

Abstract
Enhancing CRISPR-mediated site-specific transgene insertion efficiency by homology-directed repair (HDR) using high concentrations of double-stranded DNA (dsDNA) with Cas9 target sequences (CTSs) can be toxic to primary cells. Here, we develop single-stranded DNA (ssDNA) HDR templates (HDRTs) incorporating CTSs with reduced toxicity that boost knock-in efficiency and yield by an average of around two- to threefold relative to dsDNA CTSs. Using small-molecule combinations that enhance HDR, we could further increase knock-in efficiencies by an additional roughly two- to threefold on average. Our method works across a variety of target loci, knock-in constructs and primary human cell types, reaching HDR efficiencies of >80–90%. We demonstrate application of this approach for both pathogenic gene variant modeling and gene-replacement strategies for IL2RA and CTLA4 mutations associated with Mendelian disorders. Finally, we develop a good manufacturing practice (GMP)-compatible process for nonviral chimeric antigen receptor-T cell manufacturing, with knock-in efficiencies (46–62%) and yields (>1.5 × 109 modified cells) exceeding those of conventional approaches.
CRISPR–Cas9 genome edited human cellular therapies recently have entered the clinic. Cas9-based knock-outs in T cells and hematopoietic stem cells (HSCs) have demonstrated a promising safety profile and, in some cases, signs of profound efficacy1,2. Forthcoming trials are now poised to introduce Cas9-mediated knock-ins by homology-directed repair (HDR) for correction of pathogenic mutations or insertion of new therapeutic constructs3–5. In comparison to nontargeted integrations of viruses or transposon-based approaches, Cas9-stimulated HDR allows for precisely targeted genomic changes that can improve the quality, uniformity and safety of cellular products6,7. In addition to reducing potential integration risks, targeted genome editing can repurpose endogenous genetic circuits and eliminate the need for artificial promoters. This can have important functional benefits as demonstrated for targeted chimeric antigen receptor (CAR) insertion into the TRAC locus (T cell receptor (TCR) alpha chain constant region), which enhances CAR-T cell potency and persistence in preclinical studies by taking advantage of the endogenous gene regulatory elements governing normal TCR expression6. The efficiency of scarless insertion enabled by HDR in primary cells with these large multi-kilobase DNA constructs currently is not readily matched by alternative precision genome editing tools such as base editors, prime editors, transposase, recombinase or integrase approaches8–10. Targeted introduction of large DNA sequence payloads will be essential for manufacturing many future clinical products including CAR-T cells and therapeutic gene-replacement strategies, and provides the flexibility needed for the next generation of synthetic biology constructs6,11,12.
Ex vivo CRISPR genome editing of primary human T cells has been optimized extensively by our group and others, generally using electroporation of precomplexed Cas9 and guide RNA (gRNA) ribo-nucleoproteins (RNPs) to generate targeted genomic breaks6,13,14. To introduce targeted sequence insertions or replacements with HDR, an HDR template (HDRT) is included that encodes the desired genetic change in between homology arms that flank the genomic break. Several different methods are used to introduce the HDRTs including viral transduction with recombinant adeno-associated virus (rAAV) or coelectroporation with naked DNA in dsDNA, ssDNA, circular or linear formats6,13–15. Both the efficiency of HDR and the cellular toxicity vary with the concentration and format of the HDRTs. For large constructs, rAAV-based methods have thus far achieved the most impressive knock-in efficiencies while maintaining minimal toxicity16,17. While rAAV vectors have led to rapid advances, incorporation for research and clinical use has been slowed by the cost and complexity of manufacturing these reagents. Coelectroporation of naked DNA has the potential to increase the pace of innovation in gene modified cell therapies, since it can be done at a fraction of the cost and time required for viral vector development. Nonviral approaches have been applied within primary human cell types; however, further improvements are needed—especially for large templates—to reduce DNA toxicity, improve knock-in purity and cell yields, and advance toward clinical applications13,14.
We recently developed a method to enhance the knock-in efficiency of dsDNA HDRTs through incorporation of Cas9 target sequences (CTSs), allowing the coelectroporated RNPs to bind the HDRTs and facilitate their delivery13. We found that knock-in efficiencies were increased substantially but with concurrent increases in cellular toxicity. This toxicity could be attenuated, but not eliminated, by inclusion of anionic polymers such as polyglutamic acid (PGA) to improve cell yields. In comparison to dsDNA, ssDNA exhibits less toxicity14. Cas9 binds to dsDNA targets, so we set out to establish an approach to adapt CTS-based enhancement of HDR to ssDNA templates.
Here we develop a hybrid HDRT using a long ssDNA with short regions of dsDNA containing CTS sites on each end. For simplicity, we refer to these hybrid HDRTs as ssCTS templates and refer to the fully double-stranded versions as dsCTS templates. We assess knock-in efficiency and toxicity of these ssCTS templates across a range of construct sizes, genomic loci and clinically relevant cell types, including primary human T cell subsets, B cells, natural killer (NK) cells and CD34+ cells (which should include HSCs). In addition, we evaluate a panel of small molecules reported to enhance HDR in primary human T cells, identifying the optimal combinations and concentrations that work to further enhance HDR with ssCTS templates. Finally, we adapt our approach to generate a good manufacturing practice- (GMP-)compatible process for fully nonviral CAR-T cell manufacturing. This technology promises to enable efforts to model patient mutations in primary cells and flexibly engineer cellular therapies at the clinical scale.
Results
Development of ssCTS templates for high-efficiency HDR.
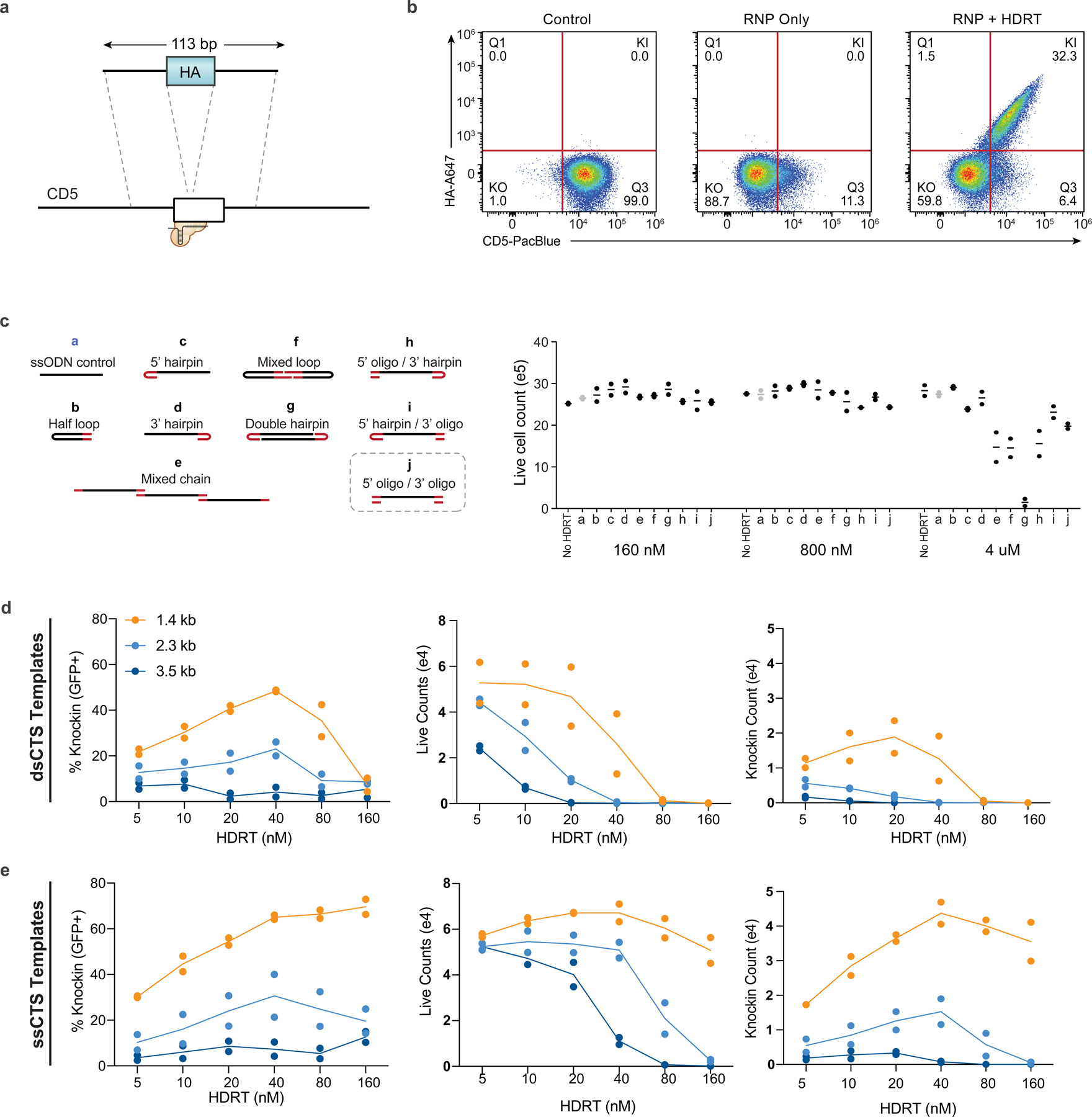
We previously developed a method to enhance delivery of dsDNA HDRTs through incorporation of CTSs that include a gRNA target sequence and an NGG protospacer-adjacent-motif (PAM) on each end of the template13. In comparison to dsDNA, ssDNA is associated with lower toxicity, which we reasoned could further improve knock-in efficiency and cell yield with large DNA templates if combined with CTS technology14. We screened a variety of hybrid structures composed predominantly of ssDNA with small stretches of dsDNA incorporating the CTS sites through hairpin loops, annealed complementary oligonucleotides or more complex secondary structures (Fig. 1a,b and Extended Data Fig. 1a–j)14. We rapidly screened to compare HDRT designs using short 113–195 nt HDRTs that generate an N-terminal CD5-HA fusion protein easily detectable by flow cytometry (Extended Data Fig. 2a,b). We found that most of these ssCTS designs increased knock-in efficiency (Fig. 1c). Improved efficiency with the ssCTS templates was apparent only at the lower two concentrations (160 and 800 nM), above which the knock-in efficiency appeared to hit a maximum of roughly 30% that was achievable with unmodified ssDNA HDRTs (Fig. 1c, gray). These results indicated that ssCTS designs would be beneficial in situations where the achievable HDRT concentration is limited, such as with large HDRTs.
Fig. 1 |. Development of ssCTS templates for high yield knock-in.
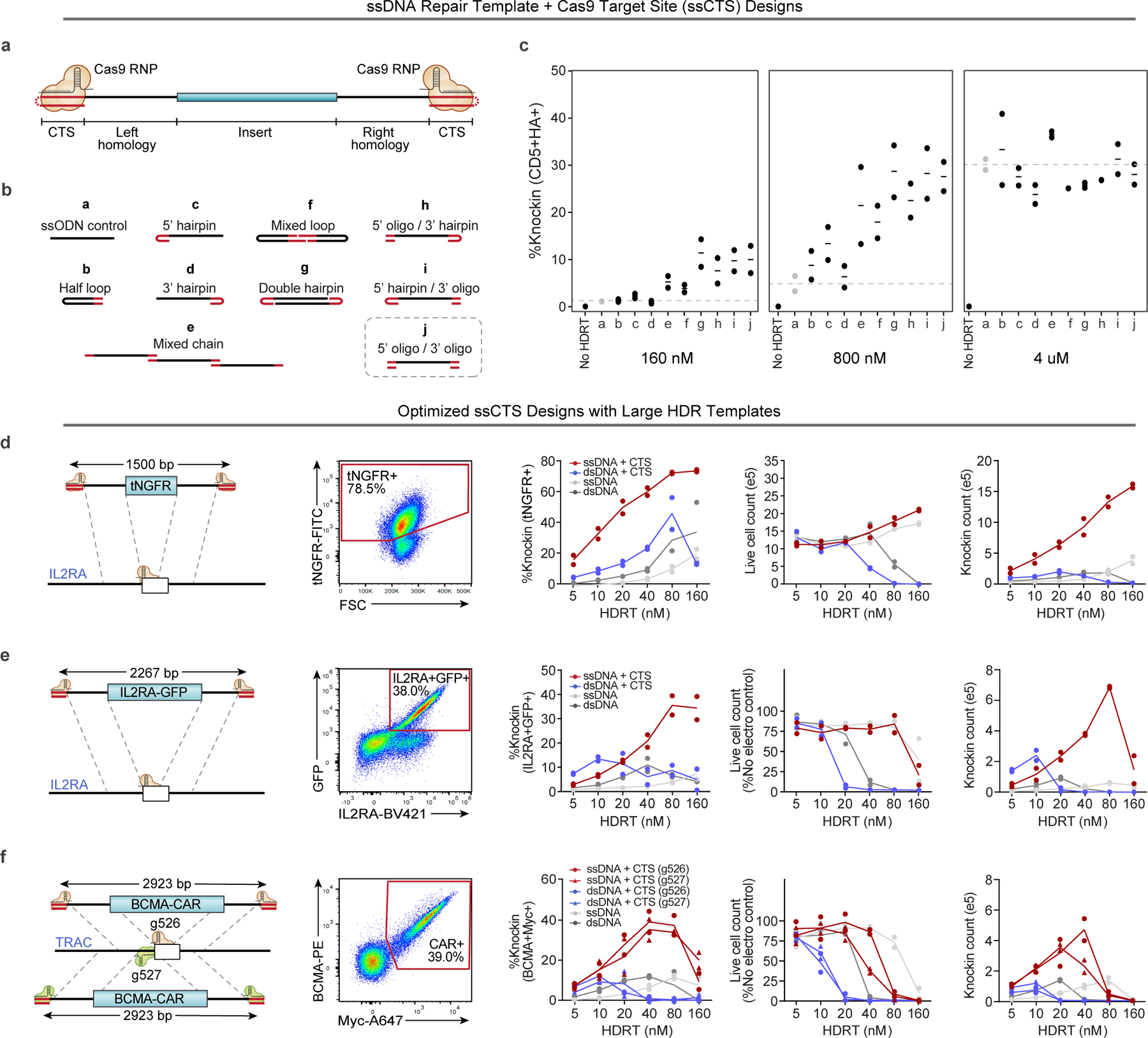
a, Diagram of hybrid ssDNA HDRT designs incorporating CTS sites. b, Panel of ssCTS designs tested. c, Knock-in efficiency for each ssCTS design using a CD5-HA knock-in construct at 160 nM–4 μM concentration assessed by flow cytometry. Dotted line represents mean knock-in percentage for control ssDNA HDRTs without CTS (construct a, gray). d–f, Knock-in strategy, gating, knock-in efficiency, live cell counts and knock-in cell counts are shown for large ssCTS templates including a tNGFR knock-in at the IL2RA locus (d), a IL2RA-GFP fusion protein knock-in to the IL2RA locus (e) or two different HDRTs inserting a BCMA-CAR construct at TRAC locus via two different gRNAs (g526 and g527) (f). Each experiment was performed with T cells from two independent healthy human blood donors represented by individual dots plus mean. CTS, Cas9 target site; FITC, fluorescein isothiocyanate; ssCTS, ssDNA HDRT + CTS sites. This figure was generated in part using graphics created by Biorender.com.
For evaluation of large HDRTs, we chose an ssCTS design that incorporates CTS sites on both the 5′ and 3′ end via annealed complementary oligonucleotides, which are easy to design for research and clinical applications. In our panel of tested ssCTS constructs, this design demonstrated maximal enhancement of knock-in efficiency (Fig. 1b,c, ‘j’), low toxicity (Extended Data Fig. 1c), and provided the most versatile process for generating CTS ends compared to hairpin loops or more complicated structures. Long ssDNA and dsDNA HDRTs ranging from 1,500 to 2,923 nt were generated with and without CTS sites (Fig. 1d–f). These templates target a knock-in detectable by flow cytometry (truncated Nerve Growth Factor Receptor (tNGFR), IL2RA-GFP fusion or BCMA-CAR) to the IL2RA or TRAC locus. We evaluated postelectroporation knock-in efficiency, yield of total live cells and yield of knock-in cells using primary T cells isolated from healthy human blood donors. Inclusion of CTS sites enhanced the knock-in efficiency of both dsDNA and ssDNA constructs across concentrations until toxic doses were reached, after which knock-in efficiency progressively decreased. ssCTS constructs demonstrated uniformly higher knock-in efficiencies and yield in comparison with dsCTS templates, generating up to sevenfold more knock-in cells at optimal concentrations. As previously reported, we observed an inverse relationship between knock-in efficiency and transgene length for both dsCTS and ssCTS templates when targeting the same locus (Extended Data Fig. 2d,e)18,19. The use of ssCTS templates allowed us to achieve up to 78.5% knock-in with a roughly 1.5 kilobase (kb) tNGFR construct, or 38% for a roughly 2.3 kb IL2RA-GFP construct targeting the IL2RA locus and up to 39% knock-in with a roughly 2.9 kb BCMA-specific CAR construct targeting the TRAC locus at HDRT concentrations compatible with high yields of live knock-in cells.
Exploration and optimization of ssCTS design parameters.
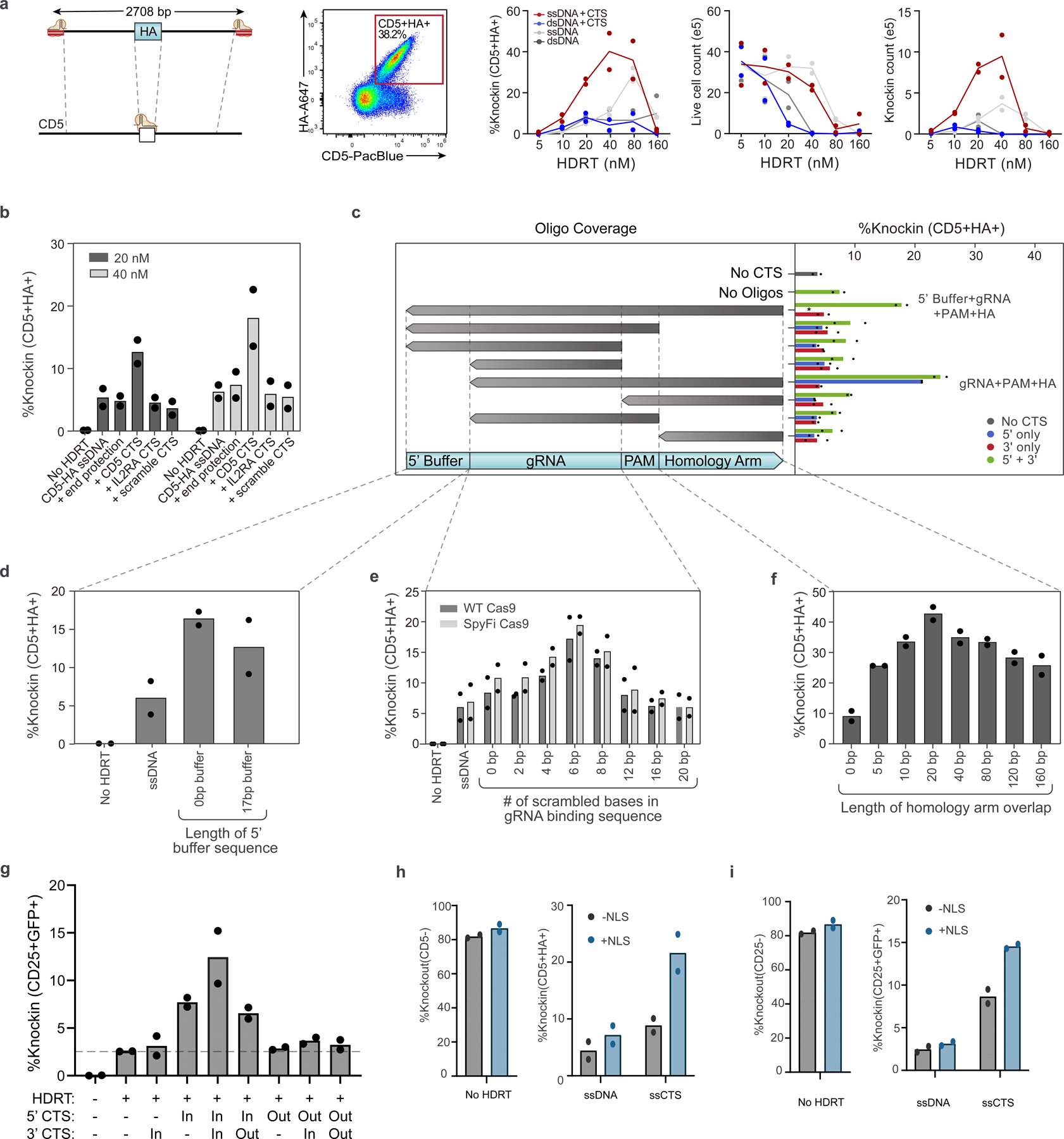
To learn rules regarding the precise sequences required for ssCTS-enhanced HDR, we evaluated variations of two constructs targeting either an IL2RA-GFP fusion to the IL2RA gene (roughly 2.3 kb, Fig. 1e) or a large version of the CD5-HA knock-in including >1 kb homology arms (roughly 2.7 kb, Extended Data Fig. 3a). We first evaluated the specificity of the CTS sequences by replacing them with a mismatched CTS site specific for the alternative RNP, an equivalent length of dsDNA within the homology arm (‘end protection’) or a CTS site with scrambled gRNA sequence (Fig. 2a and Extended Data Fig. 3b). For both constructs, only the matching CTS recognized by the cognate RNP increased knock-in efficiency, suggesting specific recognition of the gRNA sequence. We then examined closely which components of the CTS required dsDNA by annealing oligos of varied lengths and coverage (Fig. 2b and Extended Data Fig. 3c). Coverage of the gRNA sequence, PAM and a stretch of nucleotides within the homology arm downstream of the CTS site were each required for enhancement of knock-in efficiency while coverage of nucleotides upstream of the gRNA sequence in the 5′ buffer region was not. Inclusion of this additional buffer sequence upstream of the CTS was not required and appeared to reduce knock-in efficiency (Fig. 2c and Extended Data Fig. 3d). For production methods that require a 5′ buffer sequence, this region should likely be left as ssDNA. We saw that inclusion of a CTS on the 3′ end of both large ssCTS constructs provided no independent benefit and only slight benefit when combined with a 5′ CTS (Extended Data Fig. 3c,g). We further examined the requirements for gRNA recognition by generating CTS sites with a variable number of scrambled bases at the 5′ end of the 20 bp gRNA recognition sequence (Fig. 2d and Extended Data Fig. 3e). We found that for wildtype (WT) Cas9, the enhancement in knock-in efficiency was maximal with inclusion of 4–8 mismatched nucleotides. This level of mismatch likely allows the Cas9 RNP to bind without cleaving the CTS, as has been shown for truncated gRNAs20. The pattern was similar with the high-fidelity ‘SpyFi’ Cas9 variant produced by Aldevron/IDT, which has been developed to reduce off-target cuts in clinical gene editing applications21. Next, we evaluated the length of the complementary oligonucleotide coverage within the downstream homology arm, demonstrating optimal knock-in when >20–40 bp of the homology arm has complementary sequence in the corresponding oligo (Fig. 2e and Extended Data Fig. 3f). In combination, these studies define optimal sequences for CTS templates and complementary oligonucleotides.
Fig. 2 |. Evaluation of ssCTS design requirements.
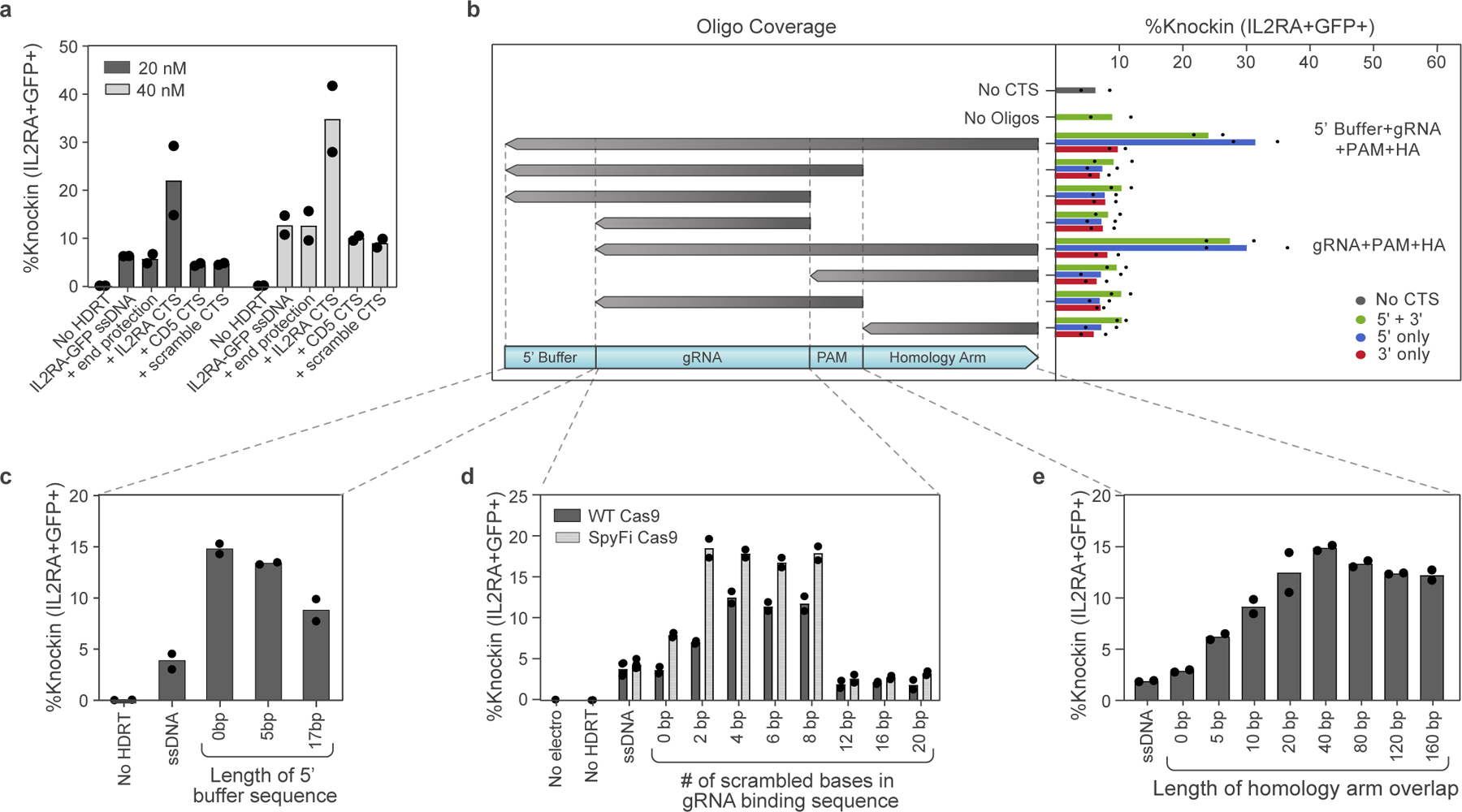
a–e, Comparison of different CTS designs with an IL2RA-GFP knock-in construct targeting IL2RA locus assessed by flow cytometry. a, Comparison of CTS with a gRNA target sequence that is specific for the cognate RNP (+IL2RA CTS), an alternative gRNA sequence (+CD5 CTS), a CTS incorporating a PAM site and scrambled gRNA sequence (+scramble CTS) or an equivalent amount of dsDNA within the 5′ end of the homology arm (+end protection). b, Comparison of complementary oligos covering varying regions of the CTS and surrounding sequences (design schematics left, knock-in results right). Constructs with CTS sites on both 5′ and 3′ end (green bars), 5′ end only (blue bars) or 3′ end only (red bars) are shown on the right panel with two best performing designs indicated (right). c, Evaluation of varied 5′ ends including different length of buffer sequence upstream of the CTS site. d, Comparison of CTS designs with varying numbers of scrambled bases at the 5′ end of the gRNA target sequence using WT or SpyFi Cas9. e, Knock-in percentages are shown with varying length of homology arm covered by the complementary oligonucleotide. Each experiment was performed with T cells from two independent healthy human blood donors represented by individual dots + mean. All comparisons except for b include complementary oligos covering the entire 5′ buffer + gRNA + PAM + homology arms. HA, homology arms.
In agreement with our previous evaluation of dsCTS templates13, we found that the full efficiency gains for ssCTS templates were also dependent on the PAM orientation facing in toward the homology arms (PAM ‘In’, Extended Data Fig. 3g), the presence of a nuclear localization sequence (NLS) on Cas9 (Extended Data Fig. 3h,i) and that anionic polymers, such as PGA or ssDNA electroporation enhancer (ssDNAenh), further increased the yield of live knock-in cells (Extended Data Fig. 4a–c). An examination of the relative RNP–HDRT molarity demonstrated no difference in optimal RNP amount for ssCTS templates relative to ssDNA controls even at high template concentration, suggesting Cas9 RNPs are not functionally sequestered by CTS sites (Extended Data Fig. 4d–f). To complement these functional studies, we examined the biophysical interaction of Cas9 RNPs with dsCTS and ssCTS templates by atomic force microscopy (AFM) (Extended Data Fig. 4g). These experiments demonstrated clear binding of Cas9 RNPs at CTS sites on the ends of dsDNA templates. ssCTS templates formed a highly compact and flexible secondary structure that was challenging to visualize and although we observed RNP interactions we could not clearly demonstrate their specificity for CTS sites (Extended Data Fig. 4g).
Taken together, these data establish design rules for introduction of CTS into large ssDNA templates to boost knock-in efficiency and yield. Optimal designs used for all further studies incorporated CTS sites on both ends of long ssDNA HDRTs using a PAM ‘In’ orientation and including four mismatched nucleotides on the 5′ end of the gRNA sequence. Complementary oligonucleotides covered the gRNA target site, the PAM sequence and roughly 20 bp of the homology arm. RNPs were formulated with Cas9-NLS proteins and either PGA or ssDNAenh anionic polymers before incubation with CTS templates. An example of this design is shown in Extended Data Fig. 1k and full sequences for each component can be found in Supplementary Table 1.
ssCTS templates broadly enhance HDR in primary human cells.
Using optimized ssCTS designs, we next assessed performance across a broad array of genomic loci, knock-in constructs and primary hematopoietic cell types. We evaluated CTS templates in a variety of clinically relevant primary cell types using an mCherry fusion construct targeting a gene not expected to affect cell fitness (Clathrin, CLTA). Knock-in at this locus demonstrated no selective growth advantage in primary human T cells (Extended Data Fig. 6a–c). ssCTS templates increased knock-in efficiency, live cell counts and absolute yield of knock-in cells across all primary human cell types evaluated here including CD4+ T cells, CD8+ T cells, regulatory T cells (Treg), NK cells, B cells, CD34− HSCs and gamma-delta T cells (γδ) (Fig. 3a–c and Extended Data Fig. 6d).
Fig. 3 |. Application of ssCTS knock-in templates across diverse target loci, knock-in constructs and primary human hematopoietic cell types.
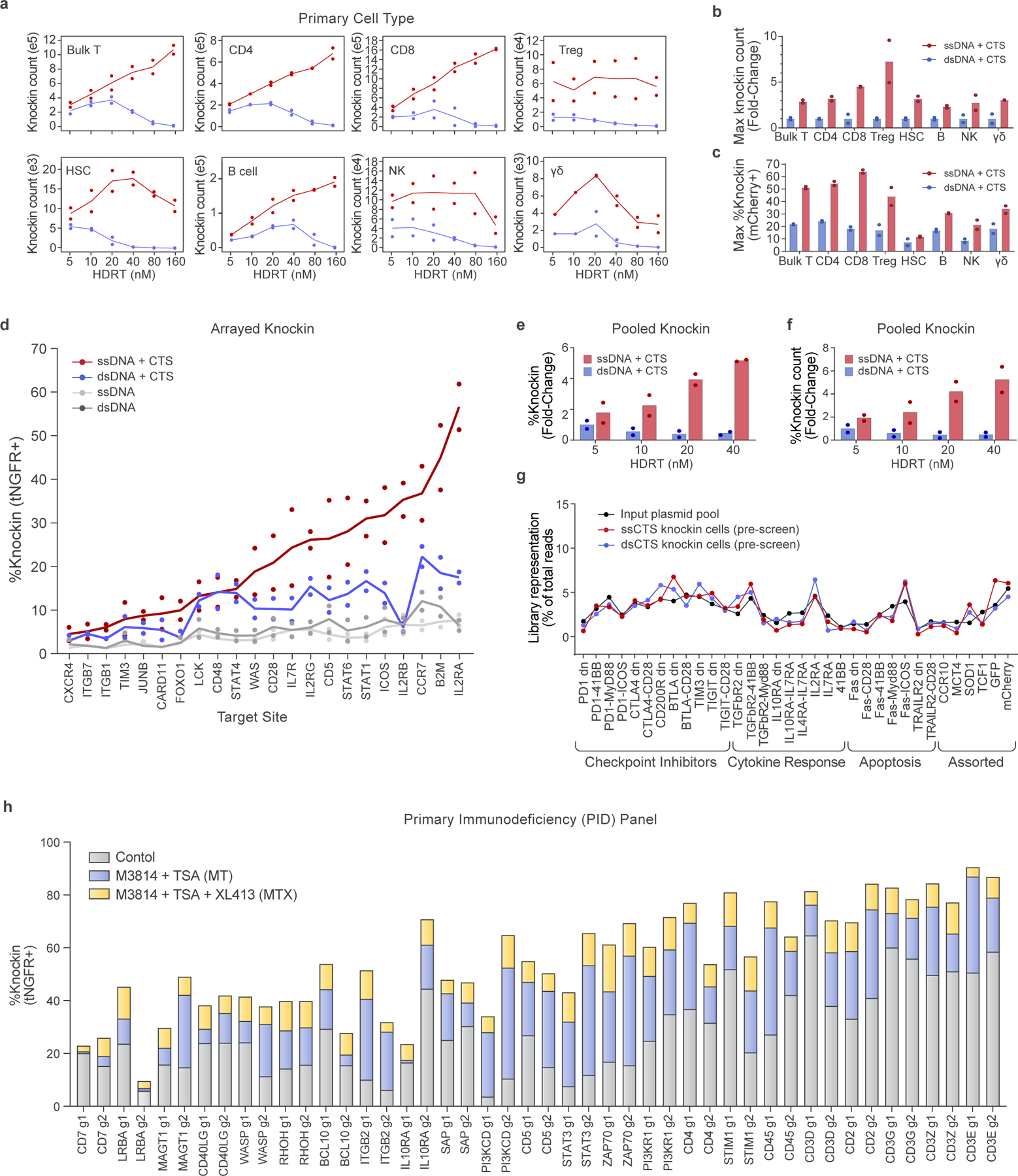
a–c, Comparison of knock-in cell yields using ssDNA (red) and dsDNA HDRTs (blue) with CTS sites across a variety of primary human hematopoietic cell types. Note that different cell type comparisons were performed with different blood donors. All comparisons were performed using a knock-in construct generating an CLTA-mCherry fusion at the CLTA locus. Shown for each cell type using HDRT concentrations from 5 to 160 nM are knock-in cell count yields (a), maximum fold-change in knock-in count yields (relative to dsCTS templates) (b) and maximum percentage knock-in (c). d, Knock-in efficiencies for constructs targeting a tNGFR marker to 22 different target genome loci. e–g, Comparison of large ssDNA and dsDNA HDRTs with CTS sites for knock-in of a pooled library of polycistronic constructs targeted to the TRAC locus (2.6–3.6 kb total size)18. Shown for each HDRT variation is relative percentage knock-in in comparison to maximum for dsDNA + CTS templates (e), relative knock-in cell count yields in comparison to maximum for dsDNA + CTS templates (f) and representation of each library member in knock-in cells postelectroporation in comparison to construct representation in the input plasmid pool (g). h, Evaluation of ssCTS templates ± MT or MTX inhibitor combinations with a panel of 44 different knock-in constructs targeting a tNGFR marker across 22 different target loci including genes implicated in PID or with potential importance for T cell engineering. Two gRNAs and corresponding ssCTS templates were used for each gene (g1 and g2). All experiments were performed with T cells from two independent healthy human blood donors represented by individual dots + mean (a–g) or mean alone (h). dsCTS, dsDNA HDRT + CTS sites.
We evaluated an arrayed panel of knock-in constructs in primary human T cells targeting a detectable tNGFR fusion at the 5′ end of 22 different genes using previously validated high-efficiency gRNAs (Fig. 3d and Extended Data Fig. 5). Most ssCTS constructs outperformed alternative HDRT variations for both knock-in efficiency and absolute knock-in counts with only a few exceptions that appeared equivalent to optimal dsCTS constructs (Fig. 3d and Extended Data Fig. 5a). To help understand variation in knock-in efficiency across these loci, we examined a variety of target characteristics with amplicon sequencing, RNA-sequencing (RNA-seq) and assay for transposase-accessible chromatin with sequencing (ATAC-seq) (Extended Data Fig. 5b–d). Multiple individual factors correlated to varying degrees with knock-in efficiency including: chromatin accessibility, target transcript expression level (particularly on the electroporation day), proximity of the gRNA cut site to the insertion site, overall cutting efficiency (assessed by indel frequency in amplicons) and frequency of characteristic microhomology-mediated end joining (MMEJ) editing outcomes in amplicons, consistent with a recent report that such sites are more amenable to HDR repair outcomes22. Further multifactorial analyses with even larger datasets may help to refine predictive models for optimized targeted knock-in strategies at diverse genome loci.
Finally, we evaluated performance with a pooled library of knock-in constructs targeting an NY-ESO-1 specific TCR and additional gene products to the endogenous TRAC locus, as previously reported by our group for use in functional knock-in screens18 (Fig. 3b–d). Knock-in pools provide a powerful approach for high-throughput screening and allowed us to assess performance with a diverse population of large knock-in templates ranging from 2.6 to 3.6 kb (ref. 18). Knock-in efficiency and yield were both increased by more than fivefold in comparison to optimal dsCTS concentrations, substantially increasing coverage for each individual construct while retaining consistent representation of the initial library in the final knock-in population (Fig. 3b–d). Altogether, these results support application of ssCTS templates across a wide variety of target loci, knock-in constructs and primary cell hematopoietic cell types.
Evaluation of small-molecule inhibitor cocktails.
We next evaluated a panel of small-molecule inhibitors that have been reported to enhance knock-in efficiency in primary human T cells including the DNA-dependent protein kinase (DNA-PK) inhibitors NU7441 and M3814, the histone deacetylase class I/II Inhibitor Trichostatin A (TSA), the CDC7 inhibitor XL413 and Integrated DNA Technologies (IDT)’s proprietary ‘Alt-R HDR enhancer,’ which is described as an nonhomologous end joining (NHEJ) inhibitor22–25. Using our short ssDNA CD5-HA knock-in construct (Extended Data Fig. 2a,b), each was titrated in isolation and then evaluated in combination to identify effects on knock-in efficiency, live cell counts and viability (Extended Data Fig. 7a,b). At optimal concentrations, M3814 showed the largest effect size (roughly 49% increase), followed by XL413 (roughly 46% increase), NU7441 (roughly 43% increase), IDT’s HDR Enhancer (roughly 29% increase) and TSA (roughly 16% increase). Live cell counts were generally unaffected at the chosen concentrations except for combinations involving XL413, which demonstrated a roughly 50% reduction in cell counts at day 4 postelectroporation that may reflect XL413’s mechanism as a transient cell cycle inhibitor rather than overt cytotoxicity (Extended Data Fig. 7b)24. Evaluation of viability and cell counts at a single time point cannot accurately distinguish between these possibilities. NHEJ inhibitor combinations (M3814, NU7441, IDT HDR Enhancer) did not demonstrate further improvements above the highest individual component, consistent with overlapping mechanisms of action. In contrast, addition of TSA or XL413 did demonstrate additional improvements in combination with NHEJ inhibitors. The M3814−TSA (MT) combination provided the largest increase in knock-in efficiency without affecting live cell counts (roughly 65% increase) and the M3814−TSA+XL413 (MTX) combination demonstrated the highest absolute increase in knock-in efficiency (roughly 134% increase) albeit with XL413-mediated reduction in total cell counts. Evaluation of Novobiocin, which has been reported to inhibit the MMEJ protein POLQ in human cells, showed no independent or additive effect when combined with other inhibitors (Extended Data Fig. 7c–e)26. Altogether, these results demonstrated several promising inhibitor cocktails with potential to enhance HDR in primary human T cells.
We further examined repair outcomes at the genetic level by amplicon sequencing of the CD5 target locus with different versions of the small CD5-HA templates and inhibitor cocktails. Sequencing and flow-based quantifications both demonstrated stepwise increases in knock-in rates with each inhibitor that was additive to the increases seen with CTS sequences in both ssDNA and dsDNA templates (Extended Data Fig. 8a,b). The ratio of perfect to imperfect HDR events was also similar across the different types of template (Extended Data Fig. 8c). Treatment combinations that included the M3814 DNA-PK inhibitor were associated with decreased frequency of indels (especially small indels characteristic of NHEJ), along with reduced frequency of imperfect HDR events (Extended Data Fig. 8b,c and Supplementary Fig. 1). Inclusion of HDRTs was associated with preferential reduction in the larger deletions characteristic of MMEJ (while smaller indels characteristic of NHEJ were more refractory in the absence of DNA-PK inhibition), in agreement with recent reports (Supplementary Fig. 1)22.
We next asked whether small-molecule inhibitors could be combined with large ssCTS templates (ranging from 1.5 to 2.7 kb) to enhance knock-in engineering (Extended Data Fig. 8d). Each ssCTS template demonstrated increased knock-in efficiencies that were enhanced further by the inclusion of MT and MTX inhibitor combinations, in some cases generating knock-in efficiencies >90%.
To evaluate these approaches more broadly at clinically relevant target sites that could lead toward diagnostic or therapeutic advances, we developed a panel of knock-in constructs for genes associated with monogenic immune disorders affecting T cell function and several controls. These diseases are part of a range of increasingly recognized genetic disorders, referred to as primary immunodeficiencies (PIDs) or inborn errors of immunity, that disrupt the healthy immune system, presenting with severe infections, autoimmune disease and malignancy27. Within this panel, we examined 44 different tNGFR constructs targeting 22 genes (two gRNA targets per gene) using ssCTS templates ±MT and MTX inhibitor combinations (Fig. 3h). This analysis demonstrated nearly universal increases in knock-in efficiency with MT that were further enhanced with the MTX combination, achieving knock-in rates >50% for these large constructs at 15/22 genes examined and >80% at 6/22 genes. The average increase in knock-in efficiency was roughly 2.1-fold for MT and roughly 2.5-fold for MTX combinations (Supplementary Table 1). The effect size of inhibitors varied among target loci, with some sites demonstrating relatively little increase (for example, CD7 g1) and others showing up to ninefold increases (for example, PI3KCD g2). Live cell counts were comparable at day 5 postelectroporation with a few notable exceptions demonstrating reduction in yield with both combinations (for example, CD7 g2, WASP g2, CD3G g2) (Extended Data Fig. 8e,f). Altogether, these findings support broad application of ssCTS templates and inhibitor combinations at relevant disease loci, in some cases demonstrating nearly pure populations of knock-in cells (>80–90%) (Fig. 3h and Extended Data Fig. 8d). This sets the stage for diagnostic and therapeutic applications of nonviral human T cell engineering that require a high purity or yield of knock-in cells at specific disease loci.
Therapeutic and diagnostic gene-replacement strategies.
To explore potential clinical applications with large nonviral templates, we chose to examine open reading frame (ORF) replacement strategies for two genes, IL2RA and CTLA4, where mutations have been identified in families with severe monogenic immune dysregulation disorders14,28–35. Although disease-causing mutations are widely distributed throughout these genes, many of these families could potentially be treated by a single complete or partial ORF replacement (Fig. 4a,e). For each construct, we included a green fluorescent protein (GFP) fusion at the 3′ end to facilitate detection of the knock-in protein. We have previously reported targeted gene corrections for a family with loss-of-function mutations in exon 4 and exon 8 of the IL2RA gene14. While we achieved knock-in efficiencies >30% with this approach, each site required a custom gRNA and HDRT that prevents extension to families with alternative IL2RA mutations. By contrast, a whole ORF knock-in at exon 1 of the IL2RA gene could potentially ameliorate any of the 11 reported mutations causing IL2RA deficiency (Fig. 4a)14,28–32. Using an ssCTS template and the MTX inhibitor combination, we achieved >80% knock-in of a roughly 2.3 kb whole ORF IL2RA-GFP fusion construct (Fig. 4b). The knock-in protein demonstrated nearly indistinguishable expression levels compared to endogenous protein.
Fig. 4 |. Whole ORF replacement at target genes for therapeutic and diagnostic human T cell editing. a–d, IL2RA exon 1–8 ORF replacement strategy.
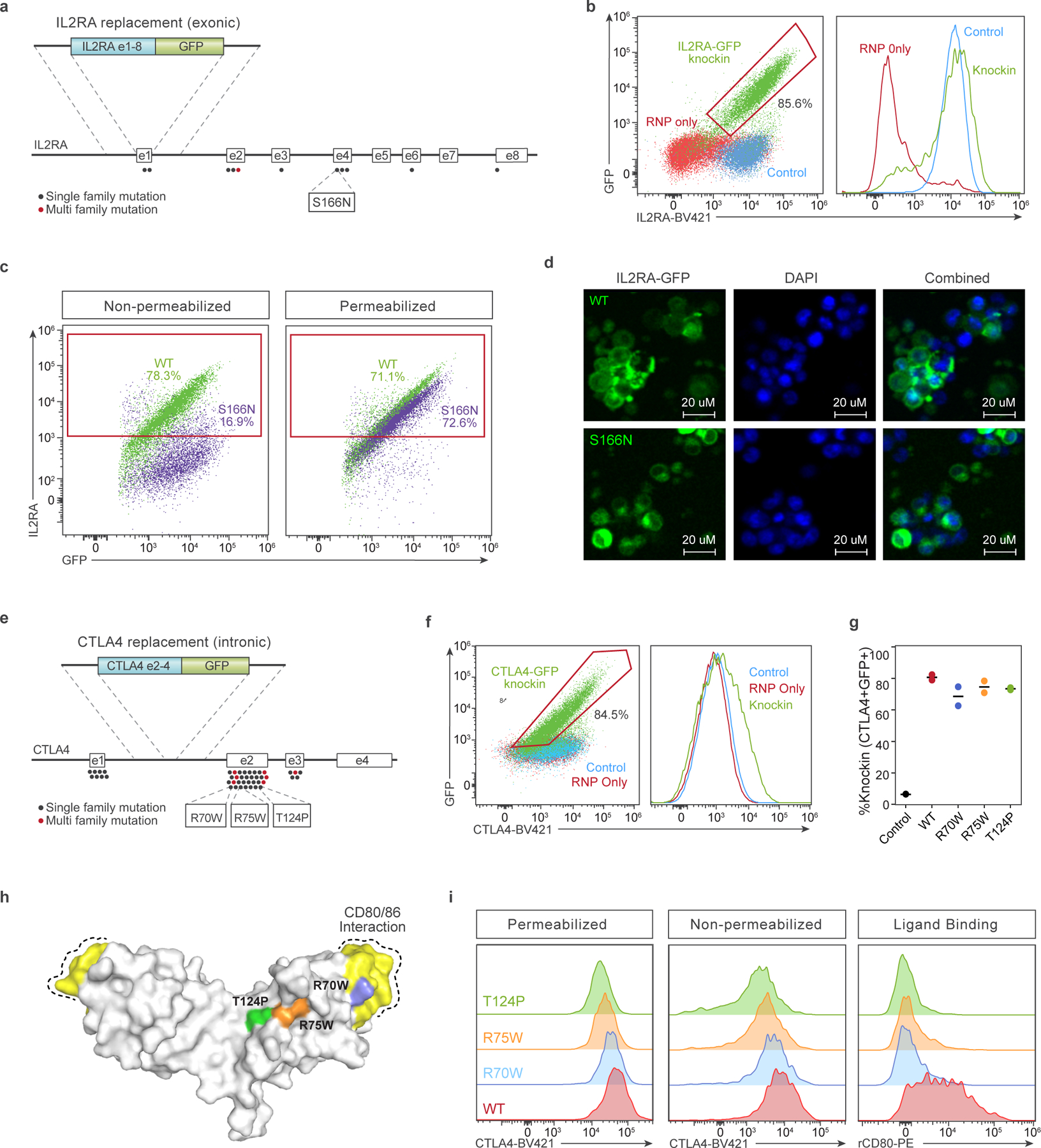
a, Diagram of the IL2RA gene with reported patient coding mutations and knock-in strategy using an IL2RA-GFP fusion protein targeted to exon 1. The S166N mutation examined in c and d is noted. b, IL2RA and GFP expression in CD4+ T cells electroporated with IL2RA-GFP ssCTS templates and cognate RNP followed by MTX inhibitor combination (green), in comparison to RNP only (red) or no electroporation control cells (blue). c, Comparison of extracellular (surface staining in nonpermeabilized cells) or total IL2RA expression (staining in permeabilized cells includes total surface and intracellular protein) with WT and S166N IL2RA-GFP knock-ins assessed by flow cytometry. Percentage IL2RA+ is shown for each panel. d, Subcellular localization of WT and S166N IL2RA-GFP protein by fluorescence microscopy. Scale bars, 20 μm. e–i, CTLA4 exon 2–4 ORF replacement strategy. e, Diagram of the CTLA4 gene with reported patient mutations and knock-in strategy using a CTLA4-GFP fusion protein targeted to intron 1. The R70W, R75W, T124P mutations examined in g–i are noted. f, CTLA4 and GFP expression in CD4+ T cells electroporated with CTLA4-GFP ssCTS templates and cognate RNP followed by MTX inhibitor combination (green), in comparison to RNP only (red) or no electroporation control cells (blue). g, Quantification of percentage knock-in for WT, R70W, R75W and T124P constructs electroporated with ssCTS templates and treated with the MTX inhibitor combination assess by flow cytometry. Mean and individual values are shown for two independent healthy human blood donors. h, Structure of CTLA4 dimer with CD80/86 interaction domain highlighted (yellow) along with location of R70W (blue), R75W (orange) and T124P (green) mutations48. i, Comparison of extracellular CTLA4 (surface staining), total CTLA4 (staining in permeabilized cells, which includes total surface and intracellular protein) and biotinylated recombinant CD80 ligand interaction stained with Streptavidin-APC in WT, R70W, R75W and T124P knock-in CD4+ T cells. DAPI, 4′,6-diamidino-2-phenylindole nuclear stain; rCD80, recombinant CD80.
This whole ORF knock-in approach also could allow for rapid functional testing and characterization of patient mutations or variants of unknown significance (VUS) in cells from healthy individuals. To demonstrate this diagnostic potential, we modified the knock-in construct to encode a previously described disease-causing mutation in exon 4 of IL2RA, c.497G>A (S166N), which was reported to eliminate surface expression while retaining cytoplasmic protein31. In agreement with what has been reported in patient cells, we found that the GFP+S166N knock-in population demonstrated a near complete absence of surface IL2RA with readily detectable intracellular IL2RA comparable to WT levels (Fig. 4c and Extended Data Fig. 9a). Fluorescence microscopy revealed that S166N protein formed distinct perinuclear aggregates consistent with intracellular retention and contrasting with the diffuse cytoplasmic and surface IL2RA seen with WT knock-ins (Fig. 4d). These results highlight the diagnostic and therapeutic potential of targeted ORF insertion within the endogenous gene, an approach that may be extended to include a number of alternative targets or additional noncoding elements.
As a further example, we examined an ORF insertion within the CTLA4 gene (Fig. 4e). CTLA4 deficiency is caused most frequently by a haploinsufficiency with a disease-causing mutation on only one of two alleles33–35. Exon-targeting strategies generate indels that could disrupt the normal allele and worsen disease. To avoid this possibility, we screened a panel of gRNA in intron 1 to identify targets that cut efficiently without disrupting protein expression (Extended Data Fig. 9b). The chosen gRNA had no detectable disruption of endogenous CTLA4 protein and the associated ORF knock-in construct generated knock-in efficiencies of 70–80% with ssCTS templates and MTX inhibitor combination (Fig. 4f,g). This intron-targeting strategy could be used to introduce or correct most reported disease-causing mutations in CTLA4 excluding those upstream of the target site (Fig. 4e). Variations in protein expression by cell type and in response to stimulation matched the endogenous protein, although basal knock-in protein levels were slightly higher, which may reflect differences between the SV40 3′ untranslated region (UTR) used in this construct and the endogenous 3′ UTR (Extended Data Fig. 9c)36. To evaluate potential functional testing of CTLA4 variants, we generated knock-in constructs with three previously reported disease-causing mutations: R70W, R75W and T124P (ref. 33). Cells were gated for the highest levels of GFP expression to enrich for homozygous knock-ins and then evaluated for surface protein, intracellular protein and ligand binding using recombinant CD80 in activated CD4+ T cells (Extended Data Fig. 9d and Fig. 4g–i). All three mutations substantially reduced ligand binding despite variable levels of surface expression, in agreement with previous reports demonstrating reduced ligand interaction in heterozygous patient cells or engineered cell lines33. Altogether, these approaches provide a powerful method for evaluating patient mutations at endogenous loci with the potential for adaptation to high-throughput screening and high-efficiency therapeutic gene-replacement strategies.
Fully nonviral and GMP-compatible T cell engineering.
Finally, we sought to generate a clinical-grade process for knock-in of large therapeutic constructs. One of the most immediate applications with demonstrated functional benefit is targeting a CAR insertion to the endogenous TRAC locus. This approach greatly enhanced the potency of CD19-specific CAR-T cells in preclinical studies and reduced T cell exhaustion through tightly regulated expression driven by the gene regulatory elements governing normal TCR expression6. To benchmark against the rAAV strategy used in these previous studies, we generated equivalent TRAC locus ssCTS templates and rAAV vectors encoding an anti-BCMA-CAR, a promising CAR to treat multiple myeloma for which lentiviral-transduced products are now US Food and Drug Administration (FDA)-approved37. Head-to-head comparisons demonstrated efficient knock-in with both approaches, although consistently higher with rAAV (average of roughly 50.7% with rAAV vectors in these experiments and 36.6% with ssCTS templates), comparable cellular immunophenotype and rapid in vivo tumor clearance by both methods (Extended Data Fig. 10a–c).
To support future clinical implementation, we then adapted our nonviral knock-in approach to use GMP-compatible reagents, equipment and processes. For electroporations, we used the Maxcyte GTx platform, which provides a GMP-compatible electroporation device with access to an FDA Master File along with sterile single-use cuvettes and assemblies that are scalable to the large numbers of cells needed for manufacturing a full patient dose. For genome editing reagents, we used research-grade equivalents that are each available at GMP-grade, including SpyFi Cas9 (a high-fidelity Cas9 variant produced at GMP-grade by Aldevron) and chemically synthesized single-guide RNA also produced at GMP-grade by Synthego21. We partnered with Genscript to develop a fully enzymatic GMP-compatible process for ssCTS template generation based on rolling circle amplification (RCA). Genscript templates encoding an anti-BCMA-CAR knock-in were able to be manufactured at large scale and consistently outperformed our internally generated HDRTs, showing lower levels of toxicity and higher knock-in efficiencies for both ssCTS and dsCTS constructs (Supplementary Fig. 2).
To demonstrate a large-scale nonviral CAR-T manufacturing process, roughly 100 × 106 primary human T cells were isolated from two healthy donors, activated on day 0 with CD3/CD28 Dynabeads along with IL-7 and IL-15, electroporated on day 2 using Maxcyte R-1000 cuvettes, then expanded in G-Rex 100M gas-permeable culture vessels to days 7 or 10 (Fig. 5a). Average knock-in efficiencies were 40.4% on day 7 and 45.8% on day 10 (Fig. 5b,c). The final yield of CAR+ cells was more than 5 × 108 by day 7 and more than 1.5 × 109 by day 10 for both donors, well within the range needed to generate the patient doses of 50–400 × 106 CAR− cells anticipated for a future clinical trial (Fig. 5d). While the addition of small-molecule inhibitors improved knock-in efficiencies to more than 60%, we observed a reduction in live cell counts such that the final yield of CAR− cells were decreased in comparison to ssCTS templates alone (Fig. 5b–d and Extended Data Fig. 10d). Most CAR− cells demonstrated an immunophenotype consistent with a T memory stem cell population on day 10 of expansion based on CD45RA/CD62L expression and confirmed with additional markers as CD45RA+CD62L+CD45RO−CCR7+CD95+ (Fig. 5e and Supplementary Fig. 3). In vitro and in vivo assays demonstrated efficient TRAC CAR-T cell killing of BCMA+MM1S myeloma cell lines in contrast to unmodified T cells expanded from the same donors (Fig. 5f and Extended Data Fig. 10e,f). Finally, targeted locus amplification sequencing demonstrated specific insertion at the TRAC locus with no detectable off-target CAR integration events (Extended Data Fig. 10g–j)38. Altogether, these results demonstrate a fully nonviral manufacturing process capable of high-efficiency and locus specific T cell engineering at clinical scale that could be transitioned to full-GMP manufacturing and quickly adapted toward additional targets.
Fig. 5 |. GMP-compatible process for nonviral CAR-T cell manufacturing.
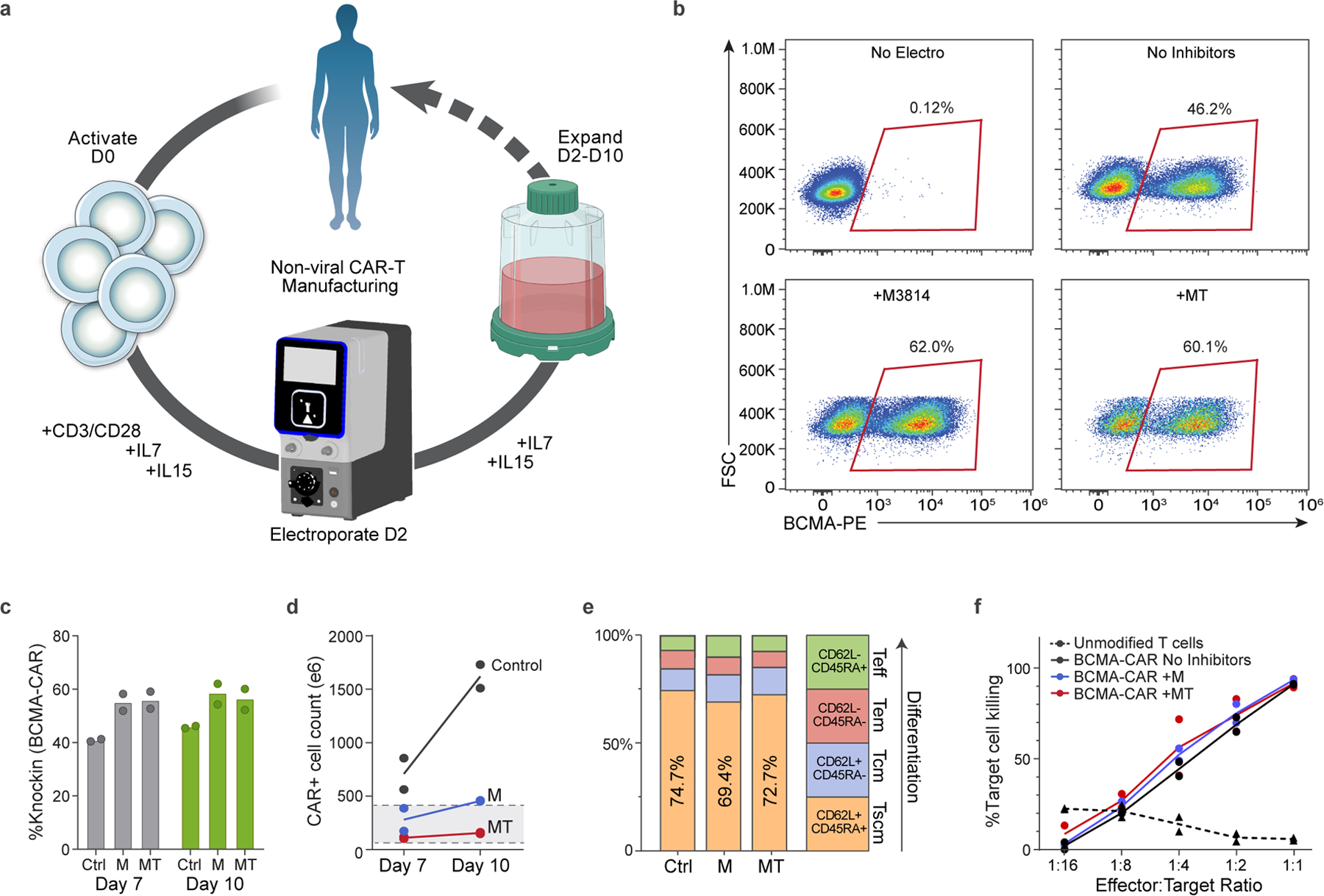
a, Diagram of nonviral CAR-T cell manufacturing process. T cells are isolated from peripheral blood and activated on day 0 with anti-CD3/anti-CD28 Dynabeads, IL-7 and IL-15. Cells are electroporated using the Maxcyte GTx electroporator on day 2 with Cas9 RNPs+ssCTS HDRTs and then expanded for a total of 7–10 days using G-Rex 100M culture vessels supplemented with IL-7+IL-15. b, Representative day 10 flow plots showing BCMA-CAR knock-in for control (no inhibitors), M3814 and MT conditions. c, BCMA-CAR knock-in rates on days 7 and 10 for each condition. d, Absolute number of CAR− cells on days 7 and 10. Gray box highlights anticipated patient doses of 50–400 × 106 CAR−T cells. e, T cell immunophenotypes on day 10 based on CD45RA and CD62L expression. f, In vitro killing of BCMA+MM1S multiple myeloma cell lines in comparison to unmodified T cells from same blood donors. Experiments performed with T cells from two independent healthy human blood donors represented by individual dots + mean (c,d,f) or mean alone (e); a was generated in part using graphics created by Biorender.com. M, M3814; Tscm, T stem cell memory; Tcm, T central memory; Tm, T effector memory; Teff, T effector.
Discussion
The ability of CRISPR genome engineering to introduce targeted sequence replacements or insertions in primary human cells holds immense promise for studying disease variants, correction of genetic disorders and reprogramming cellular therapeutics. Knock-in strategies using rAAV HDRTs are highly efficient and are enabling exciting advances in targeted cellular engineering, but require complex and resource-intensive manufacturing processes to generate viral particles, eliminate risk of adventitious agents and remove process impurities to the level of current regulatory standards39,40. As the gene and cell therapy fields continue to expand, simple, efficient and scalable manufacturing solutions are needed to reduce lead times and treatment costs; and to provide access to more patients. Nonviral genome engineering methods offer a promising alternative given that DNA templates can be produced entirely enzymatically and are facile to purify and characterize. However, clinical applications with large nonviral DNA templates have been limited by the toxicity of naked dsDNA. Here we report advances that increase both knock-in efficiency and knock-in cell yields with large DNA templates through inclusion of CTS sequences on long ssDNAs, that are less toxic than the previous generation of dsCTS templates. We applied these approaches across diverse genetic loci, knock-in constructs and primary hematopoietic cell types to demonstrate broad use for gene correction strategies, disease variant modeling and reprogrammed cell therapy development. We demonstrate a fully nonviral and GMP-compatible CAR-T manufacturing process at clinical scale supported by a simple and scalable in vitro enzymatic strategy for ssCTS template generation. We expect this platform to provide a more rapidly iterative and accessible approach for engineering future cell therapies.
We performed several optimization and mechanistic studies to understand the factors governing ssCTS performance improvements. As with dsCTS templates, we demonstrate optimal outcomes by including roughly 4 bp of mismatched sequence on the 5′ end of the gRNA target sequence13. Although this was shown by a functional readout (knock-in efficiency), this result likely reflects the capacity of these RNPs to bind but not cleave these partially mismatched sequences, as has been demonstrated with similarly truncated gRNAs20. We observed a clear biophysical interaction between RNPs and CTS sites on the ends of dsDNA templates and although we could not clearly demonstrate a specific interaction with CTS sites using ssDNA templates, they are likely to be similarly bound as recently shown for more complex DNA origami structures including identical CTS ends41. As with dsCTS templates, we observed a partial reduction in CTS effects when the NLS sequence is removed from Cas9, suggesting nuclear trafficking is one important factor driven by RNP binding. The residual benefits in the absence of an NLS have not been defined but could potentially be explained by end protection against nuclease activity, blocking interactions with DNA-sensing proteins, improved delivery during electroporation or other factors. Furthermore, why ssDNA is less toxic than dsDNA in this system remains unclear, but could be due to differential detection from DNA-sensing pathways. Furthermore, we observed greater compaction of ssDNA templates in vitro, which could be beneficial for delivery to the nucleus. Deeper understanding of template interactions with cellular pathways may point toward further improvements in Cas9 protein or DNA template design.
We also evaluated a panel of small-molecule inhibitors reported to enhance HDR in primary human T cells demonstrating potential for additive increases in knock-in efficiency when combined with ssCTS templates. We saw that imperfect HDR events—outcomes presumably mediated by NHEJ—were preferentially reduced in the presence of the DNA-PK inhibitor M3814. Despite these benefits, we also observed a variable reduction in cell yields with inclusion of inhibitors that was most apparent in our GMP scale-up experiments, reducing the yield of CAR− cells to below or only slightly above the anticipated patient doses. Future studies will be required to assess whether toxicity could be mitigated, for example, with altered treatment concentrations or with improved gRNA selection. If cell yields can be improved, several of the inhibitors tested here have been, or are currently being, evaluated clinically for in vivo administration and could potentially also be applied in the ex vivo setting42–45. M3814, in particular, has demonstrated a promising safety profile in early clinical trials for treatment of malignancy and a number of alternative clinical-grade NHEJ inhibitors could also be explored43. A wide variety of alternative strategies for HDR enhancement have been reported in research settings and could also be examined in T cells or other primary hematopoietic cell types in combination with ssCTS templates46.
Finally, the high purity of knock-in achievable with targeted insertion of large transgenes using the combination of ssCTS templates and inhibitors provides a powerful tool to probe the functions of DNA sequences in primary human cells. The most recent classification of inborn errors of immunity (also known as PIDs) from the International Union of Immunological Societies identifies >400 monogenic immune disorders with 65 new genes implicated since 2017 (ref. 27). Families with these diseases have a range of mutations scattered throughout these genes and interpretation of new VUS is a persistent challenge to diagnosis and appropriate patient management. Previous use of HDR for VUS screening has been conducted in haploid NHEJ-deficient cell lines, which facilitated functional assessment of nearly all possible single nucleotide variations for the BRCA1 gene47. Here we provide a foundation for evaluating the functional consequences of single nucleotide variations for genes expressed in primary human T cells, in some cases achieving knock-in efficiencies above 80% and facilitating biallelic editing without selection. We generate ORF replacement constructs for IL2RA and CTLA4 composed entirely of coding sequence, but alternative strategies incorporating splice variants, introns or UTRs may also be implemented to evaluate cell type specific effects of noncoding variants. We further demonstrate the capacity to use these ssCTS templates in diverse human hematopoietic cell types and in pooled knock-in approaches, which should facilitate saturation genome editing studies for a large number of genes not currently amenable to haploid cell models, including many causative genes for PIDs.
Altogether, we have developed a variety of tools and applications that improve nonviral genome editing and demonstrate the power of these methods to correct, modify and reprogram primary human cells. We have applied these approaches predominantly toward genome targets relevant for human T cell editing, demonstrating applications for functional genetic screens or therapeutic genome engineering. However, we also show the feasibility of applying ssCTS templates to a range of relevant human cell types and these approaches may be extended for many alternative applications, including targeting the >400 genes associated with a PID or incorporation of a wide variety of new synthetic biology constructs. These studies demonstrate the capacity of fully nonviral HDR to mediate complex and targeted genome modifications with high efficiency and yield, setting the stage for a number of research, diagnostic and manufacturing applications that we hope will reduce the complexity of clinical translation and streamline the development of new therapies.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41587-022-01418-8.
Methods
Cell culture.
Primary adult blood cells were obtained from healthy human donors as a leukapheresis pack purchased from StemCell Technologies, Inc. or Allcells Inc, as a Trima residual from Vitalant, or from fresh whole blood under a protocol approved by the UCSF Committee on Human Research (CHR no. 13–11950). If needed, peripheral blood mononuclear cells were isolated by Ficoll-Paque (GE Healthcare) centrifugation. Primary human cell types were then further isolated by positive and/or negative selection using EasySep magnetic cell isolation kits purchased from StemCell for CD3+ T cells (catalog no. 17951), CD4+ T cells (catalog no. 17952), CD8+ T cells (catalog no. 17953), B cells (catalog no. 17954), NK cells (catalog no. 17955) or CD4+CD127lowCD25+ regulatory T cells (catalog no. 18063) per the manufacturer’s instructions. Primary human γδ T cells were isolated using a custom γδ T cell negative isolation kit without CD16 and CD25 depletion obtained from StemCell. Primary adult peripheral blood G-CSF-mobilized CD34+ HSCs were purchased from StemExpress, LLC.
With the exception of GMP-compatible scale-up experiments (described separately below), isolated CD3+, CD4+, CD8+ and γδ T cells were activated at 1 × 106 cells per ml−1 for 2 days in complete XVivo15 medium (Lonza) (5% fetal bovine serum, 50 µM 2-mercaptoethanol, 10 mM N-acetyl L-cysteine) supplemented with antihuman CD3/CD28 magnetic Dynabeads (CTS, ThermoFisher) in a 1:1 ratio with cells, 500 U ml−1 of IL-2 (UCSF Pharmacy) and 5 ng ml−1 of IL-7 and IL-15 (R&D Systems). Regulatory T cells were activated at 1 × 106 cells per ml for 2 days in complete XVivo15 supplemented with magnetic Treg Xpander CTS Dynabeads (ThermoFisher) at a 1:1 bead to cell ratio and 500 U ml−1 of IL-2 (UCSF Pharmacy). Isolated B cells were activated at 1 × 106 cells per ml for 2 days in IMDM medium (ThermoFisher) with 10% fetal bovine serum, 50 µM 2-mercaptoethanol, 100 ng ml−1 MEGACD40L (Enzo), 200 ng ml−1 antihuman RP105 (Biolegend), 500 U ml−1 IL-2 (UCSF Pharmacy), 50 ng ml−1 IL-10 (ThermoFisher) and 10 ng ml−1 IL-15 (R&D Systems). Isolated NK cells were activated at 1 × 106 cells per ml−1 for 5 days in XVivo15 medium (Lonza) with 5% fetal bovine serum, 50 µM 2-mercaptoethanol, 10 mM N-acetyl l-cysteine, 1,000 U ml−1 IL-2 and MACSiBead Particles precoated with antihuman CD335 (NKp46) and CD2 antibodies based on manufacturer guidelines (Miltenyi Biotec). Primary adult CD34+ HSCs were cultured at 0.5 × 106 cells per ml in SFEMII medium supplemented with CC110 cytokine cocktail (StemCell). Primary cells were expanded in 96-well plates and split every 2–3 days using identical dilution factors for all experimental samples to prevent cell overgrowth and maintain comparable cell counts.
For GMP-compatible scale-up experiments, CD3+ T cells were activated with antihuman CD3/CD28 magnetic Dynabeads (CTS, ThermoFisher) in a 1:1 ratio with 100 U ml−1 of IL-7 and 10 U ml−1 IL-15 (R&D Systems) in tissue culture flasks. Postelectroporation, cells were expanded in G-Rex 100 M gas-permeable culture vessels (Wilson Wolf) supplemented with 100 U ml−1 of IL-7 and 10 U ml−1 IL-15 every 2–3 days for a total 7 or 10 days expansion as indicated.
RNP formulation.
For most experiments (excluding GMP-compatible scale-up described separately below), RNPs were produced by complexing a two-component gRNA to Cas9 with addition of either a PGA or ssDNAenh electroporation enhancer, as previously described13. Synthetic CRISPR RNA (crRNA, with guide sequences listed in Supplementary Table 1) and trans-activating crRNA (tracrRNA) were chemically synthesized (Edit-R, Dharmacon Horizon), resuspended in 10 mM Tris-HCl (pH 7.4) with 150 mM KCl or IDT duplex buffer at a concentration of 160 µM and stored in aliquots at −80 °C. The ssDNAenh electroporation enhancer (5’-TCATGTGGTCGGGGTAGCGGCTGAAGCACTGCACGCCGTAC-GTCAGGGTGGTCACGAGGGTGGGCCAGGGCACGGGCAGCTTGCC-GGTGGTGCAGATGAACTTCAGGGTCAGCTTGCCGTAGGTGGC-3’) was synthesized by IDT, resuspended to 100 µM in water and stored at −80 °C. The 15–50 kDa PGA was purchased from Sigma and resuspended to 100 mg ml−1 in water, sterile filtered and stored at −80 °C before use.
To make gRNA, aliquots of crRNA and tracrRNA were thawed, mixed 1:1 v/v and annealed by incubation at 37 °C for 30 min to form an 80 µM gRNA solution. PGA or ssDNAenh were mixed into gRNA solutions at a 0.8:1 volume ratio before adding 40 µM Cas9-NLS (Berkeley QB3 MacroLab) at a 1:1 v/v to attain a molar ratio of sgRNA–Cas9 of 2:1. Final RNP mixtures were incubated at 37 °C for 15–30 min after a thorough mix. Based on a Cas9 protein basis, 50 pmol of RNP was used for each electroporation.
For GMP-compatible scale-up experiments, synthetic single-guide RNA (sgRNA) was purchased from Synthego, resuspended to 160 µM, aliquoted and stored at −80 °C. SpyFi Cas9 nuclease was purchased from Aldevron LLC, aliquoted and stored at −20 °C. For RNP formulation, aliquots of ssDNAenh and sgRNA solutions were thawed and mixed at a 0.8:1 volume ratio before adding SpyFi Cas9 at a 2:1 molar ratio of sgRNA–Cas9. Final RNP mixtures were incubated at 37 °C for 15–30 min before electroporation.
HDRT template preparation.
Short ssDNA HDRTs (<200 bp) were synthesized (Ultramer oligonucleotides, IDT), resuspended to 100 µM in dH2O, and stored at −20 °C before use. Long dsDNA HDRTs encoding various gene insertions (Supplementary Table 1) and 300–600 bp homology arms were synthesized as gBlocks (IDT) and cloned into a pUC19 plasmid in-house or purchased directly from Genscript Biotech. These plasmids then served as a template for generating a PCR amplicon. Plasmids used in this study have been deposited at Addgene (Addgene ID 186054 to 186128). CTS sites were incorporated through additional 5′ sequence added to the base PCR primers (see Supplementary Table 1 for sequences). Amplicons were generated with KAPA HiFi polymerase (Kapa Biosystems), purified by solid phase reversible immobilization (SPRI) bead cleanup and resuspended in water to 0.5–2 µg µl−1 measured by light absorbance on a NanoDrop spectrophotometer (ThermoFisher), as previously described13,14.
For most experiments requiring long ssDNA (excluding GMP-compatible scale-up described separately below), a ssDNA isolation protocol adapted from Wakimoto et al. using biotinylated primers and streptavidin-coated magnetic beads was used49. Amplicons were generated as described above using primers that include a 5′ biotin modification (IDT) on either the forward or reverse PCR primer. Roughly 20 µl Streptavidin C1 Dynabeads (ThermoFisher, catalog no. 65001) per 1 pM of amplicon were rinsed three times with 1× Binding & Wash (B&W) buffer (prepared at 2× concentration and stored at room temperature using 10 ml of 1 M TRIS-HCl pH 7.5, 2 ml 0.5 M EDTA, 116.88 g NaCl, 1 l dH2O) using magnetic separation. The washed beads and the PCR amplicon were then resuspended in B&W buffer for 30 min at room temperature to capture the biotinylated DNA. The mixtures were washed twice with B&W buffer after which the supernatant was removed and replaced with 0.125 M NaOH Melt Solution (prepared fresh) to denature the dsDNA. The solution is placed back on the magnet for 5 min and the supernatant containing the nonbiotinylated strand is removed gently with nonstick pipettes and mixed immediately with Neutralization Buffer (100 µl of 3 M sodium acetate pH 5.2 and 4.9 ml of 1× TE Buffer, prepared fresh). Resulting ssDNA was purified and concentrated using a SPRI bead cleanup, as described previously, and quantified on a NanoDrop spectrophotometer (ThermoFisher).
To anneal complementary oligos to ssCTS templates, purified ssDNA templates and complementary oligos (IDT) were mixed at 4:1 molar ratio of oligos to ssDNA templates in nuclease-free duplex buffer (IDT). Oligo and ssDNA template solutions were heated to 95 °C and cooled gradually (that is, decreasing the temperature by 5 °C every 5 min on a thermocycler). Following annealing protocol, ssCTS templates were ready for immediate use in electroporation experiments or aliquoted for long-term storage at −20 °C.
Large-scale ssDNA production.
For GMP-compatible scale-up experiments, research-grade long ssDNA was manufactured at large scale by Genscript Biotech via a proprietary isothermal enzymatic reaction process (PCT/CN2019/128948). To be brief, sequence verified template on plasmid vector is first converted into uridine modified linear dsDNA fragments via PCR amplification. The linear dsDNA is then treated with USER Enzyme and T4 ligase (catalog nos. M5505S and M0202T, New England BioLabs) to form a self-ligated dsDNA circle with nicking sites. This nick containing dsDNA circle is used as an amplification template for RCA, which is carried out by phi29 DNA polymerase (catalog no. M0269L, New England BioLabs) in a high-fidelity and linear amplification manner. The product of RCA is ssDNA concatemers with repeats of target fragment and a palindromic adapter sequence. The annealing process is followed to let the palindromic adapter sequence form a hairpin structure, and then BspQI restriction enzyme (catalog no. R0712L, New England BioLabs) is added in the reaction system to recognize the stem part of the hairpin and digest the concatemer intermediates into target ssDNA monomers and hairpin adapters. The crude product is further purified by EndoFree Plasmid Maxi Kit (Qiagen, catalog no. 12362), to harvest the target ssDNA and remove hairpin adapters, enzymes, reaction buffer and endotoxin residues.
For production of the 2,923 nt BCMA-CAR encoding ssDNA material, amplification primers were synthesized to add specially designed adapter sequences at the 5′ and 3′ ends of the target sequence via PCR method. The uridine modified forward and reverse primer sequences manufactured by Genscript were: 5′-AACTATACUACGTCAATCGGCTCTTCACACTACTACAGTGCCAATAG-3′ and 5′-TATAGTUACGTCAATCGGC TCTTCACACCGTCTGACTAAC ATAACCTG-3′, respectively. The cycle number of the PCR reaction was set at 20, and 300 μg of linear dsDNA fragment was produced and purified by QIAquick PCR Purification Kit (Qiagen, catalog no. 28706). All of the purified 300 μg of linear dsDNA was treated with USER enzyme and T4 ligase to prepare the RCA template and, then, it was used as the template for a 100 ml RCA reaction. All of the isothermal enzymatic reactions and annealing process were done on Eppendorf ThermoMixer C. The final purified ssDNA sample was eluted with nuclease-free water (Sigma Aldrich, catalog no. W4502) from the silica column of an EndoFree Plasmid Maxi Kit, and then passed single-use 0.22-µm sterile filter (Millipore, catalog no. SLGV033RS). Before lyophilization and final packaging, the ssDNA material was quantified by NanoDrop OneC (ThermoFisher) by UV 260 nm absorbance in single-stranded DNA mode. The sequence integrity was confirmed by Sanger sequencing, and the homogeneity was measured by 2% agarose gel electrophoresis as a single band. Quality control for biosafety of the ssDNA material was also evaluated: endotoxin residue was determined as ≤10 EU per mg by an endotoxin test kit (Bioendo, catalog no. KC5028), protein residue level was below the minimum detection threshold of Micro BCA Protein Assay Kit (ThermoFisher, catalog no. 23235) and no bacterial colonies formed in bioburden detection.
Electroporation and use of small-molecule inhibitors.
Except for GMP-compatible scale-up experiments, primary cells were isolated on day 2 of culture (HSCs, CD3+, CD4+, CD8+, γδ and regulatory T cells) or day 5 (NK cells) and electroporated using the Lonza 4D 96-well electroporation system as previously described13. CD3+, CD4+, CD8+, γδ and regulatory T cells were debeaded using an EasySep magnet (StemCell). Immediately before electroporation, cells were centrifuged at 90g for 10 min and then resuspended at 0.4 × 106 HSCs, 0.5 × 106–1.0 × 106 T cells, 0.5 × 106 NK cells or 0.5 × 106 B cells per 20 µl Lonza P3 buffer. HDRT and RNP formulations were mixed and incubated for at least 5 min, then combined with cells and transferred to the Lonza 96-well electroporation shuttle. B cells, NK cells and all T cell subtypes were electroporated using pulse code EH-115 while HSCs were electroporated with pulse code ER-100. Following electroporation, cells were rescued with prewarmed growth media and incubated for at least 15 min. Cells were then transferred to fresh plates or flasks and diluted to 0.5–1.0 × 106 cells per ml in each respective growth medium as described above. Fresh cytokines and media were added every 2–3 days.
TSA (Cayman Chemical), Nedisertib (M3814) (MedKoo Biosciences), XL413 hydrochloride (XL413) (Fisher Scientific), NU7441 (Fisher Scientific) and Alt-R HDR enhancer (IDT) were prepared and stored as aliquots per the manufacturer’s guidelines. For experiments using small-molecule inhibitors, cells were incubated with the indicated concentrations on addition of fresh growth media following the 15-min rescue step and removed by media exchange after 24 h.
For GMP-compatible scale-up experiments, activated cells were separated from beads on day 2 and centrifuged for 10 min at 90g. After removing the supernatant, cells were resuspended in Maxcyte Electroporation Buffer at 200 × 106 cells per ml. HDRTs and RNPs were mixed and incubated for at least 5 min before being combined with cells. The mixture was then transferred to Maxcyte OC-1000 electroporation cuvettes. Cuvettes were filled up to roughly 60% of the total volume (roughly 600 µl) and electroporated with pulse code expanded T cell 4–2. Immediately following electroporations, roughly 400 µl of prewarmed XVivo15 media was added to the cuvette and cells were incubated for 15 min, then transferred to G-Rex culture vessels as described above.
Flow cytometry.
All flow cytometry was performed on an Attune NxT flow cytometer with a 96-well autosampler (ThermoFisher Scientific). Unless otherwise indicated, cells were collected 3–5 days postelectroporation, resuspended in fluorescence-activated cell sorting (FACS) buffer (1–2% BSA in PBS) and stained with Ghost Dye red 780 (Tonbo) and the indicated cell-surface and intracellular markers (see Supplementary Table 1 for antibodies). To obtain comparable live cell counts between conditions, events were recorded from an equivalent fixed volume for all samples. For intracellular staining, cells were stained for surface markers and then prepared for intracellular staining using True-Nuclear Transcription Factor staining kits (Biolegend). For experiments demonstrating stimulation response, cells were reactivated 24 h before analysis using ImmunoCult Human CD3/CD28/CD2 T Cell Activation reagent (StemCell). Analysis was done using FlowJo v.10 software. All gating strategies included exclusion of subcellular debris, singlet gating and live:dead stain. Final graphs were produced with Prism (GraphPad), and figures were compiled with Illustrator (Adobe).
AFM.
ssCTS and dsCTS were purified via gel extraction, using Bio-Rads Quantum PrepTM Freeze ‘N Squeeze DNA gel-extraction spin column kit according to the manufacturer’s protocol. Briefly, target bands were excised and spun down at 13,000 relative centrifugal force for 3 min at room temperature in the respective spin columns. Agarose gels were run for 75 min at 95 V (1.5% agarose in 45 mM Tris, 45 mM boric acid, 1 mM EDTA, 10 mM MgCl2, pH 8). Purified DNA constructs were imaged with a Bruker BioScope Resolve using the ScanAsyst in Air mode. Samples were prepared by applying 7 μl of sample to freshly cleaved mica (Plano GmbH) followed by 3 min of incubation before careful rinsing with ddH2O and drying under a gentle flow of air. Imaging was performed with triangular Silicon-Nitride probes (ScanAsyst-Air, Bruker), at a typical scan rate of around 1 Hz.
AAV.
AAV-ITR plasmids containing the BCMA-CAR and TRAC-targeting homology arms for HDR were used as previously described6. The AAV-ITR-containing plasmid was packaged into AAV6 using polyethylenimine-based cotransfection of HEK293T cells with pHelper and pAAV Rep-Cap plasmids. Viral particles were extracted from cells and purified using iodixanol-based density gradient ultracentrifugation. AAV titration was performed by quantitative PCR (qPCR) after treating samples with DNase I (NEB) and Proteinase K (Qiagen), using primers targeting the left homology arm (forward CTTTGCTGGGCCTTTTTCCC, reverse CCTGCCACTCAAGGAAACCT). qPCR was performed using SsoFast Eva Green Supermix (Bio-Rad) on a StepOnePlus Real-Time PCR System (Applied Biosystems). Primary human T cells were isolated, activated and electroporated with preassembled Cas9 RNPs as described above. Following electroporation, cells were then diluted into media ± serum (2 × 106 cells per ml) and incubated at 37 °C, 5% CO2. Recombinant AAV6 donor vector was added to the culture 30–60 min after electroporation at a multiplicity of infection of 5 × 104, and cells were incubated overnight. The next day, the cells were resuspended in fresh complete medium and expanded using standard culture conditions (37 °C, 5% CO2 and complete medium replenished as needed to maintain a density of 1 × 106 cells per ml every 2–3 days). Knock-out and knock-in efficiency were evaluated by staining for the TCR with an anti-TCRα/β antibody (Miltenyi Biotec) and staining for the CAR with Myc-Tag (Cell Signaling Technology), and flow cytometry was conducted on a BD LSRFortessa X-50 instrument.
In vitro killing assay.
BCMA+MM1S-luc multiple myeloma cells we cultured with BCMA TRAC CAR-T cells or unmodified T cells from same blood donors. After 24 h of coculture, luminescence was measured in a GloMax Explorer instrument (Promega) by adding d-Luceferin (Goldbio, LUCK-1G) at a final concentration of 0.375 mg ml−1 and to each well. Cytoxocity for each sample was determined by the formula: 100 × (1 − (sample-minimum)/(maximum-minimum)). The minimum value was determined by adding Tween to the tumor cell culture and the maximum value was determined by adding no treatment. GloMax Explorer software v.3 was used for acquisition.
Mouse studies.
Mice were used in accordance with ethical guidelines approved by the UCSF Institutional Animal Care and Use Committee. We used 8–12-week-old NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) male mice (in-house breeding). Mice were housed with standard 12:12 light:dark cycle, temperature 20–26 °C and humidity between 30 and 70%. Mice were inoculated with 5 × 105 MM1S-luc cells by tail vein injection, followed by 2 × 105 or 5 × 105 TRAC BCMA-CAR-T cells injected 3 weeks later. Mice were randomized to achieve similar distributions of tumor load measured with bioluminescence the day before T cells injection. Bioluminescence was measured with the Xenogen IVIS Imaging System (Xenogen) and analyzed with Living Image software v.4.7 (Xenogen).
Analysis of knock-in array targets.
Primary human T cells from six unique healthy donors were electroporated with the indicated gRNA (complexed with Cas9 to form an RNP). Genomic DNA was prepared after initial T cell isolation (day 0), immediately before electroporation (day 2) and during postelectroporation expansion (day 4) and roughly 1 × 106 CD4+ and CD8+ T cells from each donor were sorted by FACS for RNA-seq and ATAC-seq analysis at each time point. Half of the sorted cells were frozen in Bambanker freezing medium (Bulldog Bio) for ATAC-seq, and half were frozen in RNAlater (QIAGEN) for bulk RNA-seq.
Amplicon sequencing.
Roughly 100,000 cells per condition were resuspended in 20 μl of Quickextract DNA Extraction Solution (Epicenter) to a concentration of 5,000 cells per μl. Genomic DNA in Quickextract was heated to 65 °C for 6 min and then 98 °C for 2 min, according to the manufacturer’s protocol. Then 1 μl of the mixture, containing genomic DNA from 5,000 cells, was used as template in a two-step PCR amplicon sequencing approach using NEB Q5 2× Master Mix Hot Start Polymerase with the manufacturer’s recommended thermocycler conditions. After an initial 18 cycle PCR reaction with primers amplifying an approximately 200 bp region centered on the predicted gRNA cut site, a 1.0× SPRI purification was performed and a ten-cycle PCR to append P5 and P7 Illumina sequencing adapters and donor-specific barcodes was performed, followed again by a 1.0× SPRI purification. Concentrations were normalized across donor–gRNA indexes, samples pooled and the library sequenced on an Illumina Mini-Seq with a 2 × 150 bp reads run mode. Amplicons were processed with CRISPResso2, using the CRISPRessoPooled command in genome mode with default parameters. We used the hg19 human reference genome assembly. Resulting amplicon regions were matched with gRNA sites for each sample. We eliminated reads with potential sequencing errors detected as single mutated bases with no indels by CRISPResso alignment. The remaining reads were used to calculate the indel percentage, or ‘observed cutting percentage’.
Bulk RNA-seq.
Total RNA from frozen samples was extracted using RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. RNA quantification was performed using Qubit and NanoDrop 2000 and quality of the RNA was determined by the Bioanalyzer RNA 6000 Nano Kit (Agilent Technologies) for ten random samples. We confirmed that the sample had an average RNA integrity number that was more than nine and the traces revealed characteristic size distribution of intact, nondegraded total RNA. The RNA libraries were constructed with Illumina TruSeq RNA Sample Prep Kit v.2 (catalog no. RS-122-2001) according to the manufacturer’s protocol. Total RNA (500 ng) from each sample was used to establish complementary DNA libraries. A random set of the final libraries were quality checked on the High Sensitivity DNA kit (Agilent) that revealed an average fragment size of 400 bp. Samples were sequenced using the Illumina HiSeq 4000 on with 100 bp paired-end reads. RNA-seq reads were processed with kallisto using the Homo sapiens ENSEMBL GRCh37 (hg19) cDNA reference genome annotation. Transcript counts were aggregated at the gene level. Genes of interest were subsetted from the normalized gene-level counts table and analyzed as transcripts per million.
ATAC-seq.
ATAC-seq libraries were prepared following the Omni-ATAC protocol. Briefly, frozen cells were thawed and stained for live cells using Ghost Dye 710 (Tonbo Biosciences). Then, 50,000 lived cells were FACS sorted and washed once with cold PBS. Technical replicates were done for most of the samples. Cell pellets were resuspended in 50 μl cold ATAC-Resuspension buffer (10 mM Tris-HCl (Sigma Aldrich) pH 7.4, 10 mM NaCl, 3 mM MgCl2 (Sigma Aldrich)) containing 0.1% NP40 (Life Technologies), 0.1% Tween-20 (Sigma Aldrich) and 0.01% Digitonin (Promega) for 3 min. Samples were washed once in cold resuspension buffer with 0.1% Tween-20 and centrifuged at 4 °C for 10 min at 300 r.p.m. Extracted nuclei were resuspended in 50 μl of Tn5 reaction buffer (1× TD buffer (Illumina), 100 nM Tn5 Transposase (IIlumina), 0.01% Digitonin, 0.1% Tween-20, PBS and H2O) and incubated at 37 °C for 30 min at 300 r.p.m. Transposed samples were purified using MinElute PCR purification columns (Qiagen) as per the manufacturer’s protocol. Purified samples were amplified and indexed using custom Nextera barcoded PCR primers. DNA libraries were purified using MinElute columns and pooled at equal molarity. To remove primer dimers, pooled libraries were further cleaned up using AmPure beads (Beckman Coulter). ATAC libraries were sequenced on a Illumina HiSeq 4000 in paired-end 100-cycle mode. ATAC-seq reads were trimmed using cutadapt v.1.18 to remove Nextera transposase sequences, then aligned to hg19 using Bowtie2 v.2.3.4.3. Low-quality reads were removed using samtools v.1.9 view function (samtools view -F 1804 –f 2 -q 30 -h -b). Duplicates were removed using picard v.2.18.26, then reads were converted to BED format using bedtools bamtobed function and normalized to reads per million. ATAC-seq reads mapping within a 1 kb window surrounding CRISPR cut sites were counted using the bedtools intersect function.
Statistics and reproducibility.
Statistical analyses were performed with Graphpad Prism v.9. P values were calculated by unpaired two-tailed t-test, two-tailed Mann–Whitney test or log-rank Mantel–Cox test (survival) as indicated. Line-of-best-fit and R squared from a simple linear regression were calculated for normally distributed data. Spearman r was calculated for nonlinear correlations as determined by Shapiro–Wilk test. Studies using primary human cells were each performed with n = 2 independent healthy blood donors, unless otherwise indicated. Mouse studies were completed with n = 4 to n = 9 mice per treatment as indicated on each relevant figure. No statistical method was used to predetermine sample size. Sample sizes are similar to those reported in previous publications13,14,50. For mouse tumor studies, a second cohort of mice treated with cells from a second donor was excluded because tumor failed to efficiently engraft in control group. Mice were randomized to achieve similar distributions of tumor load measured with bioluminescence the day before T cells injection. Controls and treatments for studies with primary human cells were performed in matched cells from the same donors. The investigators were not blinded to allocation during experiments and outcome assessment.
Extended Data
Extended Data Fig. 1 |. Illustration of ssCTS designs.

Detailed illustrations of CTS designs evaluated in the manuscript highlighting the location and orientation of gRNA target sequences (red), 4 bp mismatch (pink), PAM interaction site (yellow), and transgene (blue). (a-j) Illustration of short CD5-HA HDRT designs evaluated in Fig. 1a–c and Extended Data Fig. 2c. (k) Representative illustration of optimal ssCTS design used for large HDRTs throughout the manuscript. CTS = Cas9 Target Site, ssCTS = ssDNA HDRT + CTS sites, HDRT = homology-directed-repair template.
Extended Data Fig. 2 |. Comparison of CTS template designs.
(a) Diagram of CD5-HA knock-in strategy and control ssDNA HDRTs. (b) Representative flow cytometry plots demonstrating CD5-HA knock-in. (c) Live cell counts for each ssCTS design using a CD5-HA knock-in construct at 160 nM – 4uM concentration. (d-e) Comparison of knock-in efficiency, live cell counts, and knockin cell counts with insertions of increasing size using (d) dsCTS or (e) ssCTS HDRTs targeting the IL2RA gene. Evaluated transgenes encode either GFP (~1.4 kb total HDRT length), an IL2RA-GFP fusion (~2.3 kb total HDRT length), or an IL2RA-GFP fusion plus separate EF1a-mCherry expression cassette (~3.5 kb total HDRT length). Each experiment was performed with T cells from 2 independent healthy human blood donors represented by individual dots + mean. RNP = Ribonucleoprotein, CTS = Cas9 Target Site, ssCTS = ssDNA, HDRT + CTS sites, HDRT = homology-directed-repair template.
Extended Data Fig. 3 |. Evaluation of ssCTS design and mechanism with large HDRTs.
(a-f) Comparison of different CTS designs with a large ~2.7 kb CD5-HA knock-in construct. (a) Diagram of long CD5-HA knock-in strategy, representative flow cytometry plot, percent knock-in, live cell counts, and knock-in cell yield counts. (b) Comparison of CTS with a gRNA target sequence that is specific for the cognate RNP ( + CD5 CTS), an alternative gRNA sequence (+ IL2RA CTS), a CTS incorporating a PAM site and scrambled gRNA sequence (+ scramble CTS), or an equivalent amount of dsDNA within the 5’ end of the homology arm (+ end protection). (c) Comparison of complementary oligos covering different regions of the CTS and surrounding sequences. Constructs with CTS sites on both 5’ and 3’ end (green bars), 5’ end only (blue bars), or 3’ end only (red bars) are shown on the right panel. (d) Evaluation of varied 5’ ends including different length of buffer sequence upstream of the CTS site. *indicates no data available for the marked column. (e) Comparison of CTS with different numbers of scrambled bases at the 5’ end of the gRNA target sequence using WT or SpyFi Cas9. (f) Length of homology arm that is covered by the complementary oligonucleotide. (g) Evaluation with and without (‘−’) CTS sites on the 5’ and 3’ end of long ssDNA IL2RA-GFP HDRTs with PAM facing inwards toward the homology arm (‘In’) or outwards away from the homology arm (‘Out’). (h-i) Comparison of knockout and knockin with large CD5-HA (h) or IL2RA-GFP (i) ssDNA and ssCTS HDRTs using RNPs formulated with Cas9 +/− NLS sequences. Each experiment was performed with T cells from 2 independent healthy human blood donors represented by individual dots + mean. RNP = Ribonucleoprotein, CTS = Cas9 Target Site, ssCTS = ssDNA HDRT + CTS sites, PAM = Protospacer Adjacent Motif, HDRT = homology-directed-repair template.
Extended Data Fig. 4 |. Additional parameters affecting ssCTS knockin and biophysical analysis of RNP interactions with HDRTs.
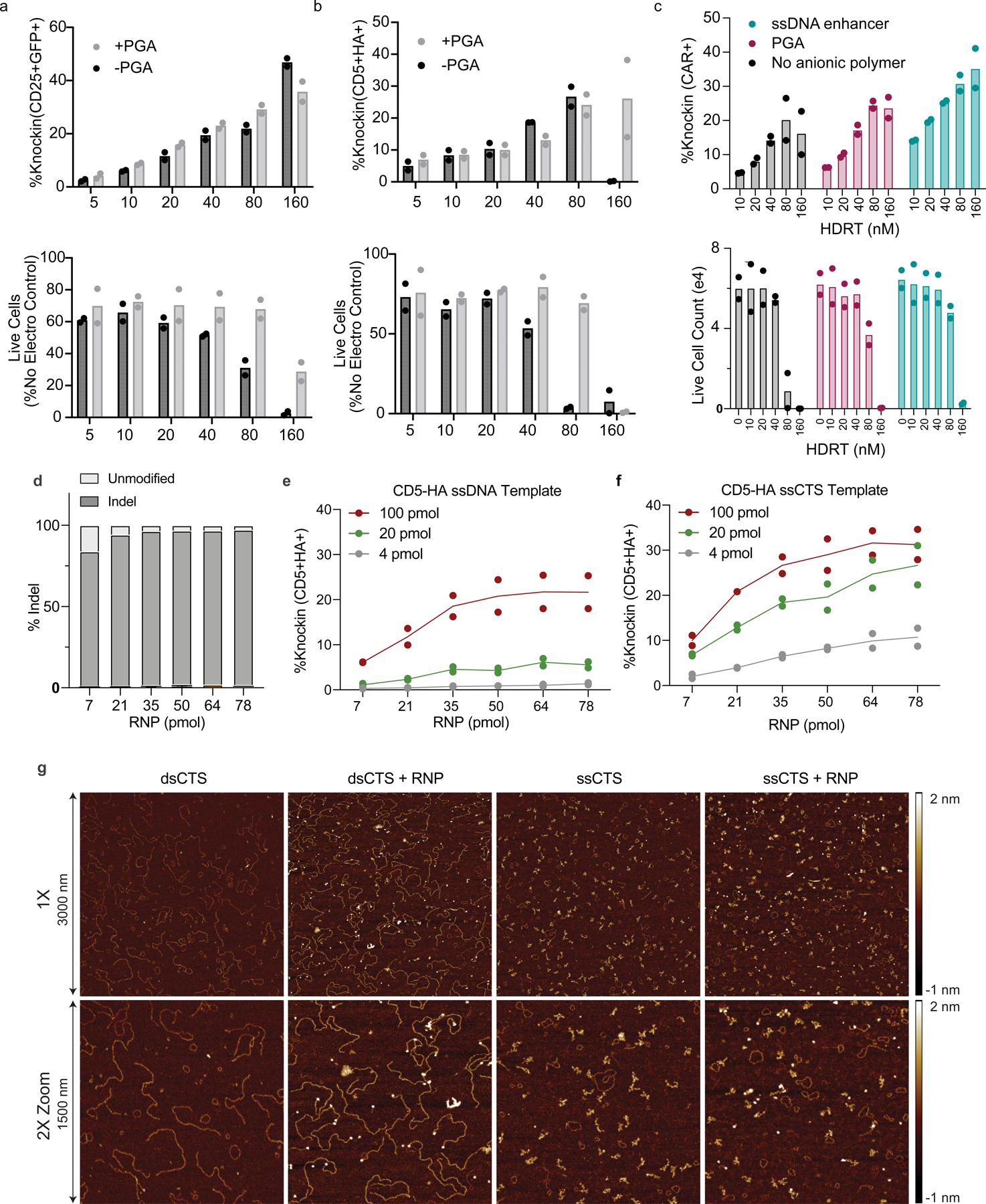
(a-b) Comparison of knockin efficiency (top) and live cell counts (bottom) +/− PGA using (a) large IL2RA-GFP ssCTS templates or (b) large CD5-HA ssCTS templates (~1.3 kb homology arms). (c) Comparison of knockin efficiency (top) and live cell counts (bottom) with PGA, ssDNA enhancer, or no anionic polymer using a BCMA-CAR ssCTS templates. (d-f) evaluation of (d) indel formation by amplicon sequencing, (e) knockin efficiency with short ssDNA CD5-HA HDRTs, or (f) knockin efficiency with short ssCTS CD5-HA HDRTs (40 nucleotide homology arms) using varied molar amounts of RNP and HDRT. (g) Representative AFM images of gel purified dsCTS or ssCTS templates +/− Cas9 RNPs. Brightness shows the relative height as indicated in by scale bars to right of figure. Background circular forms in all panels are likely residual agarose. Experiments in panels a-f were performed with T cells from 2 independent healthy human blood donors represented by individual dots + mean (a, b, c, e, f) or mean alone (d). RNP = Ribonucleoprotein, CTS = Cas9 Target Site, ssCTS = ssDNA HDRT + CTS sites, HDRT = homology-directed-repair template, AFM = atomic force microscopy.
Extended Data Fig. 5 |. Arrayed knockin analysis and target locus characteristics.
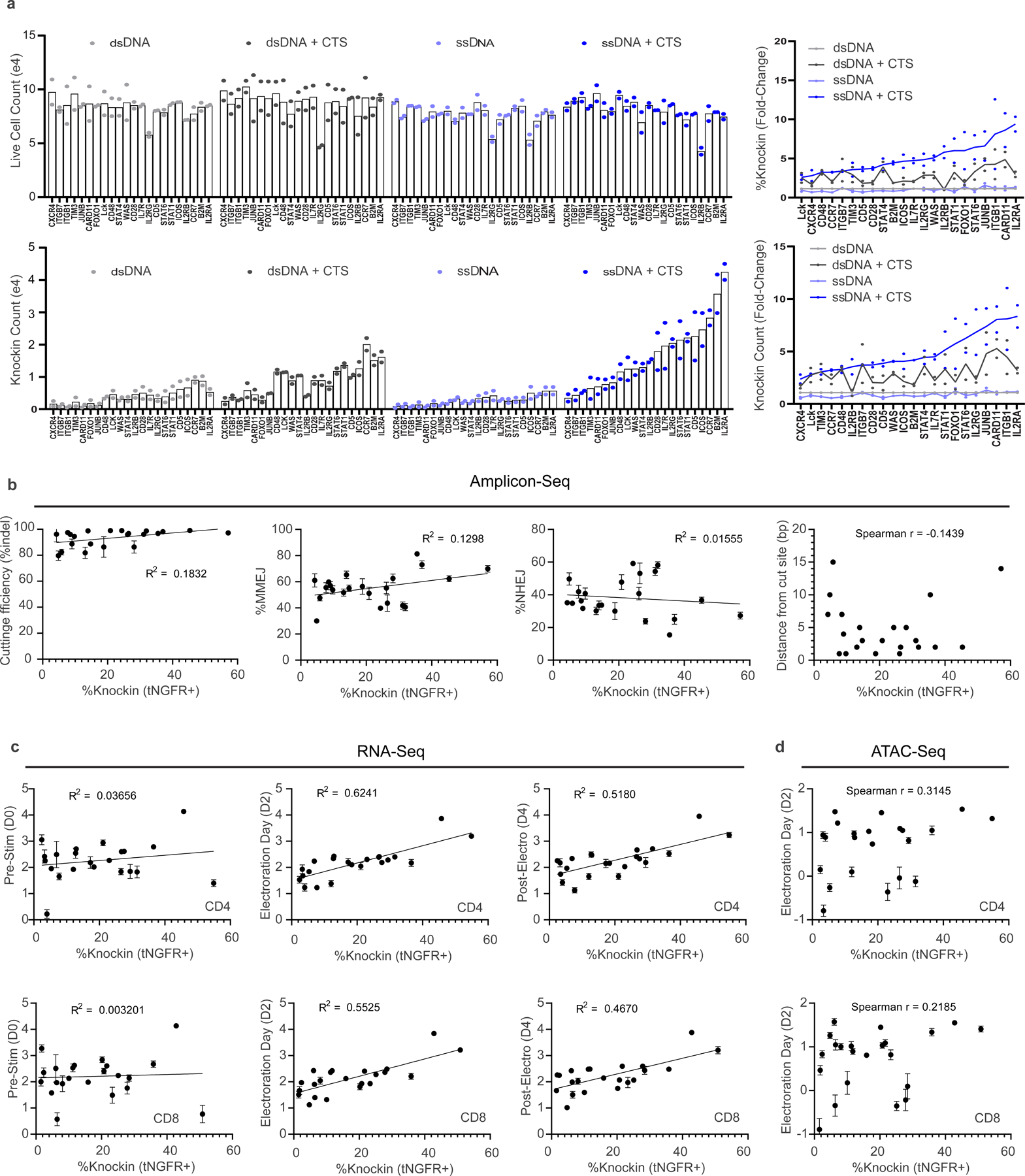
(a) Comparison of HDRT variations for knock-in constructs targeting a tNGFR marker across 22 different target loci. Shown for each construct are live cell counts, knock-in cell count yields, relative %knock-in and relative knock-in counts compared to dsDNA templates. Data show mean and individual values from 2 independent healthy human blood donors (b-d) Evaluation target locus characteristics in comparison to ssCTS knockin efficiency by (b) Amplicon-Seq, (c) RNA-Seq, and (d) ATAC-Seq methodologies. Knock-in efficiency for panels b-d shows mean from 2 independent healthy human blood donors. Amplicon-Seq, RNA-Seq, and ATAC-seq data in panels b-d were generated 6 independent healthy human blood donors presented as mean +/− SD. Line-of-best-fit and R squared from a simple linear regression are shown for normally distributed data. RNA-seq data was log-transformed prior to linear regression. Spearman r is shown for non-linear correlations as determined by Shapiro-Wilk test (‘distance from cut site’ and ATAC-seq evaluations). Y axis for panels c-d is log 10. In c and d, separate analyses were performed for CD4 + (top) and CD8 + T cells (bottom) for RNA-Seq and ATAC-Seq comparisons. tNGFR = truncated nerve growth factor, MMEJ = microhomology mediated end joining, NHEJ = non-homologous end joining, ATAC = Assay for Transposase-Accessible Chromatin.
Extended Data Fig. 6 |. Application of ssCTS to diverse primary human hematopoietic cell types.
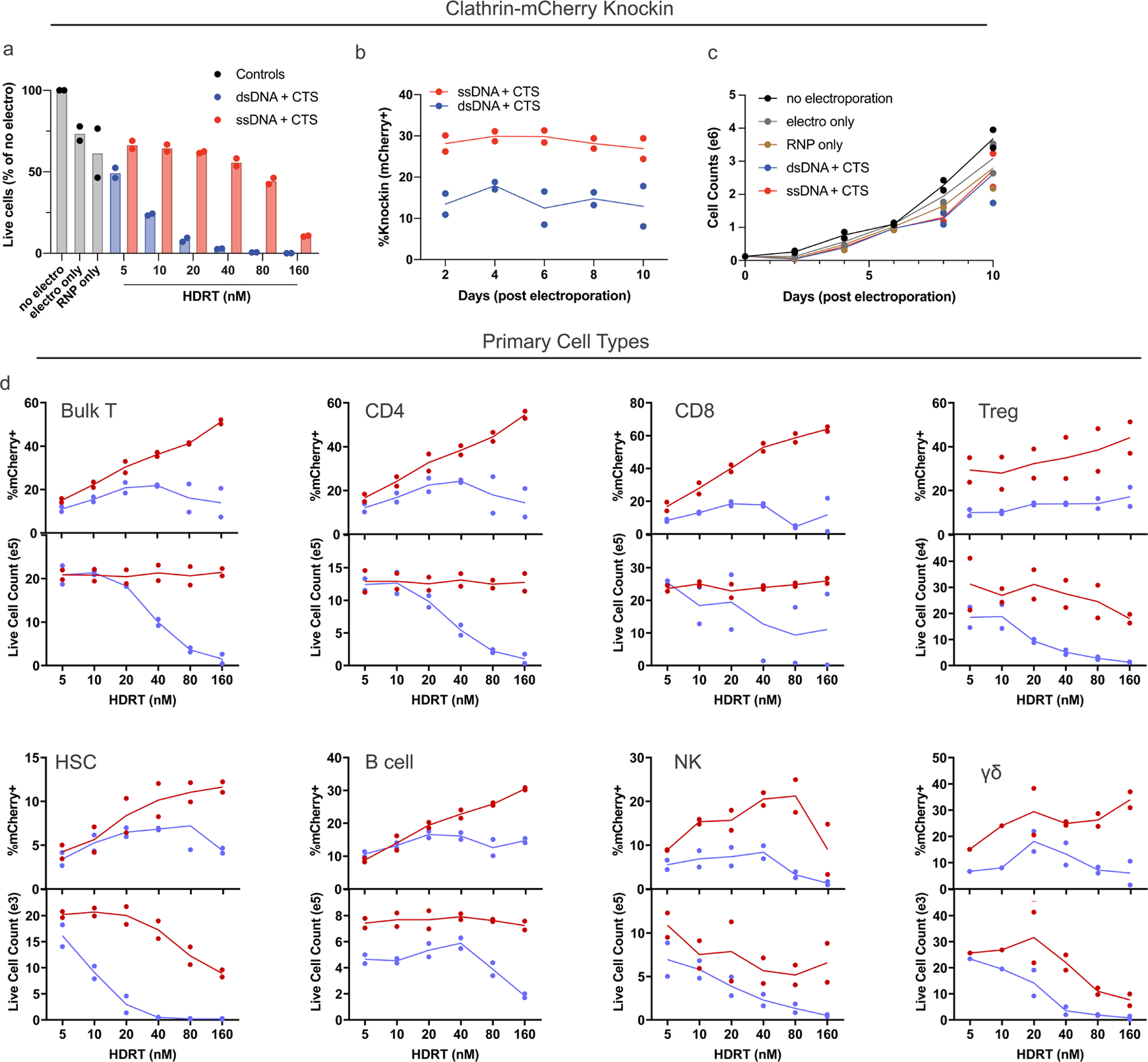
(a-c) Evaluation of CLTA-mCherry knock-in efficiency and live cell counts 0–10 days postelectroporation in primary human T cells. (a) Live cell counts represented as a percentage of the no electroporation control on day 4 post-electroporation. (b) Knock-in efficiency on day 2–10 post-electroporation. (c) Growth curves for control cells (no electroporation, electroporation only, and Cas9 RNP only) and cells edited with dsCTS or ssCTS HDRTs on day 0–10 post-electroporation. (d) Comparison of knock-in efficiency (top) and live cell counts (bottom) using ssCTS and dsCTS HDRTs (blue line) across a variety of primary human hematopoietic cell types using knockin constructs encoding a CLTA locus mCherry fusion protein. Each experiment was performed with cells from 2 independent healthy human blood donors. Each experiment was performed with T cells from 2 independent healthy human blood donors represented by individual dots + mean (c, d, f) or mean alone (e). CTS = Cas9 target site, dsCTS = dsDNA HDRT + CTS sites, ssCTS = ssDNA HDRT + CTS sites, HDRT = homology-directed repair template, RNP = ribonucleoprotein, CLTA = Clathrin, Treg = regulatory T cells, HSC = hematopoietic stem cell, NK cells = natural killer cells, γδ T cells = gamma delta T cells.
Extended Data Fig. 7 |. Evaluation of small molecule inhibitor cocktails in primary human T cells.
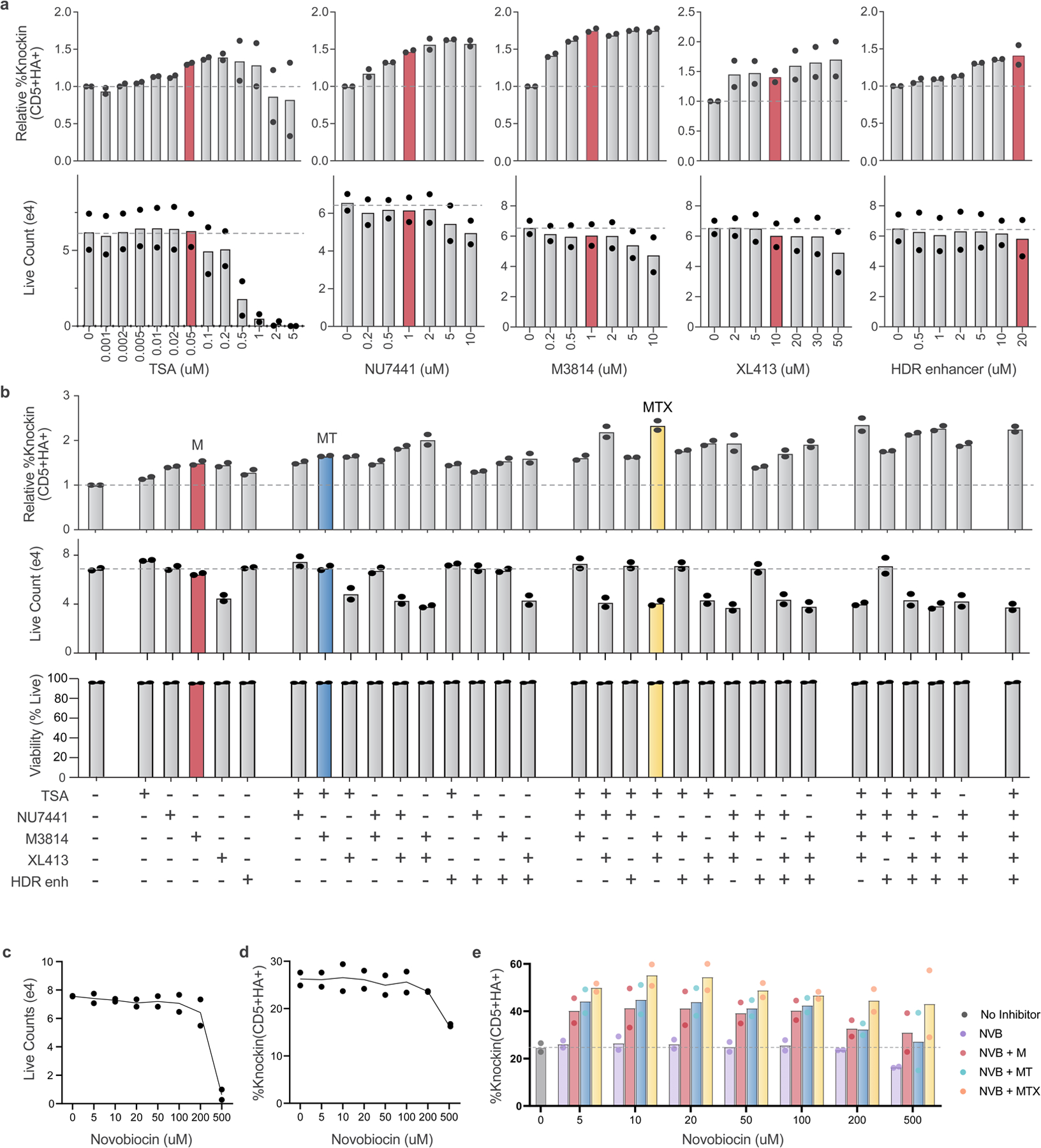
(a) Evaluation of relative increase in percent knock-in using an ssDNA CD5-HA knock-in construct over varied concentrations of 5 different small molecule inhibitors assessed by flow cytometry. Red bars indicate concentrations chosen for subsequent experiments. (b) Comparison of relative percent knock-in (top), live cell counts (middle), and viability with Ghost Dye 780 (Tonbo) (bottom) with small molecule inhibitor combinations. Cocktails chosen for subsequent experiments are highlighted in red (M3814), blue (MT) and yellow (MTX). (c-d) Evaluation of Novobiocin effects on (c) live cell counts and (d) knock-in efficiency using a small CD5-HA ssDNA HDRT. (e) Evaluation of Novobiocin effects on knockin efficiency at varied concentration using a small CD5-HA ssDNA HDRT in combination with M3814, MT, and MTX inhibitors. Each experiment was performed with T cells from 2 independent healthy human blood donors represented by individual dots + mean. M = M3814, MT = M3814 + Trichostatin A, MTX = M3814 + Trichostatin A + XL413, NVB = Novobiocin.
Extended Data Fig. 8 |. Analysis of genome editing outcomes with CTS templates and small molecule inhibitors.
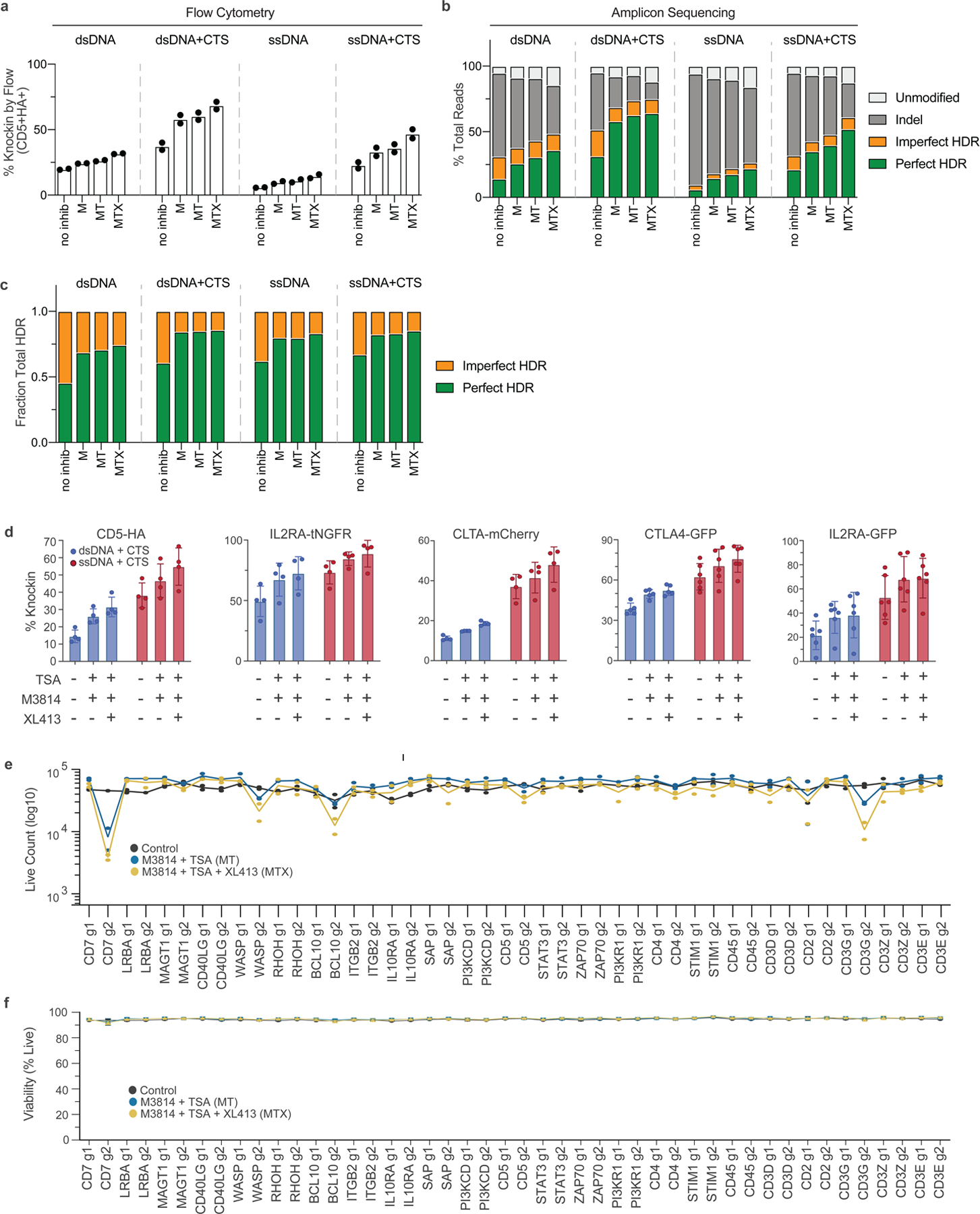
(a-c) Evaluation of genome editing outcomes by either (a) flow cytometry or (b) amplicon sequencing using small dsDNA, dsCTS, ssDNA, or ssCTS CD5-HA HDRTs at non-toxic concentrations (800 nM) with and without M, MT, and MTX inhibitor combinations. (c) Ratio of perfect:imperfect HDR events with each combination. (d) Comparison of dsCTS and ssCTS templates in combination with small molecular inhibitors in 5 different knock-in constructs using a large CD5-HA HDRT (−2.7 kb, n = 4 donors), a tNGFR knock-in to the IL2RA gene (~1.5 kb, n = 4 donors), an mCherry fusion in the clathrin gene (~1.5 kb, n = 4 donors), a near full length CTLA-4-GFP fusion to the CTLA4 gene (~2.1 kb, n = 6 donors), and a full length IL2RA-GFP fusion to the IL2RA gene (~2.3 kb, n = 6 donors). (e-f) Evaluation of (e) live cell counts and (f) viability +/− MT and MTX inhibitor combinations using 44 different knock-in constructs targeting a tNGFR marker across 22 different target loci with 2 gRNA per gene (g1 and g2). Panel a shows mean and individual values from two healthy blood donors. Panels b, c, e, and f show mean values from two healthy blood donors. Panel d shows mean +/− SD. CTS = Cas9 target site, HDRT = homology-directed repair template, dsCTS = dsDNA + CTS HDRT, ssCTS = ssDNA + CTS HDRT, M = M3814, MT = M3814 + TSA, MTX = M3814 + TSA + XL413.
Extended Data Fig. 9 |. IL2RA and CTLA4 ORF replacement strategies.
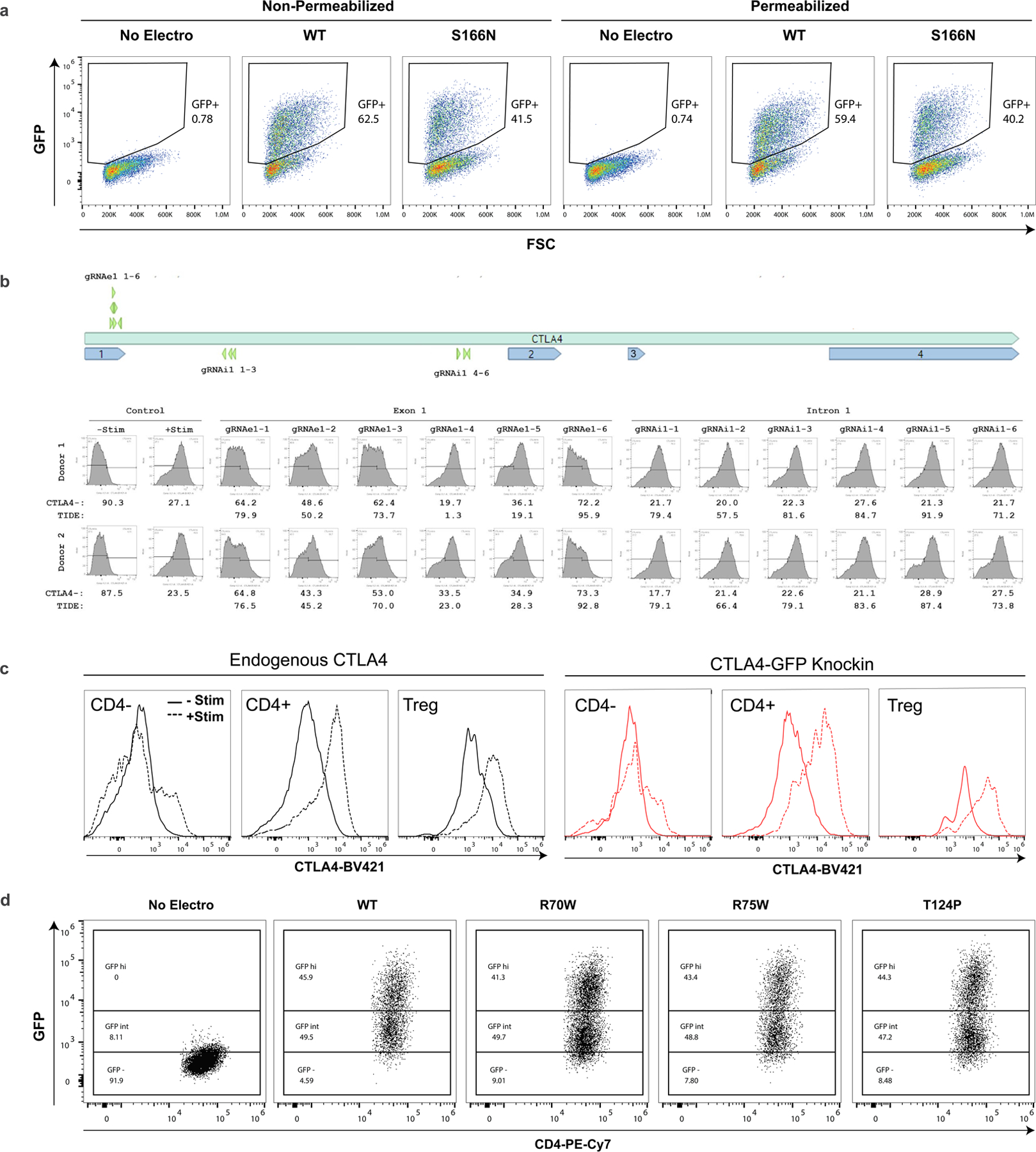
(a) Gating for GFP + cells are shown for WT and S166N IL2RA-GFP knock-in constructs. (b) Diagram of the CTLA4 gene (top), CTLA4 protein levels (bottom), and cutting efficiency (bottom) illustrating a screening panel of 12 gRNAs examined within exon 1 and intron 1. gRNAs were assessed in activated CD4 + T cells for protein disruption by CTLA4 flow cytometric analysis (flow plots and top row of numbers demonstrate the % of CTLA4-negative cells for each donor), and for cutting efficiency as determined by TIDE indel analysis51 (bottom row of numbers indicate the %indel at target locus). (c) CTLA4 expression levels assessed by flow cytometry with endogenous protein (black) and WT CTLA4-GFP knock-in protein (red) are shown for CD4- T cells, CD4 + T cells, and regulatory T cells with (dotted line) and without (solid line) stimulation. (d) Gating for GFPhi cells is shown for WT, R70W, R75W, and T124P CTLA4-GFP knock-in cells. Each experiment was performed with T cells from 2 independent healthy human blood donors. WT = Wild-Type, Treg = regulatory T cell.
Extended Data Fig. 10 |. Evaluation of a nonviral strategy for anti-BCMA CAR-T cell manufacturing.
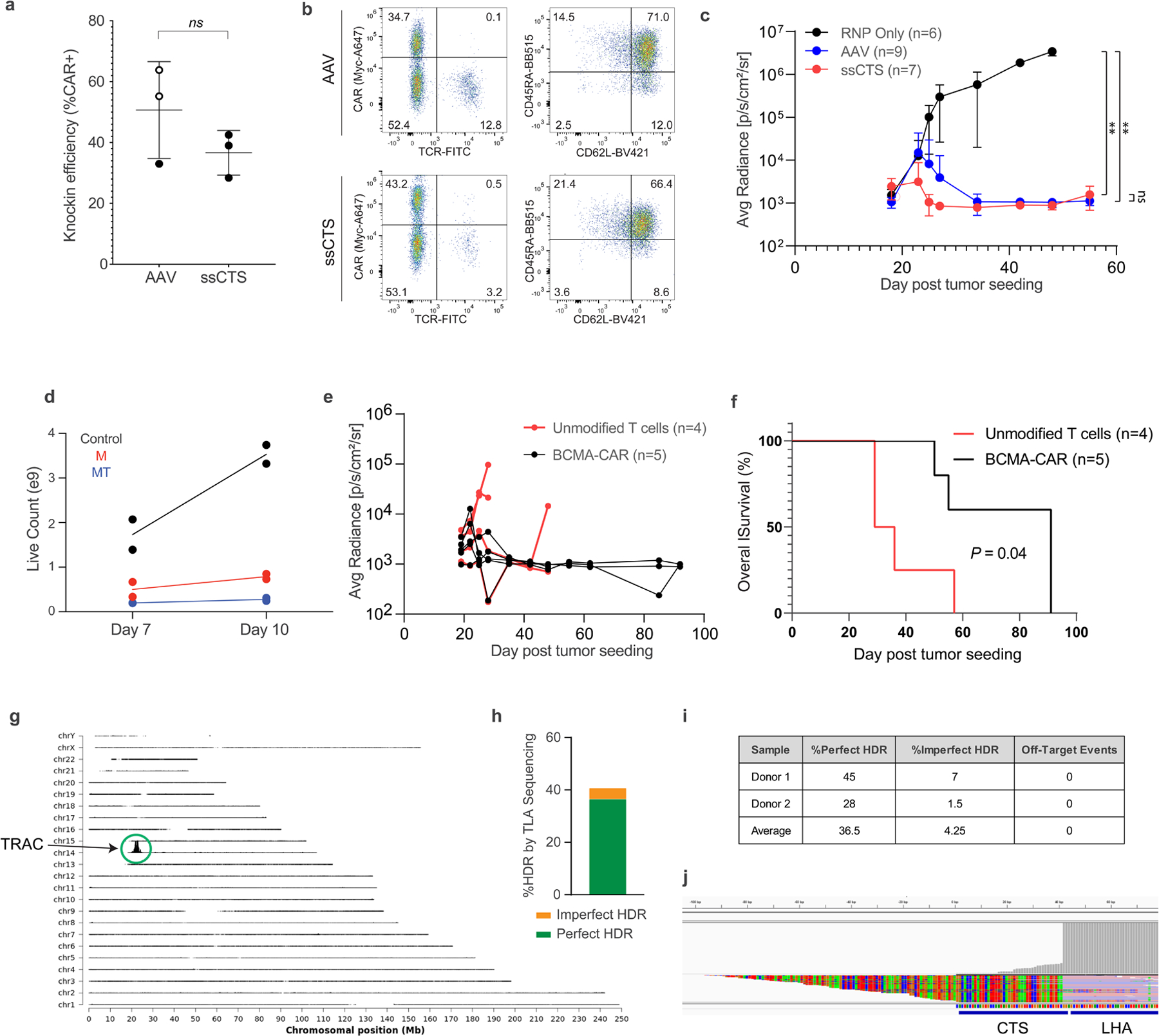
(a-c) Comparison of (a) knockin efficiency (mean +/− SD), (b) flow cytometric immunophenotypes, and (c) tumor burden of MM1S-bearing NSG mice treated with TRAC anti-BCMA CAR-T cells generated using either AAV or non-viral ssCTS HDRTs (mean +/− SD). (d) Live cell counts for large-scale GMP-compatible manufacturing process at Day 7 and Day 10 post-activation. (e) Tumor burden (average radiance) of individual MM1S-bearing NSG mice treated with Unmodified T cells and TRAC anti-BCMA CAR-T cells generated in GMP-compatible anti-BCMA-CAR T cell scaleup experiment. (f) Kaplan–Meier analysis showing overall survival of MM1S xenotransplant NSG mice treated with anti-BCMA-CAR or unmodified T cells. (g-j) Targeted Locus Amplification (TLA) analysis for anti-BCMA TRAC CAR-T cell products generated in GMP-compatible scaleup experiments. (g) Integration site analysis based on TLA sequencing demonstrating targeted insertion at the expected TRAC locus on chromosome 14. (h) Mean percentage of perfect and imperfect HDR events by TLA sequencing from 2 independent healthy human blood donors. (i) Table of perfect HDR, imperfect HDR, and off-target events for individual donors by TLA sequencing. (j) TLA sequence coverage aligned on the TRAC anti-BCMA CAR ssCTS reference construct. Grey bars on Y axis indicate sequence coverage. Low coverage across the CTS indicates relatively rare non-HDR events incorporating the indicated bases. Panel a was performed with 3 independent healthy human blood donors. Open circles represent use of serum-free media post-electroporation, closed circles represent use of serum-containing media post-electroporation. Panel c performed with the indicated number of mice using T cell products generated from a matched single healthy blood donor. Panel d performed with 2 independent healthy human blood donors. Panel e-f performed with unmodified T cells (n = 4 mice) and BCMA-CAR T cells (n = 5 mice) generated from one healthy human blood donor. A second cohort of mice treated with cells from a second donor was excluded because tumor failed to efficiently engraft in control group. TLA Analyses performed in 2 independent healthy human blood donors. **P < 0.05; ns, not significant. P values obtained by (a) unpaired two tailed t-test, (c) two tailed Mann-Whitney test, or (f) log-rank Mantel–Cox test (survival). rAAV = recombinant adeno-associated virus, HDRT = homology-directed repair templates, RNP = ribonucleoprotein, TCR = T cell receptor, CTS = Cas9 target site, ssCTS = ssDNA + CTS HDRT, CAR = chimeric antigen receptor, GMP = good manufacturing practice, TLA = targeted locus amplification, LHA = left homology arm.
Supplementary Material
Acknowledgements
We thank all members of the Marson laboratory for their thoughtful input and technical assistance. We thank S. Dodgson, S. Pyle, F. Urnov, B. Schaar, J. Woo, J. Okano and J. Sawin for their helpful suggestions and generous assistance. This research was supported by the NIAID (grant nos. P01AI138962 and P01AI155393), the UCSF Grand Multiple Myeloma Translational Initiative (MMTI), the Weill Neurohub, the Larry L. Hillblom Foundation (grant no. 2020-D-002-NET) and the Innovative Genomics Institute (IGI). The Marson laboratory has received funds from IGI, the Simons Foundation and the Parker Institute for Cancer Immunotherapy (PICI). A.M. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund, is an investigator at the Chan Zuckerberg Biohub and is a recipient of The Cancer Research Institute (CRI) Lloyd J. Old STAR grant. B.R.S. was supported by the UCSF Herbert Perkins Cellular Therapy and Transfusion Medicine Fellowship, the CIRM Alpha Stem Cell Clinic Fellowship and the NCATS (grant no. L30TR002983). D.N.N. is supported by National Institutes of Health (NIH) grant nos. L40AI140341 and K08AI153767 and the CIRM Alpha Stem Cell Clinic Fellowship. J.W. and V.S.V. were supported by the UCSF Grand MMTI. F.B. was supported by the Care-for-Rare Foundation and the German Research Foundation (DFG). T.L.R. was supported by the UCSF Medical Scientist Training Program (grant no. T32GM007618), the UCSF Endocrinology Training grant (no. T32 DK007418) and the NIDDK (grant no. F30DK120213). The Eyquem Laboratory has received funding from PICI, Mnemo Therapeutics, Takeda and Cytovia Therapeutics. M.R.M. was supported by the CRI Irvington Fellowship and the Human Vaccines Project Michelson Prize. The UCSF Flow Cytometry Core was supported by grant no. NIH S10 RR028962 and the James B. Pendleton Charitable Trust. This research was made possible by a grant from the California Institute for Regenerative Medicine (grant no. INFR-10361). W.G.P. and C.E.C. acknowledge support from National Science Federation grant no. 1933344 and acknowledge NIH grant no. 1S10OD025096-01A1, which supported the ATM.
Footnotes
Ethics Statement. This research complies with all relevant ethical regulations.
Reporting summary. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Code availability
All code used in this paper will be made available upon reasonable request.
Competing interests
A.M. is a compensated cofounder, member of the boards of directors, and a member of the scientific advisory boards of Spotlight Therapeutics and Arsenal Biosciences. A.M. is a cofounder, member of the boards of directors, and a member of the scientific advisory board of Survey Genomics. A.M. is a compensated member of the scientific advisory board of NewLimit. A.M. owns stock in Arsenal Biosciences, Spotlight Therapeutics, NewLimit, Survey Genomics, PACT Pharma and Merck. A.M. has received fees from 23andMe, PACT Pharma, Juno Therapeutics, Trizell, Vertex, Merck, Amgen, Genentech, AlphaSights, Rupert Case Management, Bernstein and ALDA. A.M. is an investor in and informal advisor to Offline Ventures and a client of EPIQ. The Marson laboratory has received research support from Juno Therapeutics, Epinomics, Sanofi, GlaxoSmithKline, Gilead and Anthem. J.E. is a compensated cofounder at Mnemo Therapeutics. J.E. is a compensated scientific advisor to Cytovia Therapeutics. J.E. own stocks in Mnemo Therapeutica and Cytovia Therapeutics. J.E. has received a consulting fee from Casdin Capital. The Eyquem laboratory has received research support from Cytovia Therapeutic and Takeda. J.E. is a holder of patents pertaining to but not resulting from this work. H.L and L.Y. are employees of Genscript Biotech Corporation. J.W. has received consulting fees from Teneobio and Adaptive Biotech. D.N.N. receives consulting fees and sits on the scientific advisory board of Navan Technologies. T.L.R. is a cofounder, holds equity in, and is a member of the Scientific Advisory Board of Arsenal Bioscience. Discounted reagents were provided by Genscript. B.R.S., V.S.V. and A.M. are inventors on patent applications based on the findings described in this paper, a subset of which have been licensed by the University of California. A.H., A.T., W.G.P., Y.Y.C., F.B., E.S., S.V., M.R.M., J.J.C., R.Y., D.W., T.G.M., C.E.C. and J.H.E. declare no competing interests.
Additional information
Extended data are available for this paper at https://doi.org/10.1038/s41587-022-01418-8.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41587-022-01418-8.
Peer review information Nature Biotechnology thanks the anonymous reviewers for their contribution to the peer review of this work.
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Data availability
RNA-seq data been deposited in the Gene Expression Omnibus (GEO) under the accession code GSE202909. ATAC-seq data been deposited in the GEO under the accession code GSE130089. Amplicon sequencing data been deposited in the GEO under the accession codes GSE202596 and GSE203349. Other relevant data are available from the corresponding authors upon reasonable request.
References
- 1.Frangoul H et al. CRISPR-Cas9 gene editing for sickle cell disease and beta-thalassemia. New Engl. J. Med 384, 252–260 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Stadtmauer EA et al. CRISPR-engineered T cells in patients with refractory cancer. Science 10.1126/science.aba7365 (2020). [DOI] [PMC free article] [PubMed]
- 3.US National Laboratory of Medicine. A safety and efficacy study evaluating ctx110 in subjects with relapsed or refractory B-cell malignancies (CARBON) ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04035434 (2019).
- 4.US National Laboratory of Medicine. CRISPR-edited allogeneic anti-CD19 CAR-T cell therapy for relapsed/refractory B cell non-Hodgkin lymphoma ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04637763 (2020).
- 5.US National Laboratory of Medicine. Transplantation of clustered regularly interspaced short palindromic repeats modified hematopoietic progenitor stem cells (CRISPR_SCD001) in patients with severe sickle cell disease ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT04774536 (2021).
- 6.Eyquem J et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543, 113–117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Micklethwaite KP et al. Investigation of product derived lymphoma following infusion of piggyBac modified CD19 chimeric antigen receptor T-cells. Blood 10.1182/blood.2021010858 (2021). [DOI] [PMC free article] [PubMed]
- 8.Anzalone AV, Koblan LW & Liu DR Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol 38, 824–844 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Ioannidi EI et al. Drag-and-drop genome insertion without DNA cleavage with CRISPR-directed integrases. Preprint at bioRxiv 10.1101/2021.11.01.466786 (2021). [DOI] [PMC free article] [PubMed]
- 10.Anzalone AV et al. Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing. Nat. Biotechnol 10.1038/s41587-021-01133-w (2021). [DOI] [PMC free article] [PubMed]
- 11.Azimi CS, Tang Q, Roybal KT & Bluestone JA NextGen cell-based immunotherapies in cancer and other immune disorders. Curr. Opin. Immunol 59, 79–87 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Goodwin M et al. CRISPR-based gene editing enables FOXP3 gene repair in IPEX patient cells. Sci. Adv 6, eaaz0571 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen DN et al. Polymer-stabilized Cas9 nanoparticles and modified repair templates increase genome editing efficiency. Nat. Biotechnol 38, 44–49 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roth TL et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 559, 405–409 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iyer S et al. Efficient homology-directed repair with circular ssDNA donors. Preprint at bioRxiv 10.1101/864199 (2019). [DOI]
- 16.Feucht J et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med 25, 82–88 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin RM et al. Highly efficient and marker-free genome editing of human pluripotent stem cells by CRISPR-Cas9 RNP and AAV6 donor-mediated homologous recombination. Cell Stem Cell 24, 821–828.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Roth TL et al. Pooled knockin targeting for genome engineering of cellular immunotherapies. Cell 181, 728–744.e4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shy BR, MacDougall MS, Clarke R & Merrill BJ Co-incident insertion enables high efficiency genome engineering in mouse embryonic stem cells. Nucleic Acids Res. 44, 7997–8010 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kiani S et al. Cas9 gRNA engineering for genome editing, activation and repression. Nat. Methods 12, 1051–1054 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vakulskas CA et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med 24, 1216–1224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tatiossian KJ et al. Rational selection of CRISPR-Cas9 guide RNAs for homology-directed genome editing. Mol. Ther 29, 1057–1069 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu YW et al. Dynamics and competition of CRISPR-Cas9 ribonucleoproteins and AAV donor-mediated NHEJ, MMEJ and HDR editing. Nucleic Acids Res 49, 969–985 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wienert B et al. Timed inhibition of CDC7 increases CRISPR-Cas9 mediated templated repair. Nat. Commun 11, 2109 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kath J et al. Pharmacological interventions enhance virus-free generation of TRAC-replaced CAR T cells. Mol. Ther. Methods Clin. Dev 25, 311–330 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou J et al. A first-in-class polymerase theta inhibitor selectively targets homologous-recombination-deficient tumors. Nat. Cancer 2, 598–610 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bousfiha A et al. Human inborn errors of immunity: 2019 update of the IUIS phenotypical classification. J. Clin. Immunol 40, 66–81 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vignoli M et al. CD25 deficiency: a new conformational mutation prevents the receptor expression on cell surface. Clin. Immunol 201, 15–19 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Bezrodnik L, Caldirola MS, Seminario AG, Moreira I & Gaillard MI Follicular bronchiolitis as phenotype associated with CD25 deficiency. Clin. Exp. Immunol 175, 227–234 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lai N et al. Effective and safe treatment of a novel IL2RA deficiency with rapamycin. J. Allergy Clin. Immunol 8, 1132–1135.e4 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Goudy K et al. Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin. Immunol 146, 248–261 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jamee M et al. Clinical, immunological, and genetic features in patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-like syndrome. J. Allergy Clin. Immunol. Pract 8, 2747–2760.e7 (2020). [DOI] [PubMed] [Google Scholar]
- 33.Schubert D et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat. Med 20, 1410–1416 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuehn HS et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science 345, 1623–1627 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwab C et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J. Allergy Clin. Immunol. Pract 142, 1932–1946 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Jong VM et al. Variation in the CTLA4 3′ UTR has phenotypic consequences for autoreactive T cells and associates with genetic risk for type 1 diabetes. Genes Immunity 17, 75–78 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Munshi NC et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. New Engl. J. Med 384, 705–716 (2021). [DOI] [PubMed] [Google Scholar]
- 38.de Vree PJ et al. Targeted sequencing by proximity ligation for comprehensive variant detection and local haplotyping. Nat. Biotechnol 32, 1019–1025 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Terrence D, Davide G, John P & Junghae S AAV manufacturing for clinical use: insights on current challenges from the upstream process perspective. Curr. Opinion Biomed. Eng 20, 100353 (2021). [Google Scholar]
- 40.Wright JF Manufacturing and characterizing AAV-based vectors for use in clinical studies. Gene Ther 15, 840–848 (2008). [DOI] [PubMed] [Google Scholar]
- 41.Lin-Shiao E et al. CRISPR-Cas9-mediated nuclear transport and genomic integration of nanostructured genes in human primary cells. Nucleic Acids Res 50, 1256–1268 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.US National Library of Medicine. Tolerability study of trichostatin A in subjects with relapsed or refractory hematologic malignancies ClinicalTrials.gov https://ClinicalTrials.gov/show/NCT03838926 (2019).
- 43.Damia G Targeting DNA-PK in cancer. Mutation Res 821, 111692 (2020). [DOI] [PubMed] [Google Scholar]
- 44.European Medicines Agency. A phase 1/2 multiple ascending dose study to evaluate the safety, pharmacokinetics and pharmacodynamics of BMS-863233 in subjects with advanced and/or metastatic solid tumors. EU Clinical Trials Register https://www.clinicaltrialsregister.eu/ctr-search/search?query=CA198002 (2009).
- 45.Chen EW, Tay NQ, Brzostek J, Gascoigne NRJ & Rybakin V A dual inhibitor of Cdc7/Cdk9 potently suppresses T cell activation. Front. Immunol 10, 1718 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yeh CD, Richardson CD & Corn JE Advances in genome editing through control of DNA repair pathways. Nat. Cell Biol 21, 1468–1478 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Findlay GM et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature 562, 217–222 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu C et al. Rigid-body ligand recognition drives cytotoxic T-lymphocyte antigen 4 (CTLA-4) receptor triggering. J. Biol. Chem 286, 6685–6696 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wakimoto Y, Jiang J & Wakimoto H Isolation of single-stranded DNA. Curr. Protoc. Mol. Biol 107, 2.15.1–2.15.9 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Mansilla-Soto J et al. HLA-independent T cell receptors for targeting tumors with low antigen density. Nat. Med 28, 345–352 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brinkman EK et al. Easy quantification of template-directed CRISPR/Cas9 editing. Nucleic Acids Res 46, e58 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data been deposited in the Gene Expression Omnibus (GEO) under the accession code GSE202909. ATAC-seq data been deposited in the GEO under the accession code GSE130089. Amplicon sequencing data been deposited in the GEO under the accession codes GSE202596 and GSE203349. Other relevant data are available from the corresponding authors upon reasonable request.