

Summary
DNA damage has long been advocated as a molecular driver of aging. DNA damage occurs in a stochastic manner, and is therefore more likely to accumulate in longer genes. The length-dependent accumulation of transcription-blocking damage, unlike that of somatic mutations, should be reflected in gene expression datasets of aging. We analyzed gene expression as a function of gene length in several single-cell RNA sequencing datasets of mouse and human aging. We found a pervasive age-associated length-dependent underexpression of genes across species, tissues, and cell types. Furthermore, we observed length-dependent underexpression associated with UV-radiation and smoke exposure, and in progeroid diseases, Cockayne syndrome, and trichothiodystrophy. Finally, we studied published gene sets showing global age-related changes. Genes underexpressed with aging were significantly longer than overexpressed genes. These data highlight a previously undetected hallmark of aging and show that accumulation of genotoxicity in long genes could lead to reduced RNA polymerase II processivity.
Subject areas: Biological sciences, Molecular biology, Molecular Genetics, Omics, Transcriptomics
Graphical abstract
Highlights
-
•
Transcription-blocking DNA damage is more likely to accumulate in longer genes
-
•
Aged tissues show a global length-dependent gene underexpression in mice and humans
-
•
The effect is also seen in murine UV-radiated skin and in airway cells from smokers
-
•
The effect is also seen in progeroid TC-NER defects Cockayne syndrome and TTD
Biological sciences; Molecular biology; Molecular Genetics ; Omics; Transcriptomics
Introduction
DNA damage has long been proposed as a primary molecular driver of aging.1,2,3,4 Aging has also been associated with a series of transcriptional changes, most of which are highly tissue- and cell type-specific.5 Even though the search for a global aging signature has been the goal of much research,6,7,8,9 meta-analyses have shown that very few genes are consistently up- or downregulated with aging across different tissues.10 It appears that, at the mRNA level, aging signatures are not defined by the overexpression of particular sets of genes, but rather an overall decay in transcription.11 In fact, the differences between the transcriptome of middle-aged and young individuals are bigger than those between young and old individuals, at least in some human tissues.12
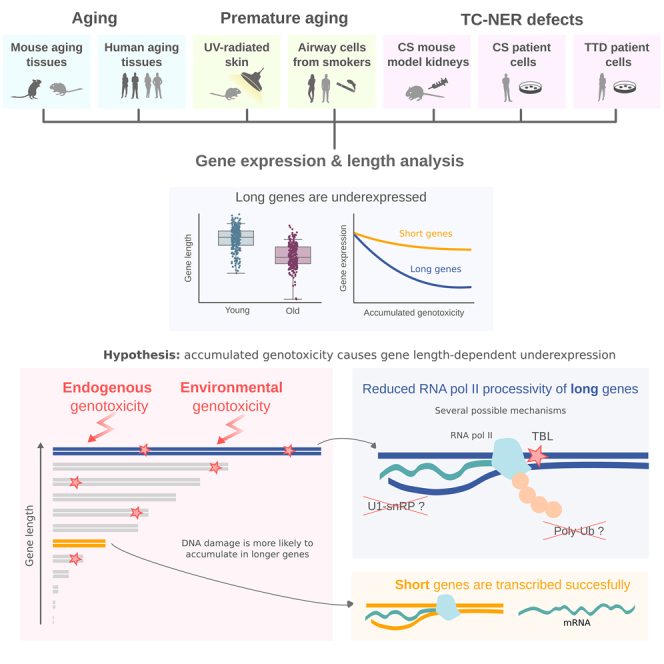
Genetic material is constantly challenged throughout the lifespan of the organism, both by endogenous and environmental genotoxins. Some of this damage happens in the form of transcription-blocking lesions (TBLs), which impede transcriptional elongation.13 Accumulation of TBLs provokes a genome-wide shutdown of transcription, which also affects undamaged genes through poorly understood mechanisms that may be related to RNA polymerase II (RNAP II) ubiquitylation and degradation.14,15 Assuming a constant TBL incidence, meaning that any base pair in the genome has a similar probability of suffering damage that results in a lesion, a greater accumulation of TBLs is to be expected in longer genes. In fact, a gene length-dependent accumulation of other forms of genetic damage, like somatic mutations, has already been reported in conditions like Alzheimer’s disease.16,17 Hence, TBLs, just like somatic mutations are expected to accumulate with aging, and their accumulation should be dependent on gene length. However, unlike somatic mutations, TBLs have a strong and direct impact on mRNA production, and their gene length-dependent effects are likely to be measurable from RNA sequencing data of aged tissues, which make single-cell RNA sequencing (scRNA-seq) atlases and datasets of aging an excellent opportunity to characterize them at the cell-type-level over a wide range of tissues.
So far, a potential relationship between age-related transcriptional changes and gene length has received relatively little attention. A recent analysis of the transcript length of 307 genes related to aging (as extracted from the GenAge database) found longer transcript lengths in these genes than that of the rest of the protein-coding genes.18 However, when they studied aging gene-expression signatures from a human, mouse, and rat meta-analysis, they found no significance regarding transcript length in overexpressed and underexpressed genes, the only exception being the brain (which downregulated long genes). Of interest, a previous analysis of gene expression profiles in the liver of mice deficient in the DNA excision-repair gene Ercc1, which present features of accelerated aging, had found specific downregulation of long genes.19 The same authors reported similar findings in naturally aged rat liver and human hippocampus, indicating that it could reflect a more generalized phenomenon. Here, we aimed to extend these early observations, which were based on bulk microarray and RNA sequencing data to the existing aging datasets based on scRNA-seq technology. We also extended our gene length analyses to mouse and human datasets of lifestyle-induced genotoxic exposure (UV, smoke) and progeroid syndromes (Cockayne syndrome and trichothiodystrophy).
Results
Age-associated shutdown of transcription preferentially affects long genes
In order to test if gene expression at the single-cell level is conserved with aging, we first analyzed 11 organs of the landmark Tabula Muris Senis (TMS) dataset of mouse aging20 on the basis of having enough experimental replicates and single cells for statistically significant analyses. Thus, we selected male animals of both young (3-month) and old (24-month) age (Figure 1). Plotting the average gene expression of aged tissues against their young counterpart’s yielded scatterplots where data presented a high linear correlation between both average expression vectors (Figure 1A). However, we observed that a large number of genes lied below the y = x line, meaning that their mean expression was lower in old mice. This was most evident in brain, heart, liver, lung, muscle, pancreas, and skin. Having established that there is an age-related decline in mRNA production, we explored the gene-length dependence of such decline. To this end, we split the whole transcriptome into four equally sized bins according to gene length and fitted a multiple linear regression model considering the interaction effect between average expression in young and the categorical variable representing the gene-length quartile. We found that the slope of the straight line that fits the gene expression data decreases with gene length, which confirms that the decay in mRNA production is strongly dependent on gene length. We graphically show this difference for the two most extreme quartiles (the 25% shortest and the 25% longest genes) in Figure 1B (gene lengths and p values for all comparisons are shown in Tables S1 and S2). The differences in gene lengths were statistically significant in all analyzed organs.
Figure 1.

Age-associated shutdown of transcription preferentially affects long genes
(A) Gene expression is highly conserved but shows a detectable decay with aging. Scatterplots showing the average gene expression in 24-month-old mice against average gene expression in 3-month-old mice in 11 tissues (12 comparisons) from the TMS FACS and the TMS droplet datasets.20 Each dot represents a gene. N: number of single cells; n: number of biological replicates. R2: coefficient of determination. The gray line represents y = x.
(B) A generalized shutdown of transcription is apparent in long genes. The scatterplots show the average gene expression of the 25% shortest (yellow) and the 25% longest (blue) genes in 24-month-old versus 3-month-old mice. βs and βl represent the slopes of the straight lines that best fit the data points corresponding to short and long genes, respectively. The number of young (ny) and old (no) biological replicates are shown.
See also Figures S1 and S2 and Tables S1 and S2.
In addition, we conducted a bootstrap-based permutation analysis for B = 200 bootstrap samples for the same TMS datasets. In each bootstrap sample, we adjusted the regression model with an interaction term considering the continuous log10 (gene length) variable. Results showed that the interaction term was statistically significant (p value < 0.0001) in all the 200 bootstrap samples considered (data not shown).
This length-dependent effect was also detected in independent scRNA-seq datasets obtained from mouse lung, kidney, spleen, and skin,20,21,22,23,24 although there were relevant experimental differences among datasets (Figure S1). Importantly, downregulation of longer genes was also evident in single-cell data of human lung, pancreas, and skin25,26,27,28 (Figure S1). Similarly, the effect was sex-independent since it was also detectable in TMS female animals (Figure S2). These results suggested a generalized underexpression of long genes associated with age, which is seen across tissues, sexes, and species, and in data extracted from several independent scRNA-seq datasets.
Differentially expressed genes between young and old individuals show a preferential bias toward long gene underexpression
A number of genes change their expression in the same direction during aging in several tissues, and the search for differentially expressed genes (DEGs) may thus, provide a molecular signature of aging.9 We next analyzed if DEGs between young and old animals from the TMS dataset showed a preferential bias toward the underexpression of long genes (Figure 2). Indeed, DEGs between young (3-month) and old (24-month) mice showed a global and strong bias for the underexpression of long genes for all tissues and comparisons, as seen in the volcano plots (Figure 2A). Differences in gene length were apparent as well in the boxplots showing the top 300 DEGs between age groups (Figure 2B). Differences in top 300 DEG lengths were statistically significant, based on a Wilcoxon-Mann-Whitney test (p values are provided in the Table S3). Once more, this effect was not specific to the TMS dataset, since it was also detected in independent scRNA-seq datasets obtained from mouse lung, kidney, spleen, and skin and human lung, pancreas, and skin (Figure S3). Finally, the effect was also detectable in TMS female animals (Figure S4). Despite the fact that inter-individual and inter-tissue differences were apparent in some cases, these data confirmed that long genes were differentially affected by the age-associated shutdown of gene expression.
Figure 2.

Differentially expressed genes between young and old individuals show a preferential bias for the downregulation of long genes
Output of the differential expression analysis between old (24 months) and young (3 months) male mice in 12 datasets from the Tabula Muris Senis (using the Wilcoxon method).
(A) Volcano plots showing the length of DEGs: -log10(p value) against log2(fold change). Each gene is colored according to its log10-transformed length.
(B) Boxplots showing the top 300 DEGs for each category. Whiskers extend to extend to the furthest datapoint within the 1.5∗IR (interquartile range). “Young”: top 300 most overexpressed genes in young cells with respect to old cells (blue). “Old”: top 300 most overexpressed genes in old cells with respect to young cells (pink). The difference in DEG length is significant in all tissues (p value < 0.001); see Table S3.
Se also Figures S3 and S4.
The age-associated decrease in the expression of long genes is not cell type-specific
Since many aging signatures are cell type-specific, a relevant open question was if the age-associated underexpression of long genes might be restricted to a particular cell type that is abundantly and ubiquitously located across tissues, such as fibroblasts or endothelial cells. To answer this question, we selected the four existing TMS heart datasets and analyzed the gene length of expressed genes (Figure 3, p values of the regression analysis are provided in Table S4). As expected, shorter genes were overexpressed in old mice as compared to those in young mice in all four datasets (Figure 3A). Compartmentalization of the analyses onto the 11 single-cell types detected in at least two datasets showed that young animals expressed longer genes in all cell types analyzed, including tissue-specific cells such as cardiomyocytes and infiltrating cell types such as B and T lymphocytes (Figure 3B). Therefore, a pervasive underexpression of long genes was detectable across aged cell types.
Figure 3.

The age-associated decrease in the expression of long genes is not cell type-specific
(A) Genes ranked according to their age-related difference in average gene expression. Genes are shown sorted according to their difference in mean expression between old and young cells. The positions of the top 200 shortest (yellow) and the top 200 longest (blue) genes are shown according to their overexpression in young (left) or in old animals (right).
(B) Genes differentially expressed between old and young murine heart cells have significantly different gene lengths. Gene length of the top 200 DEGs between young and old cells within each cell type. Whiskers extend to the furthest datapoint within the 1.5∗IR. “Young”: top 200 most overexpressed genes in young cells with respect to old cells. “Old”: top 200 most overexpressed genes in old cells with respect to young cells.
EC, endothelial cell. SMC, smooth muscle cell. Significant differences (Wilcoxon-Mann-Whitney test, p value < 0.01) are marked with an asterisk (∗). See also Table S4.
Genotoxic UV exposure of young mouse skin mimics age-associated decrease in the expression of long genes
Ultraviolet (UV) radiation of skin exposed to sunlight produces accumulation of DNA damage and photoaging.29,30 Notably, UV-induced photolesions—mainly cyclobutane pyrimidine dimers (CPDs) and pyrimidine-(6-4)-pyrimidone photoproducts (6-4 PPs)—trigger a general shutdown of transcription and are mainly fixed by the nucleotide excision repair (NER) pathways.31 The vitamin D system provides a local adaptive response to UV radiation, reducing DNA damage, inflammation, and photocarcinogenesis.32 To test if genotoxic damage to DNA (a premature aging model) also affected the transcription of long genes, we analyzed a single-cell RNAseq dataset of young (five to six-week-old) mouse skin irradiated with UVB or normal light33 (Figure 4). One of the UV-irradiated groups was injected with vitamin D before exposure (Figure 4A). A Uniform manifold approximation and projection (UMAP) plot of the merged datasets of mice skin shows the 11 cell types detected in this experiment using unsupervised cell clustering (Figure 4B). A global differential expression analysis showed that UV radiation causes long genes to be underexpressed in untreated mice. This effect was not evident on vitamin D-treated animals (Figure 4C, left and right panels, respectively). As expected, a ranking of genes according to their differential expression showed that the top 200 shortest and top 200 longest genes were located at positions consistent with a non-uniform distribution (Figure 4D). An analysis of the length of the top 300 DEGs computed between the three conditions (the genes differentially expressed in each of the conditions against the remaining two) further demonstrated that longer genes were differentially affected by UV exposure (Figures 4E and 4G). Finally, this effect was detected in all skin cell types; although not all long gene transcriptional phenotypes were rescued by vitamin D injection (Figure 4F). These results strongly suggested that environmental genotoxic damage by UV-radiation might induce a generalized shutdown of long gene transcription in young animals, which may be partially reverted by vitamin D injection.
Figure 4.

Genotoxic UV exposure of young mouse skin mimics age-associated decrease in the expression of long genes
(A) Experimental workflow: mice were sorted into three groups (n = 3 per condition). The Vit D group was injected with a vitamin D treatment. All mice were shaved and irradiated, either with UV light (Vit D and UV groups) or with visible light (healthy). Skin samples were extracted and analyzed using scRNAseq.
(B) UMAP plot showing 11 cell types in the murine skin dataset (Lin et al., 2022). The samples corresponding to the three conditions were merged into a single dataset. Diff, differentiated. EC, endothelial cell. HF, hair follicle. IFE, interfollicular epidermis. Kerat, keratinocytes. SG, sebaceous gland.
(C) Volcano plots showing UV radiation-related gene overexpression without prior vitamin D treatment (left) and with the vitamin D treatment (right): -log10(p value) against the log2(fold change). DEGs were computed using the Wilcoxon method. Each gene is colored according to its log10-transformed gene length.
(D) Position of the top 200 shortest (yellow) and top 200 longest (blue) genes in the differential expression ranking. Genes are shown ranked according to their difference in mean expression between every pair of conditions and colored according to their length.
(E) Boxplots showing the log10(length) of the DEGs between conditions. Top 300 DEGs were computed between the three conditions (those differentially expressed in each of the conditions against the remaining two). The differences were statistically significant (p values correspond to the Tukey post-hoc test after ANOVA).
(F) Boxplots showing the log10(length) of the DEGs between conditions per cell type. The DEGs were computed between the three conditions for each cell type separately. Whiskers extend to the furthest datapoint within the 1.5∗IR.
(G) Scatterplots showing the mean expression in every pair of conditions: UV-radiated vs healthy skin (top), UV-radiated vs vitamin D treated skin (middle), and vitamin D-treated vs healthy skin (bottom). βs and βl correspond to the slopes of the multiple linear regression models with interaction fitted on the first and fourth quartiles (top 25% shortest and top 25% shortest genes), respectively.
See also Table S5.
Smoke exposure of human airways mimics age-associated decrease in the expression of long genes
Chronological age of never-smokers does increase the frequency of mutations in bronchial epithelial cells at a rate of 28 mutations per cell per year. Mutation frequency in cells from smokers increased at a rate of 91 mutations per cell per year, i.e., 3.25X higher.34 In addition to somatic mutations, exposure to smoke from organic matter is known to provoke TBLs.13 This seems to be due to benzo[a]pyrene diol epoxide (BPDE) reacting with guanines to form bulky DNA adducts.15 To test if the lifestyle of smokers affected specifically the expression of long genes in airway epithelial cells, we analyzed a scRNA-seq dataset35 of human trachea of never-smokers and heavy smokers (subjects who had been smoking for > 20 years) of a similar age range (Figure 5A). A UMAP plot of the merged datasets of both never-smokers and heavy smokers detected 13 cell types in human trachea (Figure 5B). As expected by their increased accumulated genotoxicity, long gene expression significantly decreased in heavy smokers as compared to never-smokers (Figures 5C–5E and 5G, p values in Table S5). Once more, this effect was not cell-specific since it was detected in all tracheal cell types (Figure 5F). These results confirmed that environmental genotoxic damage induces a generalized shutdown of long gene transcription.
Figure 5.

Smoke exposure of human airways mimics age-associated decrease in the expression of long genes
(A) Experimental workflow: trachea samples from three non-smokers and five heavy smokers (for > 20 years) were analyzed through scRNAseq.
(B) UMAP showing the 13 detected cell types in the human trachea dataset. The samples corresponding to the two conditions (never-smokers and heavy smokers) were merged into a single dataset. Diff, differentiated. KRT8, Keratin 8. PNEC, pulmonary neuroendocrine cells. Prolif, proliferating. Prot, proteasomal. SMG, submandibular salivary glands.
(C) Volcano plot showing smoking-related gene overexpression: -log10(p value) against the log2(fold change). Each gene is colored according to its log10-transformed gene length.
(D) Position of the top 200 shortest (yellow) and top 200 longest (blue) genes in the differential expression ranking. Longest genes are underexpressed in airway cells from heavy smokers.
(E) Boxplots showing the log10(length) of the DEGs between heavy smokers and never-smokers, computed using the Wilcoxon method. “Never”: top 300 most overexpressed genes in cells from never-smokers with respect to cells from heavy smokers. “Heavy”: top 300 most overexpressed genes in cells from heavy smokers with respect to cells from never-smokers. The distributions of log10 gene length (bp) were significantly different between conditions (Mann-Whitney U test).
(F) Boxplots showing the DEGs between the conditions for each cell type. DEGs associated with heavy smoker airway cell types are significantly shorter. DEGs were computed between never-smokers and heavy smokers for each cell type separately. Whiskers extend to the furthest datapoint within the 1.5∗IR.
(G) Scatter plot showing the average gene expression in heavy smokers against the average gene expression in never-smokers. βs and βl correspond to the slopes of the linear regression models fitted on the first and fourth quartiles (top 25% shortest and top 25% shortest genes), respectively.
See also Table S5.
Transcriptional stress in progeroid diseases cockayne syndrome and trichothiodystrophy results in underexpression of long genes
A number of progeroid diseases are caused by mutations functionally linked to genome maintenance and DNA damage repair.36 Of particular interest to this work, a subset of defects in repair genes impair transcription-coupled nucleotide excision repair (TC-NER), i.e. TBLs remain unrepaired, causing RNAP II stalling and ultimately syndromic features such as Cockayne syndrome, xeroderma pigmentosum, and trichothiodystrophy.13 Of interest, increased cutaneous photosensitivity is one of the clinical features of patients suffering from these conditions, and is caused by deficiencies in genes coding for components of the TC-NER. To explore if long gene transcription is specifically affected in progeroid diseases caused by TC-NER deficiencies, we generated three independent lines of evidence: (i) a dataset of a mouse model of Cockayne syndrome, (ii) a dataset based on cells derived from a human Cockayne syndrome patient, and (iii) a list of DEGs between a trichothiodystrophy patient and her healthy mother.
Endogenous formaldehyde is abundant in the body, causing DNA crosslinks, oxidative stress, and potentially contributing to the onset of Fanconi anemia and other syndromes37 (Figure 6). On the other hand, Cockayne syndrome is caused by loss of the Cockayne syndrome A (CSA) or CSB proteins. Double knock-out mice deficient in both formaldehyde clearance (Adh5−/−) and CSB protein (Csbm/m) develop transcriptional stress in a subset of kidney cells and features consistent with human Cockayne syndrome38 (Figure 6A). To test if kidney cells of these animals undergoing formaldehyde-driven transcriptional stress specifically decreased transcription of long genes, we analyzed single-cell datasets of three knockout mice—ADH5KO (deficient in formaldehyde clearance), CSBKO (Cockayne syndrome group B knockout, also known as Ercc6), and DKO (Adh5−/− Csbm/m double knock-out)—against those of wild-type (WT) mice (Figure 6B). A UMAP plot of the merged datasets of all data showed no obvious batch effect between animal groups (Figure 6C). Interestingly, specific downregulation of long genes was already detected in ADH5KO and CSBKO single mutants (Figure 6D). Both mutations seemed to synergize causing further downregulation of long genes in the DKO animals as compared to WT mice (Figures 6D–6F, p values in Table S5).
Figure 6.

A mouse model of Cockayne syndrome mimics age-associated decrease in the expression of long genes in the kidney
(A) In mouse kidney, methanol is oxidized to formaldehyde, which causes DNA damage. Damage can be prevented by the effect of alcohol dehydrogenase (Adh5), which clears formaldehyde and is repaired by the TC-NER system. The Cockayne syndrome group B (Csb) protein is part of the TC-NER system that repairs DNA damage. Mice that have either (or both) of these two genes knocked out will suffer increased levels of DNA damage.
(B) Experimental approach: two mice of each genotype (WT: wild-type; ADH5KO: deficient in formaldehyde clearance; CSBKO: Cockayne syndrome group B knock-out, also known as Ercc6, and DKO: Adh5−/−Csbm/m double knock-out) were subjected to a genotoxic methanol treatment (1.5 g/kg via intraperitoneal injection once a week). Kidneys were harvested and gene expression analyzed using scRNAseq.
(C) UMAP plot showing the genotype of proximal tubule cells selected for the analysis.
(D) Volcano plots showing the output of differential expression analysis of each knockout against wild-type mice: -log10(p value) against the log2(fold change). DEGs were computed using the Wilcoxon method. Each gene is colored according to its log10-transformed gene length.
(E) Boxplots showing the length distribution of the top 300 DEGs obtained when comparing each of the knockouts against wild-type mice. Whiskers extend to the furthest datapoint within the 1.5∗IR.
(F) Scatterplots showing the average gene expression in each of the knockouts against wild-type mice. βs and βl correspond to the slopes of the linear regression models fitted on the first and fourth quartiles (top 25% shortest and top 25% shortest genes), respectively.
See also Table S5.
Encouraged by these results, we analyzed a bulk RNAseq dataset of human mesenchymal stromal cells (MSCs) derived from a Cockayne syndrome patient bearing a CSB/ERCC6 mutation, which are known to present marked changes in their transcriptome upon UV-radiation31 (Figure 7). In fact, skin fibroblasts from this patient were first reprogrammed to generate induced pluripotent stem cells (iPSCs), which were then gene-corrected with CRISPR-Cas9 and differentiated to MSCs. Thus, the available data included UV-radiated MSCs vs MSCs in normal conditions in both mutant (ERCCmut) and gene-corrected (ERCCGC) backgrounds (Figure 7A). First, we analyzed the baseline effect of bearing the ERCC6 mutation and observed that, while there was no significant difference in gene length between gene corrected and mutant cells in basal conditions, mutant cells expressed shorter genes than gene corrected cells upon UV-radiation (Figures 7B and 7C). We then compared the effect of UV-radiation on gene corrected and mutant cells separately. As expected, UV-radiation on ERCCmut cells induced a decrease in long gene expression as compared to normal conditions in both mutant and gene-corrected (ERCCGC) cells (Figures 7D and 7E). Overall, these results demonstrated that photosensitivity in ERCCmut cells caused underexpression of long genes.
Figure 7.

Human Cockayne syndrome and trichothiodystrophy (TTD)-derived cells mimic age-associated decrease in the expression of long genes
(A) Experimental approach: skin fibroblasts were extracted from a Cockayne syndrome (ERCC6mut) patient and reprogrammed to generate induced pluripotent stem cells (iPSCs), gene-corrected using CRISPR-Cas9 (ERCC6GC), and differentiated to mesenchymal stromal cells (MSCs). The transcriptome of ERCC6mut and ERCC6GC MSCs was analyzed using RNAseq in basal conditions and after UV-radiation exposure.
(B and C) Baseline effect of the Cockayne syndrome group B (ERCC6) mutation on length-dependent expression. (B) Boxplots showing the length of the top 300 DEGs between mutant (ERCC6mut) and gene-corrected (ERCC6GC) cells in normal conditions (control) and after UV-radiation exposure. Whiskers extend to the furthest datapoint within the 1.5∗IR. (C) Average gene expression of mutant against gene-corrected cells in basal conditions and after UV-radiation exposure.
(D and E) Effect of UV-radiation on cells carrying the ERCC6 mutation and gene-corrected cells. (D) Boxplots showing the length of the top 300 DEGs between cells with and without UV-radiation exposure. Whiskers extend to the furthest datapoint within the 1.5∗IR. (E) Average gene expression in UV-radiated cells against cells in basal conditions.
(F) Experimental approach: dermal fibroblasts from a PS-TTD patient (ERCC2mut) and her healthy mother were extracted and analyzed using RNAseq.
(G and H) Length of the DEGs (|logFC| ≥ 2 and p value ≤ 0.05) between PS-TTD cells and healthy cells in basal conditions (G) and upon UV-radiation (H). Whiskers extend to the furthest datapoint within the 1.5∗IR.
See also Table S5.
Finally, we tested if long gene expression was also affected in photosensitive trichothiodystrophy (PS-TTD), another TC-NER-deficient progeroid syndrome (Figures 7F–7H). To this end, we analyzed the length of the DEGs obtained by Lombardi et al.39 between a cancer-free PS-TTD patient carrying a mutation in the ERCC2 gene and her healthy mother, both in basal conditions and upon UV-radiation (Figure 7F). Selecting the genes that were significantly (p value ≤ 0.05) over- or underexpressed in PS-TTD and with a substantial effect size (logFC ≥ 2 in either direction) we observed that the DEGs associated with PS-TTD were significantly shorter upon UV-radiation (Figures 7G and 7H). These results suggested that other progeroid syndromes might present a similar phenotype of reduced long gene expression.
Published aging signatures are influenced by gene length-dependent transcriptional decay
A number of aging-related transcriptional signatures have been proposed for both mice and humans. A recent study identified a set of mouse global aging genes (GAGs),9 defined as genes whose expression varies substantially with age in most (> 50%) of the tissue-cell types across several tissues of the TMS dataset. These authors found that GAGs exhibited a strong bimodality, i.e., that they were either upregulated or downregulated with aging in most tissues. However, gene length was not analyzed in that study. To test if the length of GAGs influenced their up- or downregulation, we represented the distribution of log-transformed gene lengths in the two groups (Figure 8). We found that downregulated GAGs are longer than those that were found to be upregulated and that their difference in length is statistically significant (Figure 8A, Wilcoxon-Mann-Whitney test, p value < 0.001).
Figure 8.

Published aging signatures are influenced by gene length-dependent transcriptional decay
Down-regulated genes are longer than up-regulated genes in two published aging transcriptomic signatures. The length of the genes from two independent aging signatures, murine (A) and human (B), are shown as two overlapped histograms and separate boxplots. The number of down-and up-regulated genes in each signature are shown as nd and nu, respectively. The gene length is significantly different between the two categories according to the Mann-Whitney test (p values shown in the figure). Whiskers extend to the furthest datapoint within the 1.5∗IR.
In humans, the first large-scale meta-analysis (14,983 individuals) of aging-related gene expression profiles identified 1,497 genes differentially expressed with chronological age in peripheral blood mononuclear cells.40 Interestingly, long genes downregulated with aging in this human cohort, the differences in length between upregulated and downregulated genes being statistically significant (Figure 8B, Wilcoxon-Mann-Whitney test, p value = 0.0015). Overall, these data suggest that transcriptomic aging signatures are influenced by gene length.
Discussion
In this article, we report that a generalized age-related decline in gene expression is dependent on gene length. The fact that gene length affects mRNA expression levels has long been known.41 In early development, gene size and architecture influence the expression timing of specific genes.42 This is also true more generally, for instance in the immediate cellular response to external stimuli, where shorter pre-mRNA molecules are synthesized first.43 Furthermore, gene lengths appear to be compartmentalized among chromosomes and tissue-specific expression patterns may be detected.44
RNA polymerase II (RNAP II)-driven transcription can be divided into initiation, pausing, elongation, 3′ end formation, and termination stages; each step being tightly regulated.45 Once initiated, transcription pauses downstream from the transcription start site and requires specific signaling for pause-release, elongation and processivity. Cyclin-dependent kinases CDK12 and CDK13 seem to be involved in the regulation of RNAP II elongation, processivity, and selection of alternative polyadenylation sites.46 Of interest, the GC content of the initially transcribed sequence determines early RNAP II elongation rates, and recognition of a 5′ splice site (SS) by U1 snRNP promotes RNAP II elongation potential.47 This is related to a process known as telescripting, whereby U1 snRNP base pairing with 5′SS avoids premature 3′ end cleavage and polyadenylation at cryptic intronic sites.48,49 It is likely that long gene transcription is mediated by many other RNA-binding proteins (RBPs) as well, many of which have additional functions in the regulation of pre mRNA splicing.50 In fact, only about half of the introns present in newly synthesized pre-mRNA are co-transcriptionally spliced,51 further supporting alternative roles for specific RBP subsets. Although, we have no mechanistic understanding of which dysfunction mediates the apparent loss of long gene transcription associated with aging, our data may generate new avenues for aging-related research, where the relevance of pathways related to RNAP II elongation and processivity remains virtually unexplored.
Premature transcript termination by RNAP II has already been described in some contexts. An increase in elongation rate (speed) concomitant to premature termination at cryptic intronic polyadenylation signals has recently been reported during heat shock, which was mediated by inhibition of U1 telescripting.52 Interestingly, failure to target the stalled RNAP II for degradation by polyubiquitination of a single residue is enough to shutdown long gene transcription, the expression of shorter genes being unaffected.53,54 Further, the concept of long-gene transcriptopathy has been proposed as a possible mechanism underlying a number of neurological and psychiatric disorders some of which are age-associated.16,17,50,55,56 RNA-binding protein SFPQ mediates CDK9 recruitment to the transcription elongation complex, which activates RNAP II-CTD. Neuron-specific ablation of SFPQ downregulated a regulon of 135 genes, which account for less than 10 percent of the genes with a pre mRNA > 100 kb in length inducing neuronal cell death and embryonic lethality.56 Similarly, muscle-specific ablation of SFPQ induced metabolic myopathy, severe progressive muscle mass reduction, and impairment of motor function. This was shown to be mediated by downregulation of long genes regulating energy metabolism in skeletal muscle.50 While the specific mechanisms underlying the generalized age-associated downregulation of long genes that we report here remain to be determined, it seems likely that they will be related to some of the aforementioned mechanisms. For example, a longitudinal analysis of gene expression differences in a human cohort that followed 65 healthy individuals between ages 70 and 8057 found changes in the expression of the SFPQ gene among the strongest associations with age. Of note, the key importance of RNA metabolism dysregulation in human aging has long been known.58
Accumulation of genotoxic damage with chronological age is pervasive, and it may also be significantly incremented through lifestyle choices.29,34,59,60 The fact that augmented DNA damage specifically induces downregulation of long genes is of great interest. A recent study has shown that UV-mediated global transcription shutdown favored transcription restart from shorter mRNAs with less exons.61 Similarly, transcription blockage by DNA damage is known to generate neurodegenerative processes associated with human genetic syndromes deficient in nucleotide excision repair, such as Cockayne syndrome and xeroderma pigmentosum.62 Our data showing that several models of progeroid disease specifically downregulate long genes are most likely true as well for other TC-NER syndromes.
The search for aging-related gene signatures has provided relatively little advance to the field. In our opinion, the straightforward mechanism depicted here (of DNA damage-induced loss of RNAP II processivity as a molecular driver of aging) might better explain many of the age-associated features and may thus provide a fruitful research avenue for the aging field. Alternatively, other mechanisms distinct of TBL accumulation and loss of RNAP II processivity might also be conceived. For instance, an epitranscriptomic mechanism mediated by m6A-marked intronic LINE-1 elements has recently been suggested to preferentially impair long gene transcription in human neurons. This may in turn be counteracted by RNA-binding proteins SAFB and SAFB2, which are highly expressed in the hippocampus and cerebellum.63 As our knowledge of 3D chromatin topology advances, it is likely that novel potential mechanisms will arise. In another relevant example, long (> 300 kb) neuronal genes have been shown to present a “gene-decondensed” or “melted” state in mouse brain slices that results in higher levels of chromatin accessibility and gene transcription.64 The authors suggested that extensive melting of long genes was associated with the resolution of topological constraints. It will be interesting to see whether this is still the case in aged mouse brains. Finally, a third unexplored possibility is that changes in cell cycle duration act as a “transcriptional filter” that constrains transcription of long genes. Mathematical simulations of embryonic development already suggest the relevance of such mechanisms in early cell type specification.65
Importantly, while this manuscript was under review, other authors66 independently reached the conclusion that there is a strong transcript length association with aging. Of note, they reported that the age-associated transcriptome imbalance was countered by several distinct anti-aging interventions (7 out of 11 interventions tested), indicating that this phenomenon may be (at least partly) reversible, and thus amenable for pharmacologic intervention. On the other hand, another recent work in aged mouse liver by Gyenis et al.67 found that accumulation of transcription-blocking DNA damage during normal aging causes RNAP II stalling and leads to disruption of long gene transcription. While this mechanism is compatible with our findings, TBLs in principle should not be reversible. However, tissues with high turnover constantly replace damaged cells, and thus the additive effect of cellular aging may be diluted. Future work should shed light on the specific mechanisms underlying loss of long gene transcription associated with aging.
Limitations of the study
This study is mainly limited by the fact that, despite the strong evidence for a gene length-dependent decrease in mRNA production associated with aging, the underlying mechanism is yet to be fully understood and experimentally validated. Additionally, it is not possible to tell from current single-cell RNAseq data whether the length-dependent imbalance is due to underexpression of long genes and/or to overexpression of short genes. The evidence presented here is entirely based on reanalyzes of bulk and single-cell RNA sequencing datasets. Further research will be needed to determine the exact mechanisms that result in this decrease in long gene expression.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Single-cell RNAseq datasets of 12 tissues from the Tabula muris senis. Male mice aged 3 and 24 months. Organs: bladder, brain, brain myeloid, heart, kidney, liver, lung, muscle, pancreas, skin, spleen and thymus. | Almanzar et al. (2020)20 | https://doi.org/10.6084/m9.figshare.12654728.v1 |
| Single-cell RNAseq datasets of 12 tissues from Tabula muris senis. Female mice aged 3 and 18 months. Organs: muscle, brain, brain myeloid, heart, heart, thymus, skin, pancreas, mammary gland, spleen and kidney. | Almanzar et al. (2020)20 | https://doi.org/10.6084/m9.figshare.12654728.v1 |
| Single-cell RNAseq datasets of heart and aorta from the Tabula muris senis. Male and female mice aged 3, 18, 21 and 24 months. | Almanzar et al. (2020)20 | https://doi.org/10.6084/m9.figshare.12654728.v1 |
| Single-cell RNAseq dtaaset of the murine aging lung. | Angelidis et al. (2019)21 | GEO: GSE124872 |
| Single-cell RNAseq datasets of the murine lung, spleen and kidney. | Kimmel et al. (2019)22 | GEO: GSE132901 |
| Single-cell RNAseq dataset of the murine aging brain. | Ximerakis et al. (2019)23 | GEO: GSE129788 |
| Single-cell RNAseq dataset of murine aging dermal fibroblasts. | Salzer et al. (2018)24 | GEO: GSE111136 |
| Single-cell RNAseq dataset of human lungs (Human lung cell atlas). | Travaglini et al. (2020)27 | Synapse: syn21041850 |
| Single-cell RNAseq dataset of lung cells from young (21, 22, 32, 35 and 41 years old) and old (64, 65, 76 and 88 years old) male and female healthy donors. | Raredon et al. (2019)28 | GEO: GSE133747 |
| Single-cell RNAseq dataset of human pancreatic cells from 21-22 and 44–54 years old male and female healthy donors. | Enge et al. (2017)25 | GEO: GSE81547 |
| Single-cell RNAseq dataset of human whole-skin from donors aged 25–27 (young) and 53–70 years (old). | Solé-Boldo et al., (2020)26 | GEO: GSE130973 |
| Single-cell RNAseq dataset of murine skin upon UV radiation treatment with and without vitamin D treatment, and control. | Lin et al. (2022)33 | GEO: GSE173385 |
| Single-cell RNAseq datasets of human airway cells from heavy smokers and never-smokers. | Goldfarbmuren et al. (2020)35 | GEO: GSE134174, samples T101, T120, T154, T167, T85, T164, T165, T166. |
| Single-cell RNAseq dataset of murine kidney cells from WT, Adh5 KO (aldehyde clearance deficient), Csb KO (impaired TC-NER) or a double KO after methanol treatment. | Mulderrig et al. (2020)38 | GEO: GSE175792 |
| RNAseq dataset of human Cockayne Syndrome (CS) and gene corrected (GC)-MSCs upon UV treatment and in normal conditions. | Wang et al. (2020)31 | GEO: GSE124208; samples GSM3525718, GSM3525717, GSM3525714, GSM3525715, GSM3525719, GSM3525716, GSM3525713 and GSM3525720 |
| List of DEGs between a cancer-free PS-TTD patient carrying a mutated ERCC2 gene and her healthy mother in basal conditions and upon UV-radiation. | Lombardi et al. (2021)39 | supplemental information (https://doi.org/10.1073/pnas.2024502118) |
| List of Global Aging Genes (GAGs). | Zhang et al. (2021)9 | https://github.com/czbiohub/tabula-muris-senis/tree/master/2_aging_signature |
| Software and algorithms | ||
| Jupyter Notebooks and R scripts to reproduce the analyses described in the article. | Gitlab | https://gitlab.com/olgaibanez/transcription_stress |
| Jupyter Notebooks to reproduce the analyses described in the article. | Figshare | https://doi.org/10.6084/m9.figshare.22140515.v1 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ander Izeta (ander.izeta@biodonostia.org).
Materials availability
This study did not generate new unique reagents.
Method details
Data inclusion criteria
In order to analyze balanced aging datasets, samples were selected according to the following criteria: i) When sex annotations were available, same-sex datasets were generated. ii) Individuals of the same age were used to create the "young" and the "old" cohorts. iii) In datasets including samples from different sub-tissues, samples corresponding to the sub-tissues with representation in the two age cohorts were selected. In murine datasets derived from Tabula Muris Senis data, 3 month-old and 24 month old mice were used to form the young and old cohorts, respectively. In all TMS female murine aging datasets 18-month animals were used to form the old cohort. In the murine dermal fibroblast dataset (Salzer et al. 2018), samples from newborn mice were not included. Regarding human aging datasets, samples from newborn and middle-aged individuals were discarded and sex-stratified cohorts were created when possible. In the human aging pancreas dataset (Enge et al. 2017), samples from pediatric donors as well as those from a 38-year old patient were not used. Thus, only two young (21 and 22 years old) and two old (44 and 54 years old) donors were included in the aging dataset. In the human trachea of heavy smokers and never-smokers dataset (Goldfarbmuren et al. 2020) only donors aged over 50 years were included in the dataset to avoid age as a confounding variable.
General data processing pipeline
Single-cell RNA-seq datasets were preprocessed using a standard preprocessing pipeline in Scanpy (Wolf et al. 2018): normalization, log-transformation of counts, feature selection using triku (Ascensión et al. 2022), dimensionality reduction through Principal Component Analysis (PCA) and Uniform Manifold Approximation and Projection (UMAP) (McInnes et al. 2018), and community detection using Leiden68 (Traag et al. 2019). In some cases, when the original labels were too granular, some cell identities were merged into broader categories before proceeding to downstream analyses.
Data processing of each dataset
Male murine aging datasets
TMS male mice aged 3 months and 24 months were selected to create balanced datasets of aging of 11 organs (12 comparisons): bladder, brain, brain myeloid, heart, kidney, liver, lung, muscle, pancreas, skin, spleen and thymus.20
Female murine aging datasets
Due to the lack of available 24-month-old females in the TMS dataset, we chose a set of 3-month and 18-month-old mice to create 12 balanced female aging datasets: TMSF muscle, TMSF brain, TMSF brain myeloid, TMSD heart, TMSF heart, TMSF thymus, TMSF skin, TMSF pancreas, TMSD mammary gland, TMSF mammary gland, TMSF spleen and TMSF kidney.
Additional murine and human datasets
We analyzed six additional murine aging datasets of several tissues: lung cells from 3 and 24-month-old mice21 (GEO:GSE124872), lung, spleen and kidney cells from 7 and 21-months-old mice22 (GEO: GSE132901), brain cells from 2-3 and 21-23 month-old mice23 (GEO: GSE129788) and dermal fibroblasts from 2 and 18-month-old mice24 (GEO: GSE111136). We also analyzed four human datasets: lung cells from 46 and 75 years old male healthy donors27 (available at Synapse under accession syn21041850), lung cells from young (21, 22, 32, 35 and 41 years old) and old (64, 65, 76 and 88 years old) male and female healthy donors28 (GEO: GSE133747), pancreatic cells from 21-22 and 44-54 years old male and female healthy donors25 (GEO: GSE81547), and whole-skin cells from 25-27 and 53-70 years old donors26 (GEO: GSE130973). Murine lung, human lung and human pancreas datasets were processed and cell type annotated as in Ibáñez-Solé et al.69
Murine aging heart
Four aging balanced datasets were created from samples from the TMS FACS heart and the TMS droplet heart and aorta datasets. All mice aged 3, 18, 21 and 24 months were selected and combined so that all mice representing an age cohort within a dataset were of equal age and sex: TMS FACS male (3–24 months), TMS FACS female (3–18 months), TMS droplet female (3–18 months) and TMS droplet female (3–21 months).
Murine UV-radiated skin with and without vitamin D treatment
The datasets of murine UV-radiated skin33 corresponding to the three conditions (healthy, UV-radiated and vitamin D) were downloaded from the Gene Expression Omnibus (GEO: GSE173385). We checked that the age of the mice used in the study was identical between conditions. The three datasets were subjected to the standard processing pipeline described in Data processing pipeline separately. Then, the Leiden community detection algorithm was run and cell type annotations were added to the resulting clusters based on the expression of known cell type markers. The murine dermal cell type characterization by Joost et al.70 was used as a reference.
The clusters were annotated based on the following gene markers: «IFE basal » (basal keratinocytes from the interfollicular epidermis, Krt5, Krt14, Mt2); «IFE diff.» (differentiating keratinocytes, Krt1, Krt10, Ptgs1); «IFE kerat.» (terminally differentiated cells in the keratinyzed layer, Lor, Flg2); «HF» (hair follicle cells, Krt17, Krt79, Sox9); «Fibroblast» (Col1a1, Col3a1, Col1a2, Dcn, Lum, Sparc); «Myeloid» (Cd74, Lyz2); «SG» (sebaceous gland cells, Mgst1, Scd1, Krt25, Pparg); «T cell» (Cd3d, Thy1, Nkg7); «EC» (endothelial cells, Mgp, Fabp4); «Melanocyte» (Mlana, Pmel, Tyrp1); «Erythrocyte» (Hbb-bs, Hbb-bt, Hbba-a2).
The Lilliefors normality test71 was conducted on the log-transformed lengths of the differentially expressed genes for each of the conditions, using Python module statsmodel. The null hypothesis – that the log10 gene lengths follow a normal distribution – could not be rejected (cutoff: 0.05), meaning that the distribution of gene lengths within each group is normally distributed. We tested whether the mean lengths of the DEGs were significantly different across conditions using ANOVA (stats.f_oneway). The null hypothesis that the three means were equal was rejected (p value 3.67E-06). Post-hoc analysis (Tukey test, scikit_posthocs.posthoc_tukey) was run to test which of the pairwise comparisons between the three conditions yielded a statistically significant difference. Additionally, statistical significance was confirmed with non-parametric alternatives: Kruskal-Wallis (stats.kruskal) and Dunn test (scikit_posthocs.posthoc_dunn).
Human airway cells from heavy smokers
The dataset used in Goldfarbmuren et al.35 was downloaded from the Gene Expression Omnibus (GEO: GSE134174). Original cell type annotations were used, but subtypes of the same cell types were pooled into a single category. The final dataset contained 13 cell types: «Diff. basal» (differentiating basal cells), «Prolif. basal» (proliferating basal cells), «Prot. basal » (proteasomal basal cells), «ciliated» (the two mature ciliated clusters –A and B– were pooled together), «ionocytes», «PNEC» (pulmonary neuroendocrine cells), «secretory/ciliating» (hybrid secretory early ciliating cells), «KRT8 high», «secretory» (mucus secretory cells), «tuft-like» (Tuft-like cells), «SMG basal» (basal cells from the submucosal gland or SMG, the two clusters –A and B– were pooled into a single category), «SMG myoepithelial» (myoepithelial cells from the SMG), «SMG secretory» (mucus secretory cells from the SMG).
In order to control for age as a possible confounding factor, we checked the ages of the subjects in the original dataset. We discarded the youngest donors and only kept samples from donors aged >50 years. The final dataset consisted of 21,425 cells from 8 donors. Heavy smokers (T101, T120, T154, T167, T85) were aged 55–66 years, and never-smokers (T164, T165, T166) were 64–68 years old. Since the average never-smoker age is slightly higher than the average heavy-smoker age, we can safely attribute transcriptional changes between these two groups to their smoking status.
The Lilliefors test71 was used to test whether the log10 (length) of the DEGs for the two conditions ("heavy smokers" and "never-smokers") were normally distributed. The null hypothesis could be rejected (cut-off: 0.05) for the "never-smokers", meaning that DEGs associated with that condition were not normally distributed, so a Mann-Whitney U test was used to compare between the means of the two distributions.
Kidney cells from mouse model of Cockayne Syndrome
The dataset generated by Mulderrig et al.38 was downloaded from the Gene Expression Omnibus (GEO: GSE175792). Proximal tubule cells were selected on the basis of marker expression, following the annotation done by the authors (see Extended Data, Figures and Tables from Mulderrig et al.38).
Human Cockayne Syndrome-derived MSCs
The dataset by Wang et al.31 was downloaded from the Gene Expression Omnibus (GEO: GSE124208). The following samples were included in the dataset: GSM3525718, GSM3525717, GSM3525714, GSM3525715, GSM3525719, GSM3525716, GSM3525713 and GSM3525720. Those samples correspond to four experimental conditions: MSCs from Cockayne syndrome patients carrying the ERCC6 mutation, with (UV) and without (ct) UV-radiation treatment (MSC_mut_ct, MSC_mut_UV); MSCs from gene-corrected cells with and without UV radiation treatment (MSC_GC_ct and MSC_GC_UV). All samples were merged into a single dataset and expression values were log-transformed.
DEG list between human PS-TTD-derived and healthy cells
The complete list of DEGs between a cancer-free PS-TTD patient carrying a mutated ERCC2 gene and her healthy mother in basal conditions and upon UV-radiation were obtained from the Supplementary Material provided by Lombardi et al.39 From the original DEG list, we selected the genes with a log fold-change greater than 2 (either overexpressed in the sample from the PS-TTD patient or in the sample from the healthy donor). The same threshold for statistical significance (p value ≤ 0.05) as the one defined by the original authors was used.
Icons used in the Figures
The following icons were downloaded from the Noun Project (CC BY 3.0): mouse (Pedro Santos), syringe (Anconer Design), lamp (Stan Diers), test tube (Misbahul Mun) cigarette (Robert Kyriakis), scissors (Sandra).
Gene length analysis
Human and mouse gene length annotations were obtained from Biomart. The version of Ensembl used in the analysis was Ensembl 106 (released in April 2022, human: GRCh38.p13; mouse: GRCm39). Gene length was calculated by subtracting the coordinates for the gene end from the gene start: “Gene end (bp)” - Gene start (bp)”.
Length-dependent difference in expression in aging and genotoxic conditions
Two different types of analyses were run between conditions: global average gene expression and length-dependence of transcriptional decay and gene length analysis of the differentially expressed genes between conditions.
Quantification and statistical analysis
Gene length dependence in age-related transcriptional decay
Here, we computed the average gene expression across all cells for a pair of conditions (for instance, "young" and "old"). We used a scatter plot to represent each gene according to its average expression in old cells (y axis) against its average expression in young cells (x axis). This is a way of looking at how predictable the expression of each particular gene is in old cells based on the expression of the same gene in young cells. As we observed that most genes show a strong correlation between young and old cells, even though many of them show expression levels that are lower than what we would have expected from their expression in young individuals, we then looked at the role gene length plays in this transcriptional decay. We did so by splitting the transcriptome into four quartiles according to their length. We considered whole sequence length from the transcription start site to the transcription end site. Then, we fitted a linear regression model to the average gene expression in old and young cells for each of the quartiles, thus obtaining a separate linear model for each quartile, using the formula ME_old ∼ ME_young ∗ Q, where ME old and ME young are the mean expression vectors for old and young cells, and Q is the vector that assigns each gene to a length quartile, to be used as a factor by the linear model. The model included an intercept, which would correspond to the old mean expression value for a gene whose length is in the 1st quartile and young mean expression value is 0. We observed that the shorter the genes included in the linear model (for instance, Q1 genes), the greater was the slope of the resulting straight. We performed statistical analysis to compare between the slope of the Q1 model against each of the three remaining models (Q2, Q3 and Q4).
Additionally, we fitted a linear regression model to the average gene expression in old and young cells, using log10(gene length) as a continuous interaction term, using the formula ME_old ∼ ME_young ∗ L, where L is the log10(gene length). The intercept in this model would correspond to the old mean expression value when log-transformed gene length is 0. We conducted a bootstrap-based permutation analysis for B=200 bootstrap samples for each aging dataset to verify the robustness of the length-association.
The same analysis was extended to conditions other than aging, by making analogous comparisons. In the UV-radiated murine skin analysis, we compared UV-radiated skin against the healthy skin control (to test for the effect of UV-radiation), the UV-radiated skin against the vitamin D-treated and UV-radiated skin (effect of vitamin D treatment on damage caused by UV-radiation), and the vitamin D-treated skin against the healthy skin control (effect of UV-radiation after vitamin D treatment). In the analysis on the murine model for Cockayne syndrome we compared between each of the knockouts (Adh5−/−, Csbm/m, and double KO) against the wild type (WT). In the analysis of human mesenchymal stromal cells derived from Cockayne syndrome patients, we compared between the following conditions: UV-radiated cells against control (both in mutant and gene corrected cells), and ERCCmut against ERCCGC (to test for the effect of carrying the ERCC6 mutation, both in basal conditions and after UV-radiation exposure).
Gene length analysis of the differentially expressed genes between conditions
We carried out two types of differential expression analysis: overall differential expression between conditions and differential expression at the cell type level. Overall differential expression between conditions is based on the assumption that the changes in cell type composition between the conditions to be compared are negligible, so that the genes that are detected to be differentially expressed do not correspond to markers defining specific cell types that are more abundant in one of the conditions. Differential expression analysis between conditions at the cell type level identifies genes that are overexpressed in one of the conditions. Of course, DEGs can only be computed for cell types that are present in the conditions to be compared in sufficient amounts (we used 10 cells as the minimum). Its output is not directly affected by changes in cell type composition between conditions. However, if the abundance of cell type under study is very different between conditions – if one cell type is very rare in one of the conditions – the population might not be well sampled for that condition and the gene length analysis might not be reliable. We therefore used both approaches as they are complementary to one another. In either case, we used the Scanpy function sc.tl.rank_genes_groups with method = “wilcoxon” to obtain the top 300 differentially expressed genes between conditions.
In most cases, pairwise comparisons were made, as in the aging analysis ("young" vs "old") or when analyzing the effect of smoking of human airways ("never-smokers" vs "heavy smokers"). In those cases, two lists of genes were obtained: one per condition. In the analysis of murine UV-radiated skin (Figure 4), we compared between the three conditions simultaneously. In that case, each of three DEG lists corresponds to the genes that are over-expressed in one condition against the other two conditions pooled together. First, the Lilliefors test71 was used to check whether gene lengths in each of the conditions were normally distributed. In cases where the null hypothesis could be rejected (p-value < 0.05) in at least one of the conditions to be compared, a non-parametric test was used to compare between means. In order to make statistical comparisons between the mean gene length between conditions, we used the following tests: Student’s T test (two conditions, normally distributed), Mann-Whitney’s U test (two conditions, not normally distributed), ANOVA (three conditions) and Tukey’s test for post-hoc analysis.
Acknowledgments
We thank Alex M. Ascensión, Javier Cabau-Laporta, Mattin Lucu, Laura Yndriago, Sonia Alonso-Martin, Ander Matheu, David Otaegui, and Héctor Lafuente for their thorough revision of the manuscript and for useful suggestions. This work was supported by grants from Instituto de Salud Carlos III (PI22/01247 and PI19/01621), co-funded by the European Union, and Diputación Foral de Gipuzkoa. OI-S received the support of a fellowship from “Programa Investigo” of Lanbide-Servicio Vasco de Empleo, co-funded by the European Union (NextGenerationEU), la Caixa Foundation (ID 100010434; code LCF/BQ/IN18/11660065), and from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 713673. The work of IB was financially supported in part by grants from the Departamento de Educación, Política Lingüística y Cultura del Gobierno Vasco [IT1456-22] and by the Ministry of Science and Innovation through BCAM Severo Ochoa accreditation [CEX2021-001142-S/MICIN/AEI/10.13039/501100011033] and through project [PID2020-115882RB-I00/AEI/10.13039/501100011033] funded by Agencia Estatal de Investigación and acronym “S3M1P4R” and also by the Basque Government through the BERC 2022–2025 program.
Author contributions
O.I.-S. conceived and performed the experiments. I.B. conceived and performed some experiments. A.I. conceived some experiments and supervised the work. O.I.-S. and A.I. wrote the manuscript. All authors revised and approved the final submitted version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: March 9, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106368.
Supplemental information
Data and code availability
This paper analyzes existing, publicly available data. All the transcriptomics datasets used in this study were downloaded from public repositories, mainly from the Gene Expression Omnibus (GEO). The accession numbers for all these datasets are listed in the key resources table. The source of the lists of differentially expressed genes from published studies can also be found in the key resources table.
All original code, including reproducible documented Jupyter Notebooks and R scripts, has been deposited at Figshare and is publicly available as of the date of publication, and its DOI is listed in the key resources table. Code is also available at our GitLab repository (https://gitlab.com/olgaibanez/transcription_stress).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Failla G. The aging process and carcinogenesis. Ann. N. Y. Acad. Sci. 1958;71:1124–1140. doi: 10.1111/j.1749-6632.1958.tb46828.x. [DOI] [PubMed] [Google Scholar]
- 2.Szilard L. On the nature of the aging process. Proc. Natl. Acad. Sci. USA. 1959;45:30–45. doi: 10.1073/pnas.45.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schumacher B., Pothof J., Vijg J., Hoeijmakers J.H.J. The central role of DNA damage in the ageing process. Nature. 2021;592:695–703. doi: 10.1038/s41586-021-03307-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yousefzadeh M., Henpita C., Vyas R., Soto-Palma C., Robbins P., Niedernhofer L. DNA damage—how and why we age? Elife. 2021;10:e62852. doi: 10.7554/eLife.62852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frenk S., Houseley J. Gene expression hallmarks of cellular ageing. Biogerontology. 2018;19:547–566. doi: 10.1007/s10522-018-9750-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Magalhães J.P., Curado J., Church G.M. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics. 2009;25:875–881. doi: 10.1093/bioinformatics/btp073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tacutu R., Thornton D., Johnson E., Budovsky A., Barardo D., Craig T., Diana E., Lehmann G., Toren D., Wang J., et al. Human ageing genomic resources: new and updated databases. Nucleic Acids Res. 2018;46:D1083–D1090. doi: 10.1093/nar/gkx1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palmer D., Fabris F., Doherty A., Freitas A.A., de Magalhães J.P. Ageing transcriptome meta-analysis reveals similarities between key mammalian tissues. Aging (Albany NY) 2021; 13:3313-3341 doi: 10.18632/aging.202648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang M.J., Pisco A.O., Darmanis S., Zou J. Mouse aging cell atlas analysis reveals global and cell type-specific aging signatures. Elife. 2021;10:e62293. doi: 10.7554/eLife.62293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stegeman R., Weake V.M. Transcriptional signatures of aging. J. Mol. Biol. 2017;429:2427–2437. doi: 10.1016/j.jmb.2017.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yannarell A., Schumm D.E., Webb T.E. Age-dependence of nuclear RNA processing. Mech. Ageing Dev. 1977;6:259–264. doi: 10.1016/0047-6374(77)90026-4. [DOI] [PubMed] [Google Scholar]
- 12.Haustead D.J., Stevenson A., Saxena V., Marriage F., Firth M., Silla R., Martin L., Adcroft K.F., Rea S., Day P.J., et al. Transcriptome analysis of human ageing in male skin shows mid-life period of variability and central role of NF-κB. Sci. Rep. 2016;6:26846. doi: 10.1038/srep26846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lans H., Hoeijmakers J.H.J., Vermeulen W., Marteijn J.A. The DNA damage response to transcription stress. Nat. Rev. Mol. Cell Biol. 2019;20:766–784. doi: 10.1038/s41580-019-0169-4. [DOI] [PubMed] [Google Scholar]
- 14.Gregersen L.H., Svejstrup J.Q. The cellular response to transcription-blocking DNA damage. Trends Biochem. Sci. 2018;43:327–341. doi: 10.1016/j.tibs.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J., Muste Sadurni M., Saponaro M. RNAPII response to transcription-blocking DNA lesions in mammalian cells. FEBS J. 2022:In press. doi: 10.1111/febs.16561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soheili-Nezhad S. Alzheimer’s disease: the large gene instability hypothesis. bioRxiv. 2017 doi: 10.1101/189712. Preprint at. [DOI] [Google Scholar]
- 17.Soheili-Nezhad S., van der Linden R.J., Olde Rikkert M., Sprooten E., Poelmans G. Long genes are more frequently affected by somatic mutations and show reduced expression in Alzheimer’s disease: implications for disease etiology. Alzheimers Dement. 2021;17:489–499. doi: 10.1002/alz.12211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lopes I., Altab G., Raina P., de Magalhães J.P. Gene size matters: an analysis of gene length in the human genome. Front. Genet. 2021;12:559998. doi: 10.3389/fgene.2021.559998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vermeij W.P., Dollé M.E.T., Reiling E., Jaarsma D., Payan-Gomez C., Bombardieri C.R., Wu H., Roks A.J.M., Botter S.M., van der Eerden B.C., et al. Restricted diet delays accelerated ageing and genomic stress in DNA-repair-deficient mice. Nature. 2016;537:427–431. doi: 10.1038/nature19329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tabula Muris Consortium. Almanzar N., Antony J., Baghel A.S., Bakerman I., Bansal I., Barres B.A., Beachy P.A., Berdnik D., Bilen B., Brownfield D., et al. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature. 2020;583:590–595. doi: 10.1038/s41586-020-2496-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Angelidis I., Simon L.M., Fernandez I.E., Strunz M., Mayr C.H., Greiffo F.R., Tsitsiridis G., Ansari M., Graf E., Strom T.-M., et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat. Commun. 2019;10:963. doi: 10.1038/s41467-019-08831-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kimmel J.C., Penland L., Rubinstein N.D., Hendrickson D.G., Kelley D.R., Rosenthal A.Z. Murine single-cell RNA-seq reveals cell-identity- and tissue-specific trajectories of aging. Genome Res. 2019;29:2088–2103. doi: 10.1101/gr.253880.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ximerakis M., Lipnick S.L., Innes B.T., Simmons S.K., Adiconis X., Dionne D., Mayweather B.A., Nguyen L., Niziolek Z., Ozek C., et al. Single-cell transcriptomic profiling of the aging mouse brain. Nat. Neurosci. 2019;22:1696–1708. doi: 10.1038/s41593-019-0491-3. [DOI] [PubMed] [Google Scholar]
- 24.Salzer M.C., Lafzi A., Berenguer-Llergo A., Youssif C., Castellanos A., Solanas G., Peixoto F.O., Stephan-Otto Attolini C., Prats N., Aguilera M., et al. Identity noise and adipogenic traits characterize dermal fibroblast aging. Cell. 2018;175:1575–1590.e22. doi: 10.1016/j.cell.2018.10.012. [DOI] [PubMed] [Google Scholar]
- 25.Enge M., Arda H.E., Mignardi M., Beausang J., Bottino R., Kim S.K., Quake S.R. Single-cell analysis of human pancreas reveals transcriptional signatures of aging and somatic mutation patterns. Cell. 2017;171:321–330.e14. doi: 10.1016/j.cell.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Solé-Boldo L., Raddatz G., Schütz S., Mallm J.-P., Rippe K., Lonsdorf A.S., Rodríguez-Paredes M., Lyko F. Single-cell transcriptomes of the human skin reveal age-related loss of fibroblast priming. Commun. Biol. 2020;3:188. doi: 10.1038/s42003-020-0922-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Travaglini K.J., Nabhan A.N., Penland L., Sinha R., Gillich A., Sit R.V., Chang S., Conley S.D., Mori Y., Seita J., et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature. 2020;587:619–625. doi: 10.1038/s41586-020-2922-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raredon M.S.B., Adams T.S., Suhail Y., Schupp J.C., Poli S., Neumark N., Leiby K.L., Greaney A.M., Yuan Y., Horien C., et al. Single-cell connectomic analysis of adult mammalian lungs. Sci. Adv. 2019;5:eaaw3851. doi: 10.1126/sciadv.aaw3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martincorena I., Roshan A., Gerstung M., Ellis P., Van Loo P., McLaren S., Wedge D.C., Fullam A., Alexandrov L.B., Tubio J.M., et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880–886. doi: 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Passeron T., Lim H.W., Goh C.-L., Kang H.Y., Ly F., Morita A., Ocampo Candiani J., Puig S., Schalka S., Wei L., et al. Photoprotection according to skin phototype and dermatoses: practical recommendations from an expert panel. J. Eur. Acad. Dermatol. Venereol. 2021;35:1460–1469. doi: 10.1111/jdv.17242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang S., Min Z., Ji Q., Geng L., Su Y., Liu Z., Hu H., Wang L., Zhang W., Suzuiki K., et al. Rescue of premature aging defects in Cockayne syndrome stem cells by CRISPR/Cas9-mediated gene correction. Protein Cell. 2020;11:1–22. doi: 10.1007/s13238-019-0623-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gordon-Thomson C., Tongkao-on W., Song E.J., Carter S.E., Dixon K.M., Mason R.S. Skin Cancer. Springer; 2014. Protection from ultraviolet damage and photocarcinogenesis by vitamin D compounds. In: Sunlight, Vitamin D and; pp. 303–328. [DOI] [PubMed] [Google Scholar]
- 33.Lin Y., Cao Z., Lyu T., Kong T., Zhang Q., Wu K., Wang Y., Zheng J. Single-cell RNA-seq of UVB-radiated skin reveals landscape of photoaging-related inflammation and protection by vitamin D. Gene. 2022;831:146563. doi: 10.1016/j.gene.2022.146563. [DOI] [PubMed] [Google Scholar]
- 34.Huang Z., Sun S., Lee M., Maslov A.Y., Shi M., Waldman S., Marsh A., Siddiqui T., Dong X., Peter Y., et al. Single-cell analysis of somatic mutations in human bronchial epithelial cells in relation to aging and smoking. Nat. Genet. 2022;54:492–498. doi: 10.1038/s41588-022-01035-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goldfarbmuren K.C., Jackson N.D., Sajuthi S.P., Dyjack N., Li K.S., Rios C.L., Plender E.G., Montgomery M.T., Everman J.L., Bratcher P.E., et al. Dissecting the cellular specificity of smoking effects and reconstructing lineages in the human airway epithelium. Nat. Commun. 2020;11:2485. doi: 10.1038/s41467-020-16239-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rieckher M., Garinis G.A., Schumacher B. Molecular pathology of rare progeroid diseases. Trends Mol. Med. 2021;27:907–922. doi: 10.1016/j.molmed.2021.06.011. [DOI] [PubMed] [Google Scholar]
- 37.Umansky C., Morellato A.E., Rieckher M., Scheidegger M.A., Martinefski M.R., Fernández G.A., Pak O., Kolesnikova K., Reingruber H., Bollini M., et al. Endogenous formaldehyde scavenges cellular glutathione resulting in redox disruption and cytotoxicity. Nat. Commun. 2022;13:745. doi: 10.1038/s41467-022-28242-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mulderrig L., Garaycoechea J.I., Tuong Z.K., Millington C.L., Dingler F.A., Ferdinand J.R., Gaul L., Tadross J.A., Arends M.J., O’Rahilly S., et al. Aldehyde-driven transcriptional stress triggers an anorexic DNA damage response. Nature. 2021;600:158–163. doi: 10.1038/s41586-021-04133-7. [DOI] [PubMed] [Google Scholar]
- 39.Lombardi A., Arseni L., Carriero R., Compe E., Botta E., Ferri D., Uggè M., Biamonti G., Peverali F.A., Bione S., Orioli D. Reduced levels of prostaglandin I 2 synthase: a distinctive feature of the cancer-free trichothiodystrophy. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2024502118. e2024502118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peters M.J., Joehanes R., Pilling L.C., Schurmann C., Conneely K.N., Powell J., Reinmaa E., Sutphin G.L., Zhernakova A., Schramm K., et al. The transcriptional landscape of age in human peripheral blood. Nat. Commun. 2015;6:8570. doi: 10.1038/ncomms9570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chiaromonte F., Miller W., Bouhassira E.E. Gene length and proximity to neighbors affect genome-wide expression levels. Genome Res. 2003;13:2602–2608. doi: 10.1101/gr.1169203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heyn P., Kalinka A.T., Tomancak P., Neugebauer K.M. Introns and gene expression: cellular constraints, transcriptional regulation, and evolutionary consequences: prospects & Overviews. Bioessays. 2015;37:148–154. doi: 10.1002/bies.201400138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kirkconnell K.S., Magnuson B., Paulsen M.T., Lu B., Bedi K., Ljungman M. Gene length as a biological timer to establish temporal transcriptional regulation. Cell Cycle. 2017;16:259–270. doi: 10.1080/15384101.2016.1234550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown J.C. Role of gene length in control of human gene expression: chromosome-specific and tissue-specific effects. Int. J. Genomics. 2021;2021:8902428. doi: 10.1155/2021/8902428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cramer P. Organization and regulation of gene transcription. Nature. 2019;573:45–54. doi: 10.1038/s41586-019-1517-4. [DOI] [PubMed] [Google Scholar]
- 46.Fan Z., Devlin J.R., Hogg S.J., Doyle M.A., Harrison P.F., Todorovski I., Cluse L.A., Knight D.A., Sandow J.J., Gregory G., et al. CDK13 cooperates with CDK12 to control global RNA polymerase II processivity. Sci. Adv. 2020;6:eaaz5041. doi: 10.1126/sciadv.aaz5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vlaming H., Mimoso C.A., Field A.R., Martin B.J.E., Adelman K. Screening thousands of transcribed coding and non-coding regions reveals sequence determinants of RNA polymerase II elongation potential. Nat. Struct. Mol. Biol. 2022;29:613–620. doi: 10.1038/s41594-022-00785-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaida D., Berg M.G., Younis I., Kasim M., Singh L.N., Wan L., Dreyfuss G. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature. 2010;468:664–668. doi: 10.1038/nature09479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berg M.G., Singh L.N., Younis I., Liu Q., Pinto A.M., Kaida D., Zhang Z., Cho S., Sherrill-Mix S., Wan L., Dreyfuss G. U1 snRNP determines mRNA length and Regulates isoform expression. Cell. 2012;150:53–64. doi: 10.1016/j.cell.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hosokawa M., Takeuchi A., Tanihata J., Iida K., Takeda S., Hagiwara M. Loss of RNA-binding protein sfpq causes long-gene transcriptopathy in skeletal muscle and severe muscle mass reduction with metabolic myopathy. iScience. 2019;13:229–242. doi: 10.1016/j.isci.2019.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bedi K., Magnuson B.R., Narayanan I., Paulsen M., Wilson T.E., Ljungman M. Cotranscriptional splicing efficiencies differ within genes and between cell types. RNA. 2021;27:829–840. doi: 10.1261/rna.078662.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cugusi S., Mitter R., Kelly G.P., Walker J., Han Z., Pisano P., Wierer M., Stewart A., Svejstrup J.Q. Heat shock induces premature transcript termination and reconfigures the human transcriptome. Mol. Cell. 2022;82:1573–1588.e10. doi: 10.1016/j.molcel.2022.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakazawa Y., Hara Y., Oka Y., Komine O., van den Heuvel D., Guo C., Daigaku Y., Isono M., He Y., Shimada M., et al. Ubiquitination of DNA damage-stalled RNAPII promotes transcription-coupled repair. Cell. 2020;180:1228–1244.e24. doi: 10.1016/j.cell.2020.02.010. [DOI] [PubMed] [Google Scholar]
- 54.Tufegdžić Vidaković A., Mitter R., Kelly G.P., Neumann M., Harreman M., Rodríguez-Martínez M., Herlihy A., Weems J.C., Boeing S., Encheva V., et al. Regulation of the RNAPII pool is integral to the DNA damage response. Cell. 2020;180:1245–1261.e21. doi: 10.1016/j.cell.2020.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barbash S., Sakmar T.P. Length-dependent gene misexpression is associated with Alzheimer’s disease progression. Sci. Rep. 2017;7:190. doi: 10.1038/s41598-017-00250-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takeuchi A., Iida K., Tsubota T., Hosokawa M., Denawa M., Brown J.B., Ninomiya K., Ito M., Kimura H., Abe T., et al. Loss of sfpq causes long-gene transcriptopathy in the brain. Cell Rep. 2018;23:1326–1341. doi: 10.1016/j.celrep.2018.03.141. [DOI] [PubMed] [Google Scholar]
- 57.Balliu B., Durrant M., Goede O.D., Abell N., Li X., Liu B., Gloudemans M.J., Cook N.L., Smith K.S., Knowles D.A., et al. Genetic regulation of gene expression and splicing during a 10-year period of human aging. Genome Biol. 2019;20:230. doi: 10.1186/s13059-019-1840-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harries L.W., Hernandez D., Henley W., Wood A.R., Holly A.C., Bradley-Smith R.M., Yaghootkar H., Dutta A., Murray A., Frayling T.M., et al. Human aging is characterized by focused changes in gene expression and deregulation of alternative splicing: gene expression changes in human aging. Aging Cell. 2011;10:868–878. doi: 10.1111/j.1474-9726.2011.00726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lodato M.A., Rodin R.E., Bohrson C.L., Coulter M.E., Barton A.R., Kwon M., Sherman M.A., Vitzthum C.M., Luquette L.J., Yandava C.N., et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science. 2018;359:555–559. doi: 10.1126/science.aao4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mitchell E., Spencer Chapman M., Williams N., Dawson K.J., Mende N., Calderbank E.F., Jung H., Mitchell T., Coorens T.H.H., Spencer D.H., et al. Clonal dynamics of haematopoiesis across the human lifespan. Nature. 2022;606:343–350. doi: 10.1038/s41586-022-04786-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu J., Wu Z., He J., Wang Y. Cellular fractionation reveals transcriptome responses of human fibroblasts to UV-C irradiation. Cell Death Dis. 2022;13:177. doi: 10.1038/s41419-022-04634-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kajitani G.S., Nascimento L.L.S., Neves M.R.C., Leandro G.D.S., Garcia C.C.M., Menck C.F.M. Transcription blockage by DNA damage in nucleotide excision repair-related neurological dysfunctions. Semin. Cell Dev. Biol. 2021;114:20–35. doi: 10.1016/j.semcdb.2020.10.009. [DOI] [PubMed] [Google Scholar]
- 63.Xiong F., Wang R., Lee J.-H., Li S., Chen S.-F., Liao Z., Hasani L.A., Nguyen P.T., Zhu X., Krakowiak J., et al. RNA m6A modification orchestrates a LINE-1–host interaction that facilitates retrotransposition and contributes to long gene vulnerability. Cell Res. 2021;31:861–885. doi: 10.1038/s41422-021-00515-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Winick-Ng W., Kukalev A., Harabula I., Zea-Redondo L., Szabó D., Meijer M., Serebreni L., Zhang Y., Bianco S., Chiariello A.M., et al. Cell-type specialization is encoded by specific chromatin topologies. Nature. 2021;599:684–691. doi: 10.1038/s41586-021-04081-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abou Chakra M., Isserlin R., Tran T.N., Bader G.D. Control of tissue development and cell diversity by cell cycle-dependent transcriptional filtering. Elife. 2021;10:e64951. doi: 10.7554/eLife.64951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stoeger T., Grant R.A., McQuattie-Pimentel A.C., Anekalla K.R., Liu S.S., Tejedor-Navarro H., Singer B.D., Abdala-Valencia H., Schwake M., Tetreault M.-P., et al. Aging is associated with a systemic length-associated transcriptome imbalance. Nat. Aging. 2022;2:1191–1206. doi: 10.1038/s43587-022-00317-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gyenis A., Chang J., Demmers J., Bruens S.T., Barnhoorn S., Brandt R.M.C., Baar M.P., Raseta M., Derks K.W.J., Hoeijmakers J.H.J., Pothof J. Genome-wide RNA polymerase stalling shapes the transcriptome during aging. Nat. Genet. 2023;55:268–279. doi: 10.1038/s41588-022-01279-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Traag V.A., Waltman L., van Eck N.J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 2019;9:5233. doi: 10.1038/s41598-019-41695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ibañez-Solé O., Ascensión A.M., Araúzo-Bravo M.J., Izeta A. Lack of evidence for increased transcriptional noise in aged tissues. Elife. 2022;11:e80380. doi: 10.7554/eLife.80380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Joost S., Zeisel A., Jacob T., Sun X., La Manno G., Lönnerberg P., Linnarsson S., Kasper M. Single-cell transcriptomics reveals that differentiation and spatial signatures shape epidermal and hair follicle Heterogeneity. Cell Syst. 2016;3:221–237.e9. doi: 10.1016/j.cels.2016.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lilliefors H.W. On the Kolmogorov-smirnov test for normality with mean and variance unknown. J. Am. Stat. Assoc. 1967;62:399–402. doi: 10.1080/01621459.1967.10482916. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes existing, publicly available data. All the transcriptomics datasets used in this study were downloaded from public repositories, mainly from the Gene Expression Omnibus (GEO). The accession numbers for all these datasets are listed in the key resources table. The source of the lists of differentially expressed genes from published studies can also be found in the key resources table.
All original code, including reproducible documented Jupyter Notebooks and R scripts, has been deposited at Figshare and is publicly available as of the date of publication, and its DOI is listed in the key resources table. Code is also available at our GitLab repository (https://gitlab.com/olgaibanez/transcription_stress).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.