

Abstract
Successful vaccination strategies offer the potential for lifelong immunity against infectious diseases and cancer. There has been increased attention regarding the limited translation of some preclinical findings generated using specific pathogen-free (SPF) laboratory mice to humans. One potential reason for the difference between preclinical and clinical findings lies in maturation status of the immune system at the time of challenge. In this study, we used a ‘dirty’ mouse model, where SPF laboratory mice were cohoused with pet store mice to permit microbe transfer and immune system maturation, to investigate the priming of a naïve T cell response after vaccination with a peptide subunit mixed with polyI:C and agonistic anti-CD40 mAb. While this vaccination platform induced robust antitumor immunity in SPF mice, it failed to do so in microbially-experienced cohoused (CoH) mice. Subsequent investigation revealed that despite similar numbers of Ag-specific naïve CD4 and CD8 T cell precursors, the expansion, differentiation, and recall responses of these CD4 and CD8 T cell populations in CoH mice were significantly reduced compared to SPF mice after vaccination. Evaluation of the dendritic cell compartment revealed reduced IL-27p28 expression by XCR1+ DC from CoH mice after vaccination, correlating with reduced T cell expansion. Importantly, administration of recombinant IL-27:EBI3 complex to CoH mice shortly after vaccination significantly boosted Ag-specific CD8 and CD4 T cell expansion, further implicating the defect to be T cell extrinsic. Collectively, our data show the potential limitation of exclusive use of SPF mice when testing vaccine efficacy.
INTRODUCTION
Multiple studies over the past quarter century have rigorously defined the T cell response to vaccination, providing exquisite detail to the processes essential for the development and maintenance of memory T cells that provide protection to subsequent challenges (1–3). While most vaccination research falls under the ‘infectious disease umbrella’, there has been interest in recent years in using vaccination to stimulate antitumor immunity (4,5) – especially with the identification of tumor-specific immunogenic neoantigens. Current vaccines contain attenuated microorganisms (i.e, bacteria or viruses), purified proteins or toxins, synthetic peptides, or mRNA that encode for a defined protein to induce protective immunity (6). Vaccines also frequently include an adjuvant, which is designed to increase the activity of antigen (Ag) presenting cells and potency of the immunogen (7).
A variety of immunological techniques and reagents – including the extensive array of genetically altered mice – have given researchers the means to interrogate how stimulation through the TCR shapes an Ag-specific T cell response (i.e., T cell expansion, contraction, differentiation, and function) after vaccination or infection at the cellular and molecular level. Recently, there has been growing interest in understanding how environmental microbial exposure impacts the ability of the immune system to respond to new challenges (8–10). Justification for these studies stems from the fact that the human immune system is shaped and trained by the vaccinations received and pathogenic and commensal microbes encountered daily from birth. In contrast, nearly all laboratory studies use mice housed in specific pathogen-free (SPF) conditions, which keeps their immune systems in a more naïve state (8,11). The landmark study by Beura et al. (12) using SPF and microbially-experienced ‘dirty’ mice revealed key immunological parallels between SPF and dirty mice to neonatal and adult humans, respectively. Subsequent studies from our lab and others have shown the power of using dirty mice to complement experiments done in SPF mice, revealing how prior microbial exposure can better replicate clinical observations (13–16).
The objective of the present study was to define how previous microbial exposure impacts naïve T cell expansion, differentiation into memory populations, and function after immunization with a defined peptide subunit vaccine. To investigate this question, we used a vaccine platform consisting of synthetic CD8 and/or CD4 T cell epitope peptides, the TLR 3 agonist polyinosinic-polycytidylic acid (polyI:C), and agonistic anti-CD40 mAb (17), to stimulate a primary Ag-specific CD8 or CD4 T cell response. Our data show this immunization protocol induced dramatically muted T cell immunity in microbially-experienced dirty mice compared to SPF mice. The observed differences in T cell expansion, differentiation, and function did not appear to be intrinsic to the Ag-specific T cells. Instead, the attenuated T cell response in dirty mice correlated with reduced IL-27 production from XCR1+ DC, which is a known predictor of the magnitude of the T cell response following subunit vaccination (18). Overall, the data presented herein further highlight the impact of microbial exposure on T cell immune responses to new Ag encounters.
MATERIALS and METHODS
Mice
Female CD45.2+ C57BL/6N (B6; 8–10 weeks of age) and CD45.1+ B6.SJL mice were purchased from Charles River (Wilmington, MA) and Jackson Laboratories (Bar Harbor, ME), respectively. Female pet store mice were purchased from local pet stores in the Minneapolis-St. Paul, MN metropolitan area. All mice were housed in AALAC-approved animal facilities at the University of Minnesota (BSL-1 for SPF B6 mice, and BSL-3 for cohoused B6 and pet store mice). SPF B6 or B6.SJL and pet store mice were cohoused (CoH) at a ratio of 8:1 in large rat cages for 30–60 days to facilitate microbe transfer (19). Mice within each cohousing cohort were randomly assigned to experimental groups. Experimental procedures were approved by the University of Minneapolis Animal Care and Use Committee and performed following the Office of Laboratory Animal Welfare guidelines and PHS Policy on Human Cancer and Use of Laboratory Animals.
Peptide vaccinations
Mice were vaccinated i.v. with a cocktail of synthetic peptide (50 μg; Vivitide), polyI:C (50 μg; InVivogen), and agonistic anti-CD40 mAb (100 μg; BioXcel). We refer to this cocktail as “TriVax”, with the specific peptide(s) as a prefix. Alternatively, mice were immunized s.c. with peptide emulsified in Complete Freund’s Adjuvant (Sigma) or Alhydrogel (InvivoGen).
Tumor challenge
Mice were immunized with OVA257/OVA323-TriVax 30 days before being challenged with B16-ova tumor cells (2.5 × 105 cells in 0.1 ml PBS s.c.). Tumor size (mm2) was measured over time with calipers.
Tetramers
Kb-specific tetramers containing B8R20–27 (TSYKFESV), OVA257–264 (SIINFEKL), HSV peptide glycoprotein B498–505 (SSIEFARL), Plasmodium ovale glideosone-associated protein 5040–48 (GAP-50; SQLLNAKYL), and LCMV glycoprotein (gp)33–41 (KAVYNFATM) were used to identify and quantify Ag-specific CD8 T cells by flow cytometry. Biotinylated peptide:Kb monomers were obtained from the NIH tetramer core. I-Ab–specific tetramers containing 2W1S (EAWGALANWAVDSA), Ag28m (VEIHRPVPGTA), LCMV glycoprotein (gp)66–77 (DIYKGVYQFKSV), or L. monocytogenes LLO190–201 (NEKYAQAYPNVS) peptides were used to identify and quantify Ag-specific CD4 T cells by flow cytometry. Biotinylated peptide:I-Ab monomers were made into tetramers with streptavidin (SA)-phycoerythrin (PE) or SA-allophycocyanin (APC; Prozyme) as previously described (20,21).
Quantitation of endogenous Ag-specific T cell populations using p:Kb or p:I-Ab tetramer based-enrichment
To quantify the number of Ag-specific CD8 or CD4 T cells within the spleens of SPF or CoH mice, a tetramer-based enrichment protocol (20) using p:Kb or p:I-Ab tetramers was employed. Briefly, spleens were harvested for each mouse analyzed, a single-cell suspension was prepared, and APC- and PE-conjugated tetramers were added at a 1:400 dilution in tetramer staining buffer (PBS containing 5% BCS, 2mM EDTA, 1:50 normal mouse serum, and 1:100 anti-CD16/32 mAb). The cells were incubated in the dark on ice (for p:Kb tetramers) or at room temperature (for p:I-Ab tetramers) for 1 hour, followed by a wash in 10 mL cold FACS Buffer. The tetramer-stained cells were then resuspended in 0.2 mL FACS Buffer, mixed with 0.05 mL of both anti-APC and -PE mAb-conjugated magnetic microbeads (StemCell Technologies), and incubated in the dark on ice for 30 minutes. The cells were washed and resuspended in 3 mL cold FACS Buffer and passed over a magnet (StemCell Technologies) to enrich for tetramer-specific cells. The resulting enriched fractions were then stained with a cocktail of fluorochrome-labeled mAb (see below). Cell numbers for each sample were determined using AccuCheck Counting Beads (Invitrogen). Samples were then analyzed using an LSR II flow cytometer (BD) and FlowJo software (TreeStar Inc.). The percentage of tetramer-positive events was multiplied by the total number of cells in the enriched fraction to calculate the total number of Ag-specific CD8 or CD4 T cells in the spleen.
Flow cytometry
Peripheral blood leukocytes or splenocytes were evaluated by flow cytometry. RBC were removed from heparinized whole blood or single cell splenocyte suspensions using ACK lysis buffer. To assess the expression of cell surface proteins, cells were incubated with fluorochrome-conjugated mAb at 4°C for 30 minutes. The cells were then washed with FACS buffer (PBS containing 2% BCS and 0.2% NaN3). For some experiments, the cells were then fixed with PBS containing 2% paraformaldehyde. In procedures requiring intracellular staining (i.e., to detect transcription factor or intracellular cytokine presence), cells were permeabilized following surface staining using the Foxp3 transcription factor staining kit (Tonbo) for transcription factors or the BD Cytofix/Cytoperm kit (BD Biosciences), stained for 1 hour at 4°C with a second set of fluorochrome-conjugated mAb, and suspended in FACS buffer for acquisition. The fluorochrome-conjugated mAb used in both surface and intracellular stainings were: Horizon™ V500 CD90.2 (clone 53–2.1; BD Biosciences), Brilliant Violet™ (BV) 510 and FITC CD3 (clone 17A2; BioLegend), BV421 and BV605 CD4 (clone GK1.5; BioLegend), BV650 CD8 (clone 53–6.7; BioLegend), PE-CD45.1 (clone A20; eBioscience), FITC-CD45.2 (clone 104; eBioscience), AlexaFluor®700 CD44 (clone IM7; BioLegend), PE-Cy7 KLRG1 (clone 2F1; eBioscoence), PerCP-Cy5.5 CD62L (clone MEL-14; BD Horizon), BV510 CD127 (clone A7R34; BioLegend), APC and BV650 IFNγ (clone XMG1.2; BioLegend), PE-Cy7 IL-2 (clone JES6–5H4; BioLegend), PerCP-Cy5.5 B220 (clone RA3–6B2; eBioscience), PerCP-Cy5.5 CD11b (clone M1/70; eBioscience), PerCP-Cy5.5 CD11c (clone N418; eBioscience), PerCP-Cy5.5 F4/80 (clone BM8; eBioscience), FITC FoxP3 (clone FJK-15S; eBioscience), FITC TNFα (clone MP6-XT22; eBioscience), PE IL-12p40 (clone C15.6; BioLegend), and PE IL-27p28 (clone MM27–7B1; BioLegend).
CD8 T cell in vitro restimulation
Ag-specific CD8 T cell responses were measured following ex vivo peptide stimulation. Briefly, spleens were harvest from SPF and CoH mice 30 days after vaccination with B8R-TriVax. Approximately 106 splenocytes were then stimulated with B8R peptide (1 μg/mL) in media containing 3 μg/mL of brefeldin A (Tonbo Biosciences). Cells were incubated for 6 h at 37°C. Cells were then stained for surface antibodies to identify CD8 T cells, and then fixed and permeabilized (BD Biosciences Cytofix/Cytoperm Kit). Following permeabilization, cells were stained for intracellular cytokines with anti-IFNγ and anti-TNFα mAb. After staining, cells were resuspended in FACS buffer and 0.02 mL of counting beads (eBioscience) were added to each sample immediately before acquisition.
CD4 T cell in vivo restimulation
In vivo peptide stimulation was used to determine Ag-specific CD4 T cell function by intracellular cytokine production, as previously described (22–24). Briefly, SPF and CoH mice were injected i.v. with 100 μg of the 2W1S peptide 28 days after vaccination with 2W1S-TriVax. After 3 hours, spleens were harvested in media containing 10 μg/ml brefeldin A. The resulting cell suspensions were first stained with 2W1S:I-Ab tetramer and other mAb to identify Ag-specific CD4 T cells, as described above. The cells were then fixed, permeabilized, and stained with anti-IFNγ, -TNFα, and -IL-2 mAb. Cells were resuspended in FACS buffer after staining, and 0.02 mL of counting beads (eBioscience) were added to each sample immediately before acquisition.
Immune cell adoptive transfer
SPF CD45.1+ P14 donor mice were bled, and 5 × 104 P14 T cells were adoptively transferred into SPF or CoH CD45.2+ B6 mice by i.v. injection 1 day before gp33-TriVax immunization. Mice were bled on days 3, 7, 14, and 21 after immunization to identify CD45.1+ P14 T cells in the circulation by flow cytometry after staining with gp33:Kb tetramers and anti-CD45.1 mAb. Spleens were also collected on day 21-post immunization to determine the number of CD45.1+ P14 T cells and endogenous CD45.2+ gp33-specific CD8 T cells using gp33:Kb tetramers, anti-CD45.1, and anti-CD45.2 mAb. Alternatively, 4 × 106 bulk splenocytes from SPF or CoH CD45.1+ B6.SJL mice were transferred into SPF or CoH CD45.2+ B6 mice by i.v. injection 1 day before B8R-TriVax immunization. Spleens were then collected 7 days later to determine the number of CD45.1+ B8R-specific CD8 T cells using B8R:Kb tetramers, anti-CD45.1 mAb, and anti-CD45.2 mAb.
Administration of recombinant IL-27p28:EBI3 complex
CoH mice received recombinant mouse IL-27p28:EBI3 complex (250 μg/mouse; Sino Biological) i.v. 2 h after B8R/2W1S-TriVax immunization. The frequency and number of B8R-specific CD8 T cells and 2W1S-specific CD4 T cells in the spleen was determined 5 days later by flow cytometry.
Quantification and Statistical Analysis
Data shown are presented as mean values ± SEM. GraphPad Prism 9 was used for statistical analysis, where statistical significance was determined using two-tailed Unpaired, nonparametric Mann-Whitney U test (for 2 individual groups) or group-wise, one-way ANOVA analyses followed by multiple-testing correction using the Holm-Sidak method, with α = 0.05. * p < 0.05, ** p < 0.01, *** p < 0.005, **** p < 0.001.
RESULTS
TriVax-immunized CoH mice demonstrate reduced antitumor immunity compared to SPF mice.
Immunization with peptide epitopes can elicit robust priming of Ag-specific T cell populations that mediate effective protection against pathogen challenge or tumor growth. Moreover, inclusion of TLR and costimulatory receptor agonists, which are thought to mimic the multiple immunostimulatory pathways activated during true pathogen infection, can maximize the T cell response to the peptides. For example, the combination of defined antigenic peptides mixed with the TLR3 agonist polyI:C and agonistic anti-CD40 mAb ((17), hereafter referred to as “TriVax”) elicits strong, stable T cell responses in SPF mice capable of mediating protection to viral infection or tumor rejection (25,26). Because of the documented impact microbial exposure has on shaping and training the composition and function of the immune system to new challenges, we used the TriVax vaccination platform to examine the generation of antitumor immunity in SPF and microbially-experienced B6 mice that had been cohoused (CoH) with pet store mice. SPF and CoH mice were immunized with OVA257- and OVA323-TriVax to activate both OVA-specific CD8 and CD4 T cells. Mice were then challenged with B16-ova tumor cells 28 days later, and tumor growth was monitored over time (Figure 1A). Consistent with previous data (25), B16-ova tumors were reduced in size in the TriVax-immunized SPF mice compared to unimmunized SPF mice (Figure 1B). To our surprise, we saw no difference in the size of the B16-ova tumors in untreated and TriVax-immunized CoH mice. Multiple reports have demonstrated TriVax immunization elicits protective immunity (25,27,28), so we set out to further examine why this vaccination platform failed to suppress tumor growth in CoH mice.
FIGURE 1. Reduced antitumor immunity in TriVax-immunized microbially experienced cohoused (CoH) mice.
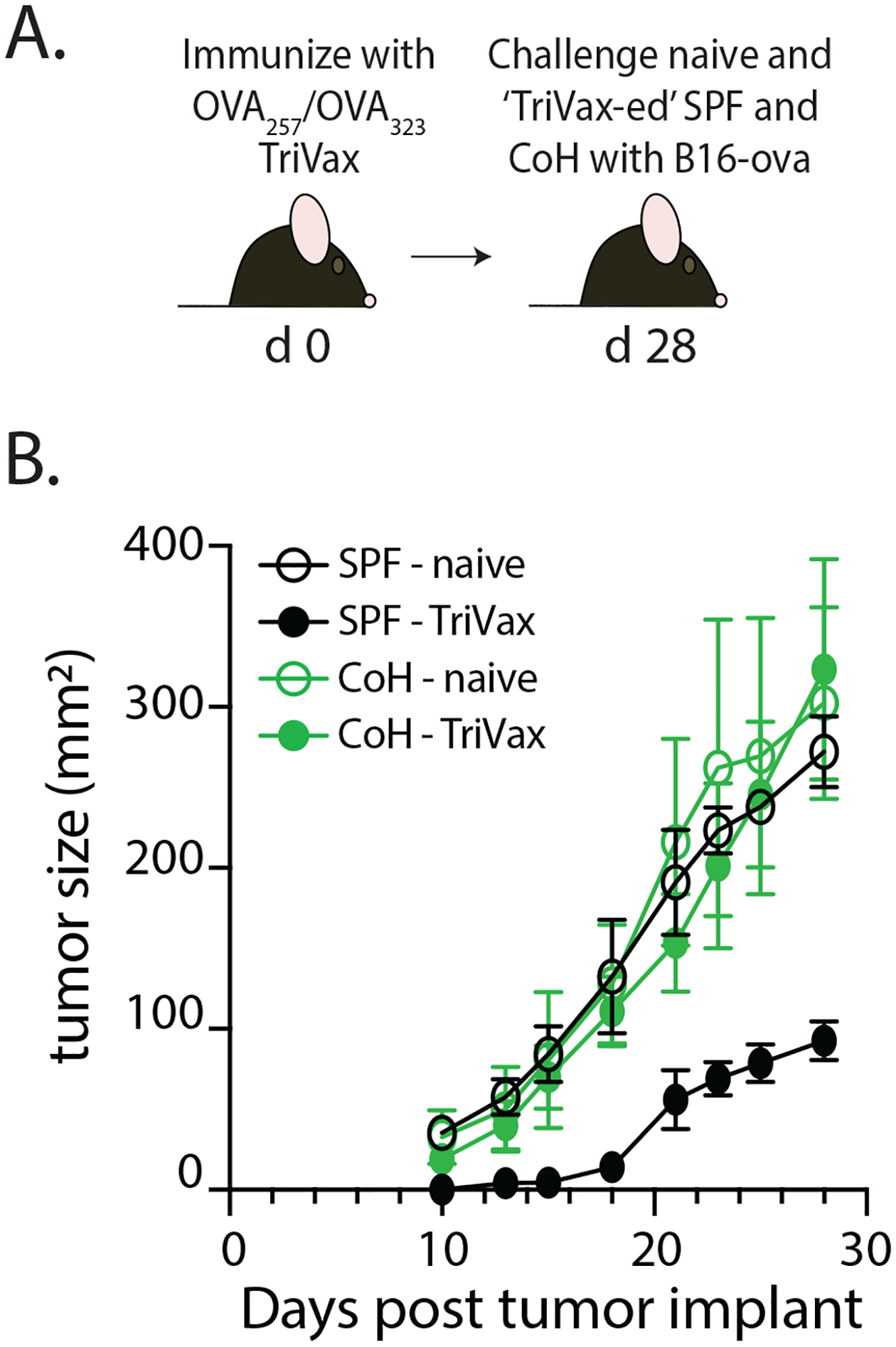
A. Experimental Design – SPF and CoH B6 mice were immunized with OVA257/OVA323-TriVax. After 28 days, mice were challenged with B16-ova cells (2.5 × 105 cells in 0.1 ml s.c.). B. Tumor growth was then monitored over the next 28 days. As a reference, cohorts of unimmunized SPF and CoH mice were also challenged with B16-ova cells. Data in B are representative of 2 replicate experiments. n = 8 mice/group.
The magnitude of naïve CD8 and CD4 T cell expansion/contraction is muted in TriVax-immunized CoH mice.
Peptide immunization, especially when combined with polyI:C and anti-CD40 mAb, can dramatically increase the number of Ag-specific T cells (25,27,28). To examine the impact of microbial experience on naïve T cell priming and expansion, we monitored the frequency and number of B8R-specific CD8 T cells or 2W1S-specific CD4 T cells in the blood and spleens of SPF and CoH mice following vaccination with either B8R- or 2W1S-TriVax, respectively (Figure 2A). Robust expansion of both B8R-specific CD8 T cells and 2W1S-specific CD4 T cells was noted in the blood of SPF mice, peaking on day 8 and followed by a contraction in population size by days 14 and 28 (Figure 2B–C). We were able to detect expansion of these Ag-specific T cell populations in the blood of CoH mice, but not to the extent seen in SPF mice. Splenic analysis of B8R-specific CD8 T cells or 2W1S-specific CD4 T cells at day 28 revealed similar significant reductions in frequency and number in the CoH mice compared to SPF mice (Figure 2D–E). The reduced T cell expansion in CoH mice was not limited to TriVax immunization, as we also noted reduced numbers of Ag-specific CD8 and CD4 T cells in CoH mice immunized with peptide mixed with either Complete Freund’s Adjuvant (CFA) or the alum vaccine adjuvant Alhydrogel (Figure 2F–G). Collectively, these data demonstrate that Ag-specific CD8 and CD4 T cell expansion and the number of cells at a memory timepoint (after contraction) are diminished in CoH mice (compared to SPF mice) after peptide immunization.
FIGURE 2. TriVax-immunized CoH mice exhibit blunted CD8 and CD4 T cell expansion.
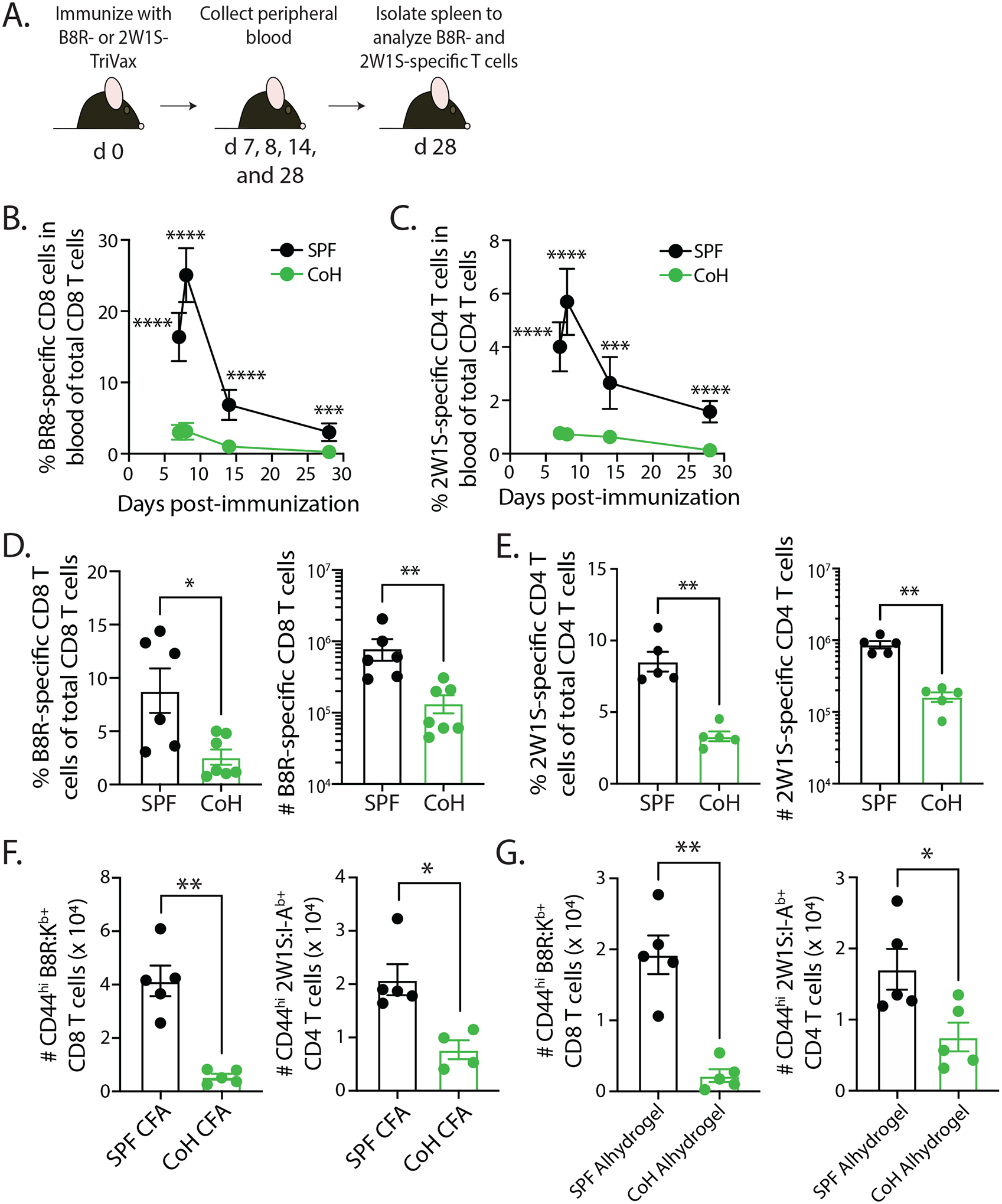
A. Experimental Design – SPF and CoH B6 mice were immunized with either B8R- or 2W1S-TriVax. Blood was collected on days 7, 8, 14, and 28 after immunization to determine the frequency of (B) B8-specific CD8 T cells or (C) 2W1S-specific CD4 T cells of total CD8 or CD4 T cells, respectively. On day 28, spleens were also isolated to determine the frequency and number of (D) B8R-specific CD8 T cells or (E) 2W1S-specific CD4 T cells. F-G. SPF and CoH mice were immunized with either B8R or 2W1S peptide mixed with either (F) Complete Freund’s Adjuvant (CFA) or (G) Alhydrogel. Spleens were collected 7 days later, and the number of B8R-specific CD8 T cells or 2W1S-specific CD4 T cells were determined. Data in B-G are representative of at least 2 technical replicates. n = 4–7 mice/group. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.005, **** p ≤ 0.001.
TriVax-immunized CoH mice display altered CD8 and CD4 T cell differentiation and function.
CD8 and CD4 T cell proliferation is accompanied by differentiation into effector cells, which is followed by a contraction phase. The T cells that survive contraction establish the memory pool of cells, displaying phenotypic and functional differences from the naïve precursor T cells (29–32). The data presented in Figure 2 showed significant reductions in Ag-specific CD8 and CD4 T cells at the peak of expansion and at a memory time point (≥ day 28) after contraction in TriVax-immunized CoH mice. Thus, we next evaluated the extent of CD8 T cell differentiation in SPF and CoH mice after TriVax immunization (Figure 3A). CD8 T cell subsets, such as effector memory (TEM), central memory (TCM), and long-lived effector cells (LLEC), can be distinguished based on the expression of CD62L and KLRG1 (33). Early (7 days) after TriVax immunization, there was decreased frequency of B8R-specific CD8 T cells among all CD8 T cells in the spleens of CoH mice compared to what was seen in SPF mice (Figure 3B), but this did not translate to a significant numerical difference. There were, however, notable reductions in the frequency and number of B8R-specific CD8 T cells in CoH mice 30 days after immunization. Interestingly, CoH mice had an increase in memory precursor cell (MPEC) frequency (KLRG1−CD127+) and a decrease in short-lived effector cell (SLEC) frequency (KLRG1+CD127−) 7 days after immunization (Figure 3C–D). Further examination of the B8R-specific CD8 T cells via CD62L confirmed a decrease in KLRG1+CD62L− and KLRG1−CD62L− cells by percentages and numbers of cells from CoH mice at 7 days post-immunization, with an increase in KLRG1−CD62L+ cells (Figure 3E–G). Intriguingly at 30 days post-TriVax, we saw a reduction by percentage and number within the central memory (TCM; CD62L+KLRG1−) and effector memory (TEM; CD62L−KLRG1−) subsets, and a reduction in number but increase in frequency of the long-lived effector cell (LLEC; CD62L−KLRG1+) subset (Figure 3E–G). Interestingly, we also noted the appearance of a CD62L+ population with low expression of KLRG1. Thus, at 7 days post-TriVax, the responding CD8 T cells in CoH mice favor MPEC generation, but by memory time points (i.e., 30 days post-immunization) the B8R-specific CD8 T cells are fewer in number and contain a larger proportion of LLECs. We then examined the ability of B8R-specific CD8 T cells to produce effector cytokines after in vitro peptide restimulation (Figure 3H). Splenocytes from TriVax-immunized CoH mice had a lower percentage of CD44hi CD8 T cells producing IFNγ or IFNγ and TNFα after restimulation compared to splenocytes from TriVax-immunized SPF mice (Figure 3I).
FIGURE 3. CD8 T cells from TriVax-immunized CoH mice display reduced differentiation and function after restimulation.
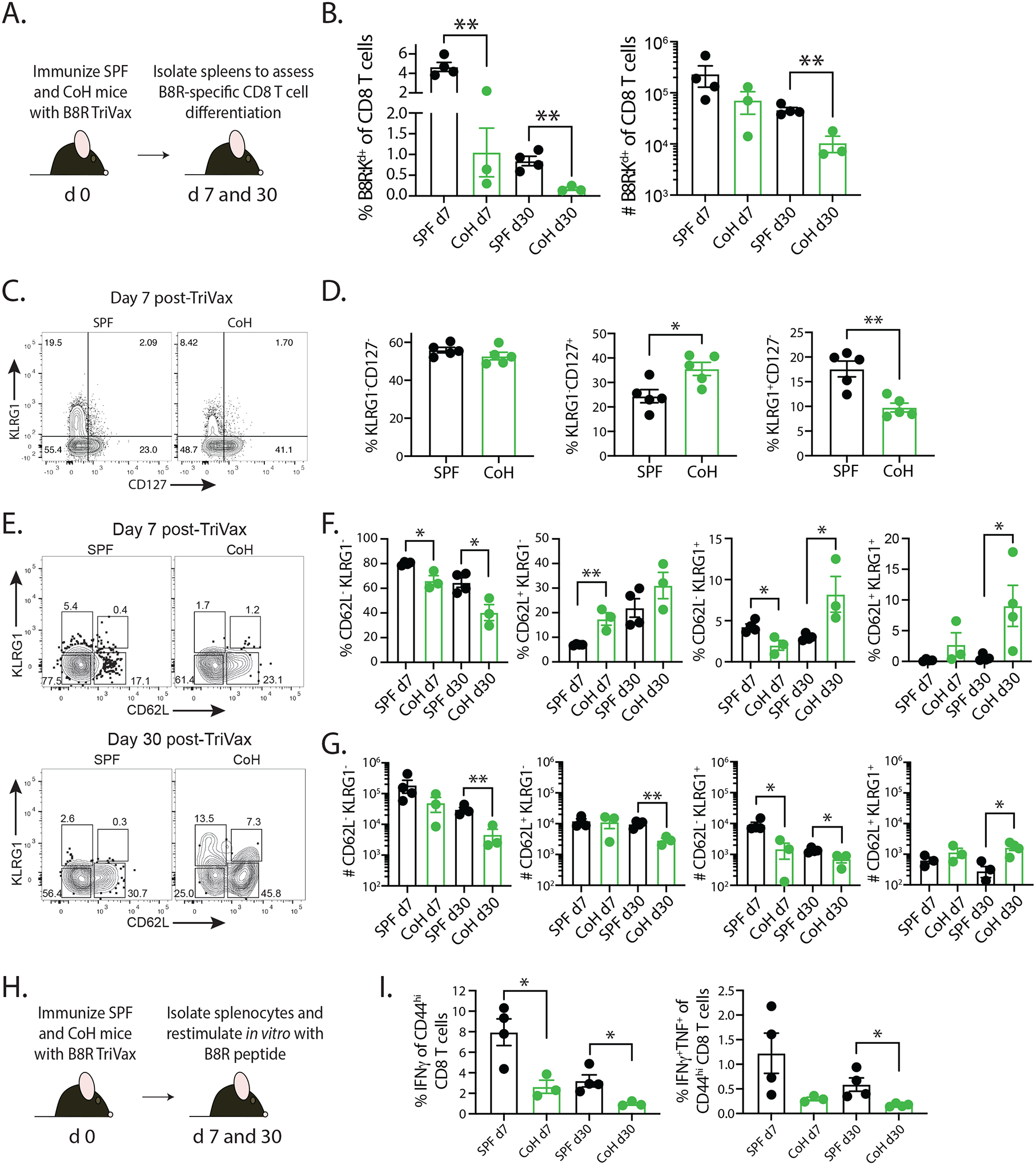
A. Experimental design – SPF and CoH B6 mice were immunized with B8R-TriVax. On days 7 and 30 after immunization, spleens were isolated and processed for flow cytometry to determine the (B) frequency and number of B8R-specific CD8 T cells. C-D. Day 7 post-TriVax samples were examined for KLRG1 and CD127 expression to define MPEC (KLRG1−CD127+) and SLEC (KLRG1+CD127−) frequency. E-G. Day 7 and 30 post-TriVax samples were examined for KLRG1 and CD62L expression to define the frequency and number of KLRG1−CD62L− (TEM), KLRG1−CD62L+ (TCM), and KLRG1+CD62L− (LLEC) subsets. Data are representative of at 3 technical replicates consisting of 3–4 mice/group/timepoint. * p ≤ 0.05, ** p ≤ 0.01. H. Experimental design – SPF and CoH B6 mice were immunized with B8R-TriVax. After 7 and 30 days, spleens were collected and processed into a single cell suspension for in vitro restimulation with B8R peptide to induce effector cytokine (IFNγ and TNFα) production. I. The frequency of IFNγ+ and IFNγ+TNFα+ cells among the CD44hi CD8 T cells within the culture was determined by flow cytometry. Data are representative of 2 technical replicates consisting of 3–4 mice/group/timepoint. * p ≤ 0.05
Naïve CD4 T cells have the potential to differentiate into multiple ‘helper’ populations depending on the cytokines present at the time of activation (34). These subsets can be defined by lineage-specific transcription factor and/or surface protein expression, such as Tbet+ TH1, Bcl6+CXCR5+ TFH, or Foxp3+ regulatory T cells (35–37). To evaluate how CD4 T cell differentiation in CoH mice after 2W1S-TriVax immunization compared to differentiation in SPF mice, we determined the frequency and number of 2W1S-specific CD4 T cells that expressed Tbet, Bcl6 and CXCR5, or Foxp3+ 7 or 28 days after immunization (Figure 4A). There was no difference in frequency of Tbet+, Bcl6+CXCR5+, or Foxp3+ 2W1S-specific CD4 T cells at the day 7 timepoint, but the frequency of 2W1S-specific CD4 T cells expressing Tbet in CoH mice 28 days after immunization was reduced compared to similarly immunized SPF mice (Figure 4B–C and Supplemental Figure 1). Moreover, there was a small (but significant) increase in the frequency of Foxp3+ 2W1S-specific regulatory CD4 T cells in the CoH mice on day 28. When applying these frequency data to total number of 2W1S-specific CD4 T cells in SPF and CoH mice after 2W1S-TriVax immunization, the CoH mice contained significantly fewer numbers of Tbet+, Bcl6+CXCR5+, and Foxp3+ 2W1S-specific CD4 T cells (Figure 4D). As with the B8R-specific CD8 T cells, we were also interested in assessing the ability of the TriVax-induced 2W1S-specific CD4 T cells in SPF and CoH mice to produce cytokine after peptide restimulation in vivo (Figure 4E; (24)). Data from this assay showed a dramatic reduction in frequency and number of IFNγ+, IFNγ+TNFα+, and IFNγ+TNFα+IL-2+ 2W1S-specific CD4 T cells in CoH mice (Figure 4F). Together, the results in Figures 3 and 4 highlight deficiencies in Ag-specific CD8 and CD4 T cell differentiation and percentage/number of cells capable of making cytokine after restimulation in TriVax-immunized CoH mice, in addition to the reduced proliferative capacity seen in Figure 2.
FIGURE 4. Reduced CD4 T cell differentiation and recall response in TriVax-immunized CoH mice.
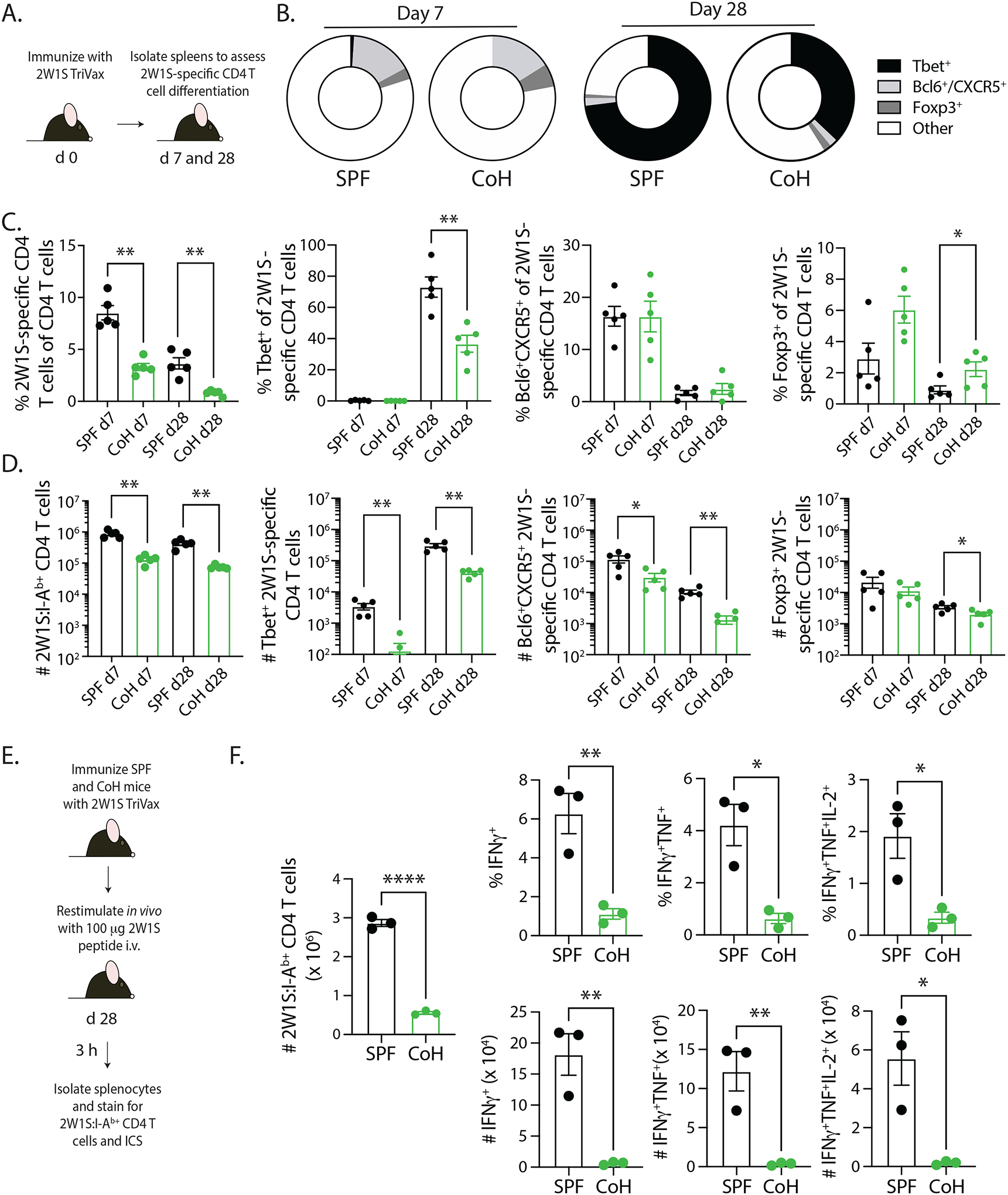
A. Experimental design – SPF and CoH B6 mice were immunized with 2W1S-TriVax. On days 7 and 28 after immunization, spleens were isolated and processed for flow cytometry to determine the extent of 2W1S-specific CD4 T cell differentiation. B-D. The frequency and number of 2W1S-specific CD4 T cells that had differentiated into Th1 (Tbet+), Tfh (Bcl6+/CXCR5+), and Treg (Foxp3+) was determined. Representative flow plots are shown in Supplemental Figure 1. Data are representative of 2 technical replicates consisting of 5 mice/group/timepoint. * p ≤ 0.05, ** p ≤ 0.01. E. Experimental design – SPF and CoH B6 mice were immunized with 2W1S-TriVax. After 28 days, the mice were injected i.v. with 100 μg 2W1S56–68 peptide to restimulate the 2W1S-specific CD4 T cells to assess the ability to produce effector cytokines. Spleens were isolated 3 h later, and the cells were processed for flow cytometry. F. The frequency and number of IFNγ+, IFNγ+TNFα+, or IFNγ+TNFα+IL-2+ CD44+ 2W1S-specific CD4 T cells were determined by flow cytometry. Data are representative of 2 technical replicates, with 3 mice/group in each experiment. **** p ≤ 0.001, ** p ≤ 0.01, and * p ≤ 0.05.
Reduced T cell expansion in CoH mice is not T cell-intrinsic, but is inherent to the CoH environment.
One possible explanation for the reduced T cell expansion in the TriVax-immunized CoH mice we considered was the presence of fewer naïve Ag-specific precursors, as the number of precursors can strongly influence the extent of expansion and number of memory T cells after contraction (20,31,38–40). Thus, we quantitated the number of T cell precursors for panel of Ag-specific CD8 and CD4 T cells. For each of the 5 Ag-specific CD8 T cell and 4 Ag-specific CD4 T cell populations examined, there was no difference in the number of naïve precursors in the spleens of SPF and CoH mice (Figure 5A–B). These data suggest the reduced naïve T cell expansion seen in CoH after TriVax immunization was not due to fewer numbers of Ag-specific precursors.
FIGURE 5. SPF and CoH B6 mice contain equivalent numbers of Ag-specific CD8 and CD4 T cell precursors.
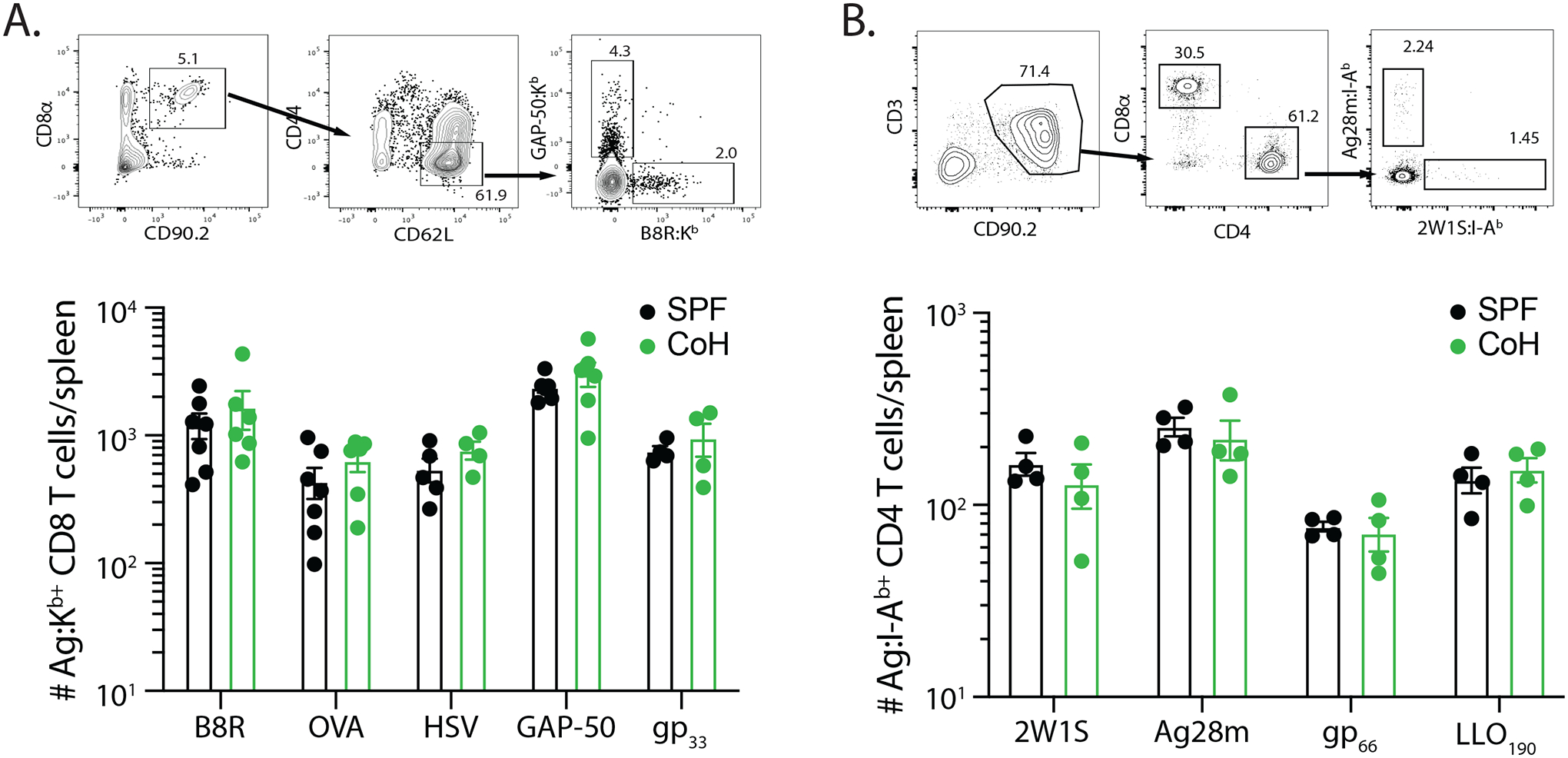
The number of Ag-specific CD8 T cells specific for B8R, OVA257, HSV, GAP-50, and LCMV gp33 and CD4 T cells specific for 2W1S, Ag28m, LCMV gp66–77, and L. monocytogenes LLO190–201 was determined using tetramer enrichment. Representative flow plots showing gating strategy used in tetramer-enriched cell fractions to detect the frequency of Ag-specific (A) CD8 and (B) CD4 T cell populations. Shown are examples used to detect B8R- and GAP-50-specific CD8 T cells and Ag28- and 2W1S-specific CD4 T cells. Graphs show the number of Ag-specific T cell precursors across the 5 MHC I- and 4 MHC II-restricted epitopes in SPF and CoH mice. Data were combined from 3 individual experiments, using 4–7 mice/group. Statistical significance was determined using group-wise, one-way ANOVA with multiple-testing correction using the Holm-Sidak method, and α = 0.05. No significant difference was noted for any of the Ag-specific T cell populations.
We next examined the impact of the host environment on naïve T cell expansion by transferring 50,000 naïve P14 TCR-transgenic CD8 T cells obtained from CD45.1+ SPF donor mice into either SPF or CoH CD45.2+ recipient mice 1 day before immunizing with gp33-TriVax (Figure 6A). The frequency and number of CD45.1+ P14 CD8 T cells in the blood was determined 3, 7, 14, and 21 days later, as well as the number of CD45.1+ P14 CD8 T cells and endogenous CD45.2+ gp33-specific CD8 T cells in the spleens 21 days after immunization. Interestingly, P14 T cell expansion on day 7 post-immunization was considerably blunted in the CoH recipients (Figure 6B–C). Moreover, the reduced number of P14 T cell numbers in the spleens of CoH recipient mice (vs. SPF recipient mice) on day 21 after immunization mirrored that for the endogenous gp33-specific CD8 T cell populations present in the same SPF and CoH mice (Figure 6D–F). Because of the inability to cohouse P14 mice, we were unable to perform the desired reciprocal transfers with the P14 T cells. Instead, we transferred 4 × 106 bulk splenocytes from SPF or CoH CD45.1+ mice and transferred them to either SPF or CoH CD45.2+ recipients 1 day before B8R-TriVax immunization (Figure 6G). This approach allowed us to use B8R:Kb tetramers to identify the transferred CD45.1+ B8R-specific CD8 T cells that responded to immunization. The results clearly showed equivalent expansion of SPF- and CoH-derived CD45.1+ B8R-specific CD8 T cells in SPF recipients, while the expansion of either donor CD45.1+ B8R-specific CD8 T cell populations was significantly reduced in CoH recipients (Figure 6H–I). Thus, data in Figures 5 and 6 together suggest the differences in T cell expansion/differentiation seen between SPF and CoH mice after TriVax immunization are not the result of intrinsic T cell differences and are instead governed by T cell-extrinsic factors.
FIGURE 6. Differential expansion of SPF-derived P14 TCR-Tg CD8 T cells in TriVax-immunized SPF and CoH mice.
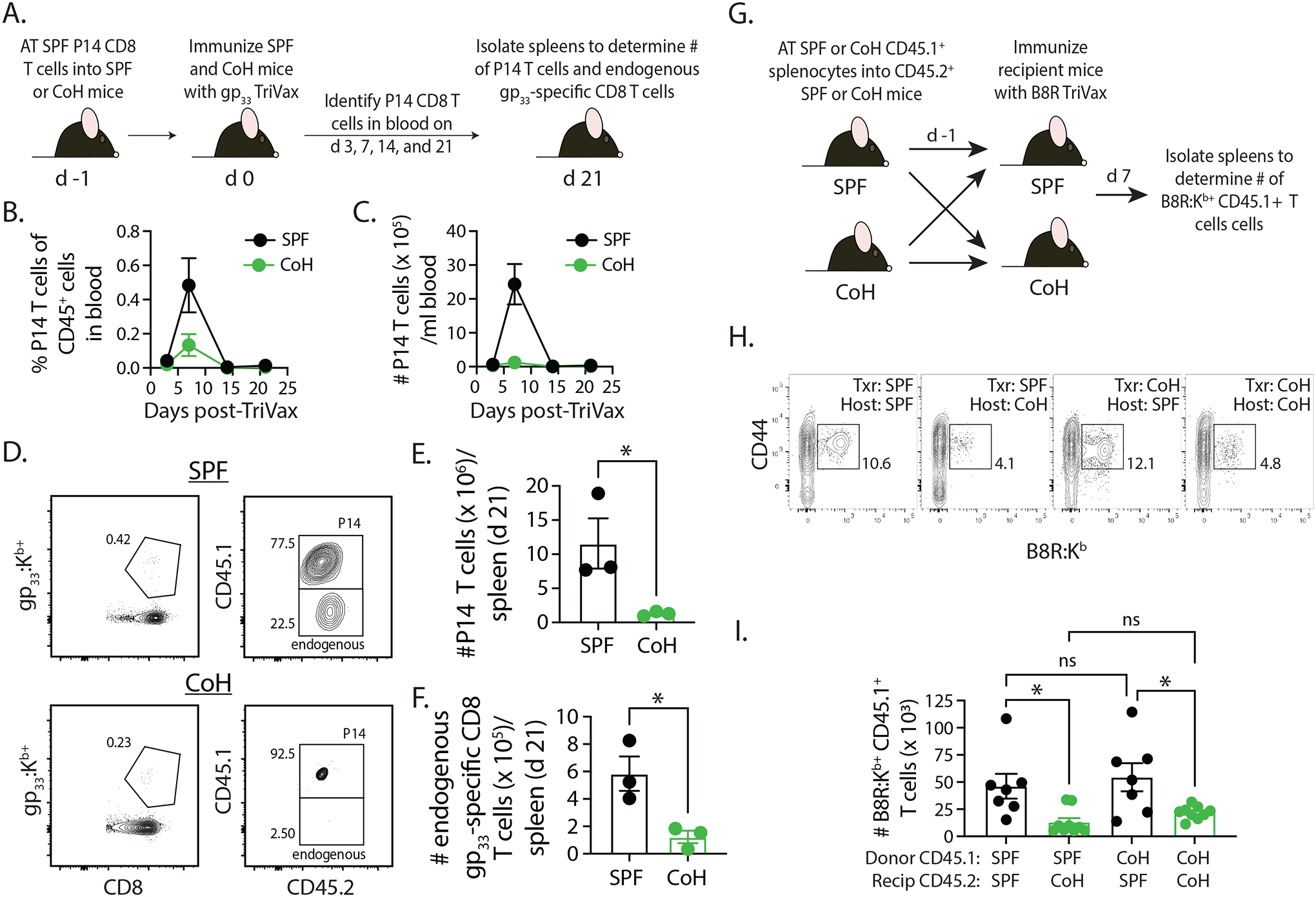
A. Experimental design – CD45.1+ P14 T cells were isolated from the spleens of P14 mice housed under SPF conditions, and then 5000 P14 CD8 T cells were adoptively transferred into SPF or CoH CD45.2+ B6 mice. The following day, the SPF and CoH recipient mice were immunized with gp33-TriVax. Blood was collected on days 3, 7, 14, and 21 post-immunization to determine the (B) frequency and (C) number of CD45.1+ P14 CD8 T cells among CD45.2+ cells. D-F. In addition, spleens were isolated on day 21 post-immunization to determine the number of (E) CD45.1+ P14 CD8 T cells and (F) endogenous CD45.2+ gp33-specific CD8 T cells. Representative flow plots are shown in panel D. * p ≤ 0.05. Data are representative of 2 technical replicates, with 3 mice/group in each experiment. G. Experimental design – 4 × 106 bulk splenocytes from SPF or CoH CD45.1+ B6.SJL mice were adoptively transferred into SPF or CoH CD45.2+ B6 mice. The following day, the SPF and CoH recipient B6 mice were immunized with B8R-TriVax. H-I. Spleens were isolated from the recipient B6 mice 7 days after immunization to determine the number of CD45.1+ B8R-specific CD8 T cells. Representative flow plots are shown in panel H. * p ≤ 0.05. Data are combined from 2 technical replicates, with a total of 7–9 mice/group.
Despite CoH mice having increased numbers of splenic dendritic cells, IL-27p28 production from XCR1+ cDC1 is dramatically reduced after TriVax immunization.
Optimal CD8 and CD4 T cell responses require the right combination of signaling pathways activated via Ag stimulation though the TCR, CD80/86-CD28 costimulation, and cytokines (41,42). To evaluate the potential impact of changes within the dendritic cell (DC) compartment in CoH mice on the reduced capacity of TriVax immunization to stimulate a naïve T cell response, we first determined the number of XCR1+ cDC1, SIRPα+ cDC2, and Siglec H+ plasmacytoid DC (pDC) in the spleens of SPF and CoH mice. Interestingly, each of these DC populations were expanded in CoH mice (Figure 7A). We also assessed CD40, CD80, and CD86 expression on these DC subsets, finding no differences on XCR1+ cDC1, increased CD86 gMFI on CoH SIRPα+ cDC2, and increased CD80 and CD86 gMFI on CoH Siglec H+ pDC (Figure 7B–D). Several recent publications from the Kedl laboratory have described the importance of IL-27p28-expressing XCR1+ cDC1 cells in defining the magnitude of T cell expansion following TriVax immunization (18,43). Thus, we next assessed the extent of IL-27p28 production by XCR1+ cDC1 cells from TriVax-immunized SPF and CoH mice. We did not detect any IL-27p28-producing XCR1+ cDC1 cells in either SPF or CoH mice at baseline, but there was a substantial increase in IL-27p28 production by these cells in SPF mice 6 h after TriVax immunization (Figure 7E–H). Strikingly, this IL-27p28 production by XCR1+ cDC1 was not seen in TriVax-immunized CoH mice. IL-27p28 is part of the IL-12 cytokine family, so we also measured the expression of IL-12p40 by the XCR1+ cDC1 cells. In contrast to the lack of IL-27p28-producing XCR1+ cDC1 cells in TriVax-immunized CoH mice, we found SPF and CoH mice had similar frequencies and numbers of IL-12p40-expressing XCR1+ cDC1 (Figure 7I–J).
FIGURE 7. TriVax-immunized CoH mice have fewer IL-27p28-producing XCR1+ cDC1 cells.
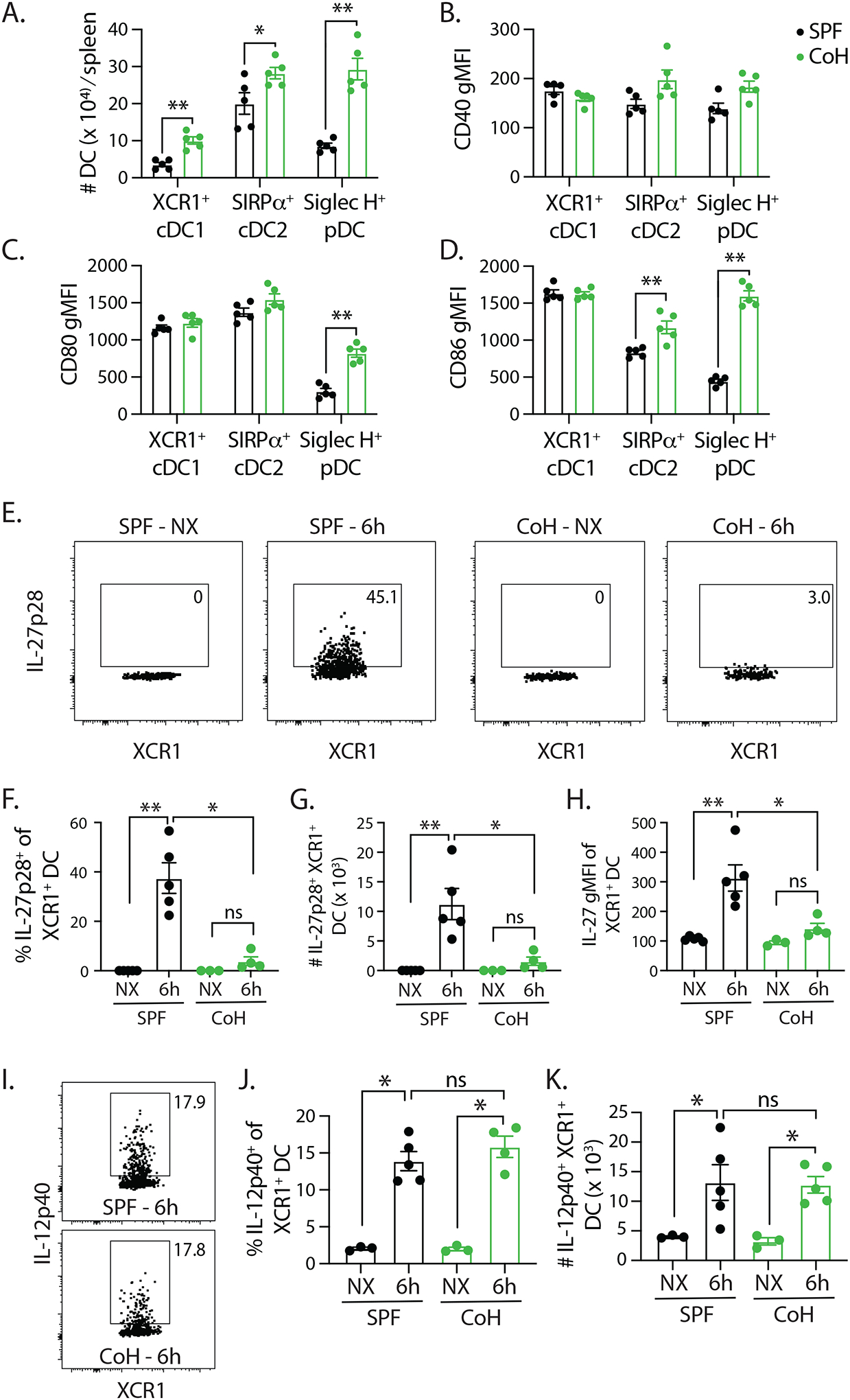
A. Baseline numbers of XCR1+ cDC1, SIRPα+ cDC2, and Siglec H+ pDC in SPF and CoH mice. B-D. CD40, CD80, and CD86 expression was measured on each of these cell population, and the geometric mean fluorescence intensity (gMFI) is graphed. ** p ≤ 0.01, and * p ≤ 0.05. Data are representative of 2 technical replicates, with 3–5 mice/group in each experiment. SPF and CoH mice were immunized with B8R TriVax. Spleens were isolated 6 h later (as well as from unimmunized (NX) mice), and the expression of (E-H) IL-27p28 or (I-K) IL-12p40 by XCR1+ cDC1 cells was examined by flow cytometry. Representative flow plots show data from unimmunized and TriVax-immunized SPF and CoH mice. These data were used to determine the frequency and number of cytokine expressing XCR1+ cDC1 cells, as well as the IL-27p28 gMFI on the XCR1+ cDC1 cells.
We were intrigued by the limited expression of IL-27p28 by the XCR1+ cDC1 cells, and wondered whether administration of recombinant IL-27p28:EBI3 complex shortly after TriVax immunization would boost T cell expansion in CoH mice. Using a protocol similar to that described by Pennock et al. (44), we saw a significant increase in the frequency and number of B8R-specific CD8 T cells and 2W1S-specific CD4 T cells in CoH mice that were given a single dose of IL-27p28:EBI3 complex 2 h after immunization compared to CoH mice that did not receive exogenous IL-27 (Figure 8). Together with the data in Figure 7, these data suggest the limited capacity of TriVax immunization to expand Ag-specific CD8 T cells in CoH mice is due (in part) to the reduced frequency/number of IL-27-producing XCR1+ cDC1, as well as the reduced amount of IL-27 produced on a per cell basis (based on gMFI). Moreover, these data further support the idea that the defect restricting vaccine-driven T cell expansion in CoH mice is T cell extrinsic.
FIGURE 8. Administration of recombinant IL-27p28:EBI3 complex boosts CD8 and CD4 T cell expansion in TriVax-immunized CoH mice.
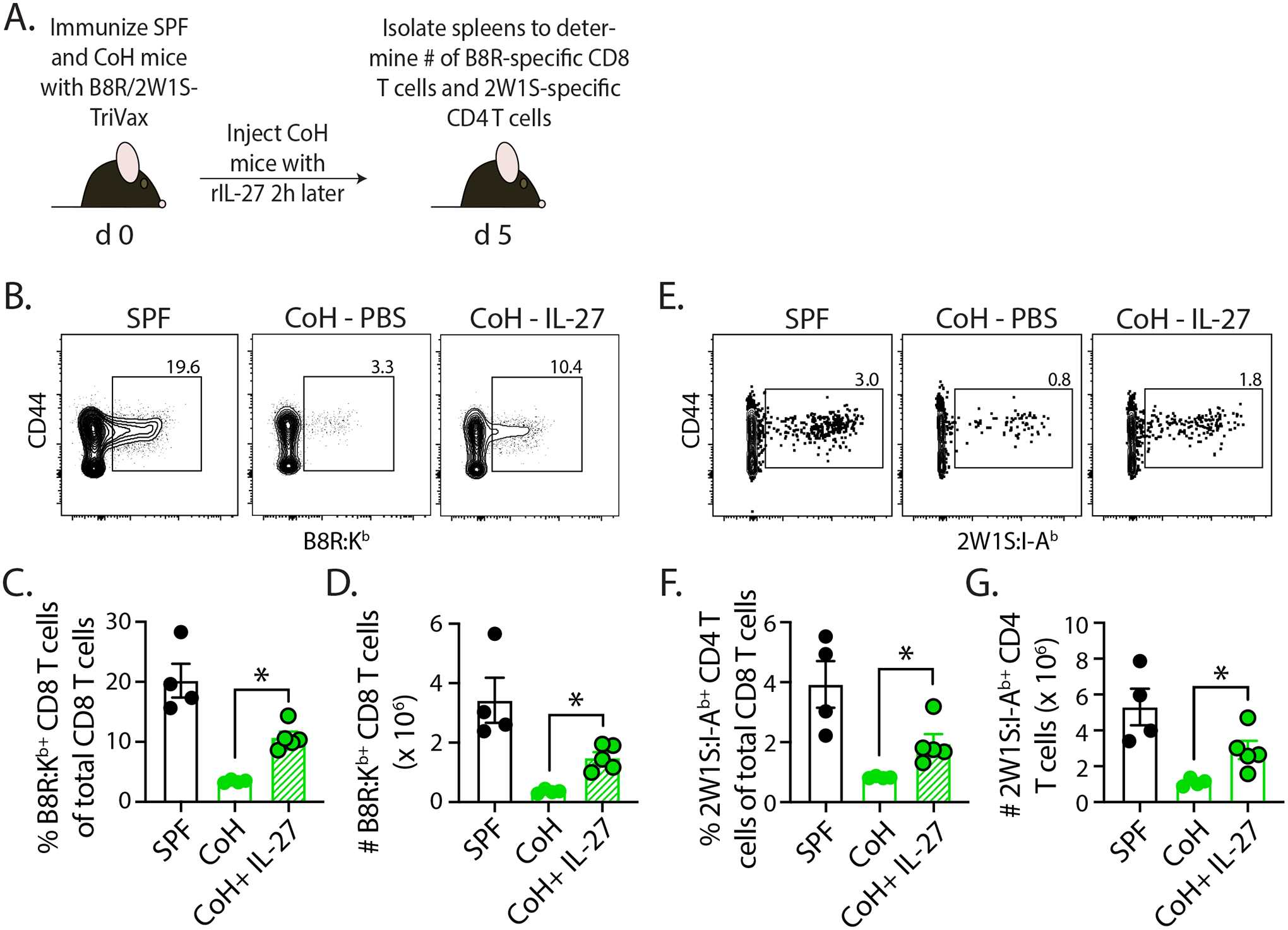
A. Experimental design – SPF and CoH B6 mice were immunized with B8R/2W1S-TriVax. Some of the CoH mice then received a single dose (250 μg i.v.) of recombinant IL-27p28:EBI3 complex 2 h later. After 5 days, spleens were collected to quantitate the frequency and number of (B-D) B8R-specific CD8 and (E-G) 2W1S-specific CD4 T cells. * p ≤ 0.05. Data are representative from 2 technical replicates, with a total of 4–5 mice/group.
DISCUSSION
Vaccines that elicit robust adaptive immune responses using synthetic peptides combined with adjuvants have generated impressive preclinical data inducing immunity against tumors and pathogens. Prophylactic vaccination for infectious disease typically stimulates the production of neutralizing antibodies specific for a conserved region of the microbe. Cancer vaccines can be given prophylactically or therapeutically to induce a robust neutralizing antibody response against immunodominant viral antigens (e.g., L1 proteins from human papillomavirus) or a tumor Ag-specific T cell response, respectively. Regardless of the target of the induced immune response, vaccine efficacy relies on many factors, including the route of delivery, adjuvant, and immunogen used. Interest in peptide subunit vaccines (especially for cancer) has increased in recent years due to detailed knowledge of immunogenic tumor Ag and translation of cancer immunology from the bench to the clinic. Yet, objective response rates have been underwhelming in cancer vaccine clinical trials, and few cancer vaccines have progressed to Phase III testing (45–48).
Recent work with peptide subunit immunization has shown differing requirements for supporting factors (such as IL-27, IL-15, and Eomes) when comparing priming of vaccine-stimulated T cells vs. infection-stimulated T cells (44). However, it is important to recall that most of the preclinical research on peptide subunit immunization has been conducted in SPF mice, which have limited circulating inflammatory cytokines and are more immunologically naïve compared to pet-shop or wild mice (12,13,15,16). With that in mind, we were interested to determine to what extent the modified immune environment in laboratory mice that had been cohoused with pet store mice to induce immune system maturation still supports robust naïve T cell responses following peptide subunit immunization. The data presented herein provide evidence to suggest reduced ability of peptide subunit immunization to prime a naïve CD8 or CD4 T cell response in microbially-experienced ‘dirty’ mice (compared to traditional SPF-housed laboratory mice) stemming, in part, from reduced IL-27p28 production from XCR1+ cDC1 cells. Consequently, antitumor immunity in peptide subunit immunized dirty mice was less effective in controlling experimental tumor growth compared to that in SPF mice, which is consistent with the limited success peptide subunit cancer vaccines in the clinical setting.
Over their lifetime, humans are exposed to numerous commensal and pathogenic microbes. This microbial exposure, along with vaccination, results in an adaptive immune system primed to respond rapidly and vigorously against subsequent microbial challenge. In contrast, the immune systems of most laboratory mice used in biomedical research are composed primarily of naïve cells, largely resulting from the SPF housing conditions used in most animal facilities (11,49). There is no question that the implementation of SPF housing has had a positive impact on preclinical research (e.g., improving experimental reproducibility), but data published in recent years showing how the maturation of the immune system in laboratory mice with a normalized microbial experience (i.e., exposure to natural pathogenic and commensal microbes present in the environment or transferred from other mice) can significantly affect responses to new challenges demonstrate the benefit of including animals with a matured immune system (like that seen in adult humans) in preclinical experimentation (12–16). With this in mind, we were very intrigued by the lack of antitumor immunity in CoH mice after peptide subunit immunization (Figure 1). Interestingly, these data are consistent with the subdued antibody responses seen in sequentially-infected or CoH mice after yellow fever vaccine or influenza virus vaccine administration, respectively (14,50). Subsequent investigation found clear reductions in CD8 and CD4 T cell expansion, differentiation into memory populations, and percentage/number of cells capable of producing cytokine after restimulation in TriVax-immunized CoH mice, even though naïve precursor T cell numbers were equivalent to that found in SPF mice. We reasoned the reduced T cell priming in cohoused mice could be due to either T cell intrinsic or extrinsic factors. Limited expansion of adoptively transferred TCR-transgenic or Ag-specific CD8 T cells obtained from SPF donor mice in CoH recipients pointed to T cell-extrinsic differences that throttled the potential for T cell expansion.
Cohousing laboratory mice with pet store mice results in a constitutive increase in many adaptive and innate immune cell populations (12–16), but the data in Figure 7 are the first (to our knowledge) to show increased numbers of multiple DC subsets in the spleen. In many ways, it makes sense for there to be an increased DC load in CoH mice to deal with the increased amount of microbial Ag presentation needed to prime the various pathogen- and commensal-specific T cell populations needed to clear these microbes. Similarly, the modest (but significant) differences in costimulatory molecule expression on the SIRPα+ cDC2 and Siglec H+ pDC from CoH mice should be expected because of the elevated steady-state inflammation present, since proinflammatory cytokines (e.g., IFNγ and TNFα) can increase CD80 and CD86 expression on DC (51). Based on these parameters – more XCR1+ cDC1, SIRPα+ cDC2, and Siglec H+ pDC in the spleens of CoH mice at baseline, and higher costimuatory molecule expression – it would have been reasonable to predict CoH mice would exhibit similar (or even more robust) naïve T cell expansion/differentiation/function compared to SPF mice. However, the most profound finding from our DC analyses was the inability of XCR1+ cDC1 to make IL-27p28 after TriVax immunization. Work from the Kedl lab has identified IL-27 as being key for supporting T cell responses following peptide subunit vaccination (18,43,44). T cell dependence on IL-27 is thought to regulate the metabolic state of vaccine-elicited T cells (52), and many factors, including several TLR agonists and IFNγ induce the production of IL-27 (53). Interestingly, we have previously noted the reduced abundance of Tlr3 transcripts in splenic adherent myeloid cells, which include dendritic cells (54), from CoH mice compared to SPF mice (13). Altered TLR3 expression could be a means to explain the reduced stimulatory capacity of the polyI:C-containing TriVax immunization in CoH mice, but it is important to recall that we also saw reduced T cell expansion in CoH mice immunized with peptide combined with CFA or Alhydrogel (Figure 2F–G). These data further demonstrate the need for additional investigation into why XCR1+ cDC1 fail to make IL-27 in CoH mice.
Increasing evidence supports the notion that mice exposed to diverse microbes exhibit substantial changes in the immune response to pathogens. Moreover, these changes in the baseline immune cell activation profiles correlate more closely with human immune profiles than that observed in SPF mice, suggesting the potential use of ‘dirty’ mice as an intermediate preclinical step between traditional SPF mice and human testing. These changes can benefit the host in some cases, but they can result in detrimental outcomes in other settings. Indeed, epidemiological data show increased vaccine efficacy in humans living in more developed parts of the world where microbial encounter/infections are less frequent compared to that observed in developing countries (55–57). Our data suggest caution in the interpretation that peptide subunit/adjuvant vaccines will induce robust T cell responses when only tested preclinically using hosts with a limited microbial history (i.e., SPF mice), and point to the need for additional analysis of vaccine efficacy in dirty mice that includes an assessment of dendritic cell populations presenting the antigen of interest.
Supplementary Material
KEY POINTS.
Vaccine-induced T cell expansion is blunted in dirty mice compared to SPF mice.
XCR1+ DC from dirty mice fail to produce IL-27p28 after vaccination.
Exogenous IL-27 restores T cell expansion in dirty mice.
ACKNOWLEDGEMENTS
We thank the members of our laboratories for technical assistance and helpful discussions. We also thank the University of Minnesota Flow Cytometry Resource Facility, CFI Dirty Mouse Colony, and BSL-3 Program for support.
This works was supported by NIH Grants AI155468 (S.E.H.), T32AI007313 (F.V.S.), GM115462 (T.S.G.), GM140881 (T.S.G.), and a Department of Veterans Affairs Merit Review Award I01BX001324 (T.S.G.). T.S.G. is the recipient of a Research Career Scientist award (IK6BX006192) from the Department of Veterans Affairs.
REFERENCES
- 1.Hand TW, and Kaech SM. 2009. Intrinsic and extrinsic control of effector T cell survival and memory T cell development. Immunol. Res 45: 46–61. [DOI] [PubMed] [Google Scholar]
- 2.Jameson SC, and Masopust D. 2018. Understanding Subset Diversity in T Cell Memory. Immunity 48: 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mueller SN, Gebhardt T, Carbone FR, and Heath WR. 2013. Memory T cell subsets, migration patterns, and tissue residence. Annu. Rev. Immunol 31: 137–161. [DOI] [PubMed] [Google Scholar]
- 4.Kumai T, Yamaki H, Kono M, Hayashi R, Wakisaka R, and Komatsuda H. 2022. Antitumor Peptide-Based Vaccine in the Limelight. Vaccines (Basel) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma M, Liu J, Jin S, and Wang L. 2020. Development of tumour peptide vaccines: From universalization to personalization. Scand. J. Immunol 91: e12875. [DOI] [PubMed] [Google Scholar]
- 6.Ghattas M, Dwivedi G, Lavertu M, and Alameh MG. 2021. Vaccine Technologies and Platforms for Infectious Diseases: Current Progress, Challenges, and Opportunities. Vaccines (Basel) 9: 1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bastola R, Noh G, Keum T, Bashyal S, Seo JE, Choi J, Oh Y, Cho Y, and Lee S. 2017. Vaccine adjuvants: smart components to boost the immune system. Arch. Pharm. Res 40: 1238–1248. [DOI] [PubMed] [Google Scholar]
- 8.Masopust D, Sivula CP, and Jameson SC. 2017. Of Mice, Dirty Mice, and Men: Using Mice To Understand Human Immunology. J. Immunol 199: 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamilton SE, Badovinac VP, Beura LK, Pierson M, Jameson SC, Masopust D, and Griffith TS. 2020. New Insights into the Immune System Using Dirty Mice. J. Immunol 205: 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huggins MA, Jameson SC, and Hamilton SE. 2019. Embracing microbial exposure in mouse research. J. Leukoc. Biol 105: 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foster HL 1959. Housing of disease-free vertebrates. Ann. N. Y. Acad. Sci 78: 80–88. [DOI] [PubMed] [Google Scholar]
- 12.Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, Thompson EA, Fraser KA, Rosato PC, Filali-Mouhim A, Sekaly RP, Jenkins MK, Vezys V, Haining WN, Jameson SC, and Masopust D. 2016. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532: 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huggins MA, Sjaastad FV, Pierson M, Kucaba TA, Swanson W, Staley C, Weingarden AR, Jensen IJ, Danahy DB, Badovinac VP, Jameson SC, Vezys V, Masopust D, Khoruts A, Griffith TS, and Hamilton SE. 2019. Microbial Exposure Enhances Immunity to Pathogens Recognized by TLR2 but Increases Susceptibility to Cytokine Storm through TLR4 Sensitization. Cell Rep 28: 1729–1743 e1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reese TA, Bi K, Kambal A, Filali-Mouhim A, Beura LK, Burger MC, Pulendran B, Sekaly RP, Jameson SC, Masopust D, Haining WN, and Virgin HW. 2016. Sequential Infection with Common Pathogens Promotes Human-like Immune Gene Expression and Altered Vaccine Response. Cell Host Microbe 19: 713–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosshart SP, Herz J, Vassallo BG, Hunter A, Wall MK, Badger JH, McCulloch JA, Anastasakis DG, Sarshad AA, Leonardi I, Collins N, Blatter JA, Han SJ, Tamoutounour S, Potapova S, Foster St Claire MB, Yuan W, Sen SK, Dreier MS, Hild B, Hafner M, Wang D, Iliev ID, Belkaid Y, Trinchieri G, and Rehermann B. 2019. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 365: eaaw4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosshart SP, Vassallo BG, Angeletti D, Hutchinson DS, Morgan AP, Takeda K, Hickman HD, McCulloch JA, Badger JH, Ajami NJ, Trinchieri G, Pardo-Manuel de Villena F, Yewdell JW, and Rehermann B. 2017. Wild Mouse Gut Microbiota Promotes Host Fitness and Improves Disease Resistance. Cell 171: 1015–1028 e1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahonen CL, Doxsee CL, McGurran SM, Riter TR, Wade WF, Barth RJ, Vasilakos JP, Noelle RJ, and Kedl RM. 2004. Combined TLR and CD40 triggering induces potent CD8+ T cell expansion with variable dependence on type I IFN. J. Exp. Med 199: 775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kilgore AM, Welsh S, Cheney EE, Chitrakar A, Blain TJ, Kedl BJ, Hunter CA, Pennock ND, and Kedl RM. 2018. IL-27p28 Production by XCR1(+) Dendritic Cells and Monocytes Effectively Predicts Adjuvant-Elicited CD8(+) T Cell Responses. Immunohorizons 2: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pierson M, Merley A, and Hamilton SE. 2021. Generating Mice with Diverse Microbial Experience. Curr. Protoc 1: e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, and Jenkins MK. 2007. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity 27: 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moon JJ, Chu HH, Hataye J, Pagan AJ, Pepper M, McLachlan JB, Zell T, and Jenkins MK. 2009. Tracking epitope-specific T cells. Nat. Protoc 4: 565–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pepper M, Pagan AJ, Igyarto BZ, Taylor JJ, and Jenkins MK. 2011. Opposing signals from the Bcl6 transcription factor and the interleukin-2 receptor generate T helper 1 central and effector memory cells. Immunity 35: 583–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pagan AJ, Pepper M, Chu HH, Green JM, and Jenkins MK. 2012. CD28 promotes CD4+ T cell clonal expansion during infection independently of its YMNM and PYAP motifs. J. Immunol 189: 2909–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nelson RW, McLachlan JB, Kurtz JR, and Jenkins MK. 2013. CD4+ T cell persistence and function after infection are maintained by low-level peptide:MHC class II presentation. J. Immunol 190: 2828–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho HI, and Celis E. 2009. Optimized peptide vaccines eliciting extensive CD8 T-cell responses with therapeutic antitumor effects. Cancer Res 69: 9012–9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee S, Stokes KL, Currier MG, Sakamoto K, Lukacs NW, Celis E, and Moore ML. 2012. Vaccine-elicited CD8+ T cells protect against respiratory syncytial virus strain A2-line19F-induced pathogenesis in BALB/c mice. J. Virol 86: 13016–13024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Assudani D, Cho HI, DeVito N, Bradley N, and Celis E. 2008. In vivo expansion, persistence, and function of peptide vaccine-induced CD8 T cells occur independently of CD4 T cells. Cancer Res 68: 9892–9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barrios K, and Celis E. 2012. TriVax-HPV: an improved peptide-based therapeutic vaccination strategy against human papillomavirus-induced cancers. Cancer Immunol. Immunother 61: 1307–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harty JT, and Badovinac VP. 2008. Shaping and reshaping CD8+ T-cell memory. Nat. Rev. Immunol 8: 107–119. [DOI] [PubMed] [Google Scholar]
- 30.Martin MD, and Badovinac VP. 2018. Defining Memory CD8 T Cell. Front. Immunol 9: 2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pepper M, and Jenkins MK. 2011. Origins of CD4(+) effector and central memory T cells. Nat. Immunol 12: 467–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor JJ, and Jenkins MK. 2011. CD4+ memory T cell survival. Curr. Opin. Immunol 23: 319–323. [DOI] [PubMed] [Google Scholar]
- 33.Renkema KR, Huggins MA, Borges da Silva H, Knutson TP, Henzler CM, and Hamilton SE. 2020. KLRG1(+) Memory CD8 T Cells Combine Properties of Short-Lived Effectors and Long-Lived Memory. J. Immunol 205: 1059–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruterbusch M, Pruner KB, Shehata L, and Pepper M. 2020. In Vivo CD4(+) T Cell Differentiation and Function: Revisiting the Th1/Th2 Paradigm. Annu. Rev. Immunol 38: 705–725. [DOI] [PubMed] [Google Scholar]
- 35.Hori S, Nomura T, and Sakaguchi S. 2003. Control of regulatory T cell development by the transcription factor Foxp3. Science 299: 1057–1061. [DOI] [PubMed] [Google Scholar]
- 36.Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, Wang YH, and Dong C. 2009. Bcl6 mediates the development of T follicular helper cells. Science 325: 1001–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, and Glimcher LH. 2000. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100: 655–669. [DOI] [PubMed] [Google Scholar]
- 38.Badovinac VP, Haring JS, and Harty JT. 2007. Initial T cell receptor transgenic cell precursor frequency dictates critical aspects of the CD8(+) T cell response to infection. Immunity 26: 827–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marzo AL, Klonowski KD, Le Bon A, Borrow P, Tough DF, and Lefrancois L. 2005. Initial T cell frequency dictates memory CD8+ T cell lineage commitment. Nat. Immunol 6: 793–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Obar JJ, Khanna KM, and Lefrancois L. 2008. Endogenous naive CD8+ T cell precursor frequency regulates primary and memory responses to infection. Immunity 28: 859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Curtsinger JM, and Mescher MF. 2010. Inflammatory cytokines as a third signal for T cell activation. Curr. Opin. Immunol 22: 333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith-Garvin JE, Koretzky GA, and Jordan MS. 2009. T cell activation. Annu. Rev. Immunol 27: 591–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kilgore AM, Pennock ND, and Kedl RM. 2020. cDC1 IL-27p28 Production Predicts Vaccine-Elicited CD8(+) T Cell Memory and Protective Immunity. J. Immunol 204: 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pennock ND, Gapin L, and Kedl RM. 2014. IL-27 is required for shaping the magnitude, affinity distribution, and memory of T cells responding to subunit immunization. Proc. Natl. Acad. Sci. U. S. A 111: 16472–16477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abd-Aziz N, and Poh CL. 2022. Development of Peptide-Based Vaccines for Cancer. J. Oncol 2022: 9749363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hollingsworth RE, and Jansen K. 2019. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 4: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumai T, Yamaki H, Kono M, Hayashi R, Wakisaka R, and Komatsuda H. 2022. Antitumor Peptide-Based Vaccine in the Limelight. Vaccines (Basel) 10: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosenberg SA, Yang JC, and Restifo NP. 2004. Cancer immunotherapy: moving beyond current vaccines. Nat. Med 10: 909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lane-Petter W 1962. Provision of pathogen-free animals. Proc. R. Soc. Med 55: 253–256. [PubMed] [Google Scholar]
- 50.Fiege JK, Block KE, Pierson MJ, Nanda H, Shepherd FK, Mickelson CK, Stolley JM, Matchett WE, Wijeyesinghe S, Meyerholz DK, Vezys V, Shen SS, Hamilton SE, Masopust D, and Langlois RA. 2021. Mice with diverse microbial exposure histories as a model for preclinical vaccine testing. Cell Host Microbe 29: 1815–1827 e1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Banchereau J, and Steinman RM. 1998. Dendritic cells and the control of immunity. Nature 392: 245–252. [DOI] [PubMed] [Google Scholar]
- 52.Klarquist J, Chitrakar A, Pennock ND, Kilgore AM, Blain T, Zheng C, Danhorn T, Walton K, Jiang L, Sun J, Hunter CA, D’Alessandro A, and Kedl RM. 2018. Clonal expansion of vaccine-elicited T cells is independent of aerobic glycolysis. Sci. Immunol 3: eaas9822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Molle C, Nguyen M, Flamand V, Renneson J, Trottein F, De Wit D, Willems F, Goldman M, and Goriely S. 2007. IL-27 synthesis induced by TLR ligation critically depends on IFN regulatory factor 3. J. Immunol 178: 7607–7615. [DOI] [PubMed] [Google Scholar]
- 54.Steinman RM, Kaplan G, Witmer MD, and Cohn ZA. 1979. Identification of a novel cell type in peripheral lymphoid organs of mice. V. Purification of spleen dendritic cells, new surface markers, and maintenance in vitro. J Exp Med 149: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Levine MM 2010. Immunogenicity and efficacy of oral vaccines in developing countries: lessons from a live cholera vaccine. BMC Biol 8: 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lopman BA, Pitzer VE, Sarkar R, Gladstone B, Patel M, Glasser J, Gambhir M, Atchison C, Grenfell BT, Edmunds WJ, Kang G, and Parashar UD. 2012. Understanding reduced rotavirus vaccine efficacy in low socio-economic settings. PLoS One 7: e41720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zimmermann P, and Curtis N. 2019. Factors That Influence the Immune Response to Vaccination. Clin. Microbiol. Rev 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.