

Abstract
A growing body of evidence suggests that dysbiosis of the human gut microbiota is associated with neurodegenerative diseases like Alzheimer’s disease (AD) via neuroinflammatory processes across the microbiota-gut-brain axis. The gut microbiota affects brain health through the secretion of toxins and short-chain fatty acids, which modulates gut permeability and numerous immune functions. Observational studies indicate that AD patients have reduced microbiome diversity, which could contribute to the pathogenesis of the disease. Uncovering the genetic basis of microbial abundance and its effect on AD could suggest lifestyle changes that may reduce an individual’s risk for the disease. Using the largest genome-wide association study of gut microbiota genera from the MiBioGen consortium, we used polygenic risk score (PRS) analyses with the “best-fit” model implemented in PRSice-2 and determined the genetic correlation between 119 genera and AD in a discovery sample (ADc12 case/control: 1278/1293). To confirm the results from the discovery sample, we next repeated the PRS analysis in a replication sample (GenADA case/control: 799/778) and then performed a meta-analysis with the PRS results from both samples. Finally, we conducted a linear regression analysis to assess the correlation between the PRSs for the significant genera and the APOE genotypes. In the discovery sample, 20 gut microbiota genera were initially identified as genetically associated with AD case/control status. Of these 20, three genera (Eubacterium fissicatena as a protective factor, Collinsella, and Veillonella as a risk factor) were independently significant in the replication sample. Meta-analysis with discovery and replication samples confirmed that ten genera had a significant correlation with AD, four of which were significantly associated with the APOE rs429358 risk allele in a direction consistent with their protective/risk designation in AD association. Notably, the proinflammatory genus Collinsella, identified as a risk factor for AD, was positively correlated with the APOE rs429358 risk allele in both samples. Overall, the host genetic factors influencing the abundance of ten genera are significantly associated with AD, suggesting that these genera may serve as biomarkers and targets for AD treatment and intervention. Our results highlight that proinflammatory gut microbiota might promote AD development through interaction with APOE. Larger datasets and functional studies are required to understand their causal relationships.
Subject terms: Genetics, Neuroscience
Introduction
Alzheimer’s disease (AD), the most common form of dementia, is a neurodegenerative disorder characterized by a multitude of pathological and clinical hallmarks such as a progressive decline in cognitive function and the buildup of toxic β-amyloid and tau proteins1,2. Due to the growing elderly population worldwide, the number of individuals with dementia is projected to reach 150 million globally by the year 20503. Despite this growing burden on world health, the mechanisms underlying the disease pathology are not fully understood, impeding the development of optimally effective treatments4. Neuroinflammation has emerged as a key feature of AD with mechanistic and treatment implications due to the central role of microglia and inflammation in brain health5,6. There remains an urgent need to understand the genetic risk factors and pathological basis of neuroinflammation in AD so that individuals with a higher risk can be identified for earlier intervention.
Recently, an association between dysbiosis of the gut microbiome and neuroinflammation has been hypothesized to drive AD. The gut microbiota comprises a complex community of microorganism species that reside in our gastrointestinal ecosystem; alterations in the gut microbiota have been reported to influence not only various gut disorders but also brain disorders such as AD7,8. The human gut microbiota has been suggested to modulate brain function and behavior via the microbiota-gut-brain axis (MGBA), a bidirectional communication system connecting neural, immune, endocrine, and metabolic pathways9. Observational studies across multiple countries show reductions in gut microbiota diversity in AD patients compared to cognitively normal controls10–12. Current research indicates that bacteria populating the gut microbiota are capable of releasing lipopolysaccharide (LPS) and amyloids, which may induce microglial activation in the brain and contribute to the production of proinflammatory cytokines associated with the pathogenesis of AD13. The secretion of these biomolecules also harms the integrity of the MGBA and blood–brain barrier (BBB), which worsens with increasing dysbiosis8,14. The composition of the human gut microbiota and risk for AD have been suggested as heritable traits2,15. Apolipoprotein E ε4 (APOE ε4), the most well-established risk gene for AD, has recently been shown to correlate with microbiome composition in humans and mouse models of AD16–18. However, few studies have explored the correlation between APOE alleles and microbiome taxa at the human genomic level. In this study, we aim to determine the genetic correlation between the abundance of gut microbial genera and AD diagnosis. We further investigate whether gut microbial genera are correlated with APOE genotyping.
One promising approach to exploring this relationship is the use of polygenic risk score (PRS) analyses. A PRS is an overall estimate of an individual’s genetic liability for a specific trait. The software PRSice-2 is designed to calculate the PRS of an individual by aggregating and quantifying the effect of many single nucleotide polymorphisms (SNPs) in their genome, which are weighted by the effect sizes of each SNP derived from genome-wide association studies (GWASs)19. This approach has previously been used to explore the genetic relationship between gut microbial abundance and complex traits like bone mineral density, rheumatoid arthritis, and depression20–22. In the present study, we used this approach to determine the genetic relationship between 119 microbial genera and AD diagnosis. With the largest GWAS of the human gut microbiota23, we first conducted PRS analyses in an AD discovery sample to identify the genera genetically correlated with AD. We then verified our results in a replication sample and meta-analysis with the two samples. The correlation between the top ten significant genera and the APOE genotypes was further analyzed by linear regression analysis.
Materials and methods
Study design overview
The overall design of our study is shown in Fig. 1. Briefly, we used PRSice-219 to calculate PRSs for individuals from our discovery sample. PRSs were calculated based on the summary statistics for 119 microbial genera from the MiBioGen consortium. The significant association between genera and AD diagnosis was determined when the “best-fit” PRS model had a Bonferroni-corrected p < 0.00042 (0.05/119 = 0.00042). We then replicated the results in an independent sample. We conducted logistic regression analyses between the PRSs of associated genera and AD diagnosis to generate relative odds ratios (ORs) for meta-analysis. The multivariate logistic regression model was used to determine whether sex, age, and APOE genotypes affected the correlation between the PRSs of the associated genera and AD diagnosis. Furthermore, we conducted a linear regression analysis to evaluate the genetic association between the PRSs of ten significant genera and the APOE genotypes of individuals in our discovery and replication samples. This study was approved by our institutional review board (IRB) at the University of Nevada Las Vegas (UNLV).
Figure 1.

Study design flowchart. In the PRS analysis, “Base” data is used to provide effect sizes for SNPs shared with individuals in the “Target” data. Using PRSice-2, 20 genera were found to be significantly genetically associated with AD diagnosis in the discovery sample. Three genera were validated in the replication sample, and ten were confirmed by a meta-analysis from discovery and replicate samples. Linear regression analyses were used to determine the genetic correlation between the PRSs for ten significant genera and APOE genotyping. Three genera were identified as genetically correlated with APOE rs429358 risk allele C.
Data sources
Microbiome GWAS summary statistics (base data)
For our “base” GWAS data, we obtained summary statistics from the MiBioGen consortium initiative (www.mibiogen.org)23, which is the largest, multi-ethnic genome-wide meta-analysis of the gut microbiome to date (Table 1). The data includes 24 multi-ethnic cohorts comprising 18340 participants. 16S rRNA sequencing profiles from each individual were utilized to characterize their gut microbiota abundance using SILVA as a reference database24. The MiBioGen cohorts used a variety of platforms for genotyping their participants, such as the Illumina OmniExpress, Affymetrix 6.0, and more, which are detailed in the supplements of the original study23. The genotyping data from 23 cohorts were imputed at the Michigan Imputation Server (https://imputationserver.sph.umich.edu)25, while another genotyping data were imputed with IMPUTE2 software (v2.3.2)23. From the phylum to genus level, 31 loci were associated with gut microbiota taxa abundance (mbQTL, n = 20) or the presence/absence of taxa (mbBTL, n = 11) at the genome-wide significant threshold (p < 5.0 × 10−8)23. The SNP effect sizes reported in the mbQTL GWAS summary statistics represent how the host genetic loci affect the relative abundance of each microbiome taxa (mbQTLs)23. In the present study, we limited our analyses to the mbQTL summary statistics from the 119 microbial genera, as 16S rRNA sequencing correlates more accurately with the functional role of gut microbiota at lower taxonomic levels26.
Table 1.
Information for studies used in our analyses.
| Variable | Consortium or study | PMID | Year | Total sample size | Ethnic groups |
|---|---|---|---|---|---|
| Gut microbiota | MiBioGen | 33462485 | 2021 | 18340 |
European (13266) Admixed (2571) Hispanic (1097) East Asian (811) Middle-East (481) African American (114) |
| Alzheimer’s disease | ADc12 | 19001172 | 2008 | 2571 |
European (2320) African American (251) |
| dbGaP phs000168.v2.p2 NIA/LOAD consent 1 and 2 | 1278 cases/1293 controls | ||||
| GenADA |
17998437 19013250 |
2008 | 1577 | European (1577) | |
| dbGaP phs000219.v1.p1 | 799 cases/778 controls |
Information for the data used in this study. The Multi-ethnic cohorts of the MiBioGen study include 16 European cohorts (n = 13266), one Middle Eastern cohort (n = 481), one East Asian cohort (n = 811), one American Hispanic/Latin cohort (n = 1097), one African-American cohort (n = 114), and four cohorts of multi-ancestry individuals (n = 2571). The Multi-ethnic cohort of the NIA/LOAD study contains African-American (n = 251) and Caucasian individuals (n = 2320).
AD genotyping data (target data): discovery and replication samples
For AD genotyping data we requested two datasets from dbGaP (https://www.ncbi.nlm.nih.gov/gap/), including the National Institute of Aging/Late-onset Alzheimer’s Disease Study (NIA/LOAD) cohort consents 1 and 2 (ADc12) (dbGaP phs000168.v2.p2)27, and the Multi-Site Collaborative Study for Genotype–Phenotype Associations in Alzheimer’s Disease Study (GenADA) (dbGaP phs000219.v1.p1)28,29. The ADc12 data were used as the discovery sample, and the GenADA data were used as the replication sample.
In this our study, AD cases were considered as any individual with dementia diagnosed with definite, probable, or possible AD at any point in their clinical course, according to the Criteria proposed in 1984 by the National Institute of Neurological and Communicative Disorders and Stroke, and the Alzheimer's Disease and Related Disorders Association (NINCDS-ADRDA)30. Included controls were neurologically evaluated individuals who were age-matched cognitively normal. Unspecified dementia, unconfirmed controls, and controls with other neurological diseases from the original studies were removed for our analyses, resulting in 1278/1293 cases/controls in the discovery sample ADc12, and 799/778 cases/controls in the replication sample GenADA. Demographic characteristics of the ADc12 and Gen/ADA samples are listed in Table 2, along with two major APOE SNP genotype information. More detailed descriptions of the data can be found in previous studies27–29.
Table 2.
Demographic characteristics of the target data (ADc12 and GenADA) with APOE SNP genotyping.
| Discovery Sample (ADc12) | Replication Sample (GenADA) | |||||||
|---|---|---|---|---|---|---|---|---|
| Cases | Controls | Total | Cases | Controls | Total | |||
| Age, mean ± SD | 1278 | 1293 | 2571 | 799 | 778 | 1577 | ||
| 76.57 ± 6.71* | 70.27 ± 10.29 | 72.24 ± 8.41** | 73.40 ± 7.92 | |||||
| Sex (Male/Female) | 443/835 | 471/822 | 914/1657 | 339/460 | 276/502 | 615/962 | ||
| APOE SNP Genotype | rs429358 | T/T, n(%) | 409 (32.0) | 847 (65.5) | 1256 (48.9) | 296 (37.0) | 589 (75.7) | 885 (56.1) |
| T/C, n(%) | 682 (53.4) | 414 (32.0) | 1096 (42.6) | 397 (49.7) | 177 (22.8) | 574 (36.4) | ||
| C/C, n(%) | 187 (14.6) | 32 (2.5) | 219 (8.5) | 106 (13.3) | 12 (1.5) | 118 (7.5) | ||
| C/C, n(%) | 1206 (94.4) | 1128 (87.2) | 2334 (90.8) | 739 (92.5) | 661 (85.0) | 1400 (88.8) | ||
| rs7412 | C/T, n(%) | 71 (5.6) | 159 (12.3) | 230 (8.9) | 60 (7.5) | 113 (14.5) | 173 (11.0) | |
| T/T, n(%) | 1 (0.0) | 6 (5.1) | 7 (0.3) | 0 (0.0) | 4 (0.5) | 4 (0.3) | ||
Both discovery (ADc12) and replication (GenADA) samples were downloaded from dbGaP. Age, mean ± SD. For each case, the “Age” was the age at onset (AAO). For each control, the “Age” was the age at examination (AAE). *p = 5.97 × 10−71 when AAO compared to AAE in the discovery sample. **p = 4.94 × 10−3 when AAO compared to AAE in the replication sample.
The ADc12 genotyping data were originally generated with the Illumina Human610 QuadV1-B platform at 601273 SNPs, and the GenADA genotyping data with the Affymetrix 500k Set (Mapping 250k_NSP and Mapping 250k STY arrays).To maximize genetic variants, we conducted imputation for both discovery and replication samples at the Michigan Imputation Server (minimac4) (https://imputationserver.sph.umich.edu)25. The 1000 Genome Phase 3v531 was used as a reference. After the imputation, standard quality control was performed with the Plink command (--maf 0.01 --hwe 1e-6 --geno 0.01 --mind 0.01)32,33. The final datasets were composed of 2571 individuals with 9997692 SNPs in the discovery sample, and 1577 individuals with 8914585 SNPs in the replication sample.
Polygenic risk score (PRS) analyses via PRSice-2 software
PRSice-2 was mainly designed to calculate PRSs for individuals based on GWAS summary statistics data using the traditional “Clumping + Thresholding” (C + T) approach19. A key assumption of the C + T approach is that the SNPs comprising the PRS are independent of each other, which is controlled by thinning SNPs in linkage disequilibrium (LD) and retaining those that are the most significant (“Clumping”)34. SNPs are then thresholded by their p-values from the summary statistics, and the PRS is calculated for individuals at each threshold (“Thresholding”).
One major application of PRSice-2 is to evaluate the genetic correlation between different traits when provided GWAS summary statistics data from a base trait (base data) and genotyping data from a target trait (target data)19. The PRS itself is a numerical approximation of genetic liability for the base trait in the individuals in the target trait, based on their number of alleles from the target data and effect sizes drawn from the base data for a set of SNPs35. As mentioned above, the base (GWAS) data were from the 119 gut microbiome genera in the MiBioGen consortium study23. The target data were the discovery sample ADc1227 and the replication sample GenADA28,29. In this study, we first calculated PRSs for the 119 gut microbiome genera in the discovery sample ADc12 to determine which genera were genetically correlated with AD diagnosis. The best PRS model for each genus was calculated using the “best-fit” model implemented in the PRSice-2 program. For this purpose, a range of p-value thresholds applied to the base data, as well as the association p-value between the PRSs of each genus and AD diagnosis. For this purpose, a range of p-value thresholds was set from 5 × 10−8 to 1 with an incremental interval of 0.00005 (--interval 0.00005 --lower 5e-08) with LD clumping (--clump-kb 250 kb --clump-p 1.0 --clump-r2 0.1)19. In the discovery sample, a genus was considered significant if its assocation p-value from the “best-fit” model was less than 4.20 × 10−4 (0.05/119 with Bonferroni correction). To validate the significantly associated genera from the discovery sample ADc12, we conducted the same PRS analyses for them in the replication sample GenADA.
Logistic regression and meta-analysis
To further evaluate the overall association of the 20 significantly associated genera from the discovery sample, we z-score normalized the "best-fit" PRSs from both the discovery sample ADc12 and replication sample GenADA. We then performed a simple logistic regression analysis for both samples between the normalized PRSs from the “best-fit” threshold for AD diagnosis using the glm function from the R package stats36.
Next, we conducted a random effects meta-analysis from both samples using the R package metafor v3.8-137. The summary effect estimate of this meta-analysis identified ten significant genera that were used for all future analyses. Forest plots were generated to visualize the overall AD protective and risk effects across the significant genera using the “forestplot” R package38. To compare the normalized PRSs for the ten significant genera between AD cases and controls in the discovery sample, we conducted the unpaired Wilcoxon Rank Sum test with the wilcox.test function in R (v4.2.0)36 and visualized the results with box plots. Box plots were generated using the R program ggplot2 v.3.3.639.
To account for potential confounding variables in our analysis, multivariate logistic regression was conducted between AD diagnosis and z-score normalized PRSs for significant microbial genera using the glm function from the R stats package36. Sex, age, and APOE genotypes (rs429358, rs7412) were used as covariates.
Linear regression analyses between APOE genotypes and PRSs for the ten significant genera
Two APOE SNPs, rs429358 minor allele C and rs7412 major allele C, are well-known risk factors for AD40,41. We performed linear regression analyses to determine the genetic correlation between the two APOE SNPs and the normalized PRSs of the ten significant genera from the meta-analysis. The association was further evaluated by linear regression analysis adjusted for sex and age. All linear regression was performed using the lm function from the R stats package. Box plots with the ANOVA test (state compare means function) were created using the R packages ggplot2 (v3.3.6), ggpubr (v0.4.0), and stats (v0.1.0)36,39.
Statistical analyses
The p-value threshold for significant association in the discovery sample and meta-analysis was set as p < 4.20 × 10−4 (0.05/119 with Bonferroni correction). For the replication sample, one-side significant level p < 0.005 (0.1/20 with Bonferroni correction) was used. For all other statistical analyses, such as linear regression analysis, the ANOVA test, and Wilcoxon Rank Sum test, p < 0.05 was considered significant. The Wilcoxon Rank Sum method, also known as the Mann–Whitney test, is a non-parametric alternative to the unpaired two-sample t-test, which can be used to compare two independent groups of samples without knowing their distribution42. The ANOVA method was utilized to test the association between the normalized PRSs for the ten significant genera and APOE genotypes43.
Ethical approval and consent to participate
We are using the existing data for this study. Informed consent was obtained from all subjects and/or their legal guardian(s) in the original studies. Contributing studies received ethical approval from their respective institutional review boards (IRB). This study was performed per the Declaration of Helsinki and approved by the IRB at the University of Nevada Las Vegas (IRB #00002305, 10/12/2021).
Results
PRSs for ten microbiome genera were significantly associated with AD diagnosis
We first calculated the PRSs for the 119 microbiome genera for each individual from the discovery sample (ADc12) using the PRSice-2 program19. We found that 20 out of the 119 genera were significantly associated with AD diagnosis using the “best-fit” model (p < 4.20 × 10−4) (Table 3). Among these top 20 significant genera, six were identified as likely risk genera and 14 potentially protective genera for AD diagnosis. Risk genera included Alistipes and Bacteroides from the Bacteroidetes phylum, Lachnospira and Veillonella from the Firmicutes phylum, and Collinsella and Sutterella from the Actinobacteria and Pseudomonadota phyla, respectively. The most significant risk genus was Bacteroides (R2 = 0.011, p = 3.32 × 10−6) at the “best-fit” p-value threshold of 0.179 with 71984 SNPs. For protective genera, eleven out of fourteen were from the Firmicutes phylum (Anaerostipes, Candidatus Soleaferrea, Catenibacterium, Eisenbergiella, Eubacterium coprostanoligenes group, Eubacterium fissicatena group, Eubacterium nodatum group, Intestinibacter, Lachnospiraceae UCG-008, Oscillibacter, and Roseburia), two were from Actinobacteria (Adlercreutzia and Gordonibacter), and one was from Bacteroidetes (Prevotella 9). The most significant protective genus was Intestinibacter (R2 = 0.015, p = 1.01 × 10−7) at the “best-fit” p-value threshold of 0.190 with 70292 SNPs.
Table 3.
Association between significant microbiome genera and AD diagnosis from “best-fit” PRSice-2 model.
| Genera | Samples | Threshold | R2 | P | Coeff | SE | SNP# | SampleSize |
|---|---|---|---|---|---|---|---|---|
| Adlercreutzia* | ADc12 | 0.3705 | 0.0068 | 3.57E−04 | − 1870.66 | 524.00 | 101798 | 2571 |
| GenADA | 0.0020 | 0.0036 | 3.91E−02 | − 224.26 | 108.71 | 1684 | 1577 | |
| Alistipes | ADc12 | 0.1676 | 0.0080 | 1.04E−04 | 2581.83 | 665.28 | 67574 | 2571 |
| GenADA | 0.3246 | 0.0023 | 1.03E−01 | − 2953.44 | 1810.61 | 99785 | 1577 | |
| Anaerostipes | ADc12 | 0.0032 | 0.0078 | 1.18E−04 | − 469.15 | 121.86 | 2834 | 2571 |
| GenADA | 0.2989 | 0.0054 | 1.18E−02 | 4134.46 | 1641.01 | 96076 | 1577 | |
| Bacteroides* | ADc12 | 0.1792 | 0.0114 | 3.32E−06 | 3930.96 | 845.35 | 71984 | 2571 |
| GenADA | 0.0009 | 0.0037 | 3.68E−02 | 221.55 | 106.10 | 922 | 1577 | |
| Candidatus Soleaferrea* | ADc12 | 0.0002 | 0.0090 | 3.48E−05 | − 71.29 | 17.22 | 177 | 2571 |
| GenADA | 0.0199 | 0.0020 | 1.28E−01 | − 404.97 | 265.96 | 12431 | 1577 | |
| Catenibacterium | ADc12 | 0.3839 | 0.0082 | 8.54E−05 | − 1276.68 | 324.95 | 71434 | 2571 |
| GenADA | 0.0831 | 0.0080 | 2.11E−03 | 1068.34 | 347.56 | 25728 | 1577 | |
| Collinsella | ADc12 | 0.0002 | 0.0073 | 1.78E−04 | 125.63 | 33.51 | 230 | 2571 |
| GenADA | 0.0003 | 0.0143 | 4.36E−05 | 229.15 | 56.06 | 335 | 1577 | |
| Eisenbergiella* | ADc12 | 0.1049 | 0.0082 | 9.64E−05 | − 858.39 | 220.13 | 42727 | 2571 |
| GenADA | 0.0439 | 0.0066 | 5.28E−03 | − 1131.73 | 405.70 | 22002 | 1577 | |
| Eubacterium coprostanoligenes | ADc12 | 0.1020 | 0.0086 | 7.19E−05 | − 1085.22 | 273.37 | 48130 | 2571 |
| GenADA | 0.0101 | 0.0038 | 3.36E−02 | 707.65 | 333.01 | 7483 | 1577 | |
| Eubacterium fissicatena | ADc12 | 0.0404 | 0.0070 | 3.00E−04 | − 418.31 | 115.71 | 19567 | 2571 |
| GenADA | 0.0008 | 0.0090 | 1.17E−03 | − 142.85 | 44.01 | 663 | 1577 | |
| Eubacterium nodatum* | ADc12 | 0.4353 | 0.0072 | 2.52E−04 | − 1240.65 | 338.95 | 95579 | 2571 |
| GenADA | 0.1259 | 0.0025 | 8.82E−02 | − 805.51 | 472.45 | 42817 | 1577 | |
| Gordonibacter* | ADc12 | 0.0330 | 0.0089 | 5.57E−05 | − 254.45 | 63.13 | 16821 | 2571 |
| GenADA | 0.0116 | 0.0039 | 3.30E−02 | − 351.65 | 164.98 | 7135 | 1577 | |
| Intestinibacter* | ADc12 | 0.1903 | 0.0154 | 1.01E−07 | − 3014.37 | 566.17 | 70292 | 2571 |
| GenADA | 0.0024 | 0.0024 | 9.22E−02 | − 246.05 | 146.10 | 2152 | 1577 | |
| Lachnospira* | ADc12 | 0.0034 | 0.0067 | 3.30E−04 | 530.79 | 147.83 | 2997 | 2571 |
| GenADA | 0.0004 | 0.0021 | 1.11E−01 | 113.71 | 71.44 | 439 | 1577 | |
| LachnospiraceaeUCG008 | ADc12 | 0.0784 | 0.0081 | 1.08E−04 | − 776.50 | 200.53 | 35344 | 2571 |
| GenADA | 0.0041 | 0.0017 | 1.57E−01 | 197.29 | 139.40 | 3224 | 1577 | |
| Oscillibacter | ADc12 | 0.0124 | 0.0068 | 3.08E−04 | − 630.36 | 174.67 | 8305 | 2571 |
| GenADA | 0.0051 | 0.0027 | 7.55E−02 | 302.62 | 170.24 | 3884 | 1577 | |
| Prevotella9* | ADc12 | 0.0084 | 0.0075 | 1.57E−04 | − 556.34 | 147.21 | 6609 | 2571 |
| GenADA | 0.0008 | 0.0031 | 5.47E−02 | − 147.32 | 76.68 | 819 | 1577 | |
| Roseburia* | ADc12 | 0.2061 | 0.0100 | 1.35E−05 | − 3564.78 | 819.18 | 78446 | 2571 |
| GenADA | 0.0037 | 0.0027 | 7.64E−02 | − 366.88 | 207.02 | 3135 | 1577 | |
| Sutterella | ADc12 | 0.4928 | 0.0071 | 2.53E−04 | 3594.16 | 982.20 | 124985 | 2571 |
| GenADA | 0.0002 | 0.0009 | 3.08E−01 | − 34.75 | 34.10 | 156 | 1577 | |
| Veillonella | ADc12 | 0.0070 | 0.0081 | 8.83E−05 | 539.94 | 137.72 | 5406 | 2571 |
| GenADA | 0.0021 | 0.0085 | 1.56E−03 | 369.43 | 116.78 | 1843 | 1577 |
Genetic association between PRSs for top 20 microbiome genera and AD diagnosis from PRSice-2 “best-fit” model. The association from the “best-fit” threshold was generated from PRSice-2 with a range of p-value thresholds from 5 × 10−8 to 1 and incremental interval of 5 × 10−5; R2: Variance explained by the PRS model; P: p-value of model fit for the association; Coeff: Coefficient of the model; SE: standard error; # of SNPs: Number of SNPs included in the model at the specified threshold. Genera in bold are three genera identified to have a genetically significant association with AD in both discovery and replicate samples. *: indicated ten genera that have the same direction in both discovery and replicate samples. Seven genera, originally identified to be significantly associated with AD in the discovery sample, did not survive the replicate analysis due to the opposite direction in the replicate sample.
To validate our findings for the top 20 genera in the discovery sample, we further conducted the PRS analysis in the independent replication sample (GenADA). Two risk-associated genera (Collinsella and Veillonella) and one protective genus (Eubacterium fissicatena) remained significantly associated with AD diagnosis in the replication sample (p < 0.005). Ten other genera did not reach significance, but had the same effect direction as in the discovery sample (Table 3). To evaluate the overall association of the original top 20 genera from the discovery sample, we conducted a meta-analysis with the discovery and replication samples. As a result, a total of ten genera, including the three genera validated from the replication sample, were significantly associated with AD diagnosis (See Fig. 2 and Table S1).
Figure 2.

Forest plots of ten genera significantly associated with AD from meta-analysis. (A) The genetically predicted abundance of six genera showed significant association (p < 0.00042) with AD diagnosis as a protective factor with ORs < 1.0. (B) Conversely, four genera showed significant association with AD as a risk factor with ORs > 1.0. OR (95%CI): Odds ratio of the respective genus with the lower and upper 95% confidence intervals.
Of the ten significant genera from the meta-analysis, six genera—Adlercreutzia, Eubacterium nodatum group, Eisenbergiella, Eubacterium fissicatena group, Gordonibacter, and Prevotella9—were identified as protective, and four genera—Collinsella, Bacteroides, Lachnospira, and Veillonella—were identified as a risk factor for AD. From the meta-analysis, Eisenbergiella was identified as the strongest protective factor for AD with p = 1.39 × 10−6 and OR = 0.857 (95% CI 0.805–0.912), and Collinsella was identified as the strongest risk factor for AD p = 4.47 × 10−8 and OR = 1.188 (95% CI 1.117–1.264).
The meta-analysis also found three genera to have a suggestive association (0.00042 < p < 0.05) with AD diagnosis, of which all were potential protective factors (Intestinibacter, Candidatus Soleaferrea, and Roseburia) (See Table S1). In addition, seven genera—Alistipes, Anaerostipes, Catenibacterium, Eubacterium coprostanoligenes group, Lachnospiraceae UCG-008, Oscillibacter, and Sutterella—originally identified to be associated with AD in the discovery sample, did not show any association in the meta-analysis due to the opposite effects in the replication sample.
Next, a multivariate logistic regression analysis, including sex, age, and two APOE genotypes (rs429358 and rs7412) as covariates, was used to determine any confounding effects on the association between the ten significant genera and AD diagnosis. As shown in Supplementary Table S2, the ten significant genera remained significantly associated with AD diagnosis in the discovery sample (p < 0.05), which suggested that the genetic association between PRSs for the ten significant genera and AD diagnosis was independent of age, sex, and APOE genotypes. As expected, age and APOE were strongly associated with AD in the multivariate logistic regression analysis. Specifically, age and rs429358 minor allele C were risk factors as shown the positive correlation with AD diagnosis, while rs7412 minor allele T was a protective factor with the negative correlation with AD diagnosis. However, sex did not show any association with AD in this study.
To better visualize the difference of PRSs from the ten significant genera between AD cases and controls, we constructed a box plot along with the Wilcoxon Rank Sum test42 in the discovery sample. As compared to cognitively normal controls, Fig. 3A showed that AD patients had lower PRSs for the six likely protective genera (Adlercreutzia, Eubacterium nodatum group, Eisenbergiella, Eubacterium fissicatena group, Gordonibacter, and Prevotella9). On the other hand, Fig. 3B showed AD patients had higher PRSs for the four risk genera (Bacteroides, Collinsella, Lachnospira, and Veillonella). These results were consistent with the PRSice-2 "best-fit" model and logistic regression analysis between PRSs and AD diagnosis.
Figure 3.

Normalized PRSs for ten significant genera between AD cases and controls in the discovery sample. (A) PRSs for six genera were relatively lower in AD cases than controls (p < 0.05), suggesting they might be a protective factor for AD. (B) PRSs for four genera were relatively higher in AD cases vs. controls (p < 0.05), suggesting they were likely be a risk factor for AD. Wilcoxon Rank Sum test was applied to generate p values. X-axis: Diagnosis (AD cases/controls). Y-axis: z-score normalized PRSs for each of the ten significant genera.
Correlation between PRSs for the top ten significant genera and APOE genotypes
APOE is a well-known genetic risk for AD40,41. Depending on the alleles of two SNPs rs429358 and rs7412, the human APOE gene has three alleles (ε2, ε3, and ε4)41. The ε4 allele is the most influential risk factor for AD beyond age; a single ε4 allele increases one’s risk by three to four folds compared with the ε2 or ε3 allele40. Several studies have been conducted for the potential links between the APOE genotypes (rs429358 and rs7412) and the gut microbiota16–18, but not at the genome-wide level. For this reason, we sought to determine whether there was a genetic link between the PRSs for the ten significant genera and the APOE genotypes. Linear regression analyses were performed between the z-score normalized best PRSs for the ten significant genera and APOE minor alleles at rs429358 and rs7412. The meta-analysis showed that four out of ten significant genera were correlated with APOE rs429358 risk allele C (p < 0.05) (Table 4). Notably, Collinsella was the only genus that was positively correlated with AD diagnosis and APOE risk allele C at rs429358 in both discovery and replication samples (p < 0.05) (Tables 3 and 4). PRSs for three genera—Adlercreutzia, Eubacterium nodatum, and Prevotella9—identified negatively correlated with AD diagnosis showed negative correlation with APOE risk allele C at rs429358.
Table 4.
Association between PRSs for ten significant gut microbiota genera and APOE rs429358.
| Trait | Samples | Coeff | SE | z-value | P | OR (95%CI) |
|---|---|---|---|---|---|---|
| Adlercreutzia | ADc12 | − 0.051 | 0.031 | − 1.643 | 0.1006 | 0.95 (0.89–1.01) |
| GenADA | − 0.093 | 0.040 | − 2.309 | 0.0211 | 0.91 (0.84–0.99) | |
| Meta-analysis | − 0.067 | 0.025 | − 2.712 | 0.0067 | 0.94 (0.89–0.98) | |
| Bacteroides | ADc12 | 0.019 | 0.031 | 0.608 | 0.5434 | 1.02 (0.96–1.08) |
| GenADA | 0.014 | 0.040 | 0.350 | 0.7263 | 1.01 (0.94–1.10) | |
| Meta-analysis | 0.017 | 0.025 | 0.695 | 0.4871 | 1.02 (0.97–1.07) | |
| Collinsella | ADc12 | 0.137 | 0.031 | 4.413 | 1.06E-5 | 1.15 (1.08–1.22) |
| GenADA | 0.118 | 0.040 | 2.930 | 0.0034 | 1.12 (1.04–1.22) | |
| Meta-analysis | 0.130 | 0.025 | 5.283 | 1.27E-7 | 1.14 (1.09–1.19) | |
| Eisenbergiella | ADc12 | − 0.105 | 0.031 | − 3.382 | 0.0007 | 0.90 (0.85–0.96) |
| GenADA | 0.007 | 0.040 | 0.180 | 0.8575 | 1.01 (0.93–1.09) | |
| Meta-analysis | − 0.052 | 0.056 | − 0.924 | 0.3553 | 0.95 (0.85–1.06) | |
| Eubacterium Fissicatena | ADc12 | − 0.111 | 0.031 | − 3.557 | 0.0004 | 0.90 (0.84–0.95) |
| GenADA | − 0.010 | 0.040 | − 0.252 | 0.8011 | 0.99 (0.91–1.07) | |
| Meta-analysis | − 0.064 | 0.050 | − 1.270 | 0.2042 | 0.94 (0.85–1.04) | |
| Eubacterium Nodatum | ADc12 | − 0.097 | 0.031 | − 3.130 | 0.0018 | 0.91 (0.85–0.96) |
| GenADA | − 0.043 | 0.040 | − 1.080 | 0.2804 | 0.96 (0.88–1.04) | |
| Meta-analysis | − 0.077 | 0.026 | − 2.912 | 0.0036 | 0.93 (0.88–0.98) | |
| Gordonibacter | ADc12 | − 0.108 | 0.031 | − 3.460 | 0.0005 | 0.90 (0.84–0.95) |
| GenADA | − 0.014 | 0.040 | − 0.352 | 0.7248 | 0.99 (0.91–1.07) | |
| Meta-analysis | − 0.064 | 0.047 | − 1.383 | 0.1668 | 0.94 (0.86–1.03) | |
| Lachnospira | ADc12 | 0.047 | 0.031 | 1.524 | 0.1277 | 1.05 (0.99–1.11) |
| GenADA | 0.047 | 0.040 | 1.169 | 0.2425 | 1.05 (0.97–1.13) | |
| Meta-analysis | 0.047 | 0.025 | 1.921 | 0.0548 | 1.05 (1.00–1.10) | |
| Prevotella9 | ADc12 | − 0.046 | 0.031 | − 1.486 | 0.1373 | 0.95 (0.90–1.01) |
| GenADA | − 0.074 | 0.040 | − 1.836 | 0.0666 | 0.93 (0.86–1.01) | |
| Meta-analysis | − 0.057 | 0.025 | − 2.299 | 0.0215 | 0.94 (0.90–0.99) | |
| Veillonella | ADc12 | 0.092 | 0.031 | 2.946 | 0.0032 | 1.10 (1.03–1.17) |
| GenADA | − 0.027 | 0.040 | − 0.664 | 0.5071 | 0.97 (0.90–1.05) | |
| Meta-analysis | 0.035 | 0.059 | 0.596 | 0.5513 | 1.04 (0.92–1.16) |
Genera in bold are four genera identified to have genetically significant correlation with APOE rs429358 minor allele C in the meta-analysis (p < 0.05). Collinsella was the only genus that showed significant correlation in both discovery and replication samples.
To illustrate the correlations between PRSs for Collinsella and APOE risk allele C at rs429358, we constructed a box plot along with ANOVA analysis. As shown in Fig. 4, a positive correlation between PRSs for Collinsella and APOE risk allele C at rs429358 was found in the discovery sample (p = 2.1 × 10−5). This positive correlation indicated that a genetic factor determining Collinsella abundance was more likely to occur in individuals with APOE minor allele C (CC and TC) as compared to individuals with two T alleles (TT) at rs429358.
Figure 4.
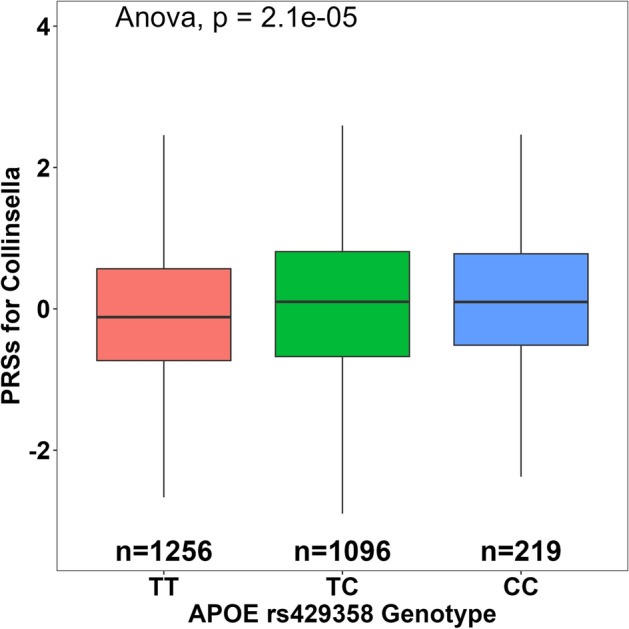
Genetic association between PRSs for Collinsella and APOE rs429358 genotype in the discovery sample. Individuals in the discovery sample were separated by their genotype at the APOE SNP rs429358. Those with the genotype of TC and CC had higher PRSs for genetically predicted Collinsella abundance than those with the TT genotype.
Overall, our results showed that Collinsella was a risk factor for AD diagnosis and that Collinsella was positively correlated with APOE risk allele C at rs429358. On the other hand, three genera identified as protective factors (Eubacterium nodatum group, Adlercreutzia, and Prevotella9) for AD diagnosis showed a negative correlation with APOE risk allele C at rs429358 (Table 4). These associations indicate that certain microbial genera and APOE may contribute to disease modulation in some similar biological pathways, synergizing in disease risk or protective effects. The associations between PRSs for the four genera and the APOE rs429358 risk allele were independent of sex and age, as the results remained significant after adjustment for these cofactors (Supplementary Table S3). For the APOE genotype at rs7412, we did not see any significant correlation with the PRSs for the ten significant genera from the meta-analysis.
Association between microbiome abundance and APOE genotypes
To further investigate the association between the abundance of all the gut microbiota genera and APOE genotypes, we retrieved summary statistics for the two APOE SNPs rs429358 and rs7412 directly from the 119 genera GWAS summary statistics in the MiBioGen consortium study. As shown in Table 5, rs429358 was marginally correlated with the abundance of ten genera, and rs7412 was marginally associated with the abundance of eight genera (p < 0.05). Together, these findings indicate that the APOE genotypes may have some impact on the microbiome abundance at the genus level and that the association may synergistically contribute to the risk for human diseases such as AD. Our results open the door for future studies to explore the role of the interaction between APOE and the gut microbiota and find a new target for treatment in human diseases.
Table 5.
List of gut microbiome genera that were nominally associated with APOE SNPs rs429358 and rs7412.
| rsID (CHR:BP:Effect Allele) | Microbiome Genera | Beta | SE | SZ | P | N | # cohorts |
|---|---|---|---|---|---|---|---|
| rs429358 (19:45411941:C) | Bacteroides | − 0.044 | 0.015 | − 2.881 | 0.0040 | 18173 | 23 |
| Butyricimonas | − 0.044 | 0.020 | − 2.241 | 0.0250 | 10657 | 23 | |
| Dorea | 0.030 | 0.015 | 2.127 | 0.0334 | 17494 | 23 | |
| Eubacterium coprostanoligenes | 0.033 | 0.015 | 2.120 | 0.0340 | 17261 | 23 | |
| Faecalibacterium | − 0.035 | 0.015 | − 2.188 | 0.0287 | 17960 | 23 | |
| Olsenella | 0.064 | 0.033 | 1.993 | 0.0462 | 3721 | 13 | |
| Parasutterella | 0.040 | 0.019 | 2.216 | 0.0267 | 11291 | 23 | |
| Senegalimassilia | 0.050 | 0.024 | 2.207 | 0.0273 | 6888 | 21 | |
| Veillonella | 0.044 | 0.021 | 2.032 | 0.0421 | 9194 | 23 | |
| rs7412 (19:45412079:T) | Butyricicoccus | 0.048 | 0.021 | 2.468 | 0.0136 | 15637 | 20 |
| Collinsella | 0.052 | 0.023 | 2.214 | 0.0268 | 12811 | 19 | |
| Coprococcus3 | 0.050 | 0.022 | 2.344 | 0.0191 | 14323 | 20 | |
| Eubacterium hallii | 0.053 | 0.021 | 2.453 | 0.0142 | 14846 | 20 | |
| Lachnospiraceae UCG001 | − 0.071 | 0.027 | − 2.534 | 0.0113 | 9357 | 20 | |
| Olsenella | − 0.103 | 0.043 | − 2.346 | 0.0190 | 3721 | 13 | |
| Ruminococcaceae UCG004 | 0.080 | 0.028 | 2.901 | 0.0037 | 9049 | 20 | |
| Senegalimassilia | 0.082 | 0.032 | 2.584 | 0.0098 | 6508 | 18 |
SNP data were extracted from GWAS summary statistics of human gut microbiome abundance conducted from 24 multi-ethnic cohorts in the MiBioGen consortium (www.mibiogen.org)24. rsID (CHR:BP:Effect Allele): the rs number of single nucleotide polymorphisms, chromosome number (CHR), base pair (BP), with the effect allele (genome assembly GRCh37/hg19). Microbiome: the genus level with significant abundance associated with APOE two SNPs. Beta: Beta coefficient. SE: Standard Error. SZ: Weighted sum of z-scores. P: p-value (p < 0.05 was considered as nominal association between the microbiome genera and APOE). N: sample count. # Cohorts: Number of cohorts involved.
Discussion
The microbiota is a complex ecosystem that comprises more than 100 trillion symbiotic microbial cells in the human body, of which 95% inhabit the human gut44. The bacteria from phylum Firmicutes and Bacteroidetes form a significant proportion (90%) of the adult gut microbiota, while Actinobacteria composes the rest45. Recently, significant evidence has shown that the gut microbiota influences normal systemic physiological homeostasis and that dysbiosis of gut microbiota may contribute to the pathogenesis of brain diseases, including AD. The gut microbiota interacts with the central nervous system (CNS) across the MGBA via microbial components, metabolic products, and neural stimulation. In this study, we leveraged extensive GWAS data to study the genetic correlation between gut microbiota genera and AD diagnosis. PRSs for 20 genera were initially found significantly associated with AD in the discovery sample, three of which were replicated in the independent replication sample. A further meta-analysis between our discovery and replication samples identified a strong genetic association between ten gut microbiota genera and AD diagnosis. Six genera were negatively associated with AD diagnosis and four genera were positively correlated with AD diagnosis. “Negative association” means that the abundance of these genera is lower in AD patients as compared to normal controls. Thus, PRSs for such genera are regarded as a protective factor for the disease. Similarly, “positive association” means that the abundance of those genera is higher in AD cases as compared to normal controls, indicating their PRSs would be seen as a risk factor against the disease. Genera identified as a protective factor were primarily from the Firmicutes phylum (Eubacterium nodatum group, Eisenbergiella, and Eubacterium fissicatena group) as well as from Actinobacteria (Adlercreutzia, Gordonibacter) and Bacteroidetes (Prevotella9). Positively correlated, or risk-associated genera were from phyla including Firmicutes (Lachnospira and Veillonella), Actinobacteria (Collinsella), and Bacteroidetes (Bacteroides).
In the discovery sample, the correlation of the ten significant genera remained statistically significant after being adjusted for sex, age, and two APOE SNPs (rs429358 and rs7412), suggesting that the genetic correlation between the ten genera and AD diagnosis was independent of age, sex, or APOE genotypes. In addition, we found that four of the ten significant genera showed a strong correlation with the APOE rs429358 risk allele C via linear regression analysis. Interestingly, the genera showing a positive correlation with APOE rs429358 risk allele C tend to have a positive (risk) association with AD, while the genera showing a negative correlation with APOE rs429358 risk allele C have a negative (protective) association with AD.
In our analyses, Collinsella from the phylum Actinobacteria was identified as a risk factor for AD in both the discovery and replication samples. Collinsella was also positively correlated with APOE rs429358 risk allele C in both samples. The abundance of Collinsella in the gut has been previously associated with rheumatoid arthritis, atherosclerosis, and Type-2 diabetes46–48. Importantly, an increased abundance of this genus has also been observed in AD transgenic mice and AD patients49,50. Our findings provide evidence at the human genome-wide level of a connection between Collinsella and AD that supports previous observational studies. At the molecular level, this connection is possibly driven by the pro-inflammatory effects of the Collinsella genus. In a human intestinal epithelial cell line, the presence of Collinsella increased the expression of inflammatory cytokines (IL-17A) and chemokines (CXCL1, CXCL5). Collinsella also increased gut permeability by reducing the expression of tight-junction proteins51. Furthermore, the strong association between Collinsella and APOE rs429358 risk allele C in our study may provide new insight into the pathogenesis of AD. For example, a study found that Collinsella correlates with higher serum levels of total cholesterol and low-density lipoprotein (LDL) cholesterol in healthy adults52, which may be correlated with the interaction between Collinsella and APOE. Functional studies that further explore the relationship between Collinsella, lipid metabolism, and inflammatory signals would help to elucidate how their interaction influences AD and other diseases.
Three genera of the Firmicutes phylum—Eubacterium nodatum group, Eisenbergiella, and Eubacterium fissicatena group—had a negative association with AD diagnosis. Eisenbergiella, Eubacterium fissicatena group, and Eubacterium nodatum group are known to contain species that metabolize the short-chain fatty acid (SCFA) butyrate from dietary carbohydrates53–56. Butyrate is a major SCFA metabolite in the colon that might be a critical mediator of the colonic inflammatory response. Alongside its anti-inflammatory properties, butyrate is also essential in maintaining tight junctions that prevent dysbiotic gut permeability57,58. Despite their production of butyrate, several studies have identified Eisenbergiella and Eubacterium nodatum group as microbial features associated with neurodegenerative diseases. A notable study of patients with AD and vascular dementia found that the gut abundance of these genera could be used to discriminate severe dementia patients against those with mild or moderate dementia59. High serum levels of the IgG antibody against oral Eubacterium nodatum were associated with lower AD risk in another study60. This suggests that oral and gut populations of the same microbial taxa may have different etiologies with the same disease, however, our base data covers only the gut abundance of microbiota. Nevertheless, we are the first to report a protective association between genetically-predicted Eisenbergiella, Eubacterium nodatum group, and Eubacterium fissicatena group abundance with AD, but more studies are needed to understand how these three genera may interact with the pathology of AD.
In addition, we identified two Firmicutes genera as risk factors for AD (Lachnospira and Veillonella), with Veillonella being validated in the replication sample. Recently, it was reported that AD patients have an abundance of Veillonella in their oral microbiome61. In the gut, it has been shown that an overabundance of species like V.parvula promotes intestinal inflammation by activating macrophages via the lipopolysaccharide-Toll-like receptor 4 (LPS-TLR4) pathway62. The dual association of oral and gut abundance of Veillonella with disease points to this genus as a target for therapeutics and a potential bridge between conditions like gut inflammation and periodontitis with AD. On the other hand, gut Lachnospira and Veillonella species have also been identified as beneficial or commensal to gut health, such as Lachnospira being protective against Crohn’s disease, or Veillonella interacting with Streptococcus species to modulate immune responses in the small intestine63,64. In an observational study from a Chinese group, patients with AD had decreased Lachnospira at the genus level compared with healthy controls65. However, this may reflect national differences in diet or the genetics of microbial abundance, as our study uses mostly Caucasian subjects from the United States in our discovery and replication samples.
The Bacteroidetes genera, Prevotella9 and Bacteroides, were identified as protective and risk factors, respectively, in our meta-analysis. There is a complex relationship between Prevotella and Bacteroides abundance and intestinal diseases66. In humans, Prevotella is more common in populations with plant-based and high-carbohydrate diets67. Conversely, Bacteroides is more abundant in those consuming “western” diets high in protein and fat68. One major study showed that Prevotella was higher in individuals with greater adherence to Mediterranean diets, which is thought to be protective against neurodegenerative diseases69–71. The protective effects of Prevotella abundance may come from the positive dietary effects on the genus. Our association of higher genetically-predicted Bacteroides abundance with AD risk supports the findings of previous observational studies11,72,73. Bacteroides species are capable of secreting LPS as an endotoxic biomolecule, which has been implicated in pathological endothelial dysfunction of the gut and can induce neuroinflammation in microglia cells74–76. However, it should be noted that a meta-analysis including Chinese studies found no risk association between Bacteroides and AD12, which may again reflect national differences in diet and microbial abundance.
Two protective genera, Gordonibacter and Adlercreutzia, are from the Actinobacteria phylum. These genera tend to produce metabolites beneficial to mitochondrial function, namely Urolithin A (UA) and Equol77,78. UA is an anti-inflammatory compound that enhances mitophagy, the removal of dysfunctional mitochondria in a cells79. Impaired mitophagy is part of the pathogenesis of AD, thus, UA and Gordonibacter species might be promising therapeutic targets against aging and AD80. Equol is an estrogen-like compound that reduces microglial inflammation when stimulated by LPS and downregulates genes in neurons related to apoptosis81. The beneficial effects of these bacterial metabolites could drive the protective association of Gordonibacter and Adlercreutzia abundance with AD that we found in this study.
The strength of our study include the use of the largest available GWASs of gut microbiota taxa to date that allow us to identify multiple genera genetically associated with AD after the strict Bonferroni correction. The use of logistic regression analysis alongside our initial PRS analyses allowed us to adjust for potential confounders, such as sex, age, and APOE alleles, and further validate that the association was independent of those confounders. Additionally, we are the first to study the genetic correlation between the gut microbiota and the APOE gene at the human genome-wide level.
Limitations
There are several limitations to our study. First, the sample size for our base microbiome GWAS may not be large enough to truly cover the effect size of the host genetic variants, even though the MiBioGen study has a larger sample size compared to other microbiome GWASs. Because of this, we may not have enough power to detect some of the associations in our meta-analysis that were considered significant in the discovery sample. Future studies with larger sample sizes would be more capable of drawing solid conclusions about the genetic connection between gut microbiota and AD. As microbiome is highly influenced by lifestyle and environmental factors, the lack of information on these confounders in our base and target data prevents the subtyping of patients. Given the phenotypes available in our genotyping data, we included age, sex, and APOE genotype as covariates in our multivariate logistic regression models to account for their confounding effects. Second, our genotyping data for AD studies were mostly drawn from European American individuals, which limits the generalization of our conclusions when applied to other ethnic groups. Although the largest ethnic cohort of the MiBioGen GWAS was European (Table 1), the inclusion of other ethnic groups in the original study’s meta-analysis may be a confounding factor in our results. More diverse genotyping datasets would enable us to capture the variability in risk for AD across different ethnicities. Third, the 16S rRNA sequencing used to generate genetic associations in the “base” GWAS only provides taxa resolution from the phylum to the genus level. Fully understanding the role of bacterial taxa that may drive the pathology of AD will require methods that can capture the abundance of individual species and their mechanistic impact on the MGBA.
Conclusions
Overall, our novel findings of ten significant genera associated with AD from the meta-analysis provide new insights into the interplay of the gut microbiota on AD. Genetic associations with the abundance of certain bacterial genera inhabiting the gut correlate with AD diagnosis in risk and protective directions. Risk-associated genera, such as Collinsella, have been previously tied to neuroinflammatory processes across the MGBA, while protection-associated genera like Gordonibacter are known to secrete metabolites that promote gut and brain health. PRSs for four genera were further identified as significant associations with the APOE genotype at rs429358. Our results advance the understanding of how gut dysbiosis may play a role in the pathology of AD. Future investigations with larger cohorts of AD patients from different ethnic backgrounds and more powerful microbiome GWASs are needed to better understand these genetic associations. Functional studies are also required to establish causality between particular gut microbiota and AD pathology.
Supplementary Information
Acknowledgements
We thank the patients for their participation in the MiBioGen consortium, NIA/LOAD ADc12 Cohort, and GenADA studies, and the original investigators who conducted these studies and made the data available.
Abbreviations
- AD
Alzheimer’s disease
- APOE
Apolipoprotein E
- BBB
blood–brain barrier
- MGBA
microbiota-gut-brain axis
- LPS
Lipopolysaccharide
- TLR4
Tol-like receptor 4
- SNP
single nucleotide polymorphism
- GWAS
genome-wide association study
- OR
odds ratio
- CI
confidence interval
- LD
linkage disequilibrium
- PRS
polygenic risk score
- CNS
central nervous system
- IRB
Institutional review board
- UNLV
University of Nevada Las Vegas
- NIA/LOAD
National Institute of Aging/Late-onset Alzheimer’s Disease Study
- GenADA
Multi-site Collaborative Study for Genotype–Phenotype Associations in Alzheimer’s Disease
- NINCDS-ADRDA
National Institute of Neurological and Communicative Disorders and Stroke, and the Alzheimer's Disease and Related Disorders Association
- LDL
low-density lipoprotein
- SCFA
short-chain fatty acid
- UA
Urolithin A
- mbQTL
microbial quantitative trait loci
- mbBTL
microbial binary trait loci
Author contributions
J.C. was responsible for the conception, sample request, data analyses, and writing the manuscript. D.C., Y.L., and M.J.C. were responsible for data analyses and writing the manuscript. M.L.Z., J.M.C, and J.D. were responsible for data analyses. X.C., E.O., J.L.C, and J.E. were responsible for writing, critiquing, and revising the manuscript. All authors have read and agreed to the content of the manuscript before sending it to the journal for publication.
Funding
This research was funded to J.C. in part by NIH grant P20GM121325, NIH grant P20GM121325-02S1, and NIH grant P20GM121325_03S2.
Data availability
The Full GWAS summary statistics of mbQTLs analyzed during this study are available at https://mibiogen.gcc.rug.nl/. Genotyping data for the NIA/LOAD (phs000168.v2.p2) and GenADA (phs000219.v1.p1) cohorts are available at https://www.ncbi.nlm.nih.gov/gap/. All data generated in this study are included in the manuscript, Supplementary Tables S1–3, and Supplementary Fig. S1.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Davis Cammann and Yimei Lu.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-023-31730-5.
References
- 1.Arvanitakis Z, Shah RC, Bennett DA. Diagnosis and management of dementia: Review. JAMA. 2019;322(16):1589. doi: 10.1001/jama.2019.4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.2022 Alzheimer’s disease facts and figures. Alzheimer's & Dementia. 18(4), 700–789. 10.1002/alz.12638 (2022). [DOI] [PubMed]
- 3.Nichols E, Steinmetz JD, Vollset SE, Fukutaki K, Chalek J, Abd-Allah F, et al. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health. 2022;7(2):e105–e125. doi: 10.1016/S2468-2667(21)00249-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014;6(4):37. doi: 10.1186/alzrt269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bachiller S, Jiménez-Ferrer I, Paulus A, Yang Y, Swanberg M, Deierborg T, et al. Microglia in neurological diseases: A road map to brain-disease dependent-inflammatory response. Front. Cell Neurosci. 2018;18(12):488. doi: 10.3389/fncel.2018.00488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021;17(3):157–172. doi: 10.1038/s41582-020-00435-y. [DOI] [PubMed] [Google Scholar]
- 7.Chen Y, Zhou J, Wang L. Role and mechanism of gut microbiota in human disease. Front. Cell Infect. Microbiol. 2021;17(11):625913. doi: 10.3389/fcimb.2021.625913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goyal D, Ali SA, Singh RK. Emerging role of gut microbiota in modulation of neuroinflammation and neurodegeneration with emphasis on Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2021;2(106):110112. doi: 10.1016/j.pnpbp.2020.110112. [DOI] [PubMed] [Google Scholar]
- 9.Carabotti, M., Scirocco, A., Maselli, M. A. & Severi, C. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 7. [PMC free article] [PubMed]
- 10.Zhuang ZQ, Shen LL, Li WW, Fu X, Zeng F, Gui L, et al. Gut microbiota is altered in patients with Alzheimer’s disease. J. Alzheimers Dis. JAD. 2018;63(4):1337–1346. doi: 10.3233/JAD-180176. [DOI] [PubMed] [Google Scholar]
- 11.Vogt NM, Kerby RL, Dill-McFarland KA, Harding SJ, Merluzzi AP, Johnson SC, et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017;7(1):13537. doi: 10.1038/s41598-017-13601-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hung CC, Chang CC, Huang CW, Nouchi R, Cheng CH. Gut microbiota in patients with Alzheimer’s disease spectrum: A systematic review and meta-analysis. Aging. 2022;14(1):477–496. doi: 10.18632/aging.203826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1β and anti-inflammatory IL-10 cytokines. Brain Behav. Immun. 2009;23(3):309–317. doi: 10.1016/j.bbi.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller AL, Bessho S, Grando K, Tükel Ç. Microbiome or infections: Amyloid-containing biofilms as a trigger for complex human diseases. Front. Immunol. 2021;26(12):638867. doi: 10.3389/fimmu.2021.638867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, et al. Human genetics shape the gut microbiome. Cell. 2014;159(4):789–799. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hou M, Xu G, Ran M, Luo W, Wang H. APOE-ε4 carrier status and gut microbiota dysbiosis in patients with Alzheimer disease. Front. Neurosci. 2021;24(15):619051. doi: 10.3389/fnins.2021.619051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zajac DJ, Green SJ, Johnson LA, Estus S. APOE genetics influence murine gut microbiome. Sci. Rep. 2022;12(1):1906. doi: 10.1038/s41598-022-05763-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tran TTT, Corsini S, Kellingray L, Hegarty C, Le Gall G, Narbad A, et al. APOE genotype influences the gut microbiome structure and function in humans and mice: Relevance for Alzheimer’s disease pathophysiology. FASEB J. 2019;33(7):8221–8231. doi: 10.1096/fj.201900071R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi SW, O’Reilly PF. PRSice-2: Polygenic risk score software for biobank-scale data. GigaScience. 2019;8(7):giz082. doi: 10.1093/gigascience/giz082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng S, Qi X, Ma M, Zhang L, Cheng B, Liang C, et al. Assessing the relationship between gut microbiota and bone mineral density. Front. Genet. 2020;31(11):6. doi: 10.3389/fgene.2020.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wells PM, Adebayo AS, Bowyer RCE, Freidin MB, Finckh A, Strowig T, et al. Associations between gut microbiota and genetic risk for rheumatoid arthritis in the absence of disease: A cross-sectional study. Lancet Rheumatol. 2020;2(7):e418–e427. doi: 10.1016/S2665-9913(20)30064-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yao Y, Qi X, Jia Y, Ye J, Chu X, Wen Y, et al. Evaluating the interactive effects of dietary habits and human gut microbiome on the risks of depression and anxiety. Psychol. Med. 2022;25:1–9. doi: 10.1017/S0033291721005092. [DOI] [PubMed] [Google Scholar]
- 23.Kurilshikov A, Medina-Gomez C, Bacigalupe R, Radjabzadeh D, Wang J, Demirkan A, et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 2021;53(2):156–165. doi: 10.1038/s41588-020-00763-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013;41((Database issue)):D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016;48(10):1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wakita Y, Shimomura Y, Kitada Y, Yamamoto H, Ohashi Y, Matsumoto M. Taxonomic classification for microbiome analysis, which correlates well with the metabolite milieu of the gut. BMC Microbiol. 2018;18(1):188. doi: 10.1186/s12866-018-1311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JH, Cheng R, Graff-Radford N, Foroud T, Mayeux R, National Institute on Aging Late-Onset Alzheimer’s Disease Family Study Group Analyses of the national institute on aging late-onset Alzheimer’s disease family study: Implication of additional loci. Arch. Neurol. 2008;65(11):1518–1526. doi: 10.1001/archneur.65.11.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Wetten S, Li L, St. Jean PL, Upmanyu R, Surh L, et al. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch. Neurol. 2008;65(1):45–53. doi: 10.1001/archneurol.2007.3. [DOI] [PubMed] [Google Scholar]
- 29.Filippini N, Rao A, Wetten S, Gibson RA, Borrie M, Guzman D, et al. Anatomically-distinct genetic associations of APOE ɛ4 allele load with regional cortical atrophy in Alzheimer’s disease. Neuroimage. 2009;44(3):724–728. doi: 10.1016/j.neuroimage.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 30.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease. Neurology. 1984;34(7):939. doi: 10.1212/WNL.34.7.939. [DOI] [PubMed] [Google Scholar]
- 31.Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verma SS, de Andrade M, Tromp G, Kuivaniemi H, Pugh E, Namjou-Khales B, et al. Imputation and quality control steps for combining multiple genome-wide datasets. Front. Genet. 2014;5:370. doi: 10.3389/fgene.2014.00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience. 2015 doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi SW, Mak TSH, O’Reilly PF. Tutorial: a guide to performing polygenic risk score analyses. Nat. Protoc. 2020;15(9):2759–2772. doi: 10.1038/s41596-020-0353-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.International Schizophrenia Consortium. Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.R Core Team. R: A Language and Environment for Statistical Computing. [Internet]. R Foundation for Statistical Computing; 2021. Available from: https://www.R-project.org/
- 37.Dewey, M., Henmi, M. & Copas, J. Meta-Analysis Package for R [Internet]. Available from: https://www.metafor-project.org/doku.php/metafor
- 38.Gordon, M. & Lumley, T. Advanced Forest Plot Using “Grid” Graphics [Internet]. CRAN; 2021. Available from: https://gforge.se/packages/
- 39.Kassambara, A. “ggplot2” Based Publication Ready Plots [Internet]. CRAN; 2020. Available from: https://rpkgs.datanovia.com/ggpubr/
- 40.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 41.Okuizumi K, Onodera O, Tanaka H, Kobayashi H, Tsuji S, Takahashi H, et al. ApoE–ε4 and early–onset Alzheimer’s. Nat. Genet. 1994;7(1):10–11. doi: 10.1038/ng0594-10b. [DOI] [PubMed] [Google Scholar]
- 42.Wilcoxon F. Individual comparisons by ranking methods. Int. Biom. Soc. 1945;1:80–83. [Google Scholar]
- 43.Fisher RA. Statistical methods for research workers. In: Kotz S, Johnson NL, editors. Breakthroughs in Statistics: Methodology and Distribution. Springer New York; 1992. pp. 66–70. [Google Scholar]
- 44.de J.R. De-Paula V, Forlenza AS, Forlenza OV. Relevance of gutmicrobiota in cognition, behaviour and Alzheimer’s disease. Pharmacol. Res. 2018;136:29–34. doi: 10.1016/j.phrs.2018.07.007. [DOI] [PubMed] [Google Scholar]
- 45.O’Toole PW, Jeffery IB. Gut microbiota and aging. Science. 2015;350(6265):1214–1215. doi: 10.1126/science.aac8469. [DOI] [PubMed] [Google Scholar]
- 46.Ruiz-Limón P, Mena-Vázquez N, Moreno-Indias I, Manrique-Arija S, Lisbona-Montañez JM, Cano-García L, et al. Collinsella is associated with cumulative inflammatory burden in an established rheumatoid arthritis cohort. Biomed. Pharmacother. Biomed. Pharmacother. 2022;153:113518. doi: 10.1016/j.biopha.2022.113518. [DOI] [PubMed] [Google Scholar]
- 47.Karlsson FH, Fåk F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, et al. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012;3(1):1245. doi: 10.1038/ncomms2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Candela M, Biagi E, Soverini M, Consolandi C, Quercia S, Severgnini M, et al. Modulation of gut microbiota dysbioses in type 2 diabetic patients by macrobiotic Ma-Pi 2 diet. Br. J. Nutr. 2016;116(1):80–93. doi: 10.1017/S0007114516001045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bello-Medina PC, Hernández-Quiroz F, Pérez-Morales M, González-Franco DA, Cruz-Pauseno G, García-Mena J, et al. Spatial memory and gut microbiota alterations are already present in early adulthood in a pre-clinical transgenic model of Alzheimer’s disease. Front. Neurosci. 2021;29(15):595583. doi: 10.3389/fnins.2021.595583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ling Z, Zhu M, Yan X, Cheng Y, Shao L, Liu X, et al. Structural and functional dysbiosis of fecal microbiota in Chinese patients with Alzheimer’s disease. Front. Cell Dev. Biol. 2021;4(8):634069. doi: 10.3389/fcell.2020.634069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen J, Wright K, Davis JM, Jeraldo P, Marietta EV, Murray J, et al. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med. 2016;8(1):43. doi: 10.1186/s13073-016-0299-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lahti L, Salonen A, Kekkonen RA, Salojärvi J, Jalanka-Tuovinen J, Palva A, et al. Associations between the human intestinal microbiota, Lactobacillus rhamnosus GG and serum lipids indicated by integrated analysis of high-throughput profiling data. PeerJ. 2013;1:e32. doi: 10.7717/peerj.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Amir I, Bouvet P, Legeay C, Gophna U, Weinberger A. Eisenbergiella tayi gen. nov., sp. Nov., isolated from human blood. Int. J. Syst. Evol. Microbiol. 2014;64((Pt 3)):907–914. doi: 10.1099/ijs.0.057331-0. [DOI] [PubMed] [Google Scholar]
- 54.Togo AH, Khelaifia S, Bittar F, Maraninchi M, Raoult D, Million M. ‘Eisenbergiella massiliensis’, a new species isolated from human stool collected after bariatric surgery. New Microbes New Infect. 2016;13:15–16. doi: 10.1016/j.nmni.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oh JK, Vasquez R, Kim SH, Lee JH, Kim EJ, Hong SK, et al. Neoagarooligosaccharides modulate gut microbiota and alleviate body weight gain and metabolic syndrome in high-fat diet-induced obese rats. J. Funct. Foods. 2022;1(88):104869. doi: 10.1016/j.jff.2021.104869. [DOI] [Google Scholar]
- 56.Uematsu H, Hoshino E. Degradation of arginine and other amino acids by Eubacterium nodatum ATCC 33099. Microb. Ecol. Health Dis. 1996;9(6):305–311. doi: 10.1002/(SICI)1234-987X(199611)9:6<305::AID-MEH437>3.3.CO;2-S. [DOI] [Google Scholar]
- 57.Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, et al. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe. 2015;17(5):662–671. doi: 10.1016/j.chom.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Silva YP, Bernardi A, Frozza RL. The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front. Endocrinol. 2020;31(11):25. doi: 10.3389/fendo.2020.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stadlbauer V, Engertsberger L, Komarova I, Feldbacher N, Leber B, Pichler G, et al. Dysbiosis, gut barrier dysfunction and inflammation in dementia: A pilot study. BMC Geriatr. 2020;20(1):248. doi: 10.1186/s12877-020-01644-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Noble JM, Scarmeas N, Celenti RS, Elkind MSV, Wright CB, Schupf N, et al. Serum IgG antibody levels to periodontal microbiota are associated with incident Alzheimer disease. PLoS ONE. 2014;9(12):e114959. doi: 10.1371/journal.pone.0114959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guo H, Li B, Yao H, Liu D, Chen R, Zhou S, et al. Profiling the oral microbiomes in patients with Alzheimer’s disease. Oral Dis. 2023;29(3):1341–1355. doi: 10.1111/odi.14110. [DOI] [PubMed] [Google Scholar]
- 62.Zhan Z, Liu W, Pan L, Bao Y, Yan Z, Hong L. Overabundance of Veillonella parvula promotes intestinal inflammation by activating macrophages via LPS-TLR4 pathway. Cell Death Discov. 2022;8(1):1–12. doi: 10.1038/s41420-022-01015-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Caparrós E, Wiest R, Scharl M, Rogler G, Gutiérrez Casbas A, Yilmaz B, et al. Dysbiotic microbiota interactions in Crohn’s disease. Gut Microbes. 2021;13(1):1949096. doi: 10.1080/19490976.2021.1949096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van den Bogert B, Meijerink M, Zoetendal EG, Wells JM, Kleerebezem M. Immunomodulatory properties of Streptococcus and Veillonella isolates from the human small intestine microbiota. PLoS ONE. 2014;9(12):e114277. doi: 10.1371/journal.pone.0114277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guo M, Peng J, Huang X, Xiao L, Huang F, Zuo Z. Gut microbiome features of Chinese patients newly diagnosed with Alzheimer’s disease or mild cognitive impairment. J. Alzheimers Dis. JAD. 2021;80(1):299–310. doi: 10.3233/JAD-201040. [DOI] [PubMed] [Google Scholar]
- 66.Fox M, Knorr DA, Haptonstall KM. Alzheimer’s disease and symbiotic microbiota: An evolutionary medicine perspective. Ann. N. Y. Acad. Sci. 2019;1449(1):3–24. doi: 10.1111/nyas.14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Larsen JM. The immune response to Prevotella bacteria in chronic inflammatory disease. Immunology. 2017;151(4):363–374. doi: 10.1111/imm.12760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tomova A, Bukovsky I, Rembert E, Yonas W, Alwarith J, Barnard ND, et al. The effects of vegetarian and vegan diets on gut microbiota. Front. Nutr. 2019;17(6):47. doi: 10.3389/fnut.2019.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Filippis FD, Pellegrini N, Vannini L, Jeffery IB, Storia AL, Laghi L, et al. High-level adherence to a Mediterranean diet beneficially impacts the gut microbiota and associated metabolome. Gut. 2016;65(11):1812–1821. doi: 10.1136/gutjnl-2015-309957. [DOI] [PubMed] [Google Scholar]
- 70.Scarmeas N, Luchsinger JA, Mayeux R, Stern Y. Mediterranean diet and Alzheimer disease mortality. Neurology. 2007;69(11):1084–1093. doi: 10.1212/01.wnl.0000277320.50685.7c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Picchianti Diamanti A, Panebianco C, Salerno G, Di Rosa R, Salemi S, Sorgi ML, et al. Impact of Mediterranean diet on disease activity and gut microbiota composition of rheumatoid arthritis patients. Microorganisms. 2020;8(12):1989. doi: 10.3390/microorganisms8121989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xia Y. Correlation and association analyses in microbiome study integrating multiomics in health and disease. Prog. Mol. Biol. Transl. Sci. 2020;171:309–497. doi: 10.1016/bs.pmbts.2020.04.003. [DOI] [PubMed] [Google Scholar]
- 73.Haran JP, Bhattarai SK, Foley SE, Dutta P, Ward DV, Bucci V, et al. Alzheimer’s disease microbiome is associated with dysregulation of the anti-inflammatory P-glycoprotein pathway. MBio. 2019;10(3):e00632-19. doi: 10.1128/mBio.00632-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lukiw WJ. Bacteroides fragilis lipopolysaccharide and inflammatory signaling in Alzheimer’s disease. Front. Microbiol. 2016;7:1544. doi: 10.3389/fmicb.2016.01544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dhaliwal G. Alistipes: The influence of a commensal on anxiety and depression. Catal. Facets Biochem. Biomed. Sci. 2019;3(1):9. [Google Scholar]
- 76.Lukiw WJ. Gastrointestinal (GI) tract microbiome-derived neurotoxins—Potent neuro-inflammatory signals from the GI tract via the systemic circulation into the brain. Front. Cell Infect. Microbiol. 2020;12(10):22. doi: 10.3389/fcimb.2020.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Selma MV, Tomás-Barberán FA, Beltrán D, García-Villalba R, Espín JC. Gordonibacter urolithinfaciens sp. nov., a urolithin-producing bacterium isolated from the human gut. Int. J. Syst. Evol. Microbiol. 2014;64((Pt_7)):2346–2352. doi: 10.1099/ijs.0.055095-0. [DOI] [PubMed] [Google Scholar]
- 78.Maruo T, Sakamoto M, Ito C, Toda T, Benno Y. Adlercreutzia equolifaciens gen. nov., sp. nov., an equol-producing bacterium isolated from human faeces, and emended description of the genus Eggerthella. Int. J. Syst. Evol. Microbiol. 2008;58(5):1221–1227. doi: 10.1099/ijs.0.65404-0. [DOI] [PubMed] [Google Scholar]
- 79.D’Amico D, Andreux PA, Valdés P, Singh A, Rinsch C, Auwerx J. Impact of the natural compound urolithin A on health, disease, and aging. Trends Mol. Med. 2021;27(7):687–699. doi: 10.1016/j.molmed.2021.04.009. [DOI] [PubMed] [Google Scholar]
- 80.Pradeepkiran JA, Hindle A, Kshirsagar S, Reddy PH. Are mitophagy enhancers therapeutic targets for Alzheimer’s disease? Biomed. Pharmacother. 2022;149:112918. doi: 10.1016/j.biopha.2022.112918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Subedi L, Ji E, Shin D, Jin J, Yeo JH, Kim SY. Equol, a dietary daidzein gut metabolite attenuates microglial activation and potentiates neuroprotection in vitro. Nutrients. 2017;9(3):207. doi: 10.3390/nu9030207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Full GWAS summary statistics of mbQTLs analyzed during this study are available at https://mibiogen.gcc.rug.nl/. Genotyping data for the NIA/LOAD (phs000168.v2.p2) and GenADA (phs000219.v1.p1) cohorts are available at https://www.ncbi.nlm.nih.gov/gap/. All data generated in this study are included in the manuscript, Supplementary Tables S1–3, and Supplementary Fig. S1.