

Abstract
Clinical diagnostic laboratories are producing next-generation sequencing-based test results that are becoming increasingly incorporated into patient care. Whole genome and exome sequencing on fetal material derived from amniocytes, chorionic villi, or products of conception is starting to be offered clinically in specialized centers, but it has not yet become routine practice. The technical, interpretation, and ethical challenges are greatest in the area of prenatal medicine because the fetus has a limited health history, and the physical examination is only indirectly available via prenatal sonography. Here, we provide an overview of these challenges and highlight the clinical utility, reporting, and counseling issues associated with prenatal DNA sequencing. Future considerations are also discussed.
INTRODUCTION
Over the past 10 years, next-generation sequencing (NGS) for genomic analysis has moved from the research setting to clinical diagnostic laboratories. Over this time, significant advances have occurred, with solutions found for many of the early technical challenges.1–3 The initially high costs of NGS continue to decrease, allowing incorporation of this powerful technology into many areas of postnatal clinical care, including oncology,4 cardiology,5 and for determining the basis of intellectual disabilities.6 While there are potentially significant benefits to NGS, there are still areas of uncertainty in the interpretation of results. The technical and ethical challenges are greatest in the area of prenatal medicine because the fetus has a limited health history, and the physical examination is available only indirectly through prenatal sonography. Here, we provide a brief overview of laboratory, counseling, and clinical considerations in the context of prenatal sequencing, while focusing on the potential successes and challenges that may be associated with its clinical implementation (Figure 1). Fetal DNA can be accessed directly from amniotic fluid or placental biopsies, or indirectly via cell-free feto-placental DNA that circulates in maternal blood. The relative simplicity of obtaining blood samples has led to the dramatic growth of maternal plasma DNA sequencing to screen for fetal aneuploidies.7 Its transformation of antenatal care has already led to questions about the next frontier, for example, whether noninvasive whole genome sequencing (WGS) of the fetus is feasible or even desirable.
Figure 1.
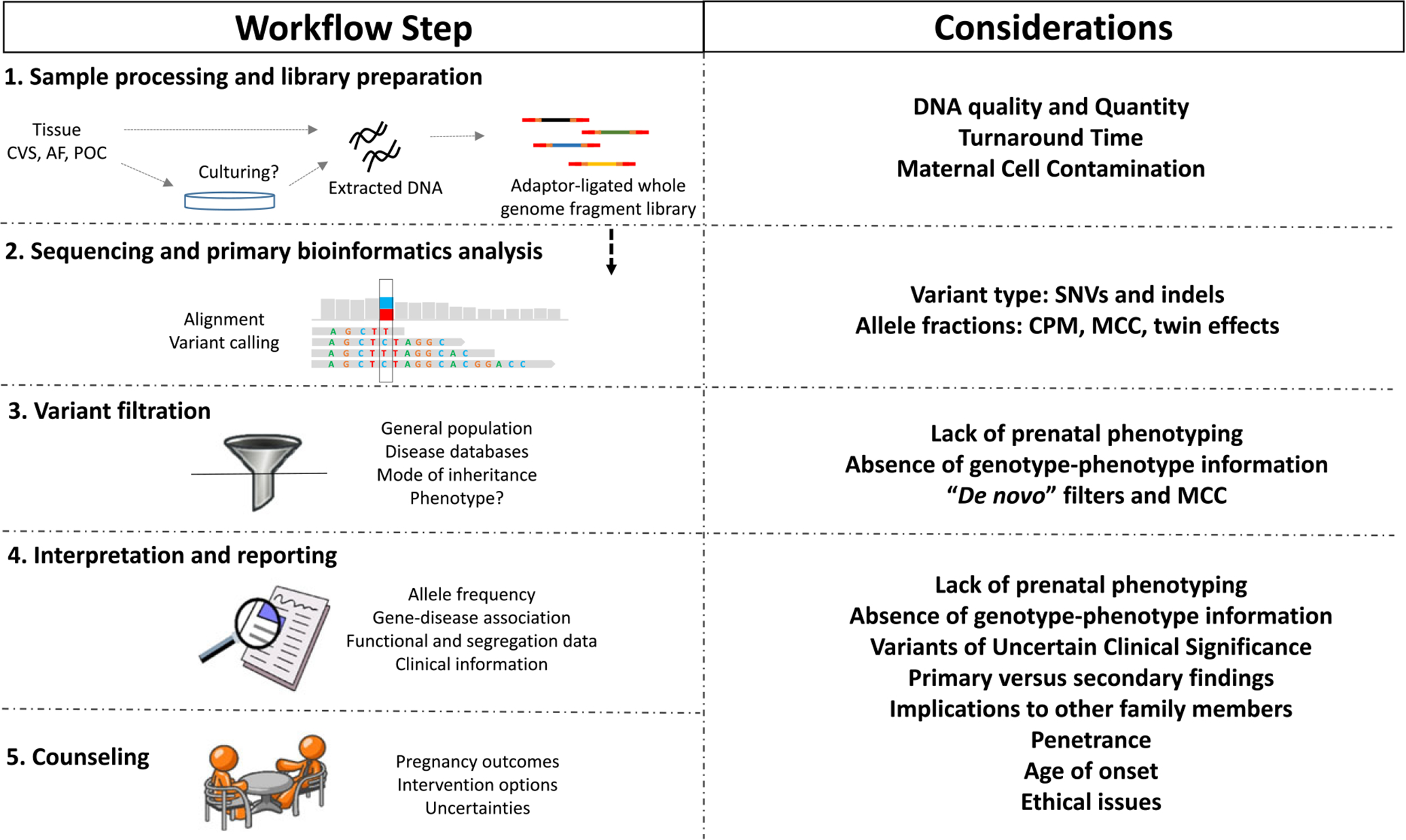
Technical, interpretation, and clinical considerations associated with prenatal sequencing. See text for detailed explanation. CVS, chorionic villus sampling; AF, amniotic fluid; POC, products of conception; MCC, maternal cell contamination; CPM, confined placental mosaicism; SNVs, single nucleotide variants; indels, small insertions and/or deletions.
In contrast to the direct analysis of intact fetal cells, noninvasive prenatal screening relies on circulating cell-free DNA that is derived from apoptotic cells in the placenta and serves as a surrogate source of fetal DNA.8 This ‘fetal’ DNA is most commonly used to screen for trisomies 13, 18, and 21 via a counting algorithm that compares the number of mapped fragments from the chromosome of clinical interest (e.g., 18) to a reference value.9,10 Although proof of concept noninvasive fetal WGS has been performed using maternal plasma cell-free DNA,11,12 separating the information in the fetal genome from the mother’s genome requires sophisticated bioinformatics analyses that are well beyond the current capabilities of most molecular diagnostic laboratories.13 The remainder of this review will therefore focus on purer sources of fetal DNA obtained through diagnostic procedures [chorionic villus sampling (CVS) and amniocentesis] and consider the challenges even when working with these direct sources of fetal DNA.
TECHNICAL CONSIDERATIONS
Sample type and sample processing
At present, depending on gestational age and the purpose of testing (screening vs diagnostic), several sample types can serve as sources of fetal DNA. For screening, maternal plasma is used. For diagnosis, invasive procedures such as chorionic villus sampling (CVS) or amniocentesis are performed to obtain pure fetal material at 11 or 15–20 gestational weeks, respectively. In the event of fetal loss, products of conception can also be used.
Several factors, including DNA yield, per cent of maternal cell contamination (MCC), and turnaround time, should be carefully considered before making a decision on the most appropriate source of DNA for sequencing.14,15 Although more DNA can be obtained from cultured cells, the turnaround time is significantly increased because of the time needed to establish adequate culture growth. With the continuous improvement of downstream whole genome library preparation protocols, less DNA – such as that obtained from the cells found in amniotic fluid or uncultured villi – can be used.
Maternal cell and DNA contamination
One consideration in the choice of sample type is the amount of anticipated MCC, which affects the purity of the sample. It is estimated that 0.3–0.7% of amniotic fluid specimens will have MCC.16,17 MCC likely stems from the presence of maternal skin cells that are carried along as the needle used to remove amniotic fluid passes across the mother’s abdomen. Discarding the initial few milliliters of fluid is recommended to decrease the presence of maternal cells. Culturing reduces contamination by selectively enhancing the growth of fetal cells.8 MCC of variable degree (up to >80% in extreme cases) occurs in 1–2.5% of CVS specimens because of the presence of maternal tissue in the sample.18 Culturing villi increases the time to receive results and does not necessarily enrich the percentage of fetal cells.19 Careful dissection of maternal tissue at the time of sample receipt has a significant impact on fetal cell purity. In all cases, MCC should be evaluated by comparing polymorphic genetic markers, most commonly short tandem repeats (STRs), between the maternal DNA (obtained from maternal blood or saliva) and the fetal DNA used for sequencing. Other laboratory quality control measures, such as culture backup and identity testing to rule out sample mix- up, should still be instituted.
Library preparation
With good quality DNA, whole genome libraries can be prepared for NGS in a manner similar to any postnatal DNA sample. Several commercial library preparation kits are currently available that, despite some differences, share two common steps: genomic DNA fragmentation – predominantly performed by ultra-sonication or enzymatic processing5 –followed by adaptor ligations. The end result is a pool of short genomic fragments – 200–600 bp, depending on the kit and downstream sequencing chemistry – with platform-specific adaptors containing sequencing primer binding sites and potentially molecular ‘barcodes’ to allow sample pooling or multiplexing. An important consideration at this step is the amount of input DNA required for the different library construction kits. Some protocols require >1–2 μg of DNA, which can be obtained from cultured prenatal specimens but might not be easily obtained directly from amniocytes or placental tissue prior to culture. Fortunately, some kits permit robust whole genome library construction from as low as 50 ng of input DNA. Alternatively, whole genome amplification can be used to enrich for genomic DNA if needed, without significantly altering the accuracy of sequencing results.20–22
Sequencing and primary bioinformatics analysis
The adaptor-ligated library can be processed into millions or billions of clonally amplified fragments that are then physically separated and sequenced in parallel using a platform such as the HiSeq (Illumina, San Diego, CA, USA).23,24 Illumina sequencing is performed by synthesis, whereby reversible fluorescently-labeled dideoxynuclotide terminators are added at each cycle to generate millions of four-color fluorescent signals (one for each base: A, T, C, and G) across all sequenced reads; such signals are imaged en masse at each cycle, and the compilation of all images across all cycles is then processed to generate base calls along each read.23 As each raw signal (image) is converted into a base call, a quality score is assigned. The file combining the sequence and quality data is referred to as the FASTQ (also called FAST-AII) file. A binary version of a sequence alignment map is called a BAM file. This file contains sequence alignment data that are generated as reads are aligned back to the reference genome. Changes relative to this reference or variants are then compiled in a text file, the variant call format file, that also contains various variant annotations. Quality metrics, including mapping quality, depth of coverage, strand bias, and quality by depth, are available to be used in filtering calls that may be inaccurate. Several bioinformatics pipelines are currently available; each has its own advantages and limitations.4,5,23
Variant calling
Careful validation is needed to establish the limit of detection of variants and to eliminate potential false positive calls that can be abundant at low allele fractions. In the postnatal setting, most germline bioinformatics pipelines deploy cutoffs that assume either heterozygous (20–60%) or homozygous (>80%) variant calls. Deviations from those ranges are more common in prenatal settings in which confined placental mosaicism (CPM), the presence of a twin, and/or MCC can be major potential contributors, especially in CVS specimens. Furthermore, with the increased postnatal clinical application of highly sensitive deep sequencing methods, hereditary disorders with genetic mosaicism are continuously being characterized.25–27 With increased resolution, if allelic fraction cutoffs are not applied too rigorously, more cases of fetal mosaicism will be identified.
In addition, although deviation from the expected ratios of base calls for biallelic variant calls might be indicative of MCC, the presence of a twin conception cannot be excluded, especially if one twin has died. In order to differentiate these possibilities, further studies should be carried out using standard assays for MCC, such as those using STR markers. However, discrepancies, due to differences in the sensitivities of NGS and STR fragment analysis, are not to be overlooked. For example, NGS might detect very low level MCC, below the limit of detection of STR assays, if high coverage sequencing is achieved.
In addition to MCC, CPM, and twin pregnancies, DNA analysis can also detect sperm or egg donors and/or surrogate mothers if trio (fetal and parental) sequencing is performed, thus underscoring the importance of good family history data and appropriate consent for the test. All such scenarios may be identified during sequence analysis, specifically, during primary variant calling in which significant deviations in read fractions from the typical biallelic configuration (~50% for heterozygous or ~100% for homozygous variants) might lead to false variant calls. In summary, the bioinformatics allele fraction filters should take into consideration the above scenarios and the presence of mosaicism, including CPM, which is observed in 1–2% of CVS specimens.28
Variant filtration
Once called, annotated variants will go through several bioinformatics filters. One specific filter uses sequencing data from the general population – such as the Exome Aggregation database or ExAC29 – to remove high allele frequency variants that are too common to associate with Mendelian disorders based on known disease prevalence, age of onset, penetrance, and mode of inheritance. This is a critical step in the process, especially for whole exome and WGS in which hundreds of thousands of variants are generated. As an initial step, it is assumed that a pathogenic variant is more likely to be absent or very rare in a general population that is not selectively enriched for the phenotype of interest.
A second strategy that is used postnatally is to select genes, and subsequently variants within these genes, that correlate with the patient’s phenotype. It is important to keep in mind, however, that known phenotypes of control (and disease) cohorts have been typically restricted to postnatal clinical manifestations. Such an approach may not be feasible in the prenatal setting, in which complete data on the fetal phenotype may not be available. An alternative strategy is to use genetic filters on the basis of assumptions about the mode of inheritance. For example, de novo variants might be prioritized to identify dominantly acting mutations if parents are unaffected, or biallelic variants might be prioritized to identify recessive conditions (providing that parental samples are available to conduct ‘trio’ analysis). A ‘trio’ consists of a set of samples that includes both biological parents and the fetus or child. However, such filters should take into consideration the possibility that maternal variants might be present in the ‘fetal genome’ as a result of MCC. Still, applying these filters can be extremely powerful in certain scenarios, such as in populations in which the consanguinity rate is high and causative homozygous variants are likely.
Finally, filtration based on the variant type or presence in the literature or in disease databases can significantly narrow down the list of potentially causative variants. For example, loss-of-function variants are often presumed to have severe effects, although this is only true for heterozygous variants when a decreased dose is disease-causing (haploinsufficient genes). ‘Healthy’ individuals have been shown to be heterozygous, and even homozygous, for this type of variant in certain genes29–31 and seemingly loss-of-function variants may not always lead to a null gene effect. In addition, it may be important not to restrict filtration so that it only reports ‘postnatal’ variants, because novel variants – in known or novel genes – that lead to severe or even lethal prenatal phenotypes may have occurred, especially in samples from fetuses that have died in utero. An interesting set of genes to target for variant filtration in prenatal populations is ones that are completely devoid of loss-of-function variants in the general population. Variation in such genes may be incompatible with life, and therefore there will be limited information about such genes in current disease databases.
Other technical limitations
Several considerations inherent to the NGS technology should be highlighted. Although NGS can accurately detect single nucleotide variants and small insertions and deletions (indels) of up to 25–50 bp, NGS is still not reliable in detecting exon- level copy number variants, structural rearrangements, or repeat expansions. Such variant types contribute significantly to numerous conditions that can be diagnosed prenatally,32 and ancillary assays might therefore be needed to attain the best chance of diagnosing a fetus. WGS has been shown to be more sensitive than whole exome sequencing for the detection of copy number variants and structural rearrangements when appropriate bioinformatics are utilized.33,34 This is attributed to the fact that WGS does not include a target enrichment step; thus, it generates more uniform coverage across sequenced coding regions. In addition, most copy number and structural events have intergenic breakpoints generating noncoding split reads that can be detected by WGS.
Another limitation of current NGS testing is the inability to detect variants in regions of high homology, as is found in genes that have almost identical sequences or pseudogenes. Such regions challenge the unique alignment of short reads, which can lead to false positive and false negative calls. Furthermore, repetitive sequence regions, such as those involved in triplet repeat expansion disorders, can be challenging to detect.
CLINICAL CONSIDERATIONS
Genotype and phenotype information
Interpreting variants obtained through whole exome and genome sequencing relies heavily on detailed phenotypic information supplied to the clinical laboratory at the time of testing. For postnatal sequencing, the clinical indication is used to prioritize variants in genes known to underlie most if not all of the patient’s features. As described previously, well-defined fetal phenotypes are often not available. Ultrasound scanning replaces the ‘physical exam’, and the ‘patient history’ is inaccessible unless there is a family history of a previously affected fetus or child. Furthermore, a compendium of ‘normal’ fetal phenotypic variation does not yet currently exist. The combination of large databases for both controls (such as ExAC)29 and affected individuals (such as ClinVar)35 has significantly improved variant interpretation in postnatal settings. However, the ClinVar database has limited variation that has been deposited to date that is associated with fetal abnormalities.
The age of disease onset and penetrance add another layer of complexity. A pathogenic variant leading to a disease of postnatal onset can be identified in a typically developing ‘presymptomatic’ fetus. Likewise, a clinically significant variant with reduced penetrance can also be carried by an apparently normal fetus, or by an abnormal fetus whose phenotype cannot be explained by this variant.
Finally, despite sequencing the whole genome, interpretation is usually limited to coding sequence variants plus or minus 2–20 bp of the exon/intron boundaries (splice sites). Aside from a few well-established regulatory and/or deep intronic variants, interpretation of noncoding variation is extremely challenging. Nonetheless, the sequencing of intergenic regions is helpful for detecting structural variation. And as more clinically phenotyped individual and fetal genomes are sequenced, our understanding of novel noncoding variants will significantly improve over time.
Reporting
Because of the lack of evidence to support classification of a large proportion of variants identified by NGS-based largescale sequencing, such variants will be of uncertain significance (VUS). In light of this, and in the absence of expert guidelines for prenatal genome sequencing, it remains a major challenge to define the criteria for reporting variants prenatally. While most molecular reports note pathogenic variants relevant to the fetal indication, the question of whether VUS should be returned to the parents is less clear. Although a VUS identified in an indication-relevant gene might have a high a priori probability of being clinically significant, the residual uncertainty can create anxiety for a pregnant couple. This adds to its complexity and the potential for termination of a fetus who would otherwise have developed into a healthy child.
Even more complex problems arise around the reporting of variants unrelated to the fetal indication. The American College of Medical Genetics and Genomics has recommended that all clinical laboratories performing postnatal exome or genome sequencing seek and report pathogenic variants in 59 secondary genes unrelated to the patient’s indication but that have medical implications for the patient.36,37 While the American College of Medical Genetics and Genomics statement included children in their recommendations, they explicitly excluded fetuses. Thus, currently, there are no guidelines for the reporting of incidental or secondary findings when prenatal sequencing is performed. If secondary findings are reported, no consensus has been reached as to whether these should be restricted to conditions affecting pregnancy management or delivery, or whether they should include pediatric or even adult-onset disorders. If the latter conditions are to be disclosed prenatally, then the potential impact of reactive pregnancy terminations without appropriate clinical understanding will need to be carefully considered. The possibility that all of these uncertainties may be discovered during prenatal testing should be discussed during pretest counseling with individuals undergoing prenatal diagnosis.
One additional challenge is that fetal testing results are normally included in the mother’s medical record, which may make it more difficult to link genetic variant data with postnatal phenotypes in the child. New systems are needed to transfer these data to the infant’s medical record after birth. Furthermore, for trio analysis, a laboratory mechanism has to be in place to facilitate the use of parental sequencing data in the infant’s data reanalysis and/or in future pregnancies for the same couple.
Counseling
Prenatal counseling is complicated by several issues, including the fact that in many parts of the world, an expectant couple may freely terminate any pregnancy for any reason. This means that genetic inferences about the health of the fetus that are communicated to the parents can be used as part of a decision to terminate a pregnancy. This raises the stakes around the disclosure of uncertain information and emphasizes the importance of pretest counseling session(s) and informed consent, including up-front discussions about the return of any incidental or secondary findings.38 If such findings will be returned, then implications for other family members should be discussed, especially if late onset conditions are included.39 Furthermore, couples should be aware that information regarding family relationships (alternate paternity or incest) can be unveiled through fetal testing. Finally, an estimate of the diagnostic yield for this type of testing should be disclosed. Recent studies using exome sequencing, performed on fetuses with sonographic abnormalities, resulted in a diagnosis in about 10–20% of cases.40,41
Turnaround time
In the prenatal setting, rapid generation of results is critical for effective clinical management, allowing timely decisions to be made regarding termination of the pregnancy or around the logistics of delivery and need for subspecialist consultation. Four factors can affect how quickly a result might be returned. First, if only variants in a defined set of genes related to the indication are evaluated, then interpretation can be slightly expedited. Second, obtaining parental samples for trio analysis can reduce interpretation time if a de novo variant is identified or the parental samples are used to clarify the phase of heterozygous variants in a single gene. Third, the Sanger confirmation step for reportable variants could be skipped if certain quality metrics for a high confidence true positive variant – as determined during up-front test validations – are satisfied and the possibility of sample mix-up has been adequately controlled. Finally, if the whole genome library preparation protocol is sensitive to small DNA amounts, then direct tissue can be used as a starting material, alleviating the need for lengthy culture times.
CLINICAL BENEFITS OF PRENATAL WHOLE GENOME SEQUENCING
There are a number of potential benefits of offering genomic sequencing prenatally. The availability of genomic sequencing results during pregnancy offers an opportunity for early prenatal intervention and potentially higher likelihood of effective treatment. A recent example is the identification of loss-of-function mutations in MAGED2 via whole exome sequencing as the underlying basis for an X-linked form of extreme polyhydramnios that results in preterm delivery and either perinatal demise or neonatal transient salt wasting and polyuria.42 MAGED2 is expressed in the ascending loop of Henle and in renal tubules. It is also critical for the maintenance of a normal gestation. An interesting aspect of this condition is that it resolves spontaneously in affected male infants. Prior to the discovery of the genetic etiology, neonates affected with salt wasting and polyuria received long-term treatment with mineral supplements and non-steroidal antiinflammatory drugs. Now that a genotype–phenotype correlation has been established in males with MAGED2 mutations, this treatment is probably unnecessary because the condition spontaneously resolves.
DNA sequencing information may also provide an early warning for perinatal complications for which delivery sites with special facilities or specialists are required to care for the newborn. Prenatal knowledge of an expected metabolic genetic condition in the neonate may allow immediate implementation of an appropriate diet to avoid metabolic decompensation, which may otherwise begin in the first few days or weeks of life before newborn screening results are available.43 For autosomal recessive conditions in which two pathogenic variants cannot always be identified (e.g., hearing loss that is due to Pendred syndrome), identification of even carrier status in the fetus could lead to more comprehensive clinical follow-up in the neonatal period, potential early diagnosis and improved clinical care.
Other potential benefits include a definitive diagnosis, even for a fetus or infant that dies in the perinatal period.34,44–46 The identification of a new autosomal recessive disorder in a family will allow accurate recurrence risk counseling and an exploration of various options in future pregnancies, including pre-implantation genetic diagnosis.
While the aforementioned examples illustrate hypothesized impacts on perinatal care, it is too early to determine the extent to which prenatal genomic sequencing results actually alter perinatal care and result in benefits or harm to families. Reports of clinical diagnostic experience in large laboratories are only beginning to appear. Amid discussion and debate about the appropriateness and medical utility of offering genomic sequencing prenatally, it will be important to gather data around these issues before commercial entities begin marketing prenatal sequencing to parents and healthcare practitioners.47
FUTURE CONSIDERATIONS AND RECOMMENDATIONS
As the genetic basis of fetal conditions is explored and the accuracy of predicting postnatal conditions through prenatal sequencing improves, NGS technology will increasingly be applied during prenatal care. If available in early pregnancy, variants can be queried throughout fetal development and perhaps combined with other factors, such as existing or emerging maternal conditions, to guide personalized fetal and pregnancy management. The possibility that a metabolic condition discovered prenatally may lead to rapid and effective treatment in the neonatal intensive care unit (NICU) can be life altering for affected neonates.48 Accumulation of fetal sequencing data will eventually lead to the proliferation of longitudinal fetal genotype–phenotype databases, ultimately leading to more effective diagnoses and overall improved prenatal care. In addition, such resources will enhance research opportunities, including novel gene discovery and development of new personalized prenatal treatments.
Nonetheless, expert guidelines are needed to carefully maximize the benefits of fetal genomic medicine. A consensus regarding the appropriateness of different types of prenatal genetic tests and reporting criteria has yet to be established, and the accompanying medical and consent issues should be carefully assessed. In particular, issues of penetrance and the uncertainties surrounding genomic results have to be clarified to minimize uninformed clinical decisions. Healthcare providers will need to be educated about this type of genetic testing, its advantages, limitations, and implications for other family members.
One approach that might alleviate the anxiety of prenatally disclosing secondary pathogenic findings of postnatal onset is to offer a tiered reporting strategy (Figure 2). While sequencing can still be performed prenatally, a preliminary report could be issued with a focus on the fetal indication and/or any conditions relevant to pregnancy management and plans for delivery. Parents could opt for a secondary report that could be released soon after birth to disclose findings relevant to postnatal care. If carefully instituted, this approach would allow families more choices for the receipt of prenatal sequencing results. In addition, if desired, this approach can offer a very fast genomic newborn screening report, bypassing the need for obtaining a newborn’s sample for sequencing and subsequent analysis.
Figure 2.

Prenatal whole genome sequencing and release of genomic results. A tiered reporting approach can be adopted whereby sequencing is performed prenatally followed by sequential prenatal, perinatal, and postnatal release of results based on indication and utility of genomic findings.
While the challenges are significant, the incorporation of NGS into the prenatal clinical diagnostic laboratory is progressing, and the associated technical issues are slowly resolving. As summarized in this paper, the potential advantages are numerous; arguably, personalized medicine starts in the mother’s womb, thus creating possibilities for therapeutic intervention long before a baby is born.49
WHAT’S ALREADY KNOWN ABOUT THIS TOPIC?
Clinical exome and genome sequencing on fetal material (amniotic fluid, chorionic villus sampling, and products of conception) is starting to be offered in specialized centers.
There are no technical or interpretation standards/guidelines for the clinical practice of prenatal sequencing.
WHAT DOES THIS STUDY ADD?
This study highlights a comprehensive overview of prenatal clinical exome and genome sequencing workflow.
This study pinpoints challenges and potential solutions at each step of the prenatal sequencing workflow including technical, bioinformatics, interpretation, counseling, and clinical challenges.
Footnotes
Conflicts of interest: A.N.A., N.B.S., H.L.R., and D.W.B. have no conflicts of interest. R.C.G. receives financial compensation for speaking or advisory services to AIA, Helix, Illumina, Invitae, and Prudential.
REFERENCES
- 1.Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 2011;12(11):745–55. [DOI] [PubMed] [Google Scholar]
- 2.Metzker ML. Sequencing technologies – the next generation. Nat Rev Genet 2010;11(1):31–46. [DOI] [PubMed] [Google Scholar]
- 3.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol 2008;26(10):1135–45. [DOI] [PubMed] [Google Scholar]
- 4.Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet 2010;11(10):685–96. [DOI] [PubMed] [Google Scholar]
- 5.Voelkerding KV, Dames S, Durtschi JD. Next generation sequencing for clinical diagnostics-principles and application to targeted resequencing for hypertrophic cardiomyopathy: a paper from the 2009 WilliamBeaumont Hospital Symposium on Molecular Pathology. J Mol Diagn 2010;12(5):539–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Ligt J, Willemsen MH, van Bon BW, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 2012;367(20):1921–9. [DOI] [PubMed] [Google Scholar]
- 7.Taylor-Phillips S, Freeman K, Geppert J, et al. Accuracy of non-invasive prenatal testing using cell-free DNA for detection of Down, Edwards and Patau syndromes: a systematic review and meta-analysis. BMJ Open 2016;6(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taglauer ES, Wilkins-Haug L, Bianchi DW. Review: cell-free fetal DNA in the maternal circulation as an indication of placental health and disease. Placenta 2014;35(Suppl):S64–S68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiu RW, Chan KC, Gao Y, et al. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc Natl Acad Sci U S A 2008;105(51):20458–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan HC, Blumenfeld YJ, Chitkara U, et al. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc Natl Acad Sci U S A 2008;105(42):16266–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kitzman JO, Snyder MW, Ventura M, et al. Noninvasive whole-genome sequencing of a human fetus. Sci Transl Med 2012;4(137):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lo YM, Chan KC, Sun H, et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med 2010;2(61):1–13. [DOI] [PubMed] [Google Scholar]
- 13.Wang E, Batey A, Struble C, et al. Gestational age and maternal weight effects on fetal cell-free DNA in maternal plasma. Prenat Diagn 2013;33(7):662–6. [DOI] [PubMed] [Google Scholar]
- 14.Frederickson RM, Wang HS, Surh LC. Some caveats in PCR-based prenatal diagnosis on direct amniotic fluid versus cultured amniocytes. Prenat Diagn 1999;19(2):113–17. [DOI] [PubMed] [Google Scholar]
- 15.Nagan N, Faulkner NE, Curtis C, et al. Laboratory guidelines for detection, interpretation, and reporting of maternal cell contamination in prenatal analyses: a report of the association for molecular pathology. J Mol Diagn 2011;13(1):7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steed HL, Tomkins DJ, Wilson DR, et al. Maternal cell contamination of amniotic fluid samples obtained by open needle versus trocar technique of amniocentesis. J Obstet Gynaecol Can 2002;24(3):233–6. [DOI] [PubMed] [Google Scholar]
- 17.Stojilkovic-Mikic T, Mann K, Docherty Z, et al. Maternal cell contamination of prenatal samples assessed by QF-PCR genotyping. Prenat Diagn 2005;25(1):79–83. [DOI] [PubMed] [Google Scholar]
- 18.Steinberg S, Katsanis S, Moser A, et al. Biochemical analysis of cultured chorionic villi for the prenatal diagnosis of peroxisomal disorders: biochemical thresholds and molecular sensitivity for maternal cell contamination detection. J Med Genet 2005;42(1):38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aina-Mumuney A, Wood ED, Corson VL, et al. Clinical consequences of an increasing trend of preferential use of cultured villi for molecular diagnosis by CVS. Prenat Diagn 2008;28(4):332–4. [DOI] [PubMed] [Google Scholar]
- 20.Korfhage C, Fisch E, Fricke E, et al. Whole-genome amplification of single-cell genomes for next-generation sequencing. Curr Protoc Mol Biol 2013;104: Unit 7 14. [DOI] [PubMed] [Google Scholar]
- 21.Li N, Wang L, Wang H, et al. The performance of whole genome amplification methods and next-generation sequencing for pre- implantation genetic diagnosis of chromosomal abnormalities. J Genet Genomics 2015;42(4):151–9. [DOI] [PubMed] [Google Scholar]
- 22.Pugh TJ, Delaney AD, Farnoud N, et al. Impact of whole genome amplification on analysis of copy number variants. Nucleic Acids Res 2008;36(13):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Voelkerding KV, Dames SA, Durtschi JD. Next-generation sequencing: from basic research to diagnostics. Clin Chem 2009;55(4):641–58. [DOI] [PubMed] [Google Scholar]
- 24.Mardis ER. Next-generation sequencing platforms. Annu Rev Anal Chem (Palo Alto Calif) 2013;6:287–303. [DOI] [PubMed] [Google Scholar]
- 25.Acuna-Hidalgo R, Bo T, Kwint MP, et al. Post-zygotic point mutations are an underrecognized source of de novo genomic variation. Am J Hum Genet 2015;97(1):67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu X, Yang X, Wu Q, et al. amplicon resequencing identified parental mosaicism for approximately 10% of “de novo” SCN1A mutations in children with Dravet syndrome. Hum Mutat 2015;36(9):861–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamaguchi K, Komura M, Yamaguchi R, et al. Detection of APC mosaicism by next-generation sequencing in an FAP patient. J Hum Genet 2015;60(5):227–31. [DOI] [PubMed] [Google Scholar]
- 28.Kalousek DK, Vekemans M. Confined placental mosaicism. J Med Genet 1996;33(7):529–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536(7616):285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacArthur DG, Balasubramanian S, Frankish A, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science 2012;335(6070):823–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Narasimhan VM, Hunt KA, Mason D, et al. Health and population effects of rare gene knockouts in adult humans with related parents. Science 2016;352(6284):474–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wapner RJ, Martin CL, Levy B, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med 2012;367(23):2175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Talkowski ME, Ernst C, Heilbut A, et al. Next-generation sequencing strategies enable routine detection of balanced chromosome rearrangements for clinical diagnostics and genetic research. Am J Hum Genet 2011;88(4):469–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Talkowski ME, Ordulu Z, Pillalamarri V, et al. Clinical diagnosis by whole-genome sequencing of a prenatal sample. N Engl J Med 2012;367(23):2226–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harrison SM, Riggs ER, Maglott DR, et al. Using ClinVar as a resource to support variant interpretation. Curr Protoc Hum Genet 2016;89:(8.16):1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 2013;15(7):565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2016;19(2):249–55. [DOI] [PubMed] [Google Scholar]
- 38.Westerfield LE, Stover SR, Mathur VS, et al. Reproductive genetic counseling challenges associated with diagnostic exome sequencing in a large academic private reproductive genetic counseling practice. Prenat Diagn 2015;35(10):1022–9. [DOI] [PubMed] [Google Scholar]
- 39.Wilfond BS, Fernandez CV, Green RC. Disclosing secondary findings from pediatric sequencing to families: considering the “benefit to families”. J Law Med Ethics 2015;43(3):552–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carss KJ, Hillman SC, Parthiban V, et al. Exome sequencing improves genetic diagnosis of structural fetal abnormalities revealed by ultrasound. Hum Mol Genet 2014;23(12):3269–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drury S, Williams H, Trump N, et al. Exome sequencing for prenatal diagnosis of fetuses with sonographic abnormalities. Prenat Diagn 2015;35(10):1010–7. [DOI] [PubMed] [Google Scholar]
- 42.Laghmani K, Beck BB, Yang SS, et al. Polyhydramnios, transient antenatal Bartter’s syndrome, and MAGED2 mutations. N Engl J Med 2016;374(19):1853–63. [DOI] [PubMed] [Google Scholar]
- 43.Rafati M, Mohamadhashem F, Hoseini A, et al. Prenatal diagnosis of tyrosinemia type 1 using next generation sequencing. Fetal Pediatr Pathol 2016;1–4. [DOI] [PubMed] [Google Scholar]
- 44.Mei L, Huang Y, Pan Q, et al. Targeted next-generation sequencing identifies novel compound heterozygous mutations of DYNC2H1 in a fetus with short rib-polydactyly syndrome, type III. Clin Chim Acta 2015;447:47–51. [DOI] [PubMed] [Google Scholar]
- 45.Olech EM, Zemojtel T, Sowinska-Seidler A, et al. Identification of a molecular defect in a stillborn fetus with perinatal lethal hypophosphatasia using a disease-associated genome sequencing approach. Pol J Pathol 2016;67(1):78–83. [DOI] [PubMed] [Google Scholar]
- 46.Zhen L, Zhang Y, Li DZ. Prenatal DNA diagnosis of Noonan syndrome in a fetus with increased nuchal translucency using next-generation sequencing. Eur J Obstet Gynecol Reprod Biol 2016;201:229–30. [DOI] [PubMed] [Google Scholar]
- 47.Dondorp W, de Wert G, Bombard Y, et al. Non-invasive prenatal testing for aneuploidy and beyond: challenges of responsible innovation in prenatal screening. Eur J Hum Genet 2015;23(11):1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tarailo-Graovac M, Shyr C, Ross CJ, et al. Exome sequencing and the management of neurometabolic disorders. N Engl J Med 2016;374(23):2246–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bianchi DW. From prenatal genomic diagnosis to fetal personalized medicine: progress and challenges. Nat Med 2012;18(7):1041–51. [DOI] [PMC free article] [PubMed] [Google Scholar]