

Summary
The appropriate regulation of telomere length homeostasis is crucial for the maintenance of genome integrity. The telomere-binding protein TZAP has been suggested to regulate telomere length by promoting t-circle and c-circle excisions through telomere trimming, yet the molecular mechanisms by which TZAP functions at telomeres are not understood. Using a system based on TZAP overexpression, we show that efficient TZAP recruitment to telomeres occurs in the context of open telomeric chromatin caused by loss of ATRX/DAXX independently of H3.3 deposition. Moreover, our data indicate that TZAP binding to telomeres induces telomere dysfunction and ALT-like activity, resulting in the generation of t-circles and c-circles in a Bloom-Topoisomerase IIIα-RMI1-RMI2 (BTR)-dependent manner.
Subject areas: Chromosome organization, Molecular biology experimental approach, Molecular interaction
Graphical abstract
Highlights
-
•
Efficient telomeric recruitment of TZAP occurs upon ATRX/DAXX loss
-
•
TZAP loading onto telomeres induces telomere dysfunction
-
•
TZAP at telomeres triggers an ALT-like pathway dependent on the BTR complex
Chromosome organization; Molecular biology experimental approach; Molecular interaction
Introduction
Telomeres are the nucleoprotein structures that protect the end of eukaryotic linear chromosomes. In human cells, telomeres consist of double-stranded (ds) DNA tandem repeats of TTAGGG ending in a 3′ single-stranded (ss) DNA overhang1,2 specifically bound by the Shelterin protein complex (constituted by the subunits TRF1, TRF2, POT1, RAP1, TIN2, and TPP1).3 Shelterin plays a central role in the protection of telomeric DNA by promoting the formation of the t-loop secondary structure through the invasion of the 3′ overhang into the telomeric dsDNA,4 as well as by repressing the activation of the DNA damage response (DDR) pathways (reviewed in detail elsewhere5).
The tight control of telomere length is essential for the maintenance of genome integrity and cell proliferation. Regulatory mechanisms contributing to both telomere shortening and telomere lengthening have been described. The best-understood mechanism of telomere shortening is the “end-replication problem”, which can be explained by the inability of canonical DNA polymerases to fully duplicate the ends of linear chromosomes,6,7 along with the post-replicative processing of telomere ends.8 As a result, telomeres shorten with each round of cell division and lose their protective nature, thus leading to the activation of DDR pathways and cellular programs that prevent additional cell divisions.9 Another mechanism contributing to telomere shortening was first discovered in budding yeast and named telomere rapid deletion (TRD). TRD employs intra-telomeric homologous recombination (HR) to catalyze deletions of telomeric DNA from excessively long telomeres, setting them back to normal length.10,11,12 HR-mediated resolution of the Holliday junction at the base of the t-loop has been proposed as the underlying mechanism of TRD.13 Telomere trimming, an analogous mechanism to TRD in yeast, was later reported to control the upper limit of telomere length in different human cell lines14 including cancer cells with excessively long telomeres,15 cells of the male germline, stimulated lymphocytes,16 and stem cells.17 Interestingly, telomere trimming was associated with the presence of ALT-associated PML bodies (APBs)15 and circular extrachromosomal telomeric repeats (cECTRs).15,16,17 The latter has been postulated to originate from the excision of the t-loop during the trimming process.15
While the number of cell divisions in somatic cells is limited due to the end-replication problem, other cells display active telomere-lengthening mechanisms and therefore possess an unlimited proliferative potential. Most cancer cells, as well as germline and stem cells, maintain their telomeres by activating the ribonucleoprotein telomerase,18,19 a reverse transcriptase that catalyzes de novo addition of telomeric repeats to chromosome ends.20 On the other hand, 4–11% of cancer cells employ an HR-dependent mechanism to maintain their telomeres, known as alternative lengthening of telomeres (ALTs).21,22 To preserve their telomeres, ALT+ cells use the break-induced replication (BIR) machinery, which orchestrates the invasion of the substrate telomere into a telomeric repeat-containing template followed by its elongation via conservative DNA synthesis.23,24 ALT+ cells present several features including the presence of APBs,25 high levels of telomeric replication stress,21 telomeric DNA synthesis in APBs,26 heterogeneous telomere length,22 abundant cECTRs,27,28 and high levels of telomere sister chromatid exchanges (t-SCE).29 Interestingly, mutations in the genes encoding the protein complex ATRX/DAXX and the histone variant H3.3 are prevalent in ALT+ cancers.30,31,32 Furthermore, ATRX expression in ALT+ cells reverses ALT markers,33,34 suggesting that ATRX is a key suppressor of the ALT mechanism. ATRX belongs to the SNF2 family of chromatin-remodeling proteins35 and, together with DAXX, promotes H3.3 deposition at telomeres.36,37 Moreover, ATRX controls telomere integrity by repressing the transcription of telomeric repeat-containing RNA (TERRA)36,38 as well as the accumulation of G4 motifs,34 both associated with telomeric replication stress.
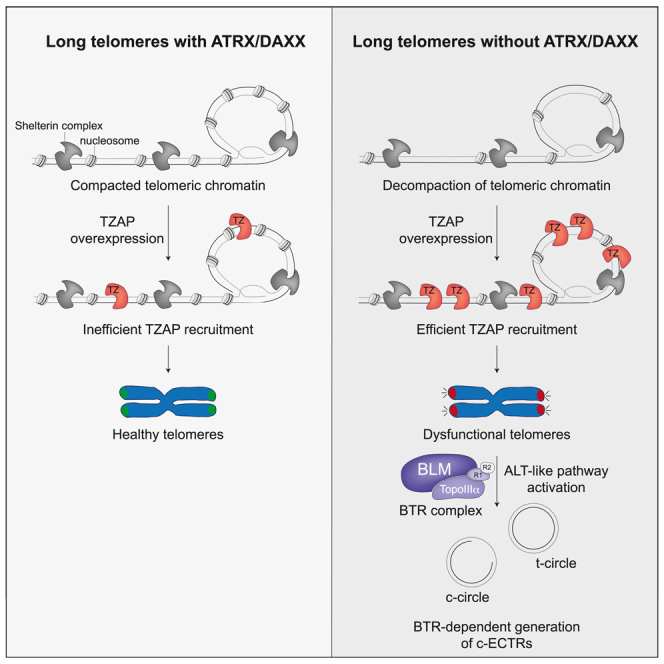
We and others identified and characterized the telomeric-associated zinc-finger protein (TZAP) (originally known as ZBTB48) as a novel telomere-binding protein implicated in telomere length regulation.39,40 We found that TZAP was preferentially associated with long telomeres.39 In addition, TZAP upregulation in ALT+ cells led to rapid telomere loss accompanied by the accumulation of cECTRs and an increase in the levels of APBs.39 Conversely, downregulation of TZAP caused a reduction in the basal levels of cECTRs in ALT+ cells.39 The present study aims to understand the molecular mechanisms of the pathway triggered by TZAP overexpression in human cancer cells. We show that the efficient telomeric recruitment of TZAP occurs in the context of open telomeric chromatin in the absence of ATRX/DAXX. Moreover, we show that TZAP loading onto telomeres induces telomere dysfunction and ALT-like features, including generation of c-circles and t-circles in a Bloom-Topoisomerase IIIα-RMI1-RMI2 (BTR)-dependent manner.
Results
TZAP overexpression in ALT+ cells results in the exacerbation of telomere dysfunction and ALT-like features
TZAP overexpression in ALT+ cells results in increased levels of cECTRs and APBs.39 Both cECTRs and APBs are well-known markers of the ALT mechanism. However, they can also be observed at lower levels in cells undergoing telomere trimming.15,16,17 Since TZAP overexpression in ALT+ cells also leads to telomere loss, it was proposed that TZAP plays a central role in the process of telomere trimming.39 To test whether TZAP overexpression indeed led to exacerbated telomere trimming, c-circle assay was performed using two ALT+ cell lines, U-2 OS and GM847, and two telomerase+ cell lines, HeLa1.2.11 and HT-1080 hTR (Figure 1A). HeLa1.2.11 is a sub-clone of the HeLa cell line with a bulk telomere length of ∼23 kb,41 while HT-1080 hTR has a bulk telomere length of ∼25 kb as a result of increased telomerase activity due to the overexpression of the telomerase RNA subunit hTR (Figure S1A). Importantly, HT-1080 hTR was previously shown to experience telomere trimming.15 While c-circle levels were apparently increased upon TZAP overexpression in the ALT+ cell lines, telomerase+ cell lines were unaffected (Figure 1A). This result indicates that the cECTRs production observed upon TZAP overexpression is specific to ALT+ cells and suggests that they are generated by an ALT-like pathway. Therefore, we investigated the presence of other ALT markers upon overexpression of TZAP in U-2 OS cells. The ATR and ATM DDR pathways, both associated with the ALT mechanism,42,43,44 were induced upon TZAP overexpression in U-2 OS cells (Figure 1B). In addition, confocal microscopy analysis revealed an increased colocalization between the RPA32 (Figures 1C and 1D) and RPA70 (Figures S1B and S1C) subunits of the ssDNA-binding protein complex RPA and the Shelterin subunit TRF1 upon TZAP overexpression in U-2 OS. RPA-coated ssDNA is known to accumulate in genomic regions experiencing replication stress, where it serves as a platform for the recruitment and activation of the ATR kinase.45 Moreover, previous studies showed that RPA localized at telomeres in ALT+ cells46 and that such localization was exacerbated upon treatments that stimulated ALT activity.44 Hence, our results (Figures 1C, 1D, S1B, and S1C) indicate that TZAP overexpression in U-2 OS cells triggers telomeric replication stress, which is also a marker of ALT.21 Finally, assessment of DNA synthesis at telomeres in the G2 phase of the cell cycle, another ALT marker, by implementation of the ALT telomere DNA synthesis in APBs (ATSA) assay,26 showed a significant induction of this phenotype upon TZAP overexpression in U-2 OS cells (Figures 1E, 1F, and S1D).
Figure 1.

TZAP overexpression exacerbates telomere dysfunction and ALT features in ALT+ cells
(A) C-circle assays (CCA) of the indicated ALT+ and telomerase+ cell lines stably overexpressing either empty vector (EV) or Myc-TZAP (TZ).
(B) Western blot analysis of the indicated proteins in U-2 OS cells overexpressing either EV or Myc-TZAP.
(C) Representative immunofluorescence maximum intensity projection images of U-2 OS cells overexpressing either EV or Myc-TZAP showing the nuclear localization of RPA32 and TRF1. White arrows are pointing to RPA32-TRF1 colocalizations. Scale bar 10 μm.
(D) Scatterplot showing quantification of experiment described in C). A total of 100 nuclei were analyzed for each sample. Data are from a single experiment. Center line, mean. n shows number of cells analyzed.
(E) Representative immunofluorescence maximum intensity projection images of G2-synchronized U-2 OS cells overexpressing either EV or Myc-TZAP showing the nuclear localization of EdU incorporated into newly synthesized DNA and TRF1. White arrows point to EdU-TRF1 colocalizations. Scale bar 10 μm.
(F) Scatterplot showing quantification of experiment described in E). A total of at least 197 nuclei were analyzed for each sample. Data are representative of three independent experiments. Center line, mean. n shows number of cells analyzed. ∗∗∗p < 0.001, ns: non-significant, t-test was used to calculate the p value.
Together these results indicate that TZAP overexpression in U-2 OS cells leads to the exacerbation of telomere dysfunction and an activity reminiscent of ALT.
ATRX/DAXX represses TZAP-induced ALT features in telomerase+ cells independently of H3.3
One difference between telomerase+ and ALT+ cancer cells is that the latter are frequently deficient for the chromatin remodeler ATRX/DAXX.30,31,32 Indeed, western blot analysis of the ATRX/DAXX status in all the cell lines used in our study revealed that, while all telomerase+ cells had detectable levels of ATRX and DAXX proteins, the ALT+ cell lines had detectable DAXX but had completely lost ATRX (Figure S2A). Therefore, we hypothesized that TZAP-induced telomere dysfunction and ALT features could be repressed by ATRX/DAXX in telomerase+ cells. To test this, TZAP was overexpressed in HeLa1.2.11 cells treated with small interfering RNAs (siRNAs) to silence ATRX, DAXX, or the histone variant H3.3 (Figure 2A). Interestingly, TZAP overexpression led to increased levels of cECTRs in the form of c-circles (Figures 2B and 2C) and t-circles (Figure 2D) in ATRX- and DAXX-silenced cells but not in control and H3.3-silenced cells. Moreover, TZAP overexpression in HeLa1.2.11 cells caused induction of G2-associated telomeric DNA synthesis in the absence of ATRX or DAXX but not in control or H3.3-deficient cells (Figures 2E and 2F). The cell-cycle profiles of HeLa1.2.11 treated with non-targeting siRNA, siATRX, siDAXX, or siH3.3 confirmed that the observed differences were not caused by changes in cell-cycle progression (Figure S2B). In addition, ATRX silencing also increased cECTRs in the form of t-circles in HT-1080 hTR overexpressing TZAP (Figures S2C and S2D).
Figure 2.

ATRX/DAXX represses ALT-like features in HeLa1.2.11 cells independently of H3.3
(A) HeLa1.2.11 cells overexpressing either empty vector (EV) or Myc-TZAP were transfected with the indicated siRNAs, and the whole-cell lysates were analyzed by western blotting using the indicated antibodies.
(B) CCA of the cells described in A).
(C) Quantification of CCA signals in B) was performed relative to si-scramble treated HeLa1.2.11 overexpressing EV. Bars represent the mean ± SD of 3 independent experiments. ∗∗p < 0.01, ns: non-significant, two-way ANOVA test was used to calculate the p values.
(D) T-circle assays (TCA) of the cells described in A). The arrow points to the products of rolling-circle amplification of TTAGGG-rich circular extrachromosomal DNA. The experiment was repeated 3 times, and one representative image is shown.
(E) Representative immunofluorescence maximum intensity projection images of G2-synchronized HeLa1.2.11 cells overexpressing Myc-TZAP and treated with the indicated siRNAs. The image shows the nuclear localization of EdU incorporated into newly synthesized DNA and TRF1. White arrows point to EdU-TRF1 colocalizations. Scale bar 10 μm.
(F) Scatterplot showing quantification of experiment described in E). A total of at least 176 nuclei were analyzed for each sample. Data are representative of two independent experiments. Center line, mean. n shows number of cells analyzed. ∗∗∗p < 0.001, ns: non-significant, one-way ANOVA test was used to calculate the p values.
These data indicate that TZAP-induced ALT features in telomerase+ cells are prevented by the ATRX/DAXX complex, independently of H3.3 deposition at telomeres.
The BTR complex is required for TZAP-induced generation of cECTRs in ATRX/DAXX-deficient cells
Taken together, our results show that TZAP overexpression in the absence of ATRX/DAXX induces telomere dysfunction and ALT-like activity, resulting in the generation of cECTRs. Since the BTR complex has been proposed as the primary regulator of ALT,44,47,48,49 we tested the requirement of the BTR complex for TZAP-induced generation of c-circles and t-cricles. The CRISPR/Cas9 system was used to generate U-2 OS knockout (KO) clones of the genes encoding the BLM or RMI1 subunits of the BTR complex,48 and TZAP was overexpressed in parental as well as in these KO clones (Figure 3A). While cECTRs production observed in c-circle (Figures 3B and 3C) and t-circle (Figure 3D) assays was increased in the parental U-2 OS cells overexpressing TZAP compared to control cells, such cECTR induction was prevented in the BLM and RMI1 KO clones. Importantly, this defect in c-circle and t-circle production was not due to excessive telomere shortening in the absence of functional BTR complex since bulk telomere length in the BLM and RMI1 KO cells was not compromised (Figure S3A).
Figure 3.

The BTR complex is required for TZAP-induced generation of cECTRs in ATRX/DAXX-deficient cells
(A) Western blot analysis of the indicated proteins in parental U-2 OS cells (WT), RMI1−/− (1A8, 1B5), and BLM−/− (2C8, 3D1) knock-out (KO) clones overexpressing either empty vector (EV) or Myc-TZAP.
(B) CCA of the cells described in A).
(C) Quantification of CCA signals in B) was performed relative to parental U-2 OS overexpressing EV. Bars represent the mean ± SD of 3 independent experiments. ∗∗∗p < 0.001, ns: non-significant, two-way ANOVA test was used to calculate the p values.
(D) TCA of the cells described in A). The arrow points to the products of rolling circle amplification of TTAGGG-rich circular extrachromosomal DNA. The experiment was repeated 3 times, and one representative experiment is shown.
(E) Western blot analysis of the indicated proteins in HeLa-LT cells overexpressing Myc-TZAP and treated with the indicated siRNAs.
(F) CCA of the cells described in E).
(G) Quantification of CCA signals in F) was performed relative to si-scramble-treated HeLa-LT cells overexpressing TZAP. Bars represent the mean ± SD of 3 independent experiments. ∗∗p < 0.01, ns: non-significant, one-way ANOVA test was used to calculate the p values.
(H) TCA of the cells described in E). The arrow points to the products of rolling-circle amplification of TTAGGG-rich circular extrachromosomal DNA. The experiment was repeated 3 times, and one representative experiment is shown.
We then asked whether the BLM helicase is also required for the TZAP-induced generation of cECTRs observed in telomerase+ cells lacking ATRX/DAXX activity. For the remaining experiments, the HeLa-LT clone was used, which is a sub-clone of HeLa1.2.11 with an average telomere length of ∼23 kb. Recruitment of the BLM helicase to telomeres in HeLa-LT cells overexpressing TZAP upon silencing of ATRX or DAXX was increased compared to control and H3.3-silenced cells (Figures S3B and S3C). In addition, BLM depletion in DAXX-deficient HeLa-LT cells overexpressing TZAP (Figure 3E) suppressed both c-circles (Figures 3F and 3G) and t-circles (Figure 3H). Finally, we assessed the requirement of other well-established factors of the ALT pathway - Rad5226 and PML26,48 -, for the TZAP-induced generation of cECTRs in DAXX-deficient HeLa-LT cells. In addition, we analyzed Rad51, which was suggested to regulate the ALT pathway by initiating the homology searches between telomeres in ALT cells50 and by alleviating replication stress at ALT telomeres.49,51 Rad51 and Rad52 were partially required for TZAP-induced c-circles in DAXX-deficient HeLa-LT cells, while PML was essential (Figures S3D and S3E).
Altogether, these data indicate that the BTR complex, Rad51, Rad52, and PML are required for the efficient induction of cECTRs production triggered by TZAP overexpression in ATRX/DAXX-deficient cells.
Efficient TZAP loading onto telomeres occurs in the context of open telomeric chromatin caused by ATRX/DAXX loss independently of H3.3 deposition
Finally, we wanted to understand how ATRX/DAXX repressed TZAP-induced telomere dysfunction and ALT features in telomerase+ cells. TZAP binds directly to dsTTAGGG repeats,39 and the zinc finger 11 of TZAP fits into the DNA major groove.52 Therefore, we reasoned that TZAP loading onto telomeric DNA, and consequently TZAP-induced telomere dysfunction, might be favored at telomeres with accessible chromatin. It has been previously shown that loss of ATRX/DAXX triggers a progressive telomere decompaction program that ultimately culminates in telomere dysfunction.53 Therefore, we hypothesized that ATRX/DAXX represses TZAP loading onto telomeres by promoting telomeric chromatin compaction. Atlantis nuclease (ATNase) digestion of chromatin followed by quantification of telomeric repeats-containing mononucleosomes was performed to visualize the chromatin status of telomeric DNA in HeLa-LT and U-2 OS cells (repressive and permissive for TZAP-induced telomere dysfunction, respectively). While the digestion pattern of bulk chromatin was indistinguishable between the two cell lines, the digestion pattern of telomeric chromatin was very different. As previously reported,54 telomeric chromatin was more compacted in telomerase+ cells (HeLa-LT) as compared to ALT+ cells (U-2 OS) as the amount of normalized mononucleosomes was significantly higher in the latter (Figures 4A and 4B). The telomeric chromatin states of ATRX-, DAXX-, or H3.3-deficient HeLa-LT cells were also analyzed. Interestingly, while the digestion pattern of bulk chromatin was comparable in all the conditions, ATRX- and DAXX-deficient HeLa-LT cells showed an increased chromatin accessibility to ATNase compared to control or H3.3-deficient cells (Figures 4C and 4D). Strikingly, while the staining of exogenously expressed Myc-TZAP often appeared as a homogeneously distributed nuclear signal in control and H3.3-depleted HeLa-LT cells, it mostly formed discrete telomeric foci in ATRX- or DAXX-depleted cells, indicating efficient telomeric recruitment of Myc-TZAP in the latest (Figures 4E and 4F).
Figure 4.

Loss of ATRX/DAXX leads to rapid decompaction of telomeric chromatin and efficient TZAP recruitment to telomeres independently of H3.3 deposition
(A) Chromatin digestion assay in HeLa-LT (telomerase+) and U-2 OS (ALT+) cells. Atlantis nuclease (ATNase) (0.125, 0.5, or 1 units) was used to digest native chromatin from 1x106 of the indicated cell lines. Left panel shows digested bulk chromatin stained with ethidium bromide. Right panel represents digested telomeric chromatin detected by southern blotting and hybridization with a 32P-end-labeled (TTAGGG)4 oligonucleotide probe. 1N: mononucleosome.
(B) Quantification of chromatin digestion assay from A). Ratios of mono-nucleosomes over total signals, for either bulk or telomeric chromatin, were calculated for the different concentrations of ATNase. Ratios of telomeric chromatin were normalized to the corresponding ratios of bulk chromatin. Finally, relative ratios of U-2 OS were normalized to their respective HeLa-LT counterparts. The experiment was performed once.
(C) Chromatin digestion assays in HeLa-LT cells treated with the indicated siRNAs. Atlantis nuclease (0.25 and 0.5 units) was used to digest native chromatin from 1x106 of the indicated cells. Left panel represents digested bulk chromatin while right panel represents digested telomeric chromatin as described in A). 1N: mononucleosome.
(D) Quantification of chromatin digestion assay from C) as described in B) Relative ratios were normalized to their respective si-scramble counterparts. Bars represent the mean ± SD of 3 independent experiments. ∗∗∗p < 0.001, ns: non-significant, two-way ANOVA test was used to calculate the p values.
(E) Representative images of Myc-TZAP immunofluorescence in combination with telomeric fluorescence in situ hybridization (FISH) in HeLa-LT cells overexpressing Myc-TZAP and treated with the indicated siRNAs. Scale bar 10 μm.
(F) Scatterplot showing quantification of experiment described in E). A total of 50 nuclei were analyzed for each sample. Data are from a single experiment. Center line, mean. n shows number of cells analyzed.
Altogether, these results indicate that telomeric chromatin becomes rapidly de-compacted upon loss of ATRX or DAXX independently of H3.3 deposition, which could be facilitating TZAP loading onto telomeres.
Discussion
TZAP overexpression in ATRX/DAXX-deficient cells induces telomere dysfunction and ALT-like activity
Our data indicate that TZAP overexpression in ALT+ cells causes telomeric replication stress, as shown by the activation of the ATR-Chk1 pathway (Figure 1B), the increased recruitment of the RPA complex to telomeres (Figures 1C, 1D, S1B, and S1C), and the exacerbation of cECTRs (Figures 3B–3D). In addition, we observe increased G2-associated telomeric DNA synthesis in ALT+ cells overexpressing TZAP (Figures 1E and 1F), which is most likely a mechanism triggered in these cells to rescue the replication problems encountered during S phase. Although all of these phenotypes are considered hallmarks of ALT, ALT+ cells overexpressing TZAP suffer from rapid telomere loss39 and cell death (data not shown). Interestingly, it was reported by others that FANCM-deficient ALT+ cells displayed similar phenotypes.55 When FANCM is absent, replication stress at ALT telomeres is exacerbated and the repair activities associated with the ALT mechanism are insufficient to maintain telomere integrity, thus causing cell death. Therefore, the authors of this work proposed that FANCM resolves replication stress at ALT telomeres by promoting replication fork remodeling and re-start.55 Based on this literature, we speculate that TZAP loading onto telomeres induces stalled replication forks that exhaust the enzymes involved in replication fork remodeling and re-start, such as FANCM. Consequently, this leads to the hyperactivation of an ALT-like pathway and telomere loss.39
The role of the BTR complex in TZAP-induced generation of cECTRs
The BTR complex has several functions in DNA replication and repair that are essential for the maintenance of genome integrity, including dissolution of double Holliday junctions into non-crossover products,56 DNA end resection,57 processing of ultrafine DNA bridges between sister chromatids in anaphase,58 and re-start of stalled replication forks.59 Moreover, many studies have demonstrated that the BTR complex is a central component of the ALT pathway. It has been reported that BLM depletion suppressed G2-associated telomeric DNA synthesis and APB formation in ALT+ cells, which led the authors to propose a model in which BLM plays a role in promoting telomere clustering at APBs upstream of telomere extension.26 However, tethering of RMI1 to telomeres in PML KO U-2 OS cells restored ALT activity, suggesting that the critical function of PML in ALT is to recruit the BTR complex to telomeres.48 In order to investigate the role of the BTR complex in ALT downstream of APB assembly, the Zou lab used an artificial system to induce APB formation by targeting PML-IV to telomeres.44 In this setting, APBs formed independently of the BTR complex. Interestingly, they found that even in the presence of APBs, anaphase telomere bridges formed in the absence of different subunits of the BTR complex. These data strongly support that BTR plays an important role in ALT by mediating D-loop migration and dissolution of recombination intermediates, which allows for efficient telomere DNA synthesis.44 Another proposed role for the BTR complex in ALT is to alleviate replication stress upstream of homology-directed repair by promoting replication fork remodeling and re-start in concert with FANCM.55
Our data show that the induction of cECTRs generated upon TZAP overexpression in ALT+ cells requires the BTR complex (Figures 3B–3D). This result supports previous literature showing that the cECTRs generated due to severe telomeric replication stress upon FANCM deficiency in ALT+ cells were also dependent on the BTR complex.55 Moreover, we observe that the cECTRs generated upon TZAP overexpression in DAXX-deficient telomerase+ cells relied on BLM (Figures 3F–3H). This observation resembles our group’s previous result where induction of telomeric replication stress in telomerase+ cells by depletion of the histone chaperone Asf1 led to a BLM-dependent increase of ALT-like phenotypes, including cECTRs.60 Altogether, we predict that one of the roles of the BTR complex in the mechanism of TZAP-induced ALT-like pathway is to resolve telomeric replication stress.
Loss of ATRX/DAXX in telomerase+ cells with long telomeres leads to telomeric chromatin decompaction and TZAP recruitment to telomeres independently of H3.3 deposition
The ATRX/DAXX complex has been established as a master regulator of chromatin state, in part due to its role in recruiting and depositing the histone variant H3.3 into GC-rich genomic regions, including pericentromeric and telomeric heterochromatin.61 However, our data show that siRNA-mediated depletion of ATRX or DAXX, but not H3.3, in telomerase+ cells leads to a rapid decompaction of telomeric chromatin (Figures 4C and 4D) and enhanced TZAP recruitment to telomeres (Figures 4E and 4F). These results suggest that ATRX/DAXX can also control telomeric chromatin state by other pathways distinct to H3.3 deposition. Another important role of ATRX/DAXX is to prevent the accumulation of G4-quadruplexes (G4) that form in GC-rich genomic regions, including telomeres, thus maintaining genome integrity.34,62 Interestingly, recent work provides evidence consistent with the idea that nucleosome positioning appears to be associated with the formation of specific DNA secondary structures, including G4s.63 Bioinformatic analysis has demonstrated that G4-forming sequences are highly enriched at nucleosome-depleted regions.64,65 Importantly, these computational predictions have been recently validated in the chromatin context by chromatin immunoprecipitation sequencing (ChIP-seq) analysis using an antibody specific to G4-motifs.66,67 We, therefore, speculate that ATRX/DAXX silencing in the telomerase+ cells with long telomeres used in our study leads to rapid accumulation of G4-motifs at telomeres, which in turn results in a nucleosome-depleted environment that allows for TZAP binding.
An overview of the consequences of TZAP overexpression
We propose a model in which overexpressed TZAP preferentially loads onto over-elongated telomeres with open chromatin in ATRX/DAXX-deficient cells. This creates impediments to the replication fork by a still-unknown mechanism, which leads to telomere dysfunction and the activation of an ALT-like pathway, resulting in BTR-dependent generation of cECTRs (Figure S4). Analysis of data provided by The Cancer Genome Atlas showed that TZAP expression is associated with different prognostic outcomes depending on the type of cancer. Importantly, the upregulation of TZAP is related with poor prognosis in adrenocortical carcinoma, colon and rectal cancer, brain lower-grade glioma, and prostate adenocarcinoma.68 Although more work needs to be done to properly understand the interplay between TZAP and ATRX/DAXX at telomeres, our data suggest that it would be interesting to study the ATRX/DAXX status in tumors with high levels of TZAP, as well as to consider studying the response of these tumors to treatments that promote telomeric chromatin relaxation.
Limitations of the study
One of the potential constraints of our study is that all of the information we report about TZAP is in the context of its overexpression. However, we find that the deleterious effects of TZAP overexpression are only observed upon conditions that promote its efficient recruitment to telomeres, such as ATRX/DAXX loss.
An important aspect that should be addressed in the future is whether efficient TZAP recruitment to telomeres requires telomeric chromatin relaxation. It will be interesting to see if telomeric chromatin relaxation induced by means different from ATRX/DAXX downregulation also facilitates TZAP recruitment to telomeres. In addition, other experiments to test this hypothesis could involve manipulation of telomeric chromatin in ALT+ cells to turn it into a close state and see if TZAP overexpression leads to exacerbation of telomere dysfunction and ALT features. Moreover, it will be of interest to further investigate the exact mechanism by which the cECTRs are generated in ATRX/DAXX-deficient cells upon TZAP overexpression.
The t-circle assay used in this study has been used in the past as an approach to detect circular ds t-circles.39,69 However, we cannot ignore that this assay can also detect partially ss telomeric TTAGGG DNA circles (G-circles).
Finally, further experiments should be conducted in the future to investigate the function of endogenous TZAP in cancer cells as well as in stem cells.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-GAPDH rabbit polyclonal antibody | Abnova | Cat# PAB17013; RRID:AB_10629928 |
| Anti-γH2A.x Ser 139 mouse monoclonal antibody | Millipore | Clone: JBW301; Cat# 05-636; RRID:AB_309864 |
| Anti-Myc mouse monoclonal antibody | Cell Signaling | Clone: 9B11; Cat# 2276S; RRID:AB_331783 |
| Anti-pChk1 Ser 345 rabbit monoclonal antibody | Cell Signaling | Clone: 133D3; Cat# 2348S; RRID:AB_331212 |
| Anti-pChk2 Thr 68 rabbit polyclonal antibody | Cell Signaling | Cat# 2661S; RRID:AB_331479 |
| Anti-pATR Thr 1989 rabbit monoclonal antibody | Abcam | Clone: EPR21991; Cat# ab223258 |
| Anti-pATM Ser 1981 rabbit monoclonal antibody | Cell Signaling | Clone: D25E5; Cat# 13050S; RRID:AB_2798100 |
| Anti-PML rabbit polyclonal antibody | Santa Cruz | Cat# sc-5621; RRID:AB_2166848 |
| Anti-DAXX rabbit polyclonal antibody | Santa Cruz | Cat# sc-7152; RRID:AB_2088784 |
| Anti-H3.3 rabbit monoclonal antibody | Abcam | Clone: EPR17899; Cat# ab176840; RRID:AB_2715502 |
| Anti-RMI1 rabbit polyclonal antibody | Novus | Cat# NB100-1720; RRID:AB_2181500 |
| Anti-FLAG mouse monoclonal antibody | Sigma | Clone: M2; Cat# F1804-1MG; RRID:AB_262044 |
| Anti-RPA32 mouse monoclonal antibody | Abcam | Clone: 9H8; Cat# ab2175; RRID:AB_302873 |
| Anti-RPA70 rabbit monoclonal antibody | Santa Cruz | Clone: B6; Cat# sc-28304; RRID:AB_628224 |
| Anti-BLM rabbit polyclonal antibody R7019 | Karlseder lab | N/A |
| Anti-TRF1 rabbit polyclonal antibody R6839 | Karlseder lab | N/A |
| Anti-TRF2 rabbit polyclonal antibody R6841 | Karlseder lab | N/A |
| Peroxidase-conjugated anti-mouse IgG | Amersham | Cat# NXA931V; RRID:AB_772209 |
| Peroxidase-conjugated anti-rabbit IgG | Amersham | Cat# NA934V; RRID:AB_772206 |
| AlexaFluor 488 anti-IgG mouse | Invitrogen | Cat# A11001; RRID:AB_2534069 |
| AlexaFluor 594 anti-IgG rabbit | Invitrogen | Cat# A21207; RRID:AB_141637 |
| AlexaFluor 568 anti-IgG mouse | Invitrogen | Cat# A-11004; RRID:AB_2534072 |
| Anti-DIG-AP antibody | Roche | Cat# 11 093 274 910; RRID:AB_2313640 |
| Chemicals, peptides, and recombinant proteins | ||
| GlutaMAX-Dulbecco’s Modified Eagle’s Medium (DMEM) | Gibco | Cat# 10569-010 |
| Fetal Bovine Serum | Avantor | Cat# 97068-085 |
| Penicillin Streptomycin Solution | Corning | Cat# 30-002-Cl |
| MEM Nonessential Amino Acids | Corning | Cat# 25-025-Cl |
| Lipofectamine RNAiMAX transfection reagent | Life Technologies | Cat# 13778-075 |
| OptiMEM | Gibco | Cat# 31985-070 |
| NuPage LDS sample buffer | Invitrogen | Cat# NP0007 |
| Benzonase | Millipore | Cat# 70746-3 |
| NuPage Bis-Tris precast electrophoresis gel | Invitrogen | Cat# NP0321 and Cat# NP0322 |
| ProLong Diamond with DAPI | Invitrogen | Cat# P36971 |
| Telomeric PNA probe | PNA Bio | Cat# F1004 |
| RO-3306 | AdipoGen Life Sciences | Cat# AG-CR1-3515-M005 CAS number: 872573-93-8 |
| Φ29 polymerase | NEB | Cat# M0269L |
| Blocking reagent | Roche | Cat# 11 096 176 001 |
| CDP star reagent | Roche | Cat# 12 041 677 001 |
| Digoxigenin-11-dUTP | Roche | Cat# 11 093 088 910 |
| 10x NEBuffer 2 | NEB | Cat# B7002S |
| T4 DNA polymerase | NEB | Cat# M0203S |
| NTC buffer | MacHerey-Nagel | Cat# 740654.100 |
| Lambda exonuclease | NEB | Cat# M0262S |
| dCTP [α-32P] | PerkinElmer | Cat# NEG513H250UC |
| Klenow fragment 3′-5′ exo- | NEB | Cat# M0212M |
| Critical commercial assays | ||
| Click-iT EdU imaging kit | Invitrogen | Cat# C10339 |
| dsDNA BR assay kit | Invitrogen | Cat# Q32850 |
| EZ Nucleosomal DNA Prep Kit | Zymo Research | Cat# E2030 |
| NucleoSpin Gel and PCR Clean-up kit | MacHerey-Nagel | Cat# 740609.50 |
| ECL kit | Prometheus | Cat# 20-302B |
| Experimental models: Cell lines | ||
| U-2 OS | ATCC | Cat# HTB-96 |
| HT-1080 | ATCC | Cat# CCL-121 |
| GM847 | Coriell Institute | Cat#GM00847 |
| HeLa 1.2.11 | 41 | N/A |
| HeLa-LT | Karlseder lab | N/A |
| RMI1KO (clone 1A8) U-2OS | 48 | N/A |
| RMI1KO (clone 1B5) U-2 OS | 48 | N/A |
| BLMKO (clone 2C8) | 48 | N/A |
| BLMKO (clone 3D1) | 48 | N/A |
| Oligonucleotides | ||
| ON-TARGETplus SMARTpool siRNA targeting ATRX | Dharmacon | Cat# L-006524-00-0005 |
| ON-TARGETplus SMARTpool siRNA targeting DAXX | Dharmacon | Cat# L-004420-00-0005 |
| ON-TARGETplus SMARTpool siRNA targeting H3FA | Dharmacon | Cat# L-011684-01-0005 |
| ON-TARGETplus SMARTpool siRNA targeting H3FB | Dharmacon | Cat# L-012051-00-0005 |
| ON-TARGETplus SMARTpool siRNA targeting BLM | Dharmacon | Cat# L-007287-00-0005 |
| ON-TARGETplus SMARTpool siRNA targeting PML | Dharmacon | Cat# L-006547-00-0005 |
| ON-TARGETplus SMARTpool siRNA targeting Rad51 | Dharmacon | Cat# L-003530-00-0005 |
| ON-TARGETplus SMARTpool siRNA targeting Rad52 | Dharmacon | Cat# L-011760-00-0005 |
| ON-TARGETplus Non-targeting Control Pool | Dharmacon | Cat# D-001810-10-20 |
| C-rich template oligonucleotide: Phospho-CCCTAACCCTAACCCTAACCCTAACCCTAAC CCTAACCCTAGATAGTTGAGAGTC |
HPLC purified from ETON Bioscience | N/A |
| Universal TRF primer: Phospho-GACTCTCAACTATC+T+A |
Biosynthesis | N/A |
| C-rich primer 3 rep: CCCTAACCCTAACCCTAA |
ETON Bioscience | N/A |
| Recombinant DNA | ||
| pLPC encoding N-terminal Myc-tagged TZAP | 39 | N/A |
| pLPC encoding N-terminal FLAG-tagged TZAP | 39 | N/A |
| pLPC enconding mCherry FLAG-tagged TZAP | Lazzerini Denchi lab | N/A |
| pBABE enconding hTR | K Collins lab | N/A |
| Software and algorithms | ||
| Imaris | OXFORD instruments | RRID:SCR_007370 |
| FlowJo | FlowJo | RRID:SCR_008520 |
| Fiji | 70 |
https://fiji.sc RRID:SCR_002285 |
| Image Quant TL | Cytiva | RRID:SCR_018374 |
| GraphPad Prism | GraphPad | RRID:SCR_002798 |
| Other | ||
| Positively charged nylon membrane | GE Healthcare | Cat# RPN203B |
| Nitrocellulose membrane | Amersham | Cat# 10600037 |
| Bio-Spin P-6 columns, Tris Buffer | BIO-RAD | Cat# 7326227 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jan Karlseder (karlseder@salk.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Cell lines
U-2 OS (#HTB-96) and HT-1080 (#CCL-121) were purchased from ATCC. GM847 (#GM00847) was purchased from the Coriell Institute. HeLa1.2.11 was generated in Titia de Lange’s lab and is a sub-clone of the HeLa cell line with a bulk telomere length of ∼23 kb.41 HeLa-LT was generated in the Karlseder lab and is a sub-clone of HeLa1.2.11 with a bulk telomere length of ∼23 kb.
Cell lines were cultured in Glutamax-Dulbecco’s modified Eagle’s medium (DMEM) (#10569-010) supplemented with 10% Fetal Bovine Serum (FBS) (#97068-085), MEM Nonessential Amino acids (#25-025-Cl) and Penicillin Streptomycin Solution (#30-002-Cl) at 37 °C, 7.5% CO2 and 3% O2. Cells have been tested free of mycoplasma.
BLM and RMI1 knockout U-2 OS clones were generated and validated as previously described.48
Method details
Plasmids and transductions
pLPC retroviral vectors encoding N-terminal Myc- or FLAG-tagged human ZBTB48 (TZAP) were described before.39 mCherry FLAG-tagged TZAP full length was cloned into pLPC retroviral vector. pBABE retroviral vector encoding hTR was obtained from K. Collins lab (University of California, Berkeley).
The production of retroviruses was achieved by calcium phosphate mediated transfection of the retrovirus producer cell line Phoenix-AMPHO with the indicated retroviral vectors in the presence of 100 μM of chloroquine. 5 hours after transfection, the media containing the transfection reagents was exchanged for fresh media. Viral supernatant was collected 24 hours later and used for transduction of the indicated cell lines in the presence of 4 μg/ml of polybrene. Cells were incubated with the viral supernatant for 24 hours, while fresh media was added to Phoenix-AMPHO cells for the second round of virus production and cells transduction. Finally, 48 hours after the second transduction, infected cells were selected by either addition of 1 μg/ml puromycin or mCherry cell-sorting.
siRNAs and transfections
The siRNAs used in our study were purchased from Dharmacon: ON-TARGETplus Human siRNA SMARTpool targeting ATRX (#L-006524-00-0005), DAXX (#L-004420-00-0005), H3F3A (#L-011684-01-0005), H3F3B (#L-012051-00-0005), BLM (#L-007287-00-0005), PML (#L-006547-00-0005), Rad52 (#L-011760-00-0005), Rad51 (#L-003530-00-0005), and ON-TARGETplus Non-targeting Control Pool (#D-001810-10-20).
Two (for Western Blotting, IF, IF-FISH, c-circle and t-circle assays) or three (for ATSA assay) consecutive siRNA transfections were carried out as follows: Cells were seeded the day before transfection. The day of transfection, cells were incubated with antibiotic-free media. Transfection solutions were prepared by mixing 250 μl of OptiMEM containing the indicated siRNAs to a final concentration of either 30 nM (when only one siRNA was included) or 25 nM (when two siRNAs were included), with 250 μl of OptiMEM containing 2.5 μg/ml final concentration of Lipofectamine RNAiMAX (#13778-075, Life technologies). Transfection solutions were incubated for 15 min at RT and then added drop-wise to the cells. 8 hours later, the media of the cells was exchanged for antibiotic-containing media and cells were either transfected again or harvested for experiments 72 hours later.
Antibody sources
α-GAPDH (#PAB17013, Abnova), α-γH2A.x Ser 139 (JBW301, #05-636, Millipore), α-Myc-tag (9B11, #2276S, Cell Signaling), α-p-Chk1 Ser 345 (133D3, #2348S, Cell Signaling), α-PML (#sc-5621, Santa Cruz) α-p-Chk2 Thr 68 (#2661S, Cell Signaling), α-p-ATR Thr 1989 (#ab223258, Abcam), α-p-ATM Ser 1981 (D25E5, #13050S, Cell Signaling), α-ATRX (#sc-15408, Santa Cruz), α-DAXX (#sc-7152, Santa Cruz), α-H3.3 (#ab176840, Abcam), α-RMI1 (#NB100-1720, Novus), α-FLAG-tag (M2, #F1804-1MG, Sigma), α-RPA32 (9H8, #ab2175, Abcam), α-RPA70 (#sc-28304, Santa Cruz). α-BLM (R7019), α-TRF1 (R6839) and α-TRF2 (R6841) were raised in our lab. All antibodies were used for Western Blotting apart from α-BLM, α-Myc and α-FLAG that were also used for IF and α-RPA32, α-RPA70, α-PML, α-TRF1 and α-TRF2 that were used for IF only. Secondary antibodies used for Western Blotting were peroxidase-conjugated anti-mouse IgG (#NXA931V, Amersham) or anti-rabbit IgG (#NA934V, Amersham). Secondary antibodies used for IF were AlexaFluor 488 anti-IgG mouse (#A11001, Invitrogen) and AlexaFluor 594 anti-IgG rabbit (#A21207, Invitrogen), and AlexaFluor 568 anti-IgG mouse (#A-11004, Invitrogen).
Western Blotting
Cell pellets were lysed in NuPage LDS sample buffer (#NP0007, Invitrogen) containing 5% benzonase (#70746-3, Millipore) at 1x104 cells per μl. Proteins were resolved using NuPage Bis-Tris gel electrophoresis (#NP0321 or #NP0322, Invitrogen) and transferred to nitrocellulose membranes (#10600037, Amersham). Membranes were incubated in blocking solution (5% milk in TBST) for 1 hour at RT, followed by incubation with primary antibodies shaking overnight at 4 °C. Membranes were then washed in TBST, incubated with peroxidase-conjugated secondary antibodies shaking for 1 hour at RT, washed again with TBST and peroxidase activity was detected using an ECL kit (#20-302B, Prometheus) and Syngene G-Box imager. All antibodies were diluted in 5% milk in TBST, with the exception of α-p-ATM, α-p-ATR, α-p-Chk1 and α-p-Chk2 that were diluted in 5% BSA in TBST.
Immunofluorescence (IF)
Cells were seeded onto glass coverslips 24 h before the experiment. Cells were fixed in 4% paraformaldehyde in PBS for 10 min, washed in PBS and incubated in permeabilization solution (0.2% Triton X-100, 30 nM glycine in PBS) for 15 min shaking at Room Temperature (RT). Cells were then incubated in blocking solution (5% BSA in PBS) for 1 hour at RT, followed by incubation with primary antibodies for 2 hours at RT or overnight at 4 °C in a humidity chamber, washed with PBS and incubated with secondary antibodies for 1 hour at RT. Cells were finally washed with PBS and mounted in ProLong Diamond with DAPI (#P36971, Invitrogen). Confocal microscopy was performed using a Zeiss LSM 880 microscope. Z-stack image acquisition was performed for the experiments in Figures 1C, 1D, 1E, 1F, 2E, 2F, S1B, and S1C, while single plane image acquisition was performed for the experiment in Figures S3B and S3C. In the experiment from Figures 1C and 1D, cells containing coverslips were pre-extracted prior to fixation by incubating them in pre-extraction buffer (25 mM HEPES pH 7.4, 50 mM NaCl, 1 mM EDTA, 3 mM MgCl2, 300 mM Sucrose, 0.5% Triton X-100) for 5 min at 4 °C. Coverslips were then fixed, stained and mounted as explained above. Raw images were processed using Zeiss ZEN (blue edition) prior to assembly of representative images.
IF-FISH
Cells were seeded onto glass coverslips 24 h before the experiment. Cells were washed in PBS, fixed in 4% paraformaldehyde in PBS for 10 min shaking at RT and permeabilized in 0.1% Triton X-100 in PBS for 15 min shaking at RT. Cells were then incubated in ABDIL (20 mM Tris pH 7.5, 2% BSA, 0.2% fish gelatin, 150 mM NaCl, 0.1% Triton X-100, 0.1% sodium azide, 100 μg/ml RNaseA) for 1 hour at 37 °C. Cells were then incubated with the primary antibody diluted in ABDIL for 1 hour at RT, washed 3x in PBST and incubated with the secondary antibody for 30 min at RT. Cells were fixed again in 4% paraformaldehyde in PBS. Cells were then dehydrated in ethanol series (70%, 90% and 100%) for 3 min each and air dried for 20 min. Cells were then covered in 0.3 ng/μl of the telomeric PNA probe (#F1004, PNA Bio) diluted in hybridization buffer (70% deionized formamide, 0.25% blocking reagent (NEN), 10 mM Tris pH 7.5) and coverslips were heated for 5 min at 80 °C, followed by incubation at RT. Afterward, cells were washed 2x in PNA wash buffer A (70% formamide, 10 mM Tris pH 7.5) and 3x in PNA wash buffer B (50 mM Tris pH 7.5, 150 mM NaCl, 0.08% Tween-20). Slides were finally mounted in ProLong Diamond with DAPI. Imaging was performed as explained in the IF method for Figures S3B and S3C.
ALT telomere DNA synthesis in APBs (ATSA) assay
The ATSA assay protocol was adapted from.26 In order to visualize G2-associated DNA synthesis at telomeres, cells were first seeded in a 10 cm dish containing glass coverslips and they were synchronized in the G2 phase of the cell cycle by treatment with 2 mM thymidine for 21 hours, release into fresh medium for 4 hours, followed by treatment with 15 μM of the CDK1 inhibitor RO-3306 (#AG-CR1-3515-M005, AdipoGen Life Sciences) for 12 hours. Cells were then incubated with 20 μM EdU for 3 hours. Cells containing coverslips were collected and fixed with 3.7% formaldehyde in PBS for 15 min at RT, and then washed twice with 3% BSA in PBS. Cells were then permeabilized by incubation of the coverslips with 0.5% Triton X-100 in PBS for 20 min shaking at RT. Cells were then subjected to the click reaction between the EdU and the azide group containing Alexa Fluor 594 following the indications from the Click-iT Edu imaging kit (#C10339, Invitrogen). TRF1 IF, imaging and EdU-TRF1 colocalization analysis were performed as explained in the IF method, with the only difference that the permeabilization step was omitted.
t-circle assay
This protocol has been adapted from the t-circle protocol described in ref.69 Genomic DNA (gDNA) was extracted from 1 million cells containing pellet by gently resuspending it into 150 μl of QCP lysis buffer pre-warmed at 56 °C (50 mM KCl (previously autoclaved); 10 mM Tris pH 8.5 (previously autoclaved); 2 mM MgCl2 (previously autoclaved); 0.5 % NP40; 0.5 % Tween20; 0.05 AU/ml QIAGEN protease). Lysates were vortexed at 2,000 rpm for 15 sec, after which they were placed in a shaker at 1,400 rpm at 56 °C for 1 h, and finally at 70 °C for 20 min. Lysates were allowed to cool down to RT, vortexed again at 2,000 rpm for 15 sec, quickly spun down and placed on ice.71 gDNA was quantitated using the Qubit fluorometer and the dsDNA BR assay kit (#Q32850, Invitrogen). DNA concentration was equalized in all samples by dilution with QCP buffer. The annealing mix was prepared by adding 400 ng of genomic DNA to 10 mM Tris pH 8 (previously autoclaved) in a final volume of 26 μl, followed by addition of 10 μl of 10 μM TelC (CCCTAA)4 oligo and 4 μl of annealing buffer (200 mM Tris-HCl pH 8, 200 mM KCl, 1 mM EDTA). The 40 μl annealing mix was incubated in a thermocycler at 96 °C for 5 min and brought gradually to 25 °C using the ramp option (0.1 °C down/s). The annealing mix was divided into two 20 μl aliquots, and each of these was mixed with 18.5 μl of master mix (2x φ29 polymerase buffer, 0.4 μM dNTP mix, 0.4 mg/ml BSA) and with 1.5 μl of either φ29 polymerase (15 U) (#M0269L, NEB) or nuclease-free water. Primer extension was achieved by incubating the reaction at 30 °C for 12 hours and φ29 polymerase was then inactivated by incubation at 65 °C for 20 min.
The extension products were separated by agarose gel electrophoresis (0.6% agarose in 1xTAE), by first setting up the voltage to 3.5 V/cm for 1 hour, followed by decreasing the voltage down to 1.3 V/cm for 18 h. The gel was then subjected to depurination (10 min incubation in 0.25 M HCl), denaturation (2 incubations of 15 min in 1.5 M NaCl, 0.5 M NaOH) and neutralization (2 incubations of 15 min in 1.5 M NaCl, 0.5 M Tris pH 7.2), and DNA was transferred onto a positively charged nylon membrane by southern blotting (#RPN203B, GE Healthcare). The DNA was crosslinked onto the membrane at a setting of 120 mJ/cm2 and membrane was incubated with prehybridization buffer (5x SSC; 0.1% sarkosyl; 0.04% SDS) for 1-2 hours at 65 °C. The t-circle extension products were finally detected by incubating the membrane with prehybridization buffer containing 1.3 nM of Digoxigenin labeled TTAGGG rich probe (generated as explained below) over night. After the incubation, the DNA bound DIG-TelG probe was detected as explained in.72 Briefly, the membrane was washed 3 times 15 min with wash buffer 1 (2x SSC 0.1% SDS) at RT and 1 time 15 min with wash buffer 2 (2x SCC) at RT. The membrane was then incubated for 30 min at RT in blocking buffer (1% blocking reagent (Roche, 11 096 176 001) in maleic acid buffer (100 mM maleic acid, 150 mM NaCl, pH7.5)). While the membrane was blocking, the anti-DIG-AP antibody (Roche, 11 093 274 910) was spun down at 13,000 g for 5 min at 4 °C. The membrane was then incubated 30 min at RT in antibody solution prepared with blocking buffer containing a 1:20,000 dilution of the anti-DIG-AP antibody. The membrane was then washed 2 times 15 min at RT in wash buffer 3 (maleic acid buffer, 0.3% (vol/vol) tween 20). The membrane was finally equilibrated in AP buffer (100 mM Tris, 100 mM NaCl, pH 9.5) for 2 min. The excess of buffer was removed from the membrane carefully, and the membrane-bound anti-DIG-AP antibody was detected by covering the membrane with the CDP star reagent (Roche, 12 041 677 001) and incubating for 5 min at RT, followed by detection of the chemiluminescent signal in the Syngene G-Box imager.
In the t-circle assay shown in Figure S2D, detection of t-circle extension products was performed slightly different. In this case, the gel was dried out at 50 °C for 2 hours and 30 min followed by denaturation and neutralization as explain above. The gel was then prehybridized with CHURCH buffer for 1 hour at 55 °C, followed by hybridization with CHURCH buffer containing a 32P-end-labeled TelG (TTAGGG)4 probe at 55 °C overnight. The gel was then washed 3x 30 min with 4xSSC and 1x 30min with 4xSSC, 0.1% SDS, exposed to a Storage Phosphor Screen (GE Healthcare) and scanned in a Thyphoon9400 Phosphorimager (Amersham, GE Healthcare).
c-circle assay
gDNA was extracted from 1 million cells containing pellets and quantitated as explained above for the t-circle assay. DNA concentration was equalized in all samples by dilution with QCP buffer. 10 and 50 ng of input DNA was added to 10 mM Tris pH 7.6 (previously autoclaved) to a final volume of 10 μl, and they were mixed with 10 μl of master mix (2x φ29 polymerase buffer, 8 μg/ml BSA, 8 mM DTT, 2 mM dNTPs, 0.2 % Tween20, 0.75 μl φ29 polymerase). A negative control reaction with 50 ng of input DNA and with nuclease-free water instead of φ29 polymerase was also set up. Rolling circle amplification reactions were performed by incubating samples in a thermocycler at 30 °C for 4 hours, then 70 °C for 20 minutes and finally temperature was cooled down to room temperature.71 The reaction products were transferred onto a positively charged nylon membrane using a dot-blot manifold and DNA was crosslinked to the membrane as explained above for the t-circle assay. The membrane was then prehybridized with CHURCH buffer (500 ml 0.5M NaPi pH 7.2, 2 ml 0.5 M EDTA pH 8, 70 g SDS, 10 g BSA, fill with miliQ water to 1 l, filtered) for 1 hour at 65 °C followed by hybridization with CHURCH buffer containing a 32P-klenow-labeled TelC probe at 65 °C overnight (the protocol for making the 32P-klenow-labeled TelC probe is explained below). The membrane was then washed briefly 2-3 times with 2xSSC to remove excess of radioactivity, followed by 2 washes with (2xSSC, 0.1% SDS) and 2 washes with (2xSSC), all of them for 5 min at 65 °C. Finally, the membrane was exposed to a Storage Phosphor Screen and scanned in a Thyphoon9400 Phosphorimager.
Telomere Restriction Fragments (TRF) assays
The TRF assay protocol was adapted from Titia de Lange’s lab website. gDNA was extracted from 5-10 x 106 cells as follows: cells were pelleted, resuspended in 1 ml TNE (10 mM Tris pH 7.4, 100 mM NaCl, 10 mM EDTA, filtered) and transferred to a 15 ml tube containing 1 ml of TENS (10 mM Tris pH 7.4, 10 mM EDTA, 100 mM NaCl, 1% SDS, filtered) with 100 μg/ml proteinase K. The mixture was gently mixed by inverting the tube 10 times, and proteinase K digestion was carried out overnight at 37 °C. The sample was then transferred to a pre-spun 15 ml phase-lock gel heavy tube containing 2 ml of Phenol:Chloroform:Isoamyl Alcohol 25:24:1, and the tube was inverted gently until the 2 phases were properly mixed. The sample was then spun down at 1,500 g for 5 min, after which the gDNA was precipitated by transferring the nucleic acid containing phase to a new 15 ml tube containing 220 μl of 2 M NaAc pH 5.5 and 2 ml of isopropanol, followed by gentle inversion of the tube until the precipitated gDNA could be observed. The precipitated gDNA was then carefully collected and gently resuspended into 300 μl of TNE containing 100 μg/ml RNAseA, followed by 2 hours and 30 min incubation at 37 °C. 300 μl of TENS with 100 μg/ml proteinase K was added to the sample, mixed and incubated for 1 hour at 37 °C. gDNA was extracted again using 2 ml phase lock gel heavy tubes and Phenol:Chloroform:Isoamyl Alcohol - NaAc - isopropanol method using similar proportions as described above. The precipitated gDNA was collected, gently resuspended in TE buffer (10 mM Tris, 0.1 mM EDTA, pH 8) and incubated for 1 hour at 37 °C. gDNA was digested overnight at 37 °C using a combination of MboI and AluI enzymes. DNA concentration was measured using the Qubit fluorometer and the dsDNA BR assay kit as explained above for the t-circle assay. 4-5 μg of digested gDNA per sample as well as 10 μl of “DNA Molecular Weight Marker II (0.12-23.1 kbp), λDNA HindIII digested” were resolved by 0.7% agarose gel electrophoresis at 1.3 V/cm for 20-24 hours in 1xTAE. Loading control was assessed by staining the gel with SYBR Gold Nucleic Acid Gel Stain (#S11494, Invitrogen) according to the manufacturer’s instructions. The gel was then subjected to depurination, denaturation, neutralization, southern blotting and detection of membrane-bound telomeric DNA as described in the t-circle assay method.
The TRF assay shown in Figure S1A was carried out in a different way. The buffer used for the agarose gel electrophoresis was 0.5xTBE instead of 1xTAE. The loading control was assessed by Ethidium Bromide staining and the gel was subsequently dried out at 50 °C for 2 hours and 30 min followed by denaturation and neutralization as explained above. The gel was then prehybridized with CHURCH buffer for 1 hour at 55 °C, followed by hybridization with CHURCH buffer containing a 32P-end-labeled TelG (TTAGGG)4 probe at 55 °C overnight. The gel was then washed 3x 30 min with 4xSSC and 1x 30min with 4xSSC, 0.1% SDS, exposed to a Storage Phosphor Screen and scanned in a Thyphoon9400 Phosphorimager as explained above in the c-circle assay method.
Generation of Digoxigenin-labeled TTAGGG rich probe
This protocol has been adapted from.73 Briefly, 40 μM of annealed template stock was generated by mixing the following reagents: 20 μl C-rich template oligonucleotide (Phospho-CCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAGATAGTTGAGAGTC, 100 μM. HPLC purified from ETON), 20 μl universal TRF primer (Phospho-GACTCTCAACTATC+T+A, 100 μM. “+” are locked bridged nucleotides. Byosynthesis), 5 μl 10x NEBuffer 2 (#B7002S, NEB) and 5 μl nuclease free H2O. The mix was incubated in a thermocycler at 95 °C for 5 min followed by gradually cooling down to RT using the ramp option 0.1 °C/second. 3′ fill in reaction was carried out by mixing the following reagents in a PCR tube: 10 μl of 40 μM annealed template stock, 5 μl of 10x dNTP mix (10 mM dATP, 10 mM dGTP, 6.5 mM dTTP), 17.5 μl DIG-11-dUTP (1 mM stock, Roche 11093088910), 4 μl 10x NEBuffer 2, 1 μl klenow exo- (50,000 U/ml stock, NEB M0212M) and 12.5 μl nuclease free H2O. The 3′ fill in mix was incubated in a thermocycler at 37 °C for 12 hours followed by heat inactivation at 75 °C for 20 min. DNA blunting reaction was then performed by mixing the inactivated mixture with 1 μl 10x NEBuffer 2, 3.3 μl T4 DNA Polymerase (3,000 U/ml stock, NEB M0203S) and 5.7 μl nuclease free H2O, and incubating it in a thermocycler at 12 °C for 15 min. The DNA was then purified with NucleoSpin Gel and PCR Clean-up kit (#740609.50, MacHerey-Nagel) according to the manufacturer’s protocol with NTC buffer (#740654.100, MacHerey-Nagel), and eluted in 30 μl 10 mM Tris pH 8. Finally, Lambda exonuclease digestion was carried out by mixing the following reagents in a PCR tube: 30 μl purified DNA, 5 μl 10x lambda exonuclease buffer, 4 μl lambda exonuclease (5,000 U/ml stock, NEB M0262S) and 11 μl nuclease free H2O. The mix was then incubated in a thermocycler at 37 °C for 1 hour. The DNA was then purified with NucleoSpin Gel and PCR Clean-up kit according to the manufacturer’s protocol for single-stranded DNA with NTC buffer. DNA was eluted in 50 μl 10 mM Tris pH 8 and DNA concentration was evaluated using Nanodrop.
Generation of 32P-labelled CCCTAA rich probe
This protocol has been adapted from Titia de Lange’s lab website. Annealed template was prepared by mixing the following reagents: 2.5 μl telomere repeats fragment (20 ng/μl) (∼ 800 bp dsDNA fragment containing TTAGGG repeats. It is cut out from a plasmid kindly provided by Titia de Lange, using EcoR1HF restriction enzyme from NEB), 1.25 μl CCCTAACCCTAACCCTAA oligo (10 μM) and 7.9 μl nuclease free H2O. The mix was then spun down and boiled for 5-10 min at 95 °C followed by gradually cooling down to RT. The following reagents were subsequently added to the annealed template: 1.5 μl 10x OLB (Oligo nucleotide labelling) buffer (0.5 M Tris pH 6.8, 0.1 M MgCl2, 1 mM DTT, 0.5 mg/ml BSA, 6 μl of 100 mM dGTP, dATP and dTTP for 1 ml of OLB buffer), 1.5 μl 32P-α-dCTP (#NEG513H250UC, PerkinElmer) and 0.5 μl klenow exo-enzyme (50,000 U/ml stock). The mix was incubated at RT for at least 90 min, after which nuclease free H2O was added up to 50 μl, and probe was finally purified using a Bio-Spin P-6 column (#7326227) according to the manufacturer’s instructions.
Chromatin digestion assays
The chromatin digestion assay protocol was adapted from.54 Nuclei isolation and digestion of native chromatin was performed using EZ Nucleosomal DNA Prep Kit from Zymo Research (#D5220). Digestion of chromatin was performed using the indicated amounts of Atlantis dsDNase (#E2030, Zymo Research) for 30 min at 42 °C. Nucleosomal DNA was purified and subsequently resolved in a 1% agarose gel. The digestion pattern of bulk chromatin was assessed by ethidium bromide staining, after which the DNA was transferred to a nylon membrane by alkaline southern blotting overnight. The DNA was then UV-crosslinked and hybridized with a 32P-end-labeled (TTAGGG)4 oligonucleotide probe to visualize the digestion pattern of telomeric nucleosomal DNA.
Cell cycle analysis
Cells were fixed with 70% ethanol overnight at 4 °C. Following wash with PBS, cells were incubated with PBS containing 4 μg/ml RNaseA and 4 μg/ml propidium iodide for 30 min at 37 °C. Cells were then analyzed by flow cytometry on BD-FACSCanto II. Data analysis was performed using FlowJo software.
Quantification and statistical analysis
IF Z-stack quantification was performed using Imaris software, while quantification of IF and IF-FISH single plane images was performed using ImageJ software. Quantification of the c-circle assay dot blot signals was done using the Image Quant TL software and analysis was performed by subtracting the average intensity of the -Φ29 sample to the respective Φ29 containing sample, and values were normalized against the indicated control sample. Quantification and analysis of telomeric mono-nucleosomes in the chromatin digestion assays was performed as explained in Figure 4 legend using ImageJ software. Statistical analysis and graphical representation were done using GraphPad prism software. Statistical test methods are described in each figure legend. Statistical significance is represented by ∗∗∗∗, ∗∗∗ and ∗∗ and indicate a computed p-value <0.0001, <0.001 and <0.01 respectively. ns = not significant.
Acknowledgments
We thank the members of the Karlseder and Lazzerini Denchi labs for constructive comments and discussion, specially Joe Nassour and Tobias Schmidt. We thank Sara Przetocka for helping with the analysis of immunofluorescence experiments. We thank Tobias Schmidt for helping with the optimization of southern blotting and detection of telomeric DNA using Digoxigenin (DIG)-labeled probes. SPM has received support from the European Molecular Biology Organization (ALTF 903-2018) and from the International Human Frontiers Science Program Organization (LT000108/2019-L). JMF has received support from the Paul F. Glenn Center for Biology of Aging Research. MK was a recipient of the Marieta Blau Grant. JN has received support from the European Molecular Biology Organization (ALTF 213-216), the Hewitt Foundation, and the National Cancer Institute (K99CA252447). JSZL is a recipient of the Damon Runyon Fellowship. JK has received support from the Salk Institute Cancer Center Core Grant (P30CA014195), the National Institutes of Health (RO1CA227934, RO1CA234047, RO1AG077324, RO1CA228211), the Donald and Darlene Shiley Chair, the Samuel Waxman Cancer Foundation, the Glenn Foundation for Medical Research, the Abney Foundation, the American Heart Association (19PABHI34610000), and the Highland Street Foundation.
Author contributions
SPM, JMF, JK, and ELD designed the experiments. SPM and JMF performed all the experiments, with the exception of Figure S2B that was performed by JN. MK helped with the experiments shown in Figures 1, 2, and 3. CH provided technical support. JSZL generated the RMI1−/− 1A8, RMI1−/− 1B5, BLM−/− 2C8, and BLM−/−3D1 KO U-2 OS cells. SPM, JMF, JK, and ELD wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper received support from a program designed to increase minority representation in their field of research. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Published: March 14, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106405.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Makarov V.L., Hirose Y., Langmore J.P. Long G tails at both ends of human chromosomes suggest a C strand degradation mechanism for telomere shortening. Cell. 1997;88:657–666. doi: 10.1016/s0092-8674(00)81908-x. [DOI] [PubMed] [Google Scholar]
- 2.McElligott R., Wellinger R.J. The terminal DNA structure of mammalian chromosomes. EMBO J. 1997;16:3705–3714. doi: 10.1093/emboj/16.12.3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palm W., de Lange T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 4.Griffith J.D., Comeau L., Rosenfield S., Stansel R.M., Bianchi A., Moss H., de Lange T. Mammalian telomeres end in a large duplex loop. Cell. 1999;97:503–514. doi: 10.1016/s0092-8674(00)80760-6. [DOI] [PubMed] [Google Scholar]
- 5.de Lange T. Shelterin-mediated telomere protection. Annu. Rev. Genet. 2018;52:223–247. doi: 10.1146/annurev-genet-032918-021921. [DOI] [PubMed] [Google Scholar]
- 6.Watson J.D. Origin of concatemeric T7 DNA. Nat. New Biol. 1972;239:197–201. doi: 10.1038/newbio239197a0. [DOI] [PubMed] [Google Scholar]
- 7.Olovnikov A.M. A theory of marginotomy: the incomplete copying of template margin in enzymatic synthesis of polynucleotides and biological significance of the phenomenon. J. Theor. Biol. 1973;41:181–190. doi: 10.1016/0022-5193(73)90198-7. [DOI] [PubMed] [Google Scholar]
- 8.Wu P., Takai H., de Lange T. Telomeric 3' overhangs derive from resection by Exo1 and apollo and fill-in by POT1b-associated CST. Cell. 2012;150:39–52. doi: 10.1016/j.cell.2012.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nassour J., Schmidt T.T., Karlseder J. Telomeres and cancer: resolving the paradox. Annu. Rev. Cell Biol. 2021;5:59–77. doi: 10.1146/annurev-cancerbio-050420-023410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kyrion G., Boakye K.A., Lustig A.J. C-terminal truncation of RAP1 results in the deregulation of telomere size, stability, and function in Saccharomyces cerevisiae. Mol. Cell Biol. 1992;12:5159–5173. doi: 10.1128/mcb.12.11.5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li B., Lustig A.J. A novel mechanism for telomere size control in Saccharomyces cerevisiae. Genes Dev. 1996;10:1310–1326. doi: 10.1101/gad.10.11.1310. [DOI] [PubMed] [Google Scholar]
- 12.Bucholc M., Park Y., Lustig A.J. Intrachromatid excision of telomeric DNA as a mechanism for telomere size control in Saccharomyces cerevisiae. Mol. Cell Biol. 2001;21:6559–6573. doi: 10.1128/MCB.21.19.6559-6573.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lustig A.J. Clues to catastrophic telomere loss in mammals from yeast telomere rapid deletion. Nat. Rev. Genet. 2003;4:916–923. doi: 10.1038/nrg1207. [DOI] [PubMed] [Google Scholar]
- 14.Pickett H.A., Reddel R.R. The role of telomere trimming in normal telomere length dynamics. Cell Cycle. 2012;11:1309–1315. doi: 10.4161/cc.19632. [DOI] [PubMed] [Google Scholar]
- 15.Pickett H.A., Cesare A.J., Johnston R.L., Neumann A.A., Reddel R.R. Control of telomere length by a trimming mechanism that involves generation of t-circles. EMBO J. 2009;28:799–809. doi: 10.1038/emboj.2009.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pickett H.A., Henson J.D., Au A.Y.M., Neumann A.A., Reddel R.R. Normal mammalian cells negatively regulate telomere length by telomere trimming. Hum. Mol. Genet. 2011;20:4684–4692. doi: 10.1093/hmg/ddr402. ddr402 [pii] 10.1093/hmg/ddr402. [DOI] [PubMed] [Google Scholar]
- 17.Rivera T., Haggblom C., Cosconati S., Karlseder J. A balance between elongation and trimming regulates telomere stability in stem cells. Nat. Struct. Mol. Biol. 2017;24:30–39. doi: 10.1038/nsmb.3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim N.W., Piatyszek M.A., Prowse K.R., Harley C.B., West M.D., Ho P.L., Coviello G.M., Wright W.E., Weinrich S.L., Shay J.W. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 19.Wright W.E., Piatyszek M.A., Rainey W.E., Byrd W., Shay J.W. Telomerase activity in human germline and embryonic tissues and cells. Dev. Genet. 1996;18:173–179. doi: 10.1002/(SICI)1520-6408(1996)18:2<173::AID-DVG10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 20.Blackburn E.H. The telomere and telomerase: nucleic acid-protein complexes acting in a telomere homeostasis system. A review. Biochemistry. 1997;62:1196–1201. [PubMed] [Google Scholar]
- 21.Zhang J.M., Zou L. Alternative lengthening of telomeres: from molecular mechanisms to therapeutic outlooks. Cell Biosci. 2020;10:30. doi: 10.1186/s13578-020-00391-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bryan T.M., Englezou A., Gupta J., Bacchetti S., Reddel R.R. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 1995;14:4240–4248. doi: 10.1002/j.1460-2075.1995.tb00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dilley R.L., Verma P., Cho N.W., Winters H.D., Wondisford A.R., Greenberg R.A. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature. 2016;539:54–58. doi: 10.1038/nature20099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roumelioti F.M., Sotiriou S.K., Katsini V., Chiourea M., Halazonetis T.D., Gagos S. Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. EMBO Rep. 2016;17:1731–1737. doi: 10.15252/embr.201643169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yeager T.R., Neumann A.A., Englezou A., Huschtscha L.I., Noble J.R., Reddel R.R. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999;59:4175–4179. [PubMed] [Google Scholar]
- 26.Zhang J.M., Yadav T., Ouyang J., Lan L., Zou L. Alternative lengthening of telomeres through two distinct break-induced replication pathways. Cell Rep. 2019;26:955–968.e3. doi: 10.1016/j.celrep.2018.12.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tokutake Y., Matsumoto T., Watanabe T., Maeda S., Tahara H., Sakamoto S., Niida H., Sugimoto M., Ide T., Furuichi Y. Extra-chromosomal telomere repeat DNA in telomerase-negative immortalized cell lines. Biochem. Biophys. Res. Commun. 1998;247:765–772. doi: 10.1006/bbrc.1998.8876. [DOI] [PubMed] [Google Scholar]
- 28.Cesare A.J., Griffith J.D. Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops. Mol. Cell Biol. 2004;24:9948–9957. doi: 10.1128/MCB.24.22.9948-9957.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Londoño-Vallejo J.A., Der-Sarkissian H., Cazes L., Bacchetti S., Reddel R.R. Alternative lengthening of telomeres is characterized by high rates of telomeric exchange. Cancer Res. 2004;64:2324–2327. doi: 10.1158/0008-5472.can-03-4035. [DOI] [PubMed] [Google Scholar]
- 30.Heaphy C.M., de Wilde R.F., Jiao Y., Klein A.P., Edil B.H., Shi C., Bettegowda C., Rodriguez F.J., Eberhart C.G., Hebbar S., et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011;333:425. doi: 10.1126/science.1207313. [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwartzentruber J., Korshunov A., Liu X.Y., Jones D.T.W., Pfaff E., Jacob K., Sturm D., Fontebasso A.M., Quang D.A.K., Tönjes M., et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [pii] [DOI] [PubMed] [Google Scholar]
- 32.Lovejoy C.A., Li W., Reisenweber S., Thongthip S., Bruno J., de Lange T., De S., Petrini J.H.J., Sung P.A., Jasin M., et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 2012;8 doi: 10.1371/journal.pgen.1002772. [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Napier C.E., Huschtscha L.I., Harvey A., Bower K., Noble J.R., Hendrickson E.A., Reddel R.R. ATRX represses alternative lengthening of telomeres. Oncotarget. 2015;6:16543–16558. doi: 10.18632/oncotarget.3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clynes D., Jelinska C., Xella B., Ayyub H., Scott C., Mitson M., Taylor S., Higgs D.R., Gibbons R.J. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun. 2015;6:7538. doi: 10.1038/ncomms8538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Picketts D.J., Higgs D.R., Bachoo S., Blake D.J., Quarrell O.W., Gibbons R.J. ATRX encodes a novel member of the SNF2 family of proteins: mutations point to a common mechanism underlying the ATR-X syndrome. Hum. Mol. Genet. 1996;5:1899–1907. doi: 10.1093/hmg/5.12.1899. [DOI] [PubMed] [Google Scholar]
- 36.Goldberg A.D., Banaszynski L.A., Noh K.M., Lewis P.W., Elsaesser S.J., Stadler S., Dewell S., Law M., Guo X., Li X., et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678–691. doi: 10.1016/j.cell.2010.01.003. [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewis P.W., Elsaesser S.J., Noh K.M., Stadler S.C., Allis C.D. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA. 2010;107:14075–14080. doi: 10.1073/pnas.1008850107. [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flynn R.L., Cox K.E., Jeitany M., Wakimoto H., Bryll A.R., Ganem N.J., Bersani F., Pineda J.R., Suvà M.L., Benes C.H., et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science. 2015;347:273–277. doi: 10.1126/science.1257216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J.S.Z., Miralles Fusté J., Simavorian T., Bartocci C., Tsai J., Karlseder J., Lazzerini Denchi E. TZAP: a telomere-associated protein involved in telomere length control. Science. 2017;355:638–641. doi: 10.1126/science.aah6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jahn A., Rane G., Paszkowski-Rogacz M., Sayols S., Bluhm A., Han C.T., Draškovič I., Londoño-Vallejo J.A., Kumar A.P., Buchholz F., et al. ZBTB48 is both a vertebrate telomere-binding protein and a transcriptional activator. EMBO Rep. 2017;18:929–946. doi: 10.15252/embr.201744095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takai K.K., Hooper S., Blackwood S., Gandhi R., de Lange T. In vivo stoichiometry of shelterin components. J. Biol. Chem. 2010;285:1457–1467. doi: 10.1074/jbc.M109.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stagno D'Alcontres M., Mendez-Bermudez A., Foxon J.L., Royle N.J., Salomoni P. Lack of TRF2 in ALT cells causes PML-dependent p53 activation and loss of telomeric DNA. J. Cell Biol. 2007;179:855–867. doi: 10.1083/jcb.200703020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koneru B., Farooqi A., Nguyen T.H., Chen W.H., Hindle A., Eslinger C., Makena M.R., Burrow T.A., Wilson J., Smith A., et al. ALT neuroblastoma chemoresistance due to telomere dysfunction-induced ATM activation is reversible with ATM inhibitor AZD0156. Sci. Transl. Med. 2021;13 doi: 10.1126/scitranslmed.abd5750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang J.M., Genois M.M., Ouyang J., Lan L., Zou L. Alternative lengthening of telomeres is a self-perpetuating process in ALT-associated PML bodies. Mol. Cell. 2021;81:1027–1042.e4. doi: 10.1016/j.molcel.2020.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gaillard H., García-Muse T., Aguilera A. Replication stress and cancer. Nat. Rev. Cancer. 2015;15:276–289. doi: 10.1038/nrc3916. [DOI] [PubMed] [Google Scholar]
- 46.Grudic A., Jul-Larsen A., Haring S.J., Wold M.S., Lønning P.E., Bjerkvig R., Bøe S.O. Replication protein A prevents accumulation of single-stranded telomeric DNA in cells that use alternative lengthening of telomeres. Nucleic Acids Res. 2007;35:7267–7278. doi: 10.1093/nar/gkm738. gkm738 [pii] 10.1093/nar/gkm738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sobinoff A.P., Allen J.A., Neumann A.A., Yang S.F., Walsh M.E., Henson J.D., Reddel R.R., Pickett H.A. BLM and SLX4 play opposing roles in recombination-dependent replication at human telomeres. EMBO J. 2017;36:2907–2919. doi: 10.15252/embj.201796889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Loe T.K., Li J.S.Z., Zhang Y., Azeroglu B., Boddy M.N., Denchi E.L. Telomere length heterogeneity in ALT cells is maintained by PML-dependent localization of the BTR complex to telomeres. Genes Dev. 2020;34:650–662. doi: 10.1101/gad.333963.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pan X., Chen Y., Biju B., Ahmed N., Kong J., Goldenberg M., Huang J., Mohan N., Klosek S., Parsa K., et al. FANCM suppresses DNA replication stress at ALT telomeres by disrupting TERRA R-loops. Sci. Rep. 2019;9 doi: 10.1038/s41598-019-55537-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cho N.W., Dilley R.L., Lampson M.A., Greenberg R.A. Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell. 2014;159:108–121. doi: 10.1016/j.cell.2014.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pan X., Drosopoulos W.C., Sethi L., Madireddy A., Schildkraut C.L., Zhang D. FANCM, BRCA1, and BLM cooperatively resolve the replication stress at the ALT telomeres. Proc. Natl. Acad. Sci. USA. 2017;114:E5940–E5949. doi: 10.1073/pnas.1708065114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao Y., Zhang G., He C., Mei Y., Shi Y., Li F. The 11th C2H2 zinc finger and an adjacent C-terminal arm are responsible for TZAP recognition of telomeric DNA. Cell Res. 2018;28:130–134. doi: 10.1038/cr.2017.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li F., Deng Z., Zhang L., Wu C., Jin Y., Hwang I., Vladimirova O., Xu L., Yang L., Lu B., et al. ATRX loss induces telomere dysfunction and necessitates induction of alternative lengthening of telomeres during human cell immortalization. EMBO J. 2019;38 doi: 10.15252/embj.201796659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Episkopou H., Draskovic I., Van Beneden A., Tilman G., Mattiussi M., Gobin M., Arnoult N., Londoño-Vallejo A., Decottignies A. Alternative Lengthening of Telomeres is characterized by reduced compaction of telomeric chromatin. Nucleic Acids Res. 2014;42:4391–4405. doi: 10.1093/nar/gku114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu R., O'Rourke J.J., Sobinoff A.P., Allen J.A.M., Nelson C.B., Tomlinson C.G., Lee M., Reddel R.R., Deans A.J., Pickett H.A. The FANCM-BLM-TOP3A-RMI complex suppresses alternative lengthening of telomeres (ALT) Nat. Commun. 2019;10:2252. doi: 10.1038/s41467-019-10180-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bizard A.H., Hickson I.D. The dissolution of double Holliday junctions. Cold Spring Harbor Perspect. Biol. 2014;6:a016477. doi: 10.1101/cshperspect.a016477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gravel S., Chapman J.R., Magill C., Jackson S.P. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22:2767–2772. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chan K.L., North P.S., Hickson I.D. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 2007;26:3397–3409. doi: 10.1038/sj.emboj.7601777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Davies S.L., North P.S., Hickson I.D. Role for BLM in replication-fork restrart and suppression of origin firing after replicative stress. Nat. Struct. Mol. Biol. 2007;14:677–679. doi: 10.1038/nsmb1267. [DOI] [PubMed] [Google Scholar]
- 60.O'Sullivan R.J., Arnoult N., Lackner D.H., Oganesian L., Haggblom C., Corpet A., Almouzni G., Karlseder J. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat. Struct. Mol. Biol. 2014;21:167–174. doi: 10.1038/nsmb.2754 nsmb.2754. [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dharmaiah S., Huse J.T. The epigenetic dysfunction underlying malignant glioma pathogenesis. Lab. Invest. 2022;102:682–690. doi: 10.1038/s41374-022-00741-7. [DOI] [PubMed] [Google Scholar]
- 62.Wang Y., Yang J., Wild A.T., Wu W.H., Shah R., Danussi C., Riggins G.J., Kannan K., Sulman E.P., Chan T.A., Huse J.T. G-quadruplex DNA drives genomic instability and represents a targetable molecular abnormality in ATRX-deficient malignant glioma. Nat. Commun. 2019;10:943. doi: 10.1038/s41467-019-08905-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robinson J., Raguseo F., Nuccio S.P., Liano D., Di Antonio M. DNA G-quadruplex structures: more than simple roadblocks to transcription? Nucleic Acids Res. 2021;49:8419–8431. doi: 10.1093/nar/gkab609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Halder K., Halder R., Chowdhury S. Genome-wide analysis predicts DNA structural motifs as nucleosome exclusion signals. Mol. Biosyst. 2009;5:1703–1712. doi: 10.1039/b905132e. [DOI] [PubMed] [Google Scholar]
- 65.Du Z., Zhao Y., Li N. Genome-wide colonization of gene regulatory elements by G4 DNA motifs. Nucleic Acids Res. 2009;37:6784–6798. doi: 10.1093/nar/gkp710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hänsel-Hertsch R., Beraldi D., Lensing S.V., Marsico G., Zyner K., Parry A., Di Antonio M., Pike J., Kimura H., Narita M., et al. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016;48:1267–1272. doi: 10.1038/ng.3662. [DOI] [PubMed] [Google Scholar]
- 67.Lago S., Nadai M., Cernilogar F.M., Kazerani M., Domíniguez Moreno H., Schotta G., Richter S.N. Promoter G-quadruplexes and transcription factors cooperate to shape the cell type-specific transcriptome. Nat. Commun. 2021;12:3885. doi: 10.1038/s41467-021-24198-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dos Santos G.A., Viana N.I., Pimenta R., de Camargo J.A., T Reis S., Moreira Leite K.R., Srougi M. Telomeric zinc-finger associated protein (TZAP) in cancer biology: friend or foe? Mol. Biol. Res. Commun. 2021;10:121–129. doi: 10.22099/mbrc.2021.40106.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zellinger B., Akimcheva S., Puizina J., Schirato M., Riha K. Ku suppresses formation of telomeric circles and alternative telomere lengthening in Arabidopsis. Mol. Cell. 2007;27:163–169. doi: 10.1016/j.molcel.2007.05.025. [DOI] [PubMed] [Google Scholar]
- 70.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Henson J.D., Lau L.M., Koch S., Martin La Rotta N., Dagg R.A., Reddel R.R. The C-Circle Assay for alternative-lengthening-of-telomeres activity. Methods. 2017;114:74–84. doi: 10.1016/j.ymeth.2016.08.016. [DOI] [PubMed] [Google Scholar]
- 72.Kimura M., Stone R.C., Hunt S.C., Skurnick J., Lu X., Cao X., Harley C.B., Aviv A. Measurement of telomere length by the Southern blot analysis of terminal restriction fragment lengths. Nat. Protoc. 2010;5:1596–1607. doi: 10.1038/nprot.2010.124. [DOI] [PubMed] [Google Scholar]
- 73.Lai T.P., Wright W.E., Shay J.W. Generation of digoxigenin-incorporated probes to enhance DNA detection sensitivity. Biotechniques. 2016;60:306–309. doi: 10.2144/000114427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.