

Abstract
Background and Objectives
We aimed to determine the population-based cumulative incidence and prevalence of developmental and epileptic encephalopathies (DEEs) and intellectual disability and epilepsy (ID+E) in children. We analyzed the cumulative incidence of specific epilepsy syndromes.
Methods
Children younger than 16 years with a DEE or ID+E were ascertained using EEG records from 2000 to 2016 in the Wellington region of New Zealand. Epilepsy syndromes were diagnosed on medical record and EEG review. Point prevalence and cumulative incidence for children with epilepsy and developmental impairment, DEE and ID+E were calculated. Cumulative incidence for each epilepsy syndrome was calculated.
Results
The cohort comprised 235 children (58% male) with developmental impairment and epilepsy, including 152 (65%) with DEE and 83 (35%) with ID+E. The median age of seizure onset was 15.4 months (range day 1–15 years). The median follow-up from seizure onset was 7.9 years (range 0–18.2 years). Point prevalence for the broad group of children with epilepsy and developmental impairment was 175/100,000 children (95% CI 149–203; DEE 112 and ID+E 63/100,000 children). Cumulative incidence for DEE was 169/100,000 children (95% CI 144–199) and that for ID+E was 125/100,000 children (95% CI 95.4–165). Cumulative incidence per 100,000 children was as follows: infantile epileptic spasms syndrome 58.2 (95% CI 45.0–75.3), epilepsy with myoclonic-atonic seizures 16.4 (95% CI 9.69–27.7), Lennox-Gastaut syndrome 13.2 (95% CI 4.1–41.9), and Dravet syndrome 5.1 (95% CI 2.1–12.2). Fifty/152 (33%) of children with DEE and 70/83 (84%) with ID+E could not be diagnosed with a known epilepsy syndrome.
Discussion
Epilepsy and developmental impairment before the age of 16 years occurs in 1 in 340 children, with 1 in 590 having a DEE and 1 in 800 having ID+E. These individuals require significant health and community resources; therefore, these data will inform complex health service and education planning. Epidemiologic studies have focused on early childhood–onset DEEs. These do not fully reflect the burden of these disorders because 27% of DEEs and 70% of ID+E begin later, with seizure onset after the age of 3 years. Understanding the cumulative incidence of specific syndromes together with the broad group of DEEs is essential for the planning of therapeutic trials. Given trials focus on specific syndromes, there is a risk that effective therapies will not be developed for one-third of children with DEE.
Epidemiologic studies have found that 17%–49% of children with epilepsy have intellectual disability (ID),1-3 and conversely, 22% of children with ID have epilepsy.4 Critically, children with epilepsy and ID fall into 2 groups: those with a static ID and epilepsy (ID+E) and children with a developmental and epileptic encephalopathy (DEE). A DEE is defined by the presence of frequent epileptiform activity that causes developmental slowing or regression; most patients have seizures that occur on a background of developmental delay.5 Epidemiologic studies have reported the incidence of DEEs with onset in those younger than 3 years,6-8 but there have been no comprehensive studies of the incidence in childhood and early adolescence. Nor has there been an epidemiologic study comparing the incidence and prevalence of DEEs and ID+E. Furthermore, there is little epidemiologic information regarding the types of seizures and epilepsy syndromes in the population of individuals with coexisting epilepsy and ID.9,10
In this study, we compared the incidence and prevalence of DEEs with those of ID+E in a population-based study of children presenting with epilepsy younger than 16 years. We then analyzed the epilepsy syndromology, epilepsy outcome, developmental course, and mortality.
Methods
Study Background and Patient Ascertainment
This is an epidemiologic study of children in the Wellington region of New Zealand, presenting with epilepsy at an age younger than 16 years and diagnosed with a DEE or ID+E. Standard of care was for all children with seizures to be seen by a pediatrician and referred for an EEG (excluding those with febrile seizures from the age of 6 months to 6 years). In the Wellington region, EEGs were performed in a single department and interpreted by a pediatric epileptologist (L.G.S.). An epilepsy syndrome code was determined using a coding system, which categorized broad epilepsy syndromes during EEG reporting (by L.G.S.), based on clinical information in the referral and the EEG findings. All children with developmental delay or ID were seen by a developmental paediatrician.
We searched for children with developmental impairment with seizures and/or an epileptiform abnormality on EEG using 2 steps (Figure 1).
All EEG reports were searched for epilepsy syndrome code or keywords associated with a DEE or ID (eTable 1, links.lww.com/WNL/C592), and
Any child seen by the child development service who had an EEG.
Figure 1. Case Ascertainment.
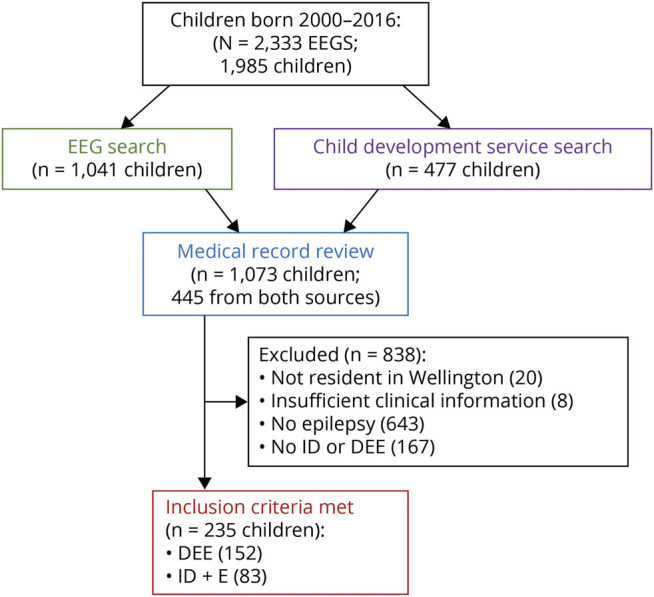
A flow diagram outlining the process used to ascertain the study cohort is shown. DEE = developmental and epileptic encephalopathy; EEG = electroencephalogram; ID = intellectual disability; ID+E = intellectual disability and epilepsy.
Medical records of all 1,073 children were then reviewed in 2020 to determine whether the child had a diagnosis of DEE or ID+E. A DEE diagnosis was made if there was evidence of developmental slowing or regression associated with frequent seizures and/or epileptiform activity on EEG.5 A diagnosis of ID+E was based on a history of epilepsy in children younger than 6 years with global developmental delay. In older children, intellectual disability was diagnosed if they had neurocognitive assessment reporting a full-scale intelligence quotient <70 or, in the absence of formal testing, an assessment of intellectual disability by a pediatrician, developmental pediatrician, or pediatric neurologist. Children were excluded if they had only nonepileptic attacks, had no evidence of development impairment or regression, or were not resident in the Wellington region during their EEG.
Clinical data were collected from the medical records of each child from birth until December 31, 2019. Medical records included clinical information from emergency visits, admissions, discharges, and all medical and allied health clinic letters. Information recorded included age at seizure onset, seizure semiology, seizure types, and seizure frequency and progression; age of developmental concerns and developmental trajectory; antiseizure medication use; investigations including EEG, brain imaging, and metabolic tests; and mortality (using mortality records updated from the Ministry of Health for deaths across New Zealand). Seizure types were classified according to the International League Against Epilepsy (ILAE) criteria,11 based on clinical records and interviews with parents when possible. Detailed developmental history was available for most children from parental reports and developmental assessments by a general or developmental pediatrician. Based on a review of the available clinical and investigational data, a diagnosis of DEE or ID+E and a specific epilepsy syndrome diagnosis was determined independently for each child by 2 authors (L.G.S. and G.P.) using the ILAE classification5 with inclusion of the recently proposed criteria.12,13 In cases where there was doubt, a consensus diagnosis was made by 2 pediatric epileptologists (L.G.S. and I.E.S.). Because children with ID can have seizure-like events that are not epileptic in nature, we took a conservative approach. If the clinical semiological descriptions were incomplete or ambiguous, an epileptic seizure was not diagnosed. DEE with spike-and-wave activation in sleep (DEE-SWAS) and epileptic encephalopathy with spike-and-wave activation in sleep (EE-SWAS) were reported together.
Children with a DEE or ID+E who did not meet the criteria for an epilepsy syndrome were classified with epilepsy based on their seizure types as focal, generalized, or combined. If localization of the seizures was not clear, they were recorded as unclassified. Evolution from one epilepsy syndrome to another was documented. When considering the incidence of each DEE epilepsy syndrome or grouping, both initial and subsequent diagnoses were included.
Epilepsy was considered resolved if the individual remained seizure-free for 10 years, with no antiseizure medication for 5 years, or they had grown past the age of an age-dependent epilepsy syndrome diagnosis, as per the ILAE definition.14
Standard Protocol Approvals, Registrations, and Patient Consents
The study was approved by the Capital and Coast District Health Board Clinical Audit and Research Committee.
Statistical Analysis
Statistical analysis was mainly performed using R 4.0 (R Institute, Vienna, Austria). Specific packages were used for the calculation of prevalence estimates (epiR package15), cumulative incidence based on the denominator framework described below (popEpi package16), and survival analysis (survival analysis package17). OpenEpi (openepi.com) was used to estimate CIs for simple proportions.
Point prevalence for DEE and ID+E was calculated based on patient data as of December 31, 2016. Denominators (person-time at risk) for children in the study region were derived from census data from Stats NZ.18 Censuses were held in 2001, 2006, 2013, and 2018, and population counts for other years were linearly interpolated from these data. The population denominators for the point prevalence calculations were the interpolated number of children aged 0–15 years (inclusive) in 2016.
Cumulative incidence estimates report the probability that an event has occurred before a given time (in this study, age 16 years). The denominator (person-years at risk [PYAR]) for this needed to reflect that cases were sampled based on date of birth (January 1, 2000–December 31, 2016) as best possible. For example, a child born at the very start of 2012 would have contributed 1 year of follow-up data to each year age bracket of 0 to <1, 1 to <2, 2 to <3, and 3 to <4 and no data in older age brackets (after 2016).
Because denominator birth cohort data were not available (with date of birth information), this counting process used census population data to calculate PYAR. Counts of individuals in the population were taken for each year to match the numerator definition (e.g., the calendar year 2000 contributes only denominator information for the first year of life, given that children will only appear in the numerator for that year if born in 2000; calendar year 2001 contributes to 0-to <1-year and 1- to <2-year age brackets; and so on). The process and resulting counts are summarized in eTables 2 and 3 (links.lww.com/WNL/C592) respectively. The PYAR for each age bracket were then summed across all the study years, giving the total number of PYAR of eligible time by 1-year age bracket (total PYAR: 874,222).
Given the even distribution of births across a calendar year, the “leading edge” of this calculation (eTable 3, links.lww.com/WNL/C592) included only half the count for that age group (for example, children born in 2000 only contribute an average of half a year's follow-up time for diagnosis in the 0- to <1-year age bracket; in 2001, those same children will contribute an average of half a year's follow-up to the 0- to <1-year age bracket and half a year's follow-up to the 1- to <2-year age bracket; and so on). This simplifies to taking half of the count for the leading-edge age bracket count and the whole amount of each other age bracket (in the last example, children born in 2001 would contribute half a year of follow-up to the 0- to <1-year age bracket from that year, which is added to the half a year of follow-up in the 0- to <1-year age bracket from those born in 2000).
The cohort ascertainment process meant that younger cases were overrepresented in the sample. This was addressed (where needed) using survival analysis methods (e.g., the Kaplan-Meier estimation) to account for censoring of observations when the outcome was not observed before the end of follow-up (e.g., a child followed up to age 10 years is censored for outcomes from age 10 years onward). These methods were used for survival (from birth) and epilepsy resolution (time from last seizure).
Measures of effect size are reported with a 95% CI and a corresponding α threshold of 0.05. Analyses were conducted for the whole cohort, DEE and ID+E groups, and specific syndromes. Mortality, resolution, and 5-year seizure freedom were plotted and compared between groups using Kaplan-Meier estimates and log-rank tests. Age of first seizure onset and first noted developmental delay are described with medians and compared between DEE and ID+E groups with the nonparametric Mann-Whitney-Wilcoxon test (MWW). Comparison of ID status (mild/moderate vs severe/profound) by epilepsy group and age of seizure onset was conducted using logistic regression, with results reported as odds ratios (ORs) with 95% CI. Two-tailed p values were reported.
Data Availability
Deidentified data that support the findings of this study are available on request from the corresponding author. Data are not publicly available due to the potential that individuals could be identified from their results, given the rarity of many conditions and the limited geographical area of the study.
Results
Ascertainment
The Wellington region of New Zealand contained 98,100 children in the 2018 census, with 108,861 births between 2000 and 2016. Of the children born between January 1, 2000, and December 31, 2016, there were 2,333 EEG studies performed in 1,985 children. The EEG code and keyword search identified 1,509 EEGs in 1,041 children, and 32 additional children were identified through child development service records (Figure 1).
Inclusion criteria were met by 235 children (58% male), of whom 152 children had a DEE (65%) and 83 ID+E (35%). The median age was 13.2 years (mean 13.1, range 3.6–19.9 years) with a median length of follow-up from seizure onset of 7.9 years (mean 8.6 years; range 4 days–18.2 years) (eFigure 1, links.lww.com/WNL/C591).
Point Prevalence
Point prevalence for children younger than 16 years on December 31, 2016 (n = 170) was 175/100,000 children (95% CI 149–203). Fourteen children had died, 6 had resolved epilepsy, 26 had moved out of the area with unknown outcome, and 19 were excluded because they were older than 16 years on that date. Point prevalence of DEE was 112/100,000 children (95% CI 92–135) and that of ID+E was 63/100,000 children (95% CI 48–80). There were more males than females in the ID+E group (OR 1.61; p = 0.03), but not in the DEE group, compared with the general population male:female sex ratio of 1.04 (eTable 4, links.lww.com/WNL/C592).
Cumulative Incidence
The cumulative incidence of the whole cohort with onset at age younger than16 years was 294/100,000 children (95% CI 253–341) (Figure 2A and Table); for DEE, 169/100,000 children (95% CI 144–199) and for ID+E 125/100,000 (95% CI 95.4–165). Focal epilepsy was the most common epilepsy type: DEE (73/152, 48%) and ID+E (50/83 children, 60%) (eFigure 2, links.lww.com/WNL/C591).
Figure 2. Cumulative Incidence of Epilepsy.
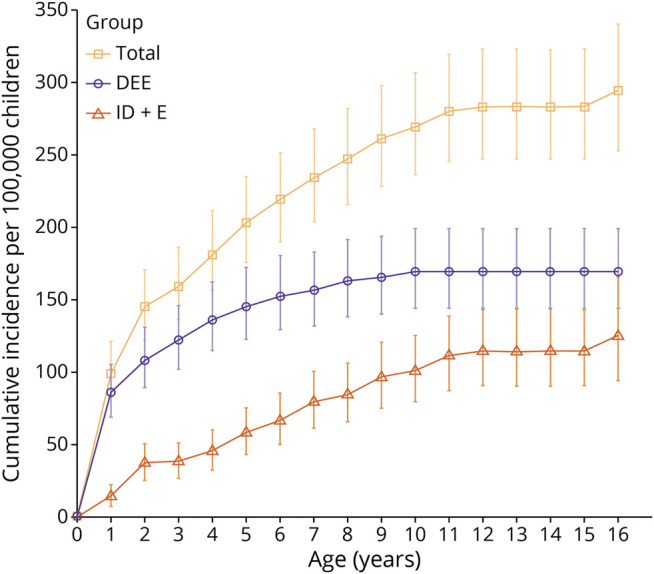
This figure shows the cumulative incidence per year up to 16 years of age, with 95% CIs. Data per 100,000 children is provided for the whole cohort and for individuals with developmental and epileptic encephalopathy and individuals with ID+E. DEE = developmental and epileptic encephalopathy; ID+E = intellectual disability and epilepsy.
Table.
Cumulative Incidence of Epilepsies and Epilepsy Syndromes

Overall, incidence was highest in the first year of life (99.3/100,000 person-years, 95% CI 81.6–121), with DEE accounting for 85.4/100,000 person-years (95% CI 68.3–106) (eFigure 3, links.lww.com/WNL/C591 and eTable 5, links.lww.com/WNL/C592). Although seizures in the DEE cohort began most often in the first year of life (51% of cumulative incident cases up to age 16 years, eTable 5), 13% of incident cases had onset in the second year of life, 9% in the third year, and 27% at ages 3 years or older (oldest 9 years). DEE syndromes presenting at 3 years or older included the following: DEE-SWAS and EE-SWAS (8/33), epilepsy with myoclonic-atonic seizures (EMAtS) (6/33), febrile infection–related epilepsy syndrome (FIRES) (3/33), and 1 child with sleep-related hypermotor epilepsy. Fifteen could not be classified with a known epilepsy syndrome: they had generalized (6/33), focal (6/33), combined (1/33), and unclassified DEEs (2/33).
For each DEE syndrome, we determined the cumulative incidence, age at onset, and syndrome evolution (Figure 3A and Table). Infantile epileptic spasms syndrome (IESS) was the most common syndrome with a cumulative incidence of 58.2/100,000 children (95% CI 45.0–75.3; 1 in 1,700 children), with 48.9/100,000 children (95% CI 37.0–64.7) initially presenting with IESS. For 9 children, IESS developed after other DEEs (Figure 4). For children with IESS, outcomes were assessed at 8 years of age (n = 43). IESS evolved to another syndrome in 27/43 (63%): 18 developed focal epilepsy, 2 Lennox-Gastaut syndrome (LGS), 4 combined DEE, 2 focal DEE, and one generalized DEE. Thirteen/43 (30%) were seizure-free for ≥3 years (median 7 years), and 6 (14%) had died.
Figure 3. Cumulative Incidence of Epilepsy Syndromes.

The cumulative incidence for each epilepsy syndrome in children to age 16 years is provided per 100,000 children with the 95% CIs in yellow. (A) Epilepsy syndromes in individuals with developmental and epileptic encephalopathy epilepsy syndromes. (B) Epilepsy syndromes in individuals with intellectual disability and epilepsy. DEE-SWAS & EE-SWAS = developmental and epileptic encephalopathy with spike-and-wave activation in sleep and epileptic encephalopathy with spike-and-wave activation in sleep; Dravet = Dravet syndrome; EME = early myoclonic encephalopathy; EEM = epilepsy with eyelid myoclonia; EOAE-DEE: early-onset absence epilepsy with DEE; FIRES = febrile infection–related epilepsy syndrome; GS-HH = gelastic seizures with hypothalamic hamartoma; FS+ = febrile seizures plus; GTCS = generalized tonic-clonic seizures; JAE = juvenile absence epilepsy; JME = juvenile myoclonic epilepsy; IESS = infantile epileptic spasms syndrome; LGS = Lennox-Gastaut syndrome; EMAtS = epilepsy with myoclonic-atonic seizures; Ohtahara = Ohtahara syndrome; PCDH19 = PCDH19 clustering epilepsy; SeLEAS = self-limited epilepsy with autonomic seizures; SHE = sleep-related hypermotor epilepsy.
Figure 4. Infantile Epileptic Spasms Syndrome.

This figure shows the syndrome progression and outcomes for children with infantile epileptic spasms syndrome. The left box shows the 9 children who presented with epilepsy before the onset of their infantile epileptic spasms syndrome. The second box shows all 49 children who had infantile epileptic spasms syndrome at some time. The third box shows the outcomes at 8 years of age for the 27 children who had at least 8 years of follow-up data. DEE = developmental and epileptic encephalopathy.
Cumulative incidences for specific DEE syndromes were as follows: EMAtS 16.4/100,000 children (95% CI 9.69–27.7), LGS 13.2/100,000 children (95% CI 4.1–41.9), and Dravet syndrome 5.1/100,000 children (95% CI 2.1–12.2). LGS had a range of onset of 5–14 years in 4 children; evolving from IESS in 2, EMAtS in 1, and generalized DEE in 1. Of importance, 59/152 (39%) children with DEEs did not present with a specific DEE syndrome; 9 evolved into a DEE syndrome (8 to ISS and 1 to LGS), leaving 50/152 (33%) that never had a specific DEE syndrome. Most of them had a focal DEE (30/50, 60%).
The median age at epilepsy onset was later in children with ID+E compared with those with a DEE (46.5 vs 9.0 months, MWW test p < 0.001) (eFigure 4, links.lww.com/WNL/C591). Seventy/83 (84%) children with ID+E did not have a specific epilepsy syndrome. Focal epilepsy was most common (72.5/100,000 children, 95% CI 54.0–97.3, 1 in 1,400 children) (Figure 3B and Table). Notably, 70% of incident cases of ID+E presented from age 3–15 years (eTable 5, links.lww.com/WNL/C592).
Development
Age at which developmental delay was first noted was based on parental and pediatricians' reports. This was slightly older in children with ID+E than those with DEE (12.0 and 7.0 months, respectively, MWW test p = 0.07) (eFigure 4, links.lww.com/WNL/C591). For children with DEE, the age at developmental delay and epilepsy onset was similar, whereas in the ID+E group, developmental delay was more likely to be noted before epilepsy onset (difference in age at developmental delay vs age of first seizure: MWW test comparing ID+E and DEE, p < 0.001).
In the 205 children aged 6 years or older, developmental outcomes were assessed using a formal cognitive assessment in 119 (58%) or clinical history in 86 (42%) (Figure 5A, eTable 6, links.lww.com/WNL/C592). One hundred eighty-three/205 had ID (89%); the remaining 22 all had DEE (14% DEE group) with their ultimate cognition in the normal range despite slowing during their encephalopathy. These individuals had EMAtS (9), IESS (6), focal DEE (3), DEE-SWAS or EE-SWAS (2), gelastic seizures with hypothalamic hamartoma (1), and generalized DEE (1). Individuals with DEEs were 3.7 times more likely to have severe/profound ID compared with mild/moderate ID than those with ID+E (Figure 5A, p < 0.001, OR 3.7, 95% CI 2.0–6.9).
Figure 5. Cognitive Outcomes.

This figure shows the cognitive outcomes for children who reached 6 years or older during the study follow-up period. (A) Comparison of the number of children with normal, mild, moderate, severe, or profound intellectual disability in the cohort between children with DEE and those with ID+E. (B) The cognitive outcomes for children with a DEE by age of seizure onset. (C) The cognitive outcomes for children with ID+E by age of seizure onset. DEE = developmental and epileptic encephalopathy; ID = intellectual disability; ID+E = intellectual disability and epilepsy.
In evaluating the children with DEEs older than 5 years, those with age of epilepsy onset at an age older than 5 years were much less likely to have severe to profound ID than those with epilepsy onset at an age younger than 3 months (OR 0.12, 95% CI 0.02–0.52; Figure 5B). Seventy-two percent (13/18) of children older than 5 years, whose DEE began at an age younger than 3 months, had severe or profound ID. For children with ID+E, 15% (2/13) with epilepsy onset before the age of 12 months had severe or profound ID (Figure 5C).
Epilepsy Resolution
Five-year seizure freedom was more often achieved by children with ID+E (59.1%) than those with DEE (27.2%) (Kaplan-Meier estimate to 18 years after first seizure, log-rank test χ2 = 6.64, p = 0.01) (eFigure 5A, links.lww.com/WNL/C591). Correspondingly, the occurrence of epilepsy resolution was higher in children with ID+E (48.1%) than in those with DEE (15.6%), though the comparison did not meet statistical significance (Kaplan-Meier estimate to 18 years after first seizure, log-rank test χ2 = 3.11, p = 0.08) (eFigure 5B). Of children with DEE, only those with EMAtS or IESS experienced epilepsy resolution. The children with EMAtS resolution all had normal intellect, while the children with IESS resolution had a variable ID (mild to profound). Of children with ID+E, all those with epilepsy resolution had mild ID.
Mortality
During the study period (2000–2019), 23 patients died, corresponding to mortality by the age of 16 years of 13.1% for children with DEE and 11.2% for children with ID+E (Kaplan-Meier estimate to age 16 years, log-rank test χ2 = 1.81, p = 0.18) (eFigure 5C, links.lww.com/WNL/C591). The children with a DEE died at age 6 days to 11 years (Ohtahara syndrome [1], early myoclonic encephalopathy [EME] [1], IESS [4], focal DEE [10], and unclassified DEE [1]). Twelve of 17 had severe to profound ID, 1 mild ID, and 4 (aged younger than 5 years) global developmental delay. The children with ID+E died at age 1–16 years (focal epilepsy [5], unclassified epilepsy [1]). Three/6 had profound ID, one mild ID, and 2 (aged younger than 5 years) global developmental delay.
Discussion
There is an urgent need for epidemiologic data on the incidence and prevalence of children with epilepsy and developmental impairment and, more specifically, DEEs and ID and epilepsy (ID+E). In this study, we present the point prevalence and cumulative incidence data for these disorders in children and adolescents up to 16 years of age (Table). These recently differentiated concepts distinguish epileptic encephalopathies, where the key characteristic is that seizures and/or abnormal interictal EEG activity adversely affect development, from ID+E. The differentiation is important because DEE and ID+E typically have different etiologies, management, and long-term outcomes. We found that 294 in 100,000 children, or 1 in 340, were diagnosed with epilepsy and developmental impairment overall, and more children were diagnosed with a DEE (169/100,000, 1 in 590) than with ID+E (125/100,000, 1 in 800).
To date, epidemiologic studies have underestimated the burden of DEEs because they have been limited to specific age groups and syndromes.6-8,19,20 In this study, we found double the cumulative incidence of the DEEs through childhood and adolescence (until 16 years), when compared with a Scottish study including DEE before the age of 3 years (86.1/100,000 live births; 95% CI 72.7–101.3).6 It is important to note that 27% of our incident DEE cases presented after 3 years of age, which contributed to our higher cumulative incidence. Of interest, the same age group in our study had a higher cumulative incidence of 122/100,000 children (95% CI 102–146), largely due to more children with IESS (58.2/100,000; CI 45.0–75.3), compared with the Scottish study (30.7/100,000; CI 22.9–40.2). Most previous studies examining the incidence of childhood and adolescent-onset epilepsy have provided only a percentage of those with DEEs, rather than definitive incidence and prevalence figures.21-24 While one population-based study of children with epilepsy aged 3–13 years reported a point prevalence for DEE of 60/100,000 children,19 they included only children with a limited number of DEE syndromes and no children without a specific DEE syndrome. Approximately half (72/152) of our DEE cohort would not have been included, using these criteria.
We found that 48.9/100,000 children (1 in 2,000) children presented with IESS as their first type of epilepsy. When we include children whose epilepsy syndrome evolved to IESS by 2 years, our cumulative incidence was 58.2/100,000 (1 in 1,700), in keeping with a meta-analysis of 39 IESS studies reporting an incidence range of 5.6–58.1/100,000 children.25 Our cumulative incidences for other DEE epilepsy syndromes with onset before the age of 3 years, such as Dravet syndrome, Ohtahara syndrome, EME, and PCDH19 clustering epilepsy, are similar to previous reports (Table).6,8,24,26
We found a 3 times higher cumulative incidence for EMAtS (16.4/100,000 children; 95% CI 9.69–27.7; 1 in 6,100) compared with that of the previous study of children younger than 3 years (5.3/100,000 live births; 95% CI 2.4–10.1).6 Our median age of onset was 2.9 years (range 0–4 years), which explains our higher incidence. If we include only children presenting under 3 years, our results are comparable (8.9/100,000 children, 95% CI 4.4–17.7).
Of interest, our cumulative incidence for LGS (13.2/100,000, 1 in 7,600 children) was half of that reported in a previous study (Table).27 We diagnosed LGS based on the triad of the following: (1) multiple seizure types including tonic seizures, (2) cognitive impairment, and (3) slow spike-wave and paroxysmal fast activity in slow sleep.12 There has been considerable debate over many years regarding the definition of LGS,28-30 often referring to any drug-resistant child with multiple seizure types, cognitive impairment, and an epileptiform EEG. Studies included patients with other DEEs, such as EMAtS (see earlier) and Dravet syndrome.31 This means that while LGS is a prototypic DEE, it has been historically overdiagnosed because of imprecise use of diagnostic criteria.
Our analysis showed that 27% of children with DEEs present between 3 and 16 years of age. Half of our cohort in this age range had well-recognized DEE syndromes (DEE-SWAS or EE-SWAS 8; EMAtS 6; and FIRES 3), while the remainder could not be classified into a specific syndrome. The group of DEE-SWAS and EE-SWAS had the highest cumulative incidence (12.8/100,000 children, 1 in 7,800). DEE-SWAS and EE-SWAS incorporate Landau-Kleffner syndrome (LKS) and epileptic encephalopathy with continuous spike-and-wave during sleep.12 The only reported data for this group of syndromes is a prevalence estimate for LKS, which was considerably lower (1/300,000–410,000 children from ages 5 to 19 years) and likely underestimated the prevalence because of its questionnaire-based methodology.32
A key point to emerge regarding the epidemiology of the DEEs is that many children cannot be classified into a known epilepsy syndrome. Sixty percentage (30/50) of our nonclassifiable children had very active multifocal epileptiform discharges, typically with multiple types of focal seizures. Although cumulative incidence was not studied, previous reports of epilepsy have also noted that many children with DEEs could not be classified.7,21,22 This highlights a critical issue because there is an increasing focus by regulatory agencies on the development of therapies for specific DEE epilepsy syndromes. This approach excludes a large number of patients with DEE who do not fall into a specific syndrome and are therefore unable to access trials and later established therapies despite their devastating diseases.
Our study differentiated between children with DEE and ID+E, which has not been performed in previous studies of children with epilepsy and developmental impairment.1,2,4,9,10 In infants younger than 3 months of age with severely impaired development, it can be difficult to distinguish whether the child has a pure epileptic encephalopathy or whether their abnormal development was due to their underlying etiology. There were 30 children in our study who had seizure onset at or before the age of 3 months, with 19 in the first month. We classified them as having a DEE if they had frequent seizures and an epileptiform EEG associated with developmental plateau or regression. In this study, we provide the first cumulative incidence for ID+E (125/100,000 children, 1 in 800), excluding children with DEEs. Eighty-four percentage (70/83) of children did not have a specific epilepsy syndrome, with 59% having unclassified focal epilepsy. In our cohort, the median age of seizure onset was considerably later in children with ID+E (46.5 months) than in those with DEEs (9 months). We acknowledge this age difference may be an underestimate because the ID+E group is more likely than the DEE group to have seizure onset after the age of 15 years, which was not captured in our series.
While severe or profound ID was over 3 times more frequent in children with a DEE than ID+E, we found no difference in the mortality rate between both groups. Most children who died had severe or profound ID, as previously reported.33 Our estimate of mortality before 16 years of age of 13% for DEE was lower than the 21% previously reported.22 This difference may relate to the previous study having a longer observed period (up to 20 years, median follow-up of 16 years) and their result being an absolute number of deaths in their cohort (12/58, 21%) rather than an estimate of mortality at a set age. Our study had a larger number of children with DEE (152 compared with 58), which affects the statistical precision of the mortality estimate, and our analysis accounted for the fact that children had different amounts of follow-up time (i.e., censoring of observations).
Strengths of our study include our relatively complete ascertainment, inclusion of all children presenting with seizure up to 16 years of age, and our detailed phenotypic analysis. The analyses allowed for robust estimates for incidence and outcomes (e.g., mortality, resolution of epilepsy) at 16 years, accounting for censoring of observations. Our larger cohort meant we could determine the cumulative incidences of epilepsy syndromes not previously ascertained.
Conversely, a potential weakness for the ID+E group relates to the ascertainment methodology (EEGs). An EEG may not be performed in this group of children for several reasons: seizures may not be recognized, particularly in children with severe or profound ID; a child may not be able to tolerate an EEG; or seizures may be infrequent and not considered of importance. The use of EEGs for ascertainment also means that our estimates of point prevalence will be conservative because children who moved into the area after they developed epilepsy and did not have an EEG in Wellington were not ascertained.
Given the recent differentiation of the DEEs from ID+E by the ILAE,5 there is a pressing need for clear epidemiologic data to inform trial design, health economic analysis, family burden, and outcome data. In this study, we provided the first cumulative incidence for DEEs with onset before the age of 16 years, finding double the incidence reported in early childhood DEEs beginning before the age of 3 years6 We highlighted that both DEEs and ID+E are not rare, affecting 1 in 590 and 1 in 800 children, respectively. Given the huge burden that these diseases pose for families and the global community, there is a desperate need for this knowledge to advocate for greater resources and novel therapies.
Acknowledgment
The authors thank the families for participating in their research.
Glossary
- DEE
developmental and epileptic encephalopathy
- DEE-SWAS
DEE with spike-and-wave activation in sleep
- EE-SWAS
epileptic encephalopathy with spike-and-wave activation in sleep
- EEM
epilepsy with eyelid myoclonia
- EME
early myoclonic encephalopathy
- EMAtS
epilepsy with myoclonic-atonic seizures
- FIRES
febrile infection–related epilepsy syndrome
- FS+
febrile seizures plus
- GTCS
generalized tonic-clonic seizures
- ID
intellectual disability
- ID+E
ID and epilepsy
- IESS
Infantile epileptic spasms syndrome
- ILAE
International League Against Epilepsy
- JAE
juvenile absence epilepsy
- JME
juvenile myoclonic epilepsy
- LKS
Landau-Kleffner syndrome
- LGS
Lennox-Gastaut syndrome
- MWW
Mann-Whitney-Wilcoxon
- OR
odds ratio
- PYAR
person-years at risk
- SeLEAS
self-limited epilepsy with autonomic seizures
Appendix. Authors
Study Funding
This study was funded by the Health Research Council of New Zealand.
Disclosure
G. Poke receives funding from the Health Research Council of New Zealand; J. Stanley receives funding from the Health Research Council of New Zealand; I.E. Scheffer has served on scientific advisory boards for BioMarin, Chiesi, Eisai, Encoded Therapeutics, GlaxoSmithKline, Knopp Biosciences, Nutricia, Rogcon, Takeda Pharmaceuticals, UCB, and Xenon Pharmaceuticals. Scheffer has received speaker honoraria from GlaxoSmithKline, UCB, BioMarin, Biocodex, Chiesi, Liva Nova, and Eisai; has received funding for travel from UCB, Biocodex, GlaxoSmithKline, Biomarin, and Eisai; has served as an investigator for Anavex Life Sciences, Cerecin Inc, Cereval Therapeutics, Eisai, Encoded Therapeutics, EpiMinder Inc, Epygenyx, ES-Therapeutics, GW Pharma, Marinus, Neurocrine BioSciences, Ovid Therapeutics, Takeda Pharmaceuticals, UCB, Ultragenyx, Xenon Pharmaceutical, Zogenix, and Zynerba; has consulted for Atheneum Partners, Care Beyond Diagnosis, Epilepsy Consortium, Ovid Therapeutics, UCB, and Zynerba Pharmaceuticals; is a Nonexecutive Director of Bellberry Ltd and a Director of the Australian Academy of Health and Medical Sciences and the Australian Council of Learned Academies Limited; may accrue future revenue on pending patent WO61/010,176 (filed: 2008): Therapeutic Compound; has a patent for SCN1A testing held by Bionomics Inc and licensed to various diagnostic companies; and has a patent molecular diagnostic/theranostic target for benign familial infantile epilepsy (BFIE) [PRRT2] 2011904493 & 2012900190 and PCT/AU2012/001,321 (TECH ID:2012-009); L.G. Sadleir receives funding from the Health Research Council of New Zealand and Cure Kids New Zealand; is a consultant for the Epilepsy Consortium; has received travel grants from Seqirus and Nutricia, has received research grants and consultancy fees from Zynerba Pharmaceuticals; and has served on an Eisai Pharmaceuticals scientific advisory panel. Go to Neurology.org/N for full disclosures.
References
- 1.Berg AT, Langfitt JT, Testa FM, et al. Global cognitive function in children with epilepsy: a community-based study. Epilepsia. 2008;49(4):608-614. doi: 10.1111/j.1528-1167.2007.01461.x [DOI] [PubMed] [Google Scholar]
- 2.Aaberg KM, Bakken IJ, Lossius MI, et al. Comorbidity and childhood epilepsy: a Nationwide Registry Study. Pediatrics. 2016;138(3):e20160921. doi: 10.1542/peds.2016-0921 [DOI] [PubMed] [Google Scholar]
- 3.Stodberg T, Tomson T, Anderlid BM, et al. Outcome at age 7 of epilepsy presenting in the first 2 years of life. A population-based study. Epilepsia. 2022; 63(8):2096-2107. doi: 10.1111/epi.17314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oeseburg B, Dijkstra GJ, Groothoff JW, Reijneveld SA, Jansen DEMC. Prevalence of chronic health conditions in children with intellectual disability: a systematic literature review. Intellect Dev Disabil. 2011;49(2):59-85. doi: 10.1352/1934-9556-49.2.59 [DOI] [PubMed] [Google Scholar]
- 5.Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia. 2017;58(4):512-521. doi: 10.1111/epi.13709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Symonds JD, Elliott KS, Shetty J, et al. Early childhood epilepsies: epidemiology, classification, aetiology, and socio-economic determinants. Brain. 2021;144(9):2879-2891. doi: 10.1093/brain/awab162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ware TL, Huskins SR, Grinton BE, et al. Epidemiology and etiology of infantile developmental and epileptic encephalopathies in Tasmania. Epilepsia Open. 2019;4(3):504-510. doi: 10.1002/epi4.12350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Howell KB, Freeman JL, Mackay MT, et al. The severe epilepsy syndromes of infancy: a population-based study. Epilepsia. 2021;62(2):358-370. doi: 10.1111/epi.16810 [DOI] [PubMed] [Google Scholar]
- 9.Nath K, Naskar S. A clinical study on seizure disorder in intellectually disabled patients in Barak Valley, North-Eastern India. Open J Psychiatry Allied Sci. 2016;7(1):46-53. doi: 10.5958/2394-2061.2015.00023.3 [DOI] [Google Scholar]
- 10.Eriksson K, Erilä T, Kivimäki T, Koivikko M. Evolution of epilepsy in children with mental retardation: five-year experience in 78 cases. Am J Ment Retard. 1997;102(5):464-472. doi: [DOI] [PubMed] [Google Scholar]
- 11.Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the International League Against Epilepsy: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):522-530. doi: 10.1111/epi.13670 [DOI] [PubMed] [Google Scholar]
- 12.Specchio N, Wirrell EC, Scheffer IE, et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: position paper by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022;63(6):1398-1442. doi: 10.1111/epi.17241 [DOI] [PubMed] [Google Scholar]
- 13.Zuberi SM, Wirrell E, Yozawitz E, et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022;63(6):1349-1397. doi: 10.1111/epi.17239 [DOI] [PubMed] [Google Scholar]
- 14.Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55(4):475-482. doi: 10.1111/epi.12550 [DOI] [PubMed] [Google Scholar]
- 15.epiR: tools for the analysis of epidemiological data. R package version 2.0.26 [computer program]. 2021. CRAN.R-project.org/package=epiR. [Google Scholar]
- 16.popEpi: functions for epidemiological analysis using population data. R package version 0.4.8 [computer program]. 2019. CRAN.R-project.org/package=popEpi. [Google Scholar]
- 17.A package for survival analysis in R. R package version 3.2-3 [computer program]. 2020. CRAN.R-project.org/package=survival. [Google Scholar]
- 18.Statistics New Zealand. Stats NZ Infoshare. Published 2021. Accessed August 8, 2021. infoshare.stats.govt.nz/.
- 19.Aaberg KM, Suren P, Soraas CL, et al. Seizures, syndromes, and etiologies in childhood epilepsy: the International League Against Epilepsy 1981, 1989, and 2017 classifications used in a population-based cohort. Epilepsia. 2017;58(11):1880-1891. doi: 10.1111/epi.13913 [DOI] [PubMed] [Google Scholar]
- 20.Stödberg T, Tomson T, Barbaro M, et al. Epilepsy syndromes, etiologies, and the use of next-generation sequencing in epilepsy presenting in the first 2 years of life: a population-based study. Epilepsia. 2020;61(11):2486-2499. doi: 10.1111/epi.16701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Camfield P, Camfield C. Long-term prognosis for symptomatic (secondarily) generalized epilepsies: a population-based study. Epilepsia. 2007;48(6):1128-1132. doi: 10.1111/j.1528-1167.2007.01072.x [DOI] [PubMed] [Google Scholar]
- 22.Berg AT, Levy SR, Testa FM. Evolution and course of early life developmental encephalopathic epilepsies: focus on Lennox–Gastaut syndrome. Epilepsia. 2018;59(11):2096-2105. doi: 10.1111/epi.14569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eltze CM, Chong WK, Cox T, et al. A population-based study of newly diagnosed epilepsy in infants. Epilepsia. 2013;54(3):437-445. doi: 10.1111/epi.12046 [DOI] [PubMed] [Google Scholar]
- 24.Symonds JD, Zuberi SM, Stewart K, et al. Incidence and phenotypes of childhood-onset genetic epilepsies: a prospective population-based national cohort. Brain. 2019;142(8):2303-2318. doi: 10.1093/brain/awz195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jia JL, Chen S, Sivarajah V, Stephens D, Cortez MA. Latitudinal differences on the global epidemiology of infantile spasms: systematic review and meta-analysis. Orphanet J Rare Dis. 2018;13(1):216. doi: 10.1186/s13023-018-0952-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu YW, Sullivan J, McDaniel SS, et al. Incidence of Dravet syndrome in a US population. Pediatrics. 2015;136(5):e1310–1315. doi: 10.1542/peds.2015-1807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rantala H, Putkonen T. Occurrence, outcome, and prognostic factors of infantile spasms and Lennox–Gastaut syndrome. Epilepsia. 1999;40(3):286-289. doi: 10.1111/j.1528-1157.1999.tb00705.x [DOI] [PubMed] [Google Scholar]
- 28.Arzimanoglou A, French J, Blume WT, et al. Lennox–Gastaut syndrome: a consensus approach on diagnosis, assessment, management, and trial methodology. Lancet Neurol. 2009;8(1):82-93. doi: 10.1016/s1474-4422(08)70292-8 [DOI] [PubMed] [Google Scholar]
- 29.Crespel A, Gelisse P, Macorig G, Nikanorova M, Ferlazzo E, Genton P. Lennox–Gastaut syndrome. In: Bureau M, Genton P, Delgado-Escueta A, Dravet C, et al., eds. Epileptic Syndromes in Infancy, Childhood and Adolescence. John Libbey; 2019:711-808. [Google Scholar]
- 30.Heiskala H. Community-based study of Lennox–Gastaut syndrome. Epilepsia. 1997;38(5):526-531. doi: 10.1111/j.1528-1157.1997.tb01136.x [DOI] [PubMed] [Google Scholar]
- 31.Epi4K Consortium, Epilepsy Phenome/Genome Project, Allen AS, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501(7466):217-221. doi: 10.1038/nature12439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaga M, Inagaki M, Ohta R. Epidemiological study of Landau–Kleffner syndrome (LKS) in Japan. Brain Dev. 2014;36(4):284-286. doi: 10.1016/j.braindev.2013.04.012 [DOI] [PubMed] [Google Scholar]
- 33.Sillanpaa M, Saarinen MM, Karrasch M, Schmidt D, Hermann BP. Neurocognition in childhood epilepsy: impact on mortality and complete seizure remission 50 years later. Epilepsia. 2019;60(1):131-138. doi: 10.1111/epi.14606 [DOI] [PubMed] [Google Scholar]
- 34.Hunter MB, Yoong M, Sumpter RE, et al. Incidence of early-onset epilepsy: a prospective population-based study. Seizure. 2020;75:49-54. doi: 10.1016/j.seizure.2019.12.020 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Deidentified data that support the findings of this study are available on request from the corresponding author. Data are not publicly available due to the potential that individuals could be identified from their results, given the rarity of many conditions and the limited geographical area of the study.