

Abstract
Objectives:
Early-onset colorectal cancer diagnosed before age 50 has been increasing. Likely reflecting the pathogenic role of the intestinal microbiome, which gradually changes across the entire colorectal length, the prevalence of certain tumor molecular characteristics gradually changes along colorectal subsites. Understanding how colorectal tumor molecular features differ by age and tumor location is important in personalized patient management.
Methods:
Using 14,004 colorectal cancer cases including 3,089 early-onset cases, we examined microsatellite instability (MSI), CpG island methylator phenotype (CIMP), and KRAS and BRAF mutations in carcinomas of the cecum, ascending colon, transverse colon, descending colon, sigmoid colon, and rectum, and compared early-onset cases to later-onset cases.
Results:
The proportions of MSI-high, CIMP-high, and BRAF-mutated early-onset tumors were lowest in the rectum (8.8%, 3.4%, and 3.5%, respectively) and highest in the ascending colon (46% MSI-high; 15% CIMP-high) or transverse colon (8.6% BRAF-mutated) (all Ptrend <0.001 across the rectum to ascending colon). Compared to later-onset tumors, early-onset tumors showed higher prevalence of MSI-high status and lower prevalence of CIMP-high status and BRAF mutations in most subsites. KRAS mutation prevalence was higher in the cecum compared to the other subsites in both early-onset and later-onset tumors (P <0.001). Notably, later-onset MSI-high tumors showed a continuous decrease in KRAS mutation prevalence from the rectum (36%) to ascending colon (9%; Ptrend <0.001) followed by an increase in the cecum (14%), while early-onset MSI-high cancer showed no such trend.
Conclusion:
Our findings support biogeographical and pathogenic heterogeneity of colorectal carcinomas in different colorectal subsites and age groups.
Keywords: colorectal continuum, colorectal neoplasm, epigenetics, mismatch repair, molecular pathological epidemiology
Introduction
There is a growing concern on early-onset colorectal cancer diagnosed before age 50, incidence of which has increased worldwide since around 1990 (1). Evidence suggests that, compared to later-onset cases, early-onset colorectal cancers occur more frequently in rectal location and less frequently in proximal colon location (2). Studies also indicate possible heterogeneity of molecular characteristics between early-onset and later-onset colorectal cancers (1–3). For instance, compared to later-onset cases, early-onset colorectal cancers are more commonly microsatellite instability (MSI)-high and less commonly CpG island methylator phenotype (CIMP)-high and BRAF-mutated (3–10). Considering these findings, it is of particular interest to examine molecular pathology of early-onset colorectal cancers in comparison to later-onset tumors according to tumor location.
Colorectal cancer consists of biologically heterogeneous neoplasms with differing sets of genetic and epigenetic alterations, influenced by the microbiome and immune system (10, 11) which may at least partly explain variable tumoral characteristics according to tumor location (12, 13). Despite the pathophysiological importance of luminal contents and the intestinal microbiota (which gradually change along the colorectal length) (14), numerous studies have used a dichotomy model of proximal (right-sided) vs. distal (left-sided) colorectum, and have shown the associations of proximal tumors with high-level MSI, high-level CIMP, and BRAF mutations (15–19). In contrast, fewer studies have examined tumor molecular features according to more detailed colorectal segments (with somewhat limited case numbers in each subsite) (12, 20–22). Likely reflecting the pathogenic role of the intestinal microbiome, which gradually changes across the entire colorectal length (14), it is conceivable that the prevalence of certain tumor molecular features of early-onset colorectal cancer might gradually change along colorectal subsites. However, molecular features of early-onset colorectal cancer according to detailed sublocations remain to be studied.
This consortium pooled analysis was conducted to test our hypotheses that the prevalence of major molecular features in early-onset colorectal cancer might change along detailed colorectal locations, and that the trend might differ from that of late-onset colorectal cancer. In addition, previous studies showed that cecal cancer had higher prevalence of KRAS-mutated tumors than all other colorectal subsites (12, 20). Hence, another hypothesis was that the association of cecal cancer with KRAS mutations might be different between early-onset and later-onset colorectal cancers. We utilized 14,004 colorectal cancer cases including, 3,089 early-onset cases, derived from The Cancer Genome Atlas (TCGA), and participating studies in the Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO).
Methods
Study Population
We use the term “early-onset” for colorectal cancer diagnosed before age 50 years and the contrasting term “later-onset” for colorectal cancer diagnosed at or after age 50 years. We pooled data for 14,004 cases of colorectal cancer with available data on tumor location and tumor molecular characteristics from the following 12 studies: the Colon Cancer Family Registry (CCFR), the Cancer Prevention Study II (CPS-II) (23), the German Darmkrebs: Chancen der Verhütung durch Screening Study (DACHS) (24), the Diet, Activity and Lifestyle Study (DALS) (25), the Early Detection Research Network (EDRN) (26), the European Prospective Investigation into Cancer - Sweden (EPIC_Sweden) (27), the Health Professionals Follow-up Study (HPFS) (28), the Melbourne Collaborative Cohort Study (MCCS) (29), the Newfoundland Familial Colorectal Cancer Registries (NFCCR) (30), the Nurses’ Health Study (NHS) (31), the Northern Sweden Health and Disease Study (NSHDS) (32), and the Cancer Genome Atlas (TCGA) (33). These studies, except TCGA, have participated in the Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO). All study participants provided informed consent, and each study was approved by their relevant research ethics committee or institutional review board. Details of the studies were described in the previous publication from this consortium (34). All colorectal cancer cases included in each study were confirmed and clinical and pathological data were extracted through review of medical records, pathological reports, and/or death certificates. Tumor location data was recorded using International Classification of Disease (ICD) across studies. To harmonize the tumor location data, we included the hepatic flexure into the transverse colon, the splenic flexure into the descending colon, and the rectosigmoid junction into the rectum. Hence, we examined six anatomical subsites (the cecum, ascending colon, transverse colon, descending colon, sigmoid colon, and rectum).
Table 1 lists pertinent clinical and pathological features in the combined dataset. Descriptive characteristics of colorectal cancer cases in each study are shown in Supplementary Table 1. In this study, patients with at least one biological parent or sibling affected with colorectal cancer (at least by self-report) were considered to have positive (present) colorectal cancer family history. As a pooled analysis, in many of the included studies, we could not obtain further information on colorectal or other cancers in family members, including age of cancer diagnosis, cancers of non-colorectal organs, and the number of affected family members.
Table 1.
Patient Characteristics of Colorectal Cancer According to Primary Tumor Location
| Clinical and molecular feature* | Total N | Cecum | Ascending colon | Transverse colon | Descending colon | Sigmoid colon | Rectum | P value |
|---|---|---|---|---|---|---|---|---|
| All cases | 14004 | 2142 | 1962 | 1390 | 1039 | 3310 | 4161 | |
| Sex | <0.001† | |||||||
| Female | 6712 (48%) | 1129 (53%) | 1088 (55%) | 704 (51%) | 505 (49%) | 1559 (47%) | 1727 (42%) | |
| Male | 7292 (52%) | 1013 (47%) | 874 (45%) | 686 (49%) | 534 (51%) | 1751 (53%) | 2434 (58%) | |
| Mean age ± SD | 61.4±13.1 | 63.4±12.8 | 64.2±13.3 | 62.7±13.6 | 60.2±13.3 | 61.2±12.3 | 58.9±13.0 | <0.001† |
| Age group | <0.001† | |||||||
| <50 (early-onset) | 3089 (22%) | 367 (17%) | 327 (17%) | 271 (19%) | 264 (25%) | 687 (21%) | 1173 (28%) | |
| Mean age ± SD | 42.4±6.0 | 42.5±5.8 | 41.6±6.7 | 41.3±6.8 | 42.3±6.3 | 42.9±5.8 | 42.7±5.6 | <0.001† |
| ≥50 (later-onset) | 10915 (78%) | 1775 (83%) | 1635 (83%) | 1119 (81%) | 775 (75%) | 2623 (79%) | 2988 (72%) | |
| Mean age ± SD | 66.7±8.9 | 67.7±9.0 | 68.7±9.0 | 67.9±9.0 | 66.3±8.9 | 66.0±8.4 | 65.2±9.0 | <0.001† |
| Year of diagnosis | <0.001† | |||||||
| Before 1995 | 2218 (16%) | 413 (20%) | 318 (17%) | 260 (19%) | 196 (19%) | 673 (21%) | 358 (8.9%) | |
| 1995–2000 | 5721 (42%) | 943 (45%) | 740 (39%) | 588 (43%) | 417 (41%) | 1325 (41%) | 1708 (42%) | |
| After 2000 | 5725 (42%) | 736 (35%) | 860 (45%) | 522 (38%) | 401 (40%) | 1235 (38%) | 1971 (49%) | |
| Family history of colorectal cancer | <0.001† | |||||||
| Absent | 10273 (76%) | 1553 (75%) | 1359 (72%) | 968 (72%) | 734 (73%) | 2504 (78%) | 3155 (79%) | |
| Present | 3222 (24%) | 512 (25%) | 522 (28%) | 375 (28%) | 269 (27%) | 690 (22%) | 854 (21%) | |
| AJCC disease stage | <0.001† | |||||||
| I | 3279 (25%) | 469 (23%) | 433 (23%) | 269 (21%) | 192 (19%) | 839 (28%) | 1077 (29%) | |
| II and III | 8228 (63%) | 1311 (65%) | 1247 (67%) | 895 (69%) | 671 (68%) | 1827 (60%) | 2277 (60%) | |
| IV | 1457 (11%) | 248 (12%) | 173 (9.3%) | 136 (10%) | 127 (13%) | 361 (12%) | 412 (11%) | |
| MSI status | <0.001‡ | |||||||
| Non-MSI-high | 10903 (83%) | 1440 (72%) | 1156 (62%) | 848 (67%) | 829 (84%) | 2942 (94%) | 3688 (95%) | |
| MSI-high | 2193 (17%) | 570 (28%) | 695 (38%) | 418 (33%) | 154 (16%) | 179 (5.7%) | 177 (4.6%) | |
| CIMP status | <0.001‡ | |||||||
| Low/negative | 7926 (84%) | 1103 (72%) | 856 (62%) | 680 (70%) | 610 (90%) | 2120 (94%) | 2557 (95%) | |
| High | 1561 (16%) | 422 (28%) | 532 (38%) | 286 (30%) | 66 (9.8%) | 128 (5.7%) | 127 (4.7%) | |
| BRAF | <0.001‡ | |||||||
| Wild-type | 10957 (89%) | 1549 (82%) | 1297 (74%) | 946 (79%) | 828 (92%) | 2791 (95%) | 3546 (97%) | |
| Mutant | 1396 (11%) | 346 (18%) | 454 (26%) | 254 (21%) | 73 (8.1%) | 145 (4.9%) | 124 (3.4%) | |
| KRAS | 0.69‡ | |||||||
| Wild-type | 6710 (67%) | 874 (55%) | 957 (67%) | 699 (71%) | 498 (67%) | 1727 (70%) | 1955 (68%) | |
| Mutant | 3369 (33%) | 710 (45%) | 479 (33%) | 282 (29%) | 248 (33%) | 726 (30%) | 924 (32%) | |
| Tumor subtype§ | ||||||||
| Type 1 | 512 (7.5%) | 157 (15%) | 222 (24%) | 91 (15%) | 17 (3.4%) | 23 (1.3%) | 2 (0.1%) | <0.001‡ |
| Type 2 | 219 (3.2%) | 50 (4.8%) | 74 (8.0%) | 40 (6.8%) | 14 (2.8%) | 29 (1.6%) | 12 (0.6%) | <0.001‡ |
| Type 3 | 2193 (32%) | 438 (42%) | 286 (31%) | 158 (27%) | 169 (34%) | 508 (29%) | 634 (32%) | 0.21‡ |
| Type 4 | 3663 (54%) | 330 (31%) | 268 (29%) | 268 (45%) | 281 (56%) | 1183 (67%) | 1333 (66%) | <0.001‡ |
| Type 5 | 257 (3.8%) | 77 (7.3%) | 78 (8.4%) | 34 (5.8%) | 19 (3.8%) | 21 (1.2%) | 28 (1.4%) | <0.001‡ |
Percentage indicates the proportion of patients with a specific patient characteristic among all patients or in strata of tumor location (cecum, ascending colon, transverse colon, descending colon, sigmoid colon, and rectum).
To compare categorical data between subgroups classified by the tumor location, the chi-square test was performed. To compare continuous variables, an analysis of variance was performed.
P value was calculated by the linear trend test across the ordinal categories of subsite location variable (from the rectum to ascending colon) in the multivariable logistic regression model adjusted for sex, family history of colorectal cancer (present vs. absent), and study (i.e., cohort).
Tumor subtypes described by Jass(38) as follows: Type 1 = MSI-high, CIMP-high, BRAF mutant, KRAS wild-type; Type 2 = non-MSI-high, CIMP-high, BRAF mutant, KRAS wild-type; Type 3 = non-MSI-high, CIMP-low/negative, BRAF wild-type, KRAS mutant; Type 4 = non-MSI-high, CIMP-low/negative, BRAF wild-type, KRAS wild-type; Type 5 = MSI-high, CIMP-low/negative, BRAF wild-type, KRAS wild-type.
Abbreviations: AJCC, American Joint Committee on Cancer; CIMP, CpG island methylator phenotype; MSI, microsatellite instability.
Evaluation of Tumor Molecular Characteristics
Molecular marker testing for MSI, CIMP, BRAF, and KRAS statuses was conducted by each study according to individual study protocols (18, 35) Details of methods and references for tumor molecular testing are described in Supplementary Materials and Supplementary Tables 2 and 3. A small fraction of the cohorts used loss of mismatch repair proteins (MLH1, MSH2, MSH6, and/or PMS2) as an acceptable surrogate of MSI-high status; we use the standardized nomenclature of genes and gene products as recommended by the expert panel (36). We compared results of MSI, BRAF, and KRAS statuses from each study with results from centralized targeted sequencing. The tumor classifications from these two approaches were highly (more than 90%) concordant (37).
We also defined tumor subtypes according to the Jass classification scheme (18, 38) as follows: Type 1 (MSI-high, CIMP-high, BRAF mutant, KRAS wild-type); Type 2 (non-MSI-high, CIMP-high, BRAF mutant, KRAS wild-type); Type 3 (non-MSI-high, CIMP-nlow/egative, BRAF wild-type, KRAS-mutant); Type 4 (non-MSI-high, CIMP-low/negative, BRAF wild-type, KRAS wild-type); Type 5 (MSI-high, CIMP-low/negative, BRAF wild-type, KRAS wild-type).
Statistical Analyses
All statistical analyses were conducted using STATA software (version 15.1, Stata Corporation, College Station, TX), and all P values were two-sided. We compared the prevalence (proportion among colorectal carcinoma cases) of a given molecular subtype (MSI-high, CIMP-high, BRAF-mutated, KRAS-mutated, or each Jass subtype) in different colorectal subsites.
Our primary hypothesis testing was an assessment of a statistically significant trend in the prevalence of MSI-high, CIMP-high, or BRAF-mutated tumors along the detailed colorectal sublocations in early-onset and later-onset colorectal cancer. We calculated the multivariable-adjusted odds ratio (OR) of molecular marker positivity (with its corresponding Ptrend) for one-subsite-unit increase from the rectum to ascending colon [the ordinal categories of the rectum (1) to ascending colon (5)] in a logistic regression model with a given molecular marker as an outcome variable, adjusted for sex (female vs. male), family history of colorectal cancer (present vs. absent), and study (i.e., cohort). Missing values for family history of colorectal cancer (N=509) were treated as separate indicator variables in the logistic regression model. We also tested another primary hypothesis that the trend of molecular marker prevalence from the rectum to ascending colon differed between early-onset and later-onset colorectal cancers. We used the Wald test for an interaction term between the subsite variable (ordinal; the rectum to ascending colon) and age (<50 vs. ≥50). Because there were 12 primary hypotheses (four marker trends in each of early-onset and later-onset groups and an interaction test between each marker (out of four markers) and age groups, we used the stringent two-sided α level of 0.005 (≒ 0.05/12) considering multiple comparisons (39).
In secondary analyses, we assessed the difference in KRAS mutation prevalence between cecum and other subsites, using multivariable logistic regression models (with the cecum location variable; yes vs. no) that adjusted for sex (female vs. male), family history of colorectal cancer (present vs. absent), and study (i.e., cohort). Other secondary analyses included comparisons of the proportions of molecular alterations between early-onset and later-onset cases (by the chi-square tests) in selected colorectal subsites. We also assessed the relationships of the tumor location with each of the other variables listed in Table 1, using the chi-square tests (or analysis of variance assuming equal variances for continuous variables).
Results
Table 1 summarizes the characteristics of 14,004 colorectal cancer cases including 3,089 early-onset (diagnosed before age 50) and 10,915 later-onset patients (diagnosed at or after age 50) according to primary tumor location. The proportion of rectal cancer was higher in early-onset cases (38%) than later-onset cases (27%).
We examined statuses of microsatellite instability (MSI), CpG island methylator phenotype (CIMP), and KRAS and BRAF mutations in early-onset and later-onset colorectal cancers according to detailed sublocations (Table 2, Figure 1). The proportions of MSI-high, CIMP-high, and BRAF-mutated early-onset tumors were lowest in the rectum (8.8%, 3.4%, and 3.5%, respectively) and highest in the ascending colon (46% MSI-high; 15% CIMP-high) or transverse colon (8.6% BRAF-mutated) (all Ptrend<0.001 across the rectum to ascending colon), followed by declines in the cecum for MSI-high (36%) and BRAF mutation (4.6%) but not much for CIMP-high (14%). Similar increasing trends in the prevalence of MSI-high, CIMP-high, and BRAF-mutated tumors from the rectum to ascending colon were observed in later-onset colorectal cancer. Further age-stratified results on later-onset tumors are shown in Figure 1 and Supplementary Table 4.
Table 2.
Molecular Characteristics of Early-onset and Later-onset Colorectal Cancers According to Primary Tumor Location
| Molecular feature* | Total N | Cecum | Ascending colon | Transverse colon | Descending colon | Sigmoid colon | Rectum | Multivariable OR (95% CI)† | Ptrend† | Pinteraction# |
|---|---|---|---|---|---|---|---|---|---|---|
| Age <50 (early-onset) | ||||||||||
| MSI status | 1.88 (1.73–2.03) | <0.001 | <0.001 | |||||||
| Non-MSI-high | 2291 (79%) | 222 (64%) | 168 (54%) | 137 (56%) | 187 (75%) | 569 (89%) | 1008 (91%) | |||
| MSI-high | 601 (21%) | 123 (36%) | 146 (46%) | 106 (44%) | 62 (25%) | 67 (11%) | 97 (8.8%) | |||
| CIMP status | 1.55 (1.28–1.88) | <0.001 | 0.002 | |||||||
| Low/negative | 1058 (94%) | 133 (86%) | 93 (85%) | 96 (93%) | 96 (97%) | 238 (96%) | 402 (97%) | |||
| High | 71 (6.3%) | 21 (14%) | 16 (15%) | 7 (6.8%) | 3 (3.0%) | 10 (4.0%) | 14 (3.4%) | |||
| BRAF | 1.29 (1.14–1.46) | <0.001 | <0.001 | |||||||
| Wild-type | 2529 (95%) | 310 (95%) | 261 (92%) | 212 (91%) | 213 (95%) | 580 (95%) | 953 (96%) | |||
| Mutant | 133 (5.0%) | 15 (4.6%) | 23 (8.1%) | 20 (8.6%) | 12 (5.3%) | 28 (4.6%) | 35 (3.5%) | |||
| KRAS | 1.10 (1.01–1.20) | 0.024 | 0.012 | |||||||
| Wild-type | 1035 (66%) | 107 (51%) | 81 (59%) | 83 (66%) | 95 (68%) | 260 (70%) | 409 (70%) | |||
| Mutant | 532 (34%) | 102 (49%) | 57 (41%) | 43 (34%) | 44 (32%) | 111 (30%) | 175 (30%) | |||
| Age ≥50 (later-onset) | ||||||||||
| MSI status | 2.15 (2.05–2.26) | <0.001 | ||||||||
| Non-MSI-high | 8612 (84%) | 1218 (73%) | 988 (64%) | 711 (70%) | 642 (87%) | 2373 (95%) | 2680 (97%) | |||
| MSI-high | 1592 (16%) | 447 (27%) | 549 (36%) | 312 (30%) | 92 (13%) | 112 (4.5%) | 80 (2.9%) | |||
| CIMP status | 1.97 (1.88–2.07) | <0.001 | ||||||||
| Low/negative | 6868 (82%) | 970 (71%) | 763 (60%) | 584 (68%) | 514 (89%) | 1882 (94%) | 2155 (95%) | |||
| High | 1490 (18%) | 401 (29%) | 516 (40%) | 279 (32%) | 63 (11%) | 118 (5.9%) | 113 (5.0%) | |||
| BRAF | 1.92 (1.82–2.02) | <0.001 | ||||||||
| Wild-type | 8428 (87%) | 1239 (79%) | 1036 (71%) | 734 (76%) | 615 (91%) | 2211 (95%) | 2593 (97%) | |||
| Mutant | 1263 (13%) | 331 (21%) | 431 (29%) | 234 (24%) | 61 (9.0%) | 117 (5.0%) | 89 (3.3%) | |||
| KRAS | 0.99 (0.96–1.03) | 0.67 | ||||||||
| Wild-type | 5675 (67%) | 767 (56%) | 876 (67%) | 616 (72%) | 403 (66%) | 1467 (70%) | 1546 (67%) | |||
| Mutant | 2837 (33%) | 608 (44%) | 422 (33%) | 239 (28%) | 204 (34%) | 615 (30%) | 749 (33%) |
Percentage indicates the proportion of patients with a specific patient molecular characteristic among all patients or in strata of tumor location (cecum, ascending colon, transverse colon, descending colon, sigmoid colon, and rectum).
Multivariable odds ratio (OR) (95% CI) (with 95% confidence interval) for molecular marker positivity(i.e., MSI-high, CIMP-high, BRAF mutation, or KRAS mutation) and Ptrend were calculated by the linear trend test across the ordinal categories of subsite location variable [from rectum (1) to ascending colon (5)] in the multivariable logistic regression model adjusted for sex (female vs. male), family history of colorectal cancer (present vs. absent), and study (i.e., cohort).
Pinteraction was calculated using the Wald test for the cross-product of the ordinal subsite variable and age (<50 vs. ≥50) in the multivariable logistic regression model that adjusted for sex (female vs. male), family history of colorectal cancer (present vs. absent), and study (i.e., cohort).
Abbreviations: CI, confidence interval; CIMP, CpG island methylator phenotype; MSI, microsatellite instability; OR, odds ratio.
Figure 1.
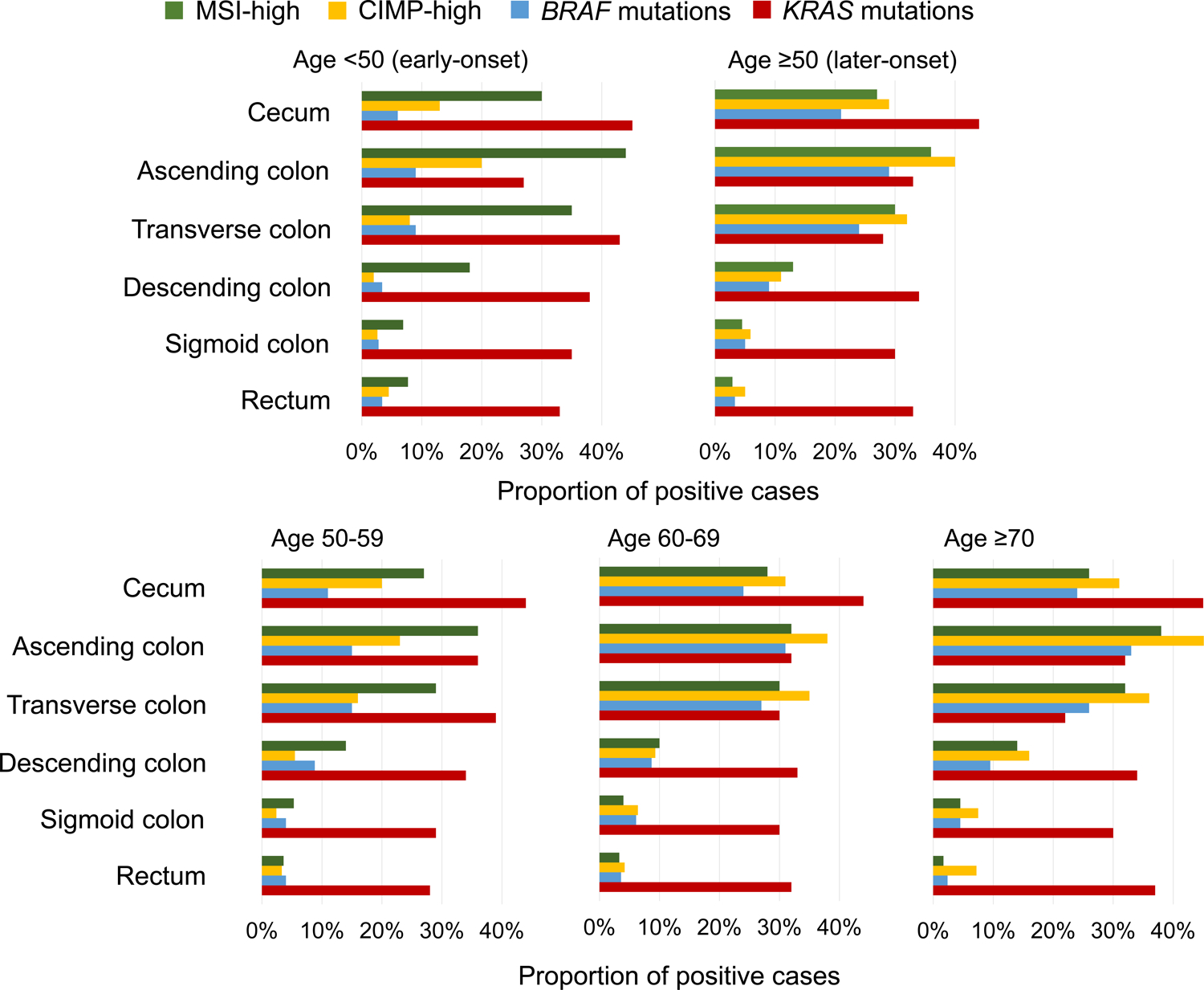
Prevalence of MSI-high status, CIMP-high status, BRAF mutations, and KRAS mutations along sublocations by age groups.
Abbreviations: CIMP, CpG island methylator phenotype; MSI, microsatellite instability.
In addition, we tested a hypothesis that the trend of molecular markers from the rectum to ascending colon differed between early-onset and later-onset tumors. The trends of the prevalence of MSI-high, CIMP-high, and BRAF-mutated tumors according to detailed sublocations significantly differed between early-onset and later-onset tumors (all Pinteraction≤0.001). Notably, compared to later-onset tumors, early-onset tumors showed higher prevalence of MSI-high and lower prevalence of CIMP-high and BRAF mutations in nearly all subsites (except for BRAF-mutated rectal tumors).
The proportion of KRAS-mutated early-onset tumors was higher in the cecum (49%) than in the other subsites (30–41%) [multivariable OR for the cecum vs. other subsites combined, 2.12 (95% CI, 1.57–2.86); P <0.001]. In later-onset tumors, cecal cancers showed higher prevalence of KRAS mutation (44%) than cancers of other subsites (28–33%) [the corresponding multivariable OR, 1.75 (95% CI, 1.56–1.97); P <0.001]. Stratified analyses by sexes and family history of colorectal cancer are shown in Table 3, Supplementary Table 5, and Supplementary Figures 1–2.
Table 3.
Molecular Characteristics of Early-onset and Later-onset Colorectal Cancers According to Primary Tumor Location in Strata of Sex
| Molecular feature* | Total N | Cecum | Ascending colon | Transverse colon | Descending colon | Sigmoid colon | Rectum | Multivariable OR (95% CI)† | Ptrend† |
|---|---|---|---|---|---|---|---|---|---|
| Age <50 (early-onset) | |||||||||
| Female | |||||||||
| MSI status | 1.90 (1.69–2.15) | <0.001 | |||||||
| Non-MSI-high | 1230 (83%) | 114 (70%) | 93 (56%) | 81 (65%) | 110 (82%) | 351 (93%) | 481 (92%) | ||
| MSI-high | 257 (17%) | 50 (30%) | 73 (44%) | 44 (35%) | 24 (18%) | 26 (6.9%) | 40 (7.7%) | ||
| CIMP status | 1.68 (1.29–2.19) | <0.001 | |||||||
| Low/negative | 549 (93%) | 65 (87%) | 43 (80%) | 49 (92%) | 50 (98%) | 151 (97%) | 191 (96%) | ||
| High | 39 (6.6%) | 10 (13%) | 11 (20%) | 4 (7.5%) | 1 (2.0%) | 4 (2.6%) | 9 (4.5%) | ||
| BRAF | 1.43 (1.19–1.71) | <0.001 | |||||||
| Wild-type | 1316 (95%) | 153 (94%) | 138 (91%) | 112 (91%) | 114 (97%) | 344 (97%) | 455 (97%) | ||
| Mutant | 64 (4.6%) | 9 (5.6%) | 14 (9.2%) | 11 (8.9%) | 4 (3.4%) | 10 (2.8%) | 16 (3.4%) | ||
| KRAS | 1.00 (0.89–1.13) | 0.94 | |||||||
| Wild-type | 524 (63%) | 50 (47%) | 54 (73%) | 36 (57%) | 47 (62%) | 146 (65%) | 191 (67%) | ||
| Mutant | 305 (37%) | 57 (53%) | 20 (27%) | 27 (43%) | 29 (38%) | 80 (35%) | 92 (33%) | ||
| Male | |||||||||
| MSI status | 1.85 (1.66–2.05) | <0.001 | |||||||
| Non-MSI-high | 1061 (76%) | 108 (60%) | 75 (51%) | 56 (47%) | 77 (67%) | 218 (84%) | 527 (90%) | ||
| MSI-high | 344 (24%) | 73 (40%) | 73 (49%) | 62 (53%) | 38 (33%) | 41 (16%) | 57 (10%) | ||
| CIMP status | 1.43 (1.06–1.94) | 0.020 | |||||||
| Low/negative | 509 (94%) | 68 (86%) | 50 (91%) | 47 (94%) | 46 (96%) | 87 (94%) | 211 (98%) | ||
| High | 32 (5.9%) | 11 (14%) | 5 (9.1%) | 3 (6.0%) | 2 (4.2%) | 6 (6.5%) | 5 (2.3%) | ||
| BRAF | 1.20 (1.01–1.42) | 0.037 | |||||||
| Wild-type | 1213 (95%) | 157 (96%) | 123 (93%) | 100 (92%) | 99 (93%) | 236 (93%) | 498 (96%) | ||
| Mutant | 69 (5.4%) | 6 (3.7%) | 9 (6.8%) | 9 (8.3%) | 8 (7.1%) | 18 (7.3%) | 19 (3.7%) | ||
| KRAS | 1.24 (1.10–1.41) | 0.001 | |||||||
| Wild-type | 511 (69%) | 57 (56%) | 27 (42%) | 47 (75%) | 48 (76%) | 114 (79%) | 218 (72%) | ||
| Mutant | 227 (31%) | 45 (44%) | 37 (58%) | 16 (25%) | 15 (24%) | 31 (21%) | 83 (28%) | ||
| Age ≥50 (later-onset) | |||||||||
| Female | |||||||||
| MSI status | 2.17 (2.03–2.33) | <0.001 | |||||||
| Non-MSI-high | 3791 (79%) | 593 (67%) | 512 (60%) | 314 (62%) | 286 (84%) | 1050 (95%) | 1036 (97%) | ||
| MSI-high | 987 (21%) | 298 (33%) | 345 (40%) | 193 (38%) | 56 (16%) | 58 (5.2%) | 37 (3.4%) | ||
| CIMP status | 2.18 (2.03–2.34) | <0.001 | |||||||
| Low/negative | 3065 (77%) | 485 (66%) | 380 (52%) | 257 (59%) | 246 (87%) | 839 (94%) | 858 (95%) | ||
| High | 921 (23%) | 247 (34%) | 357 (48%) | 176 (41%) | 36 (13%) | 56 (6.3%) | 49 (5.4%) | ||
| BRAF | 2.03 (1.89–2.18) | <0.001 | |||||||
| Wild-type | 3713 (82%) | 601 (72%) | 515 (63%) | 341 (69%) | 272 (88%) | 967 (94%) | 1017 (96%) | ||
| Mutant | 831 (18%) | 237 (28%) | 305 (37%) | 150 (31%) | 38 (12%) | 57 (5.6%) | 44 (4.1%) | ||
| KRAS | 0.90 (0.85–0.94) | <0.001 | |||||||
| Wild-type | 2703 (67%) | 436 (60%) | 538 (73%) | 312 (75%) | 193 (68%) | 637 (69%) | 587 (63%) | ||
| Mutant | 1315 (33%) | 293 (40%) | 201 (27%) | 106 (25%) | 91 (32%) | 286 (31%) | 338 (37%) | ||
| Male | |||||||||
| MSI status | 2.14 (1.99–2.31) | <0.001 | |||||||
| Non-MSI-high | 4821 (89%) | 625 (81%) | 476 (70%) | 397 (77%) | 356 (91%) | 1323 (96%) | 1644 (97%) | ||
| MSI-high | 605 (11%) | 149 (19%) | 204 (30%) | 119 (23%) | 36 (9.2%) | 54 (3.9%) | 43 (2.5%) | ||
| CIMP status | 1.76 (1.64–1.90) | <0.001 | |||||||
| Low/negative | 3803 (87%) | 485 (76%) | 383 (71%) | 327 (76%) | 268 (91%) | 1043 (94%) | 1297 (95%) | ||
| High | 569 (13%) | 154 (24%) | 159 (29%) | 103 (24%) | 27 (9.2%) | 62 (5.6%) | 64 (4.7%) | ||
| BRAF | 1.78 (1.65–1.93) | <0.001 | |||||||
| Wild-type | 4715 (92%) | 638 (87%) | 521 (81%) | 393 (82%) | 343 (94%) | 1244 (95%) | 1576 (97%) | ||
| Mutant | 432 (8%) | 94 (13%) | 126 (19%) | 84 (18%) | 23 (6.3%) | 60 (4.6%) | 45 (2.8%) | ||
| KRAS | 1.09 (1.04–1.15) | <0.001 | |||||||
| Wild-type | 2972 (66%) | 331 (51%) | 338 (60%) | 304 (70%) | 210 (65%) | 830 (72%) | 959 (70%) | ||
| Mutant | 1522 (34%) | 315 (49%) | 221 (40%) | 133 (30%) | 113 (35%) | 329 (28%) | 411 (30%) |
Percentage indicates the proportion of patients with a specific patient molecular characteristic among all patients or in strata of tumor location (cecum, ascending colon, transverse colon, descending colon, sigmoid colon, and rectum).
Multivariable odds ratio (OR) (with 95% confidence interval) for molecular marker positivity (i.e., MSI-high, CIMP-high, BRAF mutation, or KRAS mutation) and Ptrend were calculated by the linear trend test across the ordinal categories of subsite location variable [from rectum (1) to ascending colon (5)] in the multivariable logistic regression model adjusted for family history of colorectal cancer (present vs. absent) and study (i.e., cohort).
Abbreviations: CI, confidence interval; CIMP, CpG island methylator phenotype; MSI, microsatellite instability; OR, odds ratio.
We also examined Jass tumor subtype classifications (38) according to detailed sublocations (Supplementary Table 6). The proportions of type 1, 2, and 5 tumors increased from the rectum to ascending colon.
We further conducted analyses stratified by tumor characteristics (Table 4, Figure 2, and Supplementary Tables 7–9). In early-onset non-MSI-high cases, the proportion of BRAF-mutated tumors increased from the rectum to ascending colon (Ptrend <0.001). In later-onset cases of both MSI-high and non-MSI-high, we observed continuous increases in the proportions of CIMP-high and BRAF-mutated tumors from the rectum to ascending colon (all Ptrend <0.001). Remarkably, later-onset MSI-high tumors showed a continuous decrease in KRAS mutation prevalence from the rectum (36%) to ascending colon (9%; Ptrend <0.001) followed by an increase in the cecum (14%). In contrast, early-onset MSI-high tumors did not show such a trend (Pinteraction = 0.038, for the rectum-ascending colon trend in early-onset vs. later-onset MSI-high cases). Additionally, compared to later-onset MSI-high tumors, early-onset MSI-high tumors showed lower prevalence of CIMP-high in all subsites. We further conducted stratified MSI-high cases by family history of colorectal cancer (Supplementary Table 10). Although the sample size was limited, our findings tended to be consistent regardless of family history of colorectal cancer.
Table 4.
Molecular Characteristics of Early-onset and Later-onset Colorectal Cancers According to Primary Tumor Location in Strata of MSI Status
| Molecular feature* | Total N | Cecum | Ascending colon | Transverse colon | Descending colon | Sigmoid colon | Rectum | Multivariable OR (95% CI)† | Ptrend† |
|---|---|---|---|---|---|---|---|---|---|
| Age <50 (early-onset) | |||||||||
| Non-MSI-high | |||||||||
| CIMP status | 1.55 (1.23–1.96) | <0.001 | |||||||
| Low/negative | 863 (95%) | 93 (89%) | 56 (84%) | 56 (97%) | 74 (97%) | 215 (96%) | 369 (97%) | ||
| High | 48 (5.3%) | 11 (11%) | 11 (16%) | 2 (3.4%) | 2 (2.6%) | 9 (4.0%) | 13 (3.4%) | ||
| BRAF | 1.47 (1.27–1.69) | <0.001 | |||||||
| Wild-type | 1875 (95%) | 189 (95%) | 135 (88%) | 105 (89%) | 146 (94%) | 478 (95%) | 822 (97%) | ||
| Mutant | 104 (5.3%) | 11 (6%) | 18 (12%) | 13 (11%) | 9 (5.8%) | 27 (5.3%) | 26 (3.1%) | ||
| KRAS | 1.11 (0.99–1.24) | 0.060 | |||||||
| Wild-type | 813 (67%) | 68 (45%) | 42 (53%) | 48 (70%) | 78 (72%) | 224 (71%) | 353 (70%) | ||
| Mutant | 408 (33%) | 82 (55%) | 37 (47%) | 21 (30%) | 30 (28%) | 90 (29%) | 148 (30%) | ||
| MSI-high | |||||||||
| CIMP status | 1.19 (0.72–1.94) | 0.50 | |||||||
| Low/negative | 167 (89%) | 39 (81%) | 35 (88%) | 36 (90%) | 20 (95%) | 11 (92%) | 26 (96%) | ||
| High | 21 (11%) | 9 (19%) | 5 (13%) | 4 (10%) | 1 (4.8%) | 1 (8.3%) | 1 (3.7%) | ||
| BRAF | 1.05 (0.72–1.54) | 0.80 | |||||||
| Wild-type | 478 (97%) | 102 (98%) | 115 (97%) | 83 (95%) | 52 (95%) | 53 (100%) | 73 (96%) | ||
| Mutant | 15 (3.0%) | 2 (1.9%) | 3 (2.5%) | 4 (5.5%) | 3 (5.5%) | 0 (0%) | 3 (3.9%) | ||
| KRAS | 1.04 (0.83–1.30) | 0.74 | |||||||
| Wild-type | 139 (64%) | 33 (69%) | 34 (65%) | 25 (61%) | 13 (57%) | 13 (68%) | 21 (62%) | ||
| Mutant | 78 (36%) | 15 (31%) | 18 (35%) | 16 (39%) | 10 (43%) | 6 (32%) | 13 (38%) | ||
| Age ≥50 (later-onset) | |||||||||
| Non-MSI-high | |||||||||
| CIMP status | 1.63 (1.53–1.74) | <0.001 | |||||||
| Low/negative | 6121 (91%) | 802 (84%) | 614 (78%) | 468 (84%) | 460 (93%) | 1765 (96%) | 2012 (95%) | ||
| High | 632 (9.4%) | 157 (16%) | 173 (22%) | 90 (16%) | 35 (7.1%) | 80 (4.3%) | 97 (4.6%) | ||
| BRAF | 1.61 (1.50–1.73) | <0.001 | |||||||
| Wild-type | 7253 (94%) | 1011 (92%) | 763 (86%) | 551 (87%) | 537 (94%) | 2038 (96%) | 2353 (97%) | ||
| Mutant | 498 (6.4%) | 91 (8%) | 129 (14%) | 84 (13%) | 33 (5.8%) | 83 (3.9%) | 78 (3.2%) | ||
| KRAS | 1.12 (1.08–1.16) | <0.001 | |||||||
| Wild-type | 4378 (63%) | 455 (46%) | 438 (55%) | 381 (66%) | 336 (64%) | 1351 (70%) | 1417 (67%) | ||
| Mutant | 2553 (37%) | 542 (54%) | 365 (45%) | 199 (34%) | 186 (36%) | 567 (30%) | 694 (33%) | ||
| MSI-high | |||||||||
| CIMP status | 1.63 (1.41–1.88) | <0.001 | |||||||
| Low/negative | 399 (34%) | 108 (33%) | 113 (26%) | 70 (30%) | 30 (52%) | 43 (56%) | 35 (88%) | ||
| High | 768 (66%) | 217 (67%) | 319 (74%) | 165 (70%) | 28 (48%) | 34 (44%) | 5 (13%) | ||
| BRAF | 1.57 (1.38–1.78) | <0.001 | |||||||
| Wild-type | 709 (50%) | 179 (44%) | 212 (42%) | 138 (50%) | 52 (67%) | 69 (70%) | 59 (92%) | ||
| Mutant | 716 (50%) | 230 (56%) | 288 (58%) | 137 (50%) | 26 (33%) | 30 (30%) | 5 (7.8%) | ||
| KRAS | 0.67 (0.57–0.79) | <0.001 | |||||||
| Wild-type | 1017 (86%) | 288 (86%) | 401 (91%) | 198 (85%) | 52 (84%) | 53 (71%) | 25 (64%) | ||
| Mutant | 167 (14%) | 47 (14%) | 39 (9%) | 35 (15%) | 10 (16%) | 22 (29%) | 14 (36%) |
Percentage indicates the proportion of patients with a specific patient molecular characteristic among all patients or in strata of tumor location (cecum, ascending colon, transverse colon, descending colon, sigmoid colon, and rectum).
Multivariable odds ratio (OR) (with 95% confidence interval) for molecular marker positivity (i.e., CIMP-high, BRAF mutation, or KRAS mutation) and Ptrend was calculated by the linear trend test across the ordinal categories of subsite location variable [from rectum (1) to ascending colon (5)] in the multivariable logistic regression model adjusted for sex (female vs. male), family history of colorectal cancer (present vs. absent), and study (i.e., cohort).
Abbreviations: CI, confidence interval; CIMP, CpG island methylator phenotype; MSI, microsatellite instability; OR, odds ratio.
Figure 2.
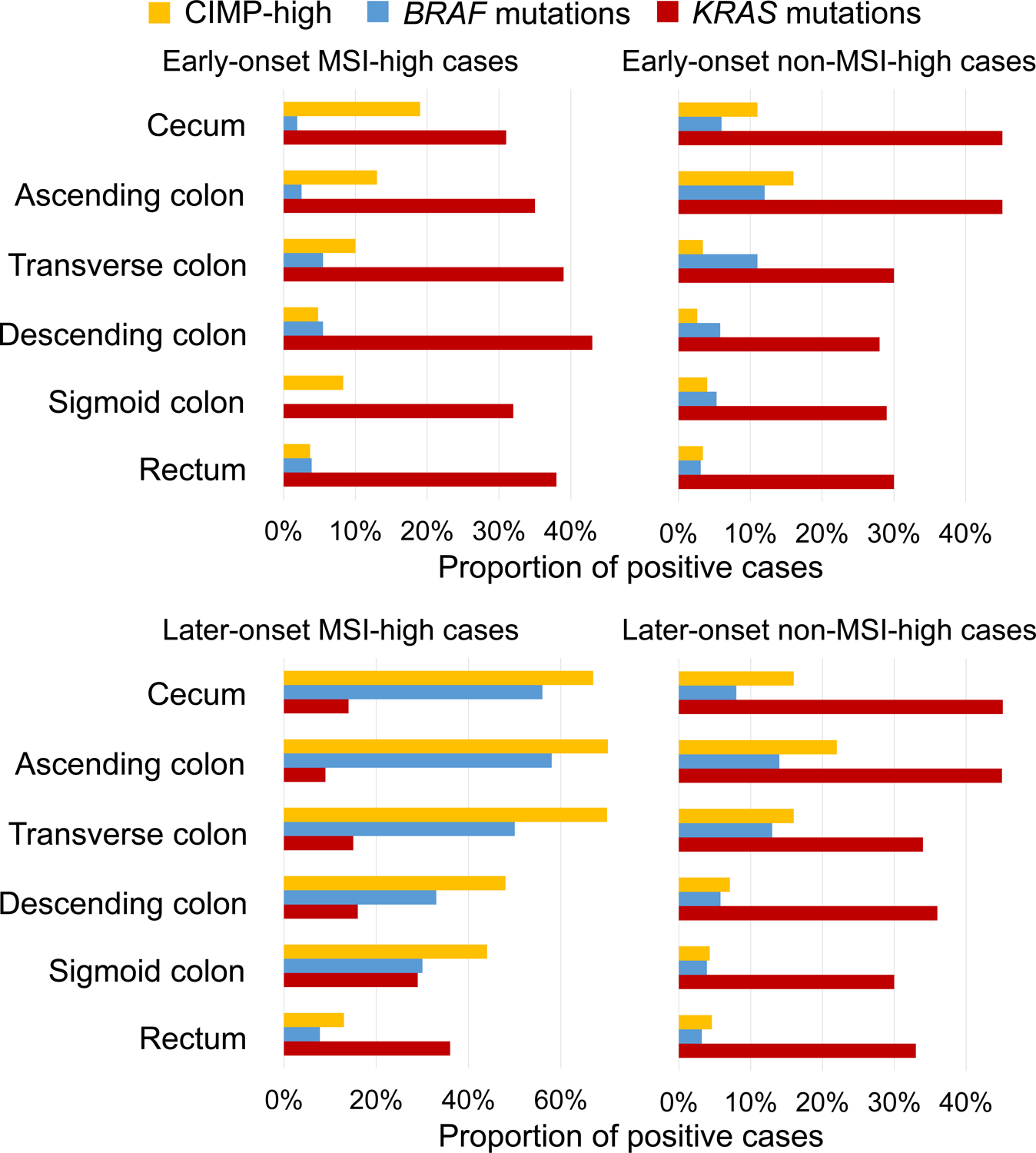
Prevalence of CIMP-high status, BRAF mutations, and KRAS mutations along sublocations by MSI status in early-onset and later-onset cancers.
Abbreviations: CIMP, CpG island methylator phenotype; MSI, microsatellite instability.
Lastly, we conducted stratified analysis by year of diagnosis (up to 2000 vs. after 2000) and observed a similar trend in both strata (Supplementary Table 11).
Discussion
Colorectal adenocarcinoma represents a heterogeneous group of complex multifactorial diseases with varying cellular molecular features influenced by local tissue microenvironment. In this consortium pooled analysis using 3,089 early-onset and 10,915 later-onset cases, we found that the proportions of MSI-high, CIMP-high, and BRAF-mutated early-onset tumors were generally highest in the transverse and ascending colon and lowest in the rectum. In addition, compared to later-onset tumors, early-onset tumors showed higher prevalence of MSI-high and lower prevalence of CIMP-high and BRAF mutations in most subsites. The prevalence of KRAS mutation in both early-onset and later-onset tumors was consistently highest in the cecum. Notably, later-onset MSI-high tumors showed a continuous decrease in KRAS mutation prevalence from the rectum to ascending colon followed by an increase in the cecum, but such a trend was not observed in early-onset MSI-high tumors. To our best knowledge, this study is the largest to date to investigate tumor molecular characteristics of early-onset and later-onset colorectal cancers along the detailed colorectal subsites.
Incidence of early-onset colorectal cancers in many organs diagnosed before age 50 years has globally been rising in recent decades for unknown reasons (40). Notably most of early-onset cancer types that have shown the recent rise relate to the digestive system, potentially implicating the etiological role of the intestinal microbiota (40). Differences in the stool microbiome have been reported between early-onset and later-onset colorectal cancer patients (41). The intestinal microbiota, which likely gradually changes across the entire colorectal length as luminal contents move toward the rectum, has been hypothetically linked with the continuous changes in the prevalence of tumor molecular features along the colorectal segments (12–14). Early-onset colorectal cancer has been associated with certain risk factor profiles, rectal location, signet ring cell histology, and specific tumor characteristics such as high-level MSI, LINE-1 hypomethylation, low/negative CIMP, BRAF wild-type, and lower tumor-infiltrating lymphocytes (1, 3–7, 42–46). Our current study attests to biogeographical (colorectal subsite) heterogeneity in molecular pathological features between early-onset and later-onset cases as well as even among early-onset cases. This study also provides rationale for the multi-segmental design in the research of early-onset and later-onset colorectal cancer.
The proximal-distal dichotomy design has prevailed in colorectal disease research for decades and provided evidence for differences in molecular pathology between proximal and distal colorectal tumors (17). However, this dichotomy approach cannot provide insight into tumor characteristics in relation to more detailed subsites. On the other hand, previous studies have demonstrated differences in etiologies, molecular pathologies, and prognostic associations between the detailed anatomical subsites (12, 20–22, 47–49). Our current findings further support the importance of the colorectal multi-segmental approach in clinical, epidemiological, and pathological research (12, 13, 50). This study also indicates that a large sample size is needed to examine tumors in each colorectal subsite with adequate statistical power.
In the colorectal tumor microenvironment, there is a dynamic interactive network that encompasses microorganisms and neoplastic, immune, and other cells, all of which are likely influenced by diet, lifestyle, environmental exposures, and other host factors (10, 51). Accumulating evidence indicates that the gut microbiota may influence the pathogenesis of colorectal cancer through cellular molecular alterations and modulation of tumor immune interactions (52, 53). An observational study has linked a certain dietary pattern with the abundance of sulfur-metabolizing bacteria in stool, and shown that the pattern is associated with an increased risk of distal and rectal cancers (54). Another study suggests that so-called western dietary pattern rich in red and processed meat is associated with an increased risk of colorectal cancer, particularly in the distal colon and rectum (55). Colonic epithelial cells are constantly in contact with bowel contents, including food debris, microorganisms, and their metabolites. Bowel contents and the gut microbiota likely vary across detailed anatomical subsites in the colorectum (14, 56). Furthermore, we also showed that tumor immune microenvironment differed by early-onset and later-onset colorectal cancers (42). Hence, available pieces of evidence underscore the importance of the multi-segmental approach in research on colorectal diseases including early-onset and later-onset colorectal cancer.
We found that later-onset MSI-high tumors exhibited a continuous decrease in KRAS mutation prevalence from the rectum to ascending colon followed by an increase in the cecum, whereas early-onset MSI-high tumors did not show such a trend. MSI-high tumors are associated with distinct clinical and pathological features such as proximal tumor location, vigorous immune response, and better prognosis (57, 58). In addition, there exists heterogeneity in clinical and pathological features between early-onset and later-onset cases (1, 3, 42–46). Early-onset MSI-high tumors tend to be related to Lynch syndrome with germline mismatch repair gene mutations, while later-onset MSI-high tumors tend to be related to CIMP-high tumors with MLH1 promoter hypermethylation (1, 59). The intriguing difference in the KRAS mutation prevalence trend across colorectal subsites between early-onset and later-onset MSI-high cancers further emphasize pathobiological heterogeneity in colorectal cancer, which necessitates further investigations.
Cecal cancers appear to represent a unique subgroup of colorectal cancer, characterized by high prevalence of KRAS mutations compared to cancers of the other sites in both early-onset and later-onset tumors, in agreement with the previous studies (12, 20, 60). Although the exact mechanism remains uncertain, the uniqueness of cecal cancers compared to cancers in the other colorectal segments may possibly reflect the following facts: (1) that the cecum is the first place where luminal contents enter into the large intestine; (2) that it has a pouch-like shape with a tendency of its content retention; and (3) that it is in the close proximity to the ileum and appendix, both of which are rich in lymphoid immune tissue. Of note, appendiceal cancers have been shown to exhibit high prevalence of KRAS mutation similar to proximal colon cancers (61, 62) (also similar to cecal cancers in the previous study (12). Thus, both cecal and appendiceal cancers are characterized by high prevalence of KRAS mutations. Further studies are needed to elucidate biogeographical characteristics of the cecum distinct from the other colorectal segments.
We acknowledge the limitations of this study. First, we relied on tumor location information derived from individual medical records. Nonetheless, recording of colorectal tumor location has been standardized through the use of the ICD (International Classification of Disease) code for each individual study. Second, tumor molecular analyses were performed using variable protocols across studies in this pooled analysis. However, all of the molecular pathological methods have been well established with good validity in the literature of colorectal cancer research. Moreover, in a subset of cases, centralized targeted sequencing has shown high concordance of molecular pathological statuses for MSI, KRAS, and BRAF. Third, our patients were predominantly non-Hispanic Whites. Therefore, future analyses need to be conducted in different populations. Fourth, although we adjusted for multiple comparisons, false positive findings could not be excluded. In addition, statistically significant but small differences may not be clinically important. Fifth, data on Lynch syndrome were not available. However, we stratified all cases and MSI-high cases by family history of colorectal cancer (Supplementary Tables 6 and 10). Our findings tended to be consistent regardless of family history of colorectal cancer. Lastly, this analysis included older cases diagnosed before 2000, which may not be the same as a contemporary cohort given the changing incidence trend of early-onset colorectal cancer. However, stratified analyses by year of diagnosis (up to 2000 vs. after 2000) yielded similar trends in both strata (Supplementary Table 11).
The current study has unprecedented strengths. First and foremost, our consortium pooled analysis design with the large sample size enabled us to robustly evaluate the prevalence of the major tumor molecular features within each of the six subsites in strata of age, sex, colorectal cancer family history, and tumor molecular biomarker status. Second, the colorectal cancer cases in this study were drawn from several countries and based on different study designs, including case-control studies, prospective cohort studies, and multi-institutional case series, which likely increased generalizability.
In conclusion, our current study showed substantial differences of tumor molecular characteristics in early-onset and later-onset colorectal carcinomas arising in different colorectal subsites. Our findings not only support biogeographical heterogeneity along colorectal length that influences carcinogenic processes, but also provide compelling rationale for designing large-scale studies to robustly investigate detailed subsite data in colorectal disease research.
Supplementary Material
Study Highlights.
WHAT IS KNOWN
The incidence of early-onset colorectal cancer diagnosed before age 50 has increased worldwide.
Early-onset colorectal cancer commonly occurs in the rectum.
Early-onset colorectal cancer has tumor characteristics different from later-onset colorectal cancer.
Tumor characteristics of colorectal cancer differ by primary tumor location.
WHAT IS NEW HERE
Molecular characteristics of early-onset colorectal cancer changed gradually along colorectal subsites.
Compared to later-onset tumors, early-onset tumors showed higher MSI-high prevalence and lower CIMP-high/BRAF mutation prevalence in most subsites.
Tumor molecular features varied by both age at diagnosis and detailed tumor location.
Acknowledgements
The Colon Cancer Family Registry (CCFR): The Colon CFR graciously thanks the generous contributions of their study participants, dedication of study staff, and the financial support from the U.S. National Cancer Institute, without which this important registry would not exist.
CPS-II: The authors thank the CPS-II participants and Study Management Group for their invaluable contributions to this research. The authors would also like to acknowledge the contribution to this study from central cancer registries supported through the Centers for Disease Control and Prevention National Program of Cancer Registries, and cancer registries supported by the National Cancer Institute Surveillance Epidemiology and End Results program.
DACHS: We thank all participants and cooperating clinicians, and Ute Handte-Daub, Utz Benscheid, Muhabbet Celik and Ursula Eilber for excellent technical assistance.
EDRN: We acknowledge all the following contributors to the development of the resource: University of Pittsburgh School of Medicine, Department of Gastroenterology, Hepatology and Nutrition: Lynda Dzubinski; University of Pittsburgh School of Medicine, Department of Pathology: Michelle Bisceglia; and University of Pittsburgh School of Medicine, Department of Biomedical Informatics.
EPIC: Where authors are identified as personnel of the International Agency for Research on Cancer/World Health Organization, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy or views of the International Agency for Research on Cancer/World Health Organization.
Harvard cohorts (HPFS, NHS): The study protocol was approved by the institutional review boards of the Brigham and Women’s Hospital and Harvard T.H. Chan School of Public Health, and those of participating registries as required. We would like to thank the participants and staff of the HPFS and NHS for their valuable contributions as well as the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. The authors assume full responsibility for analyses and interpretation of these data.
NSHDS: NSHDS investigators thank the Västerbotten Intervention Programme, the Northern Sweden MONICA study, the Biobank Research Unit at Umeå University and Biobanken Norr at Region Västerbotten for providing data and samples and acknowledge the contribution from Biobank Sweden, supported by the Swedish Research Council.
SCCFR: The authors would like to thank the study participants and staff of the Seattle Cancer Family Registry and the Hormones and Colon Cancer study (CORE Studies).
Grant Support
Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO): National Cancer Institute, National Institutes of Health, U.S. Department of Health and Human Services (U01 CA137088 and R01 CA248857). This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA015704. Scientific Computing Infrastructure at Fred Hutch funded by ORIP grant S10OD028685.
The Colon Cancer Family Registry (CCFR): The Colon Cancer Family Registry (CCFR, www.coloncfr.org) is supported in part by funding from the U.S. National Cancer Institute (NCI), National Institutes of Health (NIH) (award U01 CA167551). The Seattle Colon Cancer Family Registry was also supported in part by the National Cancer Institute (NCI) of the U.S. National Institutes of Health (NIH) under R01 CA076366 (to P.A.N.). The content of this manuscript does not necessarily reflect the views or policies of the U.S. NCI, NIH or any of the collaborating centers in the Colon Cancer Family Registry (CCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government, any cancer registry, or the CCFR.
CPS-II: The American Cancer Society funds the creation, maintenance, and updating of the Cancer Prevention Study-II (CPS-II) cohort. This study was conducted with Institutional Review Board approval.
DACHS: This work was supported by the German Research Council (BR 1704/6-1, BR 1704/6-3, BR 1704/6-4, CH 117/1-1, HO 5117/2-1, HE 5998/2-1, KL 2354/3-1, RO 2270/8-1 and BR 1704/17-1), the Interdisciplinary Research Program of the National Center for Tumor Diseases (NCT), Germany, and the German Federal Ministry of Education and Research (01KH0404, 01ER0814, 01ER0815, 01ER1505A and 01ER1505B).
DALS: U.S. National Institutes of Health (R01 CA48998 to M. L. Slattery).
EDRN: This work is funded and supported by the U.S. NCI, EDRN Grant (U01 CA 84968-06).
EPIC: The coordination of EPIC is financially supported by the European Commission (DGSANCO) and the International Agency for Research on Cancer. The national cohorts are supported by Danish Cancer Society (Denmark); Ligue Contre le Cancer, Institut Gustave Roussy, Mutuelle Générale de l’Education Nationale, Institut National de la Santé et de la Recherche Médicale (INSERM) (France); German Cancer Aid, German Cancer Research Center (DKFZ), Federal Ministry of Education and Research (BMBF), Deutsche Krebshilfe, Deutsches Krebsforschungszentrum and Federal Ministry of Education and Research (Germany); the Hellenic Health Foundation (Greece); Associazione Italiana per la Ricerca sul Cancro-AIRCItaly and National Research Council (Italy); Dutch Ministry of Public Health, Welfare and Sports (VWS), Netherlands Cancer Registry (NKR), LK Research Funds, Dutch Prevention Funds, Dutch ZON (Zorg Onderzoek Nederland), World Cancer Research Fund (WCRF), Statistics Netherlands (The Netherlands); ERC-2009-AdG 232997 and Nordforsk, Nordic Centre of Excellence programme on Food, Nutrition and Health (Norway); Health Research Fund (FIS), PI13/00061 to Granada, PI13/01162 to EPIC-Murcia, Regional Governments of Andalucía, Asturias, Basque Country, Murcia and Navarra, ISCIII RETIC (RD06/0020) (Spain); Swedish Cancer Society, Swedish Research Council and County Councils of Skåne and Västerbotten (Sweden); Cancer Research UK (14136 to EPIC-Norfolk; C570/A16491 and C8221/A19170 to EPIC-Oxford), Medical Research Council (1000143 to EPIC-Norfolk, MR/M012190/1 to EPIC-Oxford) (United Kingdom).
Harvard Cohorts (HPFS, NHS): HPFS is supported by the U.S. National Institutes of Health (P01 CA055075, UM1 CA167552, U01 CA167552, R01 CA137178, R01 CA151993, R00 CA215314, and R35 CA197735) and NHS by the U.S. National Institutes of Health (R01 CA137178, P01 CA087969, UM1 CA186107, R01 CA151993, and R35 CA197735). M.G. and S.O. were supported in part by the Cancer Research UK Grand Challenge Award (UK C10674/A27140). T.U. was supported by Prevent Cancer Foundation.
MCCS: Melbourne Collaborative Cohort Study (MCCS) cohort recruitment was funded by VicHealth and Cancer Council Victoria. The MCCS was further augmented by Australian National Health and Medical Research Council grants 209057, 396414 and 1074383 and by infrastructure provided by Cancer Council Victoria. Cases and their vital status were ascertained through the Victorian Cancer Registry and the Australian Institute of Health and Welfare, including the National Death Index and the Australian Cancer Database. New South Wales (NSW) cancer registry data were obtained via the ACD with the assistance of the NSW Ministry of Health.
NFCCR: This work was supported by an Interdisciplinary Health Research Team award from the Canadian Institutes of Health Research (CRT 43821); the National Institutes of Health, U.S. Department of Health and Human Services (U01 CA74783); and National Cancer Institute of Canada grants (18223 and 18226). The authors wish to acknowledge the contribution of Alexandre Belisle and the genotyping team of the McGill University and Génome Québec Innovation Centre, Montréal, Canada, for genotyping the Sequenom panel in the NFCCR samples. Funding was provided to Michael O. Woods by the Canadian Cancer Society Research Institute.
NSHDS: This work was supported by Biobank Sweden through funding from the Swedish Research Council (VR 2017-00650, VR 2017-01737), the Swedish Cancer Society (CAN 2017/581), Region Västerbotten (VLL-841671, VLL-833291), Knut and Alice Wallenberg Foundation (VLL-765961), and the Lion’s Cancer Research Foundation (several grants) and Insamlingsstiftelsen, both at Umeå University.
Disclosures of Potential Conflicts of Interest:
A.T.C. previously served as a consultant for Bayer Healthcare and Pfizer Inc. M.G. receives research funding from Bristol-Myers Squibb and Merck. A.T.C. is a Stuart and Suzanne Steele MGH Research Scholar. The other authors declare no potential conflicts of interest. The content is solely the responsibility of the authors and does not necessarily represent the official views of U.S. NIH. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations:
- CCFR
Colon Cancer Family Registry
- CIMP
CpG island methylator phenotype
- CPS-II
Cancer Prevention Study II
- DACHS
Darmkrebs: Chancen der Verhütung durch Screening Study
- DALS
Diet, Activity and Lifestyle Study
- EDRN
Early Detection Research Network
- EPIC_Sweden
European Prospective Investigation into Cancer_Sweden
- HPFS
Health Professionals Follow-up Study
- ICD
International Classification of Disease
- MCCS
Melbourne Collaborative Cohort Study
- MMR
mismatch repair
- MSI
microsatellite instability
- NFCCR
Newfoundland Familial Colorectal Cancer Registries
- NHS
Nurses’ Health Study
- NSHDS
Northern Sweden Health and Disease Study
- PCR
polymerase chain reaction
- TCGA
The Cancer Genome Atlas
Footnotes
Use of Standardized Official Symbols: We use HUGO (Human Genome Organisation)-approved official symbols (or root symbols) for genes and gene products, including BRAF, EGFR, KRAS, MLH1, MSH2, MSH6, and PMS2; all of which are described at www.genenames.org. Gene symbols are italicized whereas symbols for gene products are not italicized.
References
- 1.Akimoto N, Ugai T, Zhong R, et al. Rising incidence of early-onset colorectal cancer - a call to action. Nat Rev Clin Oncol 2021;18:230–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Archambault AN, Su YR, Jeon J, et al. Cumulative Burden of Colorectal Cancer-Associated Genetic Variants Is More Strongly Associated With Early-Onset vs Late-Onset Cancer. Gastroenterology 2020;158:1274–1286.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Willauer AN, Liu Y, Pereira AAL, et al. Clinical and molecular characterization of early-onset colorectal cancer. Cancer 2019;125:2002–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akimoto N, Zhao M, Ugai T, et al. Tumor Long Interspersed Nucleotide Element-1 (LINE-1) Hypomethylation in Relation to Age of Colorectal Cancer Diagnosis and Prognosis. Cancers (Basel) 2021;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antelo M, Balaguer F, Shia J, et al. A high degree of LINE-1 hypomethylation is a unique feature of early-onset colorectal cancer. PLoS One 2012;7:e45357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baba Y, Huttenhower C, Nosho K, et al. Epigenomic diversity of colorectal cancer indicated by LINE-1 methylation in a database of 869 tumors. Mol Cancer 2010;9:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lieu CH, Golemis EA, Serebriiskii IG, et al. Comprehensive Genomic Landscapes in Early and Later Onset Colorectal Cancer. Clin Cancer Res 2019;25:5852–5858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stoffel EM, Koeppe E, Everett J, et al. Germline Genetic Features of Young Individuals With Colorectal Cancer. Gastroenterology 2018;154:897–905 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pearlman R, Frankel WL, Swanson B, et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA oncology 2017;3:464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang ST, Cui WQ, Pan D, et al. Tea polyphenols and their chemopreventive and therapeutic effects on colorectal cancer. World J Gastroenterol 2020;26:562–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inamura K, Hamada T, Bullman S, et al. Cancer as microenvironmental, systemic and environmental diseases: opportunity for transdisciplinary microbiomics science. Gut 2022;71:2107–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamauchi M, Morikawa T, Kuchiba A, et al. Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colorectum. Gut 2012;61:847–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamauchi M, Lochhead P, Morikawa T, et al. Colorectal cancer: a tale of two sides or a continuum? Gut 2012;61:794–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mima K, Cao Y, Chan AT, et al. Fusobacterium nucleatum in Colorectal Carcinoma Tissue According to Tumor Location. Clin Transl Gastroenterol 2016;7:e200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ogino S, Nosho K, Kirkner GJ, et al. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut 2009;58:90–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Y, Chen Z, Huang J, et al. Clinicopathological and molecular characteristics of early-onset vs late-onset colorectal cancer according to tumor location. Int J Clin Oncol 2022;27:749–755. [DOI] [PubMed] [Google Scholar]
- 17.Lee MS, Menter DG, Kopetz S. Right Versus Left Colon Cancer Biology: Integrating the Consensus Molecular Subtypes. J Natl Compr Canc Netw 2017;15:411–419. [DOI] [PubMed] [Google Scholar]
- 18.Phipps AI, Alwers E, Harrison T, et al. Association Between Molecular Subtypes of Colorectal Tumors and Patient Survival, Based on Pooled Analysis of 7 International Studies. Gastroenterology 2020;158:2158–2168.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Domingo E, Freeman-Mills L, Rayner E, et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: a retrospective, pooled biomarker study. Lancet Gastroenterol Hepatol 2016;1:207–216. [DOI] [PubMed] [Google Scholar]
- 20.Loree JM, Pereira AAL, Lam M, et al. Classifying Colorectal Cancer by Tumor Location Rather than Sidedness Highlights a Continuum in Mutation Profiles and Consensus Molecular Subtypes. Clin Cancer Res 2018;24:1062–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosty C, Young JP, Walsh MD, et al. PIK3CA activating mutation in colorectal carcinoma: associations with molecular features and survival. PLoS One 2013;8:e65479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phipps AI, Buchanan DD, Makar KW, et al. BRAF mutation status and survival after colorectal cancer diagnosis according to patient and tumor characteristics. Cancer Epidemiol Biomarkers Prev 2012;21:1792–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Campbell PT, Deka A, Briggs P, et al. Establishment of the cancer prevention study II nutrition cohort colorectal tissue repository. Cancer Epidemiol Biomarkers Prev 2014;23:2694–702. [DOI] [PubMed] [Google Scholar]
- 24.Brenner H, Chang-Claude J, Seiler CM, et al. Protection from colorectal cancer after colonoscopy: a population-based, case-control study. Ann Intern Med 2011;154:22–30. [DOI] [PubMed] [Google Scholar]
- 25.Slattery ML, Friedman GD, Potter JD, et al. A description of age, sex, and site distributions of colon carcinoma in three geographic areas. Cancer 1996;78:1666–70. [DOI] [PubMed] [Google Scholar]
- 26.Crichton DJ, Mattmann CA, Thornquist M, et al. Bioinformatics: biomarkers of early detection. Cancer Biomark 2010;9:511–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riboli E, Kaaks R. The EPIC Project: rationale and study design. European Prospective Investigation into Cancer and Nutrition. Int J Epidemiol 1997;26 Suppl 1:S6–14. [DOI] [PubMed] [Google Scholar]
- 28.Nishihara R, Wu K, Lochhead P, et al. Long-term colorectal-cancer incidence and mortality after lower endoscopy. N Engl J Med 2013;369:1095–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giles GG, English DR. The Melbourne Collaborative Cohort Study. IARC Sci Publ 2002;156:69–70. [PubMed] [Google Scholar]
- 30.Stuckless S, Parfrey PS, Woods MO, et al. The phenotypic expression of three MSH2 mutations in large Newfoundland families with Lynch syndrome. Fam Cancer 2007;6:1–12. [DOI] [PubMed] [Google Scholar]
- 31.Liao X, Lochhead P, Nishihara R, et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med 2012;367:1596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Guelpen B, Hultdin J, Johansson I, et al. Low folate levels may protect against colorectal cancer. Gut 2006;55:1461–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peters U, Jiao S, Schumacher FR, et al. Identification of Genetic Susceptibility Loci for Colorectal Tumors in a Genome-Wide Meta-analysis. Gastroenterology 2013;144:799–807.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hidaka M, Eguchi S, Okuda K, et al. Impact of Anatomical Resection for Hepatocellular Carcinoma With Microportal Invasion (vp1): A Multi-institutional Study by the Kyushu Study Group of Liver Surgery. Ann Surg 2020;271:339–346. [DOI] [PubMed] [Google Scholar]
- 36.Fujiyoshi K, Bruford EA, Mroz P, et al. Opinion: Standardizing gene product nomenclature-a call to action. Proc Natl Acad Sci U S A 2021;118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zaidi SH, Harrison TA, Phipps AI, et al. Landscape of somatic single nucleotide variants and indels in colorectal cancer and impact on survival. Nat Commun 2020;11:3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 2007;50:113–30. [DOI] [PubMed] [Google Scholar]
- 39.Benjamin DJ, Berger JO, Johannesson M, et al. Redefine statistical significance. Nat Hum Behav 2018;2:6–10. [DOI] [PubMed] [Google Scholar]
- 40.Ugai T, Sasamoto N, Lee HY, et al. Is early-onset cancer an emerging global epidemic? Current evidence and future implications. Nat Rev Clin Oncol 2022;19:656–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kong C, Liang L, Liu G, et al. Integrated metagenomic and metabolomic analysis reveals distinct gut-microbiome-derived phenotypes in early-onset colorectal cancer. Gut 2022. [DOI] [PubMed] [Google Scholar]
- 42.Ugai T, Vayrynen JP, Lau MC, et al. Immune cell profiles in the tumor microenvironment of early-onset, intermediate-onset, and later-onset colorectal cancer. Cancer Immunol Immunother 2022;71:933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ng K, May FP, Schrag D. US Preventive Services Task Force Recommendations for Colorectal Cancer Screening: Forty-Five Is the New Fifty. JAMA 2021;325:1943–1945. [DOI] [PubMed] [Google Scholar]
- 44.O’Sullivan DE, Sutherland RL, Town S, et al. Risk Factors for Early-Onset Colorectal Cancer: A Systematic Review and Meta-analysis. Clin Gastroenterol Hepatol 2022;20:1229–1240 e5. [DOI] [PubMed] [Google Scholar]
- 45.Gu WJ, Pei JP, Lyu J, et al. The Burden of Early-Onset Colorectal Cancer and Its Risk Factors from 1990 to 2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Cancers (Basel) 2022;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eng C, Jacome AA, Agarwal R, et al. A comprehensive framework for early-onset colorectal cancer research. Lancet Oncol 2022;23:e116–e128. [DOI] [PubMed] [Google Scholar]
- 47.Phipps AI, Chan AT, Ogino S. Anatomic subsite of primary colorectal cancer and subsequent risk and distribution of second cancers. Cancer 2013;119:3140–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang L, Lo CH, He X, et al. Risk Factor Profiles Differ for Cancers of Different Regions of the Colorectum. Gastroenterology 2020;159:241–256.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jess P, Hansen IO, Gamborg M, et al. A nationwide Danish cohort study challenging the categorisation into right-sided and left-sided colon cancer. BMJ Open 2013;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Testa U, Pelosi E, Castelli G. Colorectal cancer: genetic abnormalities, tumor progression, tumor heterogeneity, clonal evolution and tumor-initiating cells. Med Sci (Basel) 2018;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mima K, Kosumi K, Baba Y, et al. The microbiome, genetics, and gastrointestinal neoplasms: the evolving field of molecular pathological epidemiology to analyze the tumor-immune-microbiome interaction. Hum Genet 2021;140:725–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ternes D, Karta J, Tsenkova M, et al. Microbiome in Colorectal Cancer: How to Get from Meta-omics to Mechanism? Trends Microbiol 2020;28:401–423. [DOI] [PubMed] [Google Scholar]
- 53.Purcell RV, Visnovska M, Biggs PJ, et al. Distinct gut microbiome patterns associate with consensus molecular subtypes of colorectal cancer. Sci Rep 2017;7:11590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nguyen LH, Ma W, Wang DD, et al. Association Between Sulfur-Metabolizing Bacterial Communities in Stool and Risk of Distal Colorectal Cancer in Men. Gastroenterology 2020;158:1313–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mehta RS, Song M, Nishihara R, et al. Dietary Patterns and Risk of Colorectal Cancer: Analysis by Tumor Location and Molecular Subtypes. Gastroenterology 2017;152:1944–1953.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.James KR, Gomes T, Elmentaite R, et al. Distinct microbial and immune niches of the human colon. Nat Immunol 2020;21:343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bai J, Chen H, Bai X. Relationship between microsatellite status and immune microenvironment of colorectal cancer and its application to diagnosis and treatment. J Clin Lab Anal 2021;35:e23810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carr PR, Alwers E, Bienert S, et al. Lifestyle factors and risk of sporadic colorectal cancer by microsatellite instability status: a systematic review and meta-analyses. Ann Oncol 2018;29:825–834. [DOI] [PubMed] [Google Scholar]
- 59.Niv Y. Microsatellite instability and MLH1 promoter hypermethylation in colorectal cancer. World J Gastroenterol 2007;13:1767–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Landau MA, Zhu B, Akwuole FN, et al. Site-specific Differences in Colonic Adenocarcinoma: KRAS Mutations and High Tumor Budding Are More Frequent in Cecal Adenocarcinoma. Am J Surg Pathol 2018;42:351–358. [DOI] [PubMed] [Google Scholar]
- 61.Holowatyj AN, Eng C, Wen W, et al. Spectrum of Somatic Cancer Gene Variations Among Adults With Appendiceal Cancer by Age at Disease Onset. JAMA Netw Open 2020;3:e2028644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tokunaga R, Xiu J, Johnston C, et al. Molecular Profiling of Appendiceal Adenocarcinoma and Comparison with Right-sided and Left-sided Colorectal Cancer. Clin Cancer Res 2019;25:3096–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.