

Abstract
Costimulatory CD40 plays an essential role in autoimmune diseases including experimental autoimmune encephalomyelitis (EAE), a murine model of human multiple sclerosis (MS). However, it is not well defined how CD40 drives autoimmune disease pathogenesis. Here, we used a conditional knockout approach to determine how CD40 orchestrates a CNS autoimmune disease induced by recombinant human MOG (rhMOG). We found that deletion of CD40 in either dendritic cells (DCs) or B cells profoundly reduced EAE disease pathogenesis. Mechanistically, CD40 expression on DCs was required for priming pathogenic T helper (Th) cells in peripheral draining lymph nodes and promoting their appearance in the CNS. By contrast, B cell CD40 was essential for class-switched MOG-specific antibody production, which played a crucial role in disease pathogenesis. In fact, passive transfer of MOG-immune serum or IgG into mice lacking CD40 on B cells but not DCs reconstituted autoimmune disease, which was associated with inundation of the spinal cord parenchyma by immunoglobulin and complement. These data demonstrate that CD40 supports distinct effector programs in B cells and DCs that converge to drive a CNS autoimmune disease and identify targets for intervention.
Introduction
We have characterized the role of the costimulatory CD40 molecule in autoimmune pathogenesis, studying experimental autoimmune encephalomyelitis (EAE), a murine model of human multiple sclerosis (MS). MS is an inflammatory demyelinating disease of the central nervous system (CNS), characterized by progressive neurological deficits and disability (1). Myelin breakdown in the white matter is accompanied by infiltrates of multiple lymphoid and myeloid cell types (2, 3). At present, the mechanism of the disease is not fully understood, and there is no curative treatment (4). Immunotherapies that block T cell growth signaling by targeting CD25 (daclizumab), impede lymphocyte trafficking to the CNS by down-regulating S1PR (fingolimod), or target B cells with anti-CD20 monoclonal antibody (rituximab) have been approved for the treatment of MS (5, 6). Additional clinical trials are targeting inflammatory cytokines with anti-IL17A (secukinumab) or anti-GM-CSF (MOR103) antibody (7, 8), collectively suggesting that multiple components of the immune response may play a role in MS pathogenesis.
Analysis of the immune mechanisms underlying MS has been informed by the murine experimental autoimmune encephalomyelitis model (EAE), which approximates the pathological features of MS. EAE can be induced by immunization with CNS antigens, including the myelin oligodendrocyte protein (MOG). In the EAE model, antigen-specific, differentiated IFN-γ-producing T helper (Th)1 (9), IL-17-producing Th17 cells (10), IFN-γ and IL-17 double positive cells (11), and GM-CSF-producing Th cells (12) are primed by antigen-presenting cells (APCs) in the peripheral lymphoid organs. These pathogenic T cells traffic across the blood brain barrier and can be re-stimulated by local APCs in the CNS to recruit additional immune cells that mediate tissue damage (13, 14). In addition to this role of pathogenic Th cells, B cell infiltration, antibodies to myelin components and activated complement are routinely detected in MS patients (15, 16), and B cell deficient mice are resistant to recombinant human myelin oligodendrocytes glycoprotein (rhMOG) induced EAE (17–19), indicating that both T and B cells are required for rhMOG induced EAE.
Induction of autoimmune diseases including EAE has also been shown to require costimulatory interactions, including those mediated by the B7-CD28 (20, 21) and CD40-CD40L pathways (22–25). In the complete absence of CD40 or when treated with CD40L-blocking antibody, mice are completely protected from EAE induction by MOG35–55 peptide (20, 26). However, the mechanisms mediating the CD40 role in EAE, including requirements for cell type-specific expression of CD40, have not been identified. To elucidate the mechanisms of CD40 involved in EAE as an informative model of autoimmune disease, we have analyzed the cellular requirements for CD40 expression in clinical EAE induction, in peripheral T cell priming to MOG, in T cell infiltration in the CNS, and in autoantibody production, using conditional CD40 deletion and bone marrow chimera strategies. We have identified distinct and complementary mechanisms mediating requirements for CD40 expression by B cells and by dendritic cells in EAE pathogenesis.
Materials and methods
Mice
Similar numbers of male and female 6–12 week old mice were used in all experiments. CD40flox (CD40fl/fl) mice were generated on a C57BL/6 background. The Bioengineering Core of the University of Colorado-Denver made the construct. In brief, the first exon of CD40 was flanked by two loxP sites. The pLFNFL cloning vector was utilized for the construction of CD40 cKO vector, which was generated through the replacement of PGK-Puro in the pFlexible (Wellcome Sanger Institute) with a PGK-neo selection cassette by restricted digestion and ligation. The left (2,808 bp) and right (2,668 bp) homologous arms of CD40 amplified by PCR from C57BL/6 genomic DNA, were cloned at the EcoRI and NotI sites of pLFNFL, respectively, so they flanked the loxP-FRT-neo-FRT-loxP cassette. The left and right homologous sequences correspond to the genomic sequence upstream and downstream of exon 1 of CD40, respectively. Next, a 594 bp genomic sequence encompassing exon 1 of CD40, generated by PCR, was cloned at the HindIII site located between the loxP and FRT sites upstream of neo. Finally, a thymidine kinase (TK) gene was cloned at the ClaI site, through blunt-end ligation, at a position proximal to the left homology arm. The resulting construct (pLFNFL-TK-Cd40) was linearized by KpnI digestion and purified by chloroform and ethanol precipitation and then introduced by electroporation into murine B6/129 hybrid EC7.1 embryonic stem cells in the Transgenic and Gene Targeting Core Laboratory in the University of Colorado-Denver. Two 96-well plates of recombinants were screened for CD40 targeting event by long-range PCR. A correctly targeted clone (A12) was identified and was microinjected into C57BL/6 blastocysts to produce chimeric founders at the Transgenic Animal Core at the University of Colorado-Denver. The Frt-flanked neo selection cassette was deleted by crossing to a Flp-deleter strain.
CD19-cre (B6.129P2(C)-Cd19tmi(cre)Cgn/J, JAX-008068) and CD11c-cre (B6.Cg-Tg(Itgax-cre)1-1Reiz/J, JAX-006785) were purchased from the Jackson Laboratory. For selective deletion of the floxed CD40 gene in B cells or DCs, CD19-cre or CD11c-cre mice were crossed with CD40fl/fl mice. Littermates heterozygous for the cre cassette (CD19-cre/CD40fl/fl, CD19-wt/ CD40fl/fl or CD11c-cre/ CD40fl/fl, CD11c-wt/ CD40fl/fl) were used in experiments. C57BL/6 mice were purchased from Charles River Laboratories (STRAIN CODE: 027). and μMT (B6.129S2-Ighmtm1Cgn/J, JAX-002288) mice were purchased from the Jackson Laboratory. BM chimera mice were prepared as previously described by reconstitution of 950 rad irradiated host mice with 6×106 total T cell–depleted BM cells i.v (27). Mixed chimeras were generated by reconstitution with 6×106 total T cell–depleted BM cells from donor mice at 1:1 ratio.
EAE induction and score
Recombinant human MOG protein extracellular domain (rhMOG30-154) was expressed in H5 insect cells in the Protein Expression Laboratory, Leidos Biomedical Research, Inc., Frederick National Laboratory for Cancer Research, Frederick, Maryland USA. EAE induction was performed by mixing rhMOG (200 μg/mouse) in complete Freund’s adjuvant (CFA, Difco Laboratories DF3114-33-8) containing Mycobacterium tuberculosis H37Ra (Difco Laboratories DF0639-60-6) and injecting subcutaneously in the abdomen bilaterally (50μl emulsion in each side). Pertussis toxin (PTX) (120 ng) (List Biological Laboratories 180) diluted in PBS was administered i.p. on days 0 and 2 post-immunization. Control mice receiving PBS in CFA were injected with the same amount of PTX. The clinical severity of EAE was scored daily by an observer who was unaware of mouse genotypes, using a grading scale of 0–5 as previously described (28): 0, asymptomatic; 1, flaccid tail; 2, hind-limb weakness and impaired righting ability; 3, hind-limb paralysis; 4, front- and hind-limb paralysis; 5, moribund or death.
Serum and IgG Transfer
Serum was collected by cardiac puncture from B6 mice three weeks after immunization with rhMOG(200 μg/mouse) /CFA or KLH(200 μg/mouse) /CFA (LGC biosearchtechnologies, N-5060) as described above. Mice from indicated groups were injected i.p. with 400 μl serum starting at the time of EAE induction and continuing every 2 days for the following weeks. IgG was purified from serum using Protein G sepharose 4 Fast Flow resin (Cytiva 17-0618-05) and Pierce IgG Elution Buffer (ThermoFisher Scientific 21009). Purified IgG was buffer-exchanged to PBS, sterile filtered and antibody integrity confirmed by SDS-PAGE analysis. Purified IgG was injected in the same amount as contained in the 400 μl serum. This protein G purification removed >95% of IgG from serum as determined by ELISA, and the IgG-depleted serum was also used in transfer experiments.
Isolation of mononuclear cells from peripheral lymph nodes and CNS parenchyma
Inguinal and axillary draining lymph nodes were harvested, mixed, and digested with 2.5 mg/ml collagenase D (Sigma 11088882001) and 1 mg/ml DNase I (Sigma 10104159001) for 30 min at 37 °C. Tissues were filtered through 70-μm mesh strainers (BD 352350) to generate single cell suspension. For isolation of CNS-infiltrating cells, brain and spinal-cord tissues were minced and digested with the same buffer as peripheral lymph nodes. Tissues were filtered through 70-μm mesh strainers and centrifuged through a Percoll (Sigma P4937) density gradient (38% and 70%). Mononuclear cells in the interphase were removed, washed and resuspended in culture medium for analysis by flow cytometry.
Flow cytometry
Cells were washed with FACS buffer (HBSS (Corning, without Calcium, Magnesium and Phenol Red) containing 0.2% BSA (Sigma A3059)and 0.05% Azide(Sigma S2002)), treated with anti-FcR (24G2, Leino C381), and then stained with specific antibodies at 4 °C for 30 min. We use anti–mouse CD4-FITC (RM4-4, Biolegend 100510), CD4-BV785(RM4-4, Biolegend 100552), CD8α-PE-Cy7 (53-6.7, Biolegend 100722), B220-AF594 (RA3-6B2, Biolegend 103254), CD11c-PE (N418, Biolegend 117308), MHCII-BV421 (M5/114.15.2, Biolegend 107632), CD40-APC (3/23, Biolegend 124612), IFN-γ-APC (XMG1.2, Biolegend 505810), anti-IL17A-BV785 (TC11-18H10.1, Biolegend 506928), GM-CSF-PE (MP1-22E9, Biolegend 505406) antibodies were purchased from Biolegend. For intracellular cytokine staining, mononuclear cells from LNs and CNS were stimulated overnight with rhMOG (10μg/ml), with monensin (Biolegend 420701) added in the last four hours, and cells were fixed and permeabilized with the BD Fix/Perm kit (BD 554715) according to the manufacturer’s instructions and then stained with intracellular antibodies for 30 min. Dead cells were excluded using Zombie Aqua™ Fixable Viability Kit (Biolegend 423102). Data were collected with a FACS LSR II or FACS Fortessa flow cytometer (BD Biosciences) and analyzed with FlowJo software.
ELISA
MOG-specific IgG was measured by ELISA. rhMOG (10 μg/ml) was coated on ELISA plates (ThermoFisher Scientific, Immulon 4HBX, 2855) overnight. The plates were then washed with ELISA wash buffer (0.5% Tween in PBS), serially diluted sera were applied to the plates, and plates were incubated 2 h at room temperature. Anti–mouse IgG HRP (Southern Biotech, 1030-05) was used to detect MOG-specific IgG. After a wash step, KPL 2,2’-Azino-di-(3-ethylbenzthiazoline-6-sulfonate) (ABTS) substrate (Seracare 5120-0041) was added to the wells, and enzyme reaction was stopped by ABTS HRP Stop Solution (Seracare 5150-0017). Optical density at 405 nm was measured with SpectraMax_iD3 plate reader.
Immunohistochemistry and image analysis
Mice were pefused transcardially with PBS followed by 2% PFA (Electron Microscopy Sciences) in PBS. The spinal column was then dissected and fixed in 2% PFA overnight at 4°C. The columns were washed twice with PBS, and spinal cords were then carefully dissected and placed in 30% sucrose overnight at 4°C. Spinal cords were then frozen in Tissue Freezing Media (VWR). Ten micron sections were cut using a Leica CM1860 cryostat. Tissue sections were blocked with 5 drops of Background Buster (Innovex NB306) per 1 mL of PBS with 0.5% Triton X-100 (Sigma T8787) at room temperature for 1 hour and then incubated with primary antibody in PBS containing 0.1% Triton X-100. Primary antibodies included anti-CD4-BV421 (RM4-5 Biolegend 100544; 1:500), anti-CD45-FITC (104; Biolegend 109806; 1:500), anti-B220-PE (RA3-6B2; ThermoFisher Scientific12-0452-82; 1:500), anti-C3 (2/11; Hycult Biotech HM1065-100UG; 1:50), and donkey anti-mouse IgG-Alexa647 (ThermoFisher Scientific A-31571; 1:500). Tissue sections were then washed 3X with PBS and incubated with secondary antibody in PBS containing 0.1% Trixon X-100 for 2 hours at room temperature if needed to detect the anti-C3 primary. Donkey anti-rat-IgG-Alexa594 (ThermoFisher Scientific A-21209; 1:500) was used as a secondary antibody. Lastly, sections were washed 3X with PBS, and Fluorosave reagent (Millipore Sigma 345789) was then added to each slide, which were coverslipped and imaged with a Olympus FV1200 confocal microscope. Images were analyzed using Imaris version 8.1. Using the Imaris ‘surface’ function, a surface was drawn around the spinal cord section, avoiding folds and tears in the section. The area of the surface was then calculated. To quantify CD4+ and B220+ cells, the ‘spots’ function was used to calculate the number of segmented CD4+ and B220+ spots within the spinal cord area. For each individual experiment, the number of spots per area was normalized to the highest value per group of mice collected, and significant outliers (2 from KLH serum and 1 from EAE serum in Fig. 7D) were removed with Grubbs’ test for outliers prior to combining the data. For C3 and IgG quantification, the ‘surface’ function in Imaris was used to calculate the total segmented C3+ or IgG+ surface within the spinal cord area. Data were normalized and combined as described above.
Figure 7.
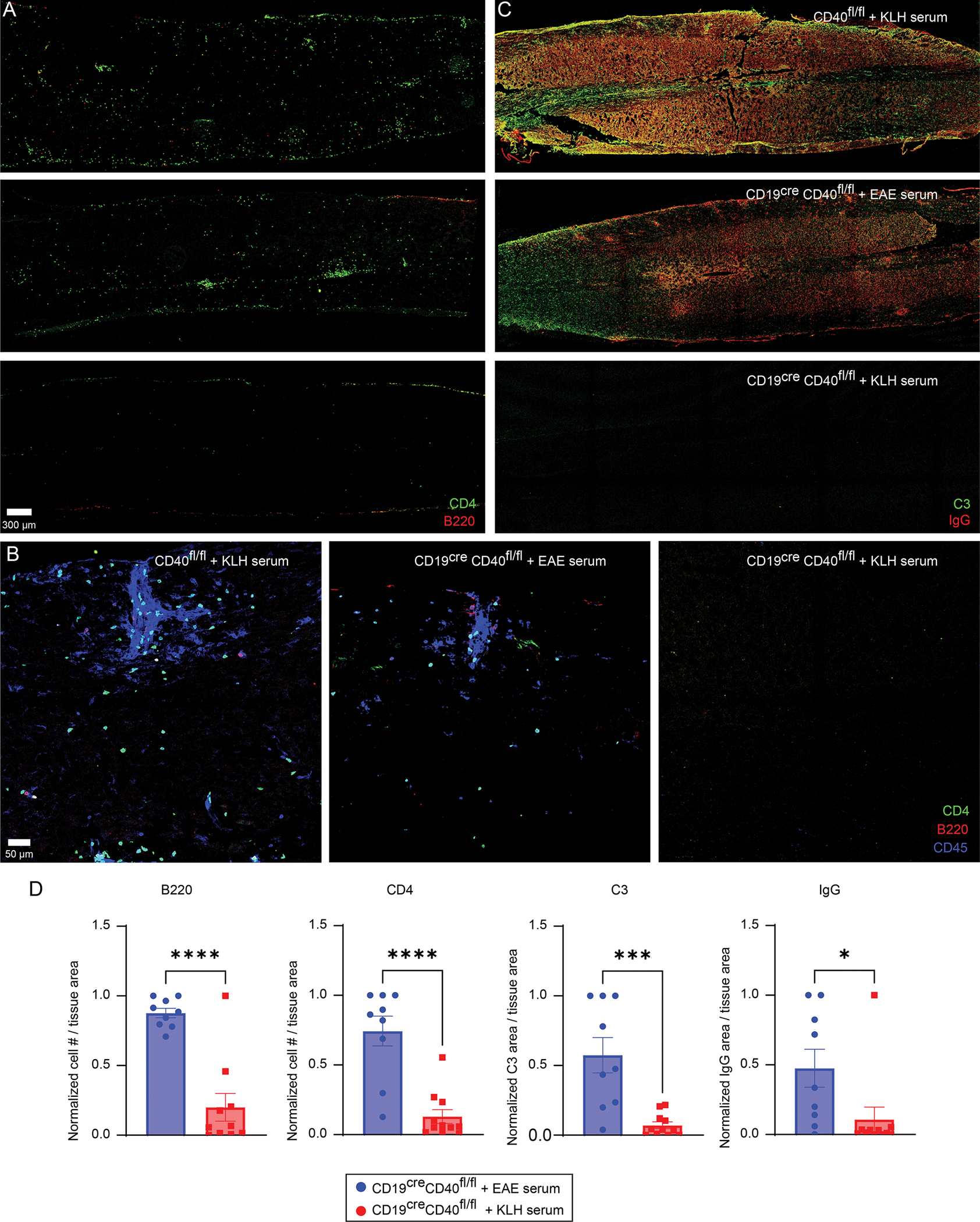
Spinal cord inflammation and immunoglobulin / complement distribution in serum treated B cell CD40 deficient mice. A. Representative confocal images of spinal cord sections stained with anti-CD4 (green) and anti-B220 (red) in the denoted groups of mice. B. Magnified confocal images from the spinal cords in A showing immune cell clustering in the denoted groups. CD4 (green), B220 (red) and CD45 (blue). C. Representative confocal images of spinal cord sections stained with anti-C3 (green) and anti-IgG (red). D. Quantification of spinal cord CD4, B220, C3, and IgG staining in CD19creCD40fl/fl mice that received EAE serum and serum from KLH immunized mice. Data are combined from three independent experiments (mean ± SEM, n = 9 for EAE serum group, n = 11 for KLH serum group) and analyzed using an unpaired t test. *p <0.05, ***p <0.001, ****p <0.0001.
Statistical analysis
Prism GraphPad 8 software was utilized. One-way ANOVA followed by Dunnett’s test against immunized CD40fl/fl group was performed for multiple comparisons. For statistical analysis of EAE, non-parametric Mann–Whitney test was performed for single comparison or One-way ANOVA followed by Dunnett’s test against immunized CD40fl/fl group was performed for multiple comparisons.
Results
Maximal induction of EAE by recombinant MOG protein requires CD40 expression on both B cells and DCs
We studied the induction of EAE by immunization of C57BL/6 (B6) mice with recombinant human MOG protein (rhMOG), previously shown to require both T and B cells (29, 30), paralleling the apparent roles of these cell types in human MS (5). This protocol induced severe disease peaking at 12–14 days in wild-type B6 mice. B cell deficient (μMT) mice were completely resistant to rhMOG-induced disease (Fig. 1A), consistent with previous report (30). CD40 knockout (CD40−/−) mice were similarly resistant to EAE induced by rhMOG (Fig. 1A), also consistent with previous studies reporting that CD40 knockout mice were resistant to MOG35-55 peptide induced EAE (31).
Figure 1.
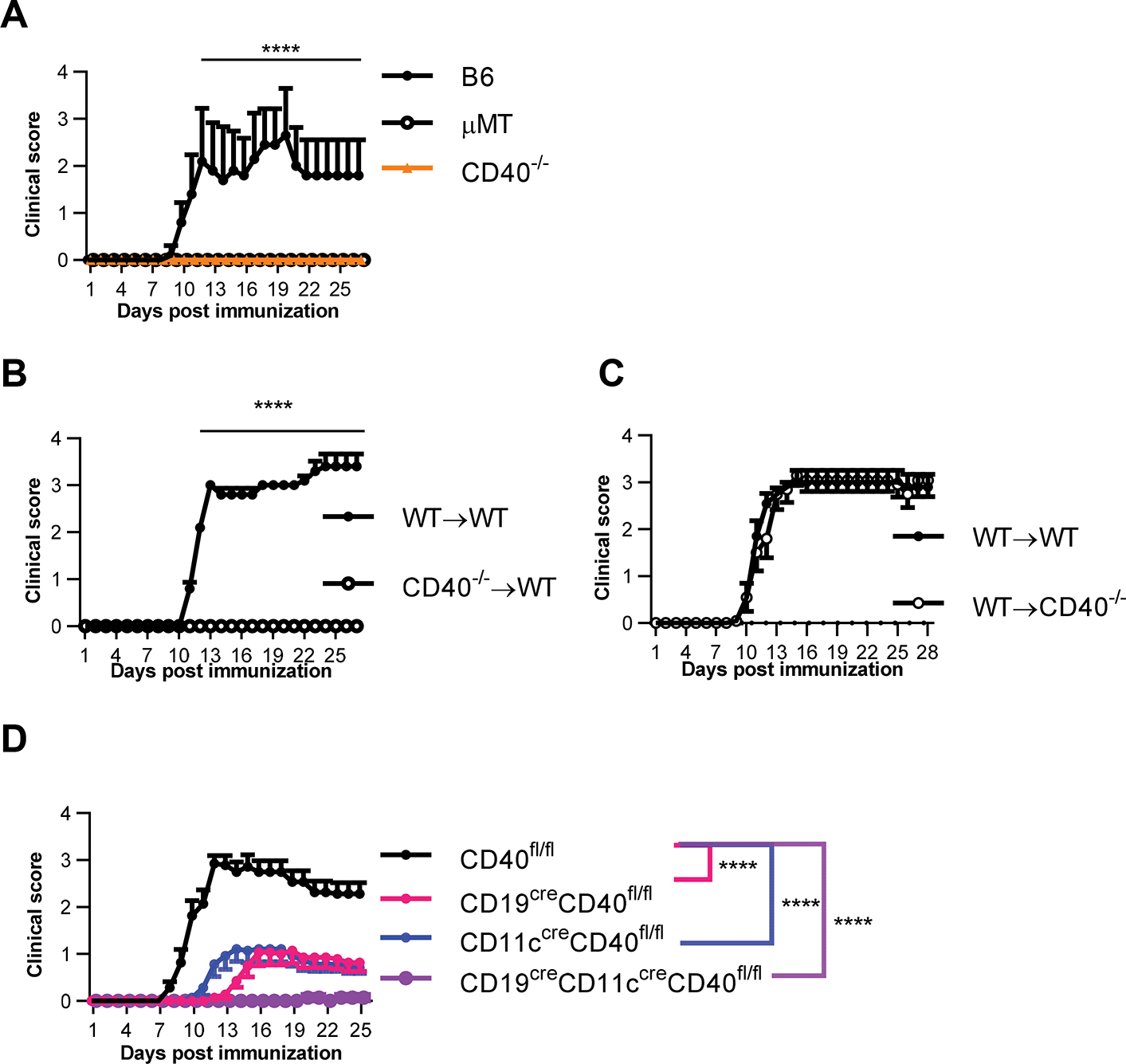
Maximal rhMOG induced EAE requires CD40 expression on both B cells and DCs. A. Clinical score of B6, B cell-deficient (mMT), or CD40-deficient (CD40−/−) mice immunized with rhMOG to induce EAE. B. BM chimera were established by injecting donor BM cells from CD40 WT(C57BL/6) or CD40 knockout (CD40−/−) mice into irradiated WT hosts. EAE was induced in the chimera mice by rhMOG immunization at 8 weeks post establishment. C. BM chimera were established by injecting donor BM cells from WT mice into irradiated WT or CD40−/− mice hosts. EAE was induced in the chimera mice by rhMOG immunization at 8 weeks post establishment. D. Clinical score of mice from CD19creCD40fl/fl, CD11ccreCD40fl/fl, CD19creCD11ccreCD40fl/fl, and no cre litter mate control (CD40fl/fl) immunized with rhMOG to induce EAE. Data in A and D are combined from three independent experiments (mean ± S.E.M. of n = 14 mice per group). Data in B and C are combined from two independent experiments (mean ± S.E.M. of n = 10 mice per group). Medians of the total clinical score during day 11–27 were compared by two-tailed non-parametric Mann–Whitney test. ****p < 0.0001.
To elucidate the role of CD40 co-stimulation in EAE, we next analyzed the identity of CD40-expressing cell types required for EAE induction. Using bone marrow (BM) chimeras, we found that WT hosts receiving CD40−/− BM donor cells were also completely resistant to rhMOG induced EAE (Fig. 1B). However, CD40−/− hosts receiving WT BM donor cells developed EAE comparable to that in WT control hosts (Fig. 1C), indicating that CD40 expression on hematopoietic derived cells but not radio-resistant cells, such as microglia, is necessary for rhMOG-induced EAE. To identify the cell types that mediate CD40 function in EAE induction, we selectively eliminated CD40 expression on either B cells or DC by crossing CD19-cre or CD11c-cre mice respectively to CD40fl/fl mice. The cell-type-specificity of conditional CD40 ablation was confirmed by flow cytometric analysis (Supplementary Fig. 1A). Strikingly, elimination of CD40 expression on either B cells or DCs resulted in significantly decreased EAE severity, indicating that CD40 expression on both B cells and DCs plays a role in EAE pathogenesis (Fig. 1D). Moreover, elimination of CD40 expression on both B cells and DCs by the combined effects of CD19-cre and CD11c-cre resulted in essentially complete elimination of EAE, indicating that CD40 expression on both B cells and DCs is required for EAE induced by rhMOG (Fig. 1D). Selective elimination of CD40 expression on B cells in vivo was also achieved by generating radiation bone marrow chimeras in which bone marrow from B cell-deficient μMT mice was mixed with bone marrow from CD40−/− mice and transferred into lethally irradiated B6 recipients. These chimeric mice, with no CD40 expression on B cells, but intact CD40 expression on all other cell types, were completely protected from rhMOG-induced EAE, confirming the requirement for CD40 on B cells (Supplementary Fig. 1B).
CD40 expression on DCs but not B cells is required for peripheral priming of CD4+ Th cells
An early event following MOG immunization is the priming and differentiation of peripheral T cells, which are thought to subsequently play a role in mediating CNS events. We therefore analyzed immune cell populations in axillary and inguinal draining lymph nodes (DLN) by flow cytometry six days post rhMOG immunization (Fig. 2A). Unimmunized naïve CD40−/− mice and WT mice had equivalent numbers of lymph node CD4 and CD8 T, B cells, and DCs (Supplementary Fig. 2), consistent with previous report (32). EAE induction by rhMOG immunization increased the total number of DLN cells including T, B and DCs in CD40fl/fl mice compared to unimmunized naïve mice. In CD40−/− mice, CD4+ and CD8+ T cell numbers post immunization were significantly reduced, with approximately 2-fold fewer than those in immunized CD40fl/fl mice. Other immune cell populations, including B220+ B cells, and CD11c+ MHCII+ DCs were present in comparable numbers in immunized WT and CD40−/− mice (Fig. 2B). We next studied the DLN responses of mice with selective deletion of CD40 on B cells or DC. Immunized mice with selective elimination of CD40 on B cells resembled immunized WT mice, while mice with deficient expression of CD40 on DCs resembled CD40−/− mice, with substantially lower numbers of CD4+ and CD8+ T cells compared to immunized non-cre control mice, demonstrating that CD40 expressed by DCs, but not CD40 expressed by B cells, is required for expansion of CD4 cell numbers in DLN following MOG immunization (Fig. 2B).
Figure 2.
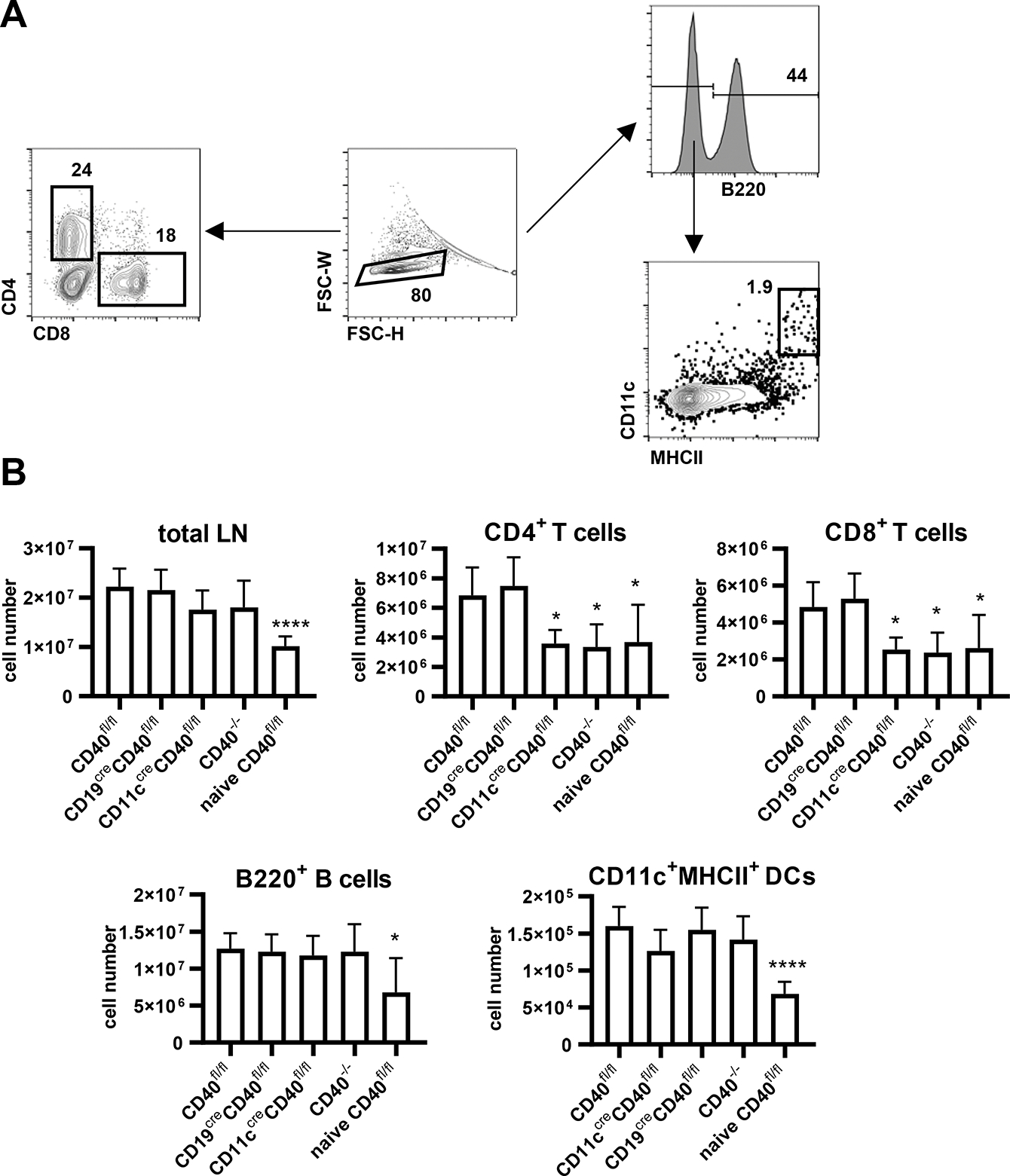
Immune cell populations in draining lymph nodes from indicated mice 6 days post EAE induction or from naïve CD40fl/fl mice. A. Gating strategy by FACS. B. Data are combined from two independent experiments (mean ± S.D. of n = 6 per group). One-way ANOVA followed by Dunnett’s test against immunized CD40fl/fl group was performed for multiple comparisons according to FACS analysis in A. *p <0.05, ****p <0.0001
IFN-γ producing Th1 and IL-17 producing Th17 cells have been shown to be pathogenic CD4+ T cells which can trigger EAE (9). In addition, several groups reported that production of the cytokine GM-CSF by CD4+ T cells is essential for their encephalitogenicity (6, 12, 33, 34). We therefore isolated DLNs from indicated mice 6 days post EAE induction or control PBS treatment, and stimulated cells in vitro with rhMOG overnight. IFN-γ, IL-17, and GM-CSF producing Th cells were analyzed by intracellular staining. These cytokines were undetectable or at very low frequency in CD4+ T cells from control PBS/CFA immunized mice (Fig. 3A). IL-17 producing, IFN-γ producing, and GM-CSF producing CD4+ T cells in DLN of rhMOG-immunized WT mice were present at frequencies substantially above those in PBS/CFA immunized WT mice, as were the numbers of what have been reported to be highly pathogenetic IL-17+ IFN-γ+ CD4 T cells (11) (Fig. 3A). The frequencies of these inflammatory cytokine-producing CD4+ T cells in DLN were markedly reduced in CD40−/− mice (Fig. 3B), showing that induction and differentiation of these Th cell populations is dependent on CD40 expression. Notably, the selective deletion of CD40 on DCs resulted in a profound decrease in cytokine-producing Th cells equivalent to that observed in complete CD40 knockouts, paralleling the failure of these mice to manifest clinical EAE. In contrast, selective deletion of CD40 on B cells, which was equally protective against clinical EAE, had no effect on the induction of these cytokines-producing Th cells (Fig. 3). Thus, CD40 expression by DC, but not B cells, is important in peripheral priming and differentiation of CD4+ Th cells that may play a role in EAE pathogenesis.
Figure 3.
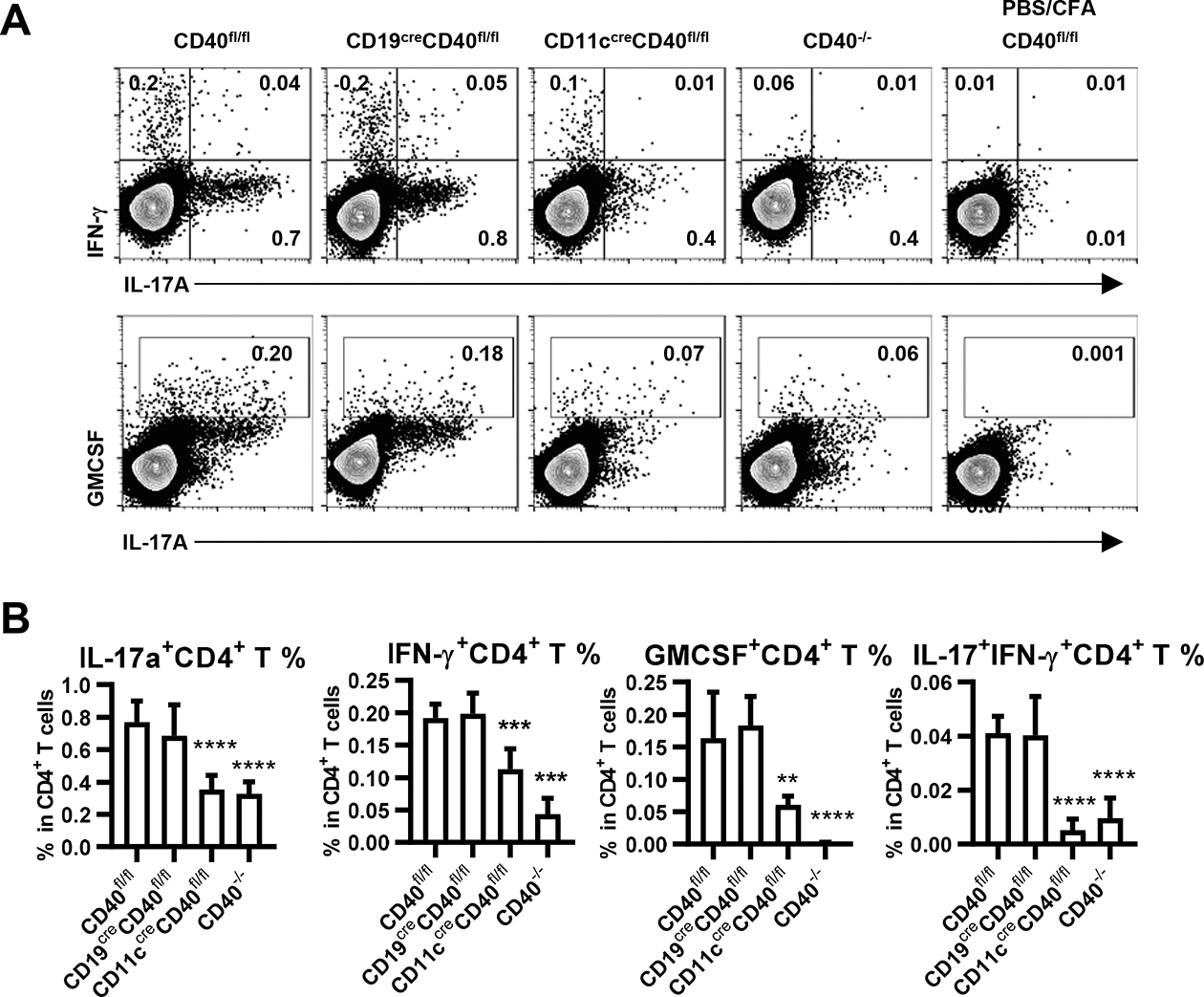
Cytokine-producing CD4+ T cells in draining lymph nodes from indicated mice 6 days post EAE induction or control PBS induction. A. CD4+ T cells were analyzed for cytokine production by intracellular staining after overnight culture with rhMOG (10μg/ml), with monensin added in the last four hours. Statistical results showing (B) frequency according to FACS analysis in A. Data are combined from two independent experiments (mean ± S.D. of n = 6 per group). One-way ANOVA followed by Dunnett’s test against immunized CD40fl/fl group was performed for multiple comparisons. *p <0.05, **p <0.01, ***p <0.001, ****p <0.0001
Maximal CNS infiltration requires CD40 expression on both B cells and DCs
We also studied the presence of immune cell populations in the CNS (Fig. 4A). At the peak of their disease, the total number of CD4+ T cells in CNS of rhMOG immunized WT mice was increased compared to unimmunized mice. MOG-immunized CD40−/− mice, which did not develop disease, correspondingly had very few CD4+ T cells recoverable from brain and spinal cord (Fig. 4B). Mice selectively deficient in CD40 on B cells or DC, which in both cases were highly protected from EAE, also had significantly reduced CD4+ T cells in the CNS when compared to WT mice, but with numbers that were greater than those in CD40−/− mice (Fig. 4B). Frequencies of IL-17, IFN-γ-and GM-CSF-producing CD4+ T cells isolated from the CNS were measured in each strain. Substantial proportions of CD4+ T cells in CD40fl/fl mice were IL-17+, IFN- γ +, IL-17+IFN- γ + and GM-CSF+ (Fig. 5A, B), while the small numbers of CD4+ T cells isolated from the CNS of CD40−/− mice had significantly lower frequencies of cytokine positive populations. Conditional deletion of CD40 from B cells or DC had similar effects in decreasing the total number of CD4+ T cells (Fig. 4B). However, CD40 deletion from DC, but not B cells, resulted in substantially reduced percentages of these cytokine-producing cells (Fig. 5A, B), paralleling the effects in DLN earlier after immunization (Fig. 3A, B). Thus, CD40 expression on DC but not on B cells is important for priming and the selective presence of pathogenic cytokine producing CD4+ T cells, suggesting that the requirement for CD40 on B cells may be mediated through a distinct mechanism in EAE pathogenesis.
Figure 4.
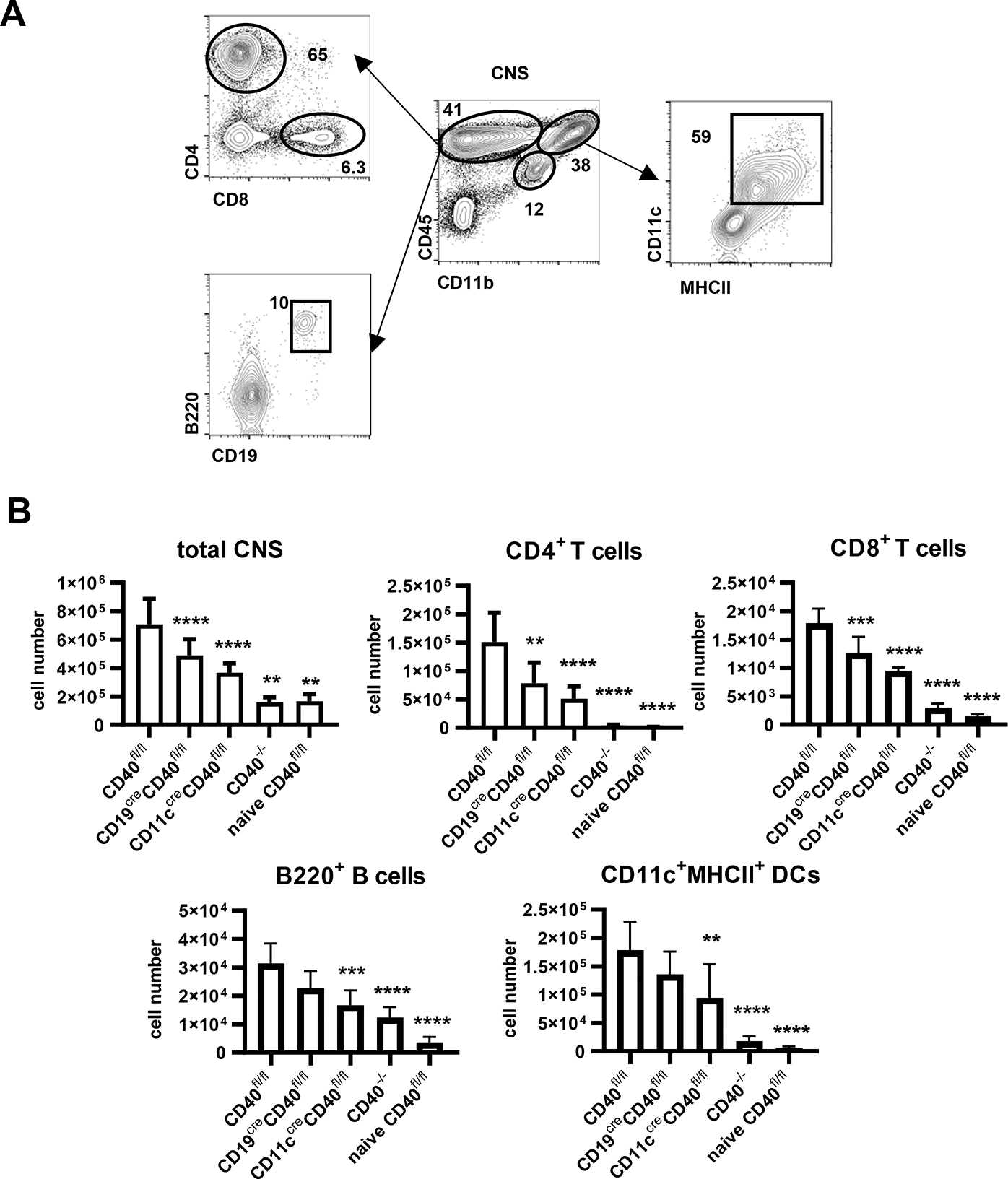
Immune cell populations in the CNS from indicated mice 14 days post EAE induction or from naïve CD40fl/fl mice. A. Gating strategy by FACS. B. Statistical analysis of results according to FACS analysis in A. Data are combined from two independent experiments (mean ± S.D. of n = 6 per group). One-way ANOVA followed by Dunnett’s test against immunized CD40fl/fl group was performed for multiple comparisons. *p <0.05, **p <0.01, ***p <0.001, ****p <0.0001.
Figure 5.
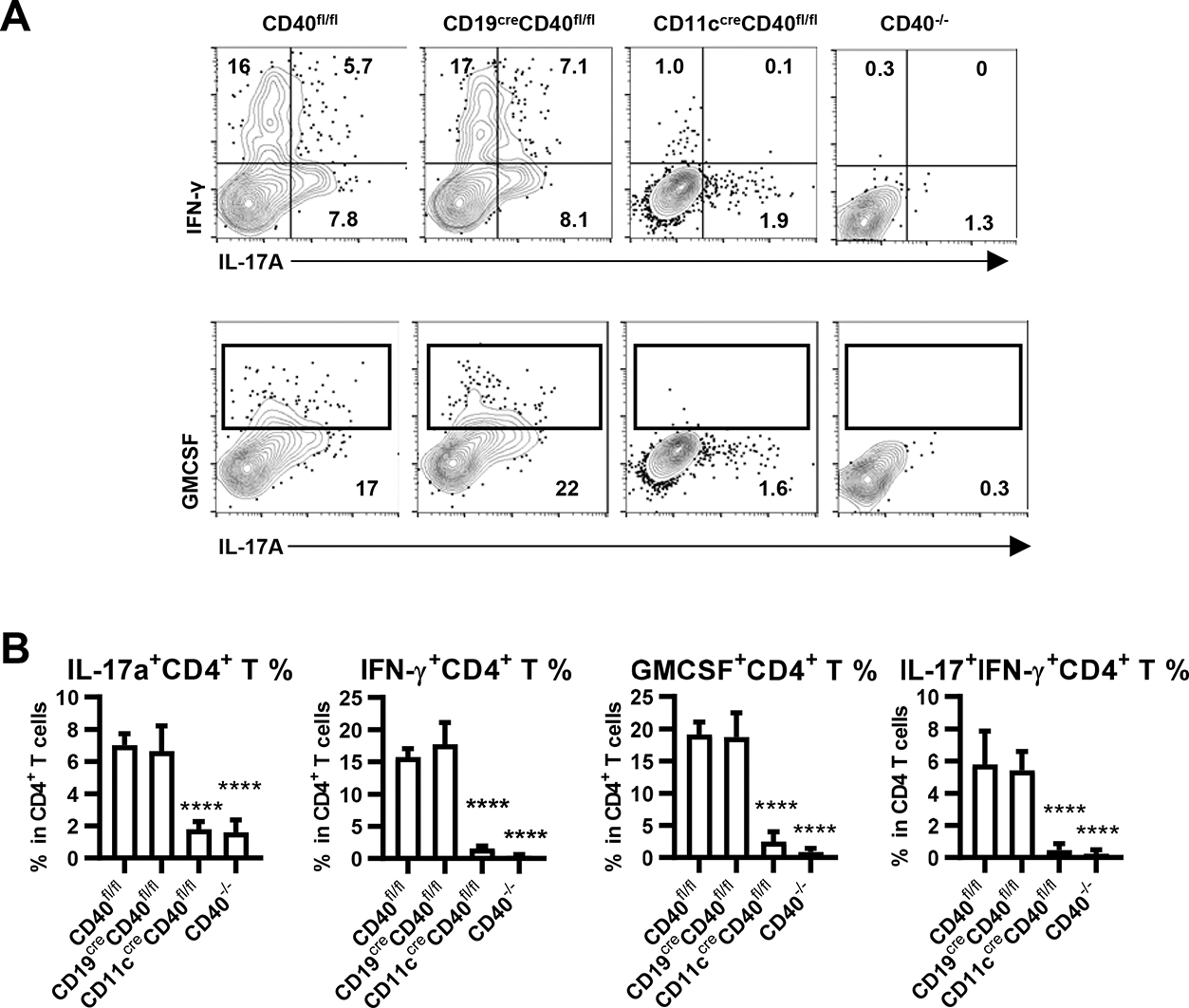
Cytokine-producing CD4+ T cells in the CNS from indicated mice 14 days post EAE induction. A. CD4+ T cells were analyzed for cytokine production by intracellular staining after overnight culture with rhMOG (10μg/ml), with monensin added in the last four hours. Statistic results showing (B) frequency according to FACS analysis in A. Data are combined from two independent experiments (mean ± S.D. of n = 6 per group). One-way ANOVA followed by Dunnett’s test against immunized CD40fl/fl group was performed for multiple comparisons. ****p <0.0001.
CD40 expression on B cells but not DCs is required for pathogenic anti-MOG antibody production
We previously identified a requirement for CD40 expression on B cells, but not on DC, in high affinity class-switched antibody responses to model antigens(35). We therefore considered that a similar function of CD40 on B cells, in generation of pathogenic antibody, might contribute to EAE pathology. We found that immunization of B6 mice with rhMOG resulted in high titers of anti-MOG IgG antibodies as detected by ELISA and that this response was absent in CD40 knockout and in B cell-deficient μMT mice (Supplementary Fig. 3A). We further observed that elimination of CD40 expression on B cells resulted in essentially complete abrogation of anti-MOG IgG production. Although elimination of CD40 expression on DCs resulted in a reduction in clinical EAE similar to that resulting from CD40 deletion on B cells, these mice mounted anti-MOG antibody responses equivalent to WT (Supplementary Fig. 3B). To determine whether the CD40 dependence of IgG anti-MOG response was reflected in an effect on germinal center (GC) B cell response, we also assessed the induction of GC B cells in DLNs following MOG immunization. MOG immunization increased the number of GC B cells in CD40fl/fl mice compared to PBS/CFA immunized or naïve CD40fl/fl mice. In parallel with what was observed for serum anti-MOG IgG levels, CD40 expression on B cells but not DCs was required for GL7+ CD38− GC B responses in the DLNs (Supplementary Fig. 3C, D).
We next tested the possibility that the pathogenic role of CD40 expression on B cells was mediated by its function in antibody production. Serum from MOG-immunized or control keyhole limpet hemocyanin (KLH)-immunized B6 mice was transferred to mice with conditional deletion of CD40 from either B cells or DC, and these mice were immunized with rhMOG. Transfer of MOG-immune serum but not KLH-immune serum, fully reconstituted the susceptibility of B cell conditional CD40 knockout mice to induction of clinical EAE. (Fig. 6A). In contrast, MOG-immune serum did not reconstitute susceptibility of mice that are deficient in CD40 expression on DC or of CD40−/− mice (Fig. 6B). To test whether the effect of immune serum was antibody mediated, we purified the IgG from the same MOG-immune serum and injected purified IgG into B cell CD40-deficient mice. Like MOG-immune serum, purified IgG reconstituted EAE susceptibility of B cell CD40 deficient mice, while mice with the same genotype receiving control IgG from KLH immunized mice, or mice receiving IgG-depleted MOG-immune serum still developed reduced EAE (Fig. 6C). These results indicate that CD40 on B cells plays a role in EAE pathogenesis through mediation of antibody response to MOG while in contrast the role of CD40 on DC is independent of antibody response.
Figure 6.
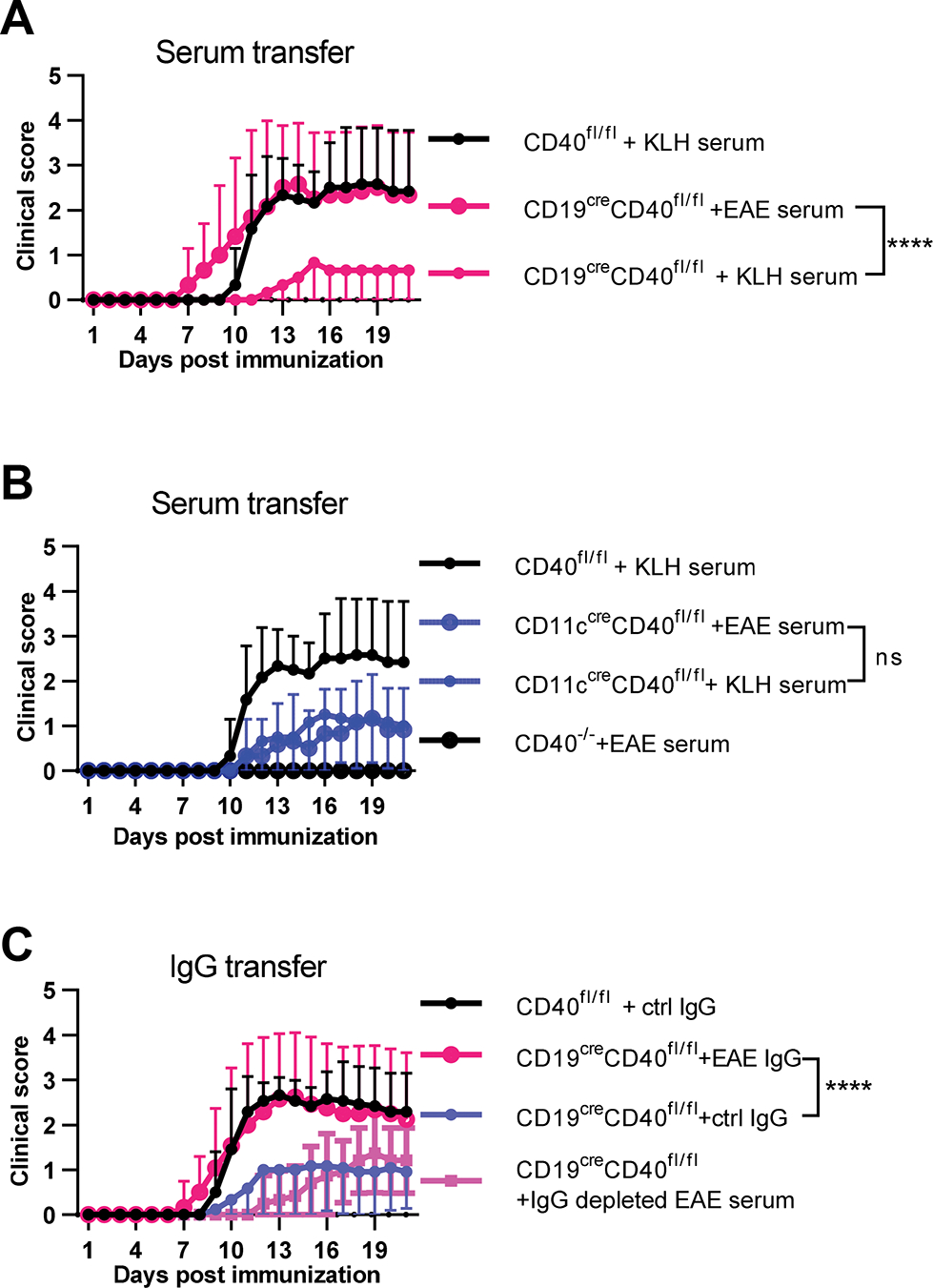
Pathogenic anti-MOG antibody reconstituted EAE susceptibility of B cell CD40 deficient mice. A, B. Serum was collected from B6 mice three weeks post EAE induction and injected into the indicated mice at day 0, 3, 7, 10 post EAE induction (400 μl serum per injection). C. IgG depleted EAE serum or IgG purified from EAE (EAE IgG) or KLH/CFA immunized mice (ctrl IgG) was injected in the same amount as contained in the 400 μl serum (1 mg). Medians of the total clinical score during day 9–21 were compared by two-tailed non-parametric Mann–Whitney test. Data are combined from three independent experiments (mean ± S.E.M. of n = 12 mice per group). ****p <0.0001.
Lastly, we examined the role of CD40-mediated antibody production from B cells in driving spinal cord inflammation during EAE. In symptomatic EAE control mice, the spinal cord parenchyma was infiltrated by both CD4+ T cells and B cells that sometimes aggregated in clusters (Fig. 7A, B). There were also massive deposits of IgG and complement in the spinal cords of these mice that would explain their observed symptoms (Fig. 7C). Importantly, removal of CD40 from B cells markedly reduced infiltration of the spinal cord parenchyma by CD4+ T cells and B cells (Fig. 7A, B). The few cells that were found in the spinal cords of these mice localized primarily to the meningeal surface. There was also very little IgG or C3 in the parenchyma following removal of CD40 from B cells despite administration of serum from KLH immunized mice (Fig. 7C). By contrast, administration of EAE serum to CD40 deficient mice reconstituted the heavy deposits of IgG and C3 observed in control EAE mice and facilitated infiltration of the spinal cord parenchyma by CD4+ T cells and B cells (Fig. 7A–D). These data demonstrate that antibody and C deposition in the spinal cord parenchyma is an important part of the pathological mechanism induced by CD40 expression on B cells.
Discussion
CD40 co-stimulation was shown to play an important role in the development of autoimmune diseases both in mice and humans (36). However, it is not fully understood how CD40 endows different disease-relevant immune cell types with their ability to elicit effector programs that ultimately cause a CNS autoimmune disease like EAE or MS. In this study, we assessed the role of CD40 on specific cell types using a conditional knockout approach to delete CD40 from two cell types, B cells and DCs, required for development of a CNS autoimmune disease initiated by myelin protein immunization. Importantly, EAE disease severity was profoundly reduced by deletion of CD40 from DCs or B cells, demonstrating that both cell types use this costimulatory protein to induce disease. The programs initiated by CD40 were, however, distinct between DCs and B cells. CD40 specifically equipped DCs with the ability to prime pathogenic, polyfunctional (IFN-γ+, IL-17+, and GM-CSF+) Th cells and promote their migration into the CNS. By contrast, B cell expression of CD40 was required for production of autoreactive, MOG-specific antibodies, which were able to reconstitute disease when passively transferred into mice with CD40-deficient B cells but not DCs. In fact, these antibodies in combination with complement were found to completely inundate the spinal cord parenchyma during the peak of disease, providing an explanation for the development of symptoms. These data support a model whereby CD40 facilitates development of distinct effector programs via B cells and DCs that converge and result in a CNS autoimmune disease. These findings identify targets for intervention to ameliorate the disease process.
A pathogenic role of Th1 and Th17 cells has been well established in mediating multiple inflammatory autoimmune diseases, including EAE (10). Adoptive transfer of in vitro cultured Th1 or Th17 cells induces EAE in recipient mice (9, 37). Th17 cells can also produce IFN-γ mediated by T-bet and Runx1 or Runx3, and this developmental flexibility has been linked to the pathogenicity of Th17 cells in multiple autoimmune disease including EAE (11). In addition to the signature proinflammatory cytokines of the Th1 and Th17 cells, both populations produce GM-CSF under polarizing conditions, and GM-CSF is required for EAE pathogenesis, as evidenced by a reduction in disease severity in mice treated with anti-GM-CSF antibodies (12). Although previous studies analyzed the in vitro requirements for induction of these pathogenic Th1, Th17 and GM-CSF-producing Th cells (38), the in vivo requirements for their induction were not elucidated. Here, we found that CD40 expression on DCs, but not on B cells, is required for optimal priming of pathogenic Th cells in peripheral draining lymph nodes during EAE, and for the appearance of these cells in the CNS. IFN-γ, IL17, and GM-CSF positive Th cells were significantly reduced in DC CD40 conditional knockout mice to a degree comparable to that observed in CD40 knockout mice. These data demonstrate an important role of CD40 on DCs in priming pathogenic Th cells, which is consistent with previous studies showing that CD40−/− DC are impaired in their ability to promote development of IFN-γ-producing Th1 (35) and IL-17 producing Th17 cells (39). Although CD11c-cre mice are widely used as a DC-specific targeting strategy, CD11c-cre may also affect macrophages or other myeloid derived cells. Activated macrophages share many features with DCs, including CD11c and CD40 expression (40), and it has been reported that myeloid-specific deletion of CD40 by LysM-cre also resulted in a significant reduction in EAE severity and reduced CNS inflammation (41, 42).
In parallel with impaired peripheral priming in response to MOG, CD4+T cells isolated from the CNS of DC CD40 conditional knockout mice expressed significantly lower levels of inflammatory cytokines that are likely important for inflaming the CNS and promoting recruitment of additional immune cells. In support of this conclusion, we detected fewer CNS mononuclear cells from DC CD40 conditional knockout mice and milder disease. Anti-IL17A antibody (secukinumab) was shown to reduce lesions detected by MRI in MS patients (7), and anti-GM-CSF antibody (MOR103) is being studied in a phase I clinical trial of patients with MS (43). Our results demonstrate that deletion of CD40 signaling on DCs significantly impairs the priming of IL-17 and GM-CSF producing Th cells in vivo and may therefore provide a promising therapeutic target in MS patients. Interestingly, treatment of mice with anti-CD154 at the time of PLP139-151/CFA immunization has been reported to inhibit clinical disease for up to 100 days after immunization(44). Moreover, although B cell depletion in mice treated with anti-CD20 antibody was shown to slightly reduce the Th1 and Th17 cell response to MOG protein immunization in vivo (45), our findings indicate that this reduction in Th cells was not regulated by CD40 on B cells. B cell CD40 conditional knockout mice developed IFN-γ-, IL-17-, and GM-CSF- producing Th cells comparable to WT mice in the periphery, but they showed much milder EAE symptoms, suggesting that CD40 on B cells has a role other than the priming of pathogenic Th cells.
In addition to immune cell infiltrates, antibodies and complement components are also found in inflammatory active lesions of MS patients (46); and the complement system has been shown to contribute to the pathology of EAE by triggering demyelination and modifying the antigen-specific T and B cell response(47–49). Previous work from our lab has explored the cellular requirements for CD40-CD40L and B7-CD28 expression for germinal center and antibody responses to model antigens. These studies demonstrated that CD40 expression on B cells is essential for the humoral immune response to T-dependent antigen in a cell intrinsic manner, whereas there is no requirement for CD40 expression by DCs for these responses (35, 50). Anti-MOG serum or antibody has been reported to have a pathogenic role in the development of EAE in mice (17, 51, 52), and monoclonal antibody against human CD40 has been reported to prevent EAE in the common marmoset(53). We found that deletion of CD40 from B cells but not DCs eliminated production of anti-MOG IgG. These data are consistent with studies in cytidine deaminase deficient mice, which are defective in Ig somatic hypermutations and class switching, showing decreased susceptibility to rhMOG-induced EAE, without a defect in MOG-specific T cells (52, 54). Importantly, we were able to fully reconstitute disease in B cell (but not DC) CD40 conditional knockout mice by passively transferring immune serum containing high titers of anti-MOG IgG antibodies. These antibodies together with complement, B cells, and CD4+ T cells were found throughout the spinal cord parenchyma of mice reconstituted with EAE serum but not control KLH serum. These data demonstrate that the major role played by CD40 on B cells is to induce a pathogenic autoantibody response. These antibodies even appear to facilitate recruitment of B cells and CD4+ T cells to the spinal cord parenchyma, as their numbers were greatly reduced in mice treated with KLH serum. Anti-CD20 monoclonal antibodies that target B cells were shown to be effective in treating MS patients(5). We propose based on our data that the therapeutic benefit of this treatment is mediated in part by inhibition of autoantibody production. In fact, antibody producing plasma cells and complement were recently found at the leading edge of chronic active MS lesions, further implicating humoral immunity as a driver of lesion development(55). Our studies do not exclude additional roles for B cells in the development of CNS autoimmune diseases (56)but do highlight the potential effectiveness of inhibiting or modulating the auto-reactive humoral response as a therapeutic option for at least some MS patients.
Collectively, our findings support a model in which CD40 induces complementary effector programs that contribute to EAE pathogenesis. Efficient priming of pathogenic proinflammatory Th cells and their eventual CNS infiltration requires CD40 expression on DCs, but not B cells. This infiltration appears to be necessary but not sufficient for EAE development. Our finding that passive transfer of EAE serum or IgG fully restores disease in B cell CD40 conditional knockout mice is an important one, especially in light of how these antibodies inundate the spinal cord parenchyma, fostering complement deposition and adaptive immune cell infiltration. CD40 maximizes development of CNS autoimmune disease via induction of complementary programs, but autoantibodies are nevertheless a major driving force in disease pathogenesis. Targeting antibody producing plasma cells may therefore be a key to stopping diseases like MS once they are underway. Our CD40 data also demonstrate that there are multiple points of potential therapeutic intervention during the development of CNS autoimmune diseases that involve distinct arms of the adaptive immune system.
Supplementary Material
Key points.
CD40 supports distinct effector programs in B cells and DCs for EAE.
CD40 expression on DCs is required for priming pathogenic T helper (Th) cells.
B cell CD40 is essential for class-switched MOG-specific antibody production.
Acknowledgements
We thank Vanja Lazarevic, Masashi Watanabe, and Karen Hathcock for their thoughtful comments and review of this manuscript. We thank Peter J Koch for ES cell injections; Christopher McNees for purified IgG quality control; Bernardo Rosa, Elena Kuznetsova, and Tarra Dumas from the NCI animal facility for assistance caring for and scoring experimental mice.
Funding
This work was supported by the Intramural Research Programs of the National Cancer Institute and the National Institute of Neurologic Disorders and Stroke, National Institutes of Health, and support to DW by National Institutes of Health grant R01DK07501-03S2
Footnotes
The authors have declared that no conflict of interest exists.
Study approval
Animal experiments were approved by the NCI Institute Animal Care and Use Committee. All mice were maintained in accordance with US National Institutes of Health guidelines.
References
- 1.Filippi M, Bar-Or A, Piehl F, Preziosa P, Solari A, Vukusic S, and Rocca MA. 2018. Multiple sclerosis. Nat Rev Dis Primers 4: 43–80. [DOI] [PubMed] [Google Scholar]
- 2.Henderson AP, Barnett MH, Parratt JD, and Prineas JW. 2009. Multiple sclerosis: distribution of inflammatory cells in newly forming lesions. Ann Neurol 66: 739–753. [DOI] [PubMed] [Google Scholar]
- 3.Ramaglia V, Rojas O, Naouar I, and Gommerman JL. 2021. The Ins and Outs of Central Nervous System Inflammation-Lessons Learned from Multiple Sclerosis. Annu Rev Immunol 39: 199–226. [DOI] [PubMed] [Google Scholar]
- 4.Torkildsen O, Myhr KM, and Bo L. 2016. Disease-modifying treatments for multiple sclerosis - a review of approved medications. Eur J Neurol 23 Suppl 1: 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hauser SL 2020. Progress in Multiple Sclerosis Research: An Example of Bedside to Bench. JAMA 324: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Segal BM 2019. The Diversity of Encephalitogenic CD4+ T Cells in Multiple Sclerosis and Its Animal Models. J Clin Med 8: 120–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Havrdova E, Belova A, Goloborodko A, Tisserant A, Wright A, Wallstroem E, Garren H, Maguire RP, and Johns DR. 2016. Activity of secukinumab, an anti-IL-17A antibody, on brain lesions in RRMS: results from a randomized, proof-of-concept study. J Neurol 263: 1287–1295. [DOI] [PubMed] [Google Scholar]
- 8.Palle P, Monaghan KL, Milne SM, and Wan ECK. 2017. Cytokine Signaling in Multiple Sclerosis and Its Therapeutic Applications. Med Sci (Basel) 5: 23–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jager A, Dardalhon V, Sobel RA, Bettelli E, and Kuchroo VK. 2009. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol 183: 7169–7177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Damsker JM, Hansen AM, and Caspi RR. 2010. Th1 and Th17 cells: adversaries and collaborators. Ann N Y Acad Sci 1183: 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y, Godec J, Ben-Aissa K, Cui K, Zhao K, Pucsek AB, Lee YK, Weaver CT, Yagi R, and Lazarevic V. 2014. The transcription factors T-bet and Runx are required for the ontogeny of pathogenic interferon-gamma-producing T helper 17 cells. Immunity 40: 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, Zhang GX, Dittel BN, and Rostami A. 2011. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol 12: 568–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hemmer B, Kerschensteiner M, and Korn T. 2015. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol 14: 406–419. [DOI] [PubMed] [Google Scholar]
- 14.Ransohoff RM, and Engelhardt B. 2012. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol 12: 623–635. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Given KS, Harlow DE, Matschulat AM, Macklin WB, Bennett JL, and Owens GP. 2017. Myelin-specific multiple sclerosis antibodies cause complement-dependent oligodendrocyte loss and demyelination. Acta Neuropathol Commun 5: 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flach AC, Litke T, Strauss J, Haberl M, Gomez CC, Reindl M, Saiz A, Fehling HJ, Wienands J, Odoardi F, Luhder F, and Flugel A. 2016. Autoantibody-boosted T-cell reactivation in the target organ triggers manifestation of autoimmune CNS disease. Proc Natl Acad Sci U S A 113: 3323–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lyons JA, Ramsbottom MJ, and Cross AH. 2002. Critical role of antigen-specific antibody in experimental autoimmune encephalomyelitis induced by recombinant myelin oligodendrocyte glycoprotein. European journal of immunology 32: 1905–1913. [DOI] [PubMed] [Google Scholar]
- 18.Marta CB, Oliver AR, Sweet RA, Pfeiffer SE, and Ruddle NH. 2005. Pathogenic myelin oligodendrocyte glycoprotein antibodies recognize glycosylated epitopes and perturb oligodendrocyte physiology. Proc Natl Acad Sci U S A 102: 13992–13997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Molnarfi N, Schulze-Topphoff U, Weber MS, Patarroyo JC, Prod’homme T, Varrin-Doyer M, Shetty A, Linington C, Slavin AJ, Hidalgo J, Jenne DE, Wekerle H, Sobel RA, Bernard CC, Shlomchik MJ, and Zamvil SS. 2013. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. The Journal of experimental medicine 210: 2921–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang TT, Jabs C, Sobel RA, Kuchroo VK, and Sharpe AH. 1999. Studies in B7-deficient mice reveal a critical role for B7 costimulation in both induction and effector phases of experimental autoimmune encephalomyelitis. The Journal of experimental medicine 190: 733–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Girvin AM, Dal Canto MC, Rhee L, Salomon B, Sharpe A, Bluestone JA, and Miller SD. 2000. A critical role for B7/CD28 costimulation in experimental autoimmune encephalomyelitis: a comparative study using costimulatory molecule-deficient mice and monoclonal antibody blockade. J Immunol 164: 136–143. [DOI] [PubMed] [Google Scholar]
- 22.Noelle RJ, Roy M, Shepherd DM, Stamenkovic I, Ledbetter JA, and Aruffo A. 1992. A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc Natl Acad Sci U S A 89: 6550–6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerritse K, Laman JD, Noelle RJ, Aruffo A, Ledbetter JA, Boersma WJ, and Claassen E. 1996. CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci U S A 93: 2499–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grewal IS, Foellmer HG, Grewal KD, Xu J, Hardardottir F, Baron JL, Janeway CA Jr., and Flavell RA. 1996. Requirement for CD40 ligand in costimulation induction, T cell activation, and experimental allergic encephalomyelitis. Science 273: 1864–1867. [DOI] [PubMed] [Google Scholar]
- 25.Xu J, Foy TM, Laman JD, Elliott EA, Dunn JJ, Waldschmidt TJ, Elsemore J, Noelle RJ, and Flavell RA. 1994. Mice deficient for the CD40 ligand. Immunity 1: 423–431. [DOI] [PubMed] [Google Scholar]
- 26.Girvin AM, Dal Canto MC, and Miller SD. 2002. CD40/CD40L interaction is essential for the induction of EAE in the absence of CD28-mediated co-stimulation. J Autoimmun 18: 83–94. [DOI] [PubMed] [Google Scholar]
- 27.Singer A, Hathcock KS, and Hodes RJ. 1979. Cellular and genetic control of antibody responses. V. Helper T-cell recognition of H-2 determinants on accessory cells but not B cells. The Journal of experimental medicine 149: 1208–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stromnes IM, and Goverman JM. 2006. Active induction of experimental allergic encephalomyelitis. Nat Protoc 1: 1810–1819. [DOI] [PubMed] [Google Scholar]
- 29.Zamvil SS, and Steinman L. 1990. The T lymphocyte in experimental allergic encephalomyelitis. Annu Rev Immunol 8: 579–621. [DOI] [PubMed] [Google Scholar]
- 30.Lyons JA, San M, Happ MP, and Cross AH. 1999. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. European journal of immunology 29: 3432–3439. [DOI] [PubMed] [Google Scholar]
- 31.Becher B, Durell BG, Miga AV, Hickey WF, and Noelle RJ. 2001. The clinical course of experimental autoimmune encephalomyelitis and inflammation is controlled by the expression of CD40 within the central nervous system. The Journal of experimental medicine 193: 967–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawabe T, Naka T, Yoshida K, Tanaka T, Fujiwara H, Suematsu S, Yoshida N, Kishimoto T, and Kikutani H. 1994. The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity 1: 167–178. [DOI] [PubMed] [Google Scholar]
- 33.Spath S, Komuczki J, Hermann M, Pelczar P, Mair F, Schreiner B, and Becher B. 2017. Dysregulation of the Cytokine GM-CSF Induces Spontaneous Phagocyte Invasion and Immunopathology in the Central Nervous System. Immunity 46: 245–260. [DOI] [PubMed] [Google Scholar]
- 34.Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, and Becher B. 2011. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol 12: 560–567. [DOI] [PubMed] [Google Scholar]
- 35.Watanabe M, Fujihara C, Radtke AJ, Chiang YJ, Bhatia S, Germain RN, and Hodes RJ. 2017. Co-stimulatory function in primary germinal center responses: CD40 and B7 are required on distinct antigen-presenting cells. The Journal of experimental medicine 214: 2795–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peters AL, Stunz LL, and Bishop GA. 2009. CD40 and autoimmunity: the dark side of a great activator. Semin Immunol 21: 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kroenke MA, Carlson TJ, Andjelkovic AV, and Segal BM. 2008. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. The Journal of experimental medicine 205: 1535–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodgers JM, and Miller SD. 2012. Cytokine control of inflammation and repair in the pathology of multiple sclerosis. Yale J Biol Med 85: 447–468. [PMC free article] [PubMed] [Google Scholar]
- 39.Iezzi G, Sonderegger I, Ampenberger F, Schmitz N, Marsland BJ, and Kopf M. 2009. CD40-CD40L cross-talk integrates strong antigenic signals and microbial stimuli to induce development of IL-17-producing CD4+ T cells. Proc Natl Acad Sci U S A 106: 876–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ponomarev ED, Shriver LP, and Dittel BN. 2006. CD40 expression by microglial cells is required for their completion of a two-step activation process during central nervous system autoimmune inflammation. J Immunol 176: 1402–1410. [DOI] [PubMed] [Google Scholar]
- 41.Aarts SA, Seijkens TT, Kusters PJ, van Tiel CM, Reiche ME, den Toom M, Beckers L, van Roomen CP, de Winther MP, Kooij G, and Lutgens E. 2019. Macrophage CD40 signaling drives experimental autoimmune encephalomyelitis. J Pathol 247: 471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aarts S, Seijkens TTP, Kusters PJH, van der Pol SMA, Zarzycka B, Heijnen P, Beckers L, den Toom M, Gijbels MJJ, Boon L, Weber C, de Vries HE, Nicolaes GAF, Dijkstra CD, Kooij G, and Lutgens E. 2017. Inhibition of CD40-TRAF6 interactions by the small molecule inhibitor 6877002 reduces neuroinflammation. J Neuroinflammation 14: 105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Constantinescu CS, Asher A, Fryze W, Kozubski W, Wagner F, Aram J, Tanasescu R, Korolkiewicz RP, Dirnberger-Hertweck M, Steidl S, Libretto SE, Sprenger T, and Radue EW. 2015. Randomized phase 1b trial of MOR103, a human antibody to GM-CSF, in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm 2: e117–e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Howard LM, Ostrovidov S, Smith CE, Dal Canto MC, and Miller SD. 2002. Normal Th1 development following long-term therapeutic blockade of CD154-CD40 in experimental autoimmune encephalomyelitis. J Clin Invest 109: 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weber MS, Prod’homme T, Patarroyo JC, Molnarfi N, Karnezis T, Lehmann-Horn K, Danilenko DM, Eastham-Anderson J, Slavin AJ, Linington C, Bernard CC, Martin F, and Zamvil SS. 2010. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann Neurol 68: 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reindl M, Di Pauli F, Rostasy K, and Berger T. 2013. The spectrum of MOG autoantibody-associated demyelinating diseases. Nat Rev Neurol 9: 455–461. [DOI] [PubMed] [Google Scholar]
- 47.Hundgeburth LC, Wunsch M, Rovituso D, Recks MS, Addicks K, Lehmann PV, and Kuerten S. 2013. The complement system contributes to the pathology of experimental autoimmune encephalomyelitis by triggering demyelination and modifying the antigen-specific T and B cell response. Clin Immunol 146: 155–164. [DOI] [PubMed] [Google Scholar]
- 48.Ramaglia V, Hughes TR, Donev RM, Ruseva MM, Wu X, Huitinga I, Baas F, Neal JW, and Morgan BP. 2012. C3-dependent mechanism of microglial priming relevant to multiple sclerosis. Proc Natl Acad Sci U S A 109: 965–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sabatino JJ Jr., Probstel AK, and Zamvil SS. 2019. B cells in autoimmune and neurodegenerative central nervous system diseases. Nat Rev Neurosci 20: 728–745. [DOI] [PubMed] [Google Scholar]
- 50.Lumsden JM, Williams JA, and Hodes RJ. 2003. Differential requirements for expression of CD80/86 and CD40 on B cells for T-dependent antibody responses in vivo. J Immunol 170: 781–787. [DOI] [PubMed] [Google Scholar]
- 51.Kinzel S, Lehmann-Horn K, Torke S, Hausler D, Winkler A, Stadelmann C, Payne N, Feldmann L, Saiz A, Reindl M, Lalive PH, Bernard CC, Bruck W, and Weber MS. 2016. Myelin-reactive antibodies initiate T cell-mediated CNS autoimmune disease by opsonization of endogenous antigen. Acta Neuropathol 132: 43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Galicia G, Lee DSW, Ramaglia V, Ward LA, Yam JY, Leung LYT, Li R, Handy M, Zhang J, Drohomyrecky PC, Lancaster E, Bar-Or A, Martin A, and Gommerman JL. 2018. Isotype-Switched Autoantibodies Are Necessary To Facilitate Central Nervous System Autoimmune Disease in Aicda(−/−) and Ung(−/−) Mice. J Immunol 201: 1119–1130. [DOI] [PubMed] [Google Scholar]
- 53.Boon L, Brok HP, Bauer J, Ortiz-Buijsse A, Schellekens MM, Ramdien-Murli S, Blezer E, van Meurs M, Ceuppens J, de Boer M, t Hart BA, and Laman JD. 2001. Prevention of experimental autoimmune encephalomyelitis in the common marmoset (Callithrix jacchus) using a chimeric antagonist monoclonal antibody against human CD40 is associated with altered B cell responses. J Immunol 167: 2942–2949. [DOI] [PubMed] [Google Scholar]
- 54.Sun Y, Peng I, Senger K, Hamidzadeh K, Reichelt M, Baca M, Yeh R, Lorenzo MN, Sebrell A, Dela Cruz C, Tam L, Corpuz R, Wu J, Sai T, Roose-Girma M, Warming S, Balazs M, Gonzalez LC, Caplazi P, Martin F, Devoss J, and Zarrin AA. 2013. Critical role of activation induced cytidine deaminase in experimental autoimmune encephalomyelitis. Autoimmunity 46: 157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Absinta M, Maric D, Gharagozloo M, Garton T, Smith MD, Jin J, Fitzgerald KC, Song A, Liu P, Lin JP, Wu T, Johnson KR, McGavern DB, Schafer DP, Calabresi PA, and Reich DS. 2021. A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature 597: 709–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rojas OL, Probstel AK, Porfilio EA, Wang AA, Charabati M, Sun T, Lee DSW, Galicia G, Ramaglia V, Ward LA, Leung LYT, Najafi G, Khaleghi K, Garcillan B, Li A, Besla R, Naouar I, Cao EY, Chiaranunt P, Burrows K, Robinson HG, Allanach JR, Yam J, Luck H, Campbell DJ, Allman D, Brooks DG, Tomura M, Baumann R, Zamvil SS, Bar-Or A, Horwitz MS, Winer DA, Mortha A, Mackay F, Prat A, Osborne LC, Robbins C, Baranzini SE, and Gommerman JL. 2019. Recirculating Intestinal IgA-Producing Cells Regulate Neuroinflammation via IL-10. Cell 177: 492–493. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.