

Abstract
Ewing Sarcoma (ES) is a cancer of bone and soft tissues affecting mostly children and young adults. Aggressive progression and poor prognosis of this malignancy call for novel and targeted treatments. CD99 is a transmembrane protein that is abundantly expressed on ES cells and is a diagnostic marker for the disease. ES cells are selectively sensitive to CD99 inhibition compared to most normal cells and other tumors. Therefore, CD99 is a good molecular target for ES treatment. Clofarabine and cladribine are two FDA approved drugs that are administered for their inhibitory acts on DNA synthesis to treat relapsed or refractory acute lymphoblastic and myeloid leukemia. They have also been shown to directly bind to CD99 and inhibit ES growth through a distinct mechanism. In the current study, we designed, synthesized and tested new ES specific derivatives of both drugs that would continue to target CD99 but with expected reduction in cellular membrane permeability and rendered unsuitable for inhibiting DNA synthesis. By using commercially available clofarabine and cladribine purine nucleoside analogs, we modified the primary alcohol moiety at the deoxyribose C-5’ terminal site to suppress phosphorylation and thus inhibition of subsequent DNA synthesis pathways. In addition, we incorporated a variety of polar groups in the ribose and purine rings to reduce membrane permeability and investigated the effects of configurational changes in the sugar moiety. Among 26 new derivatives, we identified two compounds, BK50164 and BK60106, that cause cell death specifically in ES primarily due to inhibition of CD99 but not via inhibition of DNA synthesis. These findings provide a road map for the future development selective CD99 inhibitors for targeted treatment of ES.
Keywords: Clofarabine, Cladribine, CD99 inhibitors, Ewing sarcoma, Nucleoside derivatization, Targeted Therapy
Graphical abstract
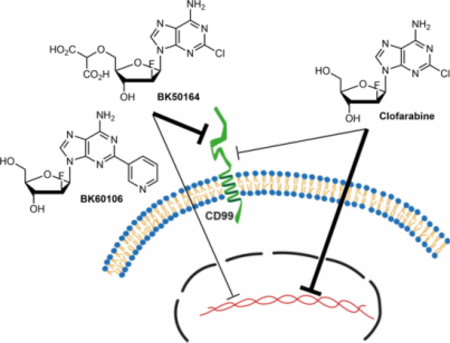
1. Introduction
CD99 is a heavily O-glycosylated, 32 kDa single pass (type I) transmembrane protein that is encoded by human pseudoautosomal MIC2 gene with limited homology to only XG (PBDX) and CD99L2 (MIC2L1) proteins.1–3 A true functional ligand for CD99 has yet to be identified. Interactions between murine CD99 and its paralog CD99-like-2 (CD99L2), paired immunoglobulin-like type 2 (PILR) and CD99 itself (as homodimers) have been reported.4–6 However, all these interactions were demonstrated with mouse CD99, which has only 45% aa homology to human CD99. None of these interactions have been confirmed in human cells yet. CD99 is primarily expressed on the plasma membrane of specific cell populations in hematopoietic system and gonads and plays a role in transendothelial migration, activation of T-cells and positive selection of thymocytes during their maturation.1 CD99 expression is specifically elevated in Ewing sarcoma (ES), lymphoma, leukemia, and myeloid malignancies with oncogenic roles.7–12
ES is a malignant tumor that grows in and around bone and soft tissues. ES affects mainly children and young adults with survival rate of ~75% for patients with localized disease and ~30% for those with metastatic or recurrent diseases.13 CD99 expression is a significant diagnostic marker for the histopathological diagnosis of ES.14–18 Besides its diagnostic value, CD99 plays an important role for the ES progression by inhibiting neuronal differentiation and by enhancing migratory abilities of ES cells.19,20 These malignancy driving properties make CD99 a potential therapeutic target for the treatment of ES. In fact, reduced expression of CD99 by genetic manipulation abrogates the tumorigenic characteristics of ES cells and they can no longer form tumors and bone metastases when xenografted into mice.19,21 Additionally, engagement of CD99 by CD99-specific antibodies delivers apoptotic or other cell death related pathway stimuli to ES cells and alleviates their malignant potential.22–24 We previously discovered two FDA-approved compounds, clofarabine and cladribine, as CD99 inhibitors by screening a library of compounds for their ability to directly bind to extracellular domain of recombinant CD99 protein.25,26 These drugs impaired CD99 dimerization and its molecular interactions with its known downstream signaling partners. Both clofarabine and cladribine inhibited the tumorigenic capability of ES cells including the growth of xenografts.
Cladribine and clofarabine are purine nucleoside analogs. They are given as prodrugs and they need to be successively phosphorylated in the cytoplasm by deoxycytidine kinase (DCK) to 5’-monophosphate metabolites, and subsequently by mono- and diphosphokinases to the active 5’-triphosphate form. Active forms of the drugs inhibit the cellular ribonucleotide reductase which results in reduction in the pools of deoxyribonucleotide triphosphates and cause impairment of DNA synthesis and repair, hence increase subsequent DNA damage triggering programmed cell death.27 Due to concerns for the stability of cladribine, clofarabine was later iteratively developed by addressing known deficiencies of the prior nucleoside analog and became the focus of further development.28,29
Inhibition of CD99 protein on the plasma membrane of Ewing sarcoma cells does not require entry in to the cytoplasm or conversion of clofarabine and cladribine to their 5’-triphosphate form. Therefore, clofarabine and cladribine analogs that cannot be phosphorylated and cannot inhibit subsequent DNA biosynthesis or that do not penetrate the plasma membrane but still maintain CD99 affinity are of considerable interest due to their promise for the development of an alternative ES treatment that exploits this relatively unexplored mechanism to overcome the inherent cell toxicity of the parent compounds. To date, relatively few drugs designed to exhibit reduced cell permeability have been studied. Examples include a cyclosporine derivative that targets extracellular cyclophilins,30,31 a small-molecule analog of geldanamycin which was used as cell-impermeable inhibitor of HSP90 and shown to reduce tumor cell migration and invasion,32 and oligopeptide derivatives of doxorubicin.33 Cell-impermeable probes or stains such as SYTOX Green, which only crosses compromised membranes characteristic of dead cells but does not penetrate intact cells, have been used for imaging purposes and as diagnostic tools.34–37
To the best of our knowledge, the potential of cell-impermeable analogs of nucleosides used in cancer treatments has not been systematically investigated. We therefore decided to selectively modify the primary alcohol moiety at the deoxyribose C-5’ terminal site in clofarabine and cladribine to suppress the phosphorylation pathway, to incorporate a variety of polar groups in the ribose and purine rings to decrease cell membrane permeability, and to investigate the effect of configurational changes in the sugar moiety (Figure 1). We identified two novel compounds, BK60106 and BK50164, as derivatives of clofarabine that retain the selective cytotoxicity on ES cells but with lessened DNA damage compared to the parent compound. These results provide promising leads for the development of specific CD99 targeted therapies for ES patients.
Figure 1.
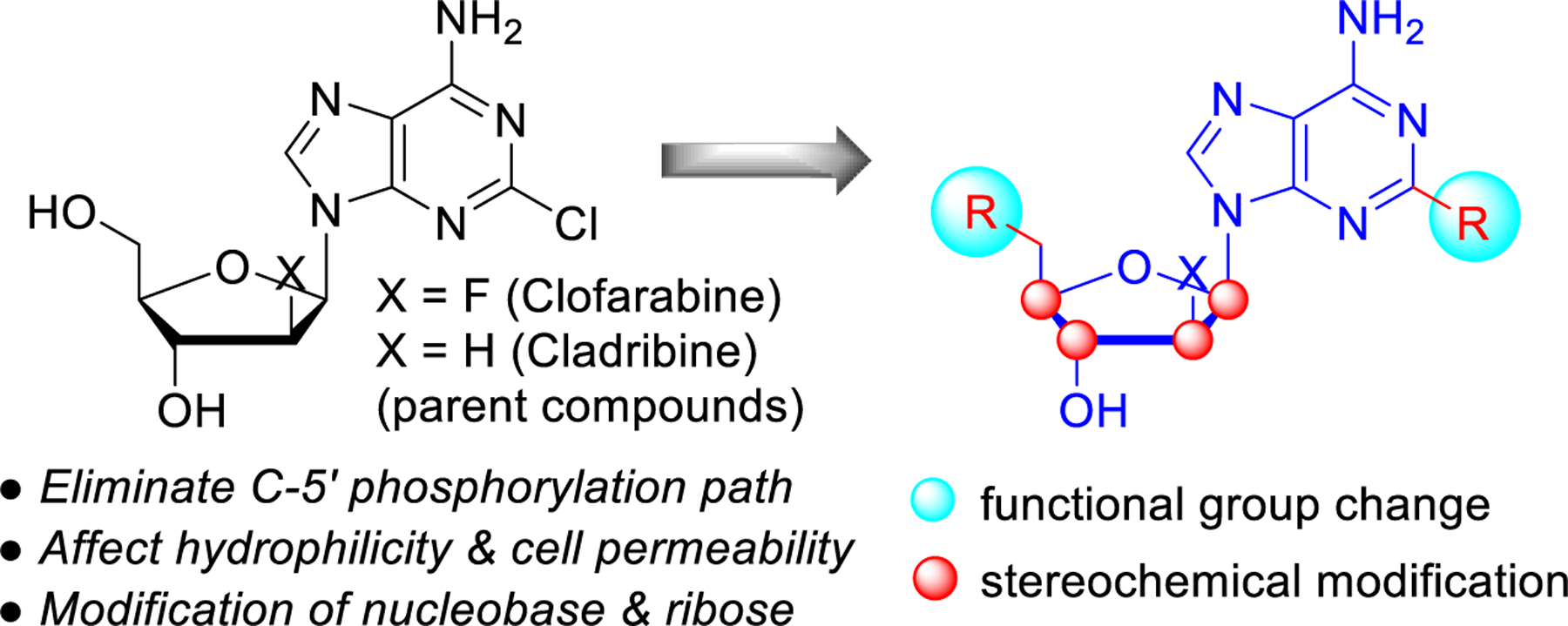
General compound design strategy.
2. Results and discussion
2.1. Synthesis of modified clofarabine and cladribine derivatives
At the beginning of this study, we identified the commercially available compounds 1-6 for the synthesis of new CD99 inhibitors and we explored the possibility of direct derivatization of the C-5’ hydroxyl group in clofarabine (1) and cladribine (2) (Figure 2). Despite several attempts, derivatization of the primary alcohol group by Mitsunobu reactions or transition metal catalyzed O–H insertion were unsuccessful and complicated product mixture were obtained due to side reactions at the C-3’ secondary hydroxyl group and at the amino group in the purine ring. Formation of a chloride, bromide, mesylate or tosylate for subsequent substitution with a carbon or heteroatom nucleophile in varying solvents also failed to produce the desired products and nucleophilic aromatic substitution of the chloride in the purine ring was observed at elevated temperatures.
Figure 2.
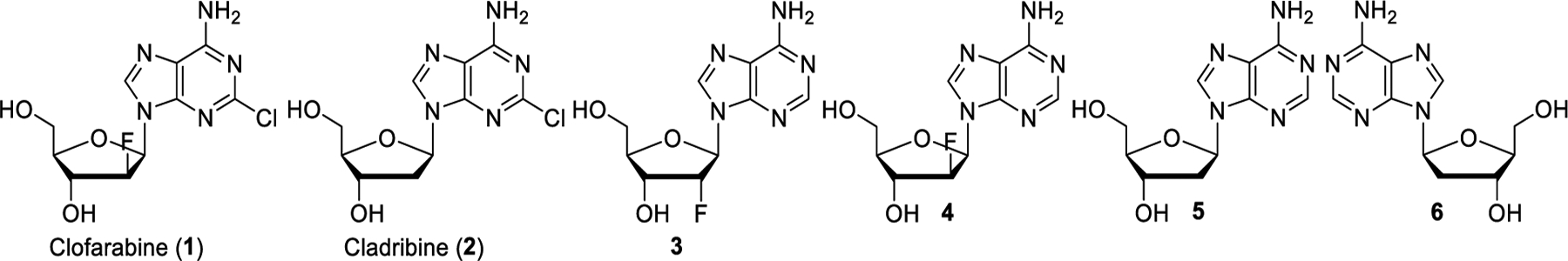
Structures of the parent drugs and commercially available stereoisomers used in this study.
We therefore resorted to protection chemistry. We expected that Boc protection of the 6-amino and the C-2’ hydroxyl groups would allow selective manipulation of the C-5’ hydroxyl group and facile deprotection under mild reaction conditions. Thus, compound 9 was prepared in three steps from commercially available clofarabine by following a literature method (Scheme 1).38 First, clofarabine was transformed to 7 using TBDPSCl in 91% yield. The amino and secondary hydroxyl groups were then treated with (Boc)2O at room temperature to give 8 in 82% yield. Silyl deprotection was carried out with TBAF to provide the Boc-protected clofarabine derivative 9 in 93% yield. Under identical reaction conditions, compound 12 was prepared from cladribine in three steps.
Scheme 1.
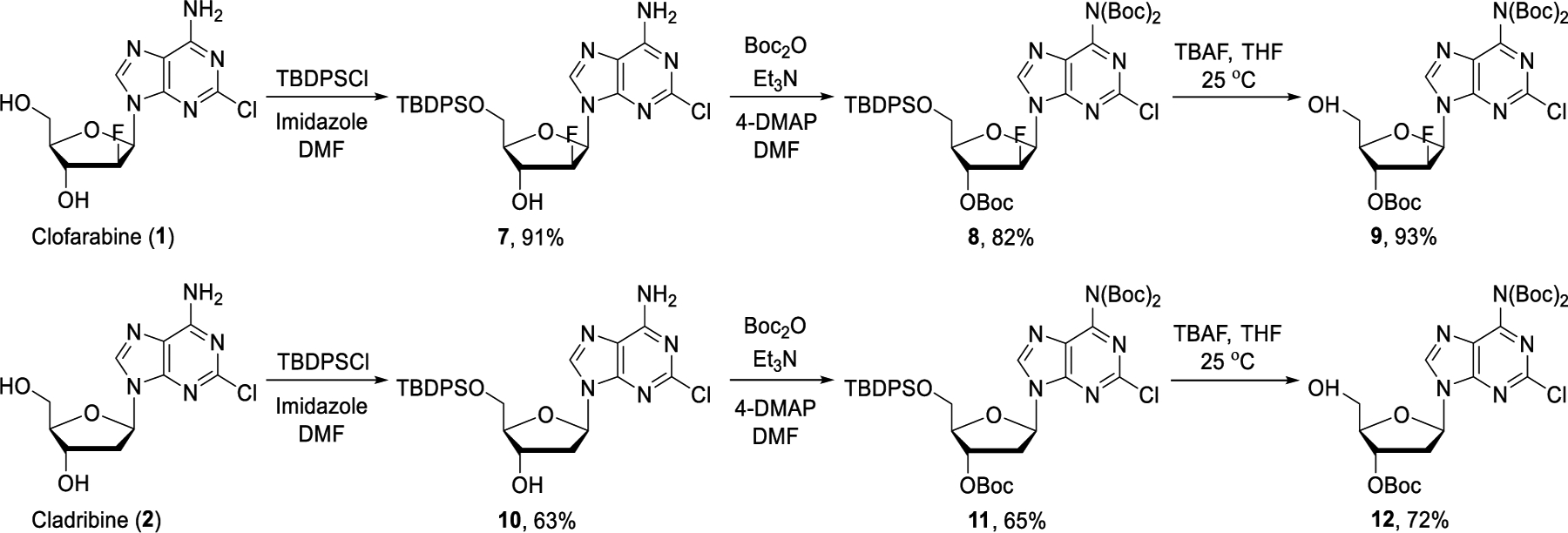
Synthesis of Boc-protected clofarabine and cladribine.
With the Boc-protected clofarabine derivative 9 in hand, we continued with introducing a dicarboxylic acid unit into the ribose ring (Scheme 2). Mitsunobu reaction of compound 9 with dimethyl 5-hydroxyisophthalate (13) gave the diester 14 in 65% yield which was then treated with trifluoroacetic acid (TFA) to afford compound 16 in 94% yield.39 The final product BK50161 was isolated in 76% yield after basic hydrolysis of 16. Identical Mitsunobu reaction conditions were applied to prepare the cladribine analog from intermediate 15 but N-glycosidic bond cleavage was observed during TFA mediated Boc deprotection. Reducing the amounts of TFA or temperature did not improve results and attempts to avoid decomposition by using other acids such as HCl and acetic acid proved fruitless. Fortunately, silica-mediated Boc-deprotection was found to cleanly convert 15 to diester 17 and subsequent hydrolysis using LiOH furnished BK50174 in 82% yield.
Scheme 2.
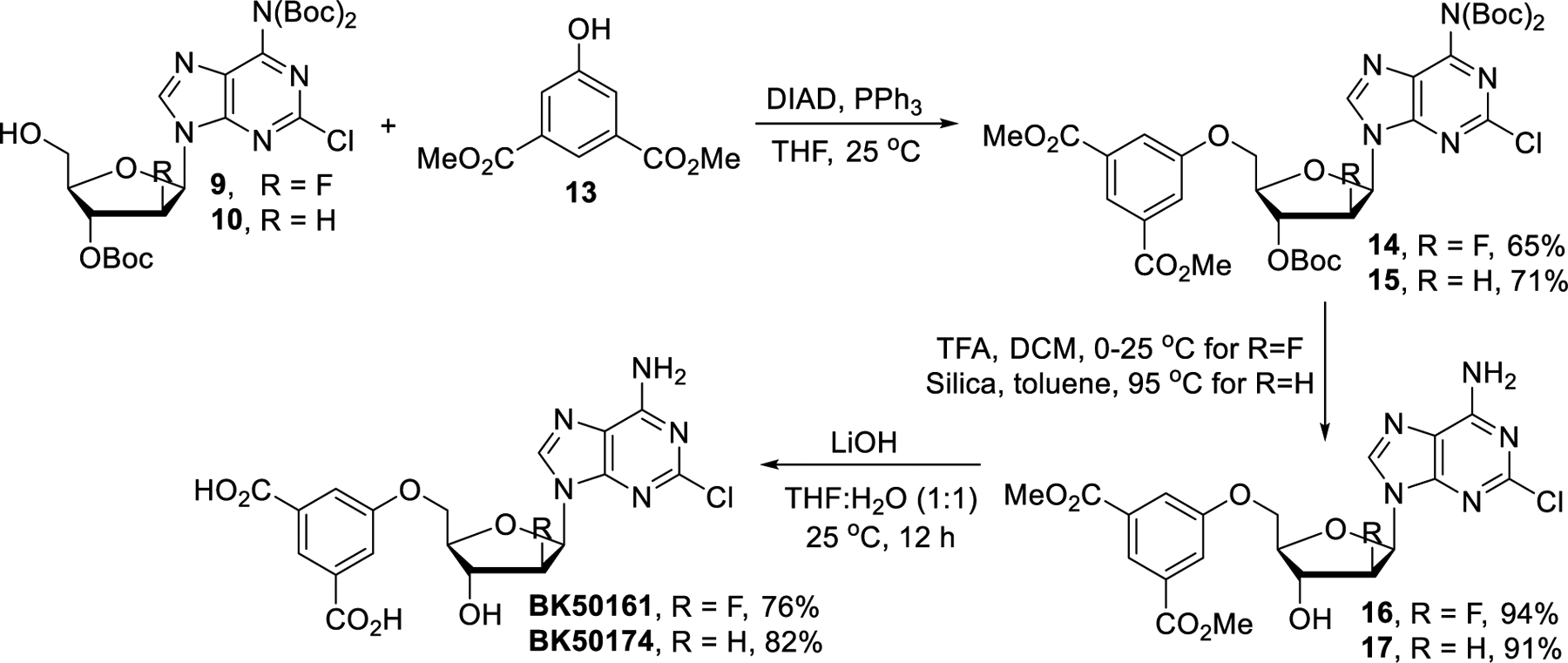
Synthesis of BK50161 and BK50174.
BK50164 was prepared by following a literature report (Scheme 3).40,41 Dimethyl 2-diazomalonate (18)42 was employed in rhodium catalyzed O-H insertion of the Boc-protected clofarabine 9. The free dicarboxylic acid BK50164 was then accessed in high yields from 19 in two steps following a sequence analogous to that employed for the synthesis of BK50161 (vide supra).
Scheme 3.
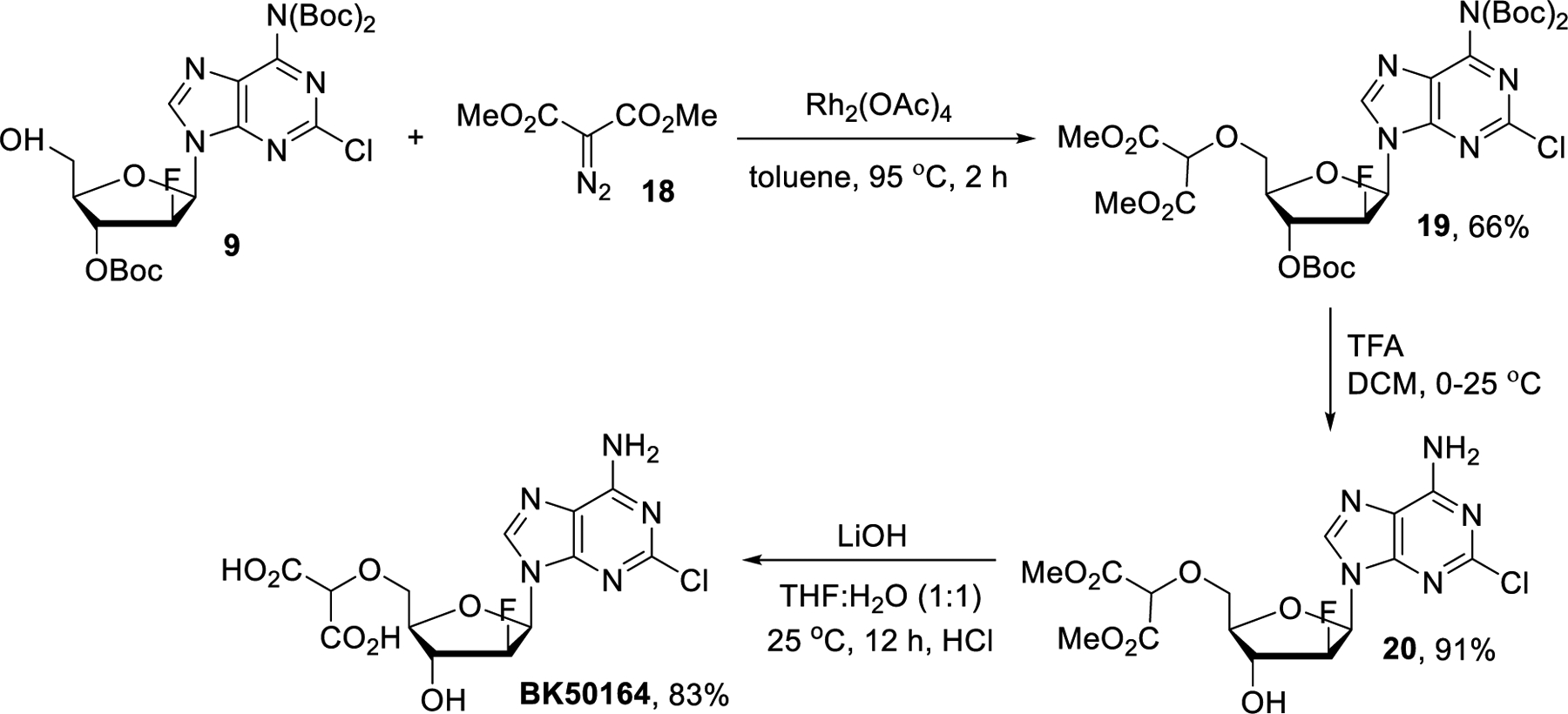
Synthesis of BK50164.
After successful preparation of the dicarboxylic acid derivatives, we turned our attention toward the synthesis of stereoisomeric clofarabine and cladribine analogs. We screened conditions for direct oxidation of the primary alcohol group and found that this is possible with TEMPO/PhI(OAc)2 upon mild heating (Scheme 4). The carboxylic acid derivatives BK5082, BK60182, BK60129 and BK60131 were thus produced directly from commercially available nucleosides in 54–71% yields without protection chemistry.
Scheme 4.
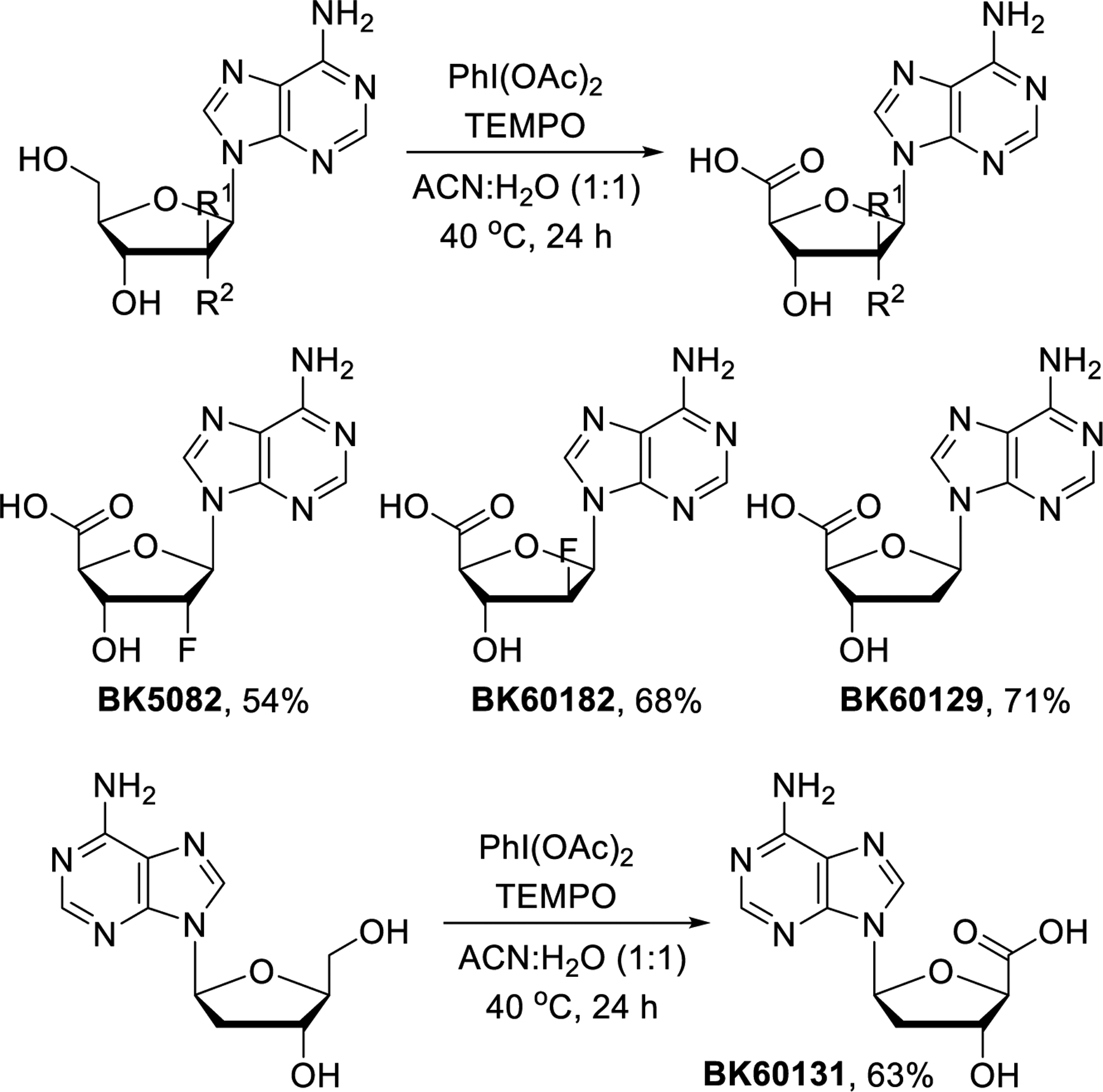
Synthesis of 5’-carboxylic acid derivatives.
The unsuccessful attempts with nucleophilic substitutions of unprotected nucleosides carrying C-5’ chloride, bromide, mesylate or tosylate leaving groups led us to revise this strategy using the corresponding iodide derivatives instead. Selective iodination of the primary alcohol group was easily achieved by following literature methods and we obtained compounds 21-26 in varying but workable yields (Scheme 5).43–47
Scheme 5.
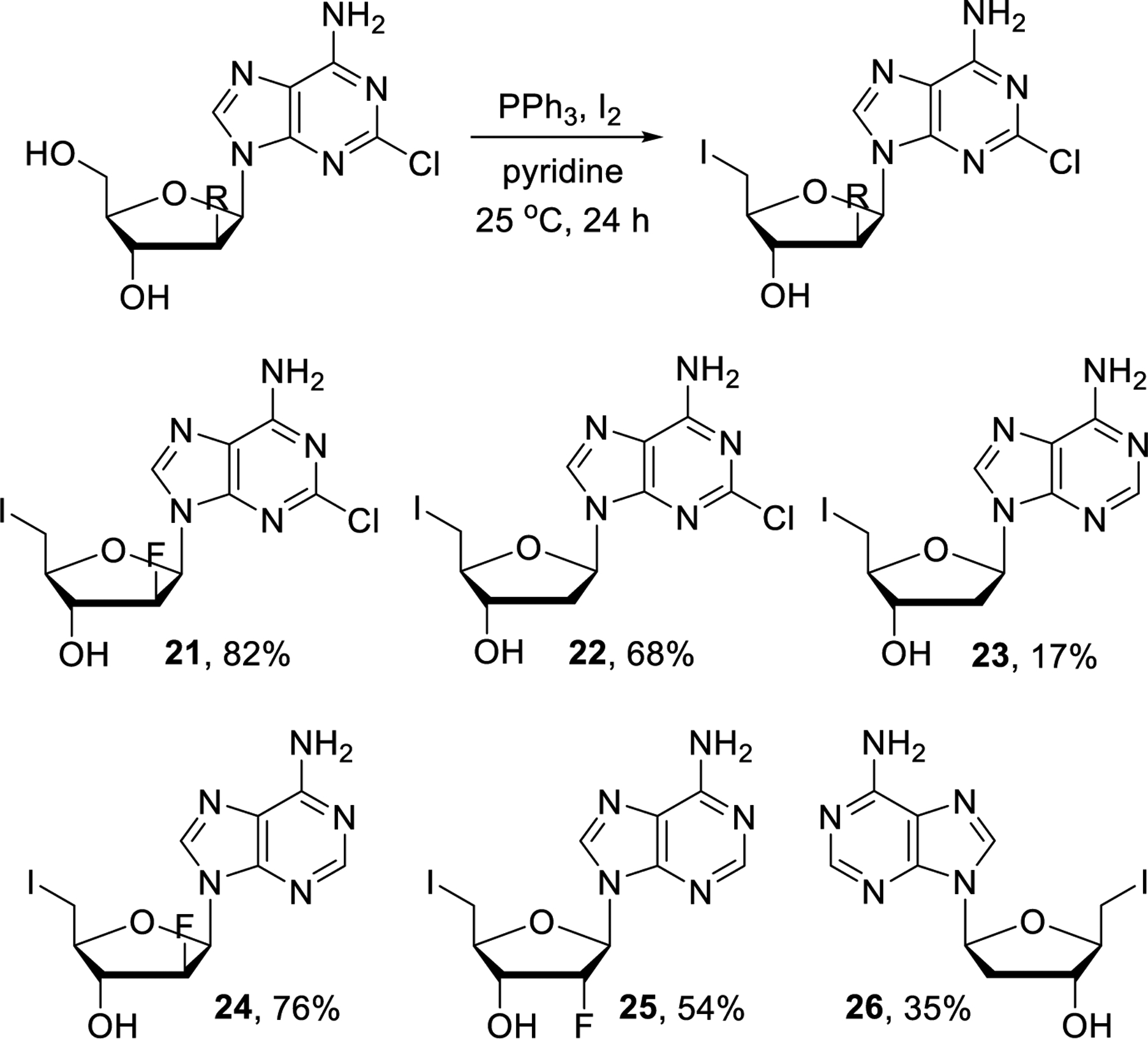
Synthesis of 5’-iodo intermediates 21-26.
Upon completion of the iodination reaction, the solvents were removed and the products were washed with cold methanol to remove excess iodine, PPh3 and Ph3PO. The light sensitivity and unstable nature of 21-26 required us to perform the nucleophilic substitutions directly and without further purification. To identify the best reaction conditions for the nucleophilic displacements, optimization experiments were carried out using 21 and morpholine (27) (Scheme 6). First, several solvent systems were tested with excess of 27 at 60 °C. Only trace amounts of product were formed when the reaction was carried out in DMF, DMSO or NMP and no reaction was observed in CH3CN. Fortunately, the reaction went to completion using pyridine as solvent and the desired product BK50142 was isolated in 40% yield while the formation of HI elimination by-product BK50138-A and of disubstituted BK50138-B was also observed. Reduction of the excess of 27 and decreasing the reaction temperature to 50 °C led to reduced amounts of the by-products and thus improved the yield of BK50142 to 78%. With optimized reaction conditions in hand, we were able to prepare the morpholine derived compounds BK50141, BK50142, BK703, BK707, BK50134 and BK705 in 68–86% yields.
Scheme 6.
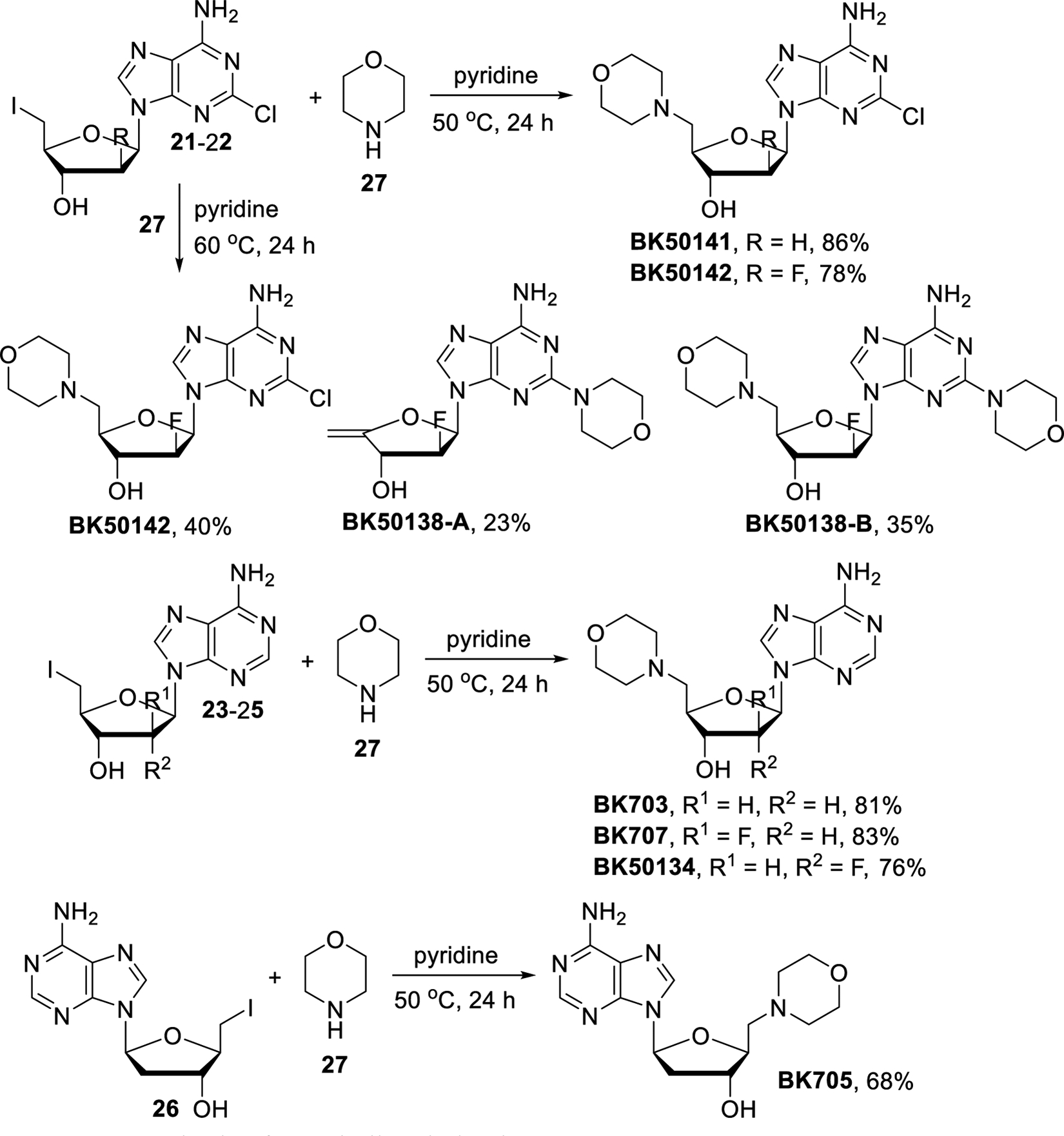
Synthesis of morpholine derivatives.
Clofarabine and cladribine analogs containing a sulfone or sulfide moiety were prepared as described in Scheme 7. The reaction of 21 with excess of sodium methanethiolate (28) went to completion and gave BK7012 in 92% yield. Unfortunately, we were not able to selectively achieve methylthiolation at the C-5’ terminus without replacing the purine chloride. By contrast, the use of less nucleophilic sodium methanesulfinate (29) avoids this problem and not even a trace of a nucleophilic aromatic substitution product was observed. Accordingly, 21 and 22 were converted to BK7013 and BK7014 in 88% and 82% yield, respectively.
Scheme 7.
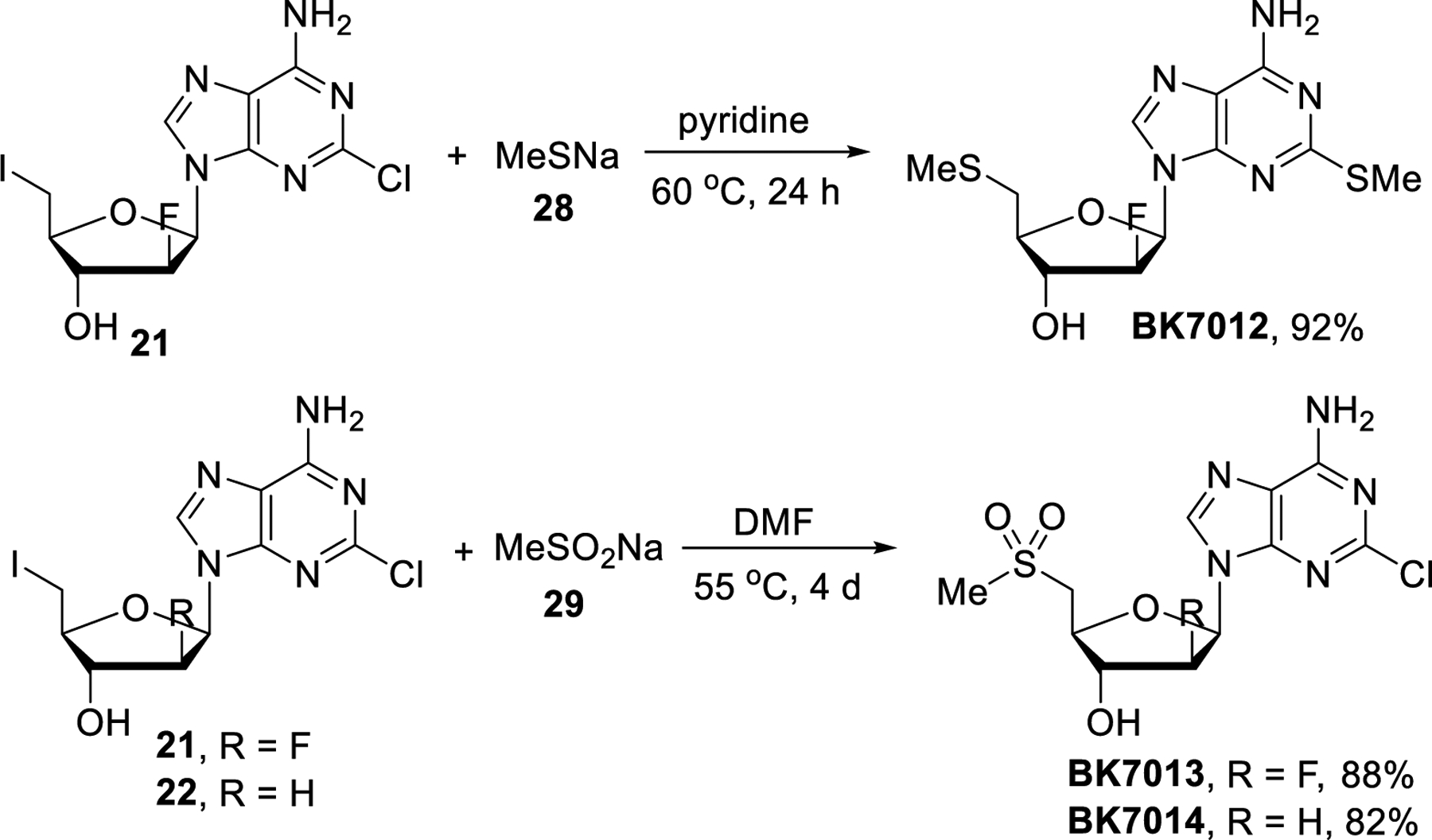
Synthesis of sulfone and sulfide derivatives.
Finally, the chlorine atom in the purine ring was replaced with heteroaryl rings by palladium catalyzed Suzuki cross-coupling reactions (Scheme 8). Compounds BK60106, BK7032, EN449 and EN450 were prepared from clofarabine with the corresponding heteroarylboronic acids using K2CO3 and Pd(PPh3)4 at 100 °C. Cladribine was also treated with 3-pyridyl, 4-pyridyl, 3-furyl and 3-thienylboronic acids to provide EN458, BK7033, EN457 and EN451, respectively. All these reactions proceeded with 55–79% yields.
Scheme 8.
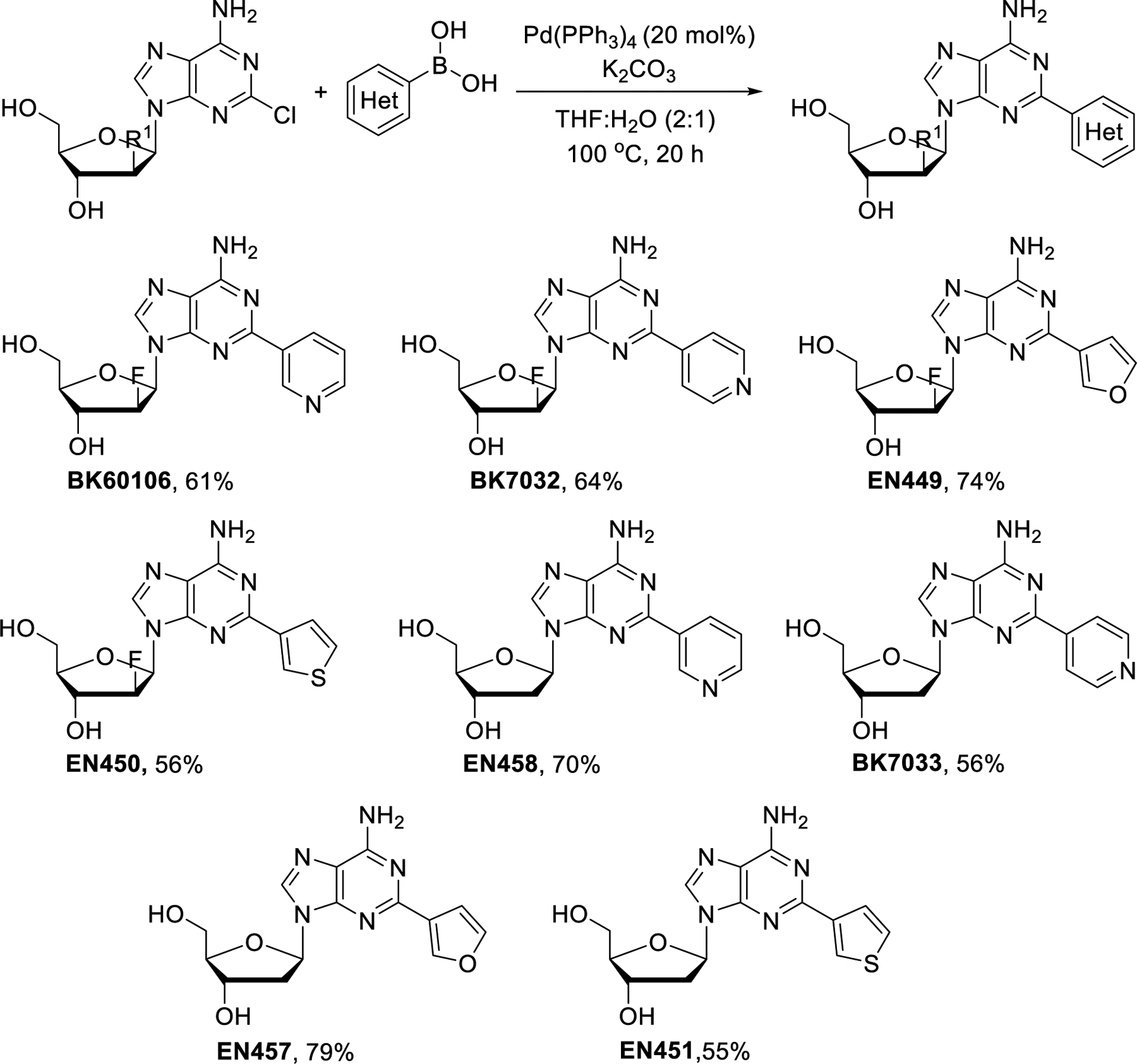
Synthesis of 2-heteroaryl derivatives of clofarabine and cladribine by Suzuki cross-coupling.
2.2. Testing the biological activity of the clofarabine and cladribine derivatives
2.2.1. Compound induced cell cytotoxicity analysis identified the hits that selectively inhibit ES cell growth
The structures of the compounds prepared in this study are shown in Figure 3. In total, we created a small library of 26 different derivatives of either clofarabine or cladribine. We initially performed a rapid screening of these compounds for their ability to maintain ES cell killing through CD99 by comparing their IC50 between ES cell lines and osteosarcoma (OS) cell lines. ES cells express high levels of CD99 and are sensitive to inhibition of CD99 by blocking antibodies, siRNA or small molecules. In contrast, OS cells have very low expression levels of CD99 and do not die when treated with CD99 blocking agents.48–50 Therefore, OS cells were used as a negative control to support CD99 specific activity. We first selected 3 different ES cell lines that have comparably high levels of CD99 protein and 2 OS cell lines that have only basal level of CD99 expression (Figure 4). For assessing the cytotoxicity of each compound in the library, we performed a cell viability assay on the cancer cell lines and calculated IC50 values of the compounds accordingly (Table 1 and Supplementary Figure 1). Clofarabine and cladribine showed selective cytotoxicity towards ES cells, which we had established previously.25 Within the concentration range used for these two drugs, they did not impact the cell viability of OS cells at all. Newly synthesized compounds were ranked depending on their selective cytotoxicity on ES cells. Among the compounds tested, 20 of them (EN449, EN450, EN451, BK703, BK705, BK707, BK7012, BK7032, BK7033, BK50134, BK60129, BK60131, BK60182, BK5082, BK5014, 1BK50142, BK50161, BK50138-A, BK50138-B, and BK50174) did not exhibit any significant cytotoxicity towards either ES or OS cell lines. These compounds were considered to have lost both their CD99 binding ability and DNA synthesis inhibition ability. Four compounds (EN457, EN458, BK7013 and BK7014) had a cytotoxic effect that was slightly stronger for at least one ES cell line. The highest priority was given to BK60106 and BK50164, because they maintained ES cell killing while sparing OS cells. There was no detectable cell death of OS cells at the highest tested concentration (400 μmol/L), therefore, proper IC50 values for them could not be calculated. The IC50 values for BK60106 and BK50164 were higher than clofarabine and cladribine but CD99 mediated cell line specificity was maintained.
Figure 3.
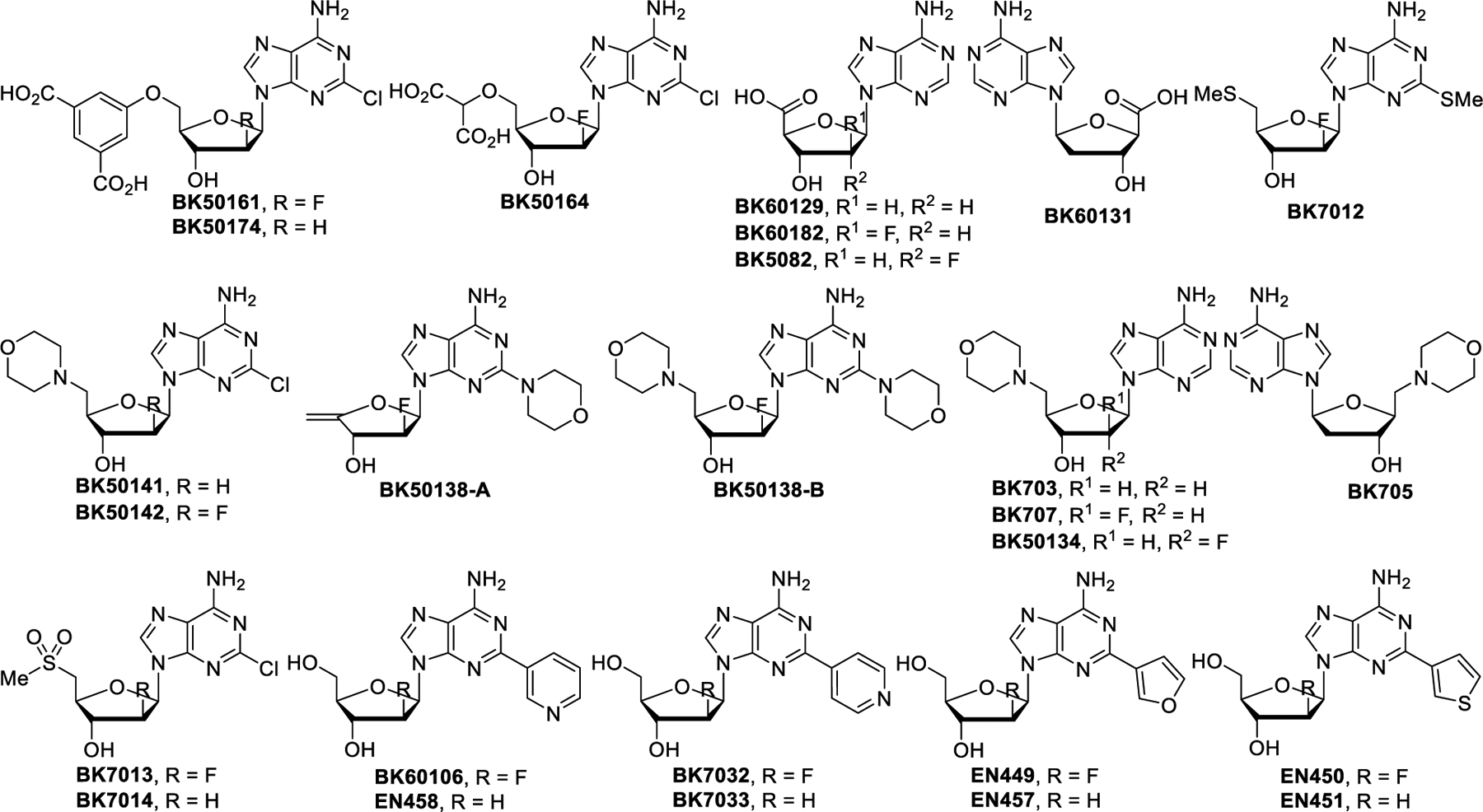
Structures of clofarabine and cladribine analogs prepared and tested in this study.
Figure 4.
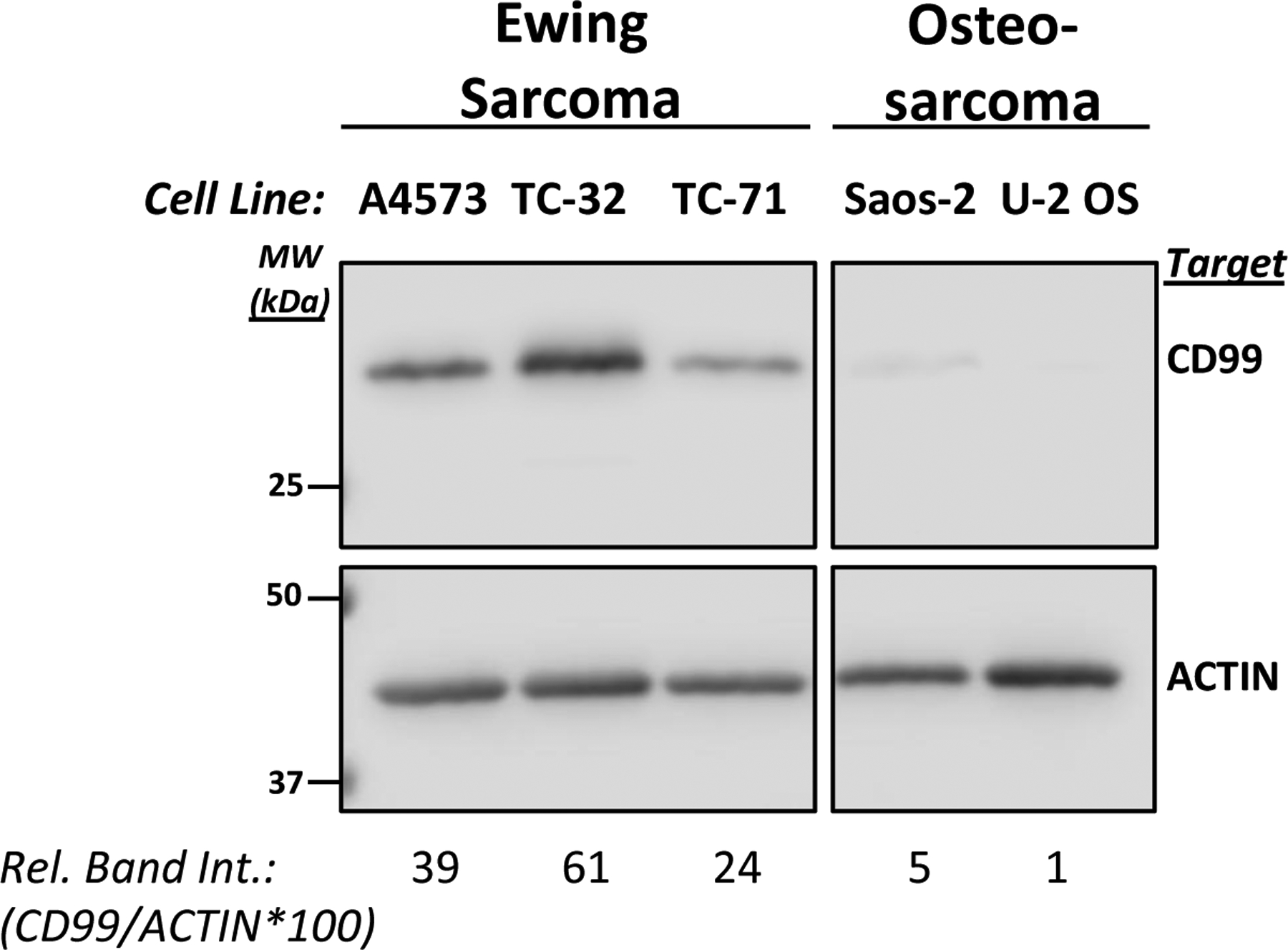
Ewing sarcoma cells express significantly more CD99 protein compared to osteosarcoma cells. CD99 protein expression was detected in total cell lysates by using western blot analyses. Actin expression was assessed for normalization purpose. Relative band intensity (Rel. Band Int.) was calculated using the densitometry data by dividing the CD99 signal to the corresponding actin signal and multiplying the resultant by 100.
Table 1.
IC50 values of the compounds on the selected Ewing Sarcoma and osteosarcoma cells. Cell viability was assessed at 48 h post-treatment by using CellTiter-Blue assay. The IC50 values were calculated by nonlinear regression analysis by using GraphPad Prism software. ND = not determined.
| IC50 in Ewing Sarcoma Cells (μmol/L) | IC50 osteosarcoma Cells (μmol/L) | |||||
|---|---|---|---|---|---|---|
| A4573 | TC-32 | TC-71 | Saos-2 | U-2 OS | ||
| Clofarabine | 0.18 | 0.13 | 0.09 | ≥ 50 | ≥ 50 | |
| Cladribine | 1.49 | 2.92 | 1.1 | ≥ 50 | ≥ 50 | |
| BK60106 | 45.15 | 58.69 | 19.91 | ≥ 400 | ≥ 400 | |
| BK50164 | 35.8 | 34.28 | 5.17 | ≥ 400 | ≥ 400 | |
| EN457 | 77.54 | 55.04 | 67.85 | 171.5 | 199.4 | |
| EN458 | 54.44 | 52.23 | 29.33 | 109.7 | 162.4 | |
| BK7013 | ≥ 50 | ≥ 50 | 31.44 | ≥ 50 | ≥ 50 | |
| BK7014 | ≥ 50 | ≥ 50 | 36 | ≥ 50 | ≥ 50 | |
| EN449 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | |
| EN450 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | |
| EN451 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | |
| BK703 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | |
| BK705 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | |
| BK707 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | |
| BK7012 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | |
| BK7032 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | |
| BK7033 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | |
| BK50134 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | |
| BK60129 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | |
| BK60131 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | |
| BK60182 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | |
| BK5082 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | ≥ 50 | |
| BK50142 | ND | ≥ 100 | ≥ 100 | ≥ 100 | ≥ 100 | |
| BK50141 | ND | ≥ 100 | ≥ 100 | 97.3 | ≥ 100 | |
| BK50138 -A | ND | 97.99 | ≥ 100 | 97.3 | 67.01 | |
| BK50138 -B | ND | ≥ 100 | ≥ 100 | ≥ 100 | ≥ 100 | |
| BK50161 | ND | ≥ 100 | 96.65 | 86.54 | ≥ 100 | |
| BK50174 | ND | ≥ 100 | ≥ 100 | ≥ 100 | ≥ 100 | |
2.2.2. BK60106 and BK50164 are highly ES selective compared to other cancers.
After identifying BK60106 and BK50164 as potential ES specific inhibitors, we performed additional cell viability screening by utilizing a larger cell line panel representing a vast variety of cancer types. We evaluated IC50 values on 7 ES (A4573, COG-E-352, MHH-ES, SKES, STA-ET-7.2, TC-32, and TC-71), 2 rhabdomyosarcoma (RD and RH30); 5 osteosarcoma (143B, HOS, MG-63.3, Saos-2, and U-2 OS), 2 glioblastoma (A172 and U87-MG), one lung cancer (A549), one prostate cancer (PC3), one cervical cancer (HeLa) cell lines and one immortalized human embryonic kidney cell line (HEK293). The IC50 values shown in Figure 5 come from replicate data sets provided in Supplementary Figure 2 and 3. The preliminary results from the initial screening study presented in Table 1 were not included for the calculation of mean IC50 values in Figure 5. The data in Table 1 were helpful for initial ranking of lead compounds but the IC50 values presented in Figure 5 are more accurate. Both BK60106 (Figure 5A and Supplementary Figure 2) and BK50164 (Figure 5B and Supplementary Figure 3) exhibited a great degree of selectivity towards all the ES cell lines used in the study compared to all non-ES cells. BK60106 showed more than 6 fold difference between average IC50 of ES cells compared to non-ES cells. BK50164 showed more than 12 fold difference between average IC50 of ES cells compared to non-ES cells. Therefore, we concluded that both BK60106 and BK50164 are ES specific cytotoxic agents that most likely function through inhibiting CD99 on ES cells.
Figure 5.
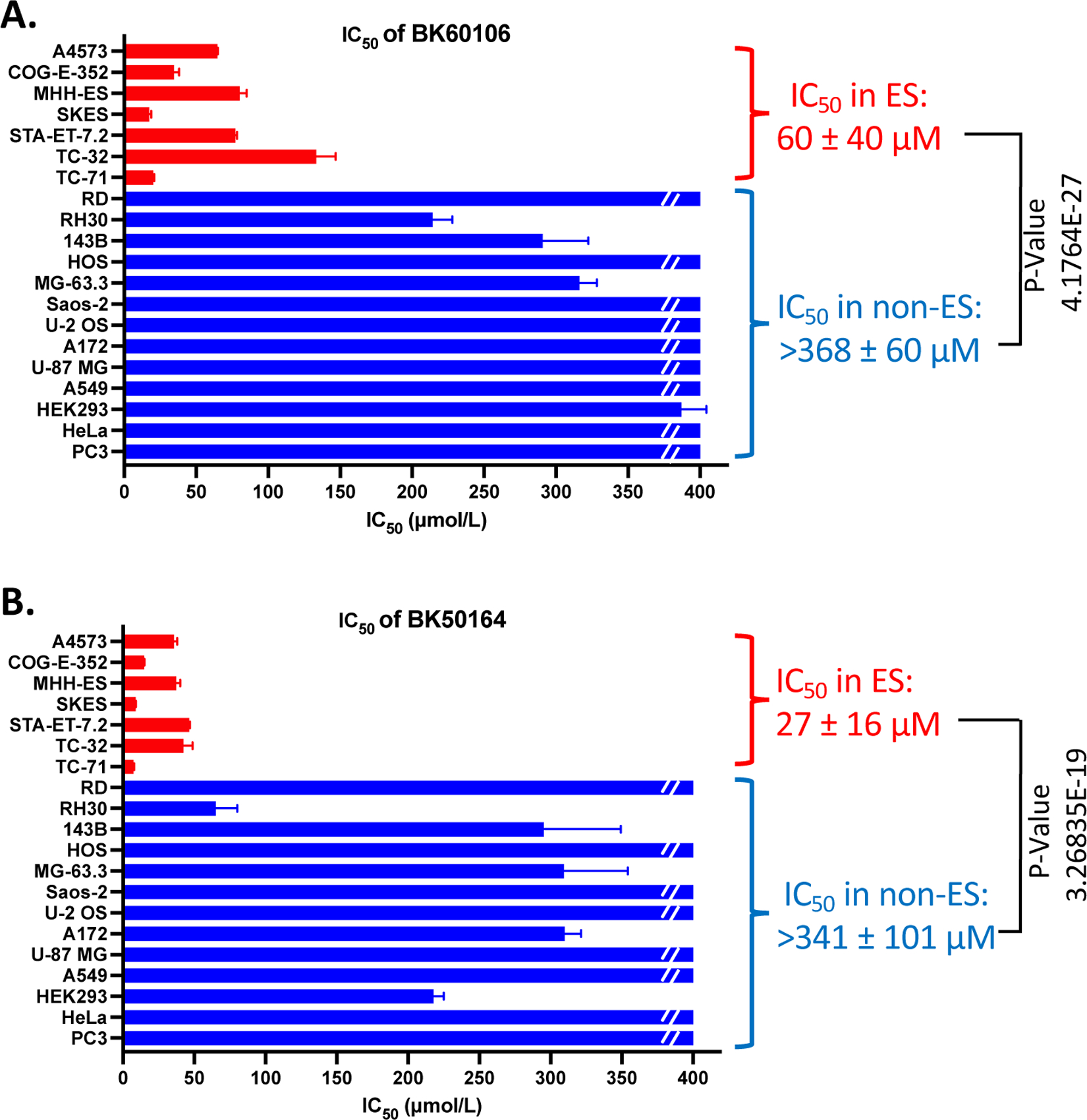
IC50 values of BK60106 and BK50164 on distinct cancer cell lines. Cell lines were treated with various concentrations of BK60106 (A) or BK50164 (B) and cell viability was assessed at 48 h post-treatment by using CellTiter-Blue assay. The IC50 values were calculated by nonlinear regression analysis (log(inhibitor) vs. normalized response) using GraphPad Prism software. Data represent the mean ± SD of replicate or triplicate determinations. Red color represents Ewing sarcoma (ES) cell lines; blue color collectively represents non-Ewing sarcoma (non-ES) cancer cell lines. Broken line indicates actual IC50 being greater than 400 μmol/L. P-values were calculated with Student’s t test.
2.2.3. BK60106 and BK50164 directly bind to CD99
We previously demonstrated that the parental drugs, clofarabine and cladribine, bind to both endogenous human CD99 protein and to purified bacterially expressed recombinant extracellular domain of CD99 protein.25,26 The ES cell specificity of BK60106 and BK50164 strongly suggested that both compounds maintained their ability to directly bind to CD99 and inhibit its activity in ES cells. In order to confirm that hypothesis, we studied the physical interaction between the two new clofarabine derived compounds and CD99. We confirmed direct physical interactions between the two compounds and CD99 by determining the binding affinities of the drugs to purified recombinant CD99 using surface plasmon resonance (SPR) technology on a Biacore T200 instrument. Recombinant CD99 protein was immobilized on a Biacore CM5 chip by amine coupling. Small molecules were injected over the protein coated surface at different concentrations while binding interaction was monitored by SPR in real time. SPR experiment demonstrated that both compounds can directly bind to recombinant CD99 protein on the chip surface. The calculated KD values for BK60106 (Figure 6A) and BK50164 (Figure 6B) for binding to CD99 were 1.8 μM and 1.5 μM, respectively. We also tested the thermal stability of endogenous CD99 in Ewing sarcoma cell lysate in the presence or absence of BK50164. In cellular thermal shift assay (CETSA), compound bound target proteins tend to remain in solution at elevated temperatures, whereas excessive heat denatures unbound proteins, resulting in their accelerated precipitation.51,52 By adding BK50164 (Figure 6C) in TC-71 cellular lysate, we quantified the presence of thermally stable CD99 protein in solution at elevated temperatures. Relative quantification of western blots of stable CD99 protein recovered from the solution showed that in the presence of BK50164 (Figure 6D), compound bound CD99 is more stable suggesting BK50164 binds endogenous CD99 protein in ES cells.
Figure 6.
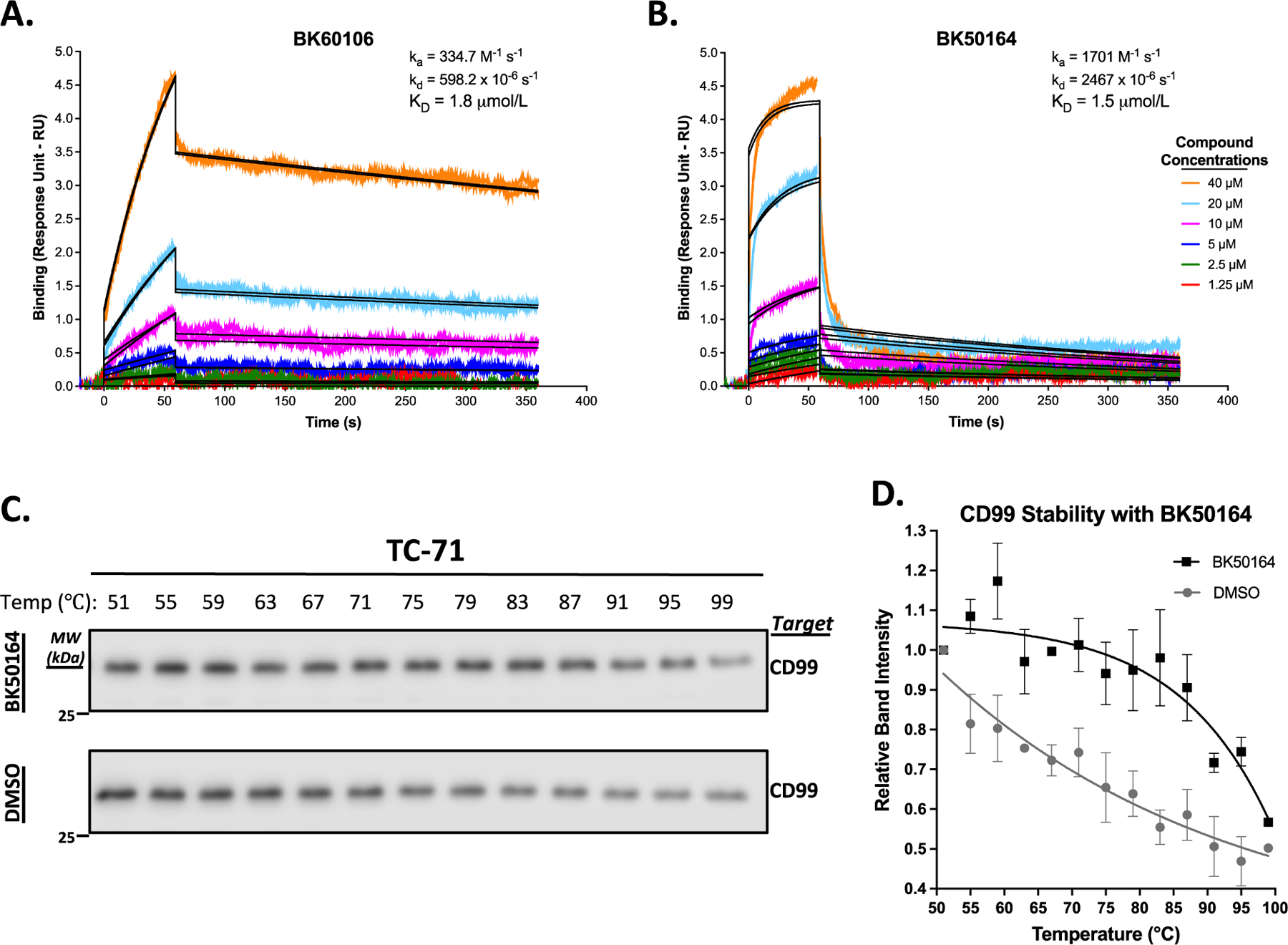
BK60106 and BK50164 directly bind CD99. The indicated various concentrations of BK60106 (A) or BK50164 (B) were flowed over the purified recombinant extracellular domain of CD99 protein, immobilized onto a CM5 chip in a Biacore T200 instrument. A kinetics interaction experiment was performed for the analytes (the compounds) binding to the ligand (CD99). The contact and dissociation times used were 60 s and 300 s, respectively. The sensorgrams were evaluated using 1:1 binding model fitting. Colored lines show actual data run in duplicates and black lines show the curve fit. The dissociation constants (KD values) of BK60106 and BK50164 on CD99 were calculated as 1.8 μM and 1.5 μM, respectively. BK50164 (C) was mixed with TC-71 cell lysate at a final concentration of 80 μM and the mixtures were subjected to various temperatures. Thermally stable CD99 protein, remaining in the lysate solution, was assessed by western blot. Relative quantification of protein amounts in BK50164 (D) treated lysates were determined by densitometry analysis of the protein bands in the western blot results in panels C. Data were analyzed by GraphPad Prism software.
2.2.4. BK60106 and BK50164 induce apoptosis in ES cells with less DNA damage than clofarabine does
We previously demonstrated CD99 mediated cell death by clofarabine and cladribine occurs trough apoptosis only in ES cells but not in OS cells.25,26 To characterize the molecular mechanisms of cell death induced by either BK60106 or BK50164, we checked for the presence of two apoptosis markers, cleaved products of PARP and Caspase 3, in the cells treated with the compounds. Increasing concentrations of BK60106 or BK50164 induced apoptosis in only TC-32 ES cells as clofarabine also did but not in U-2 OS cell line (Figure 7A and 7B). The cell cycle analysis of the compound treated cells showed that only ES cells but not OS cells had increased population in the hypodiploid sub-G1 phase of the cell cycle, another indicator that cells were undergoing apoptosis in response to drug treatment (Figure 7C).
Figure 7.
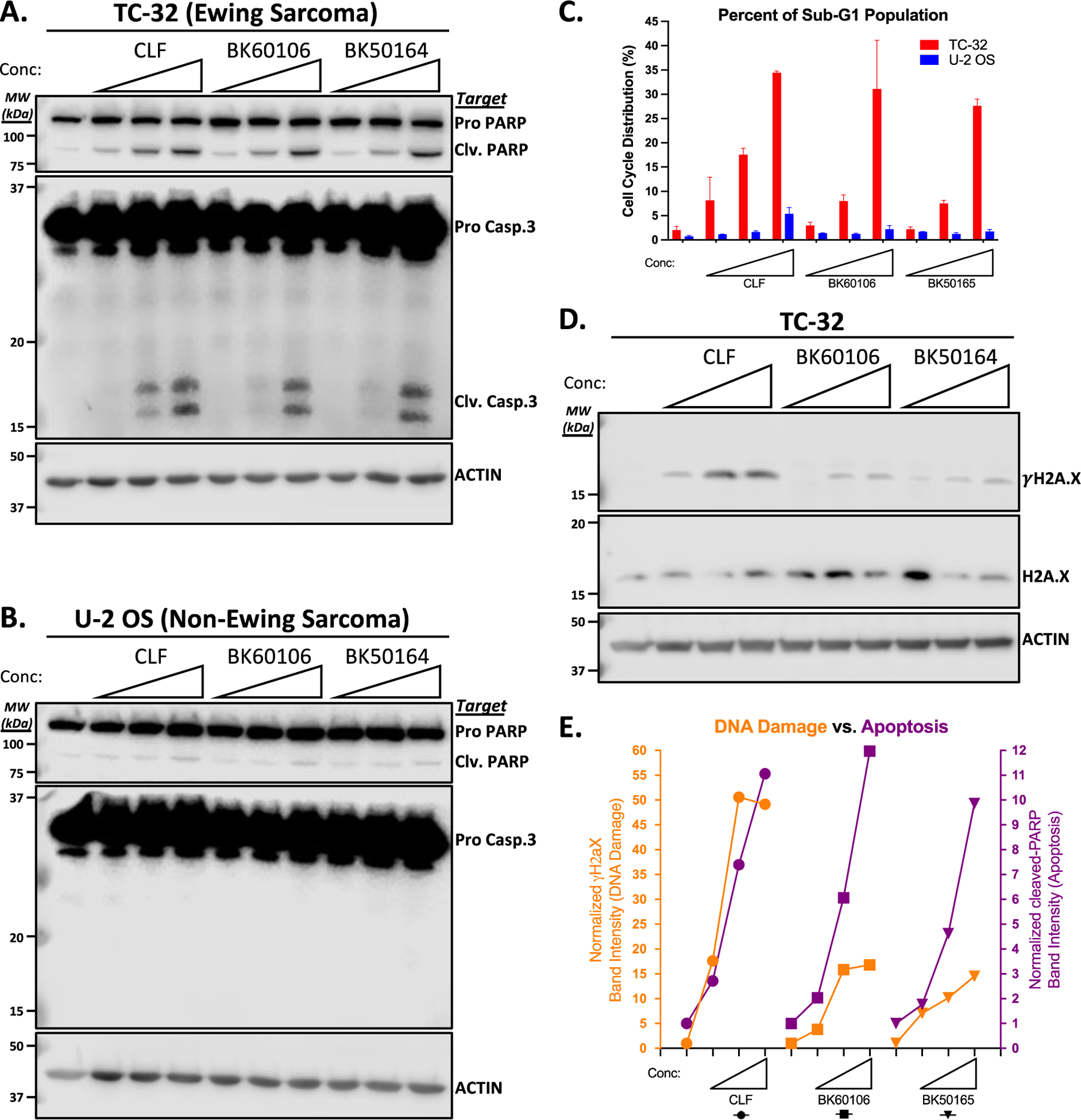
BK60106 and BK50164 selectively kill ES cells by apoptosis. TC-32 (A) or U-2 OS (B) cells were treated for 48 h with increased concentrations of the indicated drugs (for clofarabine (CLF): 0.1, 0.2, or 0.4 μM; for BK60106: 10, 20, or 40 μM; for BK50164: 4, 8, 16 μM) and total cell lysates were analyzed for the indicated apoptosis markers by using western blot. Actin expression was used as loading control and for normalization purpose. Clv is for cleaved. C. TC-32 and U-2 OS cells were treated as described in panels A and B and cell cycle distribution was assessed after propidium iodide staining. Data was analyzed by FlowJo and GraphPad Prism software. D. TC-32 cells were treated for 6 h with increased concentrations of the indicated drugs (for clofarabine (CLF): 0.2, 0.4, or 0.8 μM; for BK60106: 20, 40, or 80 μM; for BK50164: 8, 16, 32 μM) and total cell lysates were analyzed for the indicated DNA damage markers by using western blot. Actin expression was used as loading control and for normalization purpose. E. Densitometric analyses of the bands in panel A were used for the quantification of apoptosis induction (purple color) and densitometric analyses of the bands in panel D were used for the quantification of DNA damage (orange color). Data was analyzed by GraphPad Prism software.
As a nucleoside analog, clofarabine can be incorporated into DNA, leading to impaired DNA synthesis and elongation, which in turn causes DNA damage.29 Therefore, the clofarabine cytotoxicity on ES cells is likely due to combined result of DNA damage induction in addition to its physical interaction with CD99.25 We assessed the double stranded DNA damage formation in clofarabine, BK60106 or BK50164 treated TC-32 cells by determining H2A.X phosphorylation (γH2A.X) using western blot (Figure 7D). Both BK60106 and BK50164 caused less of γH2A.X accumulation compared to their parental drug, clofarabine. When doses of each drug inducing a comparable level of apoptosis were compared, BK60106 and BK50164 reduced the γH2A.X signal but clofarabine did not. BK60106 and BK50164 treatment resulting in reduced γH2A.X signal compared to clofarabine, suggests significantly less DNA damage occurring while all 3 compounds were inducing the same level of apoptosis (Figure 7E). These findings suggest that BK60106 and BK50164 assert their cytotoxic effects on ES cells through interacting with CD99 rather than by being incorporated into DNA as a nucleoside and causing DNA damage,
3. Conclusions
In addition to its diagnostic significance for ES in the clinic, CD99 is also functionally important for the pathogenesis of ES, therefore, it holds a great potential for targeted therapy in ES.53 Clofarabine and cladribine induce strong cancer cell death in ES through interacting with CD99 and significantly reduce tumorigenic capabilities of the ES cells both in vitro in cell culture and in vivo in mouse xenograph studies.25 Clofarabine and cladribine are FDA approved drugs primarily used to treat leukemias because they express high levels of deoxycytidine kinase (DCK) which is required to convert prodrugs to their 5’-triphosphate active forms. Since there is a potential for significant clinical benefit from inhibition of CD99 on ES cell surface, clofarabine and cladribine derivatives that can still bind to CD99 but not enter the cell or cannot be phosphorylated by DCK to their active forms would be ideal targeted therapy options. With the lack of plasma membrane permeability and/or 5’-triphosphate formation, new compounds will be expected to not inhibit DNA synthesis or nucleotide metabolism and therefore lack most of the myelosuppressive side effects. True CD99 inhibitors that cannot inhibit DNA synthesis will have a much better therapeutic window and hold considerable promise for treating ES patients.
A small library of 26 clofarabine and cladribine derivatives bearing a variety of functionalities instead of the primary alcohol group at the C-5’ position in the ribose ring, small heterocycles at the purine ring and stereoisomers thereof were designed and synthesized. These compounds were first screened in a cell panel consisting of 3 ES and 2 OS cell lines for their retained cytotoxicity specifically on ES cells due to their specific sensitivity to CD99 blockage. The majority of compounds that lost their cytotoxicity on ES cells or those that became toxic for OS cells were not further considered. BK60106 and BK50164 were selected as primary hits to be characterized in detail. Secondary screening of the selected compounds for their IC50 values in a larger cell line panel that represented distinct cancer types identified both compounds having at least 6- or 12-fold lower IC50 values on average in ES cells. SPR experiments showed that the selected compounds were still able to bind to CD99 protein in the similar way that their parent drug clofarabine did.25,26 Finally, both BK60106 and BK50164 induced apoptosis in ES cells at concentrations close to their corresponding IC50 values but with less DNA damage compared to that caused by clofarabine, both of which suggested the primary mechanism of cell death in ES was due to inhibition of CD99 but not inhibition of DNA synthesis.
Replacing the chlorine atom in the purine ring of the parent drugs by a small heteroaryl ring may render the nucleosides less membrane-penetrable, hence the derivatives would be expected to stay preferentially more on the extracellular side of the cell membrane preventing them being metabolized inside the cells. In this regard, BK60106 has a pyridine group built on the parent drug, clofarabine. Interestingly, EN458 has also a pyridine but on top of cladribine backbone while EN457 is also derived from clofarabine but with a furan ring at the same position in the chemical structure. In our primary screen, both EN457 and EN458 showed greater tendencies in inhibiting the growth of ES cells but with some residual cytotoxicity on osteosarcoma as well. BK50164 has a dicarboxylic acid unit replacing the C-5’ hydroxyl group on the ribose ring of clofarabine, hence it would be expected to be incapable of being phosphorylated inside the cells. In return, these various modifications may account for the alleviated DNA damage induction in derivative treated cells compared to clofarabine treated cells. It is also noteworthy that the residual DNA damage in derivative treated cells might as well be the result of cell death induction via CD99 engagement rather than the cause of DNA damage itself. Additionally, although both BK60106 and BK50164 are still able to kill ES cells, their IC50 values are greater than that of their parent drugs. It is possible that both clofarabine and cladribine act on ES cells via two mechanisms; direct interaction with CD99 and inducing DNA damage. However, new derivatives can be expected to act on ES cells more, if not solely, through CD99 binding. The results of this study provide first insights into structure activity relationships of nucleoside derived CD99 inhibitors that will be invaluable for future drug development directions and reveal that BK50164 and BK60106 are potent CD99 inhibitors. Specifically, the introduction of carboxylic acid moieties at the ribose C-5’ position and the placement of small heteroaryl rings or other functional groups into the purine ring are promising leads for further optimization.
4. Experimental section
4.1. Synthetic chemistry
Commercially available clofarabine (1), cladribine (2), nucleosides 3-6, boronic acids, catalysts, reagents and solvents were used as purchased without further purification. All chemical reactions were monitored by thin-layer chromatography (TLC) plates and visualized under UV light (254 or 365 nm). All iodides,43–47 diazo compound 18,42 intermediates 7-12, 14-17, 19, 20 and BK50164 were obtained according to previously reported procedures.38–40 NMR spectra were obtained at 400 MHz (1H NMR), 376 MHz (19F NMR) and 100 MHz (13C NMR) in deuterated solvents. The chemical shift (δ) values are expressed in ppm relative to the residual 1H signal of the solvent. All reaction products were purified by column chromatography on silica gel (particle size 40–63 μm) unless noted otherwise. HRMS data were obtained using electron spray ionization time-of-flight (ESI-TOF) spectrometry.
4.1.1. 5-(((2R,3R,4S,5R)-5-(6-Amino-2-chloro-9H-purin-9-yl)-4-fluoro-3-hydroxytetrahydrofuran-2-yl)methoxy)isophthalic acid (BK50161).
The Boc-protected intermediate 8 was obtained according to a previously reported procedure and the analytical data were in accordance with the literature.38–40 Compounds 12 and 14 were prepared by Mitsunobu reaction and Boc-deprotection from the clofarabine derivative 8 following literature methods.39 Base mediated ester hydrolysis of compound 14 to synthesize BK50161 was then carried out. To a solution of dimethyl 5-(((2R,3R,4S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-4-fluoro-3-hydroxytetrahydrofuran-2-yl)methoxy)isophthalate (14) (50.0 mg, 0.1 mmol) in THF (1.5 mL) and H2O (1.5 mL) at room temperature was added LiOH (60.0 mg). The resulting mixture was stirred overnight before it was cooled to 0 °C, acidified to pH ~6 with 1 N HCl (aq) and concentrated under reduced pressure. The white precipitate was filtered off, washed with EtOAc followed by methanol, and dried under vacuum. Compound BK50161 was obtained as a colorless solid in 76% yield (36.0 mg, 0.076 mmol). 1H NMR (400 MHz, DMSO-d6) δ = 13.39 (s, 2H), 8.27 (d, J = 2.1 Hz, 1H), 8.09 (dd, J = 1.5, 1.5 Hz, 1H), 7.91 (bs, 2H), 7.72 (d, J = 1.5 Hz, 2H), 6.41 (dd, J = 13.3, 4.8 Hz, 1H), 6.24 (bs, 1H), 5.33 (ddd, J = 52.6, 4.8, 4.4 Hz, 1H), 4.64 (ddd, J = 19.2, 4.8, 4.4 Hz, 1H), 4.50 − 4.37 (m, 2H), 4.21 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ = 166.4, 158.4, 156.8, 153.3, 150.2, 140.2 (d, JC-F = 4.3 Hz), 132.9, 122.6, 119.1, 117.4, 94.9 (d, JC-F = 192.3 Hz), 81.4 (d, JC-F = 16.4 Hz), 80.6, 73.1 (d, JC-F = 24.1 Hz), 67.8; 19F NMR (376 MHz, DMSO-d6) δ = −198.3 (ddd, J = 52.3, 17.5, 12.7 Hz); HRMS (ESI-TOF) m/z: [M+H]+ calcd for C18H16ClFN5O7 468.0722, found 468.0721.
4.1.2. 5-(((2R,3S,5R)-5-(6-Amino-2-chloro-9H-purin-9-yl)-3-hydroxytetrahydrofuran-2-yl)methoxy)isophthalic acid (BK50174).
The Boc-protected intermediate 9 was obtained according to a previously reported procedure and the analytical data were in accordance with the literature.38–40 Compounds 13 and 15 were prepared by Mitsunobu reaction and Boc-deprotection from the cladribine derivative 9 by following literature methods.39 Base mediated ester hydrolysis of compound 15 to synthesize BK50174 was carried out as follows. To a solution of dimethyl 5-(((2R,3S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-3-hydroxytetrahydrofuran-2-yl)methoxy)isophthalate (15) (48.0 mg, 0.1 mmol) in THF (1.5 mL) and H2O (1.5 mL) at room temperature was added LiOH (60.0 mg). The resulting mixture was stirred overnight before it was cooled to 0 °C, acidified with saturated NH4Cl solution. The white precipitate was filtered off, washed with EtOAc followed by methanol, and dried under vacuum. Compound BK50174 was obtained as a colorless solid in 82% yield (37.0 mg, 0.082 mmol). 1H NMR (400 MHz, DMSO-d6) δ = 8.36 (s, 1H), 8.08 (s, 1H), 7.81 (bs, 2H), 7.68 (d, J = 1.5 Hz, 2H), 7.20 (s, 2H), 6.34 (dd, J = 6.7, 6.7 Hz, 1H), 5.55 (bs, 1H), 4.58 (m, 1H), 4.40 − 4.25 (m, 2H), 4.19 (m, 1H), 2.82 (ddd, J = 13.3, 6.8, 6.4 Hz, 1H), 2.38 (ddd, J = 13.3, 6.8, 6.4 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ = 166.4, 158.4, 156.8, 153.1, 150.1, 139.8, 132.9, 122.5, 119.0, 118.2, 84.8, 83.6, 70.7, 68.4, 48.6; HRMS (ESI-TOF) m/z: [M+H]+ calcd for C18H17ClN5O7 450.0817, found 450.0813.
4.1.3. 2-(((2R,3R,4S,5R)-5-(6-Amino-2-chloro-9H-purin-9-yl)-4-fluoro-3-hydroxytetrahydrofuran-2-yl)methoxy)malonic acid (BK50164).
Compound BK50164 was obtained according to a previously reported procedure and the analytical data for BK50164 and the intermediates 10 and 11 are in accordance with the literature.38–40 Base mediated ester hydrolysis of intermediate 11 to obtain BK50164 was carried out by following the method described below. To a solution of dimethyl 2-(((2R,3R,4S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-4-fluoro-3-hydroxytetrahydrofuran-2-yl)methoxy)malonate (87.0 mg, 0.2 mmol) in THF (2.0 mL) and H2O (2.0 mL) at room temperature was added LiOH (180.0 mg). The resulting mixture was stirred overnight before it was cooled to 0 °C, acidified to pH ~6 with 1 N HCl (aq) and concentrated under reduced pressure. The white precipitate was filtered off, washed with EtOAc followed by methanol, and dried under vacuum. Compound BK50164 was obtained as a colorless solid in 83% yield (67.0 mg, 0.166 mmol). 1H NMR (400 MHz, DMSO-d6) δ = 13.27 (bs, 2H), 8.31 (d, J = 1.9 Hz, 1H), 7.88 (bs, 2H), 6.35 (dd, J = 12.8, 4.8 Hz, 1H), 6.06 (bs, 1H), 5.26 (ddd, J = 52.7, 4.8, 4.4 Hz, 1H), 4.58 (s, 1H), 4.49 (ddd, J = 19.0, 4.8, 4.8 Hz, 1H), 3.99 (m, 1H), 3.90 − 3.74 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ = 168.0, 167.9, 156.8, 153.3, 150.2, 140.1 (d, JC-F = 3.5 Hz), 117.3, 95.0 (d, JC-F = 192.4 Hz), 81.3 (d, JC-F = 18.0 Hz), 81.2 (d, JC-F = 7.6 Hz), 78.9, 72.6 (d, JC-F = 23.3 Hz), 69.0; 19F NMR (376 MHz, DMSO-d6) δ = −198.6 (ddd, J = 52.6, 18.8, 12.8 Hz); HRMS (ESI-TOF) m/z: [M+H]+ calcd for C13H14ClFN5O7 406.0566, found 406.0565.
4.1.4. General procedure for the synthesis carboxylic acid derivatives
To a solution of (2R,3R,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-fluoro-2-(hydroxymethyl)tetrahydrofuran-3-ol (500.0 mg, 1.85 mmol) in acetonitrile (5.0 mL) and H2O (5.0 mL) were added (diacetoxyiodo)benzene (1.787 g, 5.55 mmol) and TEMPO (87.0 mg, 0.55 mmol), and the mixture was stirred at 40 °C for 19 hours. The solvents were removed under reduced pressure and the residue was triturated with diethyl ether. The white precipitate was filtered off, washed with acetonitrile followed by methanol, and dried under vacuum.
4.1.4.1. (2S,3R,4R,5R)-5-(6-Amino-9H-purin-9-yl)-4-fluoro-3-hydroxytetrahydrofuran-2-carboxylic acid (BK5082).
Compound BK5082 was obtained after washing with acetonitrile (2×10.0 mL) and ethanol (2×10.0 mL) as a colorless solid in 54% yield (282.0 mg, 0.99 mmol) from (2R,3R,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-fluoro-2-(hydroxymethyl)tetrahydrofuran-3-ol (500.0 mg, 1.85 mmol) by following the general procedure described above; 1H NMR (400 MHz, DMSO-d6) δ = 13.35 (s, 1H), 8.37 (s, 1H), 8.13 (s, 1H), 7.34 (bs, 2H), 6.32 (dd, J = 16.8, 2.8 Hz, 1H), 6.19 (bs, 1H), 5.53 (m, 1H), 4.73 (m, 1H), 4.38 (d, J = 5.6 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ = 171.5, 156.6, 153.2, 149.4, 139.7, 119.3, 93.3 (d, JC-F = 189.0 Hz), 86.4 (d, JC-F = 33.0 Hz), 81.5, 72.1 (d, JC-F = 16.2 Hz); 19F NMR (376 MHz, DMSO-d6) δ = −206.7 (m, 1F); HRMS (ESI-TOF) m/z: [M+H]+ calcd for C10H11FN5O4 284.0795, found 284.0791.
4.1.4.2. (2S,3R,4S,5R)-5-(6-Amino-9H-purin-9-yl)-4-fluoro-3-hydroxytetrahydrofuran-2-carboxylic acid (BK60182).
Compound BK60182 was obtained after washing with acetonitrile (2×10.0 mL) and ethanol (2×10.0 mL) as a colorless solid in 68% yield (192.0 mg, 0.68 mmol) from (2R,3R,4S,5R)-5-(6-amino-9H-purin-9-yl)-4-fluoro-2-(hydroxymethyl)tetrahydrofuran-3-ol (269.0 mg, 1.0 mmol) by following the general procedure described above; 1H NMR (400 MHz, DMSO-d6) δ = 13.43 (bs, 1H), 8.40 (s, 1H), 8.16 (s, 1H), 7.40 (s, 2H), 6.35 (dd, J = 16.0, 3.2 Hz, 1H), 6.20 (bs, 1H), 5.56 (ddd, J = 51.8, 4.0, 3.6 Hz, 1H), 4.77 (m, 1H), 4.41 (d, J = 5.7 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ = 171.1, 156.2, 152.8, 148.9, 139.3, 118.9, 92.8 (d, JC-F = 188.8 Hz), 86.0 (d, JC-F = 32.7 Hz), 81.0, 71.7 (d, JC-F = 16.1 Hz); 19F NMR (376 MHz, DMSO-d6) δ = −206.6 (ddd, J = 51.8, 16.2, 15.8 Hz); HRMS (ESI-TOF) m/z: [M+H]+ calcd for C10H11FN5O4 284.0795, found 284.0791.
4.1.4.3. (2S,3S,5R)-5-(6-Amino-9H-purin-9-yl)-3-hydroxytetrahydrofuran-2-carboxylic acid (BK60129).
Compound BK60129 was obtained after washing with acetonitrile (2×10.0 mL) and ethanol (2×10.0 mL) as a colorless solid in 71% yield (376.0 mg, 1.42 mmol) from (2R,3S,5R)-5-(6-amino-9H-purin-9-yl)-2-(hydroxymethyl)tetrahydrofuran-3-ol (502.0 mg, 2.0 mmol) by following the general procedure described above; 1H NMR (400 MHz, DMSO-d6) δ = 13.43 (s, 1H), 8.42 (s, 1H), 8.15 (s, 1H), 7.34 (s, 2H), 6.51 (dd, J = 8.6, 5.8 Hz, 1H), 5.84 (bs, 1H), 4.61 (d, J = 4.6 Hz, 1H), 4.39 (m, 1H), 2.54 (m, 1H), 2.37 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ = 172.3, 156.1, 152.5, 149.0, 139.1, 118.8, 85.1, 84.5, 73.9, 39.2; HRMS (ESI-TOF) m/z: [M+H]+ calcd for C10H12N5O4 266.0889, found 266.0884.
4.1.4.4. (2R,3R,5S)-5-(6-Amino-9H-purin-9-yl)-3-hydroxytetrahydrofuran-2-carboxylic acid (BK60131).
Compound BK60131 was obtained after washing with acetonitrile (2×10.0 mL) and ethanol (2×10.0 mL) as a colorless solid in 63% yield (333.0 mg, 1.26 mmol) from (2S,3R,5S)-5-(6-amino-9H-purin-9-yl)-2-(hydroxymethyl)tetrahydrofuran-3-ol (502.0 mg, 2.0 mmol) by following the general procedure described above; 1H NMR (400 MHz, DMSO-d6) δ = 13.43 (s, 1H), 8.42 (s, 1H), 8.16 (s, 1H), 7.34 (s, 2H), 6.51 (dd, J = 8.6, 5.8 Hz, 1H), 5.84 (bs, 1H), 4.62 (d, J = 4.7 Hz, 1H), 4.39 (m, 1H), 2.54 (m, 1H), 2.37 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ = 172.3, 156.1, 152.5, 149.0, 139.1, 118.8, 85.1, 84.5, 73.9, 39.2; HRMS (ESI-TOF) m/z: [M+H]+ calcd for C10H12N5O4 266.0889, found 266.0883.
4.1.5. General procedure for the synthesis of morpholine derivatives
(2S,3R,4S,5R)-5-(6-Amino-2-chloro-9H-purin-9-yl)-4-fluoro-2-(iodomethyl)tetrahydrofuran-3-ol (0.2 mmol) was dissolved in anhydrous pyridine (1.0 mL). Morpholine (0.2 mL) was added and the reaction mixture was heated to 50 °C and stirred for 24 hours. Solvents were removed under vacuum at 70 °C and the crude product was purified by flash column chromatography on silica gel using ethyl acetate-methanol as mobile phase as described below.
4.1.5.1. (2R,3R,4S,5R)-5-(6-Amino-2-chloro-9H-purin-9-yl)-4-fluoro-2-(morpholinomethyl)tetrahydrofuran-3-ol (BK50142).
Compound (2S,3R,4S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-4-fluoro-2-(iodomethyl)tetrahydrofuran-3-ol (1) was prepared from clofarabine by following a literature method.43–47 After the reaction was complete the solvents were removed and the solid products were washed with cold methanol to remove excess iodine, PPh3 and Ph3PO. The crude product was used directly for the next step without further purification. Compound BK50142 was obtained after column purification using ethyl acetate/methanol (19:1) as mobile phase as a colorless solid in 78% yield (63.0 mg, 0.170 mmol) from (2S,3R,4S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-4-fluoro-2-(iodomethyl)tetrahydrofuran-3-ol (1) (90.0 mg, 0.217 mmol) and morpholine (0.2 mL) by following the general procedure described above. Rf = 0.4 (EtOAc/methanol, 9:1); 1H NMR (400 MHz, methanol-d4) δ = 8.21 (d, J = 2.3 Hz, 1H), 6.37 (dd, J = 16.7, 4.0 Hz, 1H), 5.12 (ddd, J = 52.2, 4.1, 2.9 Hz, 1H), 4.43 (ddd, J = 18.6, 5.0, 2.9 Hz, 1H), 4.13 (m, 1H), 3.70 (t, J = 4.7 Hz, 4H), 2.87 − 2.74 (m, 2H), 2.69 − 2.53 (m, 4H); 13C NMR (100 MHz, methanol-d4) δ = 158.1, 155.6, 151.6, 141.9 (d, JC-F = 5.1 Hz), 118.6, 96.5 (d, JC-F = 192.4 Hz), 84.2 (d, JC-F = 17.0 Hz), 82.4 (d, JC-F = 4.0 Hz), 77.4 (d, JC-F = 24.4 Hz), 67.6, 61.6, 55.2; 19F NMR (376 MHz, methanol-d4) δ = −199.0 (ddd, J = 52.7, 17.6, 17.6 Hz); HRMS (ESI-TOF) m/z: [M+H]+ calcd for C14H19ClFN6O3 373.1191, found 373.1190.
4.1.5.2. (2R,3S,5R)-5-(6-Amino-2-chloro-9H-purin-9-yl)-2-(morpholinomethyl)tetrahydrofuran-3-ol (BK50141).
Compound (2S,3S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-2-(iodomethyl)tetrahydrofuran-3-ol (2) was prepared from cladribine by following a literature method.43–47 After the reaction was complete the solvents were removed and the solid products were washed with cold methanol to remove excess iodine, PPh3 and Ph3PO. The crude product was used directly for the next step without further purification. Compound BK50141 was obtained after column purification using ethyl acetate/methanol (19:1) as mobile phase as a colorless solid in 86% yield (69.0 mg, 0.195 mmol) from (2S,3S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-2-(iodomethyl)tetrahydrofuran-3-ol (2) (90.0 mg, 0.227 mmol) and morpholine (0.2 mL) by following the general procedure described above. Rf = 0.4 (EtOAc/methanol, 9:1); 1H NMR (400 MHz, methanol-d4) δ = 8.23 (s, 1H), 6.33 (t, J = 6.6 Hz, 1H), 4.52 (dt, J = 6.6, 4.7 Hz, 1H), 4.11 (dt, J = 7.3, 4.7 Hz, 1H), 3.67 (t, J = 4.7 Hz, 4H), 2.87 (dt, J = 13.9, 6.3 Hz, 1H), 2.80 − 2.73 (m, 2H), 2.57 (t, J = 4.7 Hz, 4H), 2.43 (ddd, J = 13.8, 7.0, 4.9 Hz, 1H); 13C NMR (100 MHz, methanol-d4) δ = 158.1, 155.2, 151.4, 141.8, 119.6, 85.9, 85.5, 74.0, 67.5, 62.0, 55.2, 39.8; HRMS (ESI-TOF) m/z: [M+H]+ calcd for C14H20ClN6O3 355.1285, found 355.1282.
4.1.5.3. (3R,4S,5R)-5-(6-Amino-2-morpholino-9H-purin-9-yl)-4-fluoro-2-methylenetetrahydrofuran-3-ol (BK50138-A).
Compound (2S,3R,4S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-4-fluoro-2-(iodomethyl)tetrahydrofuran-3-ol (1) was prepared from clofarabine by following a literature method.43–47 After the reaction was complete the solvents were removed and the solid products were washed with cold methanol to remove excess iodine, PPh3 and Ph3PO. The crude product was used directly for the next step without further purification. Compound BK50138-A was obtained after column purification using ethyl acetate/methanol (19:1) as mobile phase as a colorless solid in 23% yield (17.0 mg, 0.05 mmol) from (2S,3R,4S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-4-fluoro-2-(iodomethyl)tetrahydrofuran-3-ol (1) (90.0 mg, 0.218 mmol) and morpholine (0.2 mL) at 60 °C by following the general procedure described above. Rf = 0.6 (EtOAc/methanol, 9:1); 1H NMR (400 MHz, methanol-d4) δ = 7.77 (d, J = 2.6 Hz, 1H), 6.59 (dd, J = 17.1, 4.0 Hz, 1H), 5.12 (ddd, J = 51.7, 4.1, 3.0 Hz, 1H), 4.98 (m, 1H), 4.57 (bs, 1H), 4.39 (bs, 1H), 3.78 − 3.65 (m, 8H); 13C NMR (100 MHz, methanol-d4) δ = 161.7 (d, JC-F = 4.3 Hz), 160.9, 157.2, 153.1, 138.3 (d, JC-F = 6.3 Hz), 113.4, 95.1 (d, JC-F = 192.8 Hz), 86.8, 84.5 (d, JC-F = 16.7 Hz), 74.4 (d, JC-F = 26.8 Hz), 68.0, 46.2; 19F NMR (376 MHz, methanol-d4) δ = −203.3 (ddd, J = 52.1, 19.9, 17.0 Hz); HRMS (ESI-TOF) m/z: [M+H]+ calcd for C14H18FN6O3 337.1424, found 337.1421.
4.1.5.4. (2R,3R,4S,5R)-5-(6-Amino-2-morpholino-9H-purin-9-yl)-4-fluoro-2-(morpholinomethyl)tetrahydrofuran-3-ol (BK50138-B).
Compound (2S,3R,4S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-4-fluoro-2-(iodomethyl)tetrahydrofuran-3-ol (1) was prepared from clofarabine by following a literature method.43–47 After the reaction was complete the solvents were removed and the solid products were washed with cold methanol to remove excess iodine, PPh3 and Ph3PO. The crude product was used directly for the next step without further purification. Compound BK50138-B was obtained after column purification using ethyl acetate/methanol (9:1) as mobile phase as a colorless solid in 35% yield (32.0 mg, 0.076 mmol) from (2S,3R,4S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-4-fluoro-2-(iodomethyl)tetrahydrofuran-3-ol (1) (90.0 mg, 0.218 mmol) and morpholine (0.2 mL) at 60 °C by following the general procedure described above. Rf = 0.2 (EtOAc/methanol, 9:1); 1H NMR (400 MHz, methanol-d4) δ = 7.93 (d, J = 2.8 Hz, 1H), 6.38 (dd, J = 18.8, 3.9 Hz, 1H), 5.08 (ddd, J = 52.1, 4.1, 3.0 Hz, 1H), 4.45 (ddd, J = 18.4, 4.7, 2.7 Hz, 1H), 4.18 (dt, J = 8.5, 4.4 Hz, 1H), 3.89 (t, J = 4.7 Hz, 4H), 3.74 (t, J = 4.7 Hz, 4H), 3.24 (t, J = 4.7 Hz, 4H), 3.15 − 2.99 (m, 2H), 2.86 (t, J = 4.7 Hz, 4H); 13C NMR (100 MHz, methanol-d4) δ = 160.9, 157.1, 152.9, 138.8 (d, JC-F = 6.0 Hz), 113.3, 96.4 (d, JC-F = 191.6 Hz), 83.6 (d, JC-F = 16.8 Hz), 81.1 (d, JC-F = 3.7 Hz), 77.6 (d, JC-F = 25.0 Hz), 66.8, 64.9, 61.4, 54.8, 44.7; 19F NMR (376 MHz, methanol-d4) δ = −198.6 (ddd, J = 52.1, 19.9, 17.0 Hz); HRMS (ESI-TOF) m/z: [M+H]+ calcd for C18H27FN7O4 424.2109, found 424.2107.
4.1.5.5. (2R,3S,5R)-5-(6-Amino-9H-purin-9-yl)-2-(morpholinomethyl)tetrahydrofuran-3-ol (BK703).
Compound (2S,3S,5R)-5-(6-amino-9H-purin-9-yl)-2-(iodomethyl)tetrahydrofuran-3-ol (3) was prepared from (2R,3S,5R)-5-(6-amino-9H-purin-9-yl)-2-(hydroxymethyl)tetrahydrofuran-3-ol by following a literature method.43–47 After the reaction was complete the solvents were removed and the solid products were washed with cold methanol to remove excess iodine, PPh3 and Ph3PO. The crude product was used directly for the next step without further purification. Compound BK703 was obtained after column purification using ethyl acetate/methanol (19:1) as mobile phase as a colorless solid in 81% yield (57.0 mg, 0.178 mmol) from (2S,3S,5R)-5-(6-amino-9H-purin-9-yl)-2-(iodomethyl)tetrahydrofuran-3-ol (3) (80.0 mg, 0.221 mmol) and morpholine (0.2 mL) by following the general procedure described above. Rf = 0.4 (EtOAc/methanol, 9:1); 1H NMR (400 MHz, Methanol-d4) δ = 8.30 (s, 1H), 8.20 (s, 1H), 6.39 (dd, J = 6.8, 6.4 Hz, 1H), 4.50 (ddd, J = 7.6, 6.9, 4.9 Hz, 1H), 4.11 (ddd, J = 7.6, 6.4, 4.4 Hz, 1H), 3.66 (t, J = 4.7 Hz, 4H), 2.86 (ddd, J = 13.7, 6.4, 4.4 Hz, 1H), 2.79 − 2.66 (m, 2H), 2.60 − 2.50 (m, 4H), 2.45 (ddd, J = 13.7, 6.8, 5.0 Hz, 1H); 13C NMR (100 MHz, Methanol-d4) δ = 157.3, 153.8, 150.3, 141.2, 120.6, 85.6, 85.5, 74.0, 67.6, 62.1, 55.2, 40.1; HRMS (ESI-TOF) m/z: [M+H]+ calcd for C14H21N6O3 321.1675, found 321.1671.
4.1.5.6. (2R,3R,4S,5R)-5-(6-Amino-9H-purin-9-yl)-4-fluoro-2-(morpholinomethyl)tetrahydrofuran-3-ol (BK707).
Compound (2S,3R,4S,5R)-5-(6-amino-9H-purin-9-yl)-4-fluoro-2-(iodomethyl)tetrahydrofuran-3-ol (4) was prepared from (2R,3R,4S,5R)-5-(6-amino-9H-purin-9-yl)-4-fluoro-2-(hydroxymethyl)tetrahydrofuran-3-ol by following a literature method.43–47 After the reaction was complete the solvents were removed and the solid products were washed with cold methanol to remove excess iodine, PPh3 and Ph3PO. The crude product was used directly for the next step without further purification. Compound BK707 was obtained after column purification using ethyl acetate/methanol (19:1) as mobile phase as a colorless solid in 83% yield (40.0 mg, 0.109 mmol) from (2S,3R,4S,5R)-5-(6-amino-9H-purin-9-yl)-4-fluoro-2-(iodomethyl)tetrahydrofuran-3-ol (4) (50.0 mg, 0.132 mmol) and morpholine (0.2 mL) by following the general procedure described above. Rf = 0.5 (EtOAc/methanol, 9:1); 1H NMR (400 MHz, Methanol-d4) δ = 8.26 (d, J = 2.4 Hz, 1H), 8.22 (s, 1H), 6.46 (dd, J = 17.7, 3.8 Hz, 1H), 5.10 (ddd, J = 52.3, 3.8, 3.2 Hz, 1H), 4.41 (ddd, J = 17.8, 6.0, 3.2 Hz, 1H), 4.13 (m, 1H), 3.70 (t, J = 4.7 Hz, 4H), 2.79 (d, J = 6.0 Hz, 2H), 2.60 (t, J = 4.7 Hz, 4H); 13C NMR (100 MHz, Methanol-d4) δ = 157.4, 154.1, 150.4, 141.7 (d, JC-F = 5.2 Hz), 119.6, 96.6 (d, JC-F = 191.9 Hz), 84.2 (d, JC-F = 16.8 Hz), 82.8 (d, JC-F = 3.4 Hz), 77.7 (d, JC-F = 24.5 Hz), 67.7, 61.7, 55.2; 19F NMR (376 MHz, methanol-d4) δ = −198.5 (ddd, J = 51.9, 18.0, 18.0 Hz); HRMS (ESI-TOF) m/z: [M+H]+ calcd for C14H20FN6O3 339.1581, found 339.1576.
4.1.5.7. (2R,3R,4R,5R)-5-(6-Amino-9H-purin-9-yl)-4-fluoro-2-(morpholinomethyl)tetrahydrofuran-3-ol (BK50134).
Compound (2S,3R,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-fluoro-2-(iodomethyl)tetrahydrofuran-3-ol (5) was prepared from (2R,3R,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-fluoro-2-(hydroxymethyl)tetrahydrofuran-3-ol by following a literature method.43–47 After the reaction was complete the solvents were removed and the solid products were washed with cold methanol to remove excess iodine, PPh3 and Ph3PO. The crude product was used directly for the next step without further purification. Compound BK50134 was obtained after column purification using ethyl acetate/methanol (19:1) as mobile phase as a colorless solid in 76% yield (50.0 mg, 0.15 mmol) from (2S,3R,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-fluoro-2-(iodomethyl)tetrahydrofuran-3-ol (5) (75.0 mg, 0.197 mmol) and morpholine (0.2 mL) by following the general procedure described above. Rf = 0.5 (EtOAc/methanol, 9:1); 1H NMR (400 MHz, Methanol-d4) δ = 8.27 (s, 1H), 8.21 (s, 1H), 6.24 (dd, J = 20.2, 1.5 Hz, 1H), 5.44 (ddd, J = 53.2, 4.8, 1.5 Hz, 1H), 4.66 (ddd, J = 21.9, 8.5, 4.8 Hz, 1H), 4.20 (m, 1H), 3.65 (t, J = 4.7 Hz, 4H), 2.87 (dd, J = 13.8, 2.4 Hz, 1H), 2.72 (dd, J = 13.8, 7.6 Hz, 1H), 2.56 (t, J = 4.7 Hz, 4H); 13C NMR (100 MHz, Methanol-d4) δ = 157.4, 154.1, 150.2, 141.6, 120.6, 94.5 (d, JC-F = 186.0 Hz), 88.8 (d, JC-F = 35.0 Hz), 81.8, 72.6 (d, JC-F = 16.6 Hz), 67.6, 61.4, 55.4; 19F NMR (376 MHz, methanol-d4) δ = −203.0 (ddd, J = 53.2, 21.4, 20.3 Hz); HRMS (ESI-TOF) m/z: [M+H]+ calcd for C14H20FN6O3 339.1581, found 339.1577.
4.1.5.8. (2S,3R,5S)-5-(6-Amino-9H-purin-9-yl)-2-(morpholinomethyl)tetrahydrofuran-3-ol (BK705).
Compound (2R,3R,5S)-5-(6-amino-9H-purin-9-yl)-2-(iodomethyl)tetrahydrofuran-3-ol (6) was prepared from (2S,3R,5S)-5-(6-amino-9H-purin-9-yl)-2-(hydroxymethyl)tetrahydrofuran-3-ol by following a literature method.43–47 After the reaction was complete the solvents were removed and the solid products were washed with cold methanol to remove excess iodine, PPh3 and Ph3PO. The crude product was used directly for the next step without further purification. Compound BK705 was obtained after column purification using ethyl acetate/methanol (19:1) as mobile phase as a colorless solid in 68% yield (48.0 mg, 0.15 mmol) from (2R,3R,5S)-5-(6-amino-9H-purin-9-yl)-2-(iodomethyl)tetrahydrofuran-3-ol (6) (80.0 mg, 0.221 mmol) and morpholine (0.2 mL) by following the general procedure described above. Rf = 0.4 (EtOAc/methanol, 9:1); 1H NMR (400 MHz, Methanol-d4) δ = 8.31 (s, 1H), 8.20 (s, 1H), 6.39 (dd, J = 6.8, 6.4 Hz, 1H), 4.50 (m, 1H), 4.10 (m, 1H), 3.66 (t, J = 4.7 Hz, 4H), 2.84 (m, 1H), 2.76 − 2.63 (m, 2H), 2.53 (t, J = 4.7 Hz, 4H), 2.45 (ddd, J = 13.6, 6.9, 5.0 Hz, 1H); 13C NMR (100 MHz, Methanol-d4) δ = 157.3, 153.8, 150.4, 141.2, 120.6, 85.7, 85.4, 74.0, 67.6, 62.2, 55.3, 40.1; HRMS (ESI-TOF) m/z: [M+H]+ calcd for C14H21N6O3 321.1675, found 321.1671.
4.1.6. Synthesis sulfone and thioether derivatives
4.1.6.1. (2S,3R,4S,5R)-5-(6-Amino-2-(methylthio)-9H-purin-9-yl)-4-fluoro-2-((methylthio)methyl)tetrahydrofuran-3-ol (BK7012).
Compound (2S,3R,4S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-4-fluoro-2-(iodomethyl)tetrahydrofuran-3-ol (1) was prepared from clofarabine by following a literature method.43–47 After the reaction was complete the solvents were removed and the solid products were washed with cold methanol to remove excess iodine, PPh3 and Ph3PO. The crude product was used directly for the next step without further purification. An 8 mL vial was charged with (2S,3R,4S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-4-fluoro-2-(iodomethyl)tetrahydrofuran-3-ol (1) (50.0 mg, 0.121 mmol), sodium methanethiolate (25.0 mg, 0.363 mmol), pyridine (2.0 mL) and sealed. The vial was then placed in a 60 °C oil bath and stirred for 24 hours. Completion of the reaction was ascertained by TLC analysis and the solvent was removed in vacuo. The crude product was purified by flash chromatography on silica gel using EtOAc/methanol (19:1) as mobile phase. Compound BK7012 was obtained as a colorless solid in 92% yield (38.0 mg, 0.111 mmol). Rf = 0.3 (EtOAc/methanol, 9:1); 1H NMR (400 MHz, methanol-d4) δ = 8.07 (s, 1H), 6.44 (dd, J = 18.7, 3.8 Hz, 1H), 5.09 (ddd, J = 51.9, 3.8, 2.5 Hz, 1H), 4.51 (ddd, J = 17.0, 3.8, 2.5 Hz, 1H), 4.12 (dt, J = 6.4, 3.8 Hz, 1H), 2.98 − 2.82 (m, 2H), 2.54 (s, 3H), 2.18 (s, 3H); 13C NMR (100 MHz, methanol-d4) δ = 167.8, 156.7, 151.4, 140.3, 116.9, 96.5 (d, JC-F = 191.8 Hz), 84.9 (d, JC-F = 2.7 Hz), 84.3 (d, JC-F = 17.0 Hz), 77.6 (d, JC-F = 24.4 Hz), 37.0 (d, JC-F = 2.0 Hz), 16.3, 14.4; 19F NMR (376 MHz, methanol-d4) δ = −198.3 (ddd, J = 51.6, 18.0, 17.0 Hz); HRMS (ESI-TOF) m/z: [M+H]+ calcd for C12H17FN5O2S2 346.0808, found 346.0805.
4.1.7. Synthesis sulfone and thioether derivatives
4.1.7.1. (2S,3R,4S,5R)-5-(6-Amino-2-chloro-9H-purin-9-yl)-4-fluoro-2-((methylsulfonyl)methyl)tetrahydrofuran-3-ol (BK7013).
Compound (2S,3R,4S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-4-fluoro-2-(iodomethyl)tetrahydrofuran-3-ol (1) was prepared from clofarabine by following a literature method.43–47 After the reaction was complete the solvents were removed and the solid products were washed with cold methanol to remove excess iodine, PPh3 and Ph3PO. The crude product was used directly for the next step without further purification. A solution of (2S,3R,4S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-4-fluoro-2-(iodomethyl)tetrahydrofuran-3-ol (1) (83.0 mg, 0.2 mmol) and sodium methanesulfinate (61.0 mg, 0.6 mmol) was stirred in 1.0 ml of DMF at 55 °C for 4 days. The reaction mixture was quenched with water and the solvents were removed in vacuo. The crude product was purified by flash chromatography on silica gel using ethyl acetate as mobile phase. Compound BK7013 was obtained as a colorless solid in 88% yield (64.0 mg, 0.176 mmol). Rf = 0.2 (EtOAc); 1H NMR (400 MHz, DMSO-d6) δ = 8.35 (d, J = 2.5 Hz, 1H), 7.90 (bs, 2H), 6.41 (dd, J = 17.5, 4.4 Hz, 1H), 6.24 (d, J = 4.4 Hz, 1H), 5.23 (ddd, J = 51.9, 3.6, 3.6 Hz, 1H), 4.52 (m, 1H), 4.33 (m, 1H), 3.86 (m, 1H), 3.55 (m, 1H), 2.98 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ = 156.8, 153.3, 150.1, 140.6 (d, JC-F = 4.6 Hz), 117.4, 94.0 (d, JC-F = 191.3 Hz), 82.6 (d, JC-F = 16.3 Hz), 78.2 (d, JC-F = 4.4 Hz), 75.8 (d, JC-F = 23.9 Hz), 56.5, 42.0; 19F NMR (376 MHz, DMSO-d6) δ = −197.9 (ddd, J = 51.9, 17.7, 17.3 Hz); HRMS (ESI-TOF) m/z: [M+H]+ calcd for C11H14ClFN5O4S 366.0439, found 366.0437.
4.1.7.2. (2S,3S,5R)-5-(6-Amino-2-chloro-9H-purin-9-yl)-2-((methylsulfonyl)methyl)tetrahydrofuran-3-ol (BK7014).
Compound (2S,3S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-2-(iodomethyl)tetrahydrofuran-3-ol (2) was prepared from cladribine by following a literature method.43–47 After the reaction was complete the solvents were removed and the solid products were washed with cold methanol to remove excess iodine, PPh3 and Ph3PO. The crude product was used directly for the next step without further purification. A solution of (2S,3S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-2-(iodomethyl)tetrahydrofuran-3-ol (2) (79.0 mg, 0.2 mmol) and sodium methanesulfinate (61.0 mg, 0.6 mmol) was stirred in 1.0 ml of DMF at 55 °C for 4 days. The reaction mixture was quenched with water and the solvents were removed in vacuo. The crude product was purified by flash chromatography on silica gel using ethyl acetate as mobile phase. Compound BK7013 was obtained as a colorless solid in 82% yield (57.0 mg, 0.170 mmol). Rf = 0.2 (EtOAc); 1H NMR (400 MHz, DMSO-d6) δ = 8.40 (s, 1H), 7.85 (bs, 2H), 6.34 (dd, J = 7.6, 6.4 Hz, 1H), 5.60 (d, J = 3.9 Hz, 1H), 4.45 (m, 1H), 4.27 (dt, J = 9.5, 2.9 Hz, 1H), 3.85 (dd, J = 14.8, 9.5 Hz, 1H), 3.47 (ddd, J = 14.8, 3.2, 1.4 Hz, 1H), 2.81 (s, 3H), 2.76 (ddd, J = 13.3, 8.1, 6.0 Hz, 1H), 2.32 (ddd, J = 13.4, 6.4, 3.2 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ = 156.8, 152.9, 149.9, 140.4, 118.4, 84.4, 81.4, 73.2, 56.9, 41.9, 37.4; HRMS (ESI-TOF) m/z: [M+H]+ calcd for C11H15ClN5O4S 348.0533, found 348.0532.
4.1.8. General procedure for the Suzuki coupling
An arylboronic acid (4.0 equiv) was added to a stirred solution of clofarabine or cladribine, potassium carbonate (2.0 equiv) and tetrakis(triphenylphosphine)palladium (0.2 equiv) in tetrahydrofuran (2.0 mL) and water (1.0 mL). The reaction mixture was heated to reflux for 20 hours. Solvents were removed under vacuum at 70 °C and the crude product was purified by flash column chromatography on silica gel using dichloromethane-methanol as mobile phase as described below.
4.1.8.1. (2R,3R,4S,5R)-5-(6-Amino-2-(pyridin-3-yl)-9H-purin-9-yl)-4-fluoro-2-(hydroxymethyl)tetrahydrofuran-3-ol (BK60106).
Compound BK60106 was obtained after column purification using dichloromethane/methanol (9:1) as mobile phase as a colorless solid in 61% yield (63.0 mg, 0.182 mmol) from clofarabine (91.0 mg, 0.3 mmol) and pyridin-3-ylboronic acid (148.0 mg, 1.2 mmol) by following the general procedure described above. Rf = 0.2 (dichloromethane/methanol, 9:1); 1H NMR (400 MHz, DMSO-d6) δ = 9.46 (s, 1H), 8.69 − 8.51 (m, 2H), 8.24 (d, J = 2.1 Hz, 1H), 7.56 − 7.31 (m, 3H), 6.51 (dd, J = 14.8, 4.4 Hz, 1H), 5.93 (d, J = 5.0 Hz, 1H), 5.25 (ddd, J = 52.5, 4.4, 4.2 Hz, 1H), 5.00 (dd, J = 6.0, 5.6 Hz, 1H), 4.49 (m, 1H), 3.86 (m, 1H), 3.78 − 3.54 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ = 156.4, 155.9, 150.3, 150.0, 149.0, 140.3 (d, JC-F = 4.2 Hz), 134.8, 133.5, 123.4, 117.7, 95.5 (d, JC-F = 192.1 Hz), 83.5 (d, JC-F = 5.3 Hz), 81.4 (d, JC-F = 16.9 Hz), 72.9 (d, JC-F = 23.3 Hz), 60.6; 19F NMR (376 MHz, DMSO-d6) δ = −197.5 (ddd, J = 52.7, 17.0, 17.0 Hz); HRMS (ESI-TOF) m/z: [M+H]+ calcd for C15H16FN6O3 347.1268, found 347.1263.
4.1.8.2. (2R,3R,4S,5R)-5-(6-Amino-2-(pyridin-4-yl)-9H-purin-9-yl)-4-fluoro-2-(hydroxymethyl)tetrahydrofuran-3-ol (BK7032).
Compound BK7032 was obtained after column purification using ethyl acetate/methanol (19:1) as mobile phase as a pale yellow solid in 64% yield (111.0 mg, 0.32 mmol) from clofarabine (152.0 mg, 0.5 mmol) and pyridin-4-ylboronic acid (246.0 mg, 2.0 mmol) by following the general procedure described above. Rf = 0.4 (EtOAc/methanol, 19:1); 1H NMR (400 MHz, DMSO-d6) δ = 8.73 (bs, 2H), 8.34 (s, 1H), 8.24 (bs, 2H), 7.57 (bs, 2H), 6.55 (dd, J = 14.7, 4.6 Hz, 1H), 6.02 (dd, J = 5.0, 2.3 Hz, 1H), 5.30 (ddd, J = 52.3, 4.0, 2.8 Hz, 1H), 5.08 (dd, J = 5.7, 2.3 Hz, 1H), 4.53 (ddd, J = 19.2, 4.8, 2.7 Hz, 1H), 3.90 (m, 1H), 3.80 − 3.60 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ = 156.1, 156.0, 150.1, 150.0, 145.4, 140.8, 121.8, 118.2, 95.5 (d, JC-F = 192.0 Hz), 83.6 (d, JC-F = 5.2 Hz), 81.4 (d, JC-F = 16.8 Hz), 72.9 (d, JC-F = 23.3 Hz), 60.6; 19F NMR (376 MHz, DMSO-d6) δ = −197.5 (ddd, J = 52.4, 17.6, 16.8 Hz); HRMS (ESI-TOF) m/z: [M+H]+ calcd for C15H16FN6O3 347.1268, found 347.1263.
4.1.8.3. (2R,3R,4S,5R)-5-(6-Amino-2-(furan-3-yl)-9H-purin-9-yl)-4-fluoro-2-(hydroxymethyl)tetrahydrofuran-3-ol (EN449).
Compound EN449 was obtained by column purification using DCM/MeOH (13:1) as mobile phase followed by recrystallization from ACN/MeOH to give a white solid in 74% yield (151.0 mg, 0.45 mmol) from clofarabine (182.0 mg, 0.6 mmol) and furan-3-ylboronic acid (403.0 mg, 3.6 mmol) following the general procedure described above. 1H NMR (400 MHz, DMSO) δ 8.15 (s, 2H), 7.70 (m, 1H), 7.24 (s, 2H), 6.94 (d, J = 1.8 Hz, 1H), 6.40 (dd, J = 14.6, 4.7 Hz, 1H), 5.92 (d, J = 5.0 Hz, 1H), 5.21 (ddd, J = 52.6, 4.2, 4.2 Hz, 1H), 4.98 (dd, J = 5.7, 5.7 Hz, 1H), 4.51 (ddd, J = 19.2, 4.8, 4.8 Hz, 1H), 3.83 (m, 1H), 3.73 − 3.57 (m, 2H); 13C NMR (100 MHz, DMSO) δ 156.3, 155.2, 150.3, 144.3, 143.9, 140.2, 128.0, 117.6, 110.3, 95.9 (d, JC-F = 192.3 Hz), 83.9 (d, JC-F = 5.8 Hz), 81.7 (d, JC-F = 17.2 Hz), 73.4 (d, JC-F = 23.1 Hz), 61.1; 19F NMR (376 MHz, DMSO) δ −197.7 (ddd, J = 52.5, 16.6, 16.6 Hz); HRMS (ESI-TOF) m/z: [M+H]+ calcd for C14H15FN5O4 336.1103, found 336.1104.
4.1.8.4. (2R,3R,4S,5R)-5-(6-Amino-2-(thiophen-3-yl)-9H-purin-9-yl)-4-fluoro-2-(hydroxymethyl)tetrahydrofuran-3-ol (EN450).
Compound EN450 was obtained by column purification using DCM/MeOH (9:1) as mobile phase followed by recrystallization from ACN to give a brown solid in 56% yield (119.0 mg, 0.34 mmol) from clofarabine (182.0 mg, 0.6 mmol) and thiophen-3-ylboronic acid (461.0 mg, 3.6 mmol) following the general procedure described above. 1H NMR (400 MHz, DMSO) δ 8.17 (d, J = 2.2 Hz, 1H), 8.12 (dd, J = 3.1, 1.2 Hz, 1H), 7.74 (dd, J = 5.1, 1.2 Hz, 1H), 7.54 (dd, J = 5.0, 3.1 Hz, 1H), 7.28 (s, 2H), 6.43 (dd, J = 14.6, 4.7 Hz, 1H), 5.93 (d, J = 5.0 Hz, 1H), 5.22 (ddd, J = 52.5, 4.2, 4.2 Hz, 1H), 5.00 (dd, J = 5.7, 5.7 Hz, 1H), 4.51 (ddd, J = 18.9, 4.8, 4.8 Hz, 1H), 3.84 (m, 1H), 3.73 − 3.57 (m, 2H); 13C NMR (100 MHz, DMSO) δ 156.3, 156.2, 150.4, 142.8, 127.9, 126.7, 126.6, 117.6, 95.9 (d, JC-F = 191.9 Hz), 84.0 (d, JC-F = 5.6 Hz), 81.6 (d, JC-F = 16.8 Hz), 73.43 (d, JC-F = 23.4 Hz), 61.1; 19F NMR (376 MHz, DMSO) δ −197.7 (ddd, J = 52.8, 16.7, 16.7 Hz) (m); HRMS (ESI-TOF) m/z: [M+H]+ calcd for C14H15FN5O3S 353.0874, found 352.0875.
4.1.8.5. (2R,3S,5R)-5-(6-Amino-2-(pyridin-3-yl)-9H-purin-9-yl)-2-(hydroxymethyl)tetrahydrofuran-3-ol (EN458).
Compound EN458 was obtained by column purification with DCM/MeOH (9:1) as mobile phase followed by recrystallization from ACN/MeOH to give an orange solid in 70% yield (137.0 mg, 0.4 mmol) from cladribine (171.0 mg, 0.6 mmol) and pyridine-4-ylboronic acid (442.0 mg, 3.6 mmol) following the general procedure described above. 1H NMR (400 MHz, DMSO) δ 9.44 (d, J = 1.3 Hz, 1H), 8.60 (dd, J = 4.8, 1.7 Hz, 1H), 8.56 (dt, J = 8.0, 2.0 Hz, 1H), 8.34 (s, 1H), 7.46 (dd, J = 8.0, 4.8 Hz, 1H), 7.38 (s, 2H), 6.41 (t, J = 6.9 Hz, 1H), 5.29 (d, J = 4.1 Hz, 1H), 4.88 (t, J = 5.6 Hz, 1H), 4.46 (m, 1H), 3.86 (m, 1H), 3.61 (m, 1H), 3.51 (m, 1H), 2.82 (m, 1H), 2.30 (ddd, J = 13.3, 6.3, 3.2 Hz, 1H); 13C NMR (100 MHz, DMSO) δ 156.5, 156.4, 150.7, 150.4, 149.4, 140.78, 135.3, 134.2, 123.8, 119.1, 88.3, 84.0, 71.4, 62.3; HRMS (ESI-TOF) m/z: [M+H]+ calcd for C15H17N6O3 329.1357, found 329.1359.
4.1.8.6. (2R,3S,5R)-5-(6-Amino-2-(pyridin-4-yl)-9H-purin-9-yl)-2-(hydroxymethyl)tetrahydrofuran-3-ol (BK7033).
Compound BK7033 was obtained after column purification using ethyl acetate/methanol (19:1) as mobile phase as a pale yellow solid in 56% yield (92.0 mg, 0.28 mmol) from cladribine (143.0 mg, 0.5 mmol) and pyridin-4-ylboronic acid (246.0 mg, 2.0 mmol) by following the general procedure described above. Rf = 0.4 (EtOAc/methanol, 19:1); 1H NMR (400 MHz, DMSO-d6) δ = 8.70 (bs, 2H), 8.43 (s, 1H), 8.21 (d, J = 4.8 Hz, 2H), 7.50 (bs, 2H), 6.45 (dd, J = 7.6, 6.2 Hz, 1H), 5.35 (d, J = 4.1 Hz, 1H), 4.93 (t, J = 5.6 Hz, 1H), 4.48 (dt, J = 6.4, 3.2 Hz, 1H), 3.89 (dt, J = 4.8, 2.8 Hz, 1H), 3.64 (m, 1H), 3.54 (m, 1H), 2.84 (ddd, J = 13.3, 7.6, 5.9 Hz, 1H), 2.33 (ddd, J = 13.1, 6.2, 3.2 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ = 156.0, 155.7, 150.0, 149.9, 145.5, 140.7, 121.7, 119.0, 87.8, 83.5, 70.9, 61.8, 39.2; HRMS (ESI-TOF) m/z: [M+H]+ calcd for C15H17N6O3 329.1362, found 329.1359.
4.1.8.7. (2R,3S,5R)-5-(6-Amino-2-(furan-3-yl)-9H-purin-9-yl)-2-(hydroxymethyl)tetrahydrofuran-3-ol (EN457).
Compound EN457 was obtained by column purification using DCM/MeOH (13:1) as mobile phase followed by recrystallization from ACN/MeOH to give a white solid in 79% yield (151.0 mg, 0.48 mmol) from cladribine (171.0 mg, 0.6 mmol) and furan-3-ylboronic acid (403.0 mg, 3.6 mmol) following the general procedure described above. 1H NMR (400 MHz, DMSO) δ 8.26 (s, 1H), 8.16 (d, J = 1.7 Hz, 1H), 7.69 (t, J = 1.8 Hz, 1H), 7.19 (s, 2H), 6.96 (d, J = 1.9 Hz, 1H), 6.34 (dd, J = 7.9, 6.1 Hz, 1H), 5.27 (d, J = 3.9 Hz, 1H), 5.00 (dd, J = 6.4, 5.0 Hz, 1H), 4.44 (m, 1H), 3.87 (m, 1H), 3.63 (m, 1H), 3.52 (m, 1H), 2.79 (m, 1H), 2.24 (ddd, J = 13.2, 6.2, 2.8 Hz, 1H); 13C NMR (100 MHz, DMSO) δ 156.3, 154.9, 150.1, 144.3, 143.8, 140.3, 128.0, 118.6, 110.3, 88.4, 84.3, 71.6, 62.5; HRMS (ESI-TOF) m/z: [M+H]+ calcd for C14H16N5O4 318.1197, found 318.1198.
4.1.8.8. (2R,3S,5R)-5-(6-Amino-2-(thiophen-3-yl)-9H-purin-9-yl)-2-(hydroxymethyl)tetrahydrofuran-3-ol (EN451).
Compound EN451 was obtained by column purification with DCM/MeOH (9:1) as mobile phase followed by recrystallization from ACN to give a tan solid in 55% yield (110.0 mg, 0.3 mmol) from cladribine (171.0 mg, 0.6 mmol) and thiophen-3-ylboronic acid (461.0 mg, 3.6 mmol) following the general procedure described above. 1H NMR (400 MHz, DMSO) δ 8.28 (s, 1H), 8.11 (dd, J = 3.1, 1.2 Hz, 1H), 7.74 (d, J = 5.0 Hz, 1H), 7.54 (dd, J = 5.1, 3.1 Hz, 1H), 7.22 (s, 2H), 6.36 (t, J = 7.0 Hz, 1H), 5.29 (d, J = 4.0 Hz, 1H), 4.93 (t, J = 5.6 Hz, 1H), 4.45 (bs, 1H), 3.86 (m, 1H), 3.62 (m, 1H), 3.51 (m, 1H), 2.80 (m, 1H), 2.26 (ddd, J = 13.2, 6.2, 2.9 Hz, 1H); 13C NMR (100 MHz, DMSO) δ 156.30, 155.9, 150.3, 142.9, 140.4, 128.0, 126.6, 118.6, 88.3, 84.1, 71.5, 62.4; HRMS (ESI-TOF) m/z: [M+H]+ calcd for C14H16N5O3S 334.0968, found 334.0969.
4.2. Drug testing experiments
4.2.1. Cell culture maintenance
MHH-ES, STA-ET-7.2, TC-71, RH30, and A549 cells were grown in RPMI (Gibco, # 11875) supplemented with 10% FBS (Sigma-Aldrich #F0926). A4573, TC-32, and PC3 cells were grown in RPMI supplemented with 10% FBS and 10 mM HEPES (Gibco # 15630). RD, 143B, HOS, MG-63.3, Saos-2, U-2 OS, A172, HEK293, and Hela cells were grown in DMEM supplemented with 10% FBS. U-87 MG cells were grown in DMEM supplemented with 10% FBS and 1X MEM NEAA (Gibco #11140). COG-E-352 cells were grown in IMDM supplemented with 15% FBS and 1X ITS (Sigma-Aldrich #I3146). SKES cells were grown in McCoy’s 5A (Gibco #16600 supplemented with 15% FBS. All of the cell lines were cultured at 37 °C in a humidified atmosphere with 5% CO2. All cell lines were regularly tested in-house for the absence of mycoplasma by using MycoAlert™ Mycoplasma Detection kit (Lonza, #75860).
4.2.2. Cell viability and IC50
After cell monolayer reached 60–80% confluency (usually 18–24 h after initial seeding) in 96-well cell culture microplate wells, the cells were treated with clofarabine (Selleck Chemicals, #S1218), cladribine (Selleck Chemicals, #S1199), or the compounds synthesized in the current study at various concentrations prepared in the corresponding media of the cell line used, for 48 h. Cell viability was determined by using CellTiter-Blue® reagent (Promega, #G8080). Briefly, at the end of the treatment, 40 μL of the reagent was added per 200 μL of cell culture media in the microplate and the plate was incubated at 37 °C in a humidified atmosphere with 5% CO2 for 3–5 h. After the incubation period, the reagent in the individual wells was excited at 560 nm and the emission was collected at 590 nm by using a Synergy H4 reader (Biotek).
4.2.3. Western Blot
Preparation of cellular lysates and subsequent immunoblotting were performed as previously described.54 The antibodies used for the immunoblotting are anti-CD99 (Abcam, # ab75858, EPR3097Y), anti-PARP (Cell Signaling Technology, #9542), anti-Caspase 3 (Cell Signaling Technology (CST), #9662), anti-H2A.X (CST, #7631, D17A3), anti-γH2A.X (CST, #9718, Ser139 - 20E3), anti-ACTIN-HRP (Abcam, #ab49900, AC-15), anti-Rabbit IgG-HRP (Cytiva, # NA934), and anti-Mouse IgG-HRP (Cytiva, # NA9311). Enhanced chemiluminescence (ECL) signal on PVDF membranes was captured by using LI-COR Odyssey XF imaging system (LI-COR Biosciences) and densitometry analysis of the protein bands were performed by using Image Studio software (LI-COR Biosciences).
4.2.4. Cell cycle analysis
Adherent cells were harvested by trypsinization and combined with the floating cells present in the media. Cells were first washed with PBS then fixed with ice-cold 70% ethanol for 1 h on ice. They were washed again and rehydrated by PBS, then treated with 100 μg/mL RNase A (Qiagen, #19101), and finally stained with propidium iodide (PI) (AnaSpec, #AS-83215) at a final concentration of 50 μg/mL in Cell Staining Buffer (Biolegend, #420201) for 1 h on ice. Cell cycle profile was acquired by FACSCalibur flow cytometry (Becton Dickinson)
4.2.5. Cellular thermal shift assay
Cellular thermal shift assay (CETSA) was performed as previously described.26 Samples were subjected to western blot and the target protein was detected by using anti-CD99 antibody.
4.2.6. Surface plasmon resonance
Surface plasmon resonance (SPR) experiments were done using a Biacore T200 instrument with a CM5 chip. The ligand, bacterially expressed recombinant extracellular domain of CD99, was immobilized onto the chip in 10 mM sodium acetate buffer at pH 4.0 to a level of ~3500 RU by using standard amine coupling chemistry. BK50164 and BK60106 were used as analytes to flow over the immobilized ligand. PBS-P (20 mM phosphate buffer pH 7.4, 137mM NaCl, 2.7 mM KCl, 0.05% surfactant P20) was used as the immobilization running buffer. Overnight kinetics were performed for all analytes binding to the ligands in the presence of PBS-P + 1% DMSO. The flow rate of all analyte solutions was maintained at 50 μL/min. The contact and dissociation times used were 60 s and 300 s, respectively. One 15 s pulse of 1:500 H3PO4:H2O (v/v) was injected for surface regeneration. All compounds were injected in duplicate. The sensorgrams from the overnight kinetics were evaluated using 1:1 kinetics model fitting.
4.2.7. Statistics
Statistical analyses were performed using GraphPad Prism software. Student’s t test was utilized for statistical comparison. P-Values that are smaller than 0.05 were taken as significant.
Supplementary Material
Highlights.
BK50164 and BK60106 selectively kill 7 Ewing sarcoma cells compared to 13 other cell lines.
BK50164 and BK60106 directly bind to the extracellular domain of CD99.
BK50164 and BK60106 function through an alternative mechanism compared to clofarabine.
Acknowledgments
These experiments were made possible by funding from the Bone Cancer Research Trust, United Kingdom and Children’s Cancer Foundation, Baltimore Maryland USA. EN thanks the Henry Luce Foundation for a Clare Boothe Luce Graduate Fellowship. We would like to acknowledge the Biacore Molecular Interaction Shared Resource (BMISR) at Georgetown University, which was supported by an NIH grant P30CA51008.
Aykut Uren reports financial support was provided by Bone Cancer Research Trust, United Kingdom. Eryn Nelson reports financial support was provided by Henry Luce Foundation. Aykut Uren reports financial support was provided by National Institutes of Health. Christian Wolf reports financial support was provided by Bone Cancer Research Trust, United Kingdom.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
Georgetown University has an issued patent (Patent Number: 11,202,792) on CD99 inhibitors where Dr. Aykut Uren is one of the inventors.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.
Data availability
Data will be made available on request.
References
- 1.Gelin C, Aubrit F, Phalipon A, Raynal B, Cole S, Kaczorek M, Bernard A, The E2 antigen, a 32 kd glycoprotein involved in T-cell adhesion processes, is the MIC2 gene product, EMBO J 8 (11) (1989) 3253–3259. 10.1002/j.1460-2075.1989.tb08485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ellis NA, Tippett P, Petty A, Reid M, Weller PA, Ye TZ, German J, Goodfellow PN, Thomas S, Banting G, PBDX is the XG blood group gene, Nat. Genet 8 (3) (1994) 285–290. 10.1038/ng1194-285. [DOI] [PubMed] [Google Scholar]
- 3.Suh YH, Shin YK, Kook M-C, Oh KI, Park WS, Kim SH, Lee I-S, Park HJ, Huh T-L, Park SH, Cloning, genomic organization, alternative transcripts and expression analysis of CD99L2, a novel paralog of human CD99, and identification of evolutionary conserved motifs, Gene 307 (2003) 63–76. 10.1016/S0378-1119(03)00401-3. [DOI] [PubMed] [Google Scholar]
- 4.Choi G, Lee SW, Jung KC, Choi EY, Detection of homodimer formation of CD99 through extracelluar domain using bimolecular fluorescence complementation analysis, Exp. Mol. Med 39 (6) (2007) 746–755. 10.1038/emm.2007.81. [DOI] [PubMed] [Google Scholar]
- 5.Nam G, Lee Y-K, Lee HY, Ma MJ, Araki M, Araki K, Lee S, Lee I-S, Choi EY, Interaction of CD99 with Its Paralog CD99L2 Positively Regulates CD99L2 Trafficking to Cell Surfaces, J. Immunol 191 (11) (2013) 5730–5742. 10.4049/jimmunol.1203062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park HJ, Ban YL, Byun D, Park SH, Jung KC, Interaction between the mouse homologue of CD99 and its ligand PILR as a mechanism of T cell receptor-independent thymocyte apoptosis, Exp. Mol. Med 42 (5) (2010) 353–365. 10.3858/emm.2010.42.5.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manara MC, Pasello M, Scotlandi K, CD99: A Cell Surface Protein with an Oncojanus Role in Tumors, Genes 9 (3) (2018) 159. 10.3390/genes9030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ambros IM, Ambros PF, Strehl S, Kovar H, Gadner H, Salzer-Kuntschik M, MIC2 is a specific marker for ewing’s sarcoma and peripheral primitive neuroectodermal tumors. Evidence for a common histogenesis of ewing’s sarcoma and peripheral primitive neuroectodermal tumors from MIC2 expression and specific chromosome aberration, Cancer 67 (7) (1991) 1886–1893. . [DOI] [PubMed] [Google Scholar]
- 9.Llombart-Bosch A, Machado I, Navarro S, Bertoni F, Bacchini P, Alberghini M, Karzeladze A, Savelov N, Petrov S, Alvarado-Cabrero I, Mihaila D, Terrier P, Lopez-Guerrero JA, Picci P, Histological heterogeneity of Ewing’s sarcoma/PNET: an immunohistochemical analysis of 415 genetically confirmed cases with clinical support, Virchows Arch 455 (5) (2009) 397–411. 10.1007/s00428-009-0842-7. [DOI] [PubMed] [Google Scholar]
- 10.Dworzak MN, Fritsch G, Fleischer C, Printz D, Fröschl G, Buchinger P, Mann G, Gadner H, CD99 (MIC2) expression in paediatric B-lineage leukaemia/lymphoma reflects maturation-associated patterns of normal B-lymphopoiesis, Brit. J. Haematol 105 (3) (1999) 690–695. 10.1046/j.1365-2141.1999.01426.x. [DOI] [PubMed] [Google Scholar]
- 11.Dworzak MN, Fröschl G, Printz D, Zen LD, Gaipa G, Ratei R, Basso G, Biondi A, Ludwig W-D, Gadner H, on behalf of the I-BFM-ALL-FCM-MRD-Study Group, CD99 expression in T-lineage ALL: implications for flow cytometric detection of minimal residual disease, Leukemia 18 (4) (2004) 703–708. 10.1038/sj.leu.2403303. [DOI] [PubMed] [Google Scholar]
- 12.Chung SS, Eng WS, Hu W, Khalaj M, Garrett-Bakelman FE, Tavakkoli M, Levine RL, Carroll M, Klimek VM, Melnick AM, Park CY, CD99 is a therapeutic target on disease stem cells in myeloid malignancies, Sci. Transl. Med 9 (374) (2017) eaaj2025. 10.1126/scitranslmed.aaj2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grünewald TGP, Cidre-Aranaz F, Surdez D, Tomazou EM, de Álava E, Kovar H, Sorensen PH, Delattre O, Dirksen U, Ewing sarcoma, Nat. Rev. Dis. Primers 4 (1) (2018) 5. 10.1038/s41572-018-0003-x. [DOI] [PubMed] [Google Scholar]
- 14.Fellinger EJ, Garin-Chesa P, Triche TJ, Huvos AG, Rettig WJ, Immunohistochemical analysis of Ewing’s sarcoma cell surface antigen p30/32MIC2, Am. J. Pathol 139 (2) (1991) 317–325. [PMC free article] [PubMed] [Google Scholar]
- 15.Perlman EJ, Dickman PS, Askin FB, Grier HE, Miser JS, Link MP, Ewing’s sarcoma—Routine diagnostic utilization of MIC2 analysis: A pediatric oncology group/children’s cancer group intergroup study, Hum. Pathol 25 (3) (1994) 304–307. 10.1016/0046-8177(94)90203-8. [DOI] [PubMed] [Google Scholar]
- 16.Weidner N, Tjoe J, Immunohistochemical profile of monoclonal antibody O13: antibody that recognizes glycoprotein p30/32MIC2 and is useful in diagnosing Ewing’s sarcoma and peripheral neuroepithelioma, Am. J. Surg. Pathol 18 (5) (1994) 486–494. [PubMed] [Google Scholar]
- 17.Lee CS, Southey MC, Waters K, Kannourakis G, Georgiou T, Armes JE, Chow CW, Venter DJ, EWS/FLI-1 Fusion Transcript Detection and MIC2 Immunohistochemical Staining in the Diagnosis of Ewing’s Sarcoma, Pediatr. Pathol. Lab. M 16 (3) (1996) 379–392. 10.1080/15513819609168678. [DOI] [PubMed] [Google Scholar]
- 18.Halliday BE, Slagel DD, Elsheikh TE, Silverman JF, Diagnostic utility of MIC-2 immunocytochemical staining in the differential diagnosis of small blue cell tumors, Diagn. Cytopathol 19 (6) (1998) 410–416. . [DOI] [PubMed] [Google Scholar]
- 19.Rocchi A, Manara MC, Sciandra M, Zambelli D, Nardi F, Nicoletti G, Garofalo C, Meschini S, Astolfi A, Colombo MP, Lessnick SL, Picci P, Scotlandi K, CD99 inhibits neural differentiation of human Ewing sarcoma cells and thereby contributes to oncogenesis, J. Clin. Invest 120 (3) (2010) 668–680. 10.1172/JCI36667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kreppel M, Aryee DNT, Schaefer K, Amann G, Kofler R, Poremba C, Kovar H, Suppression of KCMF1 by constitutive high CD99 expression is involved in the migratory ability of Ewing’s sarcoma cells, Oncogene 25 (19) (2006) 2795–2800. 10.1038/sj.onc.1209300. [DOI] [PubMed] [Google Scholar]
- 21.Ventura S, Aryee DNT, Felicetti F, De Feo A, Mancarella C, Manara MC, Picci P, Colombo MP, Kovar H, Carè A, Scotlandi K, CD99 regulates neural differentiation of Ewing sarcoma cells through miR-34a-Notch-mediated control of NF-κB signaling, Oncogene 35 (30) (2016) 3944–3954. 10.1038/onc.2015.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sohn HW, Choi EY, Kim SH, Lee I, Chung DH, Sung UA, Hwang DH, Cho SS, Jun BH, Jang JJ, Chi JG, Park SH, Engagement of CD99 Induces Apoptosis Through a Calcineurin-Independent Pathway in Ewing’s Sarcoma Cells, Am. J. Pathol 153 (6) (1998) 1937–1945. 10.1016/S0002-9440(10)65707-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scotlandi K, Baldini N, Cerisano V, Manara MC, Benini S, Serra M, Lollini P-L, Nanni P, Nicoletti G, Bernard G, Bernard A, Picci P, CD99 Engagement: An Effective Therapeutic Strategy for Ewing Tumors1, Cancer Res 60 (18) (2000) 5134–5142. [PubMed] [Google Scholar]
- 24.Manara MC, Terracciano M, Mancarella C, Sciandra M, Guerzoni C, Pasello M, Grilli A, Zini N, Picci P, Colombo MP, Morrione A, Scotlandi K, CD99 triggering induces methuosis of Ewing sarcoma cells through IGF-1R/RAS/Rac1 signaling, Oncotarget 7 (48) (2016) 79925–79942. https://www.oncotarget.com/article/13160/text/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Çelik H, Sciandra M, Flashner B, Gelmez E, Kayraklıoğlu N, Allegakoen DV, Petro JR, Conn EJ, Hour S, Han J, Oktay L, Tiwari PB, Hayran M, Harris BT, Manara MC, Toretsky JA, Scotlandi K, Üren A, Clofarabine inhibits Ewing sarcoma growth through a novel molecular mechanism involving direct binding to CD99, Oncogene 37 (16) (2018) 2181–2196. 10.1038/s41388-017-0080-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sevim H, Çelik H, Düşünceli L, Ceyhan CS, Molotkova A, Nakazawa K, Graham GT, Petro JR, Toretsky JA, Üren A, Clofarabine induces ERK/MSK/CREB activation through inhibiting CD99 on Ewing sarcoma cells, PLOS ONE 16 (6) (2021) e0253170. 10.1371/journal.pone.0253170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carson DA, Wasson DB, Kaye J, Ullman B, Martin DW, Robins RK, Montgomery JA, Deoxycytidine kinase-mediated toxicity of deoxyadenosine analogs toward malignant human lymphoblasts in vitro and toward murine L1210 leukemia in vivo, P. Natl. A. Sci. USA 77 (11) (1980) 6865–6869. 10.1073/pnas.77.11.6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lindemalm S, Liliemark J, Juliusson G, Larsson R, Albertioni F, Cytotoxicity and pharmacokinetics of cladribine metabolite, 2-chloroadenine in patients with leukemia, Cancer Lett 210 (2) (2004) 171–177. 10.1016/j.canlet.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 29.Bonate PL, Arthaud L, Cantrell WR, Stephenson K, Secrist JA, Weitman S, Discovery and development of clofarabine: a nucleoside analogue for treating cancer, Nat. Rev. Drug. Discov 5 (10) (2006) 855–863. 10.1038/nrd2055. [DOI] [PubMed] [Google Scholar]
- 30.Balsley MA, Malesevic M, Stemmy EJ, Gigley J, Jurjus RA, Herzog D, Bukrinsky MI, Fischer G, Constant SL, A cell-impermeable cyclosporine A derivative reduces pathology in a mouse model of allergic lung inflammation, J. Immunol 185 (12) (2010) 7663–7670, 10.4049/jimmunol.1001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malesevic M, Kuhling J, Erdmann F, Balsley MA, Bukrinsky MI, Constant SL, Fischer G, A cyclosporin derivative discriminates between extracellular and intracellular cyclophilins, Angew. Chem. Int. Ed 49 (1) (2010) 213–215, 10.1002/anie.200904529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsutsumi S, Scroggins B, Koga F, Lee MJ, Trepel J, Felts S, Carreras C, Neckers L, A small molecule cell-impermeant Hsp90 antagonist inhibits tumor cell motility and invasion, Oncogene 27 (17) (2008) 2478–2487, 10.1038/sj.onc.1210897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu W, Luo Y, Sun C, Liu Y, Kuo P, Varga J, Xiang R, Reisfeld R, Janda KD, Edgington TS, Liu C, Targeting cell-impermeable prodrug activation to tumor microenvironment eradicates multiple drug-resistant neoplasms, Cancer Res 66 (2) (2006) 970–980, 10.1158/0008-5472.CAN-05-2591. [DOI] [PubMed] [Google Scholar]
- 34.Michalski R, Zielonka J, Hardy M, Joseph J, Kalyanaraman B, Hydropropidine: a novel, cell-impermeant fluorogenic probe for detecting extracellular superoxide, Free Radic. Biol. Med 54 (1) (2013) 135–147, 10.1016/j.freeradbiomed.2012.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lindberg E, Mizukami S, Ibata K, Miyawaki A, Kikuchi K, Development of luminescent coelenterazine derivatives activatable by beta-galactosidase for monitoring dual gene expression, Chemistry 19 (44) (2013) 14970–14976, 10.1002/chem.201302002. [DOI] [PubMed] [Google Scholar]
- 36.Yudina A, Lepetit-Coiffe M, De Smet M, Langereis S, Grull H, Moonen C, In vivo temperature controlled ultrasound-mediated intracellular delivery of cell-impermeable compounds, J. Control Release 161 (1) (2012) 90–97, 10.1016/j.jconrel.2012.04.018. [DOI] [PubMed] [Google Scholar]
- 37.Zhang N, Chen Y, Yu M, Liu Y, Benzenesulfonamidoquinolino-beta-cyclodextrin as a cell-impermeable fluorescent sensor for Zn2+, Chem. Asian. J 4 (11) (2009) 1697–1702, 10.1002/asia.200900233. [DOI] [PubMed] [Google Scholar]
- 38.Ghoteimi R, Nguyen VT, Rahimova R, Grosjean F, Cros-Perrial E, Uttaro JP, Mathe C, Chaloin L, Jordheim LP, Peyrottes S, Synthesis of substituted 5′-aminoadenosine derivatives and evaluation of their inhibitory potential toward CD73, ChemMedChem 14 (15) (2019) 1431–1443, 10.1002/cmdc.201900348. [DOI] [PubMed] [Google Scholar]
- 39.Qiu X, Huang Y, Wu D, Mao F, Zhu J, Yan W, Luo H-B, Li J, Discovery of novel purine nucleoside derivatives as phosphodiesterase 2 (PDE2) inhibitors: Structure-based virtual screening, optimization and biological evaluation, Bioorg. Med. Chem 26 (1) (2018) 119–133, 10.1016/j.bmc.2017.11.022. [DOI] [PubMed] [Google Scholar]
- 40.Chen L, Li J, Sjogren EB, Billedeau RJ, Ectonucleotidase inhibitors and methods of use thereof. WO20180491452018.
- 41.Billedeau RJ, Ll J, Chen L (2018) Ectonucleotidase Inhibitors and Methods of Use Thereof. WO20181192842018.
- 42.Griffith AK, Vanos CM, Lambert TH, Organocatalytic carbonyl-olefin metathesis, J. Am. Chem. Soc 134 (45) (2012) 18581–18584, 10.1021/ja309650u. [DOI] [PubMed] [Google Scholar]
- 43.Perrone P, Daverio F, Valente R, Rajyaguru S, Martin JA, Lévêque V, Pogam SL, Najera I, Klumpp K, Smith DB, McGuigan C, First Example of Phosphoramidate Approach Applied to a 4’-Substituted Purine Nucleoside (4’-Azidoadenosine): Conversion of an Inactive Nucleoside to a Submicromolar Compound versus Hepatitis C Virus, J. Med. Chem 50 (22) (2017) 5463–5470, 10.1021/jm070362i. [DOI] [PubMed] [Google Scholar]
- 44.Kershaw NM, Byrne DP, Parsons H, Berry NG, Fernig DG, Eyers PA, Cosstick R, Structure-based design of nucleoside-derived analogues as sulfotransferase inhibitors, RSC Adv 9 (55) (2019) 32165–32173, 10.1039/C9RA07567D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maag H, Rydzewski RM, McRoberts MJ, Crawford-Ruth D, Verheyden JPH, Prisbe EJ, Synthesis and anti-HIV activity of 4’-azido- and 4’-methoxynucleosides, J. Med. Chem 35 (8) (1992) 1440–1451, 10.1021/jm00086a013. [DOI] [PubMed] [Google Scholar]
- 46.Beigelman L, Deval J, Smith DB, Wang G, Rajwanshi VK, Preparation of azido nucleosides and nucleotide analogs as antiviral agents. US20120070415.
- 47.The conversions were typically high and the low yields of some iodides result from loss during work-up.
- 48.Sciandra M, Marino MT, Manara MC, Guerzoni C, Grano M, Oranger A, Lucarelli E, Lollini P-L, Dozza B, Pratelli L, Di Renzo MF, Colombo MP, Picci P, Scotlandi K, CD99 Drives Terminal Differentiation of Osteosarcoma Cells by Acting as a Spatial Regulator of ERK 1/2, J. Bone Miner. Res 29 (5) (2014) 1295–1309. 10.1002/jbmr.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manara MC, Bernard G, Lollini P-L, Nanni P, Zuntini M, Landuzzi L, Benini S, Lattanzi G, Sciandra M, Serra M, Colombo MP, Bernard A, Picci P, Scotlandi K, CD99 Acts as an Oncosuppressor in Osteosarcoma, Mol. Biol. Cell 17 (4) (2006) 1910–1921. 10.1091/mbc.e05-10-0971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zucchini C, Manara MC, Pinca RS, De Sanctis P, Guerzoni C, Sciandra M, Lollini P-L, Cenacchi G, Picci P, Valvassori L, Scotlandi K, CD99 suppresses osteosarcoma cell migration through inhibition of ROCK2 activity, Oncogene 33 (15) (2014) 1912–1921. 10.1038/onc.2013.152. [DOI] [PubMed] [Google Scholar]
- 51.Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundbäck T, Nordlund P, Molina DM, The cellular thermal shift assay for evaluating drug target interactions in cells, Nat. Protoc 9 (9) (2014) 2100–2122. 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
- 52.Molina DM, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, Nordlund P, Monitoring Drug Target Engagement in Cells and Tissues Using the Cellular Thermal Shift Assay, Science 341 (6141) (2013) 84–87. 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- 53.Pasello M, Manara MC, Scotlandi K, CD99 at the crossroads of physiology and pathology, J. Cell Commun. Signal 12 (1) (2018) 55–68. 10.1007/s12079-017-0445-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Çelik H, Sajwan KP, Selvanathan SP, Marsh BJ, Pai AV, Kont YS, Han J, Minas TZ, Rahim S, Erkizan HV, Toretsky JA, Üren A, Ezrin Binds to DEAD-Box RNA Helicase DDX3 and Regulates Its Function and Protein Level, Mol. Cell. Biol 35 (18) (2015) 3145–3162. 10.1128/MCB.00332-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.