

SUMMARY
Background:
Reducing treatment burden is a priority for people with cystic fibrosis (CF) whose health has benefited from using newer modulators that substantially increase cystic fibrosis transmembrane conductance regulator (CFTR) protein function. The SIMPLIFY study included two parallel, randomized, open-label controlled trials to independently test the effects of discontinuing nebulized hypertonic saline (HS) or dornase alfa (DA) in individuals treated with the CFTR modulator elexacaftor/tezacaftor/ivacaftor (ETI). This study is registered with www.clinicaltrials.gov (NCT04378153).
Methods:
Individuals ≥12 years old using ETI and either or both nebulized therapies for ≥90 days were eligible. Participants on both HS and DA were randomized to one of the two trials, and those on a single therapy were assigned to the applicable trial. All were then randomized 1:1 to continue or discontinue therapy for 6 weeks. The primary objective for each trial was to determine whether discontinuing is non-inferior (NI) to continuing, measured by the 6-week change in % predicted forced expiratory volume in one second (ppFEV1).
Findings:
A total of 370 and 477 participants with an average ppFEV1 of 96.9% were randomized in the HS and DA trials, respectively. Discontinuing treatment was non-inferior to continuing with respect to the 6-week change in ppFEV1 in both the HS (per protocol [PP] population: n=133 discontinued, n=140 continued, difference=−0.32%, 95% CI:−1.25,0.60) and DA (PP population: n=199 discontinued, n=193 continued, difference = 0.35%, 95% CI:−0.45,1.14) trials according to a pre-specified −3% NI margin, with consistent results in the intent-to-treat populations. Secondary endpoints including lung clearance index and patient-reported symptom scores also demonstrated no meaningful differences, with few experiencing increased respiratory events.
Interpretation:
In individuals with CF on ETI with relatively well-preserved pulmonary function, discontinuing as compared to continuing daily HS or DA for six weeks did not result in clinically meaningful differences in pulmonary function or symptoms.
Keywords: Treatment withdrawal, treatment burden, CFTR modulators, non-inferiority trial
Introduction
Cystic fibrosis (CF) is an autosomal recessive disease caused by mutations in the cystic fibrosis transmembrane conductance regulator gene (CFTR), which encodes an epithelial anion channel responsible for hydrating secretions in multiple organs.1 Inadequate CFTR function in the lungs perpetuates a destructive cycle of mucociliary dysfunction, infection, and inflammation,2–4 which accelerates loss of lung function and contributes to substantial morbidity and mortality.5 Dornase alfa (DA) and hypertonic saline (HS), inhaled mucoactive therapies with unique but complementary mechanisms, facilitate clearance of airway secretions by addressing downstream manifestations of CFTR dysfunction including high amounts of extracellular DNA in mucus and airway surface dehydration, respectively.6,7 In randomized, placebo-controlled trials, these medications resulted in improvements in lung function, amelioration of respiratory symptoms, and reduced risk of pulmonary exacerbation.6–8 They are also included in CF clinical practice guidelines.9,10 However, people with CF (pwCF) dedicate considerable time and effort daily to these inhaled therapies, which contributes to high treatment burden.11
Based on the presence of at least one copy of the F508del-CFTR mutation, approximately 90% of pwCF are candidates for orally administered modulators that substantially, though incompletely, restore CFTR function. Clinical trials of the newest modulator combination, elexacaftor/tezacaftor/ivacaftor (ETI), demonstrated unparalleled improvements in lung function and respiratory symptoms and lower risk of pulmonary exacerbations,12,13 marking a watershed moment in the history of CF care. In post-approval research, ETI reduced objective indicators of airway mucus plugging,14 identifying overlap and potential redundancy between modulator-induced CFTR restoration and inhaled mucoactive therapies. The ongoing necessity of medications that improve airway mucus clearance downstream of CFTR is now unclear. As simplifying treatment burden remains a top CF community research priority,15,16 and data suggest that pwCF taking ETI are using other chronic therapies less frequently,17 it is an opportune time to rigorously evaluate the effects of discontinuing burdensome inhaled therapies such as DA and HS in pwCF on modulator drugs that markedly improve CFTR function.
The SIMPLIFY study was designed with important input from the CF community,16 and included two parallel, multicenter, open-label randomized, controlled non-inferiority trials to determine the effects of discontinuing DA or HS among pwCF established on ETI.18 For each trial, we hypothesized that discontinuing as compared to continuing inhaled therapy has no clinically meaningful impact on lung function, thereby providing evidence to support simplifying care regimens in pwCF with preserved lung function who benefit from ETI.
Role of the Funding Source
This study was funded by the CF Foundation and designed by the study PIs in collaboration with the CF Therapeutics Development Network (CF TDN). The funder of the study had no role in study design, data collection, or data analysis. Data were collected by local site investigators and analyzed by the TDN Coordinating Center at Seattle Children’s Research Institute. JPC is included as a co-author employed by the sponsor and provided insight into interpretation and writing of the manuscript along with all other co-authors. The corresponding and senior authors had final responsibility for the decision to submit for publication.
METHODS
Study Design and Participants
Details of the study design have been described18 with additional information provided in the Supplementary Appendix. SIMPLIFY is a master protocol with two concurrent 6-week randomized, open-label, controlled non-inferiority trials, each of which is designed to evaluate the independent effect of discontinuing versus continuing HS (Trial A) or DA (Trial B) (Supplement, Figure S1). The study was conducted in the United States (U.S.) at participating sites in the CF TDN. An independent review board approved the trial protocol and informed consent forms. All enrolled participants, or their legal guardians, provided written informed consent (and assent, when appropriate). Individuals with CF ages 12–17 years with percent-predicted forced expiratory volume in one second (ppFEV1) ≥70 and those ≥18 years of age with ppFEV1 ≥60 were eligible to enroll if they had been on ETI and either or both mucoactive therapies (≥3% hypertonic saline and/or dornase alfa) for at least 90 days prior to screening (Week −2). Full eligibility criteria are provided in Table S1.
Randomization and Masking
Enrolled participants were followed during a two-week screening period and were then eligible for randomization into Trial A or B conditional on reported adherence to their inhaled drug therapy (HS and/or DA), >70% compliance with electronic recording of daily medication use, no absolute decrease in ppFEV1 of ≥ 10%, clinical stability, and no acute use of antibiotics or corticosteroids for respiratory symptoms (Table S1). At Week 0, participants treated with only HS or DA were enrolled in Trial A or Trial B (as applicable) and randomized 1:1 to either continue or discontinue therapy. Concurrent users of HS and DA remained on both therapies during the screening period and were first randomized to participate in Trial A or Trial B and then randomized to continue vs. discontinue the applicable therapy in their assigned study. Individuals on both HS and DA could participate sequentially in both trials upon reassessment of individual study eligibility. Participants were randomized 1:1 to continue or discontinue therapy in each trial using permuted blocks of varying size, stratified by Week 0 ppFEV1 (≥90, <90), single or concurrent use of HS and/or DA, prior SIMPLIFY study participation (yes/no), and age (≥18 vs < 18). Further randomization details provided in the Supplement. Although there was no masking to treatment assignment for individual participants or their clinicians, aggregate study results were blinded and tightly controlled by the TDN Coordinating Center with only the study’s Data Monitoring Committee (DMC) and select unblinded study team members with access to interim study data.
Procedures
All randomized participants were followed and assessed over a 6-week period with study visits at Weeks 2 and 6 post-randomization. Sex at birth was recorded by the investigators as documented in the medical record. Protocol adherence to continue or discontinue therapy was measured using daily electronic assessments based on time-triggered ecological momentary assessment (EMA) documenting use or non-use of therapy.19–22 At the completion of each 6-week trial, participants were asked to complete electronic questionnaires every 28 days for up to 24 weeks to assess medication use patterns and use of additional antibiotics after study completion, and these data will be presented elsewhere.
Objectives and Outcomes
The primary objective of each trial determined whether discontinuing therapy is non-inferior to continuing therapy, as measured by the 6-week mean absolute change in ppFEV1.23 Secondary objectives included evaluating the safety of discontinuing therapy as measured by comparing adverse event rates between arms and proportions of participants that changed their assigned treatment regimen. Secondary outcomes included 6-week changes in the lung clearance index at 2.5% of the starting gas concentration (LCI2.5) measured by nitrogen multiple breath washout (MBW) among a sub-cohort enrolled at 27 participating sites trained and certified to perform MBW as described previously,24 and patient reported outcomes (PROs) including the Chronic Respiratory Infection Symptom Score (CRISS)25 and Cystic Fibrosis Questionnaire-Revised Respiratory Domain Score (CFQ-R).26 Additional secondary outcomes included the proportion of participants who initiated acute antibiotics courses, were hospitalized, or experienced a pulmonary exacerbation defined according to expanded Fuchs criteria.27
Statistical Analysis
The primary analysis for each trial was conducted on the per-protocol (PP) population (Supplement). The intent-to-treat (ITT) population was used for safety and sensitivity analyses with and without accounting for missing data (see Supplement for methods). Analysis of Variance (ANOVA) models adjusted for randomization strata were used to estimate the 6-week change within treatment arms and the difference between treatment arms in the 6-week change in ppFEV1 for each trial with corresponding 95% confidence intervals (CIs). Within-arm estimates of change were by least-squares means (LS means). A non-inferiority margin of −3% for the difference between arms in the 6-week change in ppFEV1 was established a priori with community input and clinical consensus during scientific review of the protocol.18 Pre-defined subgroup analyses were conducted. Differences between treatment groups in continuous secondary efficacy outcomes were estimated analogous to the primary outcome. Differences in secondary binary outcomes were estimated using logistic regression adjusting for randomization strata.
The type I error for the primary hypothesis was based on a one-sided 0.025 alpha-level test, reporting a two-sided 95% CI. A sample size of 400 enrolled per trial with assumed attrition and exclusions from the PP population less than 20% would result in at least 308 included in the primary analysis. Assuming a standard deviation for the 6-week change in ppFEV1 equal to 8.4,12,28 308 participants provided 88% power to detect non-inferiority with a margin of −3% assuming no effect of discontinuation. Interim safety data were reviewed every 6 months by a Data Monitoring Committee (DMC) from the CFF Data Safety Monitoring Board (DSMB).
RESULTS
The 6-week multi-center randomized trials in the SIMPLIFY study were conducted from September 2020 to July 2022 at 80 sites in the CF TDN in the U.S. The DA trial enrolled faster due to a higher proportion of enrollees using monotherapy with DA rather than HS, consistent with the broader U.S. CF population.29 The DA trial achieved its enrollment milestone in October 2021 but was provided DMC approval to over-enroll, with both trials completing enrollment in May 2022 just short of the enrollment milestones for the HS trial (see Supplement).
A total of 672 unique participants were assessed for eligibility across 1021 screening visits, resulting in 847 total randomizations across both studies among 594 unique participants (Figure 1). A subset of 253 (43%) of the 594 participants were using both HS and DA and participated in both trials. Upon completion of the screening period, eligible participants were randomly assigned to either discontinue (n=184 in HS trial, n=240 DA trial) or continue (n=186 HS trial, n=237 DA trial) treatment in each trial. Treatment arms within and across studies were well matched (Table 1) with a notably high average ppFEV1 across the entire study cohort of 97%. The majority (64% of 511) of participants with specific ETI start dates recorded, beyond confirmed recording of 90 days of use necessary for eligibility, were using ETI for 12 months or longer at the time of randomization into their first or only trial.
Figure 1.
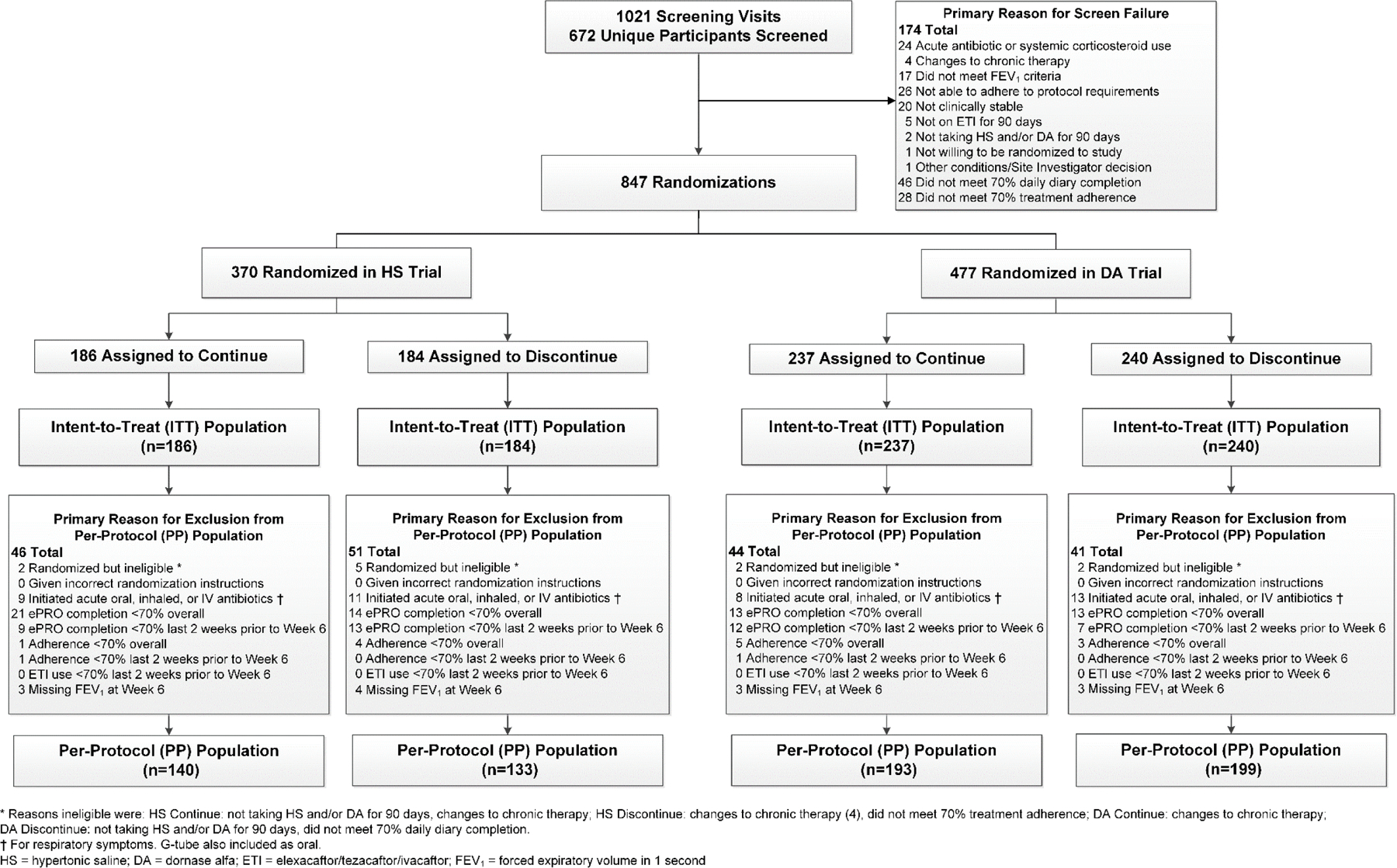
Flow of Study Participants
Table 1.
Demographic and Baseline Characteristics of Randomized Participants
| Hypertonic Saline (HS) Trial | Dornase Alfa (DA) Trial | |||||
|---|---|---|---|---|---|---|
| Continue N=186 | Discontinue N=184 | Total N=370 | Continue N=237 | Discontinue N=240 | Total N=477 | |
| Sex at Birth | ||||||
| Male | 99 (53.2%) | 97 (52.7%) | 196 (53.0%) | 117 (49.4%) | 127 (52.9%) | 244 (51.2%) |
| Female | 87 (46.8%) | 87 (47.3%) | 174 (47.0%) | 120 (50.6%) | 113 (47.1%) | 233 (48.8%) |
| Age (years) | ||||||
| N | 186 | 184 | 370 | 237 | 240 | 477 |
| Mean (SD) | 22.4 (10.73) | 21.7 (10.28) | 22.0 (10.50) | 23.2 (11.50) | 21.9 (9.19) | 22.6 (10.41) |
| Age Distribution | ||||||
| ≥12 to <18 | 92 (49.5%) | 90 (48.9%) | 182 (49.2%) | 108 (45.6%) | 110 (45.8%) | 218 (45.7%) |
| ≥18 to <24 | 39 (21.0%) | 37 (20.1%) | 76 (20.5%) | 48 (20.3%) | 51 (21.3%) | 99 (20.8%) |
| ≥24 to <30 | 18 (9.7%) | 32 (17.4%) | 50 (13.5%) | 31 (13.1%) | 32 (13.3%) | 63 (13.2%) |
| ≥30 | 37 (19.9%) | 25 (13.6%) | 62 (16.8%) | 50 (21.1%) | 47 (19.6%) | 97 (20.3%) |
| Race | ||||||
| White | 182 (97.8%) | 176 (95.7%) | 358 (96.8%) | 227 (95.8%) | 231 (96.3%) | 458 (96.0%) |
| Black or African American | 1 (0.5%) | 2 (1.1%) | 3 (0.8%) | 4 (1.7%) | 1 (0.4%) | 5 (1.0%) |
| Asian | 0 (0%) | 1 (0.5%) | 1 (0.3%) | 1 (0.4%) | 0 (0%) | 1 (0.2%) |
| American Indian or Alaskan Native | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (0.4%) | 1 (0.2%) |
| Native Hawaiian or Other Pacific Islander | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Other/Unknown* | 3 (1.6%) | 5 (2.7%) | 8 (2.2%) | 5 (2.1%) | 7 (2.9%) | 12 (2.5%) |
| Ethnicity | ||||||
| Hispanic or Latino | 6 (3.2%) | 14 (7.6%) | 20 (5.4%) | 15 (6.3%) | 19 (7.9%) | 34 (7.1%) |
| ot Hispanic or Latino | 180 (96.8%) | 170 (92.4%) | 350 (94.6%) | 222 (93.7%) | 221 (92.1%) | 443 (92.9%) |
| Genotype Group | ||||||
| F508del Homozygous | 111 (59.7%) | 111 (60.3%) | 222 (60.0%) | 140 (59.1%) | 129 (53.8%) | 269 (56.4%) |
| F508del Heterozygous | 72 (38.7%) | 71 (38.6%) | 143 (38.6%) | 91 (38.4%) | 106 (44.2%) | 197 (41.3%) |
| Other/Unknown | 3 (1.6%) | 2 (1.1%) | 5 (1.4%) | 6 (2.5%) | 5 (2.1%) | 11 (2.3%) |
| FEV1 (% predicted) | ||||||
| N | 186 | 184 | 370 | 237 | 240 | 477 |
| Mean (SD) | 97.2 (16.50) | 96.7 (17.17) | 97.0 (16.82) | 97.2 (16.32) | 96.5 (17.34) | 96.8 (16.83) |
| FEV1 (% predicted) Distribution | ||||||
| <60 | 1 (0.5%) | 1 (0.5%) | 2 (0.5%) | 3 (1.3%) | 1 (0.4%) | 4 (0.8%) |
| ≥60 to <70 | 10 (5.4%) | 13 (7.1%) | 23 (6.2%) | 11 (4.6%) | 20 (8.3%) | 31 (6.5%) |
| ≥70 to <90 | 53 (28.5%) | 46 (25.0%) | 99 (26.8%) | 63 (26.6%) | 57 (23.8%) | 120 (25.2%) |
| ≥90 to <100 | 33 (17.7%) | 45 (24.5%) | 78 (21.1%) | 51 (21.5%) | 53 (22.1%) | 104 (21.8%) |
| ≥100 | 89 (47.8%) | 79 (42.9%) | 168 (45.4%) | 109 (46.0%) | 109 (45.4%) | 218 (45.7%) |
| LCI2.5 † | ||||||
| N | 47 | 52 | 99 | 61 | 69 | 130 |
| Mean (SD) | 7.8 (2.15) | 8.2 (2.56) | 8.0 (2.37) | 7.9 (1.77) | 8.2 (2.73) | 8.1 (2.32) |
| Prior Study Enrollee | 62 (33.3%) | 62 (33.7%) | 124 (33.5%) | 64 (27.0%) | 65 (27.1%) | 129 (27.0%) |
| Current Chronic Therapy | ||||||
| DA | 156 (83.9%) | 154 (83.7%) | 310 (83.8%) | 237 (100%) | 240 (100%) | 477 (100%) |
| HS | 186 (100%) | 184 (100%) | 370 (100%) | 147 (62.0%) | 148 (61.7%) | 295 (61.8%) |
| ETI | 186 (100%) | 184 (100%) | 370 (100%) | 237 (100%) | 240 (100%) | 477 (100%) |
| Airway clearance | 178 (95.7%) | 174 (94.6%) | 352 (95.1%) | 228 (96.2%) | 226 (94.2%) | 454 (95.2%) |
| Inhaled antibiotic (Continuous) | 7 (3.8%) | 2 (1.1%) | 9 (2.4%) | 3 (1.3%) | 6 (2.5%) | 9 (1.9%) |
| Inhaled antibiotic (Cycled) | 51 (27.4%) | 43 (23.4%) | 94 (25.4%) | 50 (21.1%) | 56 (23.3%) | 106 (22.2%) |
| Inhaled antibiotic (Continuous Alternating) | 17 (9.1%) | 15 (8.2%) | 32 (8.6%) | 25 (10.5%) | 23 (9.6%) | 48 (10.1%) |
| Oral antibiotic | 85 (45.7%) | 81 (44.0%) | 166 (44.9%) | 103 (43.5%) | 104 (43.3%) | 207 (43.4%) |
| Ibuprofen | 8 (4.3%) | 15 (8.2%) | 23 (6.2%) | 20 (8.4%) | 21 (8.8%) | 41 (8.6%) |
| Systemic steroids | 1 (0.5%) | 2 (1.1%) | 3 (0.8%) | 2 (0.8%) | 3 (1.3%) | 5 (1.0%) |
| Previous Modulator Use ‡ | ||||||
| Ivacaftor | 8 (4.3%) | 1 (0.5%) | 9 (2.4%) | 7 (3.0%) | 12 (5.0%) | 19 (4.0%) |
| Lumacaftor/Ivacaftor | 49 (26.3%) | 54 (29.3%) | 103 (27.8%) | 69 (29.1%) | 62 (25.8%) | 131 (27.5%) |
| Tezacaftor/Ivacaftor | 45 (24.2%) | 43 (23.4%) | 88 (23.8%) | 53 (22.4%) | 55 (22.9%) | 108 (22.6%) |
| Positive Microbiology Culture (past year) § | ||||||
| Pseudomonas aeruginosa | 57 (30.6%) | 39 (21.2%) | 96 (25.9%) | 63 (26.6%) | 66 (27.5%) | 129 (27.0%) |
| Staphylococcus aureus | 118 (63.4%) | 137 (74.5%) | 255 (68.9%) | 163 (68.8%) | 158 (65.8%) | 321 (67.3%) |
| Methicillin-resistant staphylococcus aureus | 34 (18.3%) | 40 (21.7%) | 74 (20.0%) | 56 (23.6%) | 54 (22.5%) | 110 (23.1%) |
| Stenotrophomonas maltophilia | 9 (4.8%) | 11 (6.0%) | 20 (5.4%) | 19 (8.0%) | 9 (3.8%) | 28 (5.9%) |
| Achromobacter xylosoxidans | 1 (0.5%) | 2 (1.1%) | 3 (0.8%) | 4 (1.7%) | 4 (1.7%) | 8 (1.7%) |
| Burkholderia cepacia complex | 3 (1.6%) | 2 (1.1%) | 5 (1.4%) | 4 (1.7%) | 4 (1.7%) | 8 (1.7%) |
| Haemophilus influenzae | 9 (4.8%) | 5 (2.7%) | 14 (3.8%) | 12 (5.1%) | 8 (3.3%) | 20 (4.2%) |
| Mycobacterium abscessus | 2 (1.1%) | 1 (0.5%) | 3 (0.8%) | 1 (0.4%) | 2 (0.8%) | 3 (0.6%) |
| Mycobacterium avium complex | 1 (0.5%) | 3 (1.6%) | 4 (1.1%) | 4 (1.7%) | 3 (1.3%) | 7 (1.5%) |
Data are mean (SD) or n (%). Percentages may not sum to 100 in each category due to rounding.
Other includes participants of more than one race.
LCI result could be obtained at either the Week –2 or Week 0 Visit.
Participants may fall into more than one category of prior modulator use.
Culture results obtained clinically within 12 months prior to screening.
Randomized participants met eligibility after a two-week screening which required ≥70% compliance with electronic daily medication use reporting and ≥70% adherence to applicable therapies. Of the 1021 study screenings, 46 (5%) and 28 (3%) did not result in randomization due to lack of adequate compliance or adherence, respectively (Figure 1). Compliance with electronic daily medication usage reporting during the 6-week trial continued to be high, with 303 (82%) and 422 (89%) of 370 and 477 participants overall in the HS and DA trials, respectively, achieving ≥70% compliance. The 6-week average reported treatment regimen adherence to either discontinue or continue therapy was also high at 96% and 98% amongst the 184 and 186 participants in the HS trial, respectively, and 97% and 96% amongst the 240 and 237 participants in the DA trial, respectively. The 6-week average reported ETI adherence was even higher across treatment arms in both trials (99%), with consistent reported use of airway clearance therapy (ACT) throughout the study (Table S2).
A total of 273 of 370 (74%) participants in the HS and 392 of 477 (82%) in the DA study comprised the per-protocol population, with reasons for exclusion provided in Figure 1. Participant characteristics were comparable between the ITT and PP study populations for both trials (Tables S3-S4).
The average change in ppFEV1 during the two-week run-in period and prior to randomization was 0.12% in the HS trial (n=370, 95% CI: −0.27,0.50) and 0.11% in the DA trial (n=477, 95% CI: −0.22,0.43). The absolute change in ppFEV1 from baseline (Week 0) to Week 6 in the PP population of the HS trial was −0.19% (n=133, 95% CI: −0.85,0.48) in the discontinuation arm as compared to 0.14% (n=140, 95% CI: −0.51,0.78) in the continuation arm, reflecting a between-group difference (discontinuation minus continuation) of –0.32% (95% CI: −1.25, 0.60) (Figure 2). In the PP population of the DA trial, a mean 6-week change of 0.18% was observed in the discontinuation arm (n=199, 95% CI: −0.38,0.74) as compared to −0.16% in the continuation arm (n=193, 95% CI: −0.73,0.41), with between-group difference 0.35% (95% CI: −0.45,1.14) (Figure 2). In both trials, NI was established with the lower bounds of the 95% CI for the treatment differences exceeding the pre-defined threshold of −3%.18 Sensitivity analyses using the ITT population produced consistent treatment effect estimates (Figure 2), and estimates were additionally consistent across pre-defined subgroups (Figure 3 and S2) as well as for the change in ppFEV1 from Week 0 to Week 2 and Week −2 to Week 6 (Tables S5-S6). While the study was not powered to detect significant differences between subgroups, the lower bound of the CIs consistently exceeded the NI margin with the exception of the smallest subgroups with the widest CIs in both directions including those not using ACT, non-white study participants, and Hispanic or Latino participants. Notably, consistent results were seen between those participating in one versus both trials; additional analyses of the subgroups participating in both trials will be presented elsewhere. Graphical displays of individual participant 6-week changes in ppFEV1 are provided in Figures S4 and S5.
Figure 2.
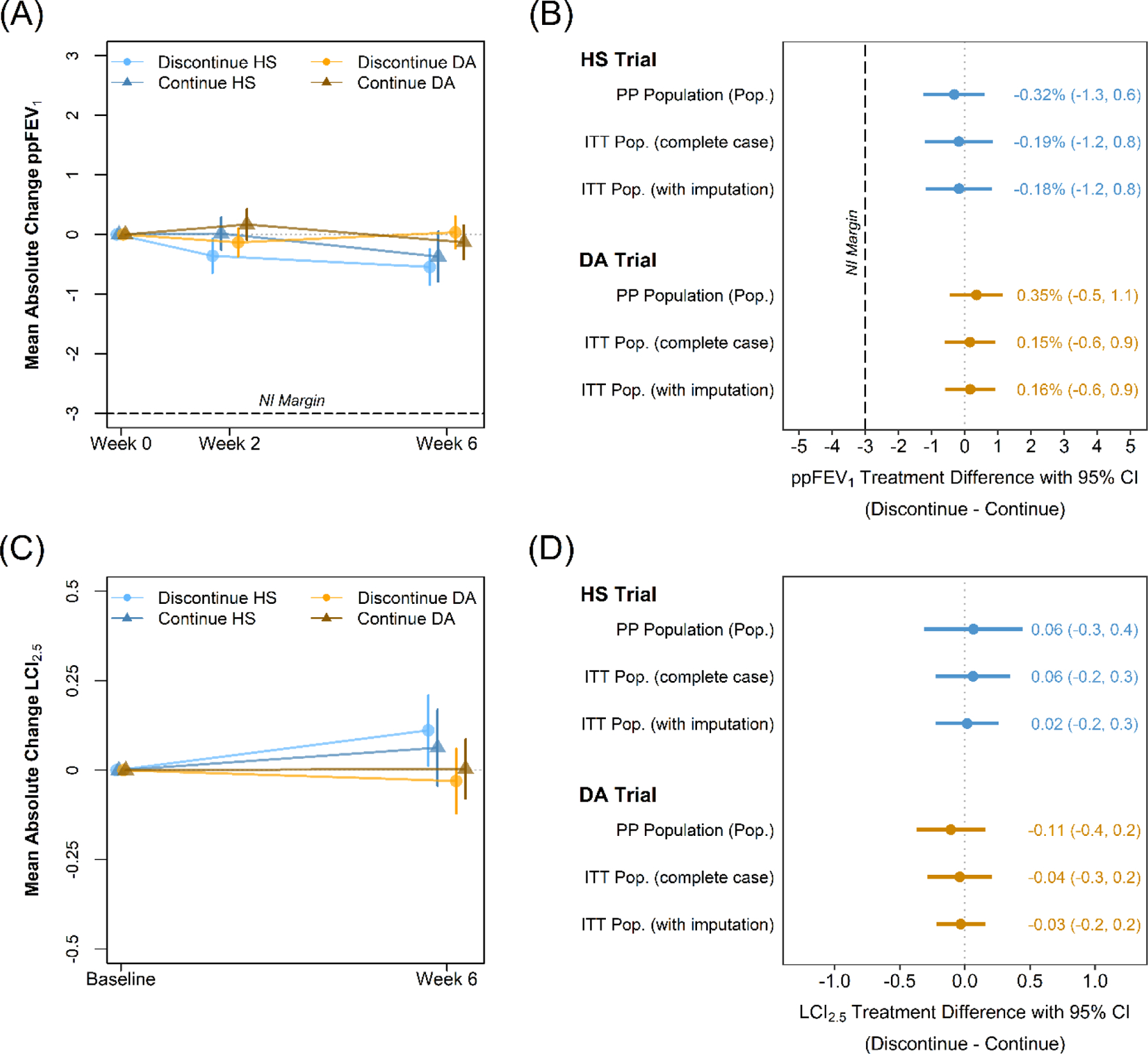
Unadjusted mean absolute changes from baseline in (A) ppFEV1 and (C) LCI2.5 among all randomized participants. Bars indicate standard errors. Estimated differences between the discontinuation and continuation arms in the 6-week change in (B) ppFEV1 and (D) LCI2.5 among the primary PP population and ITT population with and without imputation to account for missing data. Treatment differences adjusted for randomization strata.
Figure 3.
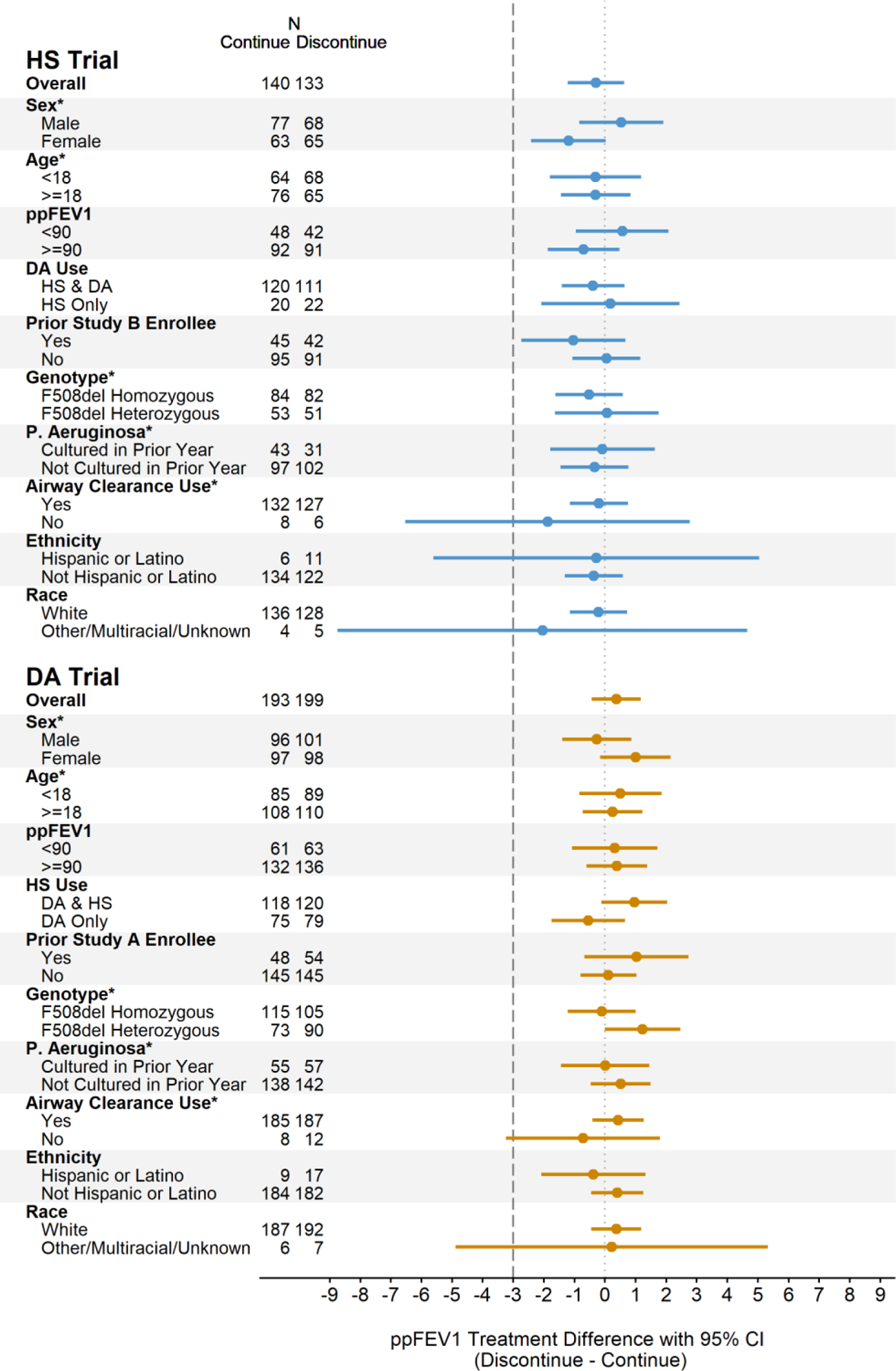
Difference between the discontinuation and continuation treatment arms in the 6-week change in ppFEV1 among subgroups in the PP population in the HS trial and DA trial. Treatment differences adjusted for randomization strata. Pre-defined subgroups noted with *. Sensitivity analyses in the ITT population provided in Figure S2.
LCI2.5 was obtained among a subset of participants with demonstrated comparability to the main trial cohort (Table S7-S8). Among participants in the PP population in the HS trial, a 6-week mean absolute change in LCI2.5 of 0.17 (n=31, 95% CI: −0.09,0.43) was observed in the discontinuation arm as compared to 0.11 in the continuation arm (n=28, 95% CI: −0.16,0.38), with associated treatment difference 0.06 (95% CI: −0.31,0.44). Among participants in the PP population in the DA trial, a 6-week mean change of −0.11 (n=40, 95% CI: −0.29,0.08) was observed in the discontinuation arm as compared to 0.00 in the continuation arm (n=42, 95% CI: −0.18,0.19), with associated treatment difference −0.11 (95% CI: −0.37,0.15). Confidence interval bounds for the treatment differences exclude the smallest published treatment-associated changes in LCI2.5 equal to 0.5 (Figure 2).30 Sensitivity analyses in the ITT population and as a relative change demonstrated consistency (Figure S5).
Baseline respiratory symptom scores measured through the CRISS and CFQ-R (respiratory domain) indicated a low prevalence of symptoms among the participants in each trial, averaging 11.5 (standard deviation [sd] 12.2) and 94.1 (sd=9.9) in the HS trial and 12.0 (sd=11.8) and 95.0 (sd=7.9) in the DA trial, respectively. Larger numbers in CRISS represent more symptoms while larger numbers in CFQ-R represent fewer symptoms. There were no significant differences between treatment arms in 6-week CRISS or CFQ-R symptom score changes in either trial or across analysis populations (Figure S6). In the PP population, the treatment difference (discontinuation minus continuation) in the 6-week change in CRISS score was −1.5 in the HS trial (95% CI: −4.0,0.9) and 1.0 in the DA trial (95% CI: −1.0,3.0). Upper bounds of the 95% CIs exclude a previously suggested minimally important difference of 11.31 Similarly, the treatment difference in the 6-week change in CFQ-R score was 0.2 in the HS trial (95% CI: −1.7,2.1) and −0.9 in the DA trial (95% CI: −2.4,0.6). The lower CI bounds exclude a previously established minimal clinically important difference of −4.32 Incidence of protocol defined pulmonary exacerbations and hospitalizations were infrequent and comparable between treatment groups for each trial (Table S9-S10).
There were no treatment-related serious adverse events reported in either trial and the majority of trial participants experienced no adverse events during the 6-week trial (Table 2). Higher relative percentages of participants experienced at least one adverse event in the discontinuation versus continuation arms in the HS trial (35%, 64 of 184 vs. 24%, 44 of 186 participants) and in the DA trial (37%, 89 of 240 vs. 23%, 55 of 237 participants), predominantly driven by respiratory AEs (Table 2). However, no single AE was observed in more than 7% of participants within a given trial arm. Post-hoc analyses demonstrated that differences in respiratory AEs were more prominent among a small subset of participants with ppFEV1 <70% (Table S11), although no systematic associations were identified between baseline ppFEV1 and change in ppFEV1 (Figure S7).
Table 2.
Overview of Safety Outcomes
| Hypertonic Saline (HS) Trial |
Dornase Alfa (DA) Trial |
|||
|---|---|---|---|---|
| Continue N=186 | Discontinue N=184 | Continue N=237 | Discontinue N=240 | |
| Participants with any SAE, n (%) | 1 (0.5%) | 2 (1.1%) | 0 (0.0%) | 0 (0.0%) |
| Abdominal Pain | 0 (0.0%) | 1 (0.5%) | 0 (0.0%) | 0 (0.0%) |
| Infective Pulmonary Exacerbation | 1 (0.5%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) |
| Suicidal ideation | 0 (0.0%) | 1 (0.5%) | 0 (0.0%) | 0 (0.0%) |
| Participants with any AE, n (%) | 44 (23.7%) | 64 (34.8%) | 55 (23.2%) | 89 (37.1%) |
| Maximum AE Severity, n (%) * | ||||
| Mild | 23 (52.3%) | 41 (64.1%) | 36 (65.5%) | 62 (69.7%) |
| Moderate | 20 (45.5%) | 18 (28.1%) | 15 (27.3%) | 24 (27.0%) |
| Severe † | 1 (2.3%) | 4 (6.3%) | 4 (7.3%) | 3 (3.4%) |
| Life Threatening ‡ | 0 (0.0%) | 1 (1.6%) | 0 (0.0%) | 0 (0.0%) |
| Participants with at least one respiratory AE, n (%) | 22 (11.8%) | 30 (16.3%) | 24 (10.1%) | 47 (19.6%) |
| Participants with AE leading to Treatment Regimen Modification, n (%) | 1 (0.5%) | 5 (2.7%) | 2 (0.8%) | 7 (2.9%) |
| Participants with AE leading to Physician Directed Modification, n (%) | 1 (0.5%) | 0 (0.0%) | 1 (0.4%) | 4 (1.7%) |
| Participants with Most Common AEs, n (%) § | ||||
| Cough | 7 (3.8%) | 7 (3.8%) | 10 (4.2%) | 16 (6.7%) |
| Nasal Congestion | 10 (5.4%) | 5 (2.7%) | 4 (1.7%) | 11 (4.6%) |
| Chest Discomfort | 1 (0.5%) | 6 (3.3%) | 1 (0.4%) | 11 (4.6%) |
| Sputum Increased | 3 (1.6%) | 3 (1.6%) | 2 (0.8%) | 9 (3.8%) |
| COVID-19 | 2 (1.1%) | 6 (3.3%) | 1 (0.4%) | 6 (2.5%) |
| Infective Pulmonary Exacerbation | 5 (2.7%) | 3 (1.6%) | 3 (1.3%) | 7 (2.9%) |
| Myalgia | 2 (1.1%) | 1 (0.5%) | 1 (0.4%) | 6 (2.5%) |
| Headache | 3 (1.6%) | 4 (2.2%) | 6 (2.5%) | 4 (1.7%) |
Data are n (%).
Percentages out of participants with any AE.
The AEs classified as severe were the following, participants could have more than one severe event: Hypertonic Saline Continue: infective pulmonary exacerbation; Hypertonic Saline Discontinue: abdominal pain, chest discomfort, juvenile idiopathic arthritis, nasal congestion, pulmonary congestion; Dornase Alfa Continue: diarrhoea, eye infection bacterial, rib fracture, upper respiratory tract infection, wrist fracture; Dornase Alfa Discontinue: chest pain, immunization reaction, infective pulmonary exacerbation.
The AE classified as life threatening was suicidal ideation.
Most common AEs defined as those occurring in at least 2.5% of participants in any treatment group across studies.
DISCUSSION
SIMPLIFY is the first study to test whether pwCF who are clinically stable on CFTR modulator drug therapy that substantially restores CFTR function, specifically ETI, can discontinue either inhaled DA or HS without experiencing a meaningful decline in lung function. Through randomized, controlled non-inferiority trials, our study demonstrated that the individual discontinuation of either inhaled therapy was not associated with diminished lung function as measured by the 6-week change in ppFEV1. These results are strengthened by lack of meaningful changes in LCI2.5 and patient-reported respiratory outcome scores. There were no significant safety concerns with overall low rates of adverse events across both treatment arms, though slightly higher respiratory adverse event rates were observed in those discontinuing, which were more prominent in the small subgroup representing 7% or less of the total trial populations with lower lung function (ppFEV1 60 to <70%). Collectively, these data point to an important opportunity to reevaluate daily treatment requirements wherein routine use of certain longstanding inhaled therapies may not be required to maintain pulmonary function in individuals likely experiencing substantial drug-induced restoration of CFTR function and relatively good pulmonary health as a result of ETI.12,17,28
SIMPLIFY was designed to test for the non-inferiority of discontinuing versus continuing therapy with respect to the 6-week change in ppFEV1, assessed using a pre-defined non-inferiority margin of −3% derived with community input.18 Notably, a study summarizing the trade-offs people with CF were willing to make to reduce treatment burden published after the establishment of the NI margin used in SIMPLIFY suggested that people were willing to accept a 4.4% reduction in ppFEV1 (95% CI: 2.6,6.3) or −2.3 years of additional life expectancy (95% CI: 1.3,3.3) to halve the time spent on inhaled medicines.33 Based on the lower bounds of the CIs for the treatment effects observed in our study, we were able to rule out declines of more than −1.3% ppFEV1 associated with discontinuing HS and −0.6% with discontinuing DA among this study population. While the PP population was the primary analysis population, consistent results were observed in the ITT population and across several relevant subgroups. Confidence in the interpretation of the primary endpoint was increased by results from the LCI2.5 endpoint, available among a subset of participants at sites that performed MBW. LCI is a more sensitive measure of change in pulmonary function—particularly in pwCF who have preserved ppFEV1.34–38 In the youngest and healthiest populations with CF studied, LCI2.5 changes as small as 0.9 were observed for a 4-week study period of DA and as small as 0.5 were observed over a 48-week study of HS.30,35,36,39 Despite a much older study population in SIMPLIFY, the mean differences in LCI2.5 change were near zero and the 95% CI bounds excluded an undesirable effect of discontinuation.
The 6-week period to continue or discontinue each inhaled therapy was carefully considered when designing this study.18 Adherence to the assigned regimen and other prescribed therapies is critically important to confirming or refuting non-inferiority. A relatively short study period undoubtedly improves adherence to enable a more robust examination of the physiological effects of discontinuing therapy. It is equally important to ask whether six weeks was sufficient to capture changes in lung function. Several studies of DA consistently show a maximum impact on ppFEV1 within 7 days of starting therapy and a return to baseline within 2–3 weeks after stopping the medication.6,40–43 Recent small studies of children in Canada and Denmark reported changes in LCI2.5 within one month after starting or stopping DA, respectively.35,44 Reports of the effects of HS on pulmonary function also find maximum effect and return to baseline forppFEV1 or LCI within 4 weeks or by the first study visit.7,36,39,45 We are not aware of data demonstrating an independent, delayed effect on lung function beyond the first several weeks when starting or stopping either inhaled therapy. Collectively, these studies indicate that the 6-week observational period in SIMPLIFY, which was necessary to maximize adherence to treatment regimen, was suitable to observe changes in ppFEV1 or LCI after discontinuing HS or DA. We recognize that longer-term changes in pulmonary function were not directly tested in this study and therefore could occur despite the lack of evidence indicating latent and/or time-dependent effects on lung function from historical trials.6,7,41,46
It is vital to recognize the study population when interpreting these findings. As the first large-scale trial to test withdrawal of pre-existing daily therapy, we intentionally recruited a healthier population based on ppFEV1 criteria of ≥70% for adolescents (ages 12–17y) and ≥60% for adults. The mean ppFEV1 in SIMPLIFY was 97% (mean age 22 years). For comparison, an ongoing real-world observational study of ETI use in the U.S., which had no baseline criteria for lung function, recently reported the mean ppFEV1 at 6 months into ETI use was just over 90% with mean age of 25 years.17 The results of SIMPLIFY may not represent what would occur in pwCF with pulmonary function significantly outside the range observed in our study population. Within this study, a small number of adults with ppFEV1 60 to <70% were enrolled, and in this group, more respiratory adverse events were reported in those randomized to stop an inhaled medication. Baseline ppFEV1 did not appear systematically associated with change in ppFEV1 and the majority of respiratory adverse events were mild or moderate. Importantly however, this may suggest greater caution when considering changes to daily therapy in individuals with more advanced CF pulmonary disease.
We carefully monitored self-reported adherence to study therapy (DA and/or HS), ETI, and mechanical airway clearance through a daily electronic questionnaire. To be randomized, participants had to complete ≥70% of daily questionnaires over 2 weeks during screening and report use of their baseline therapies ≥70% of all days reported. This design element successfully enriched for a study population with high adherence to study assignment, near 100% for ETI and >95% for adherence to their assigned treatment (i.e., discontinue or continue the inhaled medication). Our adherence rates in these 6-week trials are higher than expected in longer term studies and higher than what can be expected in real-world use of medications or other daily therapies in pwCF.47 When testing for non-inferiority of discontinuing versus continuing therapy, it is desirable to create the most extreme setting to produce the most conservative treatment effect (e.g. in this case allowing for greater declines in lung function to be observed). Lesser adherence to the assigned treatment regimens would be more likely to demonstrate non-inferiority between treatment arms as actual use would overlap more. This is particularly true for studies involving discontinuation of medications or therapies that remain available to study participants. Thus, lesser adherence to HS or DA in clinical use is unlikely to affect the generalizability of our findings under idealized conditions, but it is unknown whether such high adherence to the study protocol extends to other aspects of health maintenance and if that would impact the generalizability of our findings. Adherence to ETI was also high in this study, and it is possible that the effects of discontinuation of therapy would differ among those non-adherent to modulator therapy. Additional research into perspectives on participation in the SIMPLIFY study, including reporting daily therapy use, was conducted in a complementary qualitative study (QUEST, NCT04320381) and will be reported separately.
Respiratory symptoms were measured using the CFQ-R respiratory domain and CRISS scores, which indicated remarkably low presence of symptoms at baseline across the cohort. The observed differences in scores between those discontinuing vs. continuing inhaled DA or HS were negligible with confidence intervals excluding minimum clinically important differences for these instruments.31,32 SIMPLIFY was not designed or powered to evaluate risk of acute pulmonary exacerbation, so we cannot assess whether there is greater risk associated with discontinuing therapy independent of unobserved changes in ppFEV1, LCI, or respiratory symptoms. An ongoing pragmatic clinical trial being conducted in the United Kingdom (CF-STORM, EudraCT number 2020–005864-77) and a U.S. registry-based real-world study (HERO-2, NCT04798014) are currently considering the effects of withdrawing inhaled mucoactive therapies on lung function over longer observational periods and may provide additional important data regarding the impact of treatment withdrawal on pulmonary exacerbations.
Hospitalizations, acute antibiotic use, and serious adverse events occurred in only a few participants in each trial and did not differ between treatment arms. Overall absolute adverse event rates were low and no significant safety concerns were identified. A markedly lower proportion of participants experienced an adverse event across all arms of the SIMPLIFY study than was observed in a comparable duration (4-week) trial of ETI versus tezacaftor/ivacaftor, whereby approximately two-thirds experienced at least one adverse event across treatment groups.28 No single AE in SIMPLIFY was reported in ≥7% of participants, though a relative increased rate of respiratory related AEs was reported in the discontinuation as compared to continuation arm of both trials amidst overall low absolute rates. These AEs were mostly mild upper and lower respiratory symptoms and were found more often in participants with baseline ppFEV1 <70%. It is possible that discontinuing daily inhaled therapy resulted in increased upper or lower respiratory symptoms for some and must be considered particularly for those with lower lung function. Notably, this study could not adequately blind (i.e., placebo) these familiar inhaled medications for participants and site investigators, and it is unclear whether this fact impacted self-reported symptoms or decisions to restart therapy in a handful of participants. The SIMPLIFY study was conducted during the SARS-CoV-2 pandemic and social distancing measures that may have reduced the overall rates of acute respiratory illness. What impact this may have had on our safety data is unclear, as low rates of acute pulmonary exacerbations were demonstrated in those on ETI even prior to the pandemic reaching the U.S.12
This study has certain limitations, including the aforementioned inability to blind participants in the study, particularly for longstanding inhaled therapies with visible characteristics and distinctive taste. Further, home electronic adherence monitoring devices were unfeasible. Thus, daily self-reporting methods were used as supported in other settings and developed in collaboration with the CF Foundation’s Success with Therapies Consortium (STRC).18 Consistent and high reporting throughout the trials, small differences in adherence rates between oral ETI and other inhaled therapies, and minimal ppFEV1 change between screening and randomization visits all strengthen our confidence in the accuracy of daily reporting in this study. Further limitations include the higher baseline ppFEV1 and much lower rate of acute pulmonary exacerbation in this study population as compared with historical study populations in trials of HS and DA and the intentional 6-week observational period as discussed above. As is inherent to clinical trials, the average treatment responses reported may not predict individual experience if discontinuing either HS or DA. While there was no suggestion of differential findings across subgroups, the study was not adequately powered to test for these differences. Additional studies will also be required to address critical questions surrounding the necessity and timing of initiation of these traditional standard of care therapies, particularly in young pediatric populations with well-preserved lung function in the presence of ETI or similar CFTR restorative drugs. This study also did not examine withdrawal of regular mechanical airway clearance therapy, and additional research would be needed to understand the effects of altering such foundational therapies.
In summary, the results of SIMPLIFY indicate that among a study population of adolescents and adults with CF who have relatively good lung function established on ETI, clinically meaningful declines in pulmonary function did not occur with short-term discontinuation of daily use of inhaled medications that work on downstream manifestations of CFTR dysfunction in the airway, specifically HS and DA. It is recognized that clinicians and pwCF must partner to make individualized decisions with available evidence from SIMPLIFY and forthcoming studies regarding the continuance of chronic HS or DA in the setting of ETI use. It is reasonable to hypothesize that pwCF on potent modulator drug therapy may still benefit from inhaled DA and HS when experiencing increased respiratory symptoms or as part of a regimen to address acute pulmonary exacerbations. A substudy measuring changes in mucociliary clearance through nuclear medicine imaging was conducted in SIMPLIFY and will be published separately. These data may provide additional evidence to show whether discontinuing DA or HS for 6 weeks resulted in physiological changes in the airway. An as-necessary approach to these medications may facilitate knowledge gained from long term studies that consider impact on health stability over time. We are grateful to the CF community and study teams that contributed to SIMPLIFY and hope these results will inform shared decision making for many pwCF benefiting from CFTR restorative therapies now and in the future.
Supplementary Material
RESEARCH IN CONTEXT.
Evidence Before this Study
The highly effective modulator elexacaftor/tezacaftor/ivacaftor (ETI) substantially restores cystic fibrosis transmembrane conductance regulator (CFTR) protein function and improves mucociliary clearance, thereby reducing mucus accumulation and airway obstruction. It is unknown whether additional therapies developed to improve mucus clearance from the lungs of people with CF, specifically hypertonic saline (HS) and dornase alfa (DA), remain clinically necessary in individuals treated with ETI. Studies assessing the impact of discontinuing such therapies will contribute critical evidence towards determining whether standard of care can be modified to reduce treatment burden after establishment of a highly effective modulator. Currently there are no large randomized, controlled trials that have tested the effects of withdrawing a chronic daily therapy after establishment of CFTR modulators.
Added Value of this Study
SIMPLIFY is the first study to assess the impact of discontinuing standard of care therapy after establishment of ETI in adolescents and adults 12 years of age and older. The study was comprised of two parallel randomized, open-label, controlled trials that independently tested whether discontinuation of either HS or DA is non-inferior to continuation of therapy over a 6-week study period as measured by the primary outcome, change in percent predicted forced expiratory volume in one second (ppFEV1). The 6-week observational period is consistent with the time frame for which the effects of DA and HS on lung function have been observed in prior studies. Secondary objectives evaluated whether there were meaningful changes in lung clearance index (LCI), safety, and patient reported outcomes during the 6-week study period.
Implications of All the Available Evidence
Among adolescents and adults with CF established on ETI with relatively good lung function, discontinuation of either HS or DA did not result in clinically meaningful changes in lung function including ppFEV1 and LCI at 2.5 starting concentration (LCI2.5). No significant safety concerns were identified with overall low rates of adverse events, although slightly higher rates were observed in those discontinuing inhaled therapies and appeared more likely to occur in those with lower lung function. SIMPLIFY is the first study to provide evidence suggesting discontinuation of burdensome inhaled therapies such as HS or DA is not associated with short-term lung function changes among individuals benefiting from ETI. Collectively, the results provide an opportunity to reevaluate daily treatment requirements, wherein longstanding use of certain inhaled medications may not be required to maintain pulmonary function in individuals receiving drugs known to substantially improve CFTR function and respiratory health.
Acknowledgements
Special thanks to the individuals with CF and their families who participated in this study, and whose dedication to research made the study possible. We would also like to thank the CFF for supporting this study through the CF TDN, and the CF Data Safety Monitoring Board (DSMB) with Data Monitoring Committee (DMC) led by Dr. Lynne Quittell. Funding for the SIMPLIFY study was provided by the CFF. NMH was supported by CFF grant HAMBLE20K0 and National Institutes of Health (NIH) grants P30 DK 089507 and UL1 TR002319. KR was supported by CFF RIEKER15PE0. SD was supported by CFF SIMPLIFY-DONALD20K0 and NIH 5P30DK065988. GRB was supported by NIH UL1TR002489. CG was supported by NIH P30 DK 089507 and UL1 TR002319. DPN was supported by CFF NICHOL20K0 and SINGHH19R0 and NIH 2P30DK089507. AHG was supported by the CFF SIMPLIFY-GIFFOR20K0 and GIFFOR17Y5 and NIH P30 DK 117469.
Footnotes
Data Sharing
Upon completion of the study and publication of results for all study objectives, de-identified datasets will be available through the CFF TDN Data Archive. Researchers may apply to the CF TDN for use of de-identified data from the archive for research purposes (tdncc@seattlechildrens.org). Applicants must receive appropriate IRB approval before data is sent from the TDN Data Archive.
Declaration of Interests
GRB reports grants and contracts from Vertex Pharmaceuticals and the CFF. SD reports contracts from AstraZeneca, Calithera, CFF, NIH, Vertex Pharmaceuticals, consulting fees from Polarean, 501 Ventures, and Chiesi USA, Inc, fees for advisory boards for Enterprise Therapeutics and Gilead Sciences, and participation on a DSMB for Abbvie and Boehringer Ingleheim. JTC reports grants and contracts from CFF, Vertex Pharmaceutics, Eloxx, and 4DMT, and consulting fees from Vertex Phamaceuticals, Insmed, and 4DMT, participation on a DSMB for Abbvie, and serving on an advisory board for CFF, American Thoracic Society (ATS), Journal of CF, and Emily’s Entourage. KR reports grants from CFF, royalties from Springer Publishing, honoraria from Vertex Pharmaceuticals, and serving on an advisory board for ATS. AB reports grants from the CFF for TDN studies. BO declares no competing interests. KOD declares no competing interests. FR reports grants from Vertex Phamaceuticals and consulting fees from Vertex Phamaceuticals, Proteostasis, Translate Bio, Boehringer Ingelheim, and Calithera. AG declares no competing interests. GO declares no competing interests. JY reports grants from CFF. CW declares no competing interests. SM reports grants from CFF to support TDN studies. DN reports grants from CFF and NIH, consulting fees from BiomX, Clarametyx, Genentech, GlaxoSmithKline, Nabriva, Respirion, and Vertex Pharmaceuticals, and advisory board membership for CFF and Kither Biotechnology. DR reports grants and contracts from CFF and Vertex Pharmaceuticals. AHG reports grants and contracts from CFF, Insmed, Incorporated, AbbVie, Incorporated, 4D Molecular Therapeutics. GS reports advisory board participation for Vertex Pharmaceuticals and Gilead Sciences. RR reports no competing interests. KM declares no competing interests. CHG reports grants and contracts from CFF, NIH, and the Food and Drug Administration, consulting fees from Enterprise Therapeutics, and honoraria from Gilead Sciences, Novartis, Boehringer Ingelheim, Vertex Phamaceuticals, and stock options in Aer Therapeutics. JPC reports employment at the CFF. NMH reports grants from CFF, NIH, and FDA, consulting fees from Enterprise Therapeutics, and DSMB membership for the NIH.
REFERENCES
- 1.Shteinberg M, Haq IJ, Polineni D, Davies JC. Cystic fibrosis. Lancet 2021; 397(10290): 2195–211. [DOI] [PubMed] [Google Scholar]
- 2.Nichols DP, Chmiel JF. Inflammation and its genesis in cystic fibrosis. Pediatric pulmonology 2015; 50 Suppl 40: S39–56. [DOI] [PubMed] [Google Scholar]
- 3.Batson B, Zorn B, Radicioni G, et al. Cystic Fibrosis Airway Mucus Hyperconcentration Produces a Vicious Cycle of Mucin, Pathogen, and Inflammatory Interactions that Promote Disease Persistence. Am J Respir Cell Mol Biol 2022; 67(2):253–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blanchard AC, Waters VJ. Microbiology of Cystic Fibrosis Airway Disease. Seminars in respiratory and critical care medicine 2019; 40(6): 727–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma JT, Tang C, Kang L, Voynow JA, Rubin BK. Cystic Fibrosis Sputum Rheology Correlates With Both Acute and Longitudinal Changes in Lung Function. Chest 2018; 154(2): 370–7. [DOI] [PubMed] [Google Scholar]
- 6.Fuchs HJ, Borowitz DS, Christiansen DH, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. The New England journal of medicine 1994; 331(10): 637–42. [DOI] [PubMed] [Google Scholar]
- 7.Elkins MR, Robinson M, Rose BR, et al. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. The New England journal of medicine 2006; 354(3): 229–40. [DOI] [PubMed] [Google Scholar]
- 8.Donaldson SH, Bennett WD, Zeman KL, Knowles MR, Tarran R, Boucher RC. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. The New England journal of medicine 2006; 354(3): 241–50. [DOI] [PubMed] [Google Scholar]
- 9.Lahiri T, Hempstead SE, Brady C, et al. Clinical Practice Guidelines From the Cystic Fibrosis Foundation for Preschoolers With Cystic Fibrosis. Pediatrics 2016; 137(4). [DOI] [PubMed] [Google Scholar]
- 10.Mogayzel PJ Jr., Naureckas ET, Robinson KA, et al. Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. American journal of respiratory and critical care medicine 2013; 187(7): 680–9. [DOI] [PubMed] [Google Scholar]
- 11.Sawicki GS, Sellers DE, Robinson WM. High treatment burden in adults with cystic fibrosis: Challenges to disease self-management. Journal of Cystic Fibrosis 2009; 8(2): 91–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Middleton PG, Mall MA, Drevinek P, et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. The New England journal of medicine 2019; 381(19): 1809–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keating D, Marigowda G, Burr L, et al. VX-445–Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. New Engl J Med 2018; 379(17): 1612–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graeber SY, Renz DM, Stahl M, et al. Effects of Elexacaftor/Tezacaftor/Ivacaftor Therapy on Lung Clearance Index and Magnetic Resonance Imaging in Patients with Cystic Fibrosis and One or Two F508del Alleles. American journal of respiratory and critical care medicine 2022; 206(3):311–20. [DOI] [PubMed] [Google Scholar]
- 15.Rowbotham NJ, Smith S, Leighton PA, et al. The top 10 research priorities in cystic fibrosis developed by a partnership between people with CF and healthcare providers. Thorax 2018; 73(4): 388–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gifford AH, Mayer-Hamblett N, Pearson K, Nichols DP. Answering the call to address cystic fibrosis treatment burden in the era of highly effective CFTR modulator therapy. Journal of cystic fibrosis: official journal of the European Cystic Fibrosis Society 2020; 19(5): 762–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nichols DP, Paynter AC, Heltshe SL, et al. Clinical Effectiveness of Elexacaftor/Tezacaftor/Ivacaftor in People with Cystic Fibrosis: A Clinical Trial. American journal of respiratory and critical care medicine 2022; 205(5): 529–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mayer-Hamblett N, Nichols DP, Odem-Davis K, et al. Evaluating the Impact of Stopping Chronic Therapies after Modulator Drug Therapy in Cystic Fibrosis: The SIMPLIFY Clinical Trial Study Design. Annals of the American Thoracic Society 2021; 18(8): 1397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burke LE, Shiffman S, Music E, et al. Ecological Momentary Assessment in Behavioral Research: Addressing Technological and Human Participant Challenges. J Med Internet Res 2017; 19(3): e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shiyko MP, Perkins S, Caldwell L. Feasibility and adherence paradigm to ecological momentary assessments in urban minority youth. Psychol Assess 2017; 29(7): 926–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gaudiano BA, Ellenberg S, Price LH, Moitra E. Time-lagged predictors of daily medication nonadherence beliefs during the month post-hospital discharge in patients with psychotic-spectrum disorders. Psychiatry research 2018; 270: 253–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacDonell K, Gibson-Scipio W, Lam P, Naar-King S, Chen X. Text messaging to measure asthma medication use and symptoms in urban African American emerging adults: a feasibility study. The Journal of asthma : official journal of the Association for the Care of Asthma 2012; 49(10): 1092–6. [DOI] [PubMed] [Google Scholar]
- 23.Quanjer PH, Stanojevic S, Cole TJ, et al. Multi-ethnic reference values for spirometry for the 3–95-yr age range: the global lung function 2012 equations. The European respiratory journal 2012; 40(6): 1324–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saunders C, Jensen R, Robinson PD, et al. Integrating the multiple breath washout test into international multicentre trials. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2020; 19(4): 602–7. [DOI] [PubMed] [Google Scholar]
- 25.Goss CH, Edwards TC, Ramsey BW, Aitken ML, Patrick DL. Patient-reported respiratory symptoms in cystic fibrosis. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2009; 8(4): 245–52. [DOI] [PubMed] [Google Scholar]
- 26.Quittner AL, Sawicki GS, McMullen A, et al. Psychometric evaluation of the Cystic Fibrosis Questionnaire-Revised in a national sample. Quality of life research : an international journal of quality of life aspects of treatment, care and rehabilitation 2012; 21(7): 1267–78. [DOI] [PubMed] [Google Scholar]
- 27.VanDevanter DR, Hamblett NM, Simon N, McIntosh J, Konstan MW. Evaluating assumptions of definition-based pulmonary exacerbation endpoints in cystic fibrosis clinical trials. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2021;20(1): 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heijerman HGM, McKone EF, Downey DG, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet 2019;394(10212):1940–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.2020 Cystic Fibrosis Foundation Patient Registry Report Bethesda, MD: Cystic Fibrosis Foundation; 2020. [Google Scholar]
- 30.Tiddens H, Chen Y, Andrinopoulou ER, et al. The effect of inhaled hypertonic saline on lung structure in children aged 3–6 years with cystic fibrosis (SHIP-CT): a multicentre, randomised, double-blind, controlled trial. The lancet Respiratory medicine 2022; 10(7): 669–78. [DOI] [PubMed] [Google Scholar]
- 31.Goss CH, Caldwell E, Gries KS, et al. Validation of a novel patient-reported respiratory symptoms instrument in cystic fibrosis: CFRSD-CRISS. Pediatric pulmonology 2013; 48(S36): 295–96.22553136 [Google Scholar]
- 32.Quittner AL, Modi AC, Wainwright C, Otto K, Kirihara J, Montgomery AB. Determination of the Minimal Clinically Important Difference Scores for the Cystic Fibrosis Questionnaire-Revised Respiratory Symptom Scale in Two Populations of Patients With Cystic Fibrosis and Chronic Pseudomonas aeruginosa Airway Infection. Chest 2009; 135(6): 1610–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cameron RA, Office D, Matthews J, et al. Treatment preference amongst people with cystic fibrosis: the importance of reducing treatment burden. Chest 2022. [DOI] [PMC free article] [PubMed]
- 34.Ratjen F, Klingel M, Black P, et al. Changes in Lung Clearance Index in Preschool-aged Patients with Cystic Fibrosis Treated with Ivacaftor (GOAL): A Clinical Trial. American journal of respiratory and critical care medicine 2018; 198(4): 526–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amin R, Subbarao P, Lou W, et al. The effect of dornase alfa on ventilation inhomogeneity in patients with cystic fibrosis. The European respiratory journal 2011; 37(4): 806–12. [DOI] [PubMed] [Google Scholar]
- 36.Ratjen F, Davis SD, Stanojevic S, et al. Inhaled hypertonic saline in preschool children with cystic fibrosis (SHIP): a multicentre, randomised, double-blind, placebo-controlled trial. The lancet Respiratory medicine 2019; 7(9): 802–9. [DOI] [PubMed] [Google Scholar]
- 37.Davies J, Sheridan H, Bell N, et al. Assessment of clinical response to ivacaftor with lung clearance index in cystic fibrosis patients with a G551D-CFTR mutation and preserved spirometry: a randomised controlled trial. The lancet Respiratory medicine 2013; 1(8): 630–8. [DOI] [PubMed] [Google Scholar]
- 38.Davies G, Stanojevic S, Raywood E, et al. An observational study of the lung clearance index throughout childhood in cystic fibrosis: early years matter. The European respiratory journal 2020; 56(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amin R, Subbarao P, Jabar A, et al. Hypertonic saline improves the LCI in paediatric patients with CF with normal lung function. Thorax 2010; 65(5): 379–83. [DOI] [PubMed] [Google Scholar]
- 40.Ranasinha C, Assoufi B, Shak S, et al. Efficacy and safety of short-term administration of aerosolised recombinant human DNase I in adults with stable stage cystic fibrosis. Lancet 1993; 342(8865): 199–202. [DOI] [PubMed] [Google Scholar]
- 41.Eisenberg JD, Aitken ML, Dorkin HL, et al. Safety of repeated intermittent courses of aerosolized recombinant human deoxyribonuclease in patients with cystic fibrosis. The Journal of pediatrics 1997; 131(1 Pt 1): 118–24. [DOI] [PubMed] [Google Scholar]
- 42.Shah PL, Scott SF, Fuchs HJ, Geddes DM, Hodson ME. Medium term treatment of stable stage cystic fibrosis with recombinant human DNase I. Thorax 1995; 50(4): 333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quan JM, Tiddens HA, Sy JP, et al. A two-year randomized, placebo-controlled trial of dornase alfa in young patients with cystic fibrosis with mild lung function abnormalities. The Journal of pediatrics 2001; 139(6): 813–20. [DOI] [PubMed] [Google Scholar]
- 44.Voldby C, Green K, Philipsen L, et al. Withdrawal of dornase alfa increases ventilation inhomogeneity in children with cystic fibrosis. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2021; 20(6): 949–56. [DOI] [PubMed] [Google Scholar]
- 45.Donaldson SH, Danielle Samulski T, LaFave C, et al. A four week trial of hypertonic saline in children with mild cystic fibrosis lung disease: Effect on mucociliary clearance and clinical outcomes. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2020; 19(6): 942–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eng PA, Morton J, Douglass JA, Riedler J, Wilson J, Robertson CF. Short-term efficacy of ultrasonically nebulized hypertonic saline in cystic fibrosis. Pediatric pulmonology 1996; 21(2): 77–83. [DOI] [PubMed] [Google Scholar]
- 47.Quittner AL, Zhang J, Marynchenko M, et al. Pulmonary medication adherence and health-care use in cystic fibrosis. Chest 2014; 146(1): 142–51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.