

Abstract
Background:
Abnormal macrophage function caused by dysfunctional cystic fibrosis transmembrane conductance regulator (CFTR) is a critical contributor to chronic airway infections and inflammation in people with cystic fibrosis (PWCF). Elexacaftor/tezacaftor/ivacaftor (ETI) is a new CFTR modulator therapy for PWCF. Host-pathogen and clinical responses to CFTR modulators are poorly described. We sought to determine how ETI impacts macrophage CFTR function, resulting effector functions, and relationships to clinical outcome changes.
Methods:
Clinical information and/or biospecimens were obtained at ETI initiation and 3-, 6-, 9, and 12-months post-ETI in 56 PWCF and compared to non-CF controls. Peripheral blood monocyte-derived macrophages (MDMs) were isolated and functional assays performed.
Results:
ETI treatment was associated with increased CF MDM CFTR expression, function, and localization to the plasma membrane. CF MDM phagocytosis, intracellular killing of CF pathogens, and efferocytosis of apoptotic neutrophils was partially restored by ETI, but inflammatory cytokine production remained unchanged. Clinical outcomes including increased FEV1 (+10%) and BMI (+1.0) showed fluctuations over time and were highly individualized. Significant correlations between post-ETI MDM CFTR function and sweat chloride levels were observed. However, MDM CFTR function correlated with clinical outcomes better than sweat chloride.
Conclusions:
ETI is associated with unique changes in innate immune function and clinical outcomes.
Keywords: cystic fibrosis, macrophage biology, innate immunity
INTRODUCTION
The discovery and clinical implementation of cystic fibrosis transmembrane conductance regulator (CFTR) modulator therapy has revolutionized care regimens and disease trajectories for people with cystic fibrosis (PWCF).[1] Despite their widespread clinical use in some developed countries and association with improved clinical outcomes, our understanding of how CFTR modulators alter host-pathogen responses in CF remains limited. There is a unique opportunity to study immunologic responses to CFTR modulator initiation with the recent introduction of the highly effective triple combination CFTR modulator therapy elexacaftor/tezacaftor/ivacaftor (ETI) for PWCF with at least one copy of the CFTR variant F508del. These immunologic responses are important to our understanding of CF pathogenesis as prior studies showed that devastating chronic bacterial infections can persist despite treatment with CFTR modulators.[2–5]
We and others previously demonstrated that macrophage dysfunction is related to both the acquisition of acute and persistence of chronic infections in PWCF,[6–13] and it is dependent on appropriate CFTR function.[14] Past studies of CFTR modulator effects on CF macrophages have demonstrated variable results with overall limited efficacy.[3, 4, 15] Therefore, we sought to determine how ETI initiation impacts macrophage CFTR expression and function, resulting effector functions, and their relevance to changes in longitudinal clinical outcomes. We hypothesized that ETI treatment would be associated with sustained improvements in CFTR restoration in CF macrophages and subsequently improved phagocytic function. Further, we hypothesized that robust changes in CF macrophage CFTR function after ETI treatment would correlate with improved clinical outcomes.
Our results demonstrated distinct changes in CF macrophage CFTR expression and improved CFTR function in response to ETI treatment. Alterations in macrophage CFTR were associated with improved phagocytic capacity but did not fully restore bacterial killing capabilities. Overall, changes observed were highly individualized and correlated with clinical outcomes including lung function and nutritional recovery.
MATERIALS AND METHODS
Clinical data:
Clinical and demographic information was securely recorded in a REDCap database. Clinical information recording and/or biospecimen sampling occurred at ETI initiation, 3-, 6-, 9-, and 12-months post-ETI initiation, except sweat chloride levels measured by the clinical laboratory at baseline and 1-month post-ETI initiation. Lung function was measured as percent predicted FEV1. BMI was measured as a nutritional marker. Clinical microbiologic cultures from oropharyngeal or sputum samples were recorded as surrogates of chronic infection. Hospitalizations for pulmonary exacerbations (determined by treatment team) and the number of total antibiotic courses (outpatient and inpatient) was recoded for one year prior and one-year post-ETI initiation. Due to the COVID-19 pandemic and telehealth visits, not all clinical measures were available at each visit.
Macrophage assays:
MDM isolation, cellular toxicity, phagocytosis, bacterial killing, efferocytosis, cytokine and ROS production were performed per prior methods[4, 8, 14] and as described by Kuhns.[16] CFTR function was assessed via the MQAE halide efflux assay.[14] CFTR expression was analyzed via flow cytometry, fluorescent microscopy, and western blot with subcellular protein fractionation. Additional details on all macrophage assays and RhoA/Cdc42 methods are provided in an online data supplement.
Electrophysiology recording (patch-clamp):
MDMs were seeded on glass coverslips and treated overnight (ex vivo) with ETI (5 μM elexacaftor, 5 μM ivacaftor and 5 μM tezacaftor). Whole-cell currents were elicited by 400 msec voltage steps from −80 to +80mV in 10mV steps from a holding potential of −40mV. Additional details are provided in an online data supplement.
Ion channel array:
Total RNA was extracted from MDMs with or without ETI treatment in vitro using an RNA purification kit (Norgen Bioteck, 43400) according to the manufacturer’s instruction. Additional details are provided in an online data supplement.
SEM and TEM:
Images were obtained on a Hitachi S-4800 field-emission scanning electron microscope (Hitachi High Technologies, Schaumburg). Additional details are provided in an online data supplement.
Statistical analysis:
For macrophage assays, two sample unpaired t-tests or Fisher’s exact tests were used for comparisons between PWCF and non-CF controls, and paired t-tests were used for pre- vs post-ETI comparisons and changes in sweat chloride within the PWCF group. One-way ANOVA with post-hoc Tukey correction was used for multiple comparisons as detailed in figure legends. Depending on the distribution, either Pearson’s or Spearman’s correlation coefficients were used to assess correlations between MDM CFTR function and post-ETI sweat chloride, and between MDM CFTR function and changes in clinical outcomes between 0–3 months. A linear mixed-effect model was fit for FEV1 and BMI collected over time. Fixed effects included discrete visits and random intercepts were specified for participants. Post-hoc comparisons between neighboring visits (baseline vs 3 mo, 3 vs 6 mo, 6 vs 9 m, 0 vs 12 mo) as well as baseline vs 12 mo were conducted with multiple comparison adjustment using the Sidak method. Multiple-comparison adjusted p-values were reported following the linear mixed-effect models. Statistical analyses were completed with GraphPad Prism software (version 8.2).
Study approval:
Study participants were recruited as approved by the Institutional Review Board of Nationwide Children’s Hospital (IRB16–01020). Written informed consent and/or assent was received prior to participation. Additional details on consent and enrollment criteria are provided in an online data supplement.
RESULTS
Demographics
The demographics of the enrolled PWCF at baseline and non-CF controls are shown in Table 1. PWCF were slightly younger than the non-CF participants. Among PWCF, there was an approximately equal distribution of those homozygous or heterozygous for the F508del variant. Fifty percent were on a CFTR modulator for at least six months prior to ETI initiation. Baseline percent predicted forced expiratory volume in 1 second (FEV1, 65.5 ± 25.3) and body mass index (BMI, 22.6 ± 4.7) are shown as markers of lung function and nutrition respectively, along with hospitalizations and oral antibiotic courses in the year prior to ETI initiation. Overall, the participants had moderate lung disease, but ranged from mild to severe obstructive lung disease as defined by FEV1.
Table 1:
Participant demographics
| CF (n=56) | Non-CF (n=92) | P value | |
|---|---|---|---|
| Age (years) | 28.6 ± 13.4 | 37.0 ± 9.8 | 0.0001 |
| Female | 41.1% | 51.1% | 0.24 |
| Caucasian | 94.6% | 91.3% | 0.46 |
| CFTR genotype | |||
| Homozygous F508del (n=26) | 46.4% | --- | |
| Heterozygous F508del (n=29) | 51.2% | --- | |
| Baseline BMI (mean) | 22.6 ± 4.7 | --- | |
| Prior CFTR modulator | 50.0% | ||
| Baseline FEV1 (% predicted) | 65.5 ± 25.3 | --- | |
| Hospitalizations 1 year prior to ETI initiation | 0.6 ± 1.4 | --- | |
| Oral antibiotic courses 1 year prior to ETI initiation | 2.4 ± 2.3 | --- |
P values determined by unpaired t-test for continuous variables or Fisher’s Exact test for categorical variables.
ETI treatment improves MDM CFTR expression and localization
We used primary human monocyte-derived macrophages (MDMs) to model CF macrophage interactions as monocytes are recruited to systemic sites throughout the body, including the lungs and sinuses, where they differentiate into macrophages. MDMs are also at a higher density in the CF lung compared to alveolar macrophages,[17] lending relevance to their use as models of CF immunity. In the lungs, recruited MDMs regulate both acute and chronic responses to infection, inflammation, and other stimuli. Unless otherwise noted, MDMs were grouped according to non-CF, CF pre-ETI initiation (untreated), or CF post-ETI initiation (treated- receiving in vivo plus ex vivo supplementation during culture). Prior studies have established dosing of CFTR modulators for immune cell culture use from 1–5μM, akin to in vivo exposures.[3, 4, 15] To assess MDM tolerance of elexacaftor, we examined escalating doses from 1 to 40μM. MDMs were treated with elexacaftor ex vivo and apoptosis measured via flow cytometric detection of Annexin V+ cells. Increased apoptosis was noted with increasing doses of elexacaftor above 5μM, with significantly increased apoptosis at 40μM (Fig. 1A). We then measured MDM apoptosis in response to increasing concentrations of elexacaftor in combination with fixed dosing of tezacaftor and ivacaftor (5μM). A similar trend was noted, with significantly increased MDM apoptosis at elexacaftor concentrations greater than 30μM in combination with tezacaftor/ivacaftor (Fig. 1B). We therefore chose to use 5μM elexacaftor for all further studies in combination with tezacaftor and ivacaftor.
Figure 1: ETI treatment is associated with increased MDM CFTR expression.
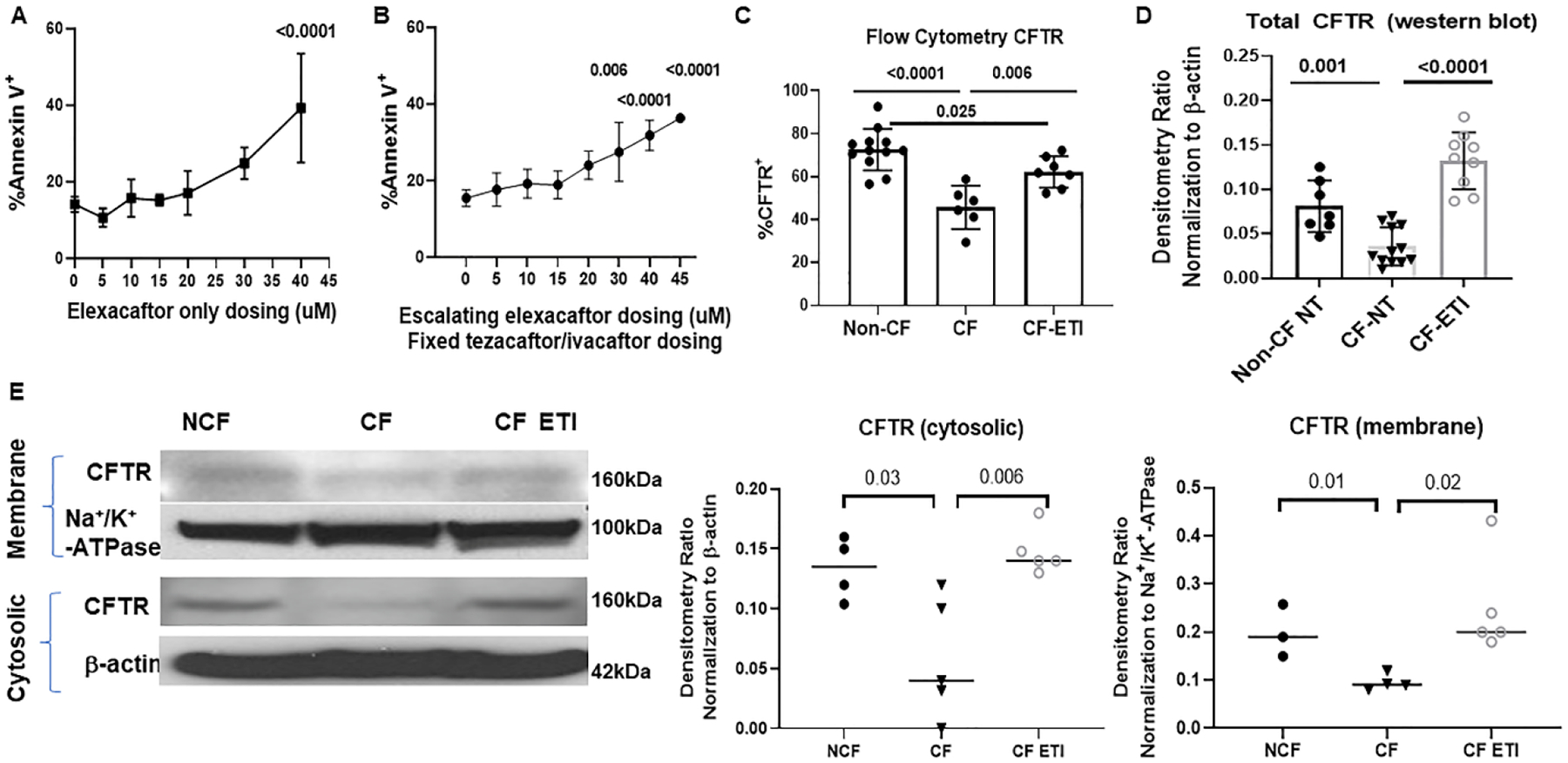
A) Flow cytometry measurements of cellular apoptosis by detection of % Annexin V positive CF MDMs in response to escalating doses of elexacaftor (0–40 μM). 40μM elexacaftor significantly increased MDM apoptosis, p <0.0001 via one-way ANOVA with Tukey’s test, n= 4–6. B) Flow cytometry % Annexin V positive CF MDMs in response to escalating doses of elexacaftor combined with fixed tezacaftor and ivacaftor dosing (5μM). Elexacaftor doses above 30μM were associated with significantly increased MDM apoptosis, p = 0.006 and <0.0001 via one-way ANOVA with Tukey’s test, n=4–7. C) Quantitation of CFTR expression via flow cytometry detection of %CFTR+ fluorescence in non-CF (n=12), CF (n=6), and CF-ETI (n=7) MDMs, detected via UNC-596 or Alamone ACL-006 antibody. P values for individual comparisons are shown, one-way ANOVA. Negative and single-color controls not shown. D) Densitometric ratios of western blot analysis of CFTR (UNC-596 antibody) Band C expression in non-CF (n=7), CF (n = 12), and CF-ETI (n=9) MDMs. Densitometry ratios normalized to the loading control β-actin. P values for individual comparisons are shown, one-way ANOVA, non-CF vs CF-ETI non-significant. E) Subcellular fractionation western blot of membrane and cytosolic fractions of CFTR in non-CF, CF, and CF-ETI MDMs. Na+/K+-ATPase was used as a membrane control, and β-actin as a cytosolic control. Representative image is shown with corresponding densitometry for cytosolic and membrane fractions from all replicates, unpaired t-tests.
We then measured CFTR expression in non-CF, untreated CF, and CF MDMs treated with ETI. CF MDMs had significantly less CFTR expression compared to non-CF MDMs when measured by a quantitative flow cytometry assay (45.7 vs 74.1%, p < 0.0001) (Fig. 1C). CF MDMs treated with ETI demonstrated a significant increase in CFTR compared to untreated CF MDMs (62.1 vs 45.7, p = 0.006), but remained below the level of expression of non-CF MDMs (62.1vs 74.1, p = 0.025) (Fig. 1C, representative flow gates in supplemental Fig. 1A). We then examined CFTR expression via western blot for total protein and subcellular protein fractionation for measurements of membrane and cytosolic concentrations of CFTR. Western blot also showed a significant increase in total CFTR expression in ETI-treated CF MDMs (Densitometry in Fig. 1D). Specifically, ETI treatment was associated with both increased cytosolic and membrane CFTR expression compared to untreated CF MDMs (Fig. 1E, all uncropped blots in supplemental Fig. 1B). Control CF MDMs with a class 1 CFTR variant did not show an increase in CFTR expression in response to ETI (supplemental Fig. 1C).
Next, we used fluorescent microscopy to determine co-localization of CFTR with surface (WGA) and trafficking/degradation markers (RAB7, EEA1, LAMP1) at rest and during LPS exposure. We found decreased expression of CFTR (green) at the plasma membrane (white) in CF MDMs compared to non-CF, with a significant increase in surface-associated CFTR in CF MDMs post-ETI with or without LPS exposure (Figs. 2A–C). CFTR was most commonly co-localized with lysosomal (LAMP1) or late endosomal markers (Rab7) in both non-CF and CF MDMs post-ETI compared to untreated CF MDMs (Figs. 2A–C). A summary of quantitative scoring of co-localization of CFTR with other fluorescent markers is displayed in Figure 2D.
Figure 2: ETI treatment is associated with altered CFTR localization.
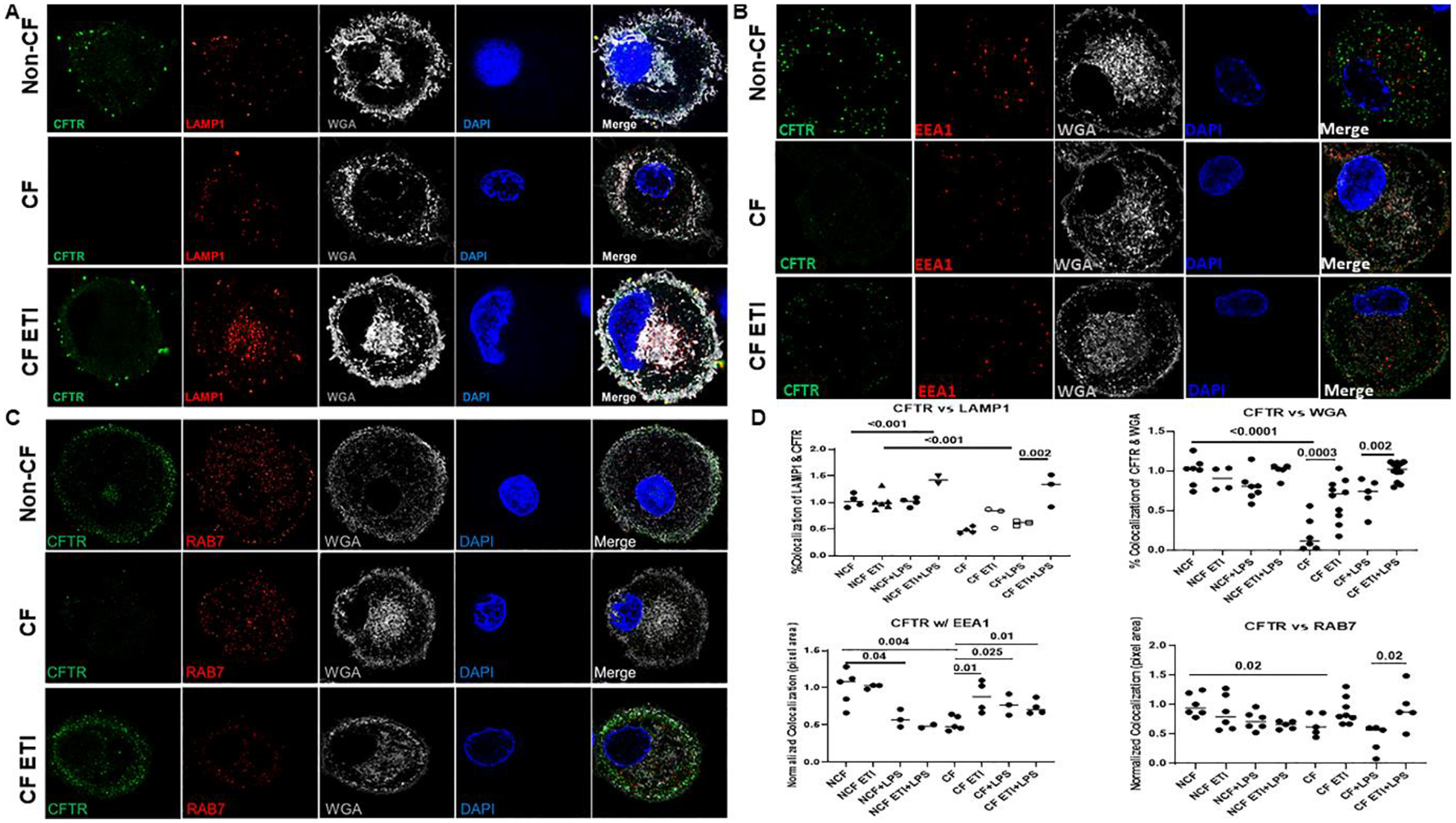
Confocal microscopy of non-CF, CF, and CF post-ETI MDMs after 16h LPS stimulation. Cells were permeabilized for detection of co-localization of CFTR (green) with A) the lysosomal marker LAMP1 (red), B) early endosomal marker EEA1 (red), and C) late endosomal marker RAB7 (red) are shown. For all comparisons the plasma membrane marker WGA (white), MDM nucleus marked with DAPI (blue), and merged images are also shown. Representative images are shown, n=3–12 unique individuals. D) Quantitative scoring of CFTR co-localization with specific markers from parts A-C was conducted using image J of random images. P values for individual comparisons are shown, one-way ANOVA.
ETI treatment alters non-CFTR ion channel expression
To specifically examine if ETI treatment is associated with changes in other macrophage ion channels besides CFTR, we performed a custom microarray analysis of ion channel expression in CF MDMs pre- and post-ETI. Untreated CF MDMs demonstrated altered expression of several sodium (SCN1A, SCN1B, SCN8A, SCN11A), potassium (KCNA3, KCNB1, KCNC1, KCNE1) and chloride (CLCNKA) channels compared to non-CF MDMs (Fig. 3A, significant values in red). However, there were not uniform directional changes in expression within each channel type (e.g., both up- and down-regulated expression was present). Post-ETI, prior differences were no longer significant except KCNB1 and KCNE1 (Fig. 3B, significant values in red). SCN4A, SCN10A, CACNA1A, TRPC1, ITPR3, and CHRND showed compensatory changes in CF MDMs post-ETI compared to non-CF MDMs (Fig. 3B). Comparing CF MDMs pre- and post-ETI, CLCN2, CACNG2, and TRPC1 were significantly increased in expression post-ETI (Supplemental Fig. 2A). Individual paired changes in ion channel expression from PWCF are shown in Figure 3C with groupings by ion channel type.
Figure 3: Taqman custom microarray ion channel relative expression ratios.
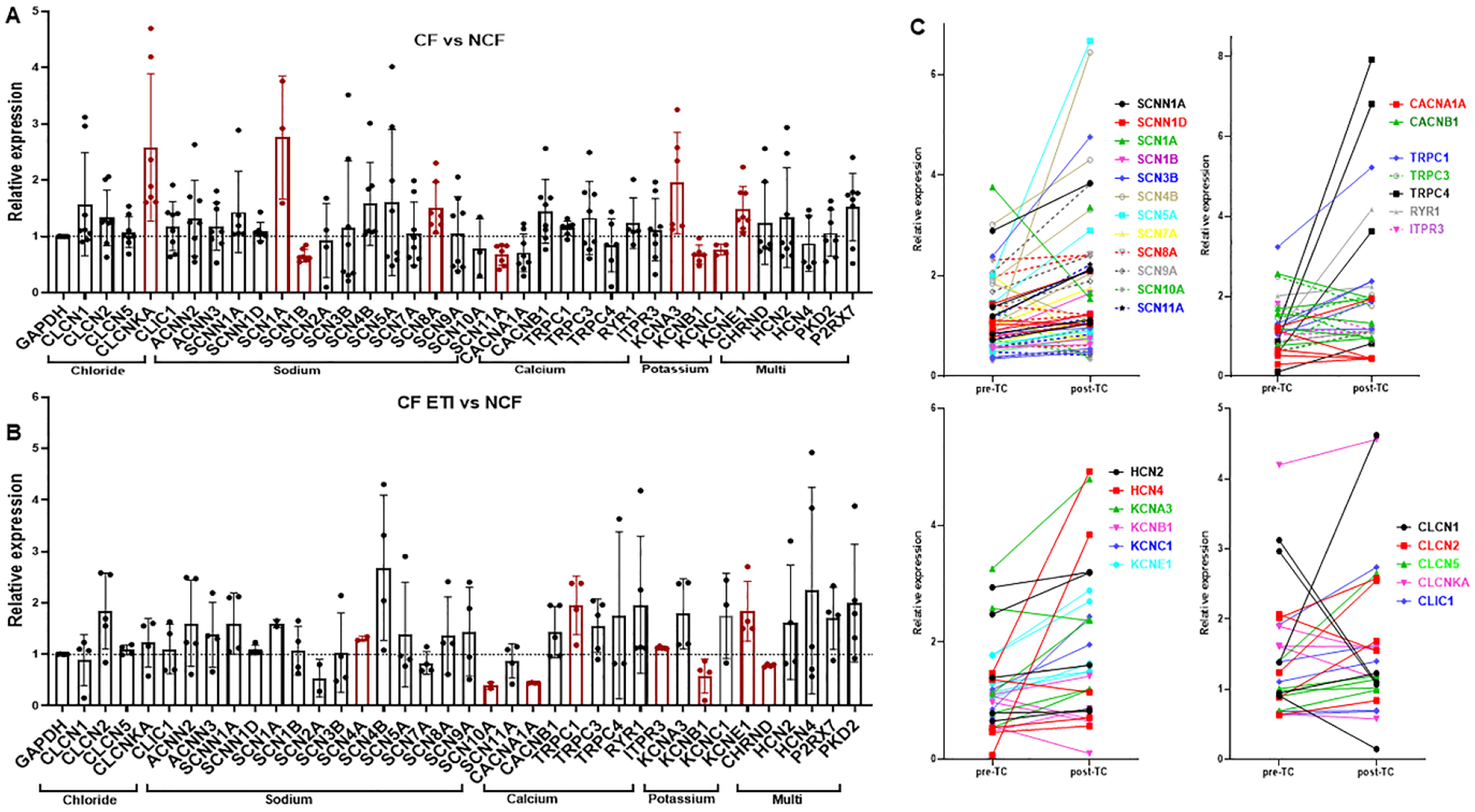
for A) CF/Non-CF MDMs at baseline and B) CF MDMs treated with ETI compared to non-CF. All values were normalized to GAPDH, n=5=8 per group, mean ± SD shown. Some channels were not expressed in all patient samples. Statistically significant comparisons via Brown-Forsythe and Welch ANOVA are displayed in red color. C) Paired changes in relative expression ratios from A & B (PWCF only) pre- and post-ETI with groupings of channels by ion type. Color coding represents individuals for each specific channel.
MDM CFTR functional responses vary by individual
Next, we examined changes in MDM CFTR channel function in response to ETI. We utilized the MQAE assay to determine quantitative changes in CFTR-dependent halide efflux in response to ETI. First, we determined the impact of a one-week washout on CFTR function. Paired monocytes from PWCF taking ETI clinically for 3–6 months were either treated (ex vivo group) or not treated (washout) with ETI daily during differentiation to MDMs. A significant increase in CFTR function was seen for each group of MDMs treated with ETI compared to its matched untreated counterparts from the same donor (Fig. 4A). These results indicate that daily treatment with ETI is necessary to maintain CFTR functional restoration in ex vivo cell culture. We then compared monocyte and MDM CFTR functional responses to ETI. Freshly isolated monocytes from PWCF 3-months post EIT who had received a morning dose of ETI were analyzed without further ETI treatment ex vivo. CF monocytes from PWCF prior to ETI initiation demonstrated negligible CFTR function and had no significant increases at 3 months post-ETI treatment (Fig. 4B). Paired individual donor monocyte responses were quite variable pre- and after 3 months of ETI treatment, with only 1 individual (male, F508del/Y1092X) demonstrating a significant response to ETI (Fig. 4C). Further, one-time ex-vivo treatment with ETI immediately after monocyte isolation did not significantly improve CFTR function compared to monocytes treated in vivo only (Fig. 4D). In contrast, CF MDMs treated with ETI during culture differentiation demonstrated a significant increase in CFTR function compared to untreated CF MDMs, but also remained with significantly less CFTR function compared to non-CF MDMs (Fig. 4E). Individual paired donor MDM responses were more consistently increased post-ETI, but again were variable from donor to donor (Fig. 4F, 2 largest increases were female, F508del/F508del and male, F508del/F508del). Overall, there were no discernible characteristics (gender, age, genotype) reflecting sample variability. A representative kinetics tracing of the halide efflux assay for untreated non-CF and CF MDMs and non-CF and CF MDMs treated with ETI is shown in Figure 4G. We confirmed these data with whole-cell patch-clamp measurements of CFTR currents. We found that stimulated CFTR function measured by patch-clamp was minimal in untreated CF MDMs (red) and CF MDMs from PWCF who had received ETI clinically for 3 months but no further supplementation during MDM differentiation (CF ETI in vivo -orange, Fig 4H). In contrast, CF MDMs treated with ETI ex vivo (purple) and non-CF MDMs (black) demonstrated robust CFTR currents in response to a forskolin cocktail stimulation (Fig. 4H). Representative whole cell patch-clamp basal and forskolin-stimulated CFTR current tracings are shown in Supplemental Fig. 2B.
Figure 4: Variable changes in human monocyte and MDM CFTR functional responses toETI treatment.
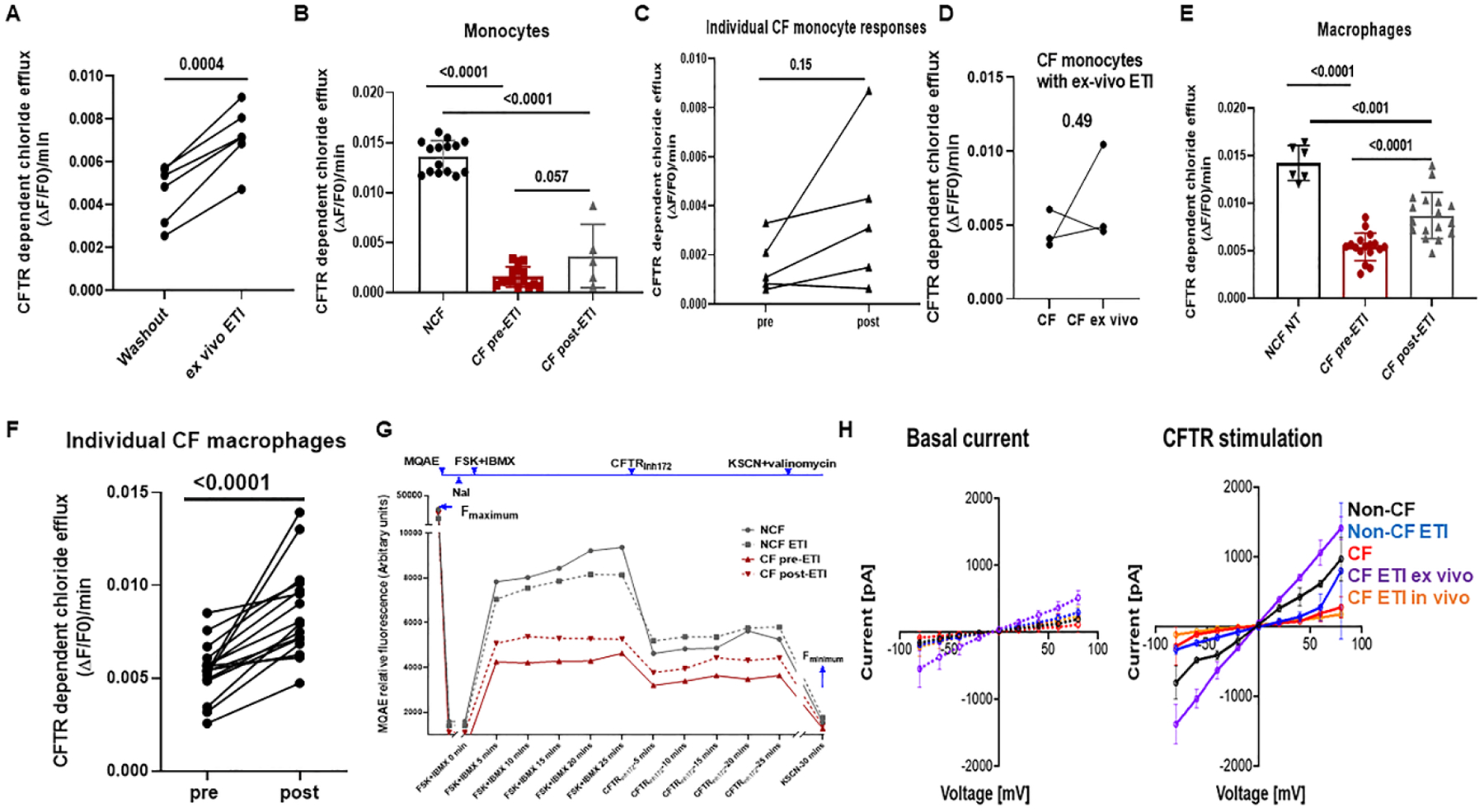
A) CFTR function as measured by maximum forskolin-stimulated halide efflux assay of CFTR-dependent chloride efflux (MQAE assay). CF monocytes were obtained from PWCF after 3 months of ETI treatment. Monocytes were differentiated into MDMs, with one experimental group receiving no drug during culture (washout) and one group receiving daily ETI ex-vivo during differentiation. A significant increase (p = 0.0004) in CFTR function was seen for MDMs receiving continued ex-vivo ETI treatment, paired t-test, n=6. B) Maximum forskolin-stimulated halide efflux (15 minutes) in non-CF, CF pre-ETI, and CF post-ETI (3 mos) monocytes. Measured as ΔF/F0/minute whereby ΔF is forskolin stimulated current – CFTRinh current, and F0 is Fmax minus Fmin. N= 5–20, p values for individual comparisons shown, one-way ANOVA, mean ±SD. C) Individual paired monocyte responses from 4B, n=5, p = 0.15, paired t-test. D) Paired monocyte halide efflux responses for monocytes exposed to ETI in vivo without culture supplementation (CF) or one-time ex-vivo ETI treatment (CF ex vivo), n=3, paired t-test. E) Max forskolin-stimulated halide efflux (15 minutes) in human non-CF, CF pre-ETI, and CF MDMs with ex vivo ETI daily during differentiation (post-ETI). N=6–20, p values for individual comparisons are shown, one-way ANOVA, mean ±SD. F) Individual paired MDM responses from 4D, n=16, p <0.0001, paired t-test. G) Representative CFTR function time course shown with annotations for MQAE dye, NaI inhibition, forskolin plus IBMX stimulation (FSK + IBMX), CFTR inhibition (CFTRinh172), and quenching with KSCN plus valinomycin. Colored lines and symbols are shown to help differentiate treatment groups. H) Non-CF and CF peripheral blood monocytes were isolated and differentiated into macrophages (MDMs). MDMs were divided into groups based on the absence (non-CF-black, CF-red) or presence of ex-vivo ETI (non-CF ETI ex vivo-blue, CF ETI ex-vivo-purple, 5 μM all ETI components for 24h prior to patch-clamp) as well as CF MDMs from PWCF who had received 3 mos of ETI clinically but no ETI added back during culture (CF ETI in vivo-orange). Whole-cell currents were elicited by 400 msec voltage steps from −80 to +80mV in 10mV steps from a holding potential of −40mV. Displayed are average current/voltage (I/V) relationships for basal and CFTR stimulated (15μM forskolin, 100μM IBMX and 2mM ATP) currents in MDMs. N=3–8 donors, data are expressed as mean ± SD.
ETI treatment improves phagocyte-mediated killing of bacteria
To determine the consequences of the observed improvements in CFTR expression and function with ETI treatment, we next measured MDM-mediated killing of clinical isolates of the CF pathogens B. cenocepacia, P. aeruginosa, and methicillin-resistant Staphylococcus aureus (MRSA). Untreated CF MDMs had increased B. cenocepacia burden compared to non-CF, similar to past studies (Fig. 5A).[4, 14] Treatment with ETI was associated with a significant decrease in B. cenocepacia burden in CF MDMs (Fig. 5A). Untreated CF MDMs also had increased P. aeruginosa and MRSA bacterial loads compared to non-CF (Fig. 5A). ETI treatment was associated with a significantly decreased P. aeruginosa and MRSA load in CF MDMs (Fig. 5A). ETI treatment did not impact bacterial load for any pathogen in non-CF MDMs (Figs. 5A). One of the proposed mechanisms for failed CF MDM clearance of bacteria is poor phagocytosis.[3, 4, 11, 14, 18–20] We measured phagocytosis of an RFP-expressing B. cenocepacia clinical isolate to see if bacterial phagocytosis was similarly impacted by ETI treatment. Surprisingly, both CF and non-CF MDMs showed increased phagocytosis of B. cenocepacia after ETI treatment (Fig. 5B). However, CF phagocytosis post-ETI remained below baseline non-CF phagocytosis (55.1% vs 74.7%, Fig. 5B). CF phagocytosis rates were also variable amongst individuals (Fig. 5B), similar to the CFTR function data. The addition of CF airway supernatants (ASN) to ETI treated cells reduced ETI-associated rescue of phagocytosis for both CF and non-CF MDMs (Fig. 5B). Representative flow cytometry gating strategy, histogram, and light microscopy are shown in Figure 5C. We then further examined MDM interactions with bacteria using scanning electron microscopy (SEM). We found distinct morphologic changes in CF MDMs compared to non-CF. CF MDMs lacked membrane protrusions and rearrangements necessary for phagocytosis of bacteria compared to non-CF (Fig. 5D). ETI treatment was associated with increased CF macrophage membrane protrusions and contact with bacteria, although overall cytoskeletal morphology was disorganized and morphologically distinct compared to non-CF (Fig. 5D).
Figure 5: ETI improves bacterial phagocytosis and intracellular killing.
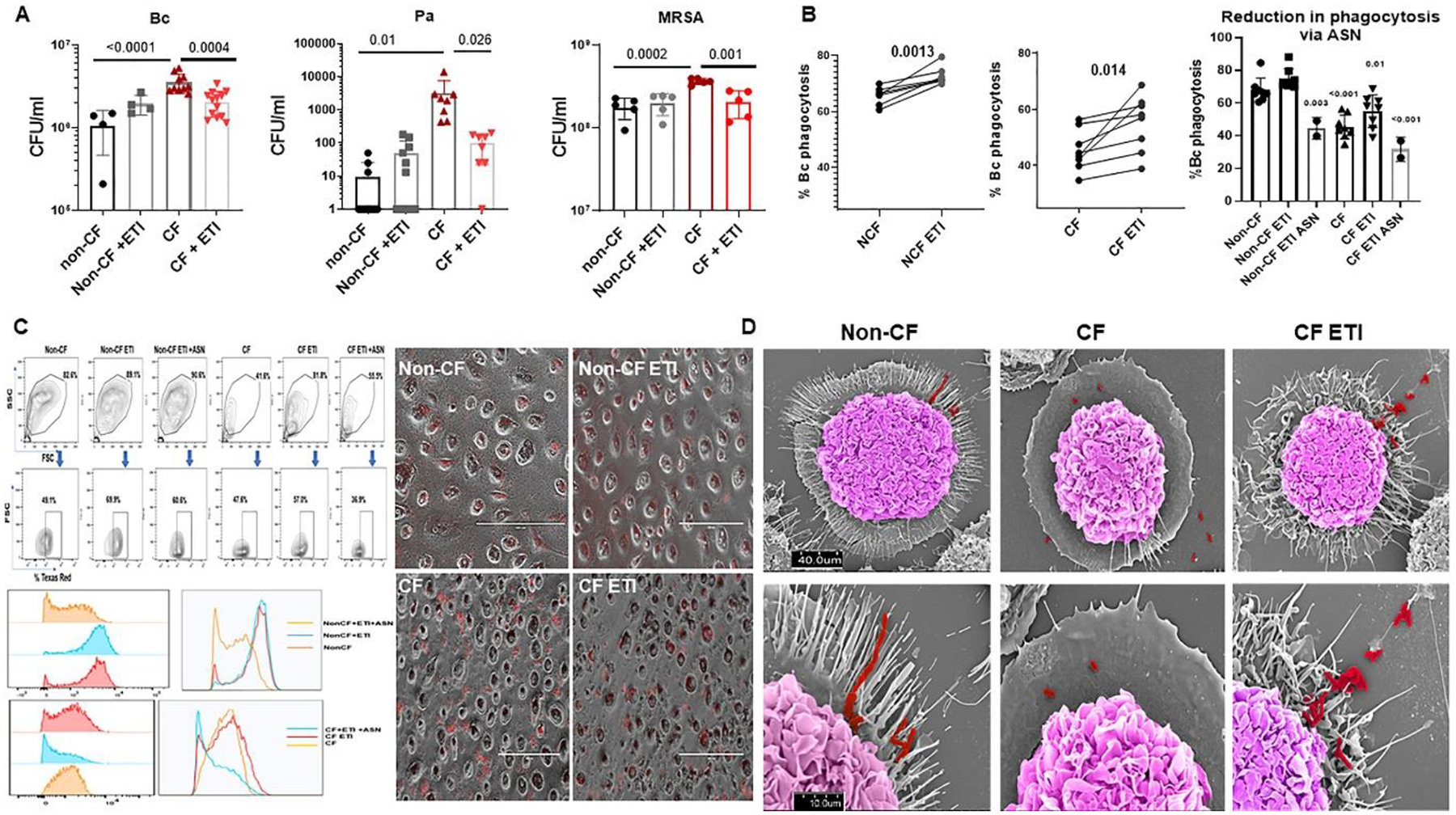
A) Colony-forming unit (CFU) assay for non-CF and CF MDMs ± ETI treatment during overnight infection with B. cenocepacia k56–2 isolate (Bc) or CF clinical isolates of P. aeruginosa and MRSA. Log scale, n=4–13 donors, mean ± SD. P values for individual comparisons shown, via one-way ANOVA. B) Paired and summed % phagocytosis of RFP-expressing B. cenocepacia in non-CF and CF MDMs ± ETI or ETI plus airway supernatant (ASN) treatment. n = 2–8/group, MOI 50, mean ±SD, p values via paired t-test or one-way ANOVA. C) Representative flow cytometry gating strategy, histogram, and light microscopy for bacterial phagocytosis from 5B. FACS gating on forward scatter (FSC), side scatter (SSC), and detection of Bc expressing Texas Red. D) SEM images of non-CF, CF, and CF ETI MDMs during infection with Bc. Non-CF ETI not shown. Scale bars displayed, top images with magnification 5.0kV 8.00mm × 1.3k SE (U) and bottom images with 5.0kV 8mm x4.00k SE(U). Bacteria are pseudocolored red and macrophages pseudocolored purple.
ETI treatment enhances MDM efferocytosis of apoptotic neutrophils
The CF airway is characterized by heavy neutrophilic infiltration and large amounts of apoptotic debris. In addition to phagocytosis and killing of bacteria, macrophages are responsible for the clearance of airway debris such as apoptotic cells through efferocytosis to avoid excess inflammation. To determine the impact of ETI on MDM efferocytosis of neutrophils, we co-cultured MDMs with apoptotic CF and non-CF neutrophils. Untreated CF MDMs had significantly less efferocytosis of neutrophils compared to non-CF MDMs (Fig. 6A–C). ETI treatment was associated with a significant increase in CF MDM efferocytosis of neutrophils, with variation in improvement on an individual basis (Fig. 6A–C, fluorescent label and apoptosis confirmation in supplemental figure 3). Non-CF MDMs treated with a CFTR inhibitor also demonstrated a significant decreased in efferocytosis, with partial rescue by ETI treatment (Fig. 6A–B). We confirmed the CF findings using transmission electron microscopy (TEM). TEM images showed increased efferocytosis in CF MDMs post-ETI (Fig. 6D with quantitative scoring). Because of the differences in phagocytosis and efferocytosis between CF and non-CF MDMs that persisted with ETI treatment and the ultrastructural characteristics identified by SEM imaging in Figure 5E, we examined 3 critical genes that encode for G proteins from the Rho family (RhoA, Rac1, Cdc42), a family of small GTPases regulating cytoskeletal dynamics and cellular motility. We found decreased relative expression of RhoA and Rac1 in CF MDMs at baseline compared to non-CF MDMs (Supplemental Fig. 4A). Further, Cdc42 was decreased in expression during infection in CF MDMs compared to non-CF (Supplemental Fig. 4A). All 3 genes had increased relative expression at baseline and during infection in CF MDMs treated with ETI (Supplemental Fig. 4A). In particular, RhoA and Rac1 demonstrated 2–3 times higher expression in some individuals with CF treated with ETI compared to non-CF (Supplemental Fig. 4A). We expanded upon the gene expression data via flow cytometry with Cdc42 and RhoA activation assays in response to an N-Formylmethionyl-leucyl-phenylalanine (fMLP) stimulus. We found decreased Cdc42 activity in CF MDMs at baseline and during fMLP stimulation compared to non-CF (Supplemental Fig. 4B). ETI treatment improved Cdc42 activation during fMLP stimulation, but the level remained about half of non-CF MDMs (Supplemental Fig. 4B). In contrast, RhoA activity was only decreased in CF MDMs in response to fMLP but approached non-CF levels after ETI treatment (Supplemental Fig. 4C). Combined with our microscopy data these results suggest that significant changes in macrophage cytoskeletal elements occur after ETI treatment.
Figure 6: ETI improves MDM efferocytosis.
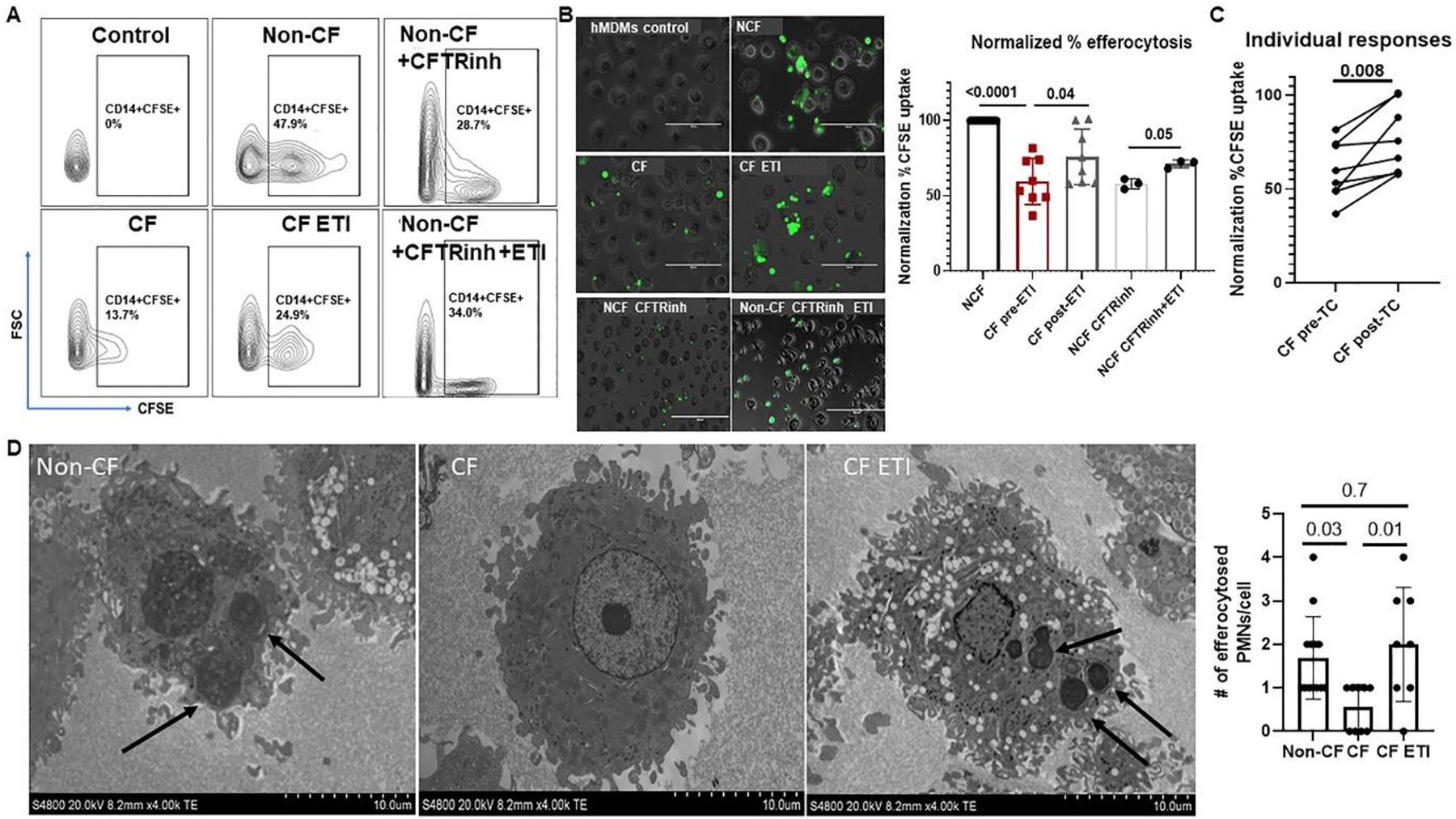
A) Gating strategy for flow cytometry-based detection of MDM efferocytosis of apoptotic neutrophils based on FSC and detection of CD14+ cells with a positive carboxyfluorescein succinimidyl ester (CFSE) signal. CFSE-labeled neutrophils underwent sterile, age-induced apoptosis for 24 hours prior to analysis. B) Representative fluorescent microscopy images of MDM efferocytosis of CFSE-labeled apoptotic neutrophils. Shown are control MDMs without apoptotic neutrophils, CF and non-CF MDMs without treatment, CF MDMs post-ETI treatment, non-CF MDMs treated with a CFTR inhibitor (CFTRinh), and non-CF MDMs treated with a CFTR inhibitor and then exposed to ETI. Also shown is a summary of % efferocytosis for all experiments, normalized to non-CF control for each sample, n= 3–12. P values are shown for individual comparisons, via one-way ANOVA with paired analysis of CFTRinh changes in non-CF. C) Paired individual CF responses from 6B, paired t-test, n=8. D) TEM images of non-CF, CF, and CF ETI MDMs co-cultured with apoptotic neutrophils. Black arrows indicate efferocytosis of apoptotic cells. Magnification shown on images. Summary of semi-quantification of # efferocytosed neutrophils per MDM also displayed, one-way ANOVA.
ETI treatment has variable effects on MDM effector functions
In addition to deficits in phagocytosis and clearance of apoptotic debris, CF MDMs have alterations in cytokine production.[4, 11, 14, 21–23] We examined pro- and anti-inflammatory cytokine secretion profiles in response to infection with B. cenocepacia. IL-10 production was significantly increased in untreated CF MDMs compared to non-CF (Fig. 7A). Interestingly, ETI treatment was associated with decreased IL-10 production in both CF and non-CF MDMs, with comparable levels of IL-10 for both CF and non-CF MDMs after ETI (Fig. 7A). ETI treatment was not associated with any significant changes in IL-1β, IL-6, IL-8, IL-12, or TNF-α production (Supplemental Figs. 5A–E). TNF-α production was increased in CF MDMs at baseline compared to non-CF, similar to IL-10 (Supplemental Fig. 5E). Because there were minimal changes in MDM polarization or cytokine production, we next measured ROS production as another measure of macrophage function. CF MDMs have a known deficit in NADPH oxidase assembly and subsequent reactive oxygen species (ROS) production measured through DCF and superoxide assays.[8] We first stimulated MDMs with PMA, a phorbol ester that stimulates ROS production. CF MDMs had a decreased oxidative burst in response to PMA compared to non-CF (Fig. 7B). ETI treatment was associated with normalized ROS production in CF MDMs stimulated with PMA (Fig.7B). There was no change in CF MDM ROS production in unstimulated cells treated with ETI, which confirms the specificity of ETI treatment for increasing ROS production during PMA stimulation (Fig. 7B). We previously demonstrated that B. cenocepacia suppresses MDM ROS production.[8] We infected MDMs with B. cenocepacia and measured ROS production during ETI treatment. ROS production was unchanged with ETI treatment during B. cenocepacia infection in both CF and non-CF MDMs (Fig. 7C). Combined, these results suggest variable changes in the inflammatory and oxidative burst potential of CF MDMs treated with ETI.
Figure 7: ETI effects on macrophage effector functions.
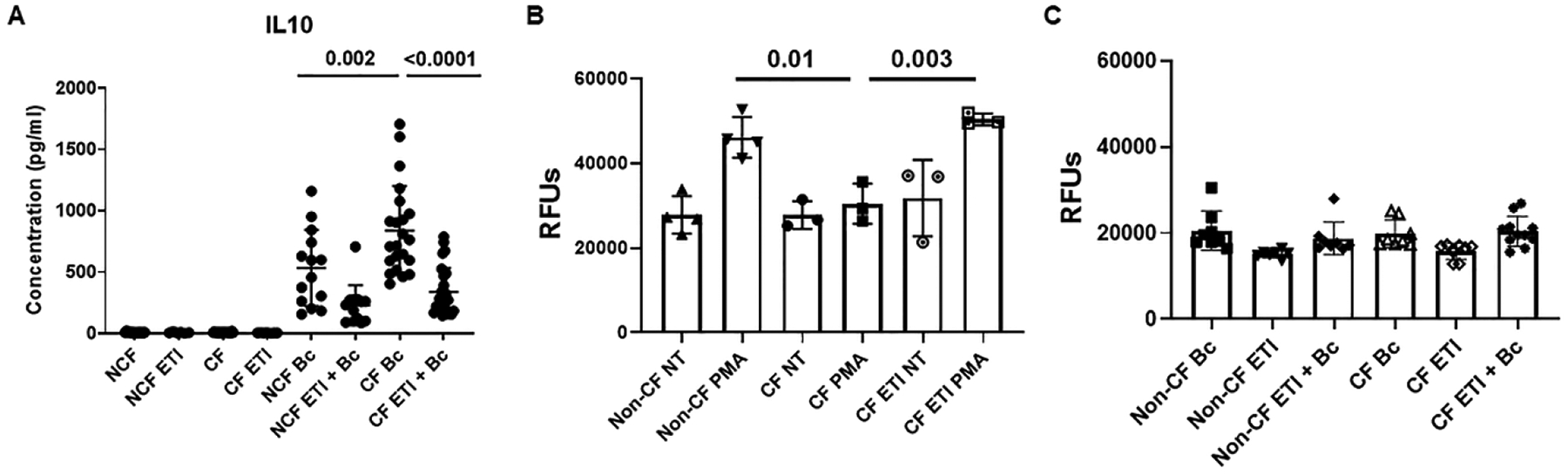
A) 24h IL-10 cytokine production (pg/ml) in non-CF and CF MDM supernatants at baseline, during ETI treatment, during B. cenocepacia (Bc) infection or combined infection and treatment. N=14–26, mean ±SD, p values for individual comparisons via one-way ANOVA. B) Summary of DCF assay end-point analysis of maximum 2h reactive oxygen species (ROS) production in non-CF and CF MDMs after no treatment (NT), PMA stimulus, ETI treatment, or PMA plus ETI treatment. N=3–4, mean ±SD, p value via one-way ANOVA. C) Summary of DCF assay end-point analysis of maximum 2h ROS production in non-CF and CF MDMs after infection with Bc, treatment with ETI, or a combination. N=8–11, mean ±SD, p value via one-way ANOVA.
Clinical outcomes post-ETI
Last, we determined changes in clinical outcomes post-ETI in our CF cohort with at least 3 months of available clinical data, and correlated outcomes with changes in MDM CFTR function. ETI treatment was associated with a significant reduction in sweat chloride levels after 1 month (99.0 ±18.5 mmol/L vs. 48.7 ±19.2, p < 0.0001, Table 2, Fig. 8A). Reductions in sweat chloride were consistently decreased for everyone, but variable in intensity of decrease (minimum reduction 15 mmol/L, max 93 mmol/L). Changes in sweat chloride were also significant for individuals when analyzed by prior CFTR modulator exposure (Table 2, Supplemental Fig. 6A). ETI was associated with significant increases in absolute % change in % predicted FEV1 (+19.7%, relative change +9.2%) and BMI (+4.6%, relative change +0.9) at 3-months post-initiation (Table 2, Figs. 8B–C). Similar effects were seen for FEV1 when analyzed by those homozygous (relative change +12.2%) or heterozygous (+9.0%) for the F508del variant. Change in BMI at 3 months was also similar between variant groups (homozygous +1.0, heterozygous +0.9).
Table 2:
Changes in clinical outcomes
| Baseline | 3-mos | 6-mos | 9-mos | 12-mos | P value | |
|---|---|---|---|---|---|---|
| Sweat chloride (ALL) (mmol/L) | 99.0 ±18.5 | 48.7 ±19.2 | --- | --- | --- | <0.0001 |
| Sweat chloride (no prior modulator) (mmol/L) | 103.0 ±18.8 | 54.0 ±20.7 | --- | --- | --- | <0.0001 |
| Sweat chloride (prior modulator) (mmol/L) | 94.9 ±17.6 | 43.3 ±16.2 | --- | --- | --- | <0.0001 |
| FEV1 (ALL) | 63.8 ±24.7 | 73.0 ±25.8 | 78.7 ±22.8 | 75.3 ±25.4 | 74.7 ±25.6 | 0.003 |
| FEV1 (no prior modulator) | 57.4 ±26.9 | 67.9 ±28.6 | 71.3 ±28.1 | 71.1 ±27.9 | 68.7 ±27.9 | 0.03 |
| FEV1 (prior modulator) | 70.3 ±22.9 | 79.4 ±22.3 | 87.8 ±12.3 | 77.7 ±24.5 | 81.0 ±21.6 | 0.001 |
| BMI (ALL) | 22.6 ± 4.5 | 23.5 ± 4.3 | 23.6 ± 4.1 | 23.3 ± 3.4 | 23.7 ± 4.1 | <0.0001 |
| BMI (no prior modulator) | 21.4 ±4.9 | 22.7 ±5.0 | 22.8 ±5.8 | 22.4 ±4.3 | 22.9 ±4.1 | 0.003 |
| BMI (prior modulator) | 23.3 ±3.3 | 24.1 ±3.2 | 24.4 ±2.9 | 23.8 ±2.6 | 24.2 ±3.2 | 0.003 |
| Hospitalizations (All) | 0.9 ± 1.4 | --- | --- | --- | 0.02 ± 0.1 | <0.0001 |
| Hospitalizations (no prior modulator) | 1.0 ± 1.1 | --- | --- | --- | 0.04 ± 0.2 | <0.0001 |
| Hospitalizations (prior modulator) | 0.7 ± 1.7 | --- | --- | --- | 0.0 ± 0.0 | <0.0001 |
| Antibiotic courses (All) | 2.4 ± 2.3 | --- | --- | --- | 0.5 ± 0.7 | <0.0001 |
| Antibiotics (no prior modulator) | 3.1 ± 2.1 | --- | --- | --- | 0.6 ± 0.8 | <0.0001 |
| Antibiotics (prior modulator) | 1.8 ± 2.4 | --- | --- | --- | 0.3 ± 0.5 | <0.0001 |
| P. aeruginosa | 45.1% | --- | --- | --- | 21.6% | |
| MRSA | 33.3% | --- | --- | --- | 15.7% | |
| MSSA | 35.3% | --- | --- | --- | 31.4% | |
| BCC | 3.9% | --- | --- | --- | 0% | |
| Achromobacter | 9.8% | --- | --- | --- | 2% | |
| Stenotrophomonas | 5.9% | --- | --- | --- | 4% | |
| Normal resp flora | 3.9% | --- | --- | --- | 29.4% |
BCC- Burkholderia cepacia complex
P values determined by paired t-test for sweat chloride, hospitalizations and antibiotic courses and linear mixed effects model with post-hoc Sidak comparison for FEV1 and BMI
Figure 8: Clinical outcome changes post-ETI.
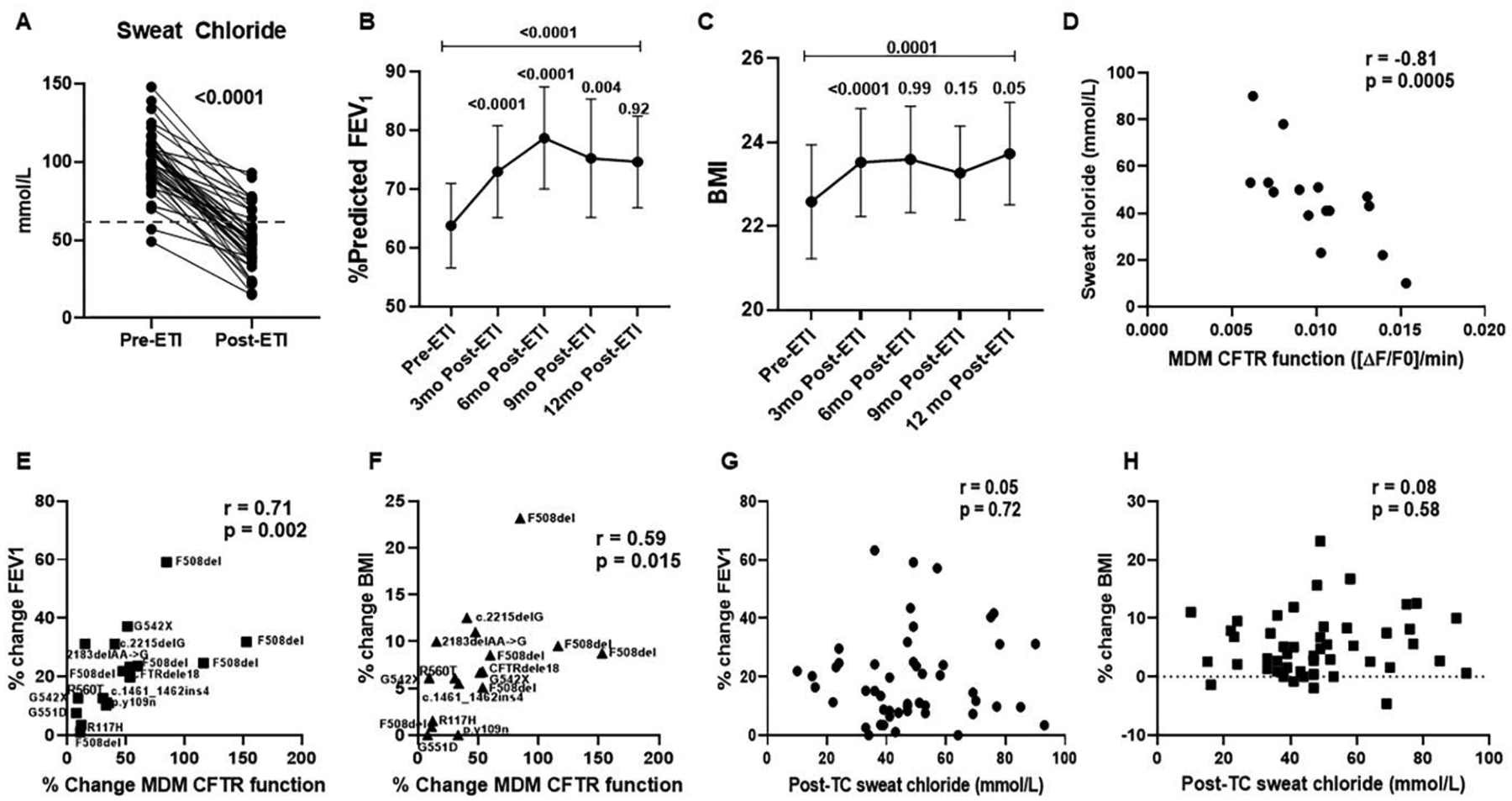
A) Changes in sweat chloride for PWCF pre- and 1-month post-ETI initiation. N=52, p <0.0001 via paired t-test. Dashed line represents the clinical threshold of 60mmol/L for a positive sweat chloride test. B) Changes in percent predicted FEV1 over 12 months for PWCF. Shown are mean values plus 95% CI at baseline (pre-ETI), 3-, 6-, 9-, and 12-months post-ETI initiation. All individuals had at least 3 months of data. P values shown for comparisons between time points, via linear mixed effects model with post-hoc comparisons between neighboring visits, as well as between baseline and 12-month using the Sidak method. C) Changes in BMI over 12 months for PWCF. Shown are mean values plus 95% CI at baseline (pre-ETI), 3-, 6-, 9-, and 12-months post-ETI initiation. All individuals had at least 3 months of data. P values shown for comparisons between time points, via linear mixed effects model with post-hoc comparisons between neighboring visits, as well as between baseline and 12-month using the Sidak method. D) Correlation plot for sweat chloride and maximal MDM CFTR function for PWCF, n=15. A significant negative correlation was observed (Spearman’s r= −0.81, p = 0.0005). E) Correlation plot for 3-month post-ETI FEV1 and change in MDM CFTR function pre- and post-ETI treatment for PWCF, n=17. A significant positive correlation was observed (Spearman’s r = 0.71, p = 0.002). The second CFTR variant is overlayed on each sample. All individuals with at least one copy of F508del. F) Correlation plot for 3-month post-ETI BMI and change in MDM CFTR function pre- and post-ETI treatment for PWCF, n=17. A significant positive correlation was observed (r = 0.59, p = 0.015). The second CFTR variant is overlayed on each sample. All individuals with at least one copy of F508del. G) Correlation plot for 3-month post-ETI FEV1 and sweat chloride for PWCF, n=49. No correlation was observed (r = 0.05, p = 0.72). H) Correlation plot for 3-month post-ETI BMI and sweat chloride for PWCF, n=49. No correlation was observed (r = 0.08, p = 0.58).
Longitudinal analysis of the entire cohort revealed that FEV1 increased from 3 to 6 months (+5%) but showed no increases thereafter and decreased from 6 to 9 months post-ETI initiation (Fig. 8B). BMI showed no significant changes between 3–12 months (Fig. 8C). Individual values for percent change in FEV1 and BMI at 3 months relative to baseline ranged from 0 – 63% and −4.6 to 23% respectively. Despite the variable changes over time, there were overall significant increases in FEV1 and BMI from baseline to 12-month follow-up measurements (Table 2, Figs. 8B–C). The longitudinal changes in FEV1 and BMI were also consistent for PWCF with and without prior CFTR modulator exposure (Supplemental Figs. 6B, C).
Hospitalizations for pulmonary exacerbations (0.88 ± 1.4 vs 0.02 ± 0.1, p <0.0001) and oral antibiotic prescriptions per year (2.4 ± 2.3 vs 0.5 ± 0.7, p <0.0001) were both significantly reduced in the year following ETI initiation compared to the prior year (Table 2). These decreases were also consistent when comparing PWCF with and without prior CFTR modulator use (Table 2). Changes in chronic infection from oropharyngeal and sputum bacterial cultures were noted over time, with 51% of individuals clearing one or more chronic pathogens after 12 months of therapy. Both individuals with a prior chronic B. multivorans infection showed negative cultures at 12 months. Of note, most 12-month cultures were oropharyngeal swabs, and we therefore did not have sufficient cultures to check non-tuberculous mycobacteria (NTM) status in the 16% of our cohort with a prior NTM infection. The percentage of individual pathogens present at the start and after 12 months of ETI treatment are shown in Table 2.
We then examined children and adults (n=17) who had an available 3-month post-ETI MDM CFTR functional assessment and analyzed for correlations with clinical outcomes. There was a significant negative correlation between post-ETI MDM CFTR function and post-ETI sweat chloride (e.g. higher MDM CFTR function correlates with lower sweat chlorides, r= −0.81, p = 0.0005, Fig. 8D). There were positive correlations between percent change FEV1 and percent change BMI with change in CFTR MDM function post-ETI ([FEV1 r = 0.71, p = 0.002] [BMI r = 0.59, p = 0.015] Figs. 8E–F). Individuals with 2 copies of the F508del variant had the largest responses. There was also a significant correlation with clearance of one or more bacterial pathogens and change in CFTR MDM function post-ETI (r=0.53, p = 0.036), including the 6 individuals with the lowest changes in CFTR function who did not demonstrate clearance of bacteria from cultures. Last, we analyzed clinical outcomes in all individuals with an available post-ETI sweat chloride. In contrast to the MDM CFTR results, post-ETI sweat chloride did not correlate with changes in FEV1 (r = 0.05, p = 0.72) or BMI (r = 0.08, p = 0.58). Overall, these data reflect robust, but highly individualized clinical responses with the first 3 months of ETI treatment. Further, changes in macrophage CFTR function may be a more sensitive indicator of individual clinical responses than sweat chloride.
DISCUSSION
The approval of highly effective CFTR modulators such as the triple combination elexacaftor/tezacaftor/ivacaftor has brought tremendous changes to the clinical care and disease trajectories for many PWCF. However, questions remain about the long-term efficacy of CFTR modulators and their impact on chronic infection and inflammation as well as responses to new infectious or inflammatory insults. The results of this study provide novel insights into alterations in innate immune responses in response to ETI, persisting phagocytic defects post-treatment, and their associations with clinical outcomes. Further, these studies build upon prior findings in a CRISPR-Cas9 CFTR knockout model in human MDMs,[14] which established several macrophage functions critically dependent upon CFTR.
The overall role of CFTR and other ion channels in CF innate immune cells remains relatively understudied. Prior human studies have elucidated low levels of expression of CFTR in monocytes, macrophages, and neutrophils, with detection of functional CFTR currents in peripheral blood monocytes and MDMs.[4, 14, 24–30] In this study, we detected changing CFTR expression and trafficking in MDMs from PWCF in response to ETI, along with improved CFTR-dependent functional currents through halide efflux and a patch-clamp assay. Biogenesis, trafficking to the cell surface, and endocytosis and recycling of CFTR are regulated by multiple trafficking pathways, including Rab7. Rab7 regulates the movement of CFTR away from the recycling pathway into late endosomes, and then participates in the transport of CFTR from late endosome to lysosomes for degradation.[31] Rab7 and other GTPases are also critical in macrophage phagosome maturation and phagolysosome formation.[32] In our study, alterations in CFTR co-localization with EEA1, Rab7, and LAMP1 in untreated CF MDMs were restored post-ETI, suggesting there exists a reciprocity between CFTR function and localization and anterograde trafficking in MDMs. CFTR localization to the plasma membrane and lysosomes was also activated by LPS, consistent with past studies where macrophage CFTR localization is activated by infectious or inflammatory stimuli.[4, 10] Further, CFTR was recently shown to regulate macrophage autophagolysosomal acidification.[33] Based on past and current studies, functional CFTR is likely important in regulating several aspects of macrophage machinery necessary for effective phagocytosis.
Importantly, we found that changes in CFTR function with ETI are short-lived and not sustained unless there is continued drug exposure. This highlights the potential importance of optimizing CFTR modulator dosing for immune cell responses and cellular distribution. Changes in CFTR function were also highly individualized, suggesting unique factors that could influence macrophage responses to CFTR modulators, such as receptor sensitivity or epigenetic modifications.[34] We did not discern gender, age, or genotype-specific responses that dictate the individualized responses, but larger multi-center studies could help achieve more nuanced testing of clinical phenotypes. Further, modifying CFTR does not appear to occur in isolation as reciprocal changes in other ion channels were observed in response to CFTR modulators as seen in our microarray data. While detailed studies on changes in individual ion channels (non-CFTR) were beyond the scope of this manuscript, reciprocal alterations in calcium channels such as TRPC1 might be beneficial in restoring aberrant macrophage inflammatory responses in CF.[35] Overall, we did not observe uniform changes across ion channel types post-ETI to easily decipher coordinated channel responses, but expression changes suggest broad normalization of chloride, potassium, and sodium conductance, with new changes in calcium signaling. Several altered ion channels that we observed have not been well described in macrophages (e.g. CLCN2, CACNG2, CHRND, etc.) and therefore have an unclear role in CF macrophage responses post-ETI. Combined, these data highlight the complex nature of macrophage ion channels and the need for further studies to monitor immune non-CFTR ion channel responses to CFTR modulators.
Concurrent with our CFTR expression and function studies, macrophage phagocytic and effector functions also changed to varying degrees after ETI exposure. Specifically, persistent deficits in phagocytosis and the macrophage cytoskeletal ultrastructure were observed post-treatment. We found decreased Cdc42 activity in macrophages pre- and post-ETI, consistent with a prior study that showed decreased Cdc42 activity in CF monocytes.[36] Monocyte adhesion was improved with CFTR modulators in the same study, however Cdc42 activity was not tested. The overall persistent deficits in phagocytosis may be related to the difficulty in clearing some pathogens such as B. cenocepacia, which have known virulence mechanisms that disrupt the actin cytoskeleton or suppress intracellular ROS production.[8, 37, 38] Prior studies of older generation CFTR modulator combinations suggested potential negative interactions between ivacaftor and lumacaftor regarding bacterial phagocytosis.[3, 4] However, in our studies ETI was associated with improved phagocytosis of B. cenocepacia in both non-CF and CF MDMs, but this effect was attenuated by the presence of CF ASN. These results suggests that the inflammatory airway milieu may modify and potentially blunt the therapeutic impact of ETI. Additionally, efferocytosis of apoptotic neutrophils was robustly enhanced post-ETI, which supports the notion that full correction of macrophage cytoskeletal signaling may not be needed for clinical effects. Further studies detailing the complex mechanisms and receptors underlying phagocytosis of bacteria and uptake of inflammatory debris after CFTR modulation are ongoing.
During inflammation, monocytes are recruited to the affected site and then differentiated into resident macrophages and activated. Interestingly, ETI normalized anti-inflammatory IL-10 expression, while pro-inflammatory cytokine production did not change. Prior studies have shown differences in CF IL-10 production based on age, sampling location (airway, blood, nasal), cell type (blood, lung lavage), and health status.[39–41] The combination of these results suggest that many aspects of CF macrophage cytokine production are likely regulated by complex local environmental signals in addition to CFTR, as has been shown in studies with CF neutrophils.[42]
As an important corollary to the observed changes in macrophage function post-ETI, we observed robust but highly variable clinical responses over time. FEV1 and BMI were markedly improved at 3 months post-treatment but leveled off over time in most individuals, with many individuals seeing a decrease in FEV1 at nine-months post-initiation. Further, although sweat chloride levels dramatically decreased on ETI, post-ETI sweat chloride was not a robust predictor of clinical responses compared to changes in MDM CFTR function, which were strongly correlated with improvements in both BMI and FEV1, as well as pathogen clearance. These data suggest that more robust improvements in immune function are linked to improved clinical responses, which opens the door for monitoring immune function as a biomarker of CF therapeutic responses. However, our findings need to be validated in large, multi-center prospective studies. Overall, a significant reduction in inpatient and outpatient pulmonary exacerbations was demonstrated in this observational cohort, supporting the clinical impact of ETI in PWCF in a real-world setting.
Our study was limited by a lack of available airway resident macrophages for comparison to MDM studies. Although MDMs are integral in innate immune and reparative functions throughout the body including their recruitment to the lungs, sinuses and GI tract, they may behave differently from resident alveolar or interstitial lung macrophages. However, new evidence suggests that alveolar macrophages can be replaced by cells of monocytic origin.[43] Post-ETI we have seen a dramatic resolution of sputum production and the need for bronchoscopy, thereby limiting any available airway resident macrophages for comparison in this study. Research bronchoscopy programs will be integral for such future comparisons of resident and recruited CF macrophages.
In conclusion, ETI was associated with unique changes in innate immune function and clinical outcomes in PWCF. Changes in macrophage CFTR function may be a more sensitive indicator of clinical responses to CFTR modulators than sweat chloride but need further validation in subsequent studies.
Supplementary Material
Take home messages:
ETI partially restores CFTR expression and function in CF macrophages resulting in improved effector functions
Macrophage CFTR restoration correlates with clinical outcomes
Results are individualized reflecting donor genotypic and phenotypic variation
ACKNOWLEDGEMENTS
Macrophages for this work were supplied by the Cure CF Columbus Immune Core (C3IC) at Nationwide Children’s Hospital. We also thank the Nationwide Children’s Hospital Morphology Core, Joseph Jurcisek, and Jennifer Edwards for assistance with electron microscopy imaging and processing. The authors thank all the participants and their families for their contributions.
SUPPORT:
This study was supported by CF Foundation grants KOPP16I0 (BTK), PARTID18P0 (SPS), HALLST18I0 (LHS), NIH R01 HL158747 (BTK, SPS, AOA, LHS), R01 HL148171 (BTK), R01 AI24121 (AOA), and R01 HL127651 (AOA). This work was supported in part by the Cure CF Columbus Research and Development Program (C3RDP) Cores including the Translational Core (C3TC) and C3IC. C3RDP is supported by the Division of Pediatric Pulmonary Medicine, the Biopathology Center Core, and the Data Collaboration Team at Nationwide Children’s Hospital. Grant support provided by The Ohio State University Center for Clinical and Translational Science (National Center for Advancing Translational Sciences, Grant UL1TR002733) and by the Cystic Fibrosis Foundation (Grant MCCOY19RO).
Footnotes
This article has an online data supplement
Conflict of interest: The authors have declared that no conflict of interest exists.
REFERENCES
- 1.Gramegna A, Contarini M, Aliberti S, Casciaro R, Blasi F, Castellani C. From Ivacaftor to Triple Combination: A Systematic Review of Efficacy and Safety of CFTR Modulators in People with Cystic Fibrosis. International journal of molecular sciences 2020: 21(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hisert KB, Heltshe SL, Pope C, Jorth P, Wu X, Edwards RM, Radey M, Accurso FJ, Wolter DJ, Cooke G, Adam RJ, Carter S, Grogan B, Launspach JL, Donnelly SC, Gallagher C, Bruce JE, Stoltz D, Welsh MJ, Hoffman LR, McKone EF, Singh PK. Restoring CFTR Function Reduces Airway Bacteria and Inflammation in People With Cystic Fibrosis and Chronic Lung Infections. Am J Respir Crit Care Med 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barnaby R, Koeppen K, Nymon A, Hampton TH, Berwin B, Ashare A, Stanton BA. Lumacaftor (VX-809) restores the ability of CF macrophages to phagocytose and kill Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol 2018: 314(3): L432–L438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang S, Shrestha CL, Kopp BT. Cystic fibrosis transmembrane conductance regulator (CFTR) modulators have differential effects on cystic fibrosis macrophage function. Sci Rep 2018: 8(1): 17066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harris JK, Wagner BD, Zemanick ET, Robertson CE, Stevens MJ, Heltshe SL, Rowe SM, Sagel SD. Changes in Airway Microbiome and Inflammation with Ivacaftor Treatment in Patients with Cystic Fibrosis and the G551D Mutation. Ann Am Thorac Soc 2020: 17(2): 212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abdulrahman BA, Khweek AA, Akhter A, Caution K, Kotrange S, Abdelaziz DH, Newland C, Rosales-Reyes R, Kopp B, McCoy K, Montione R, Schlesinger LS, Gavrilin MA, Wewers MD, Valvano MA, Amer AO. Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy 2011: 7(11): 1359–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Assani K7, Shrestha CL, Rinehardt H, Zhang S, Robledo-Avila F, Wellmerling J, Partida-Sanchez S, Cormet-Boyaka E, Reynolds SD, Schlesinger LS, Kopp BT. AR-13 reduces antibiotic-resistant bacterial burden in cystic fibrosis phagocytes and improves cystic fibrosis transmembrane conductance regulator function. J Cyst Fibros 2019: 18(5): 622–629. [DOI] [PubMed] [Google Scholar]
- 8.Assani K, Shrestha CL, Robledo-Avila F, Rajaram MV, Partida-Sanchez S, Schlesinger LS, Kopp BT. Human Cystic Fibrosis Macrophages Have Defective Calcium-Dependent Protein Kinase C Activation of the NADPH Oxidase, an Effect Augmented by Burkholderia cenocepacia. J Immunol 2017: 198(5): 1985–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Assani K, Tazi MF, Amer AO, Kopp BT. IFN-gamma stimulates autophagy-mediated clearance of Burkholderia cenocepacia in human cystic fibrosis macrophages. PLoS One 2014: 9(5): e96681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shrestha CL, Assani KD, Rinehardt H, Albastroiu F, Zhang S, Shell R, Amer AO, Schlesinger LS, Kopp BT. Cysteamine-mediated clearance of antibiotic-resistant pathogens in human cystic fibrosis macrophages. PLoS One 2017: 12(10): e0186169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bruscia EM, Bonfield TL. Cystic Fibrosis Lung Immunity: The Role of the Macrophage. Journal of innate immunity 2016: 8(6): 550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lara-Reyna S, Holbrook J, Jarosz-Griffiths HH, Peckham D, McDermott MF. Dysregulated signalling pathways in innate immune cells with cystic fibrosis mutations. Cell Mol Life Sci 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turton KB, Ingram RJ, Valvano MA. Macrophage dysfunction in cystic fibrosis: Nature or nurture? J Leukoc Biol 2020. [DOI] [PubMed] [Google Scholar]
- 14.Zhang S, Shrestha CL, Wisniewski BL, Pham H, Hou X, Li W, Dong Y, Kopp BT. Consequences of CRISPR-Cas9-Mediated CFTR Knockout in Human Macrophages. Front Immunol 2020: 11: 1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hazlett HF, Hampton TH, Aridgides DS, Armstrong DA, Dessaint JA, Mellinger DL, Nymon AB, Ashare A. Altered iron metabolism in cystic fibrosis macrophages: the impact of CFTR modulators and implications for Pseudomonas aeruginosa survival. Sci Rep 2020: 10(1): 10935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuhns DB, Priel DAL, Chu J, Zarember KA. Isolation and Functional Analysis of Human Neutrophils. Curr Protoc Immunol 2015: 111: 7 23 21–27 23 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schupp JC, Khanal S, Gomez JL, Sauler M, Adams TS, Chupp GL, Yan X, Poli S, Zhao Y, Montgomery RR, Rosas IO, Dela Cruz CS, Bruscia EM, Egan ME, Kaminski N, Britto CJ. Single-Cell Transcriptional Archetypes of Airway Inflammation in Cystic Fibrosis. Am J Respir Crit Care Med 2020: 202(10): 1419–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leveque M, Penna A, Le Trionnaire S, Belleguic C, Desrues B, Brinchault G, Jouneau S, Lagadic-Gossmann D, Martin-Chouly C. Phagocytosis depends on TRPV2-mediated calcium influx and requires TRPV2 in lipids rafts: alteration in macrophages from patients with cystic fibrosis. Sci Rep 2018: 8(1): 4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van de Weert-van Leeuwen PB, Van Meegen MA, Speirs JJ, Pals DJ, Rooijakkers SH, Van der Ent CK, Terheggen-Lagro SW, Arets HG, Beekman JM. Optimal complement-mediated phagocytosis of Pseudomonas aeruginosa by monocytes is cystic fibrosis transmembrane conductance regulator-dependent. Am J Respir Cell Mol Biol 2013: 49(3): 463–470. [DOI] [PubMed] [Google Scholar]
- 20.Di Pietro C, Zhang PX, O’Rourke TK, Murray TS, Wang L, Britto CJ, Koff JL, Krause DS, Egan ME, Bruscia EM. Ezrin links CFTR to TLR4 signaling to orchestrate anti-bacterial immune response in macrophages. Sci Rep 2017: 7(1): 10882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gharib SA, McMahan RS, Eddy WE, Long ME, Parks WC, Aitken ML, Manicone AM. Transcriptional and functional diversity of human macrophage repolarization. The Journal of allergy and clinical immunology 2019: 143(4): 1536–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tarique AA, Sly PD, Cardenas DG, Luo L, Stow JL, Bell SC, Wainwright CE, Fantino E. Differential expression of genes and receptors in monocytes from patients with cystic fibrosis. J Cyst Fibros 2019: 18(3): 342–348. [DOI] [PubMed] [Google Scholar]
- 23.Tarique AA, Sly PD, Holt PG, Bosco A, Ware RS, Logan J, Bell SC, Wainwright CE, Fantino E. CFTR-dependent defect in alternatively-activated macrophages in cystic fibrosis. J Cyst Fibros 2017: 16(4): 475–482. [DOI] [PubMed] [Google Scholar]
- 24.Shrestha CL, Zhang S, Wisniewski B, Hafner S, Elie J, Meijer L, Kopp BT. (R)-Roscovitine and CFTR modulators enhance killing of multi-drug resistant Burkholderia cenocepacia by cystic fibrosis macrophages. Sci Rep 2020: 10(1): 21700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tazi MF, Dakhlallah DA, Caution K, Gerber MM, Chang SW, Khalil H, Kopp BT, Ahmed AE, Krause K, Davis I, Marsh C, Lovett-Racke AE, Schlesinger LS, Cormet-Boyaka E, Amer AO. Elevated Mirc1/Mir17–92 cluster expression negatively regulates autophagy and CFTR (cystic fibrosis transmembrane conductance regulator) function in CF macrophages. Autophagy 2016: 12(11): 2026–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bonfield TL, Hodges CA, Cotton CU, Drumm ML. Absence of the cystic fibrosis transmembrane regulator (Cftr) from myeloid-derived cells slows resolution of inflammation and infection. J Leukoc Biol 2012: 92(5): 1111–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Del Porto P, Cifani N, Guarnieri S, Di Domenico EG, Mariggio MA, Spadaro F, Guglietta S, Anile M, Venuta F, Quattrucci S, Ascenzioni F. Dysfunctional CFTR alters the bactericidal activity of human macrophages against Pseudomonas aeruginosa. PLoS One 2011: 6(5): e19970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sorio C, Buffelli M, Angiari C, Ettorre M, Johansson J, Vezzalini M, Viviani L, Ricciardi M, Verze G, Assael BM, Melotti P. Defective CFTR expression and function are detectable in blood monocytes: development of a new blood test for cystic fibrosis. PLoS One 2011: 6(7): e22212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Painter RG, Marrero L, Lombard GA, Valentine VG, Nauseef WM, Wang G. CFTR-mediated halide transport in phagosomes of human neutrophils. J Leukoc Biol 2010: 87(5): 933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Painter RG, Valentine VG, Lanson NA Jr., Leidal K, Zhang Q, Lombard G, Thompson C, Viswanathan A, Nauseef WM, Wang G, Wang G. CFTR Expression in human neutrophils and the phagolysosomal chlorination defect in cystic fibrosis. Biochemistry 2006: 45(34): 10260–10269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farinha CM, Matos P, Amaral MD. Control of cystic fibrosis transmembrane conductance regulator membrane trafficking: not just from the endoplasmic reticulum to the Golgi. FEBS J 2013: 280(18): 4396–4406. [DOI] [PubMed] [Google Scholar]
- 32.Fountain A, Inpanathan S, Alves P, Verdawala MB, Botelho RJ. Phagosome maturation in macrophages: Eat, digest, adapt, and repeat. Adv Biol Regul 2021: 82: 100832. [DOI] [PubMed] [Google Scholar]
- 33.Badr A, Eltobgy M, Krause K, Hamilton K, Estfanous S, Daily KP, Abu Khweek A, Hegazi A, Anne MNK, Carafice C, Robledo-Avila F, Saqr Y, Zhang X, Bonfield TL, Gavrilin MA, Partida-Sanchez S, Seveau S, Cormet-Boyaka E, Amer AO. CFTR Modulators Restore Acidification of AutophagoLysosomes and Bacterial Clearance in Cystic Fibrosis Macrophages. Front Cell Infect Microbiol 2022: 12: 819554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lopes-Pacheco M CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front Pharmacol 2019: 10: 1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nascimento Da Conceicao V, Sun Y, Zboril EK, De la Chapa JJ, Singh BB. Loss of Ca(2+) entry via Orai-TRPC1 induces ER stress, initiating immune activation in macrophages. J Cell Sci 2019: 133(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sorio C, Montresor A, Bolomini-Vittori M, Caldrer S, Rossi B, Dusi S, Angiari S, Johansson JE, Vezzalini M, Leal T, Calcaterra E, Assael BM, Melotti P, Laudanna C. Mutations of Cystic Fibrosis Transmembrane Conductance Regulator Gene Cause a Monocyte-Selective Adhesion Deficiency. Am J Respir Crit Care Med 2016: 193(10): 1123–1133. [DOI] [PubMed] [Google Scholar]
- 37.Rosales-Reyes R, Skeldon AM, Aubert DF, Valvano MA. The Type VI secretion system of Burkholderia cenocepacia targets multiple Rho family GTPases disrupting the actin cytoskeleton and the assembly of NADPH oxidase complex in macrophages. Cell Microbiol 2011. [DOI] [PubMed] [Google Scholar]
- 38.Walpole GFW, Plumb JD, Chung D, Tang B, Boulay B, Osborne DG, Piotrowski JT, Catz SD, Billadeau DD, Grinstein S, Jaumouille V. Inactivation of Rho GTPases by Burkholderia cenocepacia Induces a WASH-Mediated Actin Polymerization that Delays Phagosome Maturation. Cell Rep 2020: 31(9): 107721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Armstrong DS, Hook SM, Jamsen KM, Nixon GM, Carzino R, Carlin JB, Robertson CF, Grimwood K. Lower airway inflammation in infants with cystic fibrosis detected by newborn screening. Pediatr Pulmonol 2005: 40(6): 500–510. [DOI] [PubMed] [Google Scholar]
- 40.Paats MS, Bergen IM, Bakker M, Hoek RA, Nietzman-Lammering KJ, Hoogsteden HC, Hendriks RW, van der Eerden MM. Cytokines in nasal lavages and plasma and their correlation with clinical parameters in cystic fibrosis. J Cyst Fibros 2013: 12(6): 623–629. [DOI] [PubMed] [Google Scholar]
- 41.Hauber HP, Beyer IS, Meyer A, Pforte A. Decreased interleukin-18 expression in BAL cells and peripheral blood mononuclear cells in adult cystic fibrosis patients. J Cyst Fibros 2004: 3(2): 129–131. [DOI] [PubMed] [Google Scholar]
- 42.Forrest OA, Ingersoll SA, Preininger MK, Laval J, Limoli DH, Brown MR, Lee FE, Bedi B, Sadikot RT, Goldberg JB, Tangpricha V, Gaggar A, Tirouvanziam R. Frontline Science: Pathological conditioning of human neutrophils recruited to the airway milieu in cystic fibrosis. J Leukoc Biol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arafa EI, Shenoy AT, Barker KA, Etesami NS, Martin IM, Lyon De Ana C, Na E, Odom CV, Goltry WN, Korkmaz FT, Wooten AK, Belkina AC, Guillon A, Forsberg EC, Jones MR, Quinton LJ, Mizgerd JP. Recruitment and training of alveolar macrophages after pneumococcal pneumonia. JCI Insight 2022: 7(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.