

Abstract
Mas-related G protein coupled receptors (MRGPRs) family members play important roles in the sensation of noxious stimuli and represent novel targets for the treatment of itch and pain. MRGPRs recognize a diversity of agonists and display complicated downstream signaling profiles, high sequence diversity across species and many polymorphisms in humans. The recent structural advances on MRGPRs reveals unique structural features and diverse agonist recognition modes of this receptor family, which should facilitate the structure-based drug discovery at MRGPRs. In addition, the newly discovered ligands also provide valuable tools to explore the function and the therapeutic potential of MRGPRs. In this review, we discuss these progresses in our understanding of MRGPRs and highlight the challenges and potential opportunities for the future drug discovery at these receptors.
Keywords: MRGPRs, function, polymorphism, structure, ligand recognition, drug discovery
Classification and recent structural advances of MRGPRs
Mas-related G protein-coupled receptors (MRGPRs) represent a family of Class A GPCRs which mediate a variety of noxious sensations including pain and itch [1, 2]. The first member of MRGPRs was discovered in 1986 [3] and since then they have been found to encompass a ~40-member family of GPCRs [4, 5], which are localized to primary sensory ganglia and some immune cells. Phylogenetically, MRGPRs could be divided into 9 major families (viz. MRGPRA-H and MRGPRX). Mice have the most discovered MRGPR proteins so far [6]. In human, there are only eight MRGPR members, MRGPRD, MRGPRE, MRGPRF, MRGPRG and MRGPRX1-X4. Of these, the MRGPRX-family of receptors was initially identified as a ‘primate-exclusive’ group although it is present in several non-rodent species.
Previous functional studies of human MRGPRs mainly focused on MRGPRD, MRGPRX1 and MRGRPX2 [6] and most of the MRGPRs family proteins are classified as understudied orphan receptors with little information on its endogenous ligand, signaling and biological function. Recently, MRGPRs have emerged as novel drug targets to treat itch, allergy, neuroinflammation and pain [7–14]. The MRGPR family members have complicated downstream signaling pathways and display diverse sequences across species [6, 15]. In addition, MRGPRs are highly polymorphic, and many natural occurring mutations have been reported [15–17]. Structural studies of MRGPRs have long been difficult due to its non-canonical structural motifs and intrinsic high basal activity [18]. The recent advancement of cryogenic electron microscopy (cryoEM) technique leads to the successful determination of MRGPRX2, MRGPRX4, MRGPRD and MRGPRX1 structures [18–21]. These high-resolution MRGPRs structures shed lights on the ligand recognition mechanisms and facilitate our understanding of the natural occurring mutations of MRGPRs. Moreover, the MRGPRs structures could be used as templates for the future structure-based drug discovery. In this review, we summarize the current functional, structural and pharmacological findings of MRGPRs and highlight how this information can accelerate the drug discovery at MRGPRs family receptors.
MRGPRs in itch and pain
The MRGPR-family of receptors is essential for itch sensations
Itch and pain are ubiquitous noxious stimuli that cause considerable distress and disability [22, 23] Many G protein-coupled receptors (GPCRs) and ion channels have been discovered to be involved in the sensations of itch and pain [7, 24, 25]. With respect to the sensation of itch, there are no specific antipruritic drugs that are uniformly effective for itch, especially those caused by metabolic disorders and chronic skin diseases. MRGPR-family receptors were initially identified in sensory neurons [4, 26] and in the past two decades, substantial studies have revealed that MRGPRs are essential for the sensation of nonhistaminergic itch [6, 27]. For instance, MRGPRX1 was reported to mediate the itch sensation induced by the antimalarial drug chloroquine [26], whereas MRGPRD was shown to be involved in the itch evoked by β-alanine [28]. Recently, MRGPRX4 was identified as bile acid receptor which mediates the cholestatic itch caused by the accumulation of bile acids and bilirubin due to liver dysfunction [10, 11, 29]. Of all the MRGPRs family receptors, MRGPRX2 is the most well-studied itch receptor. MRGPRX2 mediates the itch sensation of a variety of endogenous peptide agonists as well as a large number of cationic drugs including vancomycin, morphine and several other U.S. Food and Drug Administration (FDA)-approved drugs [30–33]. Notably, the ‘Red Man Syndrome’ caused by both vancomycin morphine are likely mediated by MRGPRX2 [32, 34]. Studies have also suggested that the expression of MRGPRX2 is upregulated in patients with severe chronic urticaria, allergic contact dermatitis and asthma [35–37] and the number of MRGPRX2-expressing cells are reported to correlate with itch severity [38]. These recent findings suggest that MRGPR family receptors are promising targets for the treatment of itch caused by marketed drugs, liver dysfunction and many chronic skin diseases.
The MRGPR-family of receptors is essential for pain sensations
In addition to itch, several MRGPRs have been reported to be involved in pain sensation. There is clear evidence that the activation of MRGPRX1 at the central terminals of primary sensory neurons by the endogenous opioid peptide fragment BAM8-22 could inhibit chronic pain [39]. There is also strong evidence that the substance P elicited neuroinflammatory pain is mediated by MRGPRX2 and not due to its high affinity target neurokinin 1 receptor (NK1R) [9]. Considering there is an urgent need for non-opioid medications to treat both acute and chronic pain conditions [7], these results indicate the clinical significance of potentially targeting MRGPRs.
Diverse G protein signaling pathways of MRGPRs
Multiple down-stream signaling effectors, especially Gi and Gq, have been reported for MRGPR family receptors[15], including MRGPRX1, MRGPRX2, MRGPRX4 and MRGPRD. For instance, both Gi and Gq signaling pathways have been reported and validated by several groups for MRGPRX2 [18, 34, 40]. Interestingly, the current literature suggests that Gi and Gq signaling pathways are synergistic in MRGPRX2 as both are required to stimulate a maximum MRGPRX2-mediated mast cell degranulation [17]. Recent research shows that the free Gβγ subunit released by the activated Gi protein could recruit the Phospholipase C-β to cell membrane to accelerate Gq signaling [41], which may explain the synergistic effect of Gi and Gq in MRGPRX2.
Using a newly developed bioluminescence resonance energy transfer (BRET)-based G protein heterotrimer dissociation assay [42], we found that MRGPRX2 can activate all the four major G protein subfamilies including the Gs, Gi, Gq/11 and G12/13, revealing a promiscuous signaling profile (Figure 2A) [18]. However, the Gs and G12/13 pathways have not been examined in MRGPRX2-mediated mast cell degranulation assays. Thus, further study might be required to reveal the potential role(s) of Gs and G12/13 in the biological function of MRGPRX2.
Figure 2. G protein signaling of MRGPRs.
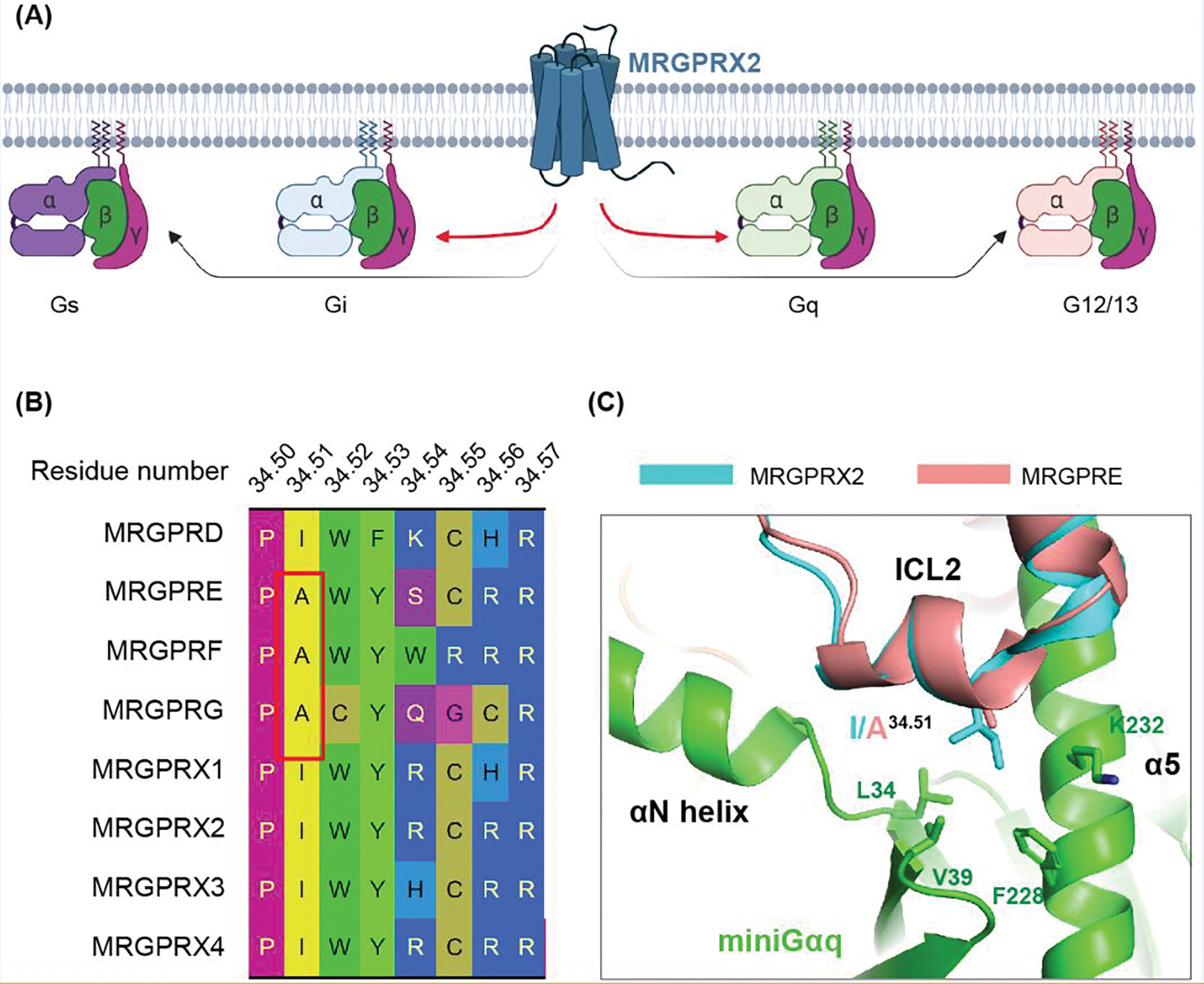
(A) MRGPRX2 couple to all the G protein subfamilies in the in vitro G protein dissociation assay. The activation of Gi and Gq by MRGPRX2 are stronger than Gs and G12/13. Figure created with Biorender.com. (B) Sequence alignment generated by GPCRDB [81] shows that MRGPRE, MRGPRF and MRGPRG have distinct ICL2 residue composition, especially the residue 34.51, compared with other MRGPRs. (C) Structural comparison of human MRGPRE AlphaFold model with MRGPRX2-Gq complex (PDB: 7S8N), showing that A34.51 in MRGPRE may weaken the interactions between the receptor and Gq.
To our knowledge, the downstream signaling effectors for several other MRGPRs including MRGPRE, MRGPRF, MRGPRG and MRGPRX3 have not been reported due to unavailability of selective, high affinity and high efficacy agonists. Nonetheless, MRGPRX3 could have a similar downstream signaling profile to MRGPRX2 as their G protein interface residues are identical [20]. Sequence analysis suggests that the intracellular loop 2 (ICL2) residue compositions of MRGPRE, MRGPRF and MRGPRG are different from other MRGPRs (Figure 2B–2C). Notably, alanine, which is small in size, is presented at the ICL2 residue 34.51 (Ballesteros–Weinstein number [43]). Previous studies suggested that large hydrophobic residues at position 34.51 of family A GPCRs may facilitate the initial coupling to Gq protein, but not Gi [44–46]. In addition, alanine mutations of residue 34.51 greatly reduced the agonist stimulated Gq signaling in MRGPR and several other GPCRs [20, 44–46]. Thus, the presence of alanine in residue 34.51 of MRGPRE, MRGPRF and MRGPRG suggest these MRGPR receptors might not mainly rely solely on Gq protein for signaling. Drug screening at these receptors may still be performed though G15, a permissive G protein that could bind many kinds of GPCRs, mediated Ca2+ flux [10]. In addition to the Gi and Gq signaling, several MRGPRs, such as MRGPRA3, MRGPRC11 and MRGPRX1, could also mediate the Gβγ-dependent modulation of ion channels including the TRP channel and N-type HVA calcium channels [39, 47–49], further indicating a potential wealth of diverse signaling pathways downstream of MRGPRs.
Sequence diversity and polymorphism of MRGPRs
MRGPRs display diverse sequences across species.
From an evolutionary perspective, MRGPR family receptors are highly variable across different species [50]. Notably, mice have 27 MRGPRs with intact open reading frames, which include members from MRGPRA to MRGPRG [4, 6]. By contrast, humans lack members in MRGPRA, B and C, with only eight MRGPRs including the primate-exclusive MRGPRX1-4 [5]. The MRGPRs members have relatively diverse primary amino acid sequences across different species (Figure 3A). For example, the rhesus macaque (colloquially known as the rhesus monkey) MRGPRX4 shares only 81% sequence identity with the human MRGPRX4. This sequence difference is also reflected in the predicted AlphaFold models of rhesus macaque MRGPRX4 and human MRGPRX4 as large deviations around the extracellular pocket are observed (Figure 3B). Notably, some of the divergent residues are located in the ligand binding pocket (Figure 3C), which raises the question as to whether rhesus macaque MRGPRX4 would respond to bile acids. This large sequence difference is likely unique to MRGPRs as many other GPCRs, such as the human 5-HT1A serotonin receptor (5-HT1AR), show almost identical sequence and structure (Figure 3D) across closely related species. The lack of conservation between MRGPRs orthologs makes it difficult to use mice and monkey to study the function of human MRGPRs, raising challenges for drug discovery. As an alternative method, humanized mice are frequently employed to test the ligand and function of selected human MRGPRs [6, 10, 39].
Figure 3. Sequence diversity of MRGPRs.
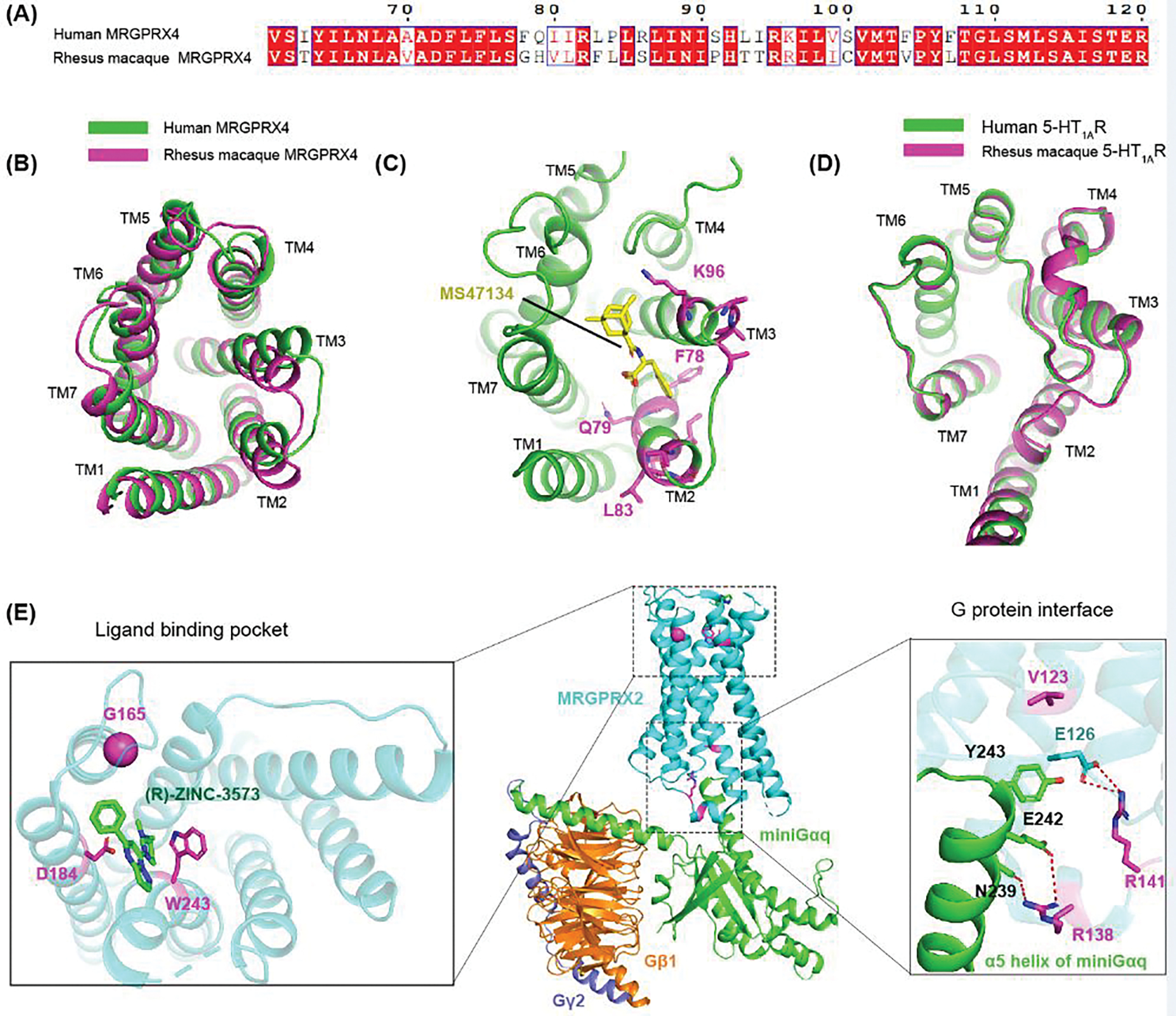
(A) Sequence alignment between human MRGPRX4 and rhesus macaque MRGPRX4, showing large residue divergence from residue 78 to residue 107. (B) Structural comparison between the predicted AlphaFold models of human MRGPR4 and rhesus macaque MRGPRX4. Extracellular view is shown to highlight the structural differences in the binding pocket. (C) Structure of MRGPRX4 (PDB: 7S8P) with the divergent residues from F78 to K96 shown in panel A colored in magenta. As these residues are located near the agonist binding site, rhesus macaque MRGPRX4 may not respond to the agonist of human MRGPRX4. (D) The predicted AlphaFold models of human 5-HT1AR and rhesus macaque 5-HT1AR overlaid well with each other, indicating 5-HT1AR is super conserved across different species. (E) MRPGRX2 loss-of-function mutations highlighted in MRGPRX2-Gq signaling complex (PDB: 7S8N). Polar interactions are depicted by red dashed lines.
Polymorphism of MRGPRs and their potential clinical relevance.
In addition to the observed sequence diversity in different species, MRGPRs display many polymorphisms of which some are quite common. According to the data in Genome Aggregation Database (gnomAD) [51], the allele frequencies of the missense mutations in human MRGPRX1-MRGPRX4 are 50–100 times higher than that of 5-HT1AR (Table 1). This high mutation rate of human MRGPRs could suggest, among other explanations, that they are continuously evolving to respond to noxious stimuli. The gnomAD data also shows that 8% of Africans carry a N245S mutation in MRGPRX4 that may lead to 5–8 fold increase in the preference over mentholated cigarettes, whereas Asians do not carry this mutation [51, 52]. It should also be noted that some of the naturally occurring mutations may affect the ligand binding and signaling of MRGPRs. In our previous study, we found that L83S variant of MRGPRX4 is relatively not sensitive to nateglinide and the synthetic agonist MS47134 [18]. Intriguingly, residue 83 is not located in the agonist binding pocket in MRGPRX4 (Figure 3C), which suggests that the L83S variant may affect the overall protein conformation that dampen the agonist recognition process of MRGPRX4.
Table 1.
Variant and allele frequency of MRGPRX family receptors.
| MRGPRX1 | MRGPRX2 | MRGPRX3 | MRGPRX4 | 5-HT1AR |
|---|---|---|---|---|
| A46T (1.72e-1) | N62S (2.79e-1) | N169D (7.72e-1) | L83S (9.90e-1) | I28V (9.21e-3) |
| F273L (3.52e-2) | N16H (6.74e-2) | C82R (1.68e-1) | Y54C (3.17e-1) | R220L (3.82e-3) |
| I36V (1.36e-2) | S313R (4.01e-3) | W236R (7.34e-2) | F8L (3.03e-1) | P16L (1.73e-3) |
| R131S (6.81e-3) | W243R (3.32e-3) | W307R (5.64e-2) | N25K (3.03e-1) | G273D (1.37e-3) |
| R55L (3.01e-3) | S284P (3.17e-3) | A70V (1.45e-2) | A128V (2.27e-1) | |
| H133R (1.61e-3) | V43I (2.66e-3) | T298P (9.13e-3) | V142M (1.06e-2) | |
| L51V (1.26e-3) | F78L (1.03e-3) | L198R (6.72e-3) | N245S (7.75e-3) | |
| P269L (5.97e-3) | ||||
| S150F (2.76e-3) | ||||
| I65L (1.67e-3) | ||||
| V124I (1.53e-3) | ||||
| A182S (1.40e-3) |
Data obtained from gnomAD. Only variants with an allele frequency (shown in brackets) higher than 1e-3 are listed.
Many MRGPRX2 naturally occurring mutations, such as V123F, R138C, R141C, D184H, G165E, W243R, H259Y and V282M, have been reported to abolish substance P stimulated MRGPRX2 activation [16, 17]. The recent high-resolution structures of MRGPRX2 shed light on how these mutations may affect the MRGPRX2 function [18, 19]. Structurally, D184H, G165E and W243R are located in the agonist binding pocket of MRGPRX2 (Figure 3E) [18, 19]. These mutations can change both the size and the charge property of the ligand binding pocket. Thus, they are unlikely to bind to substance P as well as other MRGPRX2 agonists. By contrast to the binding pocket mutations, V123F, R138C and R141C are located in the receptor-G protein interface. The abolished MRGPRX2 activation of these variants is probably due to an impaired G protein coupling. H259Y and V282M are located in neither the ligand binding pocket nor the G protein coupling interface, suggesting they affect the overall protein conformation. Other MRGPRX2 variants, such as F31V, V43I, F78L, Y137H, R140C, Q305R and D311H, are likely neutral and respond to agonist similarly to wild-type (WT) receptor [16, 17]. As some of the naturally occurring variants could dramatically affect the ligand response and signaling process of MRGPRs, drugs developed for normal people may not be effective for people carrying certain MRGPRs mutations.
Structure and ligand recognition of MRGPRs
Distinct structural features of MRGPRs
MRGPRs are unique among family A GPCRs as they lack most of the canonical motifs that are important for receptor activation, including the CWxP motif, PIF motif and the semi-conserved DRY motif [18, 20]. The recent advances in the determination of MRGPRs structures reveal distinct structural features and diverse agonist recognition modes of MRGPRs [18–21]. Firstly, MRGPRs have a unique TM4-TM5 disulfide bond (Figure 4B). The TM3-ECL2 disulfide bond observed in other family A GPCRs does not exist in MRGPRs. In most of the class A GPCRs, the TM3-ECL2 disulfide bond helps to stabilize the ECL2 conformation on the top of ligand binding pocket, which increases the resident time and decreases the dissociation rate of their corresponding endogenous agonists (Figure 4A) [53, 54]. Without this disulfide bond, the ECL2 of MRGPRs flips away from the center and does not cover the agonist binding pocket, which results in relatively low binding affinities and potencies of many endogenous MRGPR agonists [18, 20]. In addition, the toggle switch residue W6.48 in the CWxP motif is replaced with G6.48 or S6.48 in MRGPRs, which leads to a closer interaction between TM6 and TM3 (Figure 4C). This helical movement of TM6 toward TM3 closes the canonical orthosteric pocket resulting in a shallow solvent-exposed ligand binding pocket that is unique to MRGPRs (Figure 4D) [18, 20]. Compared to the canonical family A GPCRs, MRGPRs also have an unusual TM6 kink below the ligand pocket. The kink is partly stabilized by the hydrogen bond between the side chain of Y3.36 and the backbone carbonyl group of G/S6.48 [19, 21] (Figure 4C). Alanine substitution of Y3.36 resulted in reduced receptor activations in both MRGPRX2 and MRGPRD, suggesting this TM6 kink together with the Y3.36-G/S6.48 hydrogen bond are important for receptor activation of MRGPRs [19, 21]. Moreover, MRGPRs lack a key TM3 residue S3.39 for sodium binding. Numerous studies have shown that the sodium ion stabilizes GPCRs in inactive state [55] and mutations of sodium pocket residues could increase the receptor basal activity [56, 57]. Thus, the non-conserved sodium binding pocket could partly explain the high basal activity of several MRGPRs [18, 21, 58], which enables them to be activated by a gentle “push” of their shallowly bound low-affinity agonists.
Figure 4. The shallow agonist binding mode of MRGPRs.
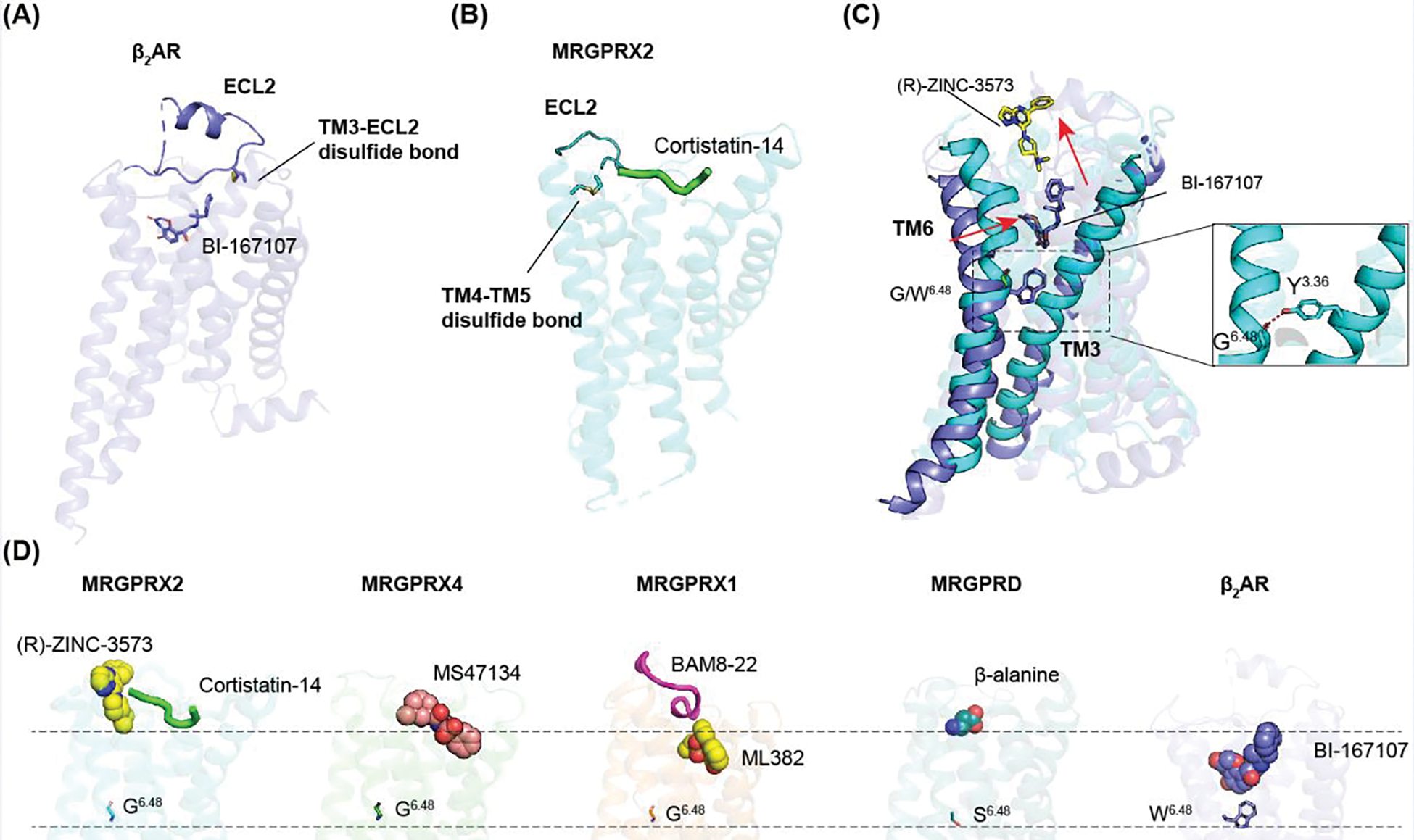
(A) The canonical TM3-ECL2 observed in β2 adrenergic receptor (β2AR) helps stabilize the conformation of ECL2 on the top of the agonist binding pocket (PDB: 3SN6). (B) MRGPRX2 (PDB: 7S8N) only contains a TM4-TM5 disulfide bond. Thus, its ECL2 does not cover the ligand binding pocket. (C) Replacement of large residue W6.48 in β2AR (PDB: 3SN6) to small residue G6.48 in MRGPRX2 (PDB: 7S8N) leads to a close contact between TM6 with TM3, which results in a shallow agonist binding pocket. MRGPRX2 and β2AR are colored by cyan and blue, respectively. The inset shows the interaction between Y3.36 and G6.48, which is important to maintain the active state of MRGPRs. (D) MRGPRs have shallow agonist binding pockets that are far away from the residue 6.48. PDB 7S8N, 7S8L, 7S8P, 8DWG, 7Y12 and 3SN6 are used for MRGPRX2-(R)-ZINC3573, MRGPRX2-cortistatin-14, MRGPRX4, MRGPRX1, MRGPRD and β2AR, respectively.
Currently, there are no available inactive state MRGPRs structures. Thus, it’s unclear how these distinct structural features transit from inactive state to activate state. The functional studies of MRGPRX2 reveal an inward movement of TM6 as well as large conformational changes of extracellular loops upon agonist binding [19]. Moreover, the agonist-bound MRGPRD displays inward displacement of the extracellular part of TM6 compared to the G protein-bound apo state [21]. Collectively, these observations indicate MRGPRs have considerable conformational plasticity that might be important for both agonist recognition and the future drug discovery.
Diverse ligand recognition motifs for MRGPRs
Many of the previously reported GPCR subfamilies are activated by shared endogenous agonist (e.g. serotonergic, adrenergic, muscarinic and so on). However, MRGPRs do not respond to a common agonist. Indeed, the currently known MRGPRs respond to a variety of structurally different agonists ranging from small molecules to large peptides, indicating they likely have distinct extracellular ligand binding pockets. In our recent studies on MRGPRX1, MRGPRX2 and MRGPRX4, we found that the residue composition, pocket size and charge distribution of the extracellular ligand binding pockets are highly diverse across the MRGPRX family receptors [18, 20].
The structure of MRGPRX2 reveals a wide-open ligand binding pocket that could be divided to two different sub-pockets: the negatively charged sub-pocket 1 formed by TM3-6 and the hydrophobic sub-pocket 2 formed by TM1-3 and TM6-7 (Figure 5A) [18]. The small molecule agonist (R)-ZINC-3573 binds to sub-pocket 1 (Figure 5B) and forms charge interactions with two acidic residues D1845.36 and E1644.60. By contrast, large peptide agonists bind to both sub-pocket 1 and sub-pocket 2 (Figure 5C) [18, 19]. Despite extensive hydrophobic interactions are observed in sub-pocket 2, the activation of MRGPRX2 is mainly triggered by the charge interactions in sub-pocket 1. Notably, all the peptides have a positively charged residue (either lysine or arginine) bound to the sub-pocket 1 and form charge interactions with D1845.36 and E1644.60. Alanine substitution of D1845.36 and E1644.60 greatly reduced the agonist stimulated MRGPRX2 activation, indicating the charge interactions with these two acidic residues are critical for receptor activation [18, 19]. This highly negatively charged surface of sup-pocket 1 explains MRGPRX2’s preference for positively charged agonist. Additionally, residue D254ECL3 of MRGPRX2 is also involved in agonist recognition by interacting with a second amide group in several polycationic compounds which contains two basic nitrogens that is separated by 8~13 Å in distance[19]. Based on the agonist-receptor interaction mode, a conserved peptide motif consisted of separated charged and hydrophobic residues has been proposed for MRGPRX2 [19], which might be helpful for searching novel peptide agonist.
Figure 5. Diverse agonist recognition mechanisms in MRGPRs.
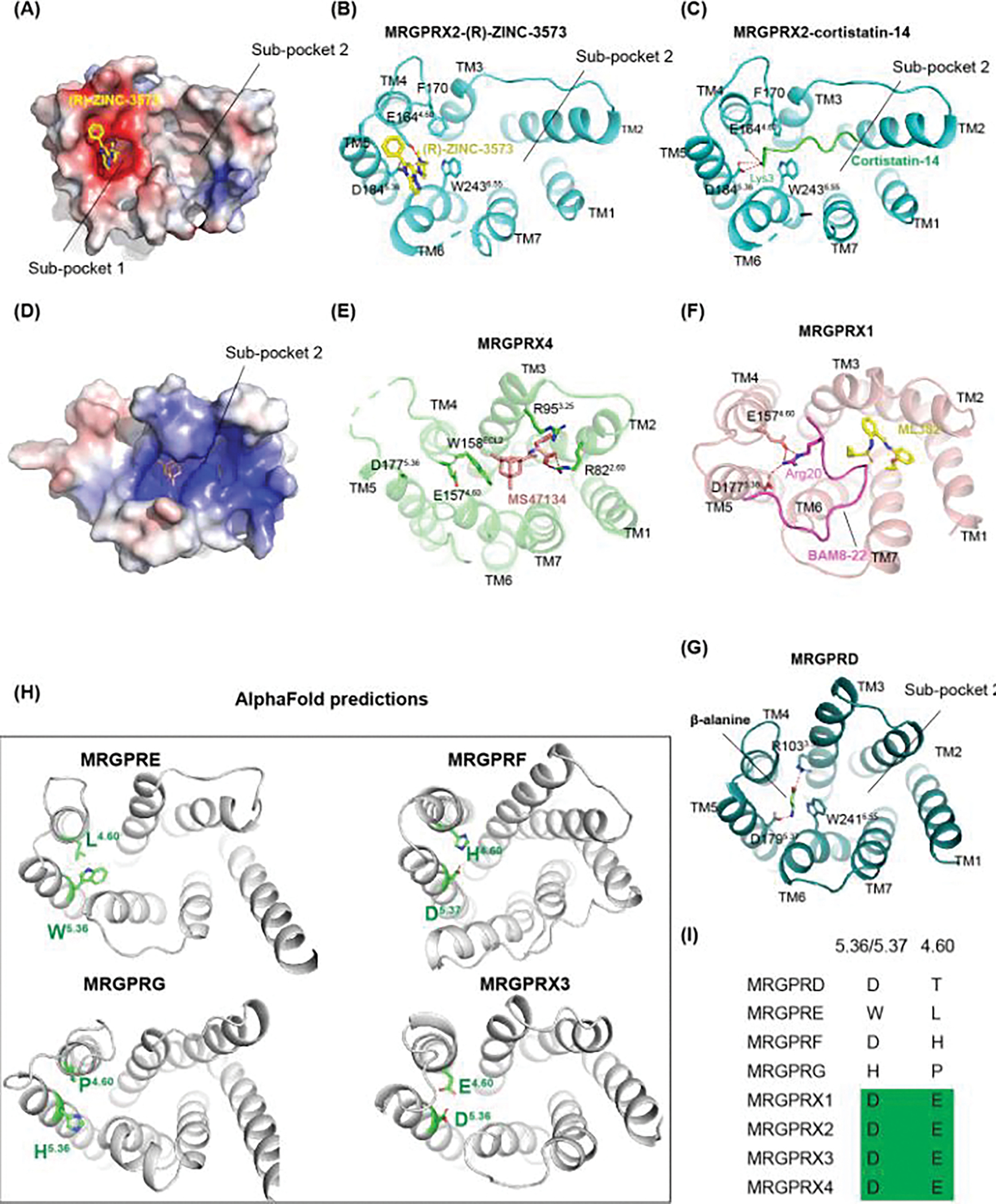
(A) Electrostatic surface representation of MRGPRX2 (PDB: 7S8N), highlighting the negatively charged (colored in red) sub-pocket 1 for agonist recognition. (B) Agonist binding mode of (R)-ZINC-3573 with key MRGPRX2 residues involving in interactions shown in sticks (PDB: 7S8N). (C) Agonist binding mode of cortistatin-14 (PDB: 7S8L), showing corstatin-14 binds to both sub-pocket 1 and sub-pocket 2. (D) Electrostatic surface representation of MRGPRX4 (PDB: 7S8P), highlighting the positively charged (colored in blue) sub-pocket 2 for agonist recognition. (E-G) Agonist binding mode of MRGPRX4 (PDB: 7S8P), MRGPRX1 (PDB: 8DWG) and MRGPRXD (PDB: 7Y12). (H) Predicted AlphaFold models of human MRGPRE, MRGPRF, MRGPRG and MRGPRX3 with residues 5.36/5.37 and 4.60 shown in green sticks. (I), Sequence alignment of the key residues 5.36/5.37 and 4.60 in TM5 and TM4 that are critical for MRGPRs activation.
The orthosteric pocket of MRGPRX4 is strikingly different from MRGPRX2 (Figure 5D and 5E). The ECL2 of MRGPRX4 inserts into a cavity between TM3 and TM6, making the sub-pocket 1 seen in MRGPRX2 inaccessible (Figure 5E) [18]. As a result, MRGPRX4 does not have a negatively charged ligand binding pocket and its agonist MS47134 could only bind to the sup-pocket 2. Additionally, several basic residues make the sub-pocket 2 positively charged (Figure 5D), which facilitates the binding of negatively charged bile acids [18]. Notably, the two acidic residues, i.e. D5.36 and E4.60, critical for the cationic agonist recognition in MRGPRX2 are also existed in MRGPRX4. The distinct structural features observed in MRGPRX4 explains why it responds to charged agonists that are opposed to that of MRGPRX2.
Although the two conserved acidic residues D1775.36 and E1574.60 are exposed to the extracellular solvent, MRGPRX1 does not have a “side chain only” pocket for the promiscuous binding of positively charged peptides [20]. The Arg20 of the MRGPRX1 peptide agonist BAM8-22 adopts a flat conformation to interact with D1775.36 and E1574.60 (Figure 5F). Intriguingly, the binding of BAM8-22 to MRGPRX1 is very sensitive to the peptide conformation as truncations of BAM8-22 peptides totally abolishes the activation of MRGPRX1 [20], which suggests MRGPRX1 is highly selective for its peptide agonist. The MRGPRX1 structures also reveal a unique binding mode of its positive allosteric modulator (PAM) ML382 [20]. In the previously solved GPCR structures, the PAM molecules bind to receptor at a position that is distal from the agonist. In MRGPRX1, however, ML382 binds into the sub-pocket 2 of MRGPRX1 (Figure 5F) and form direct interactions with the peptide agonist BAM8-22. The PAM assay showed strongly improved potencies of the MRGPRX1 agonist BAM8-22 in presence of ML382, suggesting it may potentiate the receptor activation by strengthen the BAM8-22’s affinity to MRGPRX1 [20]. Notably, PAM alone can’t activate the MRGPRX1 receptor, indicating the interactions in sub-pocket 2 is not enough to trigger the receptor activation.
Compared to the MRGPRX family members, MRGPRD, MRGPRE, MRGPRF and MRGPRG do not have the two conserved acidic residues, namely D5.36 and E4.60, that are critical for MRGPRX1 and MRGPRX2 activation. Instead, residues D1795.37 and R1033.30 are essential for MRGPRD to recognize its agonist β-alanine through charge interactions (Figure 5G). Thus, these receptors probably respond to their endogenous or exogenous agonists with distinct charge properties. Despite the differences, the sub-pocket 1 of MRGPRD overlaps well with that of MRGPRX1 and MRGPRX2, indicating the ligand-receptor interactions in this region are critical for MRGPRs activation [21]. Although the structures of MRGPRE, MRGPRF, and MRGPRG have not been experimentally determined, their distinct orthosteric pockets predicted by AlphaFold (Figure 5H) as well as the residue divergence at position 5.36/5.37 and 4.60 (Figure 5I) [59] suggest they could respond to different ligands.
Drug discovery opportunities for MRGPRs
Current drug development of MRGPRs
Considering many MRGPRs are involved in itch or pain sensation, drugs that inhibit the MRGPRs activation have substantial therapeutic potential [2]. However, as most of the MRGPRs are understudied receptors, only a few MRGPR-preferring antagonist have been reported and none of them have been approved by FDA for therapeutic use. Ogasawara et al. reported two MRGPRX2 antagonists, namely compound 1 and compound 2, which could inhibit mast cell activation by MRGPRX2 agonist [12]. Structurally, the side chain only sub-pocket 1 of MRGPRX2 indicates it probably binds to small-size antagonist. Thus, we searched low molecular weight analogs of compound 2 and identified two new MRGPRX2 antagonists, i.e. compound C9 and C9-6, with improved inhibition efficacy [18]. Both C9 and C9-6 could efficiently block the MGRPRX2 activation stimulated by a variety of peptide agonists and the small molecule agonist (R)-ZINC-3573. In addition, they also inhibited the basal activity of MRGPRX2 as inverse agonists and displayed high selectivity over NK1R, MRGPRX4 and many other receptors [18]. Previous studies of MRGPRX2 or its mice homolog MRGPRB2 usually requires knockout mice [9, 30] or RNA knockdown techniques [60]. Thus, the discovery of highly selective MRGPRX2 antagonists provides a convenient tool to assess the MRGPRX2 function.
In addition to these antagonists, several other low-affinity compounds have been reported as inhibitors of MRGPRX2 [61]. Considering the role of MRGPRX2 in mast cell degranulation, these antagonists may have clinical benefit to treat IgE-independent pseudoallergic reactions, itch and neuroinflammation [9]. Intriguingly, activation of a subset of MRGPRD neurons has been reported to inhibit MRGPRB2, which is the mice ortholog of MRGPRX2, mediated mast cell degranulation [14], suggesting potential use of MRGPRD agonist to suppress mast cell response. For MRGPRX4, there is no antagonist reported except for the EP547 compound developed by investigators at Escient Pharmaceuticals [62].
Compared to the currently identified MRGPRs, MRGPRX1 is an unusual MRGPR member which has dual roles. Essentially, peripheral activation of MRGPRX1 elicits itch, whereas the central activation of MRGPRX1 in the spinal cord by BAM8-22 inhibits pain [39]. The expression of MRGPRX1 is restricted to dorsal root ganglion neurons, which makes it a non-opioid pain-relieving target that could potentially avoid the severe side effects associated with drugs targeting opioid receptors. Thus, the drug discovery of MRGPRX1 mainly focuses on its inhibitory role in chronic inflammatory pain [7]. Currently, the MRGPRX1 agonist compound-16 [63] and several positive allosteric Modulators (PAMs) of MRGPRX1, including ML382 and compound 1t, have been developed [8, 64]. As the endogenous peptide BAM8-22 is mainly expressed in the spinal cord, these PAMs could specifically activate the MRGPRX1 in the central terminals of sensory neurons to inhibit nociceptive transmission [64]. Indeed, both ML382 and compound 1t display inhibition of neuropathic pain without causing obvious side effects, demonstrating the therapeutic potential of MRGPRX1 PAMs [8, 64].
Perspective for the structure-guide drug discovery at MRGPRs
In silico drug discovery could explore orders of magnitude more compounds than the traditional experimental screening methods [65]. In recent years, structure-based large-scale docking has facilitated the drug discovery for many GPCRs, including the mu-opioid receptor [66], D4-dopamine receptor [65], cannabinoid receptors[67], α2A adrenergic receptor [68] and 5-HT2A serotonin receptor [69]. Although the recent progress on the structure determination of MRGPRs-G protein complexes could provide templates for the large-scale docking [18–21], inactive GPCR structures are still preferred for antagonist discovery. This is especially true for MRGPRs as their shallow ligand binding sites usually consist of dynamic extracellular loops that may undergo conformation changes upon activation [19, 21]. However, high-resolution cryoEM structure determination of inactive state family A GPCRs are still challenging due to the small size of GPCRs. Considering this, fiducial markers or fusion proteins are usually required to both increase the molecule weight and facilitate particle alignment for cryoEM reconstruction [70–73]. Remarkably, by replacing the ICL3 with that of kappa opioid receptor (KOR), many of the class A GPCRs could bind to the KOR nanobody Nb6, which stabilize the receptor in an inactive state for high-resolution structure determination (Figure 6) [73, 74]. Alternatively, the intracellular loop 3 (ICL3) of GPCRs can be replaced with a soluble protein Bril. Then, inactive structure of the engineered ICL3-Bril GPCR protein could be theoretically obtained with the assist of anti-Bril Fabs by increasing its molecular weight for cryoEM structural determination (Figure 6) [75, 76]. Currently, no antagonist-bound inactive structure of MRGPRs has been reported. Thus, the future success on MRGPRs inactive structure determination using the method mentioned above would be very helpful for the structure-based drug discovery at MRGPR family receptors.
Figure 6. Inactive structural determination for the structure-based drug discovery at MRGPRs.
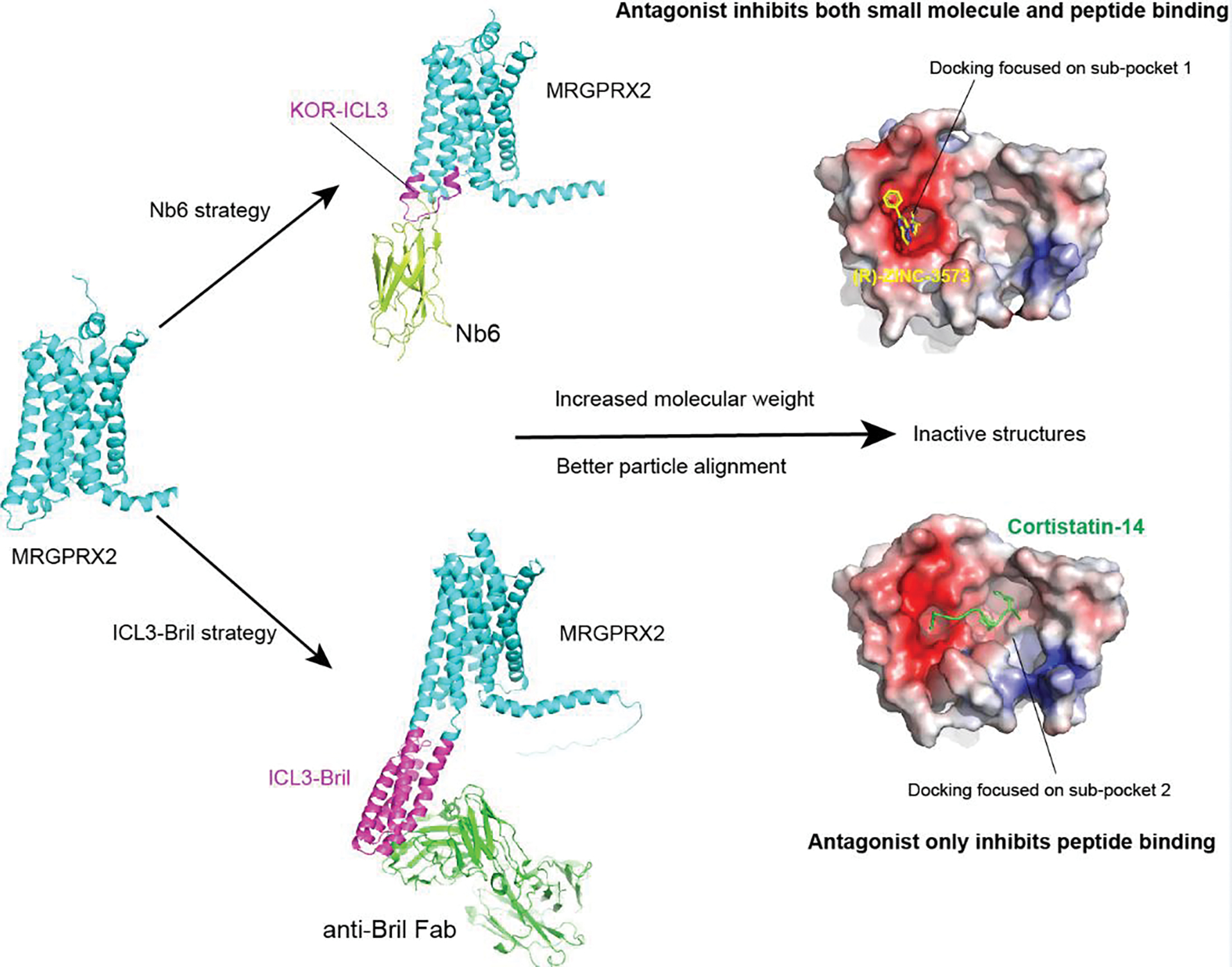
MRGPRX2 is shown here as an example. The inactive MRGPRX2 structure could be obtained though construct engineering to bind to Nb6 or anti-Bril Fab, both of which increase the molecular weight and facilitate the particle alignment in cryoEM structural determination. The obtained MRGPRX2 inactive structures then could be used as a template for structure-based docking. Both sub-pocket 1 and sub-pocket 2 of MRGPRX2 could be used for docking. However, the antagonist binds to sub-pocket 2 could only inhibit the peptide stimulated MRGPRX2 activation.
Typically, orthosteric pockets are the primary docking site for GPCRs as ligands bound at this position directly modulate receptor activity. Thus, the structure-based drug discovery at MRGPRs should mainly focus on the sub-pocket 1 for all the MRGPRs, except for MRGPRX4 whose orthosteric pocket is sub-pocket 2. Alternatively, the drug discovery at MRGPRs could also focus on other regions of their large extracellular pockets. For example, the sub-pocket 2 of MRGPRX2 is much larger than its sub-pocket 1. While the charge interactions in sub-pocket 1 is critical for MRGPRX2 activation, its small size and variable pocket shape introduced by the flexibility of ECL2 make it difficult for the structure-based docking to discovery novel antagonist. Theoretically, the relatively bigger and rigid sub-pocket 2 might be much more easily targeted. Although it may not be effective for small molecule agonist, compounds that bind to sub-pocket 2 should be able to block peptide stimulated MRGPRX2 activation (Figure 6). In addition, the binding of ML382 to sub-pocket 2 as an allosteric ligand in MRGPRX1 suggests that the sub-pocket 2 of MRGPRX2 and MRGPRD might be also used as an allosteric site for drug discovery.
Concluding Remarks and Future Perspectives
Over the past two years, significant progress has been made in the structure determination of MRGPRs [18–21]. High-resolution structures of MRGPRs-G protein complexes have provided valuable information to understand the downstream signaling and diverse agonist recognition mechanisms of this understudied receptor family. These structures also facilitate the understanding of the potential clinical effects of MRGPRs natural occurring variants and could be used as templates to perform structure-based drug discovery at MRGPRs.
Nonetheless, several key questions remain to be addressed in the future to further understand the function and therapeutic potential of MRGPRs (see Outstanding questions). Current drug discovery efforts are mainly focused on antagonists that could block itch and pain sensations. Thus, inactive MRGPRs structures are needed to act as a better template for antagonist discovery.
Outstanding questions:
Does MRGPRD, MRGPRE, MRGPRF, MRGPRG and MRGPRX3 have endogenous agonist?
What’s the function of other understudied MRGPRs?
Why MRGPRs have such high mutation rates compared to other GPCRs?
Can we obtain antagonist hits through in silico drug discovery based on the inactive MRGPRs structures?
Can we find a suitable animal model to test MRGPR drugs prior to clinical trials?
Although the functions of MRGPRD, MRGPRX1, MRGPRX2 and MRPGRX4 have been partly explored, the remaining MRGPR family members are still understudied receptors. For example, both the ligands and downstream signaling pathway of MRGPRE, MRGPRF, MRGPRG and MRGPRX3 are still incompletely elucidated. The lack of ligands for MRGPRE, MRGPRF, MRGPRG and MRGPRX3 restrict the current functional studies of these receptors. Thus, identifying the agonist or antagonist for these understudied MRGPRs is of urgent need. However, as MRGPRs usually have high basal activities, the efficacy window for agonists will be small compared to receptors with low basal activity, confounding the results of future studies. Generating stable cell lines with low MRGPR receptor expression levels may be required for efficient agonist discovery.
Moreover, it is still unknown whether MRGPRE, MRGPRF, MRGPRG and MRGPRX3 also respond to potential itch or pain stimuli from either endogenous metabolites or exogenous environment. Recently, the GPCR-based cpGFP sensors have been shown to be able to detect endogenous agonists of GPCRs [77–80]. The future development of MRGPRs fluorescent sensors might be helpful to detect their potential endogenous agonists.
Figure 1. Human MRGPRs in the sensation of itch and pain.
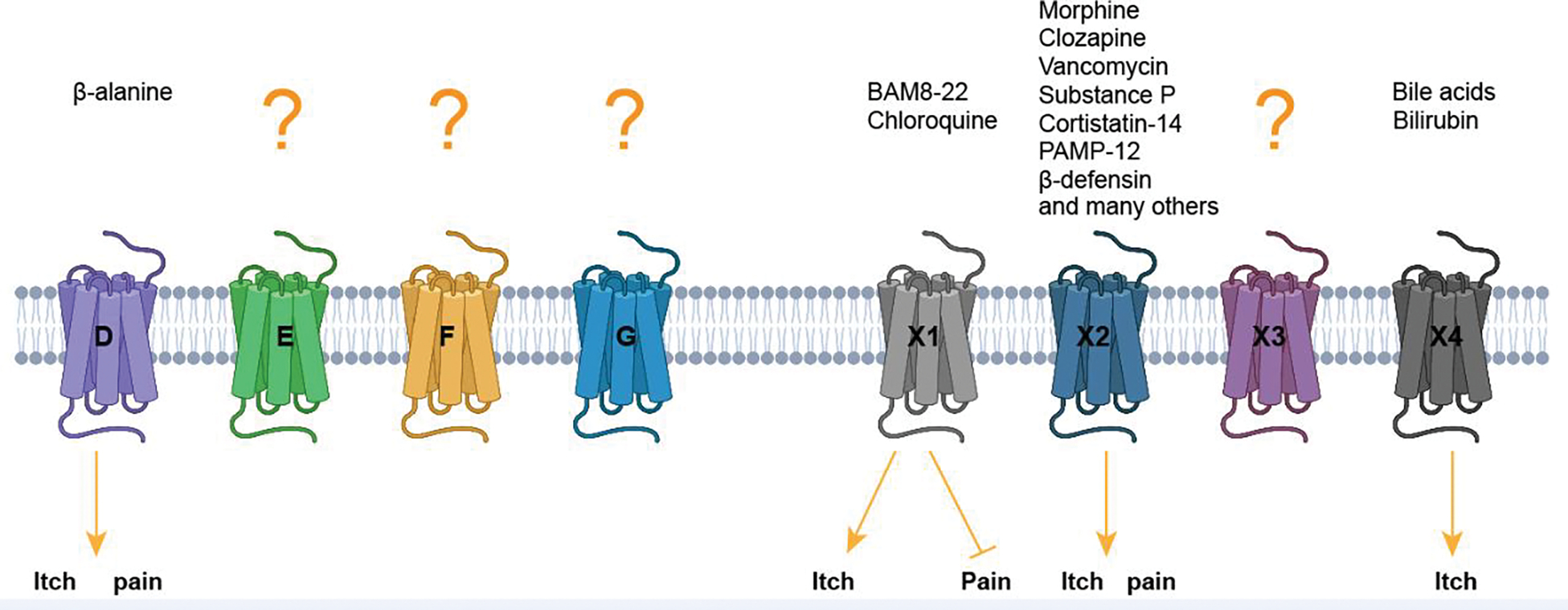
Key agonists that have been reported to elicit itch or pain signals are shown. Figure created with Biorender.com.
Highlights:
MRGPRs are primarily functioned as itch and pain receptor which display many other functions through neuroimmune communication.
MRGPRs are fast-evolving proteins with highly diverse amino acid sequences across different species and display high allele frequencies of the missense mutations in human.
MRGPRs have shallow ligand binding pockets with distinct residue composition, pocket size and charge distribution for the recognition of structurally distinct endogenous or exogenous agonists.
Technical advances in GPCR inactive structure determination may facilitate the pursuing of inactive MRGPR structures, which could be used as important templates for structure-based drug discovery of novel MRGPR antagonists.
Acknowledgement
This work was supported by NIH grants U24DA116195 to B.L.R. and by the Michael Hooker Distinguished Professorship to B.L.R.
Glossary:
- Basal activity
The ability of GPCRs to recruit and activate G protein without adding any agonist. It’s the intrinsic activity of the receptor
- Chloroquine
A medicine primarily used to treat malaria since 1940s, which could induce severe itch in some patients, especially black Africans
- Cholestatic itch
An extreme and unrelenting itching sensation caused by many liver diseases. Unlike the itch caused by histamine, cholestatic itch is multifactorial and difficult to treat. Several bile acids and bilirubin have been identified as pruritogens for cholestatic itch
- Substance P
A 11 amino acid neuropeptide secreted from the terminals of specific sensory neurons. The release of substance P is associated with inflammatory processes and pain. It has a high affinity to its endogenous receptor NK1R, whereas its affinity to MRGPRX2 is relatively low
- Mast cell degranulation
A process in mast cell that is characterized by the extrusion of granules containing many pro-inflammatory mediators, including histamine, interleukins, prostaglandins, cytokines and chemokines. The mast cell degranulation is involved in allergic reactions and can be triggered by both IgE-dependent and IgE-independent pathways
- Nateglinide
An FDA-approved drug that is used for the treatment of type 2 diabetes. Its main target is Kir6.2/SUR1 potassium channel of the pancreatic β-cell with off-target activity at MRGPRX4
- Orthosteric pocket
The agonist binding pocket that is essential for the direct activation of a GPCR. By contrast, allosteric pocket usually locates on a different side and doesn’t directly activate the receptor
- Positive allosteric modulator
Ligands that don’t have agonist activity but could strengthen the receptor activation caused by agonist. Positive allosteric modulators bind to the receptor at the allosteric site that is different from the orthosteric agonist
- Pseudoallergic reactions
IgE-independent reactions that produce the same or similar clinical symptoms as the true allergic reactions mediated by IgE antibody
Footnotes
Declaration of interests
B.L.R. and C.C. are listed as inventors on a patent application related to MRGPRX2 antagonists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- 1.Qu L et al. (2014) Enhanced excitability of MRGPRA3- and MRGPRD-positive nociceptors in a model of inflammatory itch and pain. Brain 137 (Pt 4), 1039–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klein A et al. (2021) Pruriception and neuronal coding in nociceptor subtypes in human and nonhuman primates. Elife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Young D et al. (1986) Isolation and characterization of a new cellular oncogene encoding a protein with multiple potential transmembrane domains. Cell 45 (5), 711–9. [DOI] [PubMed] [Google Scholar]
- 4.Dong X et al. (2001) A diverse family of GPCRs expressed in specific subsets of nociceptive sensory neurons. Cell 106 (5), 619–32. [DOI] [PubMed] [Google Scholar]
- 5.Zylka MJ et al. (2003) Atypical expansion in mice of the sensory neuron-specific Mrg G protein-coupled receptor family. Proc Natl Acad Sci U S A 100 (17), 10043–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meixiong J and Dong X (2017) Mas-Related G Protein-Coupled Receptors and the Biology of Itch Sensation. Annu Rev Genet 51, 103–121. [DOI] [PubMed] [Google Scholar]
- 7.Che T and Roth B, L. (2021) Structural Insights Accelerate the Discovery of Opioid Alternatives. Annu Rev Biochem 90. [DOI] [PubMed] [Google Scholar]
- 8.Berhane I et al. (2022) Thieno[2,3-d]pyrimidine-Based Positive Allosteric Modulators of Human Mas-Related G Protein-Coupled Receptor X1 (MRGPRX1). J Med Chem 65 (4), 3218–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green DP et al. (2019) A Mast-Cell-Specific Receptor Mediates Neurogenic Inflammation and Pain. Neuron 101 (3), 412–420 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meixiong J et al. (2019) MRGPRX4 is a G protein-coupled receptor activated by bile acids that may contribute to cholestatic pruritus. Proc Natl Acad Sci U S A 116 (21), 10525–10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu H et al. (2019) MRGPRX4 is a bile acid receptor for human cholestatic itch. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ogasawara H et al. (2019) Novel MRGPRX2 antagonists inhibit IgE-independent activation of human umbilical cord blood-derived mast cells. J Leukoc Biol 106 (5), 1069–1077. [DOI] [PubMed] [Google Scholar]
- 13.Chen E et al. (2021) Inflamed Ulcerative Colitis Regions Associated With MRGPRX2-Mediated Mast Cell Degranulation and Cell Activation Modules, Defining a New Therapeutic Target. Gastroenterology 160 (5), 1709–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang S et al. (2021) Nonpeptidergic neurons suppress mast cells via glutamate to maintain skin homeostasis. Cell 184 (8), 2151–2166 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steele HR and Han L (2021) The signaling pathway and polymorphisms of Mrgprs. Neurosci Lett 744, 135562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alkanfari I et al. (2018) Naturally Occurring Missense MRGPRX2 Variants Display Loss of Function Phenotype for Mast Cell Degranulation in Response to Substance P, Hemokinin-1, Human beta-Defensin-3, and Icatibant. J Immunol 201 (2), 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chompunud Na Ayudhya C et al. (2019) Identification of Gain and Loss of Function Missense Variants in MRGPRX2’s Transmembrane and Intracellular Domains for Mast Cell Activation by Substance P. Int J Mol Sci 20 (21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao C et al. (2021) Structure, function and pharmacology of human itch GPCRs. Nature 600 (7887), 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang F et al. (2021) Structure, function and pharmacology of human itch receptor complexes. Nature 600 (7887), 164–169. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y et al. (2022) Ligand recognition and allosteric modulation of the human MRGPRX1 receptor. Nat Chem Biol. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki S et al. (2022) Structural insight into the activation mechanism of MrgD with heterotrimeric Gi-protein revealed by cryo-EM. Commun Biol 5 (1), 707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collaborators U.S.B.o.D. et al. (2018) The State of US Health, 1990–2016: Burden of Diseases, Injuries, and Risk Factors Among US States. JAMA 319 (14), 1444–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kulkarni V et al. (2022) Global epidemiology of itch from 1990 to 2017: gender, age, sanitation, and air pollution as risk factors. Itch (Phila) (1), e60. [Google Scholar]
- 24.Basbaum AI et al. (2009) Cellular and molecular mechanisms of pain. Cell 139 (2), 267–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Braz J et al. (2014) Transmitting pain and itch messages: a contemporary view of the spinal cord circuits that generate gate control. Neuron 82 (3), 522–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Q et al. (2009) Sensory neuron-specific GPCR Mrgprs are itch receptors mediating chloroquine-induced pruritus. Cell 139 (7), 1353–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meixiong J et al. (2019) Activation of Mast-Cell-Expressed Mas-Related G-Protein-Coupled Receptors Drives Non-histaminergic Itch. Immunity 50 (5), 1163–1171 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Q et al. (2012) Mechanisms of itch evoked by beta-alanine. J Neurosci 32 (42), 14532–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meixiong J et al. (2019) Identification of a bilirubin receptor that may mediate a component of cholestatic itch. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McNeil BD et al. (2015) Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature 519 (7542), 237–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grimes J et al. (2019) MrgX2 is a promiscuous receptor for basic peptides causing mast cell pseudo-allergic and anaphylactoid reactions. Pharmacol Res Perspect 7 (6), e00547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Azimi E et al. (2017) Brief communication: MRGPRX2, atopic dermatitis and red man syndrome. Itch (Phila) 2 (1), e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wolf K et al. (2021) A group of cationic amphiphilic drugs activates MRGPRX2 and induces scratching behavior in mice. J Allergy Clin Immunol 148 (2), 506–522 e8. [DOI] [PubMed] [Google Scholar]
- 34.Lansu K et al. (2017) In silico design of novel probes for the atypical opioid receptor MRGPRX2. Nat Chem Biol 13 (5), 529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fujisawa D et al. (2014) Expression of Mas-related gene X2 on mast cells is upregulated in the skin of patients with severe chronic urticaria. J Allergy Clin Immunol 134 (3), 622–633 e9. [DOI] [PubMed] [Google Scholar]
- 36.Cao TBT et al. (2021) Elevated MRGPRX2 Levels Related to Disease Severity in Patients With Chronic Spontaneous Urticaria. Allergy Asthma Immunol Res 13 (3), 498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trier AM and Kim BS (2022) Structural insights into MRGPRX2: A new vision of itch and allergy. J Allergy Clin Immunol 149 (4), 1221–1222. [DOI] [PubMed] [Google Scholar]
- 38.Kolkhir P et al. (2022) Mast cells, cortistatin, and its receptor, MRGPRX2, are linked to the pathogenesis of chronic prurigo. J Allergy Clin Immunol 149 (6), 1998–2009 e5. [DOI] [PubMed] [Google Scholar]
- 39.Li Z et al. (2017) Targeting human Mas-related G protein-coupled receptor X1 to inhibit persistent pain. Proc Natl Acad Sci U S A 114 (10), E1996–E2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Subramanian H et al. (2011) Mas-related gene X2 (MrgX2) is a novel G protein-coupled receptor for the antimicrobial peptide LL-37 in human mast cells: resistance to receptor phosphorylation, desensitization, and internalization. J Biol Chem 286 (52), 44739–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Falzone ME and MacKinnon R (2022) Gβγ Activates PIP2 Hydrolysis by Recruiting and Orienting PLCβ on the Membrane Surface. bioRxiv, 2022.12.20.521270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olsen RHJ et al. (2020) TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat Chem Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ballesteros JA and Weinstein H (1995) Integrated Methods for the Construction of Three-Dimensional Models and Computational Probing of Structure-Function Relations in G Protein-Coupled Receptors. METHODS IN NEUROSCIENCES 25, 366. [Google Scholar]
- 44.Maeda S et al. (2019) Structures of the M1 and M2 muscarinic acetylcholine receptor/G-protein complexes. Science 364 (6440), 552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang S et al. (2022) Molecular basis for selective activation of DREADD-based chemogenetics. Nature 612 (7939), 354–362. [DOI] [PubMed] [Google Scholar]
- 46.Kim K et al. (2020) Structure of a Hallucinogen-Activated Gq-Coupled 5-HT(2A) Serotonin Receptor. Cell 182 (6), 1574–1588 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dussor G et al. (2008) Cutaneous sensory neurons expressing the Mrgprd receptor sense extracellular ATP and are putative nociceptors. J Neurophysiol 99 (4), 1581–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rau KK et al. (2009) Mrgprd enhances excitability in specific populations of cutaneous murine polymodal nociceptors. J Neurosci 29 (26), 8612–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang C et al. (2019) Facilitation of MrgprD by TRP-A1 promotes neuropathic pain. FASEB J 33 (1), 1360–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bader M et al. (2014) MAS and its related G protein-coupled receptors, Mrgprs. Pharmacol Rev 66 (4), 1080–105. [DOI] [PubMed] [Google Scholar]
- 51.Karczewski KJ et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581 (7809), 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kozlitina J et al. (2019) An African-specific haplotype in MRGPRX4 is associated with menthol cigarette smoking. PLoS Genet 15 (2), e1007916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wacker D et al. (2017) How Ligands Illuminate GPCR Molecular Pharmacology. Cell 170 (3), 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wacker D et al. (2017) Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell 168 (3), 377–389 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Katritch V et al. (2014) Allosteric sodium in class A GPCR signaling. Trends Biochem Sci 39 (5), 233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Massink A et al. (2015) Sodium ion binding pocket mutations and adenosine A2A receptor function. Mol Pharmacol 87 (2), 305–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Che T et al. (2018) Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor. Cell 172 (1–2), 55–67 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arora R et al. (2021) Constitutive, Basal, and beta-Alanine-Mediated Activation of the Human Mas-Related G Protein-Coupled Receptor D Induces Release of the Inflammatory Cytokine IL-6 and Is Dependent on NF-kappaB Signaling. Int J Mol Sci 22 (24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jumper J et al. (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596 (7873), 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu Y et al. (2017) LL-37-induced human mast cell activation through G protein-coupled receptor MrgX2. Int Immunopharmacol 49, 6–12. [DOI] [PubMed] [Google Scholar]
- 61.Ogasawara H and Noguchi M (2021) Therapeutic Potential of MRGPRX2 Inhibitors on Mast Cells. Cells 10 (11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.pharmaceuticals E (2021) MRGPRX4 ANTAGONIST PROGRAM. https://www.escientpharma.com/programs/mrgprx4/, (accessed).
- 63.Prchalova E et al. (2019) Discovery of Benzamidine- and 1-Aminoisoquinoline-Based Human MAS-Related G-Protein-Coupled Receptor X1 (MRGPRX1) Agonists. J Med Chem 62 (18), 8631–8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wen W et al. (2015) Discovery and characterization of 2-(cyclopropanesulfonamido)-N-(2-ethoxyphenyl)benzamide, ML382: a potent and selective positive allosteric modulator of MrgX1. ChemMedChem 10 (1), 57–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lyu J et al. (2019) Ultra-large library docking for discovering new chemotypes. Nature 566 (7743), 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Manglik A et al. (2016) Structure-based discovery of opioid analgesics with reduced side effects. Nature 537 (7619), 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sadybekov AA et al. (2022) Synthon-based ligand discovery in virtual libraries of over 11 billion compounds. Nature 601 (7893), 452–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fink EA et al. (2022) Structure-based discovery of nonopioid analgesics acting through the alpha2A-adrenergic receptor. Science 377 (6614), eabn7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kaplan AL et al. (2022) Bespoke library docking for 5-HT2A receptor agonists with antidepressant activity. Nature 610 (7932), 582–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mukherjee S et al. (2020) Synthetic antibodies against BRIL as universal fiducial marks for single-particle cryoEM structure determination of membrane proteins. Nat Commun 11 (1), 1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tsutsumi N et al. (2020) Structure of human Frizzled5 by fiducial-assisted cryo-EM supports a heterodimeric mechanism of canonical Wnt signaling. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xu J et al. (2022) Calcineurin-fusion facilitates Cryo-EM Structure Determination of a Family A GPCR. bioRxiv, 2022.03.27.485993. [Google Scholar]
- 73.Robertson MJ et al. (2022) Structure determination of inactive-state GPCRs with a universal nanobody. Nature Structural & Molecular Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Che T et al. (2020) Nanobody-enabled monitoring of kappa opioid receptor states. Nat Commun 11 (1), 1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang K et al. (2022) Fusion protein strategies for cryo-EM study of G protein-coupled receptors. Nat Commun 13 (1), 4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen H et al. (2022) Structures of oxysterol sensor EBI2/GPR183, a key regulator of the immune response. Structure 30 (7), 1016–1024 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dong C et al. (2021) Psychedelic-inspired drug discovery using an engineered biosensor. Cell 184 (10), 2779–2792 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Patriarchi T et al. (2018) Ultrafast neuronal imaging of dopamine dynamics with designed genetically encoded sensors. Science 360 (6396). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sun F et al. (2018) A Genetically Encoded Fluorescent Sensor Enables Rapid and Specific Detection of Dopamine in Flies, Fish, and Mice. Cell 174 (2), 481–496 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wan J et al. (2021) A genetically encoded sensor for measuring serotonin dynamics. Nat Neurosci 24 (5), 746–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pandy-Szekeres G et al. (2018) GPCRdb in 2018: adding GPCR structure models and ligands. Nucleic Acids Res 46 (D1), D440–D446. [DOI] [PMC free article] [PubMed] [Google Scholar]