

Abstract
Background
Cystic fibrosis (CF), which is caused by mutations in the CF transmembrane conductance regulator (CFTR), is characterised by chronic bacterial lung infection and inflammation. In CF, monocytes and monocyte-derived macrophages have been shown to display defective phagocytosis and antimicrobial activity against relevant lung pathogens, including Pseudomonas aeruginosa. Thus, we addressed the effect of CFTR triple modulator therapy (elexacaftor/tezacaftor/ivacaftor (ETI)) on the activity of CF monocytes against P. aeruginosa.
Methods
Monocytes from people with CF (PWCF) before and after 1 and 6 months of ETI therapy were isolated from blood and infected with P. aeruginosa to assess phagocytic activity and intracellular bacterial killing. The oxidative burst and interleukin-6 secretion were also determined. Monocytes from healthy controls were also included.
Results
Longitudinal analysis of the clinical parameters confirmed an improvement of lung function and lung microbiology by ETI. Both the phagocytic and microbicidal deficiencies of CF monocytes also improved significantly, although not completely. Furthermore, we measured an exuberant oxidative burst in CF monocytes before therapy, which was reduced considerably by ETI. This led to an improvement of reactive oxygen species-dependent bactericidal activity. Inflammatory response to bacterial stimuli was also lowered compared with pre-therapy.
Conclusions
PWCF on ETI therapy, in a real-life setting, in addition to clinical recovery, showed significant improvement in monocyte activity against P. aeruginosa, which may have contributed to the overall effect of ETI on pulmonary disease. This also suggests that CF monocyte dysfunctions may be specifically targeted to ameliorate lung function in CF.
Short abstract
In people with cystic fibrosis, elexacaftor/tezacaftor/ivacaftor ameliorates the antimicrobial activity of monocytes against Pseudomonas aeruginosa by lowering their exuberant oxidative burst, thus contributing to the improvement of lung disease http://bit.ly/3hL2Z11
Introduction
Cystic fibrosis (CF) is a life-limiting genetic disease caused by mutations in the CF transmembrane conductance regulator (CFTR) gene, with the F508del CFTR mutation being the most common. In the lung, dysfunctional CFTR causes an imbalance in ion transport across the airway epithelia, which in turn favours chronic bacterial infections associated with a decline in lung function [1]. Extensive structural and functional studies of wild-type and mutated CFTR proteins led to the identification of small molecules, which by interacting with mutated CFTR, restore its activity. CFTR modulators comprise correctors, such as elexacaftor, lumacaftor and tezacaftor, designed to improve misfolded and mistracked CFTR, and potentiators, which target gating mutations characterised by the defective opening of the channel pore [2–4]. The potentiator, ivacaftor, was the first CFTR modulator to be approved for clinical use, in 2013, in subjects with gating mutations [5, 6]. Subsequently, based on the possible synergistic activity of modulators, not only on F508del CFTR but also on other mutant proteins, these drugs were used in combination [7–9] in homozygous and heterozygous people with CF (PWCF) with at least one F508del mutation.
Randomised controlled trials that compared the efficacy of CFTR modulators in PWCF with class II mutations, including the F508del CFTR allele, have recently been evaluated [10] revealing an improvement in quality of life (QoL) and lung function for the triple therapy (elexacaftor/tezacaftor/ivacaftor (ETI)), little improvement for the dual therapy (one corrector and one potentiator), whereas no relevant effects on lung function were observed for monotherapy [10]. Based on the encouraging results obtained with ETI, in October 2019 the US Food and Drug Administration approved this combined therapy for PWCF ≥12 years old, with at least one F508del, followed by the European Medicines Agency in August 2020. Since then, ETI use has significantly increased to the extent that a rapid extension to nearly all eligible PWCF is expected [11–13]. It is therefore necessary to extend the study of the effects of ETI to other cellular districts, other than the epithelia of the airways, which have already been extensively studied.
Monocytes and monocyte-derived macrophages play a key role in maintaining lung sterility and in regulating the inflammatory response. In CF, these immune cells have been shown to display defective phagocytosis and antimicrobial activity against relevant lung pathogens, including Staphylococcus aureus, Pseudomonas aeruginosa and Burkholderia cenocepacia [14–19]. In vitro and ex vivo studies showed some improvement of CF macrophage function following treatment with mono (ivacaftor) or dual (ivacaftor/lumacaftor) CFTR modulators [20, 21] as well as a rapid modulation of the transcriptional activity of genes related to inflammation and immune response in peripheral blood cells [22, 23]. However, very little is known about the effect of triple therapy, which is the most efficient from a clinical point of view, on the function of phagocytic cells. Very recently, recovery of CFTR expression and correction of ATP/P2X7 receptor (P2X7R) signalling, with a consequential reduction of inflammasome activation and circulating pro-inflammatory markers, was shown in monocytes isolated from PWCF under ETI therapy [24].
Here we investigated the impact of ETI therapy on the ability of monocytes to phagocytose bacteria and to kill them when internalised. Additionally, we analysed the extent to which ETI contributes to the fine balance of the oxidative burst, which has recently been shown as the leading microbicidal mechanism of human macrophages [25].
Methods
Peripheral blood mononuclear cell (PBMC) infection with P. aeruginosa, phagocytosis, killing activity and reactive oxygen species (ROS) measurements were performed according to Cavinato et al. [26]. Western blotting and interleukin (IL)-6 quantification were carried out as previously reported [27, 28]. A detailed description of the materials and methods is provided in the supplementary material.
Study subjects
The PWCF (n=41) examined in this study attended the Cystic Fibrosis Regional Center, Region Lazio (Rome, Italy). At the time of the study, Trikafta (Vertex Pharmaceuticals, Boston, MA, USA) was yet to be commercialised. Therefore, PWCF were admitted to the compassionate use of Trikafta based on their clinical conditions. The study was conducted entirely during the regular care of PWCF, including the collection of blood samples obtained for routine evaluations. Informed written consent was obtained from all of the participants before the start of Trikafta. All of the methods were carried out in accordance with relevant guidelines and regulations. Buffy coats, which were not usable for other purposes, were used to isolate cells from healthy donors.
Statistics
All statistical test results were discussed with a biostatistician and analyses were performed with Prism software (GraphPad, La Jolla, CA, USA). Tests for normality distribution of data were performed, then parametric or non-parametric tests were selected. Details are reported in the figure legends. Differences were significant for a p-value cut-off of 0.05.
Results
ETI therapy rapidly improves the clinical characteristics of PWCF
The clinical efficacy of ETI in real-life was evaluated in homozygous (F508del/F508del) and heterozygous (F508del/other) Caucasian PWCF (table 1), the latter with the second minimal function mutation, as defined in Zemanick et al. [13]. At baseline, no difference in percentage predicted forced expiratory volume in 1 s (FEV1 % pred), sweat chloride concentration (SSC), body mass index (BMI) and QoL between the two groups was observed (table 1). After 1 (M1) and 6 (M6) months from the start of therapy, FEV1 % pred significantly increased, while SSC decreased (figure 1). In particular, the average increase of FEV1 % pred in homozygotes was +8.43±6.65% at M1 with a further +4.77±3.32% at M6, while in heterozygotes it was +11.81±11.61% at M1 with a further +4.89±6.84% at M6. A similar trend was observed in the SSC decrease with a stronger average effect at M1 (−46.29±26.37 and −44.37±25.25 mmol·L−1 in homozygotes and heterozygotes, respectively) compared with M6 (−7.21±11.87 and −3.81±10.81 mmol·L−1). These results, which were similar to previous clinical trial studies [29, 30] and more recent studies in real-life settings [11–13], highlight the rapid improvement of lung function and CFTR channel activity after ETI therapy. Importantly, this improvement is maintained with a slower increase in the subsequent 5 months. In agreement with FEV1 % pred and SSC data, BMI and QoL both improved in the study groups (supplementary figure S1). No significant differences were observed in these measurements between homozygous and heterozygous PWCF. Collectively, the clinical data showed that all of the subjects included in this study responded positively to ETI.
TABLE 1.
Characteristics of people with cystic fibrosis (PWCF) at initiation of elexacaftor/tezacaftor/ivacaftor therapy
| F508del/F508del | F508del/other | |||
| Patients | 14 | 27 | ||
| Caucasian | 14 | 27 | ||
| Female | 7 (50) | 18 (66.6) | ||
| Age (year) | 31.43±15.81 | 28.89±14.47 | ||
| FEV1 % pred | 41.66±10.34 | 51.37±14.92 | ||
| SSC (mmol·L−1) | 89±15.24 | 94.43±13.29 | ||
| BMI (kg·m−2) | 19.43±3.39 | 18.74±2.29 | ||
| Lung microbiology | Pre | M1/M6 | Pre | M1/M6 |
| P. aeruginosa | 12 (85.7) | −1/+1 | 25 (92.6) | −3/−1 |
| P. aeruginosa+B. cepacia | 1 (7.1) | NC/−1# | 1 (3.7) | NC/−1# |
| P. aeruginosa+S. aureus | 1 (7.1) | −1/0¶ | ||
| S. aureus | +1/NC | 1 (3.7) | NC/NC | |
Data are presented as n, n (%) or mean±sd; changes in lung microbiology are reported as n PWCF with the indicated pathogen. FEV1: forced expiratory volume in 1 s; SSC: sweat chloride concentration; BMI: body mass index; P. aeruginosa: Pseudomonas aeruginosa; B. cepacia: Burkholderia cepacia; S. aureus: Staphylococcus aureus; Pre: before therapy; M1: month 1; M6: month 6; NC: no change. #: negative for B. cepacia at M6, P. aeruginosa still present; ¶: negative for P. aeruginosa at M1, S. aureus still present.
FIGURE 1.

Improvement of forced expiratory volume in 1 s and sweat chloride concentration (SSC) after elexacaftor/tezacaftor/ivacaftor therapy. a) FEV1 % pred and b) SSC in F508del homozygous (n=14) and heterozygous (n=27) people with cystic fibrosis before (Pre) and after 1 (M1) and 6 (M6) months of therapy. Box plots with median and whiskers pointing to minimum and maximum values. One-way ANOVA with Tukey's multiple comparison test. *: p<0.05; **: p<0.01; ****: p<0.0001.
Lung microbiology showed P. aeruginosa infection in 37 out of 41 (90.2%) PWCF before therapy (table 1). After ETI, five PWCF (four at M1 and one at M6) tested negative for P. aeruginosa, resulting in an overall reduction of P. aeruginosa infection of 13.5%. Multispecies infections, although very limited, were also positively affected by ETI. In particular, B. cenocepacia was not detected in two PWCF with P. aeruginosa+B. cenocepacia, while one patient with P. aeruginosa+S. aureus resulted negative for P. aeruginosa (table 1). Collectively, it appeared that ETI, by improving lung function, also contributed to reducing lung infection.
ETI rapidly improves phagocytosis and killing activity of P. aeruginosa by CF monocytes
To determine the impact of ETI on the activity of CF monocytes, P. aeruginosa phagocytosis and killing before and after therapy were evaluated. Phagocytosis was assessed by determining the number of intracellular viable bacteria recovered from PBMCs infected by P. aeruginosa. PBMCs freshly isolated from blood samples were infected with the PAO1 strain of P. aeruginosa expressing Green Fluorescent Protein (PAO1-GFP) and, after the removal of the non-phagocytosed bacteria by antibiotic treatment, the engulfed bacteria were enumerated (CFU). PBMCs isolated from buffy coats of healthy donors were included as controls. As reported in figure 2a, at baseline, the CFU recovered from CF cells were significantly lower than those recovered from healthy donors, but the CFU recovered from ETI samples increased significantly. In particular, the average increase of intracellular bacteria compared with baseline was 2-fold at M1 and 2.5-fold at M6 (figure 2a). The uptake of P. aeruginosa by monocytes of PWCF was further evaluated by flow cytometry after infection of PBMCs with PAO1-GFP and identification of monocytes by forward and side scatter (supplementary figure S2 and figure 2b). Before ETI, the mean percentage of GFP+ monocytes was 1.66-fold lower in PWCF than in healthy donors (figure 2c). After 1 month of ETI treatment (M1), GFP+ monocytes increased significantly, while no significant difference was observed at M6 compared with M1 (figure 2c). In particular with respect to before therapy, increased phagocytosis of P. aeruginosa was detected in 10 out of 14 PWCF at M1 and in 12 out of 17 at M6 (supplementary table S3). In these analyses, no differences between F508del homozygous and heterozygous CF samples were detected. Control experiments performed in the presence of cytochalasin D, an inhibitor of phagocytosis, failed to reveal GFP+ monocytes, confirming the specificity of the assay (supplementary figure S3). Moreover, the lack of GFP+ cells in blood lymphocytes, identified by forward and side scatter, confirmed that P. aeruginosa uptake was mainly mediated by monocytes (supplementary figure S2b).
FIGURE 2.

Elexacaftor/tezacaftor/ivacaftor improves the phagocytic activity of cystic fibrosis (CF) monocytes. P. aeruginosa uptake by peripheral blood mononuclear cells (PBMCs) isolated from healthy donors (HD) and people with CF before (Pre) and after 1 (M1) and 6 (M6) months of therapy. a) CFU recovered after the end of infection. Left panel: scattered dot plot with mean±sem. Kruskal–Wallis test with Dunn's multiple comparison. Right panel: longitudinal analysis of data. Friedman test with Dunn's multiple comparison. b) Representative overlaid histograms of monocytes infected with PAO1-GFP. c) Bacterial uptake. Left panel: scattered dot plot with mean±sem. Brown–Forsythe ANOVA with t-test for multiple comparison. Right panel: longitudinal analysis of data. Mixed effect model (restricted maximum likelihood) with Tukey's multiple comparison test. GFP: Green Fluorescent Protein. *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001.
Collectively, these results demonstrate that 1 month of ETI therapy significantly improves the phagocytic capacity of CF monocytes against P. aeruginosa, thus contributing to recovering the phagocytosis defect of these cells, although only partially. The improvement gained at M1 was maintained at least for 6 months in eight out of 10 PWCF, although five of them experienced a slight reduction of phagocytosis at the later time-point (supplementary table S3). Among the remaining PWCF, three showed a further increase of phagocytosis.
Subsequently, the impact of ETI on the bactericidal activity of CF monocytes was assessed. This was done by monitoring the survival of intracellular bacteria 60 min (t60) after the end of infection (t0). Based on the identification of monocytes as the major phagocytic component in PBMCs (supplementary figure S2) and in agreement with previously published data [31–33], these cells were used to perform this analysis. As expected, the CFU recovered from healthy donors cells at t60 were significantly lower than those recovered at t0, with an average decrease in viable bacteria of 56.5%, confirming effective killing of the bacteria (figure 3a and b). At variance, the number of bacteria recovered from CF cells before ETI did not decrease, rather a very small, but significant, increase was observed (figure 3a). However, at M1 and M6 after ETI therapy the CFU recovered at t60 decreased significantly compared with those recovered at t0 (figure 3a). Accordingly, the slope of the killing curves of CF PBMCs changed from an average +2.3 before ETI to −24.8 and −17.9 at M1 and M6, respectively (figure 3a). The killing activity, defined as the percentage decrease in CFU, was almost absent in CF cells before therapy (−11.77%), but increased to 32.5% and 24.3% at M1 and M6, respectively (figure 3b and c). In particular, the improvement of bactericidal activity was observed in all PWCF at M1 and in eight out of 11 PWCF at M6 (supplementary table S4). Notably, although ETI therapy clearly improved both the phagocytic and microbicidal deficiencies, CF PBMCs did not recover completely as they still appeared less active against P. aeruginosa compared with non-CF cells, at least under the conditions examined here.
FIGURE 3.

Improvement of microbicidal activity by elexacaftor/tezacaftor/ivacaftor. a) Pseudomonas aeruginosa survival in peripheral blood mononuclear cells isolated from healthy donors (HD) and people with cystic fibrosis before (Pre) and after 1 (M1) and 6 (M6) months of therapy. CFU recovered at the end of infection (t0) and 60 min after infection (t60). The average slopes are reported above the t0–t60 connecting lines. Slopes were calculated with the linear regression model. Wilcoxon test. b) Killing activity at t60. Scattered dot plot with mean±sem. Kruskal–Wallis test with Dunn's multiple comparison. c) Longitudinal analysis of data. Friedman test with Dunn's multiple comparison. *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001.
To account for possible differences due to the infecting strain, we compared the microbicidal activity of PBMCs infected with clinical or reference P. aeruginosa strains. Taking into consideration that P. aeruginosa undergoes continuous adaptation to the CF lung environment, for this analysis we selected the clonal strains AA2 and AA44, representative of early and late CF isolates, respectively [34]. As shown in figure 4, CF PBMCs prior to therapy showed no difference in the ability to kill clinical isolates or PAO1. This result demonstrates that CF PBMCs are similarly deficient against clinical isolates, which supports a possible positive effect of ETI on their antimicrobial activity against the clinical P. aeruginosa strains.
FIGURE 4.

Microbicidal activity of peripheral blood mononuclear cells (PBMCs) against Pseudomonas aeruginosa PAO1 and clinical isolates. a) PBMCs from healthy donors (HD) and people with cystic fibrosis (PWCF) before elexacaftor/tezacaftor/ivacaftor (ETI) therapy (Pre) infected with PAO1. Demographic and clinical characteristics of PWCF are reported in supplementary table S3. t-test. b) PBMCs from PWCF before ETI therapy infected with PAO1 or the clinical isolates AA2 and AA44. One-way ANOVA test with Tukey's multiple comparison test. Scattered dot plot with mean±sem. *: p<0.05.
ETI corrects the exuberant oxidative burst of CF monocytes
It has been recently shown that the optimal bactericidal activity by macrophages against P. aeruginosa requires a fine balance between the oxidative and non-oxidative mechanisms, and that excessive and sustained oxidative burst compromises the killing of engulfed bacteria [25]. These findings led us to hypothesise a dysregulation of the oxidative burst in CF monocytes as the cause of their defective bactericidal activity. To test this hypothesis, the oxidative burst generated by PAO1-infected PBMCs was determined by measuring superoxide anion O−2 production with the luminol assay. In infected cells, the assay revealed a rapid increase of O−2 production followed by a decrease to basal level in ∼120 min, while no signal was detected in cells pre-treated with the NADPH oxidase inhibitor diphenyleneiodonium (DPI), confirming the specificity of the assay (supplementary figure S4). Additionally, we identified monocytes as the major ROS producers in the PBMC population (supplementary figure S5). Quantitative analysis of the chemiluminescence signals showed that the oxidative burst of CF cells before ETI was significantly higher than that of healthy donor controls, but it was significantly reduced at M1 (figure 5a). The higher level of ROS produced by CF cells compared with the controls was further confirmed by the 2′,7′-dichlorodihydrofluorescein diacetate (H2DFCDA) probe (supplementary figure S6).
FIGURE 5.

Elexacaftor/tezacaftor/ivacaftor reduces the exuberant oxidative burst and improves microbicidal activity. a) Superoxide anion O−2 production by infected peripheral blood mononuclear cells (PBMCs) as detected by the luminol assay. i) Representative kinetics of O−2 production (relative luminescence units (RLU)). ii) Quantitative analysis of O−2 produced by the indicated samples (area under the curve (AUC)). Scattered dot plot with mean±sem. Kruskal–Wallis test with Dunn's multiple comparison. iii) Longitudinal analysis of data. Friedman test with Dunn's multiple comparison. b) CFU recovered from untreated or diphenyleneiodonium (DPI)-treated PBMCs. i) CFU recovered from PAO1-infected cells. Wilcoxon test. ii) Fold increase of CFU in DPI-treated cells with respect to untreated cells. Scattered dot plot with mean±sem. Kruskal–Wallis test with Dunn's multiple comparison. iii) Longitudinal analysis of data. Friedman test with Dunn's multiple comparison. HD: healthy donors; Pre: before therapy; M1: month 1; M6: month 6. *: p<0.05; **: 0.01; ***: p<0.001.
Next, we evaluated the contribution of the oxidative mechanism to the microbicidal activity of monocytes. For this, bacterial survival was determined in NADPH oxidase 2 (NOX2)-inhibited cells by DPI treatment and compared with untreated cells. As expected, blocking O−2 production caused an increase in CFU recovered in both non-CF and CF cells (figure 5b, i). However, the increase was higher in healthy donor PBMCs (4.43-fold) than in CF cells (1.6-fold) and it improved after therapy (figure 5b, ii and iii).
These results strongly suggest that the microbicidal deficiency of CF monocytes may be due, at least in part, to excessive oxidative burst. This hypothesis was further corroborated by evaluating the activation of NADPH oxidase at the protein level. For this, the p47phox subunit and its phosphorylated form, phospo-p47phox (p-p47phox), which accounts for NOX2 activation, were analysed in monocyte protein lysates. p47phox was higher in CF monocytes before ETI compared with healthy donor monocytes and significantly reduced by 1 month of ETI therapy (M1) (figure 6a and b). As expected, p47phox was similar in uninfected and infected monocytes in both non-CF and CF cells (figure 6a and b). Differently, p-p47phox at baseline, although higher in CF with respect to healthy donor monocytes, did not change after ETI therapy (figure 6c). However, following infection a higher level of p-p47phox was detected in CF monocytes compared with non-CF cells, which was lowered by ETI (figure 6c). At variance, analysis of p40phox and p-p40phox failed to reveal significant differences between CF and non-CF monocytes (supplementary figure S7). In summary, ETI partially reversed this imbalance as p47phox decreased in CF monocytes, while a significant decrease of p-p47phox was limited to infected cells.
FIGURE 6.

Elexacaftor/tezacaftor/ivacaftor rapidly reduces the high level of NADPH oxidase 2 (NOX2) activation. Analysis of the p47phox and phosphorylated p47phox (p-p47phox) NOX2 subunits in monocytes from healthy donors (HD; n=6) and people with cystic fibrosis before (Pre; n=6) and after 1 month of therapy (M1; n=6). a) Representative blots of cell lysates from non-infected and PAO1-infected monocytes. b, c) Quantitative analysis of b) p47phox and c) p-p47phox. Data are presented as mean±sem. Brown–Forsythe ANOVA test with t-test for multiple comparison. GAPDH: glyceraldehyde 3-phosphate dehydrogenase (loading control). *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001.
Collectively, these results show a clear link between the recovered bactericidal activity and the reduced oxidative burst in monocytes from PWCF under ETI therapy, suggesting that ETI improves the bactericidal activity of monocytes by reducing the excessive oxidative burst of these cells.
ETI reduces the production of IL-6 by CF monocytes
Based on the aforementioned findings, we wondered whether ETI could also impact the inflammatory response of phagocytic cells. To address this issue, PBMCs, before and after therapy, were challenged with lipopolysaccharide (LPS) from P. aeruginosa, and the pro-inflammatory cytokine IL-6, whose production was reported to be dysregulated in CF innate immune cells, was determined in culture supernatants [28]. First, we confirmed that monocytes contributed mainly to the IL-6 produced by PBMCs (supplementary figure S8). Next, while similar levels of IL-6 were observed in non-stimulated CF cells, at 1 month after ETI LPS-treated cells showed reduced levels of IL-6, although differences did not reach statistical significance (figure 7). No difference was observed between homozygous and heterozygous subjects (supplementary figure S9).
FIGURE 7.
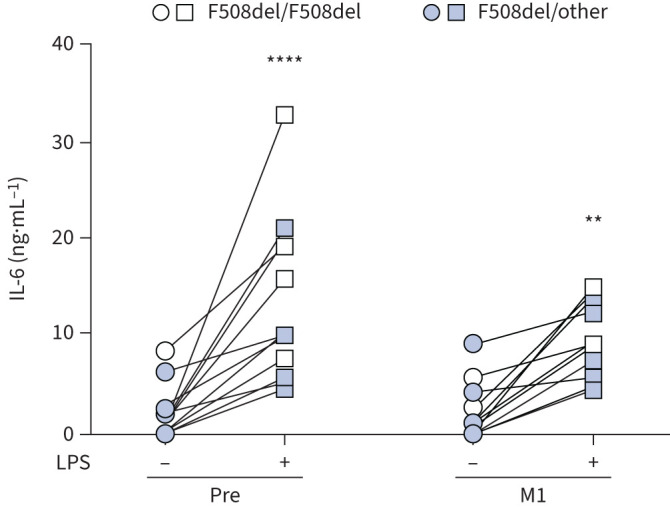
Elexacaftor/tezacaftor/ivacaftor decreases interleukin (IL)-6 production by lipopolysaccharide (LPS)-treated cells. IL-6 in supernatants of cystic fibrosis peripheral blood mononuclear cells (PBMCs) before (Pre) and after 1 month of treatment (M1) at baseline (untreated) and after LPS induction. Two-way ANOVA with Bonferroni multiple comparison test. **: p<0.01; ****: p<0.0001.
Discussion
A recent evaluation of clinical trials with CFTR modulators has confirmed the superior efficacy of ETI triple therapy compared with the previously developed mono and dual therapy [10]. Accordingly, the study group herein, analysed in a real-life setting and consisting of subjects in urgent need of therapy, responded positively to ETI therapy as demonstrated by a significant improvement of CF clinical parameters including FEV1 % pred, SSC, BMI and QoL. Thus, the clinical benefit of ETI supports the validity of the cellular models selected, i.e. freshly isolated PBMCs and purified monocytes, to study the impact of ETI on the function of phagocytic cells, whose activity is very relevant for lung homeostasis and defence against invading pathogens. Having confirmed that monocytes are the major phagocytic cells and cytokine producers in the PBMC populations, they were used for most of the tests reported here, while pure monocytes were used for protein level analysis. Previous data showed that monocytes from PWCF are impaired in their phagocytic and microbicidal activity compared with healthy controls [31, 32]. Here we report that ETI significantly improved both activities, with the highest effect being observed after 1 month of therapy (M1) and subsequent maintenance of the effect over time, up to 6 months (M6). Although we could not assess the ETI effect on CF cells infected with clinical P. aeruginosa isolates, which is a limit of our study, having observed similar microbicidal defects against PAO1 or clinical strains, it is reasonable to assume that ETI might also improve monocyte-mediated clearance of the clinical isolates. This is also supported by a previous study showing that ivacaftor improves the activity of in vitro differentiated CF macrophages infected with a P. aeruginosa strain isolated from CF sputum [21].
Collectively, although both phagocytosis and killing of P. aeruginosa by CF monocytes did not fully recover in ETI-treated subjects, the observed improvement of their activity might have contributed to the overall recovery of CF disease. This is also supported by lung microbiology which showed a decrease of PWCF positive for P. aeruginosa and B. cenocepacia.
In order to investigate the molecular mechanism underlying the improved activity of CF monocytes after ETI therapy, we focused our attention on the oxidative burst which has recently been proposed to be the conditio sine qua non for the subsequent activation of non-oxidative microbicidal mechanisms [25]. Starting from the observation of an exuberant oxidative burst in CF compared with non-CF cells, ETI samples showed a significant reduction of this response down to a similar level as non-CF cells, which in turn appears to positively impact the microbicidal activity of CF monocytes. Accordingly, the microbicidal activity of the high ROS producing PBMCs (CF Pre) was significantly lower than that of low ROS producing PBMCs (CF M1 and M6). This is in agreement with very recent investigations, at the single-cell level, showing that exaggerated ROS production by macrophages inhibits the overall bactericidal activity [25]. This inverse relationship between ROS production and killing efficiency is also confirmed by healthy controls, which displayed low ROS levels and high killing activity when compared with CF cells before ETI.
An exuberant oxidative burst by CF monocytes, compared with healthy cells, was also reported in response to Aspergillus fumigatus, which correlated with a worsening of disease severity as assessed by exacerbations and lung function [33]. Conversely, a more disparate effect of dysfunctional CFTR on the oxidative burst of macrophages has been reported. Although ROS produced by monocyte-derived macrophages in response to P. aeruginosa infection did not differ between CF and healthy controls, higher ROS production was observed in CF alveolar macrophages compared with CF monocyte-derived macrophages [16]. A further variation was observed in B. cenocepacia-infected CF monocyte-derived macrophages, which produced lower levels of ROS compared with healthy cells, a defect that was assigned to defective activation of NADPH [17]. Collectively, these results suggest that the oxidative burst response by monocytes and macrophages could be affected differently by the absence of a functional CFTR and by the infecting pathogen.
Based on the current view of the existence of a strict link between oxidative and non-oxidative microbicidal mechanisms which must be finely tuned for the optimal killing of invading pathogens [25], our results strongly suggest that ETI, by reducing the NOX2 NADPH oxidase, improves the overall bactericidal activity of monocytes. This improvement might be due to a more general effect of ETI on protein trafficking to the membrane, which in turn contributes to normalising the assembly and activity of NADPH oxidase and/or as an indirect effect mediated by the rescued CFTR. In particular, it appears that this applies to p47phox, which is consistently elevated in CF monocytes and reduced by ETI, irrespective of P. aeruginosa infection. Conversely, NOX2 activation as detected by p-p47phox [35], which appeared highly variable in CF cells and thus requiring further investigation, was reduced by ETI only in infected cells. Other NOX2 subunits, such as p40phox, do not appear to be affected by CFTR dysfunction in monocytes, which is in agreement with a recent demonstration of its irrelevance for NOX2 activity in these cells [36]. The possibility that CFTR correctors, by improving CFTR trafficking to the membrane, contribute to rescuing other proteins, whose trafficking and activity is linked to CFTR, has been well demonstrated by the recovery of membrane-bound PTEN (phosphatase and tensin homologue) in CF monocytes treated with mutation-specific CFTR correctors [32]. More recently, the effect of CFTR modulators was also demonstrated in the recovery of ATP-induced P2X7R-mediated inflammasome activation in monocytes isolated from PWCF undergoing ETI therapy [24]. Additionally, it might be speculated that CFTR modulators, by decreasing the respiratory signs and symptoms, might contribute to normalising the lung micro-environment, thus interfering with the epigenetic reprogramming of immune cells [37].
CF lungs are dominated by high levels of pro-inflammatory cytokines, which significantly drop after transplantation, suggesting that CFTR expression and function are required to control inflammation in the lung environment [38]. At present, the recovery of the hyper-inflammatory responses of CF monocytes by ETI treatment has been documented for LPS/ATP-induced IL-1β secretion [24]. Our data, which show a reduction, although not significant, of LPS-induced IL-6 secretion by monocytes after ETI therapy, support a broader anti-inflammatory property of ETI therapy whose characterisation needs further investigations [24]. This and the previous published works allow us to hypothesise that the molecular pathways recognised as responsible for specific dysfunctions of CF cells, such as the exuberant inflammatory response or defective microbicidal activity, can be considered for the development of new therapeutic strategies aimed at improving CF disease. This opportunity appears to be particularly important for the treatment of PWCF who are unresponsive to CFTR modulators and who may benefit from surrogate therapies that improve host defence against invading pathogens and the management of the inflammatory response.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-00725-2022.Supplement (809.3KB, pdf)
Shareable PDF
Acknowledgement
The authors thank Jacopo Savastano (Department of Epidemiology, Lazio Regional Health Service, Rome, Italy) for his support with the statistical analysis.
Footnotes
Author contributions: P. Del Porto, G. Cimino and F. Ascenzioni conceptualised the study. G. Cimino, E. Ferrara, P. Baiocchi and G. Mandarello recruited subjects and collected samples. G. Cimino and E. Ferrara provided clinical data. L. Cavinato, F.R. Luly, V. Pastore, D. Chiappetta and G. Sangiorgi performed the experiments. L. Cavinato, P. Del Porto and F. Ascenzioni analysed the data and wrote the manuscript. All authors reviewed the final version of the manuscript.
Conflict of interest: All authors have nothing to disclose.
Support statement: This work was supported by the Sapienza University of Rome, Italy, grant numbers RM11916B88E57B66, RM120172B1634AA0 and RP12117A86637295. Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.Elborn JS. Cystic fibrosis. Lancet 2016; 388: 2519–2531. doi: 10.1016/S0140-6736(16)00576-6 [DOI] [PubMed] [Google Scholar]
- 2.Mall MA, Mayer-Hamblett N, Rowe SM. Cystic fibrosis: emergence of highly effective targeted therapeutics and potential clinical implications. Am J Respir Crit Care Med 2020; 201: 1193–1208. doi: 10.1164/rccm.201910-1943SO [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amaral MD. How to determine the mechanism of action of CFTR modulator compounds: a gateway to theranostics. Eur J Med Chem 2021; 210: 112989. doi: 10.1016/j.ejmech.2020.112989 [DOI] [PubMed] [Google Scholar]
- 4.Kleizen B, Hunt JF, Callebaut I, et al. . CFTR: new insights into structure and function and implications for modulation by small molecules. J Cyst Fibros 2020; 19 Suppl. 1: S19–S24. doi: 10.1016/j.jcf.2019.10.021 [DOI] [PubMed] [Google Scholar]
- 5.Van Goor F, Yu H, Burton B, et al. . Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J Cyst Fibros 2014; 13: 29–36. doi: 10.1016/j.jcf.2013.06.008 [DOI] [PubMed] [Google Scholar]
- 6.Ramsey BW, Davies J, McElvaney NG, et al. . A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011; 365: 1663–1672. doi: 10.1056/NEJMoa1105185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor-Cousar JL, Munck A, McKone EF, et al. . Tezacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N Engl J Med 2017; 377: 2013–2023. doi: 10.1056/NEJMoa1709846 [DOI] [PubMed] [Google Scholar]
- 8.Taylor-Cousar JL, Mall MA, Ramsey BW, et al. . Clinical development of triple-combination CFTR modulators for cystic fibrosis patients with one or two F508del alleles. ERJ Open Res 2019; 5: 00082-2019. doi: 10.1183/23120541.00082-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wainwright CE, Elborn JS, Ramsey BW, et al. . Lumacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med 2015; 373: 220–231. doi: 10.1056/NEJMoa1409547 [DOI] [PubMed] [Google Scholar]
- 10.Southern KW, Murphy J, Sinha IP, et al. . Corrector therapies (with or without potentiators) for people with cystic fibrosis with class II CFTR gene variants (most commonly F508del). Cochrane Database Syst Rev 2020; 12: CD010966. doi: 10.1002/14651858.CD010966.pub3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nichols DP, Paynter AC, Heltshe SL, et al. . Clinical effectiveness of elexacaftor/tezacaftor/ivacaftor in people with cystic fibrosis: a clinical trial. Am J Respir Crit Care Med 2022; 205: 529–539. doi: 10.1164/rccm.202108-1986OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burgel P-R, Durieu I, Chiron R, et al. . Rapid improvement after starting elexacaftor–tezacaftor–ivacaftor in patients with cystic fibrosis and advanced pulmonary disease. Am J Respir Crit Care Med 2021; 204: 64–73. doi: 10.1164/rccm.202011-4153OC [DOI] [PubMed] [Google Scholar]
- 13.Zemanick ET, Taylor-Cousar JL, Davies J, et al. . A phase 3 open-label study of elexacaftor/tezacaftor/ivacaftor in children 6 through 11 years of age with cystic fibrosis and at least one F508del allele. Am J Respir Crit Care Med 2021; 203: 1522–1532. doi: 10.1164/rccm.202102-0509OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di A, Brown ME, Deriy LV, et al. . CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat Cell Biol 2006; 8: 933–944. doi: 10.1038/ncb1456 [DOI] [PubMed] [Google Scholar]
- 15.Del Porto P, Cifani N, Guarnieri S, et al. . Dysfunctional CFTR alters the bactericidal activity of human macrophages against Pseudomonas aeruginosa. PLoS One 2011; 6: e19970. doi: 10.1371/journal.pone.0019970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cifani N, Pompili B, Anile M, et al. . Reactive-oxygen-species-mediated P. aeruginosa killing is functional in human cystic fibrosis macrophages. PLoS One 2013; 8: e71717. doi: 10.1371/journal.pone.0071717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Assani K, Shrestha CL, Robledo-Avila F, et al. . Human cystic fibrosis macrophages have defective calcium-dependent PKC activation of the NADPH oxidase, an effect augmented by Burkholderia cenocepacia. J Immunol 2017; 198: 1985–1994. doi: 10.4049/jimmunol.1502609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li C, Wu Y, Riehle A, et al. . Staphylococcus aureus survives in cystic fibrosis macrophages, forming a reservoir for chronic pneumonia. Infect Immun 2017; 85: e00883-16. doi: 10.1128/IAI.00883-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Assani K, Tazi MF, Amer AO, et al. . IFN-γ stimulates autophagy-mediated clearance of Burkholderia cenocepacia in human cystic fibrosis macrophages. PLoS One 2014; 9: e96681. doi: 10.1371/journal.pone.0096681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barnaby R, Koeppen K, Nymon A, et al. . Lumacaftor (VX-809) restores the ability of CF macrophages to phagocytose and kill Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol 2018; 314: L432–L438. doi: 10.1152/ajplung.00461.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang S, Shrestha CL, Kopp BT. Cystic fibrosis transmembrane conductance regulator (CFTR) modulators have differential effects on cystic fibrosis macrophage function. Sci Rep 2018; 8: 17066. doi: 10.1038/s41598-018-35151-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hisert KB, Birkland TP, Schoenfelt KQ, et al. . CFTR modulator therapy enhances peripheral blood monocyte contributions to immune responses in people with cystic fibrosis. Front Pharmacol 2020; 11: 1219. doi: 10.3389/fphar.2020.01219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kopp BT, Fitch J, Jaramillo L, et al. . Whole-blood transcriptomic responses to lumacaftor/ivacaftor therapy in cystic fibrosis. J Cyst Fibros 2020; 19: 245–254. doi: 10.1016/j.jcf.2019.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gabillard-Lefort C, Casey M, Glasgow AMA, et al. . Trikafta rescues CFTR and lowers monocyte P2X7R-induced inflammasome activation in cystic fibrosis. Am J Respir Crit Care Med 2022; 205: 783–794. doi: 10.1164/rccm.202106-1426OC [DOI] [PubMed] [Google Scholar]
- 25.Riazanski V, Sui Z, Nelson DJ. Kinetic separation of oxidative and non-oxidative metabolism in single phagosomes from alveolar macrophages: impact on bacterial killing. iScience 2020; 23: 101759. doi: 10.1016/j.isci.2020.101759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cavinato L, Genise E, Luly FR, et al. . Escaping the phagocytic oxidative burst: the role of SODB in the survival of Pseudomonas aeruginosa within macrophages. Front Microbiol 2020; 11: 326. doi: 10.3389/fmicb.2020.00326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mecocci S, Gevi F, Pietrucci D, et al. . Anti-inflammatory potential of cow, donkey and goat milk extracellular vesicles as revealed by metabolomic profile. Nutrients 2020; 12: 2908. doi: 10.3390/nu12102908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luly FR, Lévêque M, Licursi V, et al. . MiR-146a is over-expressed and controls IL-6 production in cystic fibrosis macrophages. Sci Rep 2019; 9: 16259. doi: 10.1038/s41598-019-52770-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keating D, Marigowda G, Burr L, et al. . VX-445–tezacaftor–ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med 2018; 379: 1612–1620. doi: 10.1056/NEJMoa1807120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Middleton PG, Mall MA, Dřevínek P, et al. . Elexacaftor–tezacaftor–ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med 2019; 381: 1809–1819. doi: 10.1056/NEJMoa1908639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van de Weert-van Leeuwen PB, Van Meegen MA, Speirs JJ, et al. . Optimal complement-mediated phagocytosis of Pseudomonas aeruginosa by monocytes is cystic fibrosis transmembrane conductance regulator-dependent. Am J Respir Cell Mol Biol 2013; 49: 463–470. doi: 10.1165/rcmb.2012-0502OC [DOI] [PubMed] [Google Scholar]
- 32.Riquelme SA, Hopkins BD, Wolfe AL, et al. . Cystic fibrosis transmembrane conductance regulator attaches tumor suppressor PTEN to the membrane and promotes anti Pseudomonas aeruginosa immunity. Immunity 2017; 47: 1169–1181. doi: 10.1016/j.immuni.2017.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brunel SF, Willment JA, Brown GD, et al. . Aspergillus-induced superoxide production by cystic fibrosis phagocytes is associated with disease severity. ERJ Open Res 2018; 4: 00068-2017. doi: 10.1183/23120541.00068-2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cigana C, Curcurù L, Leone MR, et al. . Pseudomonas aeruginosa exploits lipid A and muropeptides modification as a strategy to lower innate immunity during cystic fibrosis lung infection. PLoS One 2009; 4: e8439. doi: 10.1371/journal.pone.0008439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Begum R, Thota S, Abdulkadir A, et al. . NADPH oxidase family proteins: signaling dynamics to disease management. Cell Mol Immunol 2022; 19: 660–686. doi: 10.1038/s41423-022-00858-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van de Geer A, Nieto-Patlán A, Kuhns DB, et al. . Inherited p40phox deficiency differs from classic chronic granulomatous disease. J Clin Invest 2018; 128: 3957–3975. doi: 10.1172/JCI97116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hey J, Paulsen M, Toth R, et al. . Epigenetic reprogramming of airway macrophages promotes polarization and inflammation in muco-obstructive lung disease. Nat Commun 2021; 12: 6520. doi: 10.1038/s41467-021-26777-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patella M, Anile M, Del Porto P, et al. . Role of cytokine profile in the differential diagnosis between acute lung rejection and pulmonary infections after lung transplantation. Eur J Cardiothorac Surg 2015; 47: 1031–1036. doi: 10.1093/ejcts/ezu395 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-00725-2022.Supplement (809.3KB, pdf)
This one-page PDF can be shared freely online.
Shareable PDF ERJ-00725-2022.Shareable (283.7KB, pdf)