried for 3.5 min, using an apparatus similar to that previously reported,35 and then immediately analyzed.
Instrumentation.
An elevated pressure (EP) MALDI source with an atmospheric pressure ionization (API) inlet (Spectroglyph LLC, Kennewick, WA) was mounted on a Q Exactive HF Orbitrap MS;36 this instrument was upgraded with Q Exactive UHMR boards and operated under custom privileges licenses.37,38 The UHMR Q Exactive HF MS with the MALDI source mounted is depicted in Supporting Figure S1, and a more detailed operation and parameter list is included in the Supporting Information. Briefly, spectra were acquired over the m/z range of 3,500 to 20,000 and these analyses were performed with a custom noise thresholding setting of 1.5 (vs default 4.54). Acquisitions were carried out at a preset resolution of 240k at m/z 200 (corresponding to a 0.512 s transient) with 250 laser shots per pixel; post acquisition, the laser spot size was measured as 12 μm × 15 μm by light microscopy. External calibration was performed using direct infusion electrospray ionization of 2 mg/mL solution of cesium iodide in isopropanol/water (50% v/v); exemplary spectra from the calibration solution are shown with default low and high m/z tuning in Supporting Figure S2.
MALDI Data Processing.
The reduced profile .RAW files were viewed with FreeStyle software by Thermo Scientific (version 1.4) and imported with positional data (.xml) from the Spectroglyph, LLC source to form ion images in SCiLS Lab software by Bruker Daltonics (version 2021c). Each dataset resulted in more than 40,000 pixels. Images were processed after root-mean-square normalization within SCiLS Lab. High-resolution bright-field imaging was completed before MALDI-MSI and after the MALDI matrix was washed off and periodic acid–Schiff (PAS) staining was completed using a standard protocol. These images were used to guide the pathology of the segments and a manual segmentation of regions of interest, which was completed within SCiLS Lab. Mean intensities were extracted, exported, and then normalized to the relative surface area (mm2) of each region for comparison of proteoform levels across tissues. Proteoform mass errors were reported from a single isotopologue from the average spectrum. Simulations of theoretical isotopic distributions at various transient lengths for several MALDI-capable instruments were completed within Peak-by-Peak software by Spectroswiss (Base Edition, version 2021.8.1.b4).39
RESULTS AND DISCUSSION
Benchmarking Proteoform Mapping within the Human Kidney by UHMR Q Exactive HF Orbitrap MS.
While no further sample pretreatment is required after lipid depletion washes to detect core histone proteoforms, additional washes can greatly influence the adducts and detection of protein fingerprints in different tissue types and organisms.32,33,40,41 Traditionally, optimization includes varied timing or additional polar aprotic solvent washes (i.e., Carnoy’s solution, chloroform, etc.) for removing higher concentrations of lipids and small-molecule interferents as well as various degrees of aqueous washes, which can be acidic, to improve desalting.41−45 While small molecules (e.g., lipids) are the first biomolecules to be removed through this tissue fixation and sample preparation, peptides and small proteins are also heavily influenced, where many water-soluble peptides and proteins are commonly depleted with extended exposure to aprotic solvents and aqueous solutions. Evaluation and slight modification of existing protocols resulted in a multistep lipid depletion protocol optimized for the detection of core histones within human kidney, as described in the Experimental Section. Since histones are also highly soluble in acidic aqueous solutions, as noted by several bulk preparation strategies,46 an additional step of tissue acidification for pre-extraction prior to matrix deposition was applied to boost the sensitivity of these analyses. Often tissue blocks and sections undergo long-term storage, and this has been observed to degrade signal intensity. However, after implementation of tissue acidification, we were still able to complete a sensitive analysis. Tissue acidification, completed by directly spraying 5% acetic acid (v/v) prior to matrix deposition, yielded a twofold enhancement in the total histone signal, as highlighted for doubly charged histone H2A and other proteins in Supporting Figure S3.
The entire protocol, including lipid depletion and histone enhancement through tissue acidification, was used to optimize instrument and source parameters and then applied to map histone proteoforms across several sections of human kidney. Overall, it was found that many default settings were optimal except when called out within the additional Supporting Information, and one of the most important tunable parameters was desolvation voltage within in-source trapping (IST), as demonstrated within Supporting Figure S4. A representative average mass spectrum post optimization from this imaging analysis is shown in Figure 1, highlighting the observation of several dozen overlapping isotopic distributions of core histone proteoforms with baseline resolution. Increasing complexity is observed ranging from histone H4 to histone H3 proteoforms as expected, with histone H3 having several more methylation (Me) and acetylation (Ac) sites. This range in complexity together with varied transmission efficiency is a putative explanation for varied intensities of histone families detected via MALDI-MSI.
Figure 1.

Full mass spectrum from the proteoform mapping of human kidney at 40 μm obtained by averaging a total of 86,189 pixels highlighting both the singly and doubly charged core histone proteoform ion clusters. Expanded views of each singly charged histone (H2A, H2B, H3, H4) proteoform family are also shown with isotopic resolution throughout the entire m/z range. The only other near isobaric species other than histone proteoforms are located within the histone H3 proteoform envelope, where several hemoglobin subunits are also presently detected.
Currently, MALDI-MSI with UHMR Q Exactive HF Orbitrap detection allows for the acquisition of protein signals up to approximately m/z 16,500 directly from kidney tissues. Data was acquired with a mass-resolution setting of 240k (at m/z 200); the 0.512 s transient resulted in an observed experimental resolution of 35k at m/z 11,306, which is in good agreement with the theoretically calculated mass resolution. Full magnitude mode (mFT) and absorption mode (aFT) processings of simulated transients within Peak-by-Peak are shown in Supporting Figure S5 and Supporting Table 1. The experimentally observed resolution was found to be consistent with the expected improvement for enhanced FT (eFT) within the entire mass range.47 It should be noted that a lowered threshold for noise rejection used in this work resulted in a noticeable baseline (Figure 1), and beat pattern and signal decay of protein ions over the duration of the transient lead to imperfect balancing of absorption peak shape in eFT,43 revealing in valleys between isotopes that sag below the baseline. Despite this observation, baseline resolution was achieved for all detected doubly and singly charged proteoforms while maintaining a low ppm mass accuracy with external calibration.
The data obtained from this benchmarking is also highly complementary to reports of high-resolution protein MALDI-MSI by 15T FTICR, which has been demonstrated to provide mass resolution up to 70k at m/z 11,000. However, this requires a 6.71 s transient,34 and more commonly a transient of roughly half this length is utilized. Simulations of isotopic distributions were completed within Peak-by-Peak for 15T FTICR, as shown in Supporting Figure S6 and Supporting Table 1. To achieve a resolution comparable to the performance demonstrated by the UHMR Q Exactive HF for the acetylated and dimethylated histone H4, a 3.4 s transient per pixel (0.3 Hz acquisition) is required for 15T FTICR MS with standard mFT processing. Specifically, observed UHMR Q Exactive HF performance within a 0.512 s transient (2 Hz acquisition) signifies a 6.6-fold improvement in the duty cycle for proteoform MALDI-MSI. Future studies using this platform could either benefit from the use of more sensitive and less vacuum stable matrixes due to the increased throughput, profile larger regions of interest, further spatial resolution to cellular and subcellular levels, or increase of the mass resolution with no significant losses in throughput. This significant boost in throughput while maintaining resolving power in extended mass ranges is among the most important benefit of this platform.
Advanced aFT signal processing and/or frequency multiple detections could significantly boost throughput FTICR-MSI.48,49 However, further examination of experimental and simulated spectra demonstrates the well-known slower reduction in resolution at increasing m/z with Orbitrap MS compared to FTICR MS. This comparison and reduction observed within the two FTMS platforms have previously been thoroughly explained,50 however, for higher field FTICR instrumentation this is less significant especially when combined with advanced detection schemes. More specifically, Orbitrap resolution diminishes proportional to 1/√m/z, whereas in FTICR, resolution is proportional to 1/m/z.50 This has also been observed previously within single-ion experiments of ultralarge protein complexes.51 Furthermore, with an extended transient of 1.024 s (Supporting Figure S7), the UHMR Q Exactive HF has an observable mass resolution of 70k at m/z 11,306, allowing for preservation of high mass resolution and near-cellular resolution for entire sections of tissues and further demonstrating the exquisite utility of this platform.52
High-Throughput MALDI-MSI for Near Single-Cell Resolution of Whole Samples.
Human kidney is an incredibly complex organ with several distinct cell types and functional tissue units, and hundreds of different diseases have various pathophysiological effects throughout the cortex and the medulla. Hence, several methodologies are typically used concurrently with both high spatial and high mass-resolution imaging for confident, clinically relevant analyses.53 Pathogenesis of disease and trauma has broad-ranging consequences spanning hypertension, metabolic stress and injury, and endothelial apoptosis. This occurs within the glomeruli, distal and proximal tubules and crosses the interfacial region into the medullary regions systemically affecting the entire renal system.54,55 We have applied MALDI-MSI at near single-cellular resolution to account for this complexity and facilitate proteoform-informed mapping of the functional tissue units. The profiling of the intrinsic heterogeneity within distinct kidney regions and tissue units allows for the extraction of clinically relevant information.
Due to the high mass resolution being maintained with shorter transients, 20 μm spatial resolution was feasible with relatively high throughput. The resultant 39,078 pixels across the tissue section required a calculated 5.56 h to complete (accounting for the detection period only), in comparison to an estimated 36.9 h that would have been required using mFT processing of 15T FTICR transients for comparable resolution at m/z 11,306. In a relatively high-throughput manner, unique histone proteoform distributions were observed within tubules in the cortical regions, which can be noted within the expanded ion images in Figure 2. This figure highlights the putatively annotated histone H4 N-Ac K20me2 and H4 N-Ac/Ac proteoforms. Dimethylation at K20 has been previously linked to double-stranded DNA damage response. Similarly, several readers of K20me2 are known to recruit various repair proteins with links to cell cycle regulation, which aligns with chronic damage.56–58
Figure 2.

Ion images produced from an expanded view of the MALDI-MSI analysis of a tissue section from a cortical nephrectomy block. The tissue was imaged at 20 μm and ion images were obtained by averaging 25 ppm windows for an acetylated and dimethylated histone H4 proteoform at m/z 11,306.3939 (−1.12 ppm) and a diacetylated histone H4 proteoform at m/z 11,320.3661 (−1.74 ppm). Images are all scaled to 200 μm each with separate relative ion abundance. The corresponding microscopic image from the PAS staining post MALDI-MSI is shown at the bottom to highlight atrophic tubular regions with no significant enhancement in signal intensity (orange) and areas of tubular atrophy with the enhancement of the signal (white). Glomeruli are highlighted in yellow.
The PAS staining of this fresh-frozen nephrectomy section confirmed pre-existing chronic kidney damage including tubular atrophy and accompanying interstitial fibrosis, indicated through thickened tubular basement membranes and a simplified tubular epithelium due to tubular injury resultant in stenosis and other afflictions.59 Histological staining also allowed for identification of glomeruli, which were observed to have lower signal intensities for all histone proteoforms relative to the tubular regions. Histone proteoforms, at large, were observed to be colocalized within this analysis, with no distinct observed localizations apparent until a more in-depth profiling was completed. After extraction of proteoform intensities through manual segmentation, significant differential abundances of proteoforms were noted. These results are presented in Figure 3 for H4 N-Ac K20me2 and H4 N-Ac/Ac for the averaged normalized mean intensity for several exemplary tubular regions.
Figure 3.

Microscopy image of the PAS-stained section of the healthy nephrectomy post MALDI-MSI. Highlighted within the tissue section are tubular regions identified from MALDI-MSI with differential levels of histone proteoforms, showing higher abundance (ET/yellow) and lower abundance (DT/teal) for an acetylated and dimethylated histone H4 proteoform at m/z 11,306.3939 (−1.12 ppm) and a diacetylated histone H4 proteoform at m/z 11,320.3661 (−1.74 ppm). The bar chart shows extracted mean intensities normalized to the area (mm2), with the calculated ratio for each region averaged below. Error bars represent one standard deviation.
Utilizing this strategy, a general trend was observed that signified certain tubules having differential abundances of histone proteoforms. After normalization of the mean intensities to the area of the extracted regions of interest, the average was taken for several enhanced atrophic regions (ET, n = 4) and several depleted atrophic tubular regions (DT, n = 4). The H4 N-Ac K20me2 proteoform showed increased abundance within a subset of ET regions, as shown in Figure 3. Differential abundance within the average of these various regions of atrophy is a unique observation for this damage-marking proteoform. In ET regions, proteoform intensities of putative H4 N-Ac K20me2 were found to be significantly greater (p-value of 0.0056; two-sample t-test, one tail). Diacetylated histone H4 displayed similar differential abundances (p-value of 0.023; two-sample t-test, one tail). These results can provide putative links to varying degrees of tubular atrophy. Further study is warranted for increased statistical power and to further differentiate tubular regions (i.e., distal and proximal tubules) within the cortex.
While histone H4 N-Ac K20me2 has been noted in several reports as the most abundant H4 proteoform,33,41,60 sole annotations by HR/AM measurements are tentative. This lends credence to multiplexed LCM-based proteomics and alternative TD-MS strategies61 such as implementation of MALDI in-source decay (ISD) or ultraviolet photodissociation (UVPD) both of which have been demonstrated within lower mass ranges on MALDI-enabled Orbitrap MS.62 Routine TD fragmentation of proteoforms has not yet been demonstrated directly from tissues for MALDI analyses, but dissociation methods are more common for ambient ionization techniques, which create multiply charged protein ions.21,23,63 Nonetheless, the variance noted within the proteoforms detected by MALDI-MSI highlights the stability and promise of this platform for the mapping of distinct (histone) proteoform signatures, as exemplified above within regions of tubular atrophy.
Case Study: MALDI-MSI on a Section of Kidney from a Renal Cell Carcinoma Nephrectomy.
Hereditary disorders, as well as external risk factors such as smoking and exposure to toxins, play roles in the pathogenesis of cancer, suggesting a strong link to the epigenetics and histone profiles.4 Several studies noted the importance of epiproteomic profiling of clinical samples and implicated histone proteoforms as potential biomarkers of disease utilizing techniques such as MALDI-FTICR and MALDI-TOF imaging for clinical research.33,60 Renal cell carcinoma (RCC) is the most common type of kidney cancer in adults and typically results in a nephrectomy. As the disease progresses from within the linings of the tubules, discrete tumors are often formed.64 While the RCC tissue section presented herein was stored for an extended period (2 months), histone proteoforms were directly detected from the tissue through the optimized MALDI-MSI protocol.
Interestingly, bulk histone distributions within the healthy region were found to be consistent with several other profiled kidney sections. However, distinct areas of increased abundance of core histone proteoforms were found within the RCC tumor segment, which exists within confined borders within this section (outlined in Figure 4A,B). Distinct enhancements of several proteoforms are noted within this border region of the RCC segment at the interface of the healthy tissue, as shown in Supporting Figure S8. Note that if bulk proteomics measurements were made, or even spatially resolved LCM-based LC/MS analyses, without a priori knowledge of the sample, these enhanced abundances at the edge of the tumor would have been missed. This finding highlights the importance of spatially resolved proteomic measurements in clinical and translational research.
Figure 4.

(A) Bright microscopy image of the RCC section prior to MALDI-MSI analysis. (B) PAS-stained section post MALDI-MSI. Ion images are produced from singular isotopic peak distributions and show an expanded view of the MALDI-MSI analysis at 30 μm spatial resolution. Spatial distribution of (C) acetylated and dimethylated histone H4 proteoforms at m/z 11,306.4130 (0.57 ppm); (D) diacetylated histone H4 proteoform at m/z 11,320.3657 (0.42 ppm); and (E) diacetylated and dimethylated histone H4 proteoforms at m/z 11,348.4344 (1.78 ppm). Scale bars within the ion images are 600 μm, and each image has a separate ion intensity scale.
Ion images from distributions of putative H4 N-Ac K20me2, H4 N-Ac/Ac, and H4 N-Ac/Ac K20me2 proteoforms are highlighted in Figure 4C–E, respectively. These results warrant further studies for the determination of the epigenetic relevance to RCC progression, as normal levels of the histone proteoforms within the healthy region are significantly lower when compared to the RCC region. As before, the base peak of the analysis corresponds to an annotated histone H4 N-ac K20me2 proteoform (Figure 4C), but interestingly majority of detected H4 histone proteoforms show unique distributions within gradients in the RCC tissue segment.
An early MALDI-TOF-MSI analysis with tandem immune-histostaining showed high levels of enhancement of K20me2 through microvascular invasion after hepatocellular carcinoma (HCC) removal. This PTM was identified as a putative biomarker of post-operative recurrence of HCC.60 As K20me2 is an indicator of damage and trauma, the enhancement within the border region of the RCC segments is also logical and could be utilized within additional studies as a potential biomarker once validated through supplementary methods and clinically relevant sample sizes. Additionally, colocalizations of 40S ribosomal protein S30 at m/z 6647.9 have broad links to cell plasticity.65,66 This annotated protein was enhanced within the healthy cortical region containing a subset of tubules and showed increased intensity within the RCC segment. This demonstration of proteoform-informed MALDI-MSI by UHMR Q Exactive HF further expands upon previous MALDI-TOF imaging completed on clear cell RCC, which demonstrated the complexities of annotation of proteins without sufficient resolution.67 This illustrates the utility of MALDI-MSI for broad profiling of proteoforms within entire tissue sections enabled by high-resolution FTMS.
CONCLUSIONS
We present the first implementation of MALDI-MSI on an UHMR-Orbitrap MS resulting in the detection of proteoforms of up to 16.5 kDa directly from human kidney tissue sections at near-cellular resolution. Through the optimization of lipid depletion washes, and the addition of a novel tissue pre-extraction step, a sensitive spatial analysis at 20 μm spatial resolution without oversampling of the tissue was routine. The sample preparation was optimized for enhanced detection of core histone proteoforms; however, over a dozen peptides and smaller proteins were also detected from fresh-frozen human tissues, which have been in storage for various periods. The UHMR Q Exactive HF also provided sufficient mass resolution for the isotopically resolved measurements at 2 Hz sampling rate, comparable at all levels to 15T FTICR for MALDI-MSI of proteoforms within an extended mass range. In fact, this new platform outperforms FTICR with a reported 6.6-fold increase in duty cycle per pixel for equivalent observed resolution in normal modes of operation. These gains within extended mass proteoform mapping by MALDI align well with the goals of clinical and translational studies, which require large cohorts for biological relevance.
This platform also permitted the annotation of proteoforms based upon HR/AM measurements matched against theoretically calculated isotopic distributions. The UHMR Q Exactive HF exhibited exceptional stability with the mass error below 3 ppm within the calibrated mass range using external calibration. Since on-tissue tandem MS of low-charge state ions is challenging by traditional dissociation methods, subsequent TD-MS experiments are still required to confidently identify various proteoforms. To further address this challenge and facilitate proteoform-aware mapping of tissues with high-throughput and near-cellular resolution, we are currently integrating MALDI-MSI with LC-MS microproteomics (i.e., microPOTS or nanoPOTS) and concurrently developing advanced dissociation techniques such as UVPD for proteoforms desorbed and ionized by MALDI. Overall, the utility of MALDI on a UHMR Q Exactive HF is well positioned to provide an ultrahigh mass resolution within extended ranges at 1–2 Hz sampling rate for significant throughput enhancement. This level of performance will also enable future developmental pushes to expand the detectable mass range to larger proteins and proteoforms. Future studies with large cohorts will also be feasible within several days (versus several weeks currently), making proteoform-informed FTMS-MSI more competitive to faster TOF-MSI and/or enabling future expansion and development of cellular and subcellular imaging modalities.
Supplementary Material
ACKNOWLEDGMENTS
The authors would also like to acknowledge Dr. Marija Veličković at EMSL for assistance with the histological staining and microscopy and Dr. Mikhail Belov at Spectroglyph, LLC, as well as Dr. Gordon Anderson and Chris Anderson at GAA Custom Electronics, LLC, for technical support for the MALDI source. The authors would also like to thank Drs. Yury Tsybin, Konstantin Nagornov, and Anton Kozhinov at Spectroswiss for support within the Peak-By-Peak software. This work was performed at the Environmental Molecular Science Laboratory (EMSL), a Department of Energy (DOE) Office of Science User Facility sponsored by the Office of Biological and Environmental Research. This research was funded by the National Institutes of Health (NIH) Common Fund, Human Biomolecular Atlas Program (HuBMAP) grant UG3CA256959-01, doi.org/10.46936/staf.proj.2020.51770/60000309 (L.P.T.), and by the intramural program on project award doi.org/10.46936/intm.proj.2019.51159/60000152 (W.K) at EMSL (grid.436923.9) operated under Contract No. DE-AC05-76RL01830.
Footnotes
The authors declare the following competing financial interest(s): K.L.F, M.R.S., and A.A.M. declare the following competing financial interest(s): these authors are employees of Thermo Fisher Scientific, manufacturer of Orbitrap mass spectrometers.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.2c01034.
Additional experimental details including extended instrument parameters and notes, a digital photograph of the instrument, additional spectra highlighting the optimization of the intact protein imaging, additional ion images from the MALDI imaging of the RCC section, as well as transient-mediated simulations of intact proteins for various Orbitrap and FTICR transient lengths (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.2c01034
Contributor Information
Kevin J. Zemaitis, Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, Washington 99352, United States
Dušan Veličković, Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, Washington 99352, United States.
William Kew, Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, Washington 99352, United States.
Kyle L. Fort, Thermo Fisher Scientific (Bremen) GmbH, 28199 Bremen, Germany
Maria Reinhardt-Szyba, Thermo Fisher Scientific (Bremen) GmbH, 28199 Bremen, Germany.
Annapurna Pamreddy, Center for Renal Precision Medicine, Department of Medicine, University of Texas Health, San Antonio, Texas 78284, United States.
Yanli Ding, Department of Pathology and Laboratory Medicine, University of Texas Health, San Antonio, Texas 78284, United States.
Dharam Kaushik, Department of Urology, University of Texas Health, San Antonio, Texas 78284, United States.
Kumar Sharma, Center for Renal Precision Medicine, Department of Medicine, University of Texas Health, San Antonio, Texas 78284, United States; Audie L. Murphy Memorial VA Hospital, South Texas Veterans Health Care System, San Antonio, Texas 78284, United States.
Alexander A. Makarov, Thermo Fisher Scientific (Bremen) GmbH, 28199 Bremen, Germany; Biomolecular Mass Spectrometry and Proteomics, Bijvoet Center for Biomolecular Research and Utrecht Institute for Pharmaceutical Sciences, University of Utrecht, Utrecht 3584, The Netherlands
Mowei Zhou, Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, Washington 99352, United States.
Ljiljana Paša-Tolić, Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, Washington 99352, United States.
REFERENCES
- (1).Strahl BD; Allis CD Nature 2000, 403, 41–45. [DOI] [PubMed] [Google Scholar]
- (2).Gillette TG; Hill JA Circ. Res. 2015, 116, 1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Lusser A; Kölle D; Loidl P Trends Plant Sci. 2001, 6, 59–65. [DOI] [PubMed] [Google Scholar]
- (4).Zhao Z; Shilatifard A Genome Biol. 2019, 20, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Jenuwein T; Allis CD Science 2001, 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- (6).Smith LM; Kelleher NL; Linial M; Goodlett D; Langridge-Smith P; Ah Goo Y; Safford G; Bonilla L; Kruppa G; Zubarev R; Rontree J; Chamot-Rooke J; Garavelli J; Heck A; Loo J; Penque D; Hornshaw M; Hendrickson C; Pasa-Tolic L; Borchers C; Chan D; Young N; Agar J; Masselon C; Gross M; McLafferty F; Tsybin Y; Ge Y; Sanders I; Langridge J; Whitelegge J; Marshall A; The Consortium for Top Down. Nat.Methods 2013, 10, 186–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Smith LM; Kelleher NL Science 2018, 359, 1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lu C; Coradin M; Porter EG; Garcia BA Mol. Cell. Proteomics 2021, 20, No. 100006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bludau I; Frank M; Dörig C; Cai Y; Heusel M; Rosenberger G; Picotti P; Collins BC; Röst H; Aebersold R Nat. Commun. 2021, 12, No. 3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Toby TK; Fornelli L; Kelleher NL Annu. Rev. Anal. Chem. 2016, 9, 499–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chen B; Brown KA; Lin Z; Ge Y Anal. Chem. 2018, 90, 110–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhou M; Uwugiaren N; Williams SM; Moore RJ; Zhao R; Goodlett D; Dapic I; Pasa-Tolič L; Zhu Y Anal. Chem. 2020, 92, 7087–7095. [DOI] [PubMed] [Google Scholar]
- (13).Lubeckyj RA; Sun L Mol. Omics 2022, 18, 112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Xu K; Liang Y; Piehowski PD; Dou M; Schwarz KC; Zhao R; Sontag RL; Moore RJ; Zhu Y; Kelly RT Anal. Bioanal. Chem. 2019, 411, 4587–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhu Y; Piehowski PD; Zhao R; Chen J; Shen Y; Moore RJ; Shukla AK; Petyuk VA; Campbell-Thompson M; Mathews CE; Smith RD; Qian W-J; Kelly RT Nat. Commun. 2018, 9, No. 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Buchberger AR; DeLaney K; Johnson J; Li L Anal. Chem. 2018, 90, 240–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Aichler M; Walch A Lab. Invest. 2015, 95, 422–431. [DOI] [PubMed] [Google Scholar]
- (18).Aebersold R; Mann M Nature 2016, 537, 347–355. [DOI] [PubMed] [Google Scholar]
- (19).Hsu C-C; Chou P-T; Zare RN Anal. Chem. 2015, 87, 11171–11175. [DOI] [PubMed] [Google Scholar]
- (20).Towers MW; Karancsi T; Jones EA; Pringle SD; Claude EJ Am. Soc. Mass Spectrom. 2018, 29, 2456–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Griffiths RL; Konijnenberg A; Viner R; Cooper HJ Anal.Chem. 2019, 91, 6962–6966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hale OJ; Cooper HJ Anal. Chem. 2021, 93, 4619–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Su P; McGee JP; Durbin KR; Hollas MAR; Yang M; Neumann EK; Allen JL; Drown BS; Butun FA; Greer JB; Early BP; Fellers RT; Spraggins JM; Laskin J; Camarillo JM; Kafader JO; Kelleher NL bioRxiv 2021, DOI: 10.1101/2021.12.07.471638. [DOI] [Google Scholar]
- (24).Yang M; Hu H; Su P; Thomas PM; Camarillo JM; Greer JB; Early BP; Fellers RT; Kelleher NL; Laskin J Proteoform-Selective Imaging of Tissues Using Mass Spectrometry. Angew. Chem., Int. Ed. n/a e202200721. DOI: 10.1002/anie.202200721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).McDonnell LA; Corthals GL; Willems SM; van Remoortere A; van Zeijl RJM; Deelder AM J. Proteomics 2010, 73, 1921–1944. [DOI] [PubMed] [Google Scholar]
- (26).Zavalin A; Todd EM; Rawhouser PD; Yang J; Norris JL; Caprioli RM J. Mass Spectrom. 2012, 47, 1473–1481. [DOI] [PubMed] [Google Scholar]
- (27).Zavalin A; Yang J; Hayden K; Vestal M; Caprioli RM Anal. Bioanal. Chem. 2015, 407, 2337–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).van Remoortere A; van Zeijl RJ; van den Oever N; Franck J; Longuespée R; Wisztorski M; Salzet M; Deelder AM; Fournier I; McDonnell LA J. Am. Soc. Mass Spectrom. 2010, 21, 1922–9. [DOI] [PubMed] [Google Scholar]
- (29).Liu H; Han M; Li J; Qin L; Chen L; Hao Q; Jiang D; Chen D; Ji Y; Han H; Long C; Zhou Y; Feng J; Wang X Anal. Chem. 2021, 93, 11920–11928. [DOI] [PubMed] [Google Scholar]
- (30).Han J; Permentier H; Bischoff R; Groothuis G; Casini A; Horvatovich P TrAC, Trends Anal. Chem. 2019, 112, 13–28. [Google Scholar]
- (31).Spraggins JM; Rizzo DG; Moore JL; Noto MJ; Skaar EP; Caprioli RM Proteomics 2016, 16, 1678–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lahiri S; Sun N; Solis-Mezarino V; Fedisch A; Ninkovic J; Feuchtinger A; Götz M; Walch A; Imhof A Proteomics 2016, 16, 437–447. [DOI] [PubMed] [Google Scholar]
- (33).Dilillo M; Ait-Belkacem R; Esteve C; Pellegrini D; Nicolardi S; Costa M; Vannini E; Graaf E. L. d.; Caleo M; McDonnell LA Sci. Rep. 2017, 7, No. 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Prentice BM; Ryan DJ; Van de Plas R; Caprioli RM; Spraggins JM Anal. Chem. 2018, 90, 5090–5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Yang J; Caprioli RM Anal. Chem. 2011, 83, 5728–5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Belov ME; Ellis SR; Dilillo M; Paine MRL; Danielson WF; Anderson GA; de Graaf EL; Eijkel GB; Heeren RMA; McDonnell LA Anal. Chem. 2017, 89, 7493–7501. [DOI] [PubMed] [Google Scholar]
- (37).Fort KL; van de Waterbeemd M; Boll D; Reinhardt-Szyba M; Belov ME; Sasaki E; Zschoche R; Hilvert D; Makarov AA; Heck AJR Analyst 2018, 143, 100–105. [DOI] [PubMed] [Google Scholar]
- (38).van de Waterbeemd M; Tamara S; Fort KL; Damoc E; Franc V; Bieri P; Itten M; Makarov A; Ban N; Heck AJR Nat. Commun. 2018, 9, No. 2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Nagornov KO; Kozhinov AN; Gasilova N; Menin L; Tsybin YO J. Am. Soc. Mass Spectrom. 2020, 31, 1927–1942. [DOI] [PubMed] [Google Scholar]
- (40).Spraggins JM; Rizzo DG; Moore JL; Rose KL; Hammer ND; Skaar EP; Caprioli RM J. Am. Soc. Mass Spectrom. 2015, 26, 974–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Piga I; Heijs B; Nicolardi S; Giusti L; Marselli L; Marchetti P; Mazzoni MR; Lucacchini A; McDonnell LA Int. J. Mass Spectrom. 2019, 437, 10–16. [Google Scholar]
- (42).Lemaire R; Wisztorski M; Desmons A; Tabet JC; Day R; Salzet M; Fournier I Anal. Chem. 2006, 78, 7145–7153. [DOI] [PubMed] [Google Scholar]
- (43).Seeley EH; Oppenheimer SR; Mi D; Chaurand P; Caprioli RM J. Am. Soc. Mass Spectrom. 2008, 19, 1069–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Deutskens F; Yang J; Caprioli RM J. Mass Spectrom. 2011, 46, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Rešetar Maslov D; Svirkova A; Allmaier G; MarchettiDeschamann M; Kraljević Pavelić S Food Chem. 2019, 283, 275–286. [DOI] [PubMed] [Google Scholar]
- (46).Shechter D; Dormann HL; Allis CD; Hake SB Nat. Protoc. 2007, 2, 1445–1457. [DOI] [PubMed] [Google Scholar]
- (47).Lange O; Damoc E; Wieghaus A; Makarov A Int. J. Mass Spectrom. 2014, 369, 16–22. [Google Scholar]
- (48).Shaw JB; Gorshkov MV; Wu Q; Paša-Tolić L Anal. Chem. 2018, 90, 5557–5562. [DOI] [PubMed] [Google Scholar]
- (49).Park S-G; Anderson GA; Bruce JE J. Am. Soc. Mass Spectrom. 2020, 31, 719–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Zubarev RA; Makarov A Anal. Chem. 2013, 85, 5288–5296. [DOI] [PubMed] [Google Scholar]
- (51).McGee JP; Melani RD; Yip PF; Senko MW; Compton PD; Kafader JO; Kelleher NL Anal. Chem. 2021, 93, 2723–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Denisov E; Damoc E; Makarov A Int. J. Mass Spectrom. 2021, 466, No. 116607. [Google Scholar]
- (53).Kruse ARS; Spraggins JM Front. Physiol. 2022, 13, No. 837773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Dumas SJ; Meta E; Borri M; Luo Y; Li X; Rabelink TJ; Carmeliet P Nat. Rev. Nephrol. 2021, 17, 441–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Matovinović MS EJIFCC 2009, 20, 2–11. [PMC free article] [PubMed] [Google Scholar]
- (56).Paquin KL; Howlett NG Mol. Cancer Res. 2018, 16, 1335–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Bannister AJ; Schneider R; Kouzarides T Cell 2002, 109, 801–806. [DOI] [PubMed] [Google Scholar]
- (58).Wang T; Holt MV; Young NL Epigenet. Chromatin 2018, 11, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Lusco MA; Fogo AB; Najafian B; Alpers CE Am. J. Kidney Dis. 2016, 67, e33–e34. [DOI] [PubMed] [Google Scholar]
- (60).Poté N; Alexandrov T; Le Faouder J; Laouirem S; Léger T; Mebarki M; Belghiti J; Camadro J-M; Bedossa P; Paradis V Hepatology 2013, 58, 983–994. [DOI] [PubMed] [Google Scholar]
- (61).Smith LM; Thomas PM; Shortreed MR; Schaffer LV; Fellers RT; LeDuc RD; Tucholski T; Ge Y; Agar JN; Anderson LC; Chamot-Rooke J; Gault J; Loo JA; Paša-Tolić L; Robinson CV; Schlüter H; Tsybin YO; Vilaseca M; Vizcaíno JA; Danis PO; Kelleher NL Nat. Methods 2019, 16, 939–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Dilillo M; de Graaf EL; Yadav A; Belov ME; McDonnell LA J. Proteome Res. 2019, 18, 557–564. [DOI] [PubMed] [Google Scholar]
- (63).Garza KY; Feider CL; Klein DR; Rosenberg JA; Brodbelt JS; Eberlin LS Anal. Chem. 2018, 90, 7785–7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Petejova N; Martinek A Biomed. Pap. Med. Fac. Univ Palacky Olomouc Czech Repub. 2016, 160, 183–94. [DOI] [PubMed] [Google Scholar]
- (65).Ogata K; Ohno R; Terao K; Endo YJ Biochem. 2000, 128, 1–9. [DOI] [PubMed] [Google Scholar]
- (66).Trakman L; Hewson C; Burdach J; Morris KV PLoS One 2016, 11, No. e0152424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Jones EE; Powers TW; Neely BA; Cazares LH; Troyer DA; Parker AS; Drake RR Proteomics 2014, 14, 924–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.