

Abstract
Background and Purpose
No current treatments target microvascular reperfusion after stroke, which can contribute to poor outcomes even after successful clot retrieval. The G protein-coupled receptor GPR39 is expressed in brain peri-capillary pericytes, and has been implicated in microvascular regulation, but its role in stroke is unknown. We tested the hypothesis that GPR39 plays a protective role after stroke, in part due to preservation of microvascular perfusion. We generated GPR39 knockout (KO) mice and tested whether GPR39 gene deletion worsens capillary blood flow and exacerbates brain injury and functional deficit after focal cerebral ischemia.
Methods
Stroke was induced in male and female GPR39 KO and WT littermates by 60-minute middle cerebral artery occlusion (MCAO). Microvascular perfusion was assessed via capillary red blood cell (RBC) flux in deep cortical layers in vivo using optical microangiography (OMAG). Brain injury was assessed by measuring infarct size by 2,3,5-triphenyltetrazolium chloride staining at 24 hours or brain atrophy at 3 weeks after ischemia. Pole and cylinder behavior tests were conducted to assess neurological function deficit at 1 and 3 weeks post-stroke.
Results
Male but not female GPR39 KO mice exhibited larger infarcts and lower capillary RBC flux than WT controls after stroke. Male GPR39 KO mice also exhibited worse neurologic deficit at 1 week post-stroke, though functional deficit disappeared in both groups by 3 weeks.
Conclusions
GPR39 deletion worsens brain injury, microvascular perfusion, and neurological function after experimental stroke. Results indicate that GPR39 plays a sex-dependent role in re-establishing microvascular flow and limiting ischemic brain damage after stroke.
Keywords: G protein-coupled receptor 39, capillary flux, pericytes, no-reflow, OMAG, sex difference
INTRODUCTION
Stroke is the second leading cause of death globally, with ischemic stroke accounting for 87% of all strokes.1 Globally, strokes account for 34% of global total healthcare expenditure.2 While t-PA clot lysis and endovascular clot retrieval have revolutionized stroke treatment, only a small percentage of patients qualify for these procedures and have access to centers that perform them.3 Patients who undergo clot retrieval have variable functional recovery, with a third to half of patients having poor outcome despite undergoing endovascular therapy.4 Success at large vessel recanalization may not be accompanied by the expected restoration of downstream microvascular reperfusion.5 This “no-reflow” phenomenon across microvascular capillary beds may be secondary to perivascular edema, pericapillary cells compressing the delicate vessels, continued occlusion by small irretrievable clots, or inadequate blood flow or blood pressure to reopen small vessels.5 Neurons have high metabolic demand, utilizing near 1:1 capillary:neuron neurovascular coupling to maintain optimal oxygenation and waste removal. Persistent capillary dysfunction would consequently lead to neural starvation and death in the affected watershed.6 Factors contributing to poor outcome after clot retrieval include vascular comorbidities such as atherosclerosis or diabetes mellitus.6
GPR39 is a member of the ghrelin family of G protein-coupled receptors. It plays a homeostatic role in vascular regulation, neuronal excitability and inflammation.7 Expressed in human brain peri-capillary pericytes and microglia,8 GPR39 has been implicated in moderating vascular inflammation and preventing excitotoxicity.7,9 GPR39 has been implicated in several neurological disorders, including cerebral small vessel disease-related dementia.8 The endogenous ligand for GPR39 is uncertain, but it has been shown to sense the balance between vasodilator and vasoconstrictor eicosanoids, which contribute to the dynamic regulation of capillary blood flow.10,11 GPR39 can also be activated by high concentrations of zinc which are known to occur during cerebral ischemia and may contribute to neurotoxicity.7,12,13 These converging lines of reasoning suggest that GPR39 may play a role in the response to acute ischemic stroke. In this study, we used CRISPR/Cas9-mediated GPR39 gene deletion to generate GRP39 KO mice. We next used these mice to assess effect of GPR39 loss on post-ischemic microvascular flow, tissue injury and functional deficits in the setting of focal, transient cerebral ischemia. Our findings indicate that GPR39 KO mice exhibit persistently impaired microvascular blood flow after reperfusion despite restored macrocirculation, leading to exacerbated ischemic brain injury and concomitantly worse neurological deficits.
METHODS
Data are available from the corresponding author on reasonable request. This article adheres to the American Heart Association Journals Implementation of Transparency and Openness Promotion Guidelines, including the ARRIVE 2.0 guidelines for reporting animal research. Detailed methods are available in the Supplemental Material.
Generation of GPR39 knockout mice
Mice with targeted GPR39 deletion were generated via CRISPR targeting technology.14,15 Multiple guide RNAs (gRNAs) were designed that flank GPR39 exon 1 and each screened for DNA cleavage efficiency in murine Neuro 2A (N2A) cells (Invitrogen). A mixture containing 30 ng/μl of the two optimal gRNAs (Supplemental Table S1) and 100 ng/μl of the Cas9 mRNA (Trilink Biotech) was prepared, injected into zygotes of C57BL/6NJ mice (Jackson Laboratory, Stock #005304). The zygotes were transferred into oviducts of pseudopregnant CD-1 females (Charles River Strain #022). The deletion event was confirmed using PCR primers that flank the predicted deletion (Supplemental Table 1). Germline founders were then backcrossed to C57BL/6 for at least 6 generations to eliminate off-target mutations, and subsequent progeny were used to establish the GPR39-null line. Mice were genotyped using ear biopsies using RT-PCR with specific probes designed for each gene (Transnetyx, Cordova, TN, USA; Supplemental Table 1). Western blot and RT-PCR analysis demonstrated missing GPR39 product in KO mice (Fig 1). Homozygous GPR39 KO mice are viable, fertile, normal in size, and do not display any overt physical or behavioral abnormalities.
Figure 1. Creation and confirmation of mouse GPR39 knockout model.
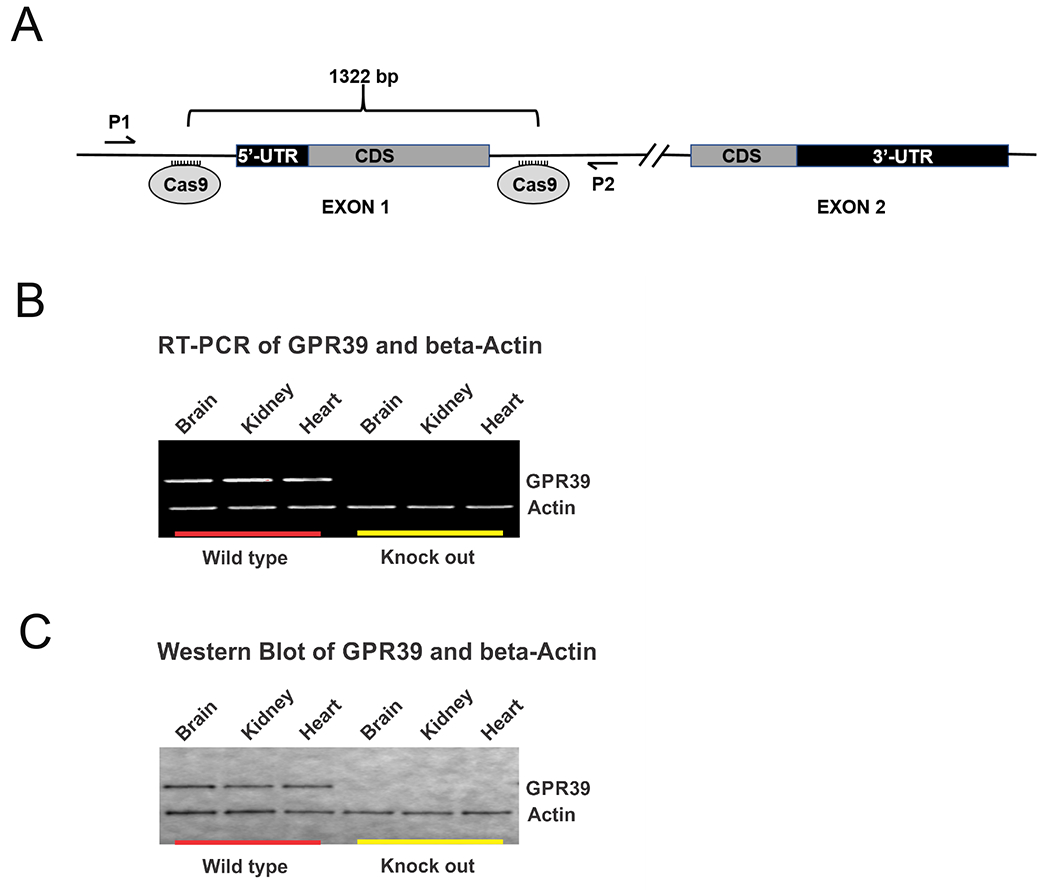
A. Schematic of mouse GPR39 gene locus targeting strategy. Guide RNAs were designed to cleave at sites flanking exon 1 of Gpr39 resulting in a 1322 base pair deletion. Exon 2 was left intact. B. RT-PCR and C. Western blot of GPR39 (top row) and beta-Actin (bottom row) confirming presence of GPR39 mRNA (B) and protein (C) in brain, heart and kidneys from wild type mice (left) but absent in GPR39 knockout mice (right).
Animals
Male and female GPR39 KO and WT littermates (n=165 mice; Supplemental Table 2) were housed in temperature-controlled rooms on a 12-hour light and 12-hour dark cycle with water and food ad libitum. Animals were randomized to experimental groups, and investigators were blinded to group assignment until datasets were complete and analyzed. All animal procedures were conducted in accordance with National Institute of Health guidelines for use of animals in research, and protocols were approved by the Animal Care and Use Committee at Oregon Health & Science University.
Middle cerebral artery occlusion (MCAO)
Transient focal cerebral ischemia was induced in male (25-32 g; n=9 WT, N=14 KO) and female (20-25 g; n=9 WT, n=12 KO) mice at the age of 12-16 weeks using the intraluminal MCAO technique, as previously described.16 Briefly, isoflurane-anesthetized mice were subjected to 60-min MCAO using a silicone-coated 6-0 monofilament (Doccol Corporation, Sharon, MA, USA). The filament was inserted through the right external carotid artery and advanced into the internal carotid artery until laser-Doppler signal dropped to less than 30% of baseline. Mice were kept anesthetized throughout MCAO with 1 % isoflurane and kept warm with water pads and heat lamp. A small laser-Doppler metal probe (Model DRT4, Moor Instruments Inc., Wilmington, DE, USA) was used to monitor cortical perfusion and verify occlusion and reperfusion. After 60 minutes, the filament was withdrawn to allow for reperfusion. Mice that did not exhibit a drop in laser-Doppler signal by at least 70% or did not have an increase upon reperfusion to at least 70% of baseline were excluded from the study. Animals were also excluded if they died during surgery or before the time point for tissue or behavioral measurement. Mice were allowed to recover from anesthesia and were subsequently housed in individual cages with free access to food and water until the end of the study. Mice were given daily 0.3 mL intraperitoneal injections of normal saline until they resumed eating solid food (usually within first 3 days). Physiological variables were measured in separate groups of non-survival WT and GPR39 KO mice (n=5 each). Arterial blood pressure was measured continuously throughout MCAO procedure via a femoral arterial catheter. Arterial blood (150 µL) was collected via a femoral artery catheter immediately before and 15 min after MCAO for blood gas measurements using the iSTAT Chem 8+ cartridge (Abbott Point of Care, Princeton, NJ, USA). Mice used for blood analysis and measurement of blood pressure were sacrificed at the end of measurements.
Cranial window
Cranial window creation was adapted from in-vivo multiphoton imaging studies.18 In brief, the cranial window was performed under isoflurane anesthesia (1.0%-1.5%) using a custom-built stereotaxic apparatus to secure the mouse head. A 5-mm-diameter circular craniotomy 1 mm posterior and 3 mm lateral to bregma was created (Supplemental Fig 1), closed with 5 mm cover glass (#0 small round cover glass, 5 mm small round, model no. CS-5R-0, Warner Instruments, Holliston, MA, USA), and a custom-built aluminum fixation headplate cemented into place. The cranial window dimensions and location were selected to cover the boundary between infarcted and non-infarcted tissue on the dorsolateral surface of the brain, based on previous histological staining and OCT-based imaging.17,18
Optical Microangiography (OMAG)
OMAG imaging was performed in WT and GPR39 KO mice (n=10 and 11, respectively) using a previously described system.18,19,20 The animal was immobilized on a custom-made stereotaxic stage and was lightly anesthetized with isoflurane (0.2 L/min O2, 0.8 L/min air). Body temperature was monitored by a rectal thermal probe throughout the experiment, and kept at 36.5± 0.5°C by hot water pad and warming blanket. The parietal cranial window was positioned under the OMAG scanning probe and tilted so that it was perpendicular to the light beam with a 10x objective lens. A large field-of-view (FOV) OMAG scan (x-y-z dimensions 2.4 mm x 2.4 mm x 2mm) was taken to ensure landmark positioning, then a smaller OMAG FOV scan (x-y-z 0.5 mm x 0.5 mm x 0.4 mm) was taken at the center for RBC flux measurements. The z dimension (0.4 mm), which represent depth below the cortical surface, was divided into 4 equal layers (100 μm each). OMAG imaging was then performed to assess the total number of RBCs passing through the imaging voxel cross-section per unit time in order to determine RBC flux (Supplemental Fig S1). After removing projection artefact in the scanned volume dataset, the maximum projected en-face image was analyzed separately for each layer to determine average flux within each layer. Flux images were processed to separate dynamic pixels (moving red blood cells within vessels) from static pixels (structural tissue such as vessel walls). Only capillaries were assessed for flux changes using a size filter, excluding all vessels >10 μm in diameter. At the end of the 24-hour scan, animals were sacrificed and infarct size was measured in these brains as described below. Separate animals were also used for infarct size measurement that did not go through OMAG scanning.
Measurement of infarct size
Mice were euthanized and brains collected and stained with 2,3,5-triphenyltetrazolium chloride (TTC; Sigma, St. Louis, MO, USA) at 24 hours after MCAO.16 Brain was sectioned into 2-mm thick coronal sections, incubated in TTC, fixed in formalin and digitally analyzed to obtain infarct volume. Infarcted and uninfarcted areas were measured with ImageJ (National Institute of Health, Bethesda, MD, USA). To account for edema, infarct size in each region (cortex, caudate-putamen (CP; striatum) and total hemisphere; Supplemental Fig S3) was measured as the difference between the contralateral tissue volume and the ipsilateral uninfarcted tissue. Infarct size was measured in male and female WT (n=9 each) and GPR39 KO (n=14 and 12, respectively) mice.
Measurement of brain atrophy
Separate groups of mice were survived for 21 days after MCAO to assess behavioral deficit as described below. Brain damage was evaluated in these mice at 21 days by measuring tissue atrophy (Supplemental Fig S3) rather than infarct because at this time point, liquefaction, clearing and scarring after stroke eliminates infarcted tissue.21 Brains were harvested after behavioral testing in post-operative week 3 (POW3), and cut into 2-mm thick coronal brain sections. Slices were photographed, and the remaining size of each ipsilateral region and its contralateral counterpart were quantified using ImageJ. The ipsilateral region volume was divided by contralateral volume to determine the percentage volume remaining and atrophied.
Behavior testing after MCAO
Locomotor function (“PRE”) was assessed 24 hours before MCAO surgery (day −1), and locomotor deficit was evaluated in post-operative week 1 (POW1) and 3 (POW3) using the Cylinder and Pole tests.22 Details of testing are provided in Supplemental Material. After testing, animals were sacrificed and brains collected to measure brain atrophy, as described above.
Statistical Analysis
Sample sizes were chosen for adequate statistical power (0.8) based on results of prior data sets. Data are presented as mean ±SEM. Analysis was performed using the PRISM 8.0 software. Differences among groups in infarct size, locomotor deficit, atrophy, microvascular perfusion and physiological variables were evaluated by two-way ANOVA with Sidak’s multiple comparisons post hoc test. Separate t-test and ANOVA were performed to assess differences in flux in the deepest cortical layer and overall flux among 4 layers, respectively. Survival and success rates were compared using Gehan-Breslow-Wilcoxon test. Statistical significance was set at p<0.05.
RESULTS
GPR39 deletion increases infarct size after MCAO in male mice
We used a dual guide RNA (gRNA)/CRISPR strategy to generate a GPR39 KO mouse model with a targeted 1322-bp deletion that encompasses exon 1 (Fig 1A). Loss of GPR39 gene product is documented by RT-PCR (Fig 1B) and Western blot analysis (Fig 1C). Fig 2 shows the effect of GPR39 knockout on laser-Doppler perfusion (LDP) and infarct size in male and female GPR3 KO and WT controls. LDP over the MCAO territory was significantly reduced by MCAO, and returned to baseline upon reperfusion, with no difference among groups at any time before, during or after MCAO (Fig 2A). Despite a similar ischemic insult, male GPR39 KO mice sustained larger infarcts than WT counterparts (55% larger in cortex, 71% in caudate-putamen and 64% in whole hemisphere, Fig 2B, all p<0.03). In contrast, brain infarct size in female KO mice did not differ from WT female controls in any brain region (Fig 2C). To further investigate male-specific vulnerability of GPR39 KO to MCAO, male mice were studied further by evaluating the effect of GPR39 knockout on capillary flux in deep brain tissue and on long-term behavioral outcome. Because infarct size was not different between WT and KO females, we did not measure capillary flux or assess long-term behavior in females.
Figure 2. GPR39 deletion increases infarct size in male mice after MCAO.
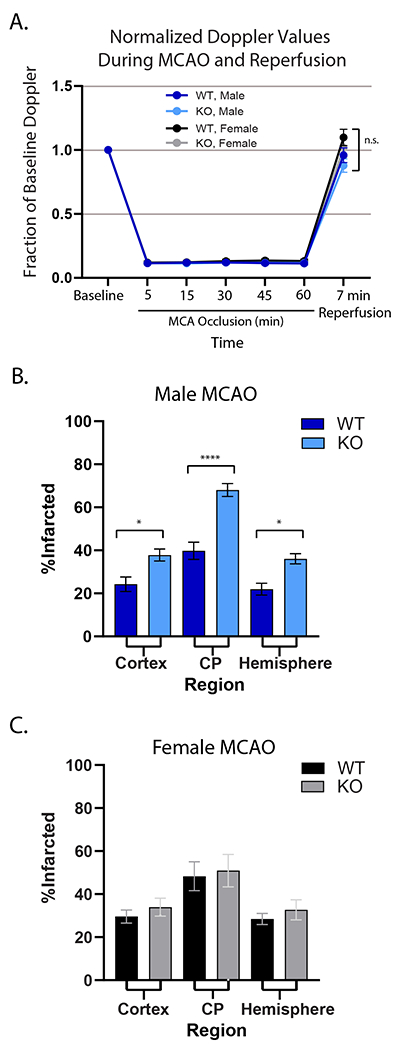
A. Relative laser Doppler perfusion of MCA territory. There were no differences among groups. B. Infarct size was significantly larger in male GPR39 KO mice (n=14) compared to WT littermates (n=9) in cerebral cortex, caudate putamen (CP) and whole hemisphere. C. There was no difference in infarct size between WT (n=9) and GPR39 KO (n=12) in females. GPR39 KO vs. WT. n.s., not significant. *p<0.05, ** p<0.001. Cortex and CP are components of the hemisphere.
GPR39 KO decreases capillary flux after MCAO in deep cortical layers in male mice
OMAG was employed to assess RBC flux through capillary beds below the cortical surface within the MCA watershed area (penumbra; area highlighted in Fig 3A and Supplemental Fig 1). Concomitant with the larger cortical and CP infarcts in male mice, there was progressive microcirculatory hypoperfusion in the KO group, which was especially evident in deeper layers 3 and 4 (Fig 3B, p<0.03). Compared to WT controls, post-ischemic RBC flux in layer 3 was halved in the KO group and decreased by 96% in the deepest layer (layer 4, Fig 3C, p< 0.05). This observation is consistent with the dependence of deeper brain tissue on the microcirculation for effective perfusion.
Figure 3. Optical microangiography (OMAG) demonstrates worsened microvascular brain tissue perfusion in GPR39 KO vs WT males after stroke.
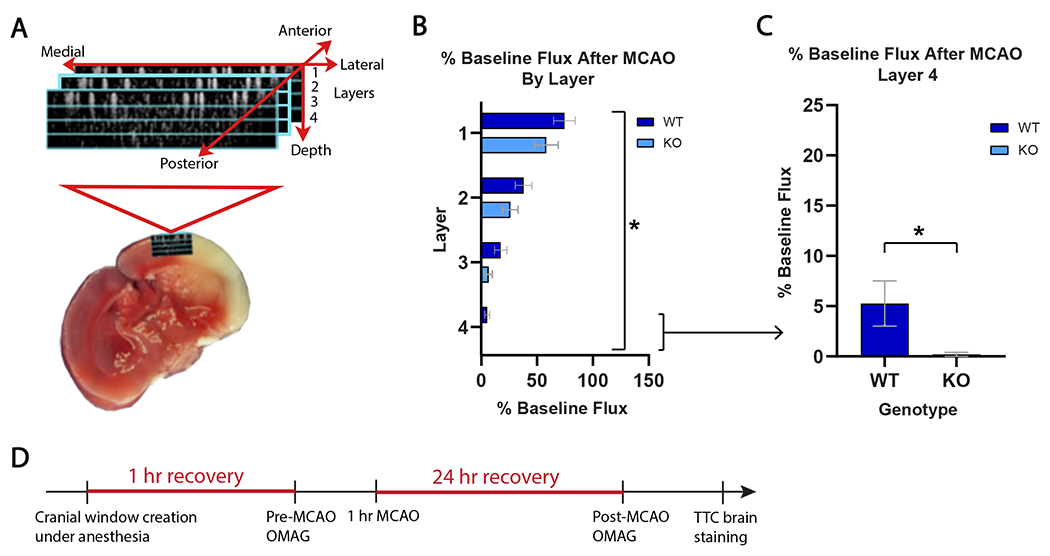
A. Schematic of OMAG setup for capillary blood flow measurement in 4 cortical layers within ischemic penumbra. B. Post-stroke cortical capillary flux in in layers 1-4 (% baseline) in GPR39 KO (n=10) and WT littermates (n=11). (Genotype F1,76 = 5.36; *p<0.0234). C. Post-stroke capillary flux in GPR39 KO vs. WT littermates in deepest cortical layer only (#4). (n=10 KO, n=11 WT, *p<0.05). D. Experimental timeline illustrating OMAG timing relative to MCAO. Mice were allowed 1 hour of recovery after cranial window creation under anesthesia prior to baseline OMAG scan. Mice were then subjected to MCAO, and recovered for 24 hours when they underwent a second post-stroke OMAG scan. Brains were collected, sectioned and stained with TTC to measure infarct size.
GPR39 KO exacerbates locomotor deficit at 1 week after MCAO
To determine whether increased ischemic damage in GPR39 KO mice affected neurological function, we assessed locomotor behavior at 1 and 3 weeks after MCAO using the Pole and Cylinder tests, and evaluated survival and ipsilateral brain tissue atrophy at these time points (Fig 4). In the first 7 days after MCAO, GPR39 KO mice died at a higher rate than WT controls (29% vs. 19%, respectively, p<0.04), but mice in both groups who survived to 7 days also survived for the full 21-day study period (Fig 4A). Using atrophy to index brain injury in mice surviving the full 3 weeks, GPR39 KO mice had significantly more tissue atrophy than WT mice in caudate/putamen (16.2%, p<0.05), with trends for more atrophy in cerebral cortex (27.6%, p=0.084) and whole hemisphere (23.1%, p=0.068). GPR39 KO mice suffered greater disability than WT controls in both the Cylinder and Pole tests at postoperative week 1 (POW1). In the Cylinder test (Fig 4C), GPR39 KO mice favored their unaffected paw (positive asymmetry index, where more positivity is worse) than WT controls in POW1, consistent with greater disability. The exaggerated disability was transient, however, disappearing in POW3. Similarly, in the pole test (Fig 4D), GPR39 KO mice took longer to complete the task than controls at POW1, a deficit also not sustained at POW3.
Figure 4. GPR39 KO decreases survival and motor function 1 week after stroke, and increases brain atrophy 3 weeks after stroke.
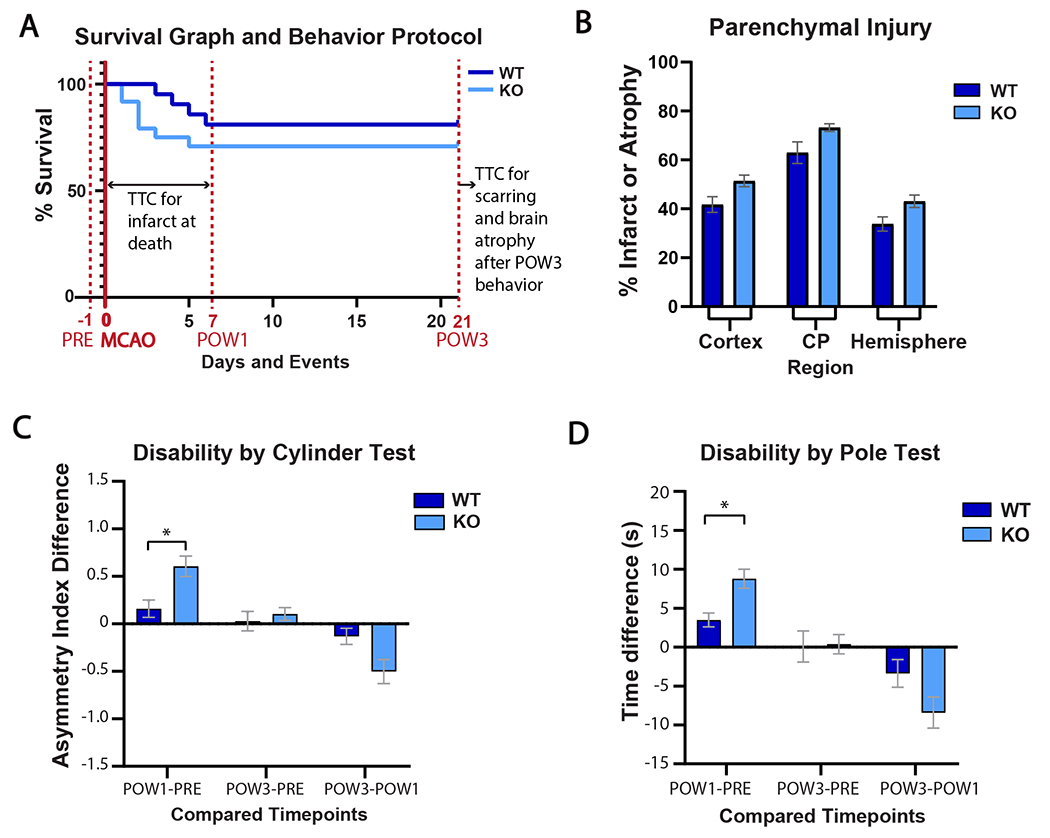
A. Timeline of behavior testing, MCAO, infarct size and brain atrophy measurement, superimposed on survival curve. GPR39 KO decreased survival at 1 week compared to WT (*p<0.05). B. Ipsilateral atrophy in cortex, caudate putamen (CP) and hemisphere in KO and WT mice at 3 weeks after stroke. C. KO mice show greater forepaw disability in the Cylinder test 1 week after stroke. Higher asymmetry indicates greater disability. D. KO mice show decreased coordination in the Pole test 1 week after stroke. Longer time indicates greater disability. Total number of KO and WT mice at beginning of protocol was 24 and 21, respectively. Number of mice at full survival to 21 days was 17 in each group (*P <0.05).
Hemodynamic and hematological parameters during MCAO
The impact of GPR39 deletion on the hemodynamic and hematological parameters during MCAO was evaluated in separate cohorts of non-surviving mice. As shown in Supplemental Fig S2, no differences were observed between WT and KO mice in arterial blood gases (ABG), base excess, pH, hematocrit and glycemic status before or after MCAO. GPR39 KO mice exhibited a mild hyperkalemic response to MCAO, although baseline kalemia did not differ from WT controls. As in Fig 2A, no difference in laser-Doppler perfusion over the MCA territory was observed between WT and KO mice in this non-survival cohort at any time point (Fig 5A). Interestingly, we observed an exaggerated drop in blood pressure to blood withdrawals in GPR39 KO mice compared to WT controls (Fig 5B).
Figure 5. Hemodynamic measures in GPR39 KO and WT mice during and after stroke.
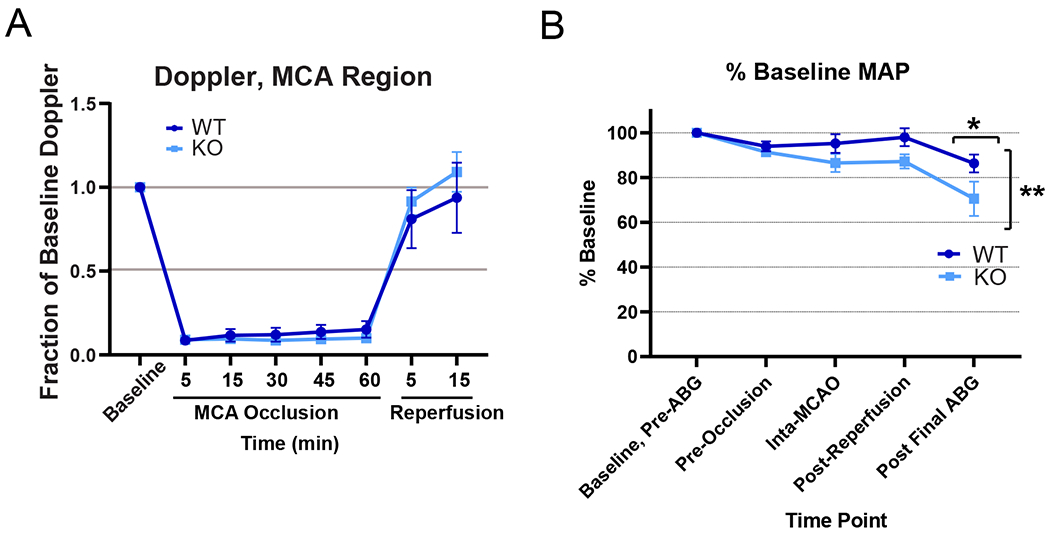
A. Relative laser-Doppler perfusion over the MCA territory during and 15 min after MCAO, n.s. not significant. B. Mean arterial pressure (MAP), as % baseline, in cohort used for arterial blood gas measurement. (Genotype F1,32 = 10.05; **p<0.0034; post final ABG adjusted p value for multiple comparisons *p<0.05; n=5 per group).
DISCUSSION
Our study reveals several new findings: 1) Targeted deletion of the GPR39 worsens ischemic damage in a sex-specific manner, especially in deeper brain regions in male; 2) Larger infarct in GPR39 KO male mice is associated with impaired capillary reperfusion at 24 hours after stroke, particularly in deeper cortical layers; 3) GPR39 KO male mice sustain larger locomotor deficit 1 week after stroke than WT male mice; 4) GPR39 KO mice experience increased mortality in the first week after focal cerebral ischemia; and 5) GPR39 KO animals are less able to maintain blood pressure under anesthesia during blood withdrawals. We conclude that the GPR39 gene deletion decreases microvascular reperfusion, exacerbates ischemic brain injury and impairs functional recovery after MCAO. GPR39 may also play a role in preserving systemic blood pressure during blood loss. Taken together, our findings are consistent with a role of GPR39 in control of the microcirculation, such that loss of GPR39 impairs the brain’s microvascular capillary response to ischemia and the body’s systemic response to hypotension. Thus, preservation or enhancement of GPR39 function during ischemic stroke and systemic hypotension may be protective.
Clinical studies provided evidence that microvascular dysfunction after stroke is associated with poor stroke outcome. In a large prospective cohort from 2 multicenter clinical trials, persistent tissue hypoperfusion 24 hours after reperfusion therapy was associated with worse cerebral edema even among patients with apparently successful angiographic reperfusion.23 It has been proposed that hypoperfusion after successful thrombectomy is related to impaired microvascular tissue reperfusion at the capillary level, the so called “no-reflow” phenomenon,24 although the presence of no-reflow remain controversial and possibly rare in human stroke.25 Few human studies investigated the sex difference in post-stroke microvascular dysfunction. In one study, women suffer worse microvascular injury in white matter after ischemic stroke than men,26 which may contribute to poorer functional outcomes in women compared to age-matched men with similar risk factors and stroke severity.27,28 Other studies have demonstrated higher efficacy of thrombolytic therapy in women compared to men, presumably due to a greater tendency for recanalization of small cardioembolic clots.29,30,31 However, some studies reported worse functional outcomes in women after thrombolysis compared to men.32,33 Conclusions are similarly equivocal regarding endovascular thrombectomy (EVT), with some studies finding better outcomes in women after adjusting for age and stroke severity,34 while others finding no sex difference in EVT outcomes.35 In experimental stroke, we have previously shown that female animals experience smaller stroke lesions than male animals36 This effect was eliminated by ovariectomy and restored by estrogen, which protects against stroke in part by improving post-stroke tissue hypoperfusion.37
The present study extends previous reports by demonstrating a male-specific increase in vulnerability to focal cerebral ischemia as a result of GPR39 KO. The differential effect of GPR39 deletion in males vs. females suggests that differences in GPR39 expression, distribution, activity or signaling may exist between male and female mice. The increase in ischemic damage in males is likely linked to poor microvascular perfusion after stroke, as male KO mice had significantly lower capillary flux in deep cortical layers than corresponding WT mice. Poor capillary perfusion in deep cortical layers coincided with the observation that the largest and most consistent regional damage in our study, assessed by measuring either infarct or atrophy, occurred in the deep caudate/putamen (striatum) region. Coexistence of poor microvascular perfusion and larger ischemic damage in the striatum fits the concept that deep brain structures are highly dependent on microvascular flow and are prone to lacunar infarcts due to lack of collaterals connecting penetrating end-arterioles.38 Moreover, at progressively deeper subcortical layers, capillary RBC flux slows and oxygen extraction increases and becomes more homogenous, again leading to higher dependence of deeper brain tissue on microvascular blood flow.39 Reduced capillary blood flow after MCAO has been linked to multiple mechanisms, including pericyte contraction40 and death,41 capillary plugging by blood cells, especially neutrophils,42,43 and interstitial edema and swelling of endothelial cells and astrocyte end feet.5 The location of GPR39 in peri-capillary pericytes supports a role for pericytes in the exaggerated decrease in post-MCAO capillary flux in GPR39 KO mice.8
It is important to note that the failure of capillaries to reperfuse deep brain tissue is observed in mice that had complete restoration of laser-Doppler blood flow on the surface of the brain. This is reminiscent of the clinical observation of incomplete tissue perfusion despite complete large-vessel recanalization after endovascular therapy or t-PA. Our data suggest that GPR39 is an endogenous microvascular protective mechanism, and that enhancing GPR39 activity may protect against microvascular dysfunction after stroke and may serve as a complementary approach to enhance efficacy of endovascular therapy or t-PA.
It is also important to note that the drop in blood pressure laser-Doppler cortical perfusion was preserved despite the drop in blood pressure, and, importantly, that animals undergoing MCAO procedure without blood withdrawal did not exhibit a blood pressure. Therefore, reduced capillary flux in deep cortical layers is unlikely related to a change in systemic blood pressure. Similarly, it is unlikely that changes in the blood composition were responsible for the difference in capillary flux between WT and KO mice, as there was no indication in blood analysis results to suggest that physical characteristics of the blood are different between WT and KO mice.
Our experimental approach allowed us to observe the effect of GPR39 KO on the evolution of infarction over time; from early reperfusion (20 min, in mice used for physiological monitoring), to 24 hours (TTC) and 21 days post-reperfusion (in animals used to measure atrophy). Longitudinal examination shows that maximum CP/striatum damage occurs acutely in GPR39 KO mice, consistent with the role of microcirculation in the acute phase of ischemic injury; whereas more generalized hemispheric damage continues to evolve over time, in part due to secondary inflammation and neurodegeneration. In WT mice, both CP/striatum and generalized hemispheric damage tend to increase more slowly over time, presumably due to intact microvascular protective mechanisms.
In addition to its role in the microcirculation, several, possibly interdependent molecular mechanisms have been proposed by which GPR39 may protect against stroke. First, GPR39 has been shown to reduce excitation of hippocampal neurons by increasing KCC2 expression and chloride efflux44 and by increasing synthesis of the endocannabinoid 2-arachidonoylglycerol in postsynaptic hippocampal neurons, which moderates presynaptic glutamate release.45 Furthermore, GPR39 has been proposed to act as a receptor for zinc, which is released during brain ischemia and has been proposed to play protective and neurotoxic roles.46,47 It is possible that receptor activation by zinc could moderate ischemic brain damage. GPR39 has constitutive activity through the Gα/serum responsive element (SRE) cascade, which decreases oxidative, endoplasmic reticulum and mitochondrial stress in hippocampal cell lines,48 potentially limiting evolving infarct size in penumbral tissues. In addition to neurons, GPR39 is expressed in microglia and peri-capillary cells affected by vascular cognitive impairment.8 GPR39 may limit inflammation and augment capillary blood flow; thus limiting secondary expansion of infarct during reperfusion. Finally, GPR39 has been proposed to sense the ratio of the vasodilator and protective eicosanoid 14,15-epoxyeicosatrienoate (14,15-EET) to the vasoconstrictor and pro-inflammatory eicosanoid 15-hydroxyeicosatetraeonate (15-HETE).10 It is possible that increased ischemic injury in GPR39 KO mice is linked to altered interplay between the two eicosanoids. If protective EETs signaling dominates in the setting of ischemia-reperfusion, then loss of GPR39 would be expected to increase injury. On the other hand, if HETEs signaling dominates during blood loss to maintain systemic blood pressure, then loss of GPR39 may explain the exaggerated drop in blood pressure upon blood withdrawal in GPR39 KO mice.
Our results are in agreement with the study by Xie S. et al.,49 which found that a GPR39 agonist reduced the infarct size, improved neurological deficit and attenuated neuroinflammation in neonatal hypoxia-ischemia, suggesting that GPR39 plays a protective role in ischemic brain injury. However, another study50 reported that a GPR39 antagonist reduced infarct size and improved tissue perfusion in a mouse model of myocardial ischemia, suggesting that GPR39 plays a detrimental role in ischemic injury in the heart. The discrepancy may be related to lack of specificity of the pharmacological compounds used in these studies to GPR39 when administered in vivo, or to tissue-specific roles of GPR39 in heart vs. brain.
In summary, GPR39 appears to play a beneficial role in recovery from ischemic stroke, and contributes through several pathways that could interact to preserve brain viability and function after stroke. Future studies are required to elucidate if and how pharmacologic activation of GPR39 can decrease infarct size and improve functional outcome after stroke.
Supplementary Material
Funding
Supported by NIH grant NS108501 to NJA and Foundation of Anesthesia Education and Research (FAER) grant RFG-08-15-2019 to YX.
Footnotes
Ethical Approval and Consent to participate
Not applicable. No human subjects were involved.
Human and Animal Ethics
No studies were performed on human subjects. Studies on animals were conducted in accordance with National Institute of Health guidelines for use of animals in research, and protocols were approved by the Animal Care and Use Committee at Oregon Health & Science University.
Consent for publication
All authors have reviewed the final manuscript, agreed with the content and gave explicit consent to publish it.
Availability of supporting data
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Competing interests
Dr. Alkayed is co-inventor of technology related to GPR39 that has been licensed to Vasocardea. This potential conflict of interest has been reviewed and managed by Oregon Health and Science University. All other authors have nothing to disclose.
REFERENCES
- 1.Virani SS et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation. 2020;141:e139–e596. [DOI] [PubMed] [Google Scholar]
- 2.Rochmah TN, Rahmawati IT, Dahlui M, Budiarto W, Bilqis N. Economic Burden of Stroke Disease: A Systematic Review. Int J Environ Res Public Health. 2021;18:7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Evans MRB, White P, Cowley P, Werring DJ. Revolution in acute ischaemic stroke care: a practical guide to mechanical thrombectomy. Pract Neurol. 2017;17:252–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goyal M, Menon BK, van Zwam WH, et al. Endovascular thrombectomy after large-vessel ischaemic stroke: a meta-analysis of individual patient data from five randomised trials. Lancet. 2016;387:1723–31. [DOI] [PubMed] [Google Scholar]
- 5.Kloner RA, King KS, Harrington MG. No-reflow phenomenon in the heart and brain. Am J Physiol Heart Circ Physiol. 2018;315:H550–H562. [DOI] [PubMed] [Google Scholar]
- 6.Shabir O, Berwick J, Francis SE. Neurovascular dysfunction in vascular dementia, Alzheimer’s and atherosclerosis. BMC Neurosci. 2018;19:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu Y, Barnes AP, Alkayed NJ. Role of GPR39 in Neurovascular Homeostasis and Disease. Int J Mol Sci. 2021;22:8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis CM, Bah TM, Zhang WH, et al. GPR39 localization in the aging human brain and correlation of expression and polymorphism with vascular cognitive impairment. Alzheimers Dement (N Y). 2021;7:e12214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu Y, Wang M, Xie Y, et al. Activation of GPR39 with the agonist TC-G 1008 ameliorates ox-LDL-induced attachment of monocytes to endothelial cells. Eur J Pharmacol. 2019;858:172451. [DOI] [PubMed] [Google Scholar]
- 10.Alkayed NJ, Cao Z, Qian ZY, et al. Control of coronary vascular resistance by eicosanoids via a novel GPCR. Am J Physiol Cell Physiol. 2022;322:C1011–C1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang W, Davis CM, Zeppenfeld DM, et al. Role of endothelium-pericyte signaling in capillary blood flow response to neuronal activity. J Cereb Blood Flow Metab. 2021;41:1873–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chorin E, Vinograd O, Fleidervish I, et al. Upregulation of KCC2 activity by zinc-mediated neurotransmission via the mZnR/GPR39 receptor. J Neurosci. 2011;31:12916–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Besser L, Chorin E, Sekler I, et al. Synaptically released zinc triggers metabotropic signaling via a zinc-sensing receptor in the hippocampus. J Neurosci. 2009;29:2890–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. [DOI] [PubMed] [Google Scholar]
- 15.Qin W, Kutny PM, Maser RS, et al. Generating Mouse Models Using CRISPR-Cas9-Mediated Genome Editing. Curr Protoc Mouse Biol. 2016;6:39–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang W, Koerner IP, Noppens R, Grafe M, Tsai HJ, Morisseau C, Luria A, Hammock BD, Falck JR, Alkayed NJ. Soluble epoxide hydrolase: a novel therapeutic target in stroke. J Cereb Blood Flow Metab. 2007;27:1931–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Srinivasan VJ, Mandeville ET, Can A, Blasi F, Climov M, Daneshmand A, Lee JH, Yu E, Radhakrishnan H, Lo EH, Sakadžić S, Eikermann-Haerter K, Ayata C. Multiparametric, longitudinal optical coherence tomography imaging reveals acute injury and chronic recovery in experimental ischemic stroke. PLoS One. 2013. Aug 7;8(8):e71478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baran U, Zhu W, Choi WJ, et al. Automated segmentation and enhancement of optical coherence tomography-acquired images of rodent brain. J Neurosci Methods. 2016;270:132–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang W, Davis CM, Zeppenfeld DM, Golgotiu K, Wang MX, Haveliwala M, Hong D, Li Y, Wang RK, Iliff JJ, Alkayed NJ. Role of endothelium-pericyte signaling in capillary blood flow response to neuronal activity. J Cereb Blood Flow Metab. 2021. Aug;41(8):1873–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Choi WJ, Wei W, et al. Aging-associated changes in cerebral vasculature and blood flow as determined by quantitative optical coherence tomography angiography. Neurobiol Aging. 2018;70:148–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brait VH, Wright DK, Nategh M, et al. Longitudinal hippocampal volumetric changes in mice following brain infarction. Sci Rep. 2021;11:10269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balkaya M, Kröber JM, Rex A, Endres M. Assessing post-stroke behavior in mouse models of focal ischemia. J Cereb Blood Flow Metab. 2013;33:330–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ng FC, Churilov L, Yassi N, Kleinig TJ, Thijs V, Wu TY, Shah DG, Dewey HM, Sharma G, Desmond PM, Yan B, Parsons MW, Donnan GA, Davis SM, Mitchell PJ, Leigh R, Campbell BCV; EXTEND-IA TNK Part 1 and 2 Investigators. Microvascular Dysfunction in Blood-Brain Barrier Disruption and Hypoperfusion Within the Infarct Posttreatment Are Associated With Cerebral Edema. Stroke. 2022. May;53(5):1597–1605. [DOI] [PubMed] [Google Scholar]
- 24.Dalkara T Pericytes: a novel target to improve success of recanalization therapies. Stroke. 2019; 50:2985–2991. [DOI] [PubMed] [Google Scholar]
- 25.Ter Schiphorst A, Charron S, Hassen WB, Provost C, Naggara O, Benzakoun J, Seners P, Turc G, Baron JC, Oppenheim C. Tissue no-reflow despite full recanalization following thrombectomy for anterior circulation stroke with proximal occlusion: A clinical study. J Cereb Blood Flow Metab. 2021. Feb;41(2):253–266. doi: 10.1177/0271678X20954929. Epub 2020 Sep 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Etherton MR, Wu O, Cougo P, Lorenzano S, Li H, Cloonan L, Bouts MJRJ, Lauer A, Arai K, Lo EH, Feske SK, Furie KL, Rost NS. Sex-specific differences in white matter microvascular integrity after ischaemic stroke. Stroke Vasc Neurol. 2019. Sep 3;4(4):198–205. doi: 10.1136/svn-2019-000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reeves MJ, Bushnell CD, Howard G, Gargano JW, Duncan PW, Lynch G, Khatiwoda A, Lisabeth L. Sex differences in stroke: epidemiology, clinical presentation, medical care, and outcomes. Lancet Neurol. 2008. Oct;7(10):915–26. doi: 10.1016/S1474-4422(08)70193-5. Epub 2008 Aug 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phan HT, Blizzard CL, Reeves MJ, Thrift AG, Cadilhac D, Sturm J, Heeley E, Otahal P, Konstantinos V, Anderson C, Parmar P, Krishnamurthi R, Barker-Collo S, Feigin V, Bejot Y, Cabral NL, Carolei A, Sacco S, Chausson N, Olindo S, Rothwell P, Silva C, Correia M, Magalhães R, Appelros P, Kõrv J, Vibo R, Minelli C, Gall S. Sex Differences in Long-Term Mortality After Stroke in the INSTRUCT (INternational STRoke oUtComes sTudy): A Meta-Analysis of Individual Participant Data. Circ Cardiovasc Qual Outcomes. 2017. Feb;10(2):e003436. doi: 10.1161/CIRCOUTCOMES.116.003436. Epub 2017 Feb 22. [DOI] [PubMed] [Google Scholar]
- 29.Kent DM, Price LL, Ringleb P, et al. Sex-based differences in response to recombinant tissue plasminogen activator in acute ischemic stroke: a pooled analysis of randomized clinical trials. Stroke. Epub ahead of print 29 November 2005. [DOI] [PubMed] [Google Scholar]
- 30.Savitz SI, Schlaug G, Caplan L, et al. Arterial occlusive lesions recanalize more frequently in women than in men after intravenous tissue plasminogen activator administration for acute stroke. Stroke. Epub ahead of print 9 June 2005. [DOI] [PubMed] [Google Scholar]
- 31.Lorenzano S, Ahmed N, Falcou A, et al. Does sex influence the response to intravenous thrombolysis in ischemic stroke?: answers from safe implementation of treatments in stroke-international stroke thrombolysis register. Stroke 2013; 44: 3401–3406. [DOI] [PubMed] [Google Scholar]
- 32.Spaander FH, Zinkstok SM, Baharoglu IM, et al. Sex differences and functional outcome after intravenous thrombolysis. Stroke 2017; 48: 699–703. [DOI] [PubMed] [Google Scholar]
- 33.Wang X, Carcel C, Wang R, et al. Worse prognosis in women, compared with men, after thrombolysis: an individual patient data pooling study of Asian acute stroke registries. Int J Stroke. Epub ahead of print 8 July 2020. [DOI] [PubMed] [Google Scholar]
- 34.Sheth SA, Lee S, Warach SJ, et al. Sex differences in outcome after endovascular stroke therapy for acute ischemic stroke. Stroke 2019; 50: 2420–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chalos V, De Ridder IR, Lingsma HF, et al. Does sex modify the effect of endovascular treatment for ischemic stroke?: a subgroup analysis of 7 randomized trials. Stroke 2019; 50: 2413–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alkayed NJ, Harukuni I, Kimes AS, London ED, Traystman RJ, Hurn PD. Gender-linked brain injury in experimental stroke. Stroke. 1998. Jan;29(1):159–65; discussion 166. doi: 10.1161/01.str.29.1.159. [DOI] [PubMed] [Google Scholar]
- 37.McCullough LD, Alkayed NJ, Traystman RJ, Williams MJ, Hurn PD. Postischemic estrogen reduces hypoperfusion and secondary ischemia after experimental stroke. Stroke. 2001. Mar;32(3):796–802. [DOI] [PubMed] [Google Scholar]
- 38.Feekes JA, Cassell MD. The vascular supply of the functional compartments of the human striatum. Brain. 2006;129(Pt 8):2189–201. [DOI] [PubMed] [Google Scholar]
- 39.Li B, Esipova TV, Sencan I, Kılıç K, et al. More homogeneous capillary flow and oxygenation in deeper cortical layers correlate with increased oxygen extraction. Elife. 2019;8:e42299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yemisci M, Gursoy-Ozdemir Y, Vural A, Can A, Topalkara K, Dalkara T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med. 2009. Sep;15(9):1031–7. [DOI] [PubMed] [Google Scholar]
- 41.Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O’Farrell FM, Buchan AM, Lauritzen M, Attwell D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014. Apr 3;508(7494):55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Erdener ŞE, Tang J, Kılıç K, Postnov D, Giblin JT, Kura S, Chen IA, Vayisoğlu T, Sakadžić S, Schaffer CB, Boas DA. Dynamic capillary stalls in reperfused ischemic penumbra contribute to injury: A hyperacute role for neutrophils in persistent traffic jams. J Cereb Blood Flow Metab. 2021. Feb;41(2):236–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.El Amki M, Glück C, Binder N, Middleham W, Wyss MT, Weiss T, Meister H, Luft A, Weller M, Weber B, Wegener S. Neutrophils Obstructing Brain Capillaries Are a Major Cause of No-Reflow in Ischemic Stroke. Cell Rep. 2020. Oct 13;33(2):108260. [DOI] [PubMed] [Google Scholar]
- 44.Gilad D, Shorer S, Ketzef M, et al. Homeostatic regulation of KCC2 activity by the zinc receptor mZnR/GPR39 during seizures. Neurobiol Dis. 2015;81:4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perez-Rosello T, Anderson CT, Schopfer FJ, et al. Synaptic Zn2+ inhibits neurotransmitter release by promoting endocannabinoid synthesis. J Neurosci. 2013;33:9259–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science. 1996;272:1013–6. [DOI] [PubMed] [Google Scholar]
- 47.Galasso SL, Dyck RH. The role of zinc in cerebral ischemia. Mol Med. 2007;13:380–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dittmer S, Sahin M, Pantlen A, et al. The constitutively active orphan G-protein-coupled receptor GPR39 protects from cell death by increasing secretion of pigment epithelium-derived growth factor. J Biol Chem. 2008;283:7074–81. [DOI] [PubMed] [Google Scholar]
- 49.Xie S, Jiang X, Doycheva DM,et al. Activation of GPR39 with TC-G 1008 attenuates neuroinflammation via SIRT1/PGC-1α/Nrf2 pathway post-neonatal hypoxic-ischemic injury in rats. J Neuroinflammation. 2021;18:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Methner C, Cao Z, Mishra A, Kaul S. Mechanism and potential treatment of the “no reflow” phenomenon after acute myocardial infarction: role of pericytes and GPR39. Am J Physiol Heart Circ Physiol. 2021;321:H1030–H1041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.