

Abstract
Levels of neutrophil extracellular traps (NETs) were measured in plasma of healthy controls (HC, n=30) and patients with granulomatosis with polyangiitis (GPA, n=123), microscopic polyangiitis (MPA, n=61), Takayasu’s arteritis (TAK, n=58), and giant cell arteritis (GCA, n=68), at times of remission or activity and correlated with levels of the platelet-derived thrombospondin-1 (TSP-1).
Levels of NETs were elevated during active disease in patients with GPA (p<0.0001), MPA (p=0.0038), TAK (p<0.0001), and GCA (p<0.0001), and in remission for GPA, p<0.0001, MPA, p=0.005, TAK, p=0.03, and GCA, p=0.0009. All cohorts demonstrated impaired NET degradation. Patients with GPA (p=0.0045) and MPA (p=0.005) had anti-NET IgG antibodies. Patients with TAK had anti-histone antibodies (p<0.01), correlating with presence of NETs. Levels of TSP-1 were increased in all patients with vasculitis, and associated with NET formation.
NET formation is a common process in vasculitides. Targeting NET formation or degradation could be potential therapeutic approaches for vasculitides.
Keywords: Anti-neutrophil cytoplasmic antibody associated vasculitis, large vessel vasculitis, neutrophils, neutrophil extracellular traps, histone, platelet activation, thrombospondin-1
1. Introduction
Neutrophils and neutrophil extracellular traps (NETs) are important mediators of the innate immune system and play a significant role in host defense [1]. NETs are extruded networks of decondensed chromosomal DNA, citrullinated histones, granule proteins, including proteinase 3 (PR3), neutrophil elastase (NE), calprotectin (also known as S100A8/A9) and myeloperoxidase (MPO), and other components such as lactoferrin, cathepsin G and LL37 [2,3]. NETs appear to play a significant role in the pathogenesis of many autoimmune diseases, including vasculitis [4].
Vasculitis is a group of systemic autoimmune diseases of unknown etiology, classified into small-, medium- and large-vessel vasculitis based on the size of the affected blood vessels [5]. Small-vessel vasculitis includes three different subtypes of anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV), namely granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis [6]. AAV is characterized by small blood vessel inflammation and presence of ANCA in the circulation [7]. Large-vessel vasculitis (LVV), including Takayasu’s arteritis (TAK) and giant cell arteritis (GCA), is characterized by vascular inflammation and subsequent damage of the aorta and/or major branch arteries [8].
In AAV, ANCA can activate neutrophils that are primed by tumor necrosis factor-alpha (TNF-α), and other stimuli including lipopolysaccharide (LPS), or complement factor 5a (C5a), which leads to externalization of MPO, and PR3 to the cell surface where ANCA can bind to them. Immune complexes then of these antibodies and their antigens bind and crosslink FcγRs on the neutrophil cell surface, resulting in neutrophil activation, neutrophil adhesion to endothelium, NET formation, and eventually inflammatory damage to the endothelium [9,10]. Interestingly, NET formation can also occur upon interaction of platelets with neutrophils via TLR9-dependent CXCL4 release as recently demonstrated in AAV [11], with platelet aggregation on the NET scaffold resulting in activation of the coagulation cascade and immune thrombosis [12]. However, the role of neutrophils, including NETs, in LVV pathogenesis is not fully known.
Histones are part of the NET scaffold that are released to the extracellular space during NET formation and undergo posttranslational modifications, with histone epitopes serving as modified auto-antigens that can be targeted by autoantibodies in several autoimmune diseases such as systemic lupus erythematosus (SLE), and rheumatoid arthritis (RA) [13, 14]. Presence of anti-histone antibodies has been described in several autoimmune diseases, including Sjögren’s syndrome, systemic sclerosis and polymyositis/dermatomyositis [15,16]. Similarly, antibodies against NETs have been reported in patients with RA and SLE [17,18]. Another study suggested that presence of anti-NET antibodies contributed to impaired NET degradation in patients with RA [19]. In patients with lupus, high levels of anti-NET antibodies protected NETs from degradation by DNase 1 [20]. Presence of antibodies targeting NET components has also been demonstrated in patients with MPA [21].
In this current study, we primarily investigated whether patients with AAV and LVV have increased levels of NETs in their plasma. Secondly, we assessed whether levels of NETs were associated with disease activity and inflammatory markers in vasculitis. We also sought to explore whether patients with AAV and LVV have impaired capacity to degrade NETs and evaluated the presence of anti-NET and anti-histone antibodies in these patients. Finally, we explored whether thrombospondin 1 (TSP-1), a platelet activation marker, is elevated in patients with AAV and LVV and associated with NETs and thromboembolism (VTE).
2. Materials and Methods
2.1. Patient characteristics and source of biospecimens
Plasma samples from patients enrolled in the Vasculitis Clinical Research Consortium (VCRC) Longitudinal Studies were utilized in this study, including patients with GPA (123 patients in remission, 73 paired samples with flare), MPA (61 patients in remission, 11 paired samples with flare), TAK (58 patients in remission, 8 paired samples with flare), and GCA (68 patients in remission, 18 paired samples with flare). Demographic data, including disease subgroups, gender, age at diagnosis, disease duration, and treatment for the study populations in remission and active disease were also recorded (Tables 1 and 2). For the GPA and MPA cohorts, the prevalence of anti-MPO and anti-PR3 antibodies is also detailed.
Table 1.
Demographic characteristics of study populations in remission
| Diagnosis | GPA | MPA | TAK | GCA |
|---|---|---|---|---|
|
| ||||
| Number of subjects | 123 | 61 | 58 | 68 |
| Gender (female, %) | 64 (52%) | 31 (51%) | 53 (91%) | 49 (72%) |
| Age at diagnosis, years (median, range) | 47 (10–80) | 59 (17–82) | 30(12–58) | 69 (54–90) |
| Disease duration (mean ± SD, years) | 8.2 ± 7.5 | 4.8 ± 5.2 | 10.4 ± 7.2 | 3.4 ± 3.6 |
| ESR (mean ± SD, mm/h) | 12.8 ± 10.8 | 18.9 ± 15.3 | 15.2 ± 15.0 | 14.1 ± 13.1 |
| CRP (mean ± SD, mg/dl) | 6.7 ± 8.8 | 4.6 ± 5.4 | 6.9 ± 9.1 | 6.3 ± 7.6 |
| Creatinine (mean ± SD, mg/dl) | 1.11 ± 0.60 | 1.61 ± 0.96 | 0.82 ± 0.20 | 0.92 ± 0.20 |
| Anti-MPO antibodies (%) | 20 (16%) | 54 (89%) | ND | ND |
| Anti-PR3 antibodies (%) | 86 (70%) | 8 (13%) | ND | ND |
| Prednisone (%) | 12 (10%) | 19 (31%) | 8 (14%) | 43 (63%) |
| Methotrexate (%) | 28 (23%) | 1 (2%) | 19 (33%) | 26 (38%) |
| Mycophenolate (%) | 9 (7%) | 14 (23%) | 7 (12%) | 0 (0%) |
| Cyclosphosphamide (%) | 6 (5%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Rituximab (%) | 60 (49%) | 16 (26%) | 0 (0%) | 0 (0%) |
| TNF inhibitors (%) | 0 (0%) | 0 (0%) | 25 (43%) | 0 (0%) |
| Azathioprine (%) | 1 (1%) | 1 (2%) | 2 (3%) | 0 (0%) |
| Cyclosporine (%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| BVAS1 score (median, range) | 0 | 0 | 0 | 0 |
| BVAS2 score (median, range) | 0 | 0 | 0 | 0 |
| PGA (mean ± SD) | 0 | 0 | 0 | 0 |
Table 2.
Demographic characteristics of study populations in active disease
| Diagnosis | GPA | MPA | TAK | GCA |
|---|---|---|---|---|
|
| ||||
| Number of subjects | 73 | 11 | 8 | 18 |
| Gender (female, %) | 38 (52%) | 6 (55%) | 8 (100%) | 12 (67%) |
| Age at diagnosis, years (median, range) | 47 (10–80) | 67 (28–75) | 28 (14–44) | 69 (58–83) |
| Disease duration (mean ± SD, years) | 6.8 ± 7.6 | 4.0 ± 5.2 | 6.9 ± 4.1 | 1.6 ± 1.8 |
| ESR (mean ± SD, mm/h) | 17.2 ± 13.9 | 31.1 ± 37.3 | 27.7 ± 22.3 | 16.2 ± 18.6 |
| CRP (mean ± SD, mg/dl) | 11.7 ± 22.7 | 17.2 ± 39.1 | 11.6 ± 13.3 | 11.7 ± 18.3 |
| Creatinine (mean ± SD, mg/dl) | 1.12 ± 0.77 | 1.98 ± 1.12 | 0.76 ± 0.13 | 0.92 ± 0.23 |
| Anti-MPO antibodies (%) | 6 (8%) | 11 (100%) | ND | ND |
| Anti-PR3 antibodies (%) | 55 (75%) | 0 (0%) | ND | ND |
| Prednisone (%) | 22 (30%) | 6 (55%) | 2 (25%) | 16 (89%) |
| Methotrexate (%) | 21 (29%) | 0 (0%) | 5 (63%) | 6 (33%) |
| Mycophenolate (%) | 3 (4%) | 2 (18%) | 0 (0%) | 0 (0%) |
| Cyclosphosphamide (%) | 4 (5%) | 1 (9%) | 0 (0%) | 0 (0%) |
| Rituximab (%) | 25 (34%) | 3 (27%) | 0 (0%) | 0 (0%) |
| TNF inhibitors (%) | 0 (0%) | 0 (0%) | 2 (25%) | 0 (0%) |
| Azathioprine (%) | 5 (7%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Cyclosporine (%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| BVAS1 score (median, range) | 5(0–35) | 8 (0–24) | 0 (0–4) | 1 (0–10) |
| BVAS2 score (median, range) | 0(0–6) | 0 (0–7) | 0 (0–4) | 0 (0–2) |
| PGA (mean ± SD) | 3.30 ± 1.73 | 4.09 ± 1.92 | 3.88 ± 1.46 | 2.78 ± 1.22 |
Disease activity was assessed using physician global assessment (PGA) in all patients with vasculitis. PGA correlates well with the Birmingham Vasculitis Activity Score (BVAS) [22]. BVAS1 score measuring new or worsening symptoms due to active vasculitis in the 28 days prior to evaluation or on the day of evaluation, and BVAS2 score which measures persistent symptoms within the last three months, were also recorded (Tables 1 and 2). For all patients, laboratory data such as creatinine, and markers of systemic inflammation (CRP and ESR) were collected.
Healthy individuals (n=30), were recruited through the University of Washington from Bloodworks in Seattle. For the healthy controls (HC), blood was drawn into EDTA tubes and centrifuged at 3000 rpm for 15 minutes within 30 minutes of the blood collection. Aliquoted plasma was stored at −80°C until used. 17/30 (57%) of HC were women. The median (range) age at the time of the plasma collection was 35 (23–70) years of age. 20/30 (67%) were White, 4/30 (13%) were Hispanic, 4/30 (13%) were Asian, whereas 2/30 (7%) were of unknown race.
The study was approved by the appropriate local institutional review boards at VCRC centers and University of Washington, Seattle, WA (#3100) and informed consent was obtained from all participants in accordance with the Helsinki Declaration.
2.2. ELISA assays
Quantification of circulating NETs was performed by utilizing neutrophil elastase (NE)-DNA ELISA as described in the literature with some modifications [23]. In brief, high binding 96-well ELISA microplates were coated with rabbit anti-human NE antibody (4 μg/ml; Calbiochem) in PBS overnight at 4°C, and then blocked with 1% bovine serum albumin (BSA) in PBS for 2 hours at room temperature (RT). Then, plasma samples diluted 1:10, were added in 1% BSA in PBS with 2mM EDTA, and incubated overnight at 4°C. Anti-DNA antibody conjugated to HRP from Cell Death Detection ELISA kit (clone MCA-33; Roche) was added as secondary antibody to detect bound NETs for 2 hours at RT. The reaction was developed with 3,3′,5,5′ tetramethylbenzidine (TMB; BD Biosciences) for 20 min and stopped by the addition of 2N sulfuric acid. Known concentrations of NE-DNA complexes (Innovative Research Inc; Calf thymus DNA, Trevigon) were utilized to construct a standard curve.
Quantification of anti-histone antibodies was performed by using a mixture of histones H1, H2A, H2B, H3, and H4, isolated from the calf thymus (Sigma-Aldrich H9250). Briefly, a 96-well plate was coated with 10 μg/ml of a mixture of histones diluted in PBS and incubated overnight, followed by blocking with 1% BSA in PBS with 0.05% Tween for 2 hours at RT. After blocking, plasma samples that had been diluted 1:10,000 in 1% BSA in PBS with 0.05% Tween, were added to the wells and incubated overnight at 4°C. HRP-conjugated anti-human IgG (Peroxidase Affini Pure Goat Anti-human IgG, Jackson ImmunoResearch Inc, West Grove, PA, USA) was added as a secondary antibody for 2 hours at RT. The reaction was developed with TMB and halted by using 2N sulfuric acid. Known concentrations of anti-histone levels from patients with SLE were used to design the standard curve. Plasma levels of human TSP-1, (R&D Systems, Minneapolis MN, USA)) were measured by ELISA following the manufacturer’s instructions. Absorbance was measured by a plate reader at 450 nm (Synergy 2, BioTek).
2.3. Neutrophil isolation
Heparinized blood from healthy individuals was layered on Polymorphprep (Axis-Shield, Dundee, UK) density gradient, according to the manufacturer’s instructions, or as described previously [24–26]. Red blood cells were lysed using RBC lysis buffer (BioLegend, San Diego, CA, USA). Neutrophils were re-suspended in serum-free RPMI-1640 medium (Life Technologies, Waltham, MA) for in vitro assays.
2.4. NET and DNA degradation assay
NET degradation was assessed using a previously published protocol [27], with some modifications. Briefly, neutrophils were isolated through density gradient (PolymorphPrep; Axis-Shield) and seeded at 1 ×106 cells/ml in a poly-l-lysine–coated black 96-well microtiter plate. Neutrophils were induced to undergo NET formation by addition of 20 nM phorbol myristate acetate (PMA) for 4 hours.
Upon washing, attached NETs were stained with Sytox green (1:5,000; Life Technologies) followed by subsequent wash steps. After recording of baseline fluorescence, plasma (2.5%, diluted in nuclease buffer [10 mM Tris HCl, pH 7.5, 10 mM MgCl2,2 mM CaCl2, 50 mM NaCl]) were added and incubated for 90 minutes at 37°C. Micrococcal nuclease was used as a positive control. After the incubation, the wells were washed, and residual NETs analyzed by plate reader at 480 nm. NET degradation was calculated as the relative loss of NETs (Sytox green signal) in each well, using the standard curve as a reference value. For DNA degradation, Sytox green–labeled DNA (20 μg/ml) was bound to poly-L-lysine–coated plates, and the capacity to degrade the DNA was assessed in the presence of 2.5% plasma in nuclease buffer. Known concentrations of DNase I were used to construct the standard curve.
2.5. Anti-NET assay
At first, 96-well MaxiSorp plates were coated with 100 ul of poly-L-lysine per well. After wash with PBS, isolated neutrophils were seeded at a concentration of 106 cells/mL per well in RPMI, and incubated for 30 min at 37°C with 5% CO2 to allow adherence to the wells. Neutrophils were subsequently activated with 25 uM of calcium ionophore A23187 (Sigma-Aldrich), and incubated for 3 hours to allow maximum NET formation, and then washed with PBS, and incubated with plasma samples diluted 1/100 in 10% FBS with 0.5 M EDTA for 90 min at 37°C with 5% CO2. The secondary Fluorescein (FITC)-conjugated anti-human IgG antibody (Jackson ImmunoResearch Inc, West Grove, PA) diluted at 1/100 in 10% FBS with 0.5 M EDTA was then added for 90 min at RT, and fluorescence was measured at 480 nm in a plate reader. Known concentrations of anti-NET antibodies from patients with SLE were used to design the standard curve.
2.6. NET visualization
A 48 well cell culture plate was coated with 100 ul of poly-L-lysine per well. Human neutrophils from a healthy individual (106 cells/mL per well in RPMI) were then seeded, and incubated for 30 min at 37°C with 5% CO2 to allow adherence to the wells. Neutrophils were subsequently activated with 25 uM of calcium ionophore A23187 (Sigma-Aldrich), incubated for 3 hours to allow maximum NET formation, and washed with PBS. The NETs were then incubated with plasma samples diluted 1/20 in 10% FBS with 0.5 M EDTA for 60 min at 37°C with 5% CO2. Subsequently, the samples were incubated with anti-human IgG APC diluted 1/100 (Clone HP6017, Biolegend), and Sytox Green (5 uM) in 10% FBS with 0.5 M EDTA for 60 minutes at room temperature. NETs were visualized by immunofluorescence (IF) microscopy (EVOS cell imaging system; Life Technology).
2.7. Statistics
Mann-Whitney U test, Wilcoxon signed-rank test, and Spearman’s correlation were used as applicable. The 95th percentile of healthy controls was used to define the cut-off value for high levels of biomarkers. All analyses were performed in GraphPad Prism (GraphPad Software Inc., San Diego, CA, USA) or SPSS Statistics software (v.27.0 IBM Corp, Armonk, NY, USA) and considered statistically significant at p < 0.05. No adjustments were done for multiple testing.
3. Results
3.1. Demographic characteristics of the study populations
The median age (range) at diagnosis of vasculitis for the patients in remission at study entry was as follows: 47 years of age (10–80) for the GPA cohort, 59 years (17–82) for MPA, 30 (12–58) for TAK, and 69 (54–90) years for GCA cohort. 64/123 (52%) patients with GPA, 31/61 (51%) with MPA, 53/58 (91%) with TAK and 49/68 (72%) with GCA were female (Tables 1 and 2).
3.2. Levels of NETs are elevated in patients with vasculitis
Levels of NE-DNA complexes were significantly elevated at flare in patients with GPA (p<0.0001), MPA (p=0.0038), TAK (p<0.0001), and GCA (p<0.0001), and in remission in patients with GPA, p<0.0001; MPA, p=0.005; TAK, p=0.03; and GCA, p=0.0009, when compared to healthy individuals (Figure 1A).
Figure 1. Levels of neutrophil elastase (NE)-DNA complexes in patients with AAV and LVV.
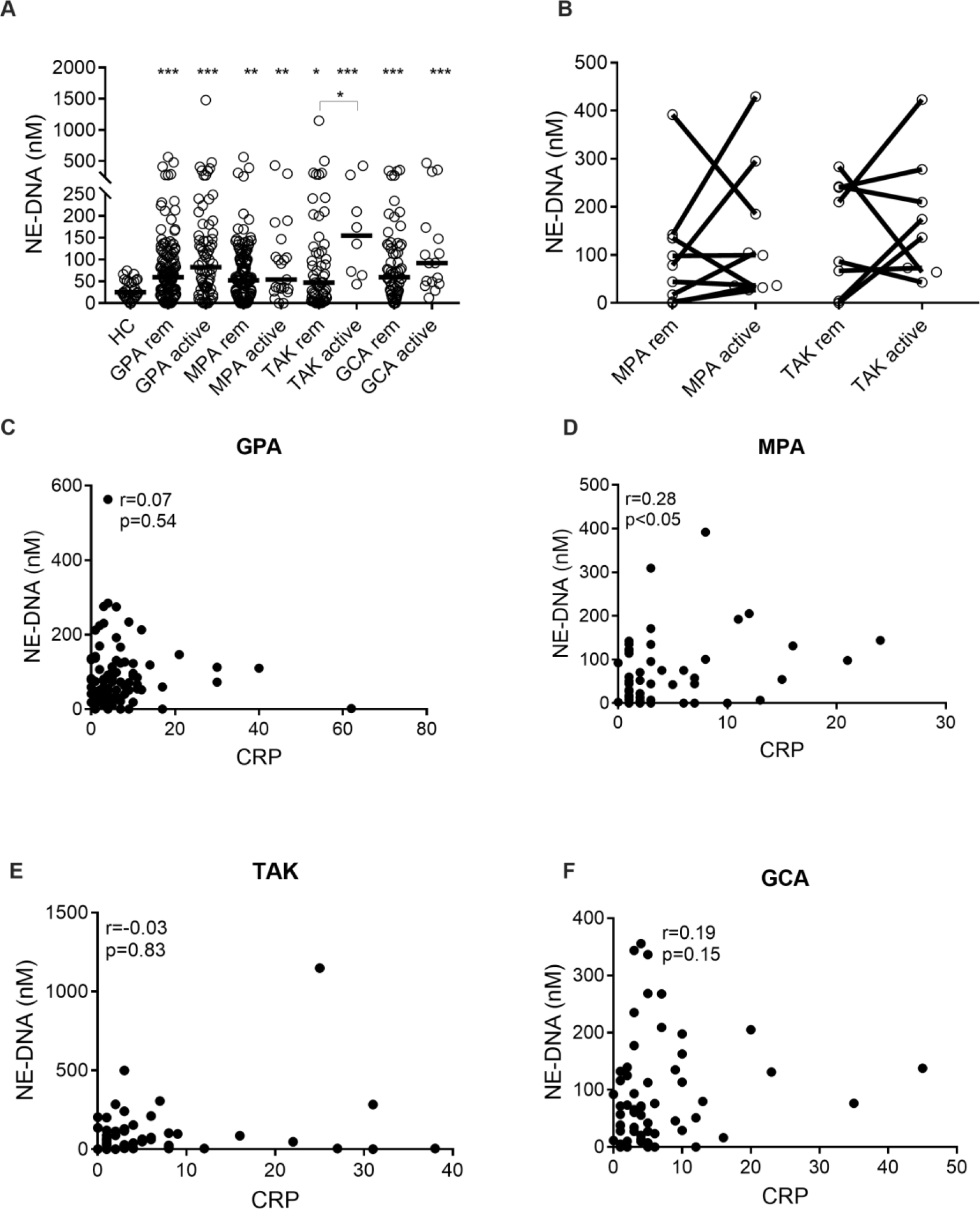
Plasma levels of NE-DNA complexes (A) were measured by ELISA in healthy controls (HC), and patients with granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), Takayasu’s arteritis (TAK), and giant cell arteritis (GCA), both in remission (rem) and at a time of active disease (active). Plasma levels of NE-DNA complexes (B) were related to disease activity in patients in remission (rem) and matching patients with active disease (active) as assessed by physician global assessment (PGA) in MPA and TAK. Plasma levels of NE-DNA complexes correlated with CRP in GPA (C), MPA (D), TAK (E), and GCA (F). Statistical analyses were done using Mann-Whitney U test (A), Wilcoxon signed-rank test (B), Spearman’s correlation test (C-F) with * p < 0.05, **p < 0.01, ***p < 0.001. Unless otherwise indicated, all analyses are compared to healthy controls. Each circle represents an individual sample, with the bar representing the median of the group.
Patients with TAK with active disease had higher levels of NE-DNA complexes compared to patients in remission (p=0.02). The median concentrations (range) of NE-DNA complexes in GPA (n=121), MPA (n=61), TAK (n=58), GCA (n=66) and HC (n=30) were 59.77 (0.00–563.2), 52.20 (0.00–391.8), 46.96 (0.00–1148), 59.66 (0.00–355.8) and 25.13 (0.00–74.46) nM, respectively.
3.3. Association of NET formation with disease activity in patients with vasculitis
No differences in levels of NE-DNA complexes were found between patients in remission and flare for any of the vasculitis groups investigated (Figure 1B). No association was observed between NET formation and CRP levels in GPA (r=0.07, p=0.54, Figure 1C), TAK (r=−0.03, p=0.83, Figure 1E), and GCA (r=0.19, p=0.15, Figure 1F), except for the MPA group (r=0.28, p<0.05, Figure 1D).
3.4. Capacity for NET degradation in patients with vasculitis
Degradation of NETs is a normal process that occurs by the enzyme DNase-I (26). Both impaired degradation and excessive NET formation may contribute to high levels of circulating NETs. We measured the ability of plasma from patients with vasculitis to degrade NETs at the time of remission and compared it to plasma from healthy subjects. As illustrated in Figure 2A, there was an overall impaired capacity for NET degradation in plasma from patients with GPA (p<0.0001), MPA (p=0.009), GCA (p<0.0001), and TAK (p<0.0001) as compared to HC. However, in contrast, plasma from patients with GPA (p=0.0001), MPA (p<0.0001), GCA (p<0.0001), and TAK (p=0.0081) demonstrated increased capacity to degrade DNA as compared to HC (Figure 2B).
Figure 2. Plasma-mediated NET degradation in patients with AAV and LVV.
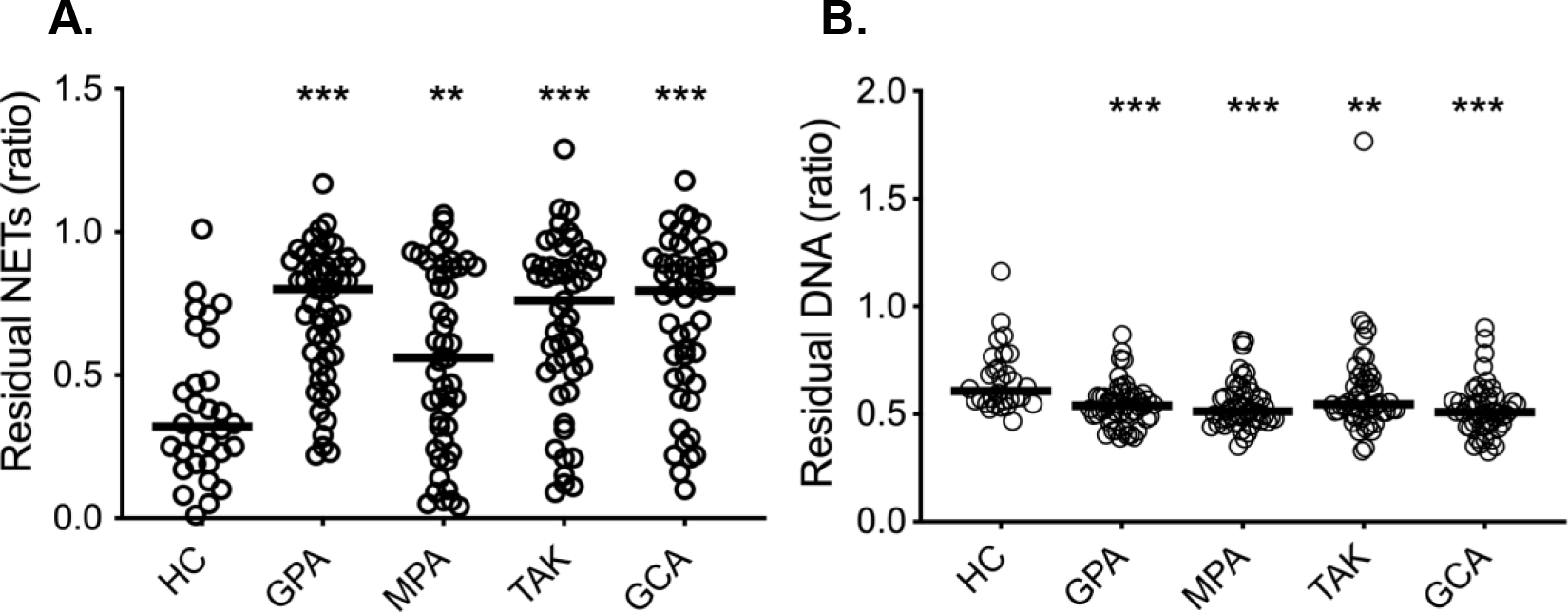
(A) NETs were incubated with plasma from either healthy controls (HC, n=30) or patients with granulomatosis with polyangiitis (GPA, n=53), microscopic polyangiitis (MPA, n=49), Takayasu’s arteritis (TAK, n=51), and giant cell arteritis (GCA, n=48) in remission. Residual NETs were determined by dividing DNA content in plasma-treated NETs with untreated NETs (B) DNA degradation ability was analyzed in plasma from healthy controls and patients with vasculitis. Statistical analyses were done using Mann-Whitney U test, with * p < 0.05, **p < 0.01, ***p < 0.001. All analyses are compared to healthy controls. Each circle represents an individual sample, with the bar representing the median of the group.
3.5. Recognition of NET-associated antigens among patients with vasculitis
Only patients with GPA (p=0.0045) and MPA (p=0.005) showed significant reactivity against NETs as shown in Figure 3A, consistent with presence of anti-PR3 and anti-MPO antibodies binding to their target antigens on NETs. The median concentrations (range) of anti-NET antibodies in GPA (n=50), MPA (n=50), TAK (n=51), GCA (n=49), and HC (n=30) were 0.61 (0.00–106.4), 1.1 (0.00–45.70), 0.35 (0.00–3.53), 0.00 (0.00–17.07), and 0.19 (0.00–3.38) U/ml, respectively.
Figure 3. Levels of anti-NET and anti-histone antibodies in patients with AAV and LVV.
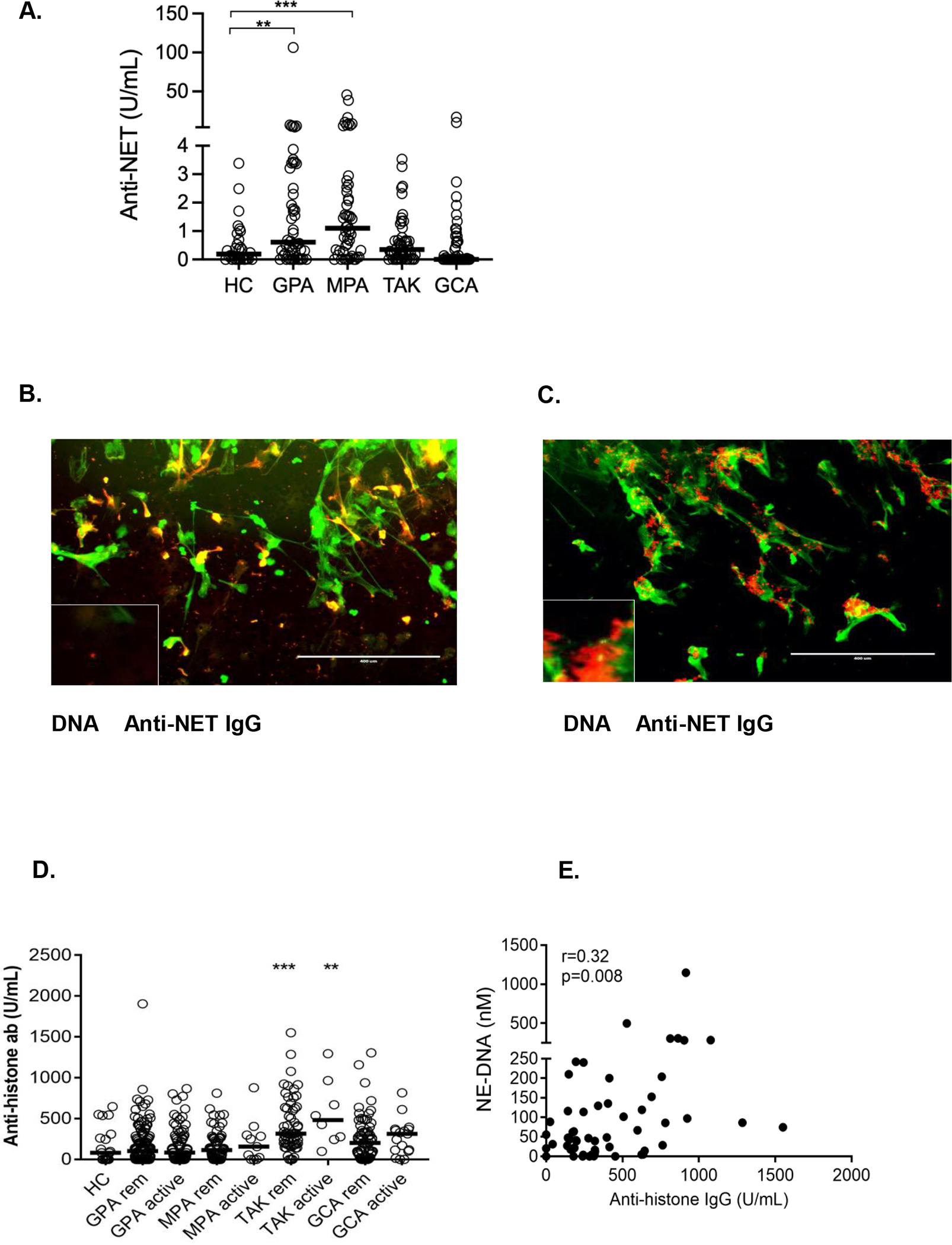
Plasma levels of anti-NET antibodies (A) were measure by ELISA in healthy controls (HC, n=30), in patients with granulomatosis with polyangiitis (GPA, n=50), microscopic polyangiitis (MPA, n=50), Takayasu’s arteritis (TAK, n=51) and giant cell arteritis (GCA, n=49). Immunofluorescence microscopic images showing DNA (green) and IgG (red) staining of stimulated neutrophils incubated with plasma from patients with MPA (B) and TAK (C). Circulating levels of anti-histone antibodies (D) were assessed by ELISA in HC, in patients with GPA, MPA, TAK, and GCA both in remission and at a time of active disease. Plasma levels of anti-histone antibodies were associated with NE-DNA levels in patients with TAK (E). Statistical analyses were done using Mann-Whitney U test (A, D), and Spearman’s correlation test (E) with * p < 0.05, **p < 0.01, ***p < 0.001. All analyses are compared to healthy controls. Each circle represents an individual sample, with the bar representing the median of the group. Scale bars, 400 μm.
Of note, using immunofluorescence microscopy, selecting patients with high levels of anti-NET antibodies, there was binding of IgG from a patient with MPA (Figure 3B) and a patient with TAK (Figure 3C) to distinct NET structures. The presence of anti-neutrophil antibodies, and more specifically anti-NET IgG antibodies, was investigated under microscopy in two patients from each of the GPA, MPA, TAK and GCA cohorts to validate the findings from the anti-NET assay.
3.6. Levels of anti-histone antibodies are elevated in patients with TAK
Given that histones are a prominent immunogenic component of NETs, we further studied whether patients with AAV and LVV have antibodies targeting histones. As depicted in Figure 3D, only patients with TAK had significantly higher levels of anti-histone antibodies both at flare (p=0.0057) and in remission (p=0.0004) compared to HC. There was also a trend towards statistical significance in the GCA group with active disease (p=0.06). Levels of anti-histone antibodies did not differ significantly in patients with AAV (GPA and MPA) compared to HC (Figure 3D). Importantly, levels of anti-histone antibodies correlated with presence of NETs in TAK (r=0.32, p=0.008, Figure 3E).
3.7. Association of platelet activation with NET formation in patients with vasculitis
As depicted in Figure 4A, plasma levels of TSP-1 were elevated compared to HC in patients with GPA (p<0.0001), MPA (p<0.0001), TAK (p<0.0001), and GCA (p<0.0001) in remission, and at flare for GPA (p<0.0001), MPA (p=0.007), TAK (p<0.0001), and GCA (p<0.0001). In GPA (p=0.01), but not in the other vasculitides, levels of TSP-1 were associated with active disease (Figure 4B). In all four subgroups of vasculitis, levels of TPS-1 were correlated with NE-DNA complexes (Figures 4C–4F). Lastly, only in GPA, levels of TSP-1 were significantly elevated among patients with a history of thromboembolism (VTE) compared to those without these events (p=0.04, Figure 4G).
Figure 4. Levels of thrombospondin-1 in patients with AAV and LVV.
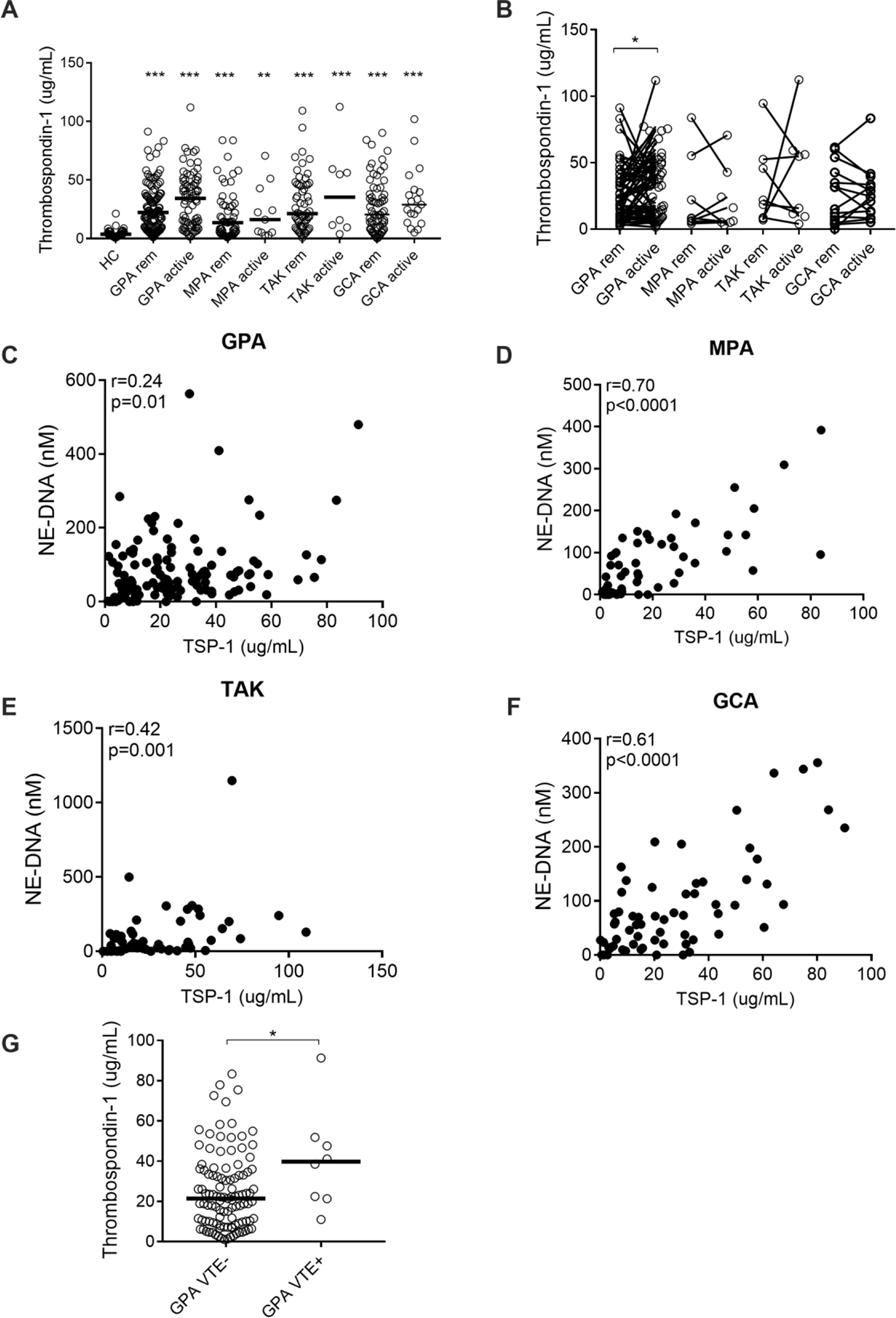
Plasma levels of thrombospondin-1 (TSP-1) (A) were measured by ELISA in healthy controls (HC), and patients with granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), Takayasu’s arteritis (TAK), and giant cell arteritis (GCA) both in remission and at a time of active disease. Plasma levels of TSP-1 (B) were related to disease activity in patients in remission (rem) and matching patients with active disease (active) as assessed by physician global assessment (PGA) in GPA, MPA, TAK and GCA. Plasma levels of TSP-1 correlated with NETs in GPA (C), MPA (D), TAK (E), and GCA (F). Plasma levels of TSP-1 (G) were compared among patients with GPA with and without thromboembolism (VTE). Statistical analyses were done using Mann-Whitney U test (A, G), Wilcoxon signed-rank test (B), Spearman’s correlation test (C-F) with * p < 0.05, **p < 0.01, ***p < 0.001. Unless otherwise indicated, all analyses are compared to healthy controls. Each circle represents an individual sample, with the bar representing the median of the group.
Discussion
Neutrophil activation appears to be a prominent process in patients with systemic vasculitis as evidenced by the presence of markedly elevated levels of neutrophil activating factors in their circulation [28]. Although NET formation has extensively been investigated in AAV [29], very little is known about the presence of NETs in LVV. In this study we made the seminal observation of levels of NETs in the peripheral blood of patients with LVV. Consistent with those findings, presence of NETs has also recently been identified in temporal artery biopsies from patients with GCA [30]. Interestingly, levels of NETs did not correlate with disease activity in any of the subgroups of vasculitis. Other studies have found mixed results regarding levels of circulating NETs in AAV with one study also finding no correlation with disease activity [31] and one finding such an association [32]. Differences in patient populations, sample sizes, and ANCA positivity status most likely could explain the discordant results in these studies. Taken together, levels of NETs do not seem to be a reliable indicator of disease activity in systemic vasculitis, but they may indicate chronic low-grade inflammation that could lead to organ damage, similarly to what we found in SLE [33].
The elevated levels of circulating NETs could be due to either excessive NET formation and/or dysfunctional clearance of NETs. The trigger(s) of NET induction in systemic vasculitis are not fully known. Previous studies implicated ANCA IgG and IgA in driving NET formation [34, 35]. However, excessive serum-mediated ex vivo NET formation has been reported in patients with AAV in an ANCA-independent process [36]. It is possible that the presence of serum factors other than ANCA IgG prevents NET degradation and/or augments NET formation in both AAV and LVV.
Our study found reduced NET degradation (but not DNA degradation) in all subgroups of vasculitis, similarly to what has been described in SLE [18,20], RA [19] and juvenile dermatomyositis [37]. In SLE, impaired NET degradation has been attributed to the presence of DNase-I inhibitor and anti-NET antibodies that enhance tissue C1q deposition [38]. Patients with AAV were found to have significantly higher levels of DNase I as compared to healthy controls [31]. This finding is compatible with our findings of the increased capacity for plasma samples from patients with systemic vasculitis to degrade DNA. However, it does not explain the reduced NET clearance in both patients with AAV and LVV, suggesting the presence of other mechanisms operating in the imbalance between NET formation and degradation.
Notably, in our study patients with AAV had higher levels of circulating anti-NET IgG antibodies compared to healthy objects. The presence of these anti-NET antibodies may not only impair the intrinsic capacity of DNase I to degrade NETs, but also result in formation of local immune complexes with NET-derived autoantigens protecting further NET clearance and amplifying the whole inflammatory process in systemic vasculitis. In our study, some TAK patients also showed significant reactivity against distinct NET structures as observed by microscopy. However, there was no significant correlation between circulating levels of NETs and antibodies against NETs in patients with both AAV and LVV (data not shown) similarly to what has been reported in patients with RA [17]. The identity of the antigen of these anti-NET antibodies is yet unknown. A recent study identified a neutrophilic 17-kDa protein that could be considered as a candidate for the anti-NET antigen in MPA (21).
The cargo of NETs can be modified by distinct stimuli, therefore exposing a variety of autoantigens that could be the target of posttranslational and/or epigenetic modifications in different autoimmune conditions. For example, in cocaine-induced midline destructive lesions [39] as well as in cocaine levamisole autoimmunity associated syndrome [40, 41], NETs are enriched with NE. One of the known inflammatory mediators that are released from NETs includes histones, that when misplaced to the extracellular space can cause organ damage such as kidney injury [42]. Histones in NETs can be deiminated, and citrullinated, but only individuals with genetic predisposition to develop antibodies to deiminated, and citrullinated proteins identified as anti-citrullinated proteins/peptides antibodies (ACPA) will eventually develop seropositive RA [43]. In our study autoantibodies targeting histones were also identified in patients with TAK and less frequently in GCA, without any association with disease activity. Mechanisms driving reactivity to histone antigens in LVV are unknown but may relate to NET formation based on the results of our study and could be the focus of future translational studies.
Our study also presents evidence of increased platelet activation as measured by the platelet marker TSP-1, and its association with neutrophil cell death in patients with AAV and LVV, supporting platelets’ potential interaction and contribution to the pathogenesis of vasculitis. Mechanistically, platelets have been suggested to be involved in GCA, with elevated levels of platelet activation, as well as formation of platelet-neutrophil complexes contributing to systemic inflammation [44]. Both platelet activation and NET formation are thought to be involved in the development of thromboembolic disease [45]. Of note TSP-1 levels were significantly higher in GPA patients with a history of thromboembolic events compared to those without. Interestingly, in another study plasma levels of TSP-1 were found to be the highest among patients with anti-phospholipid syndrome (APS) that had arterial thrombotic events [46]. We also demonstrated that levels of TSP-1 were associated with active disease in GPA. TSP-1 has known pro-apoptotic properties, with dysregulation in cell death pathways in the neutrophils of patients with GPA to have been reported in the literature [47]. Both AAV and LVV are associated with an increased risk of venous thrombosis, especially during periods of active disease [48–50]. The source of TSP-1 could not be established in this study. Although platelets appear to be the major source of TSP-1, TSP-1 can also be stored in Weibel-Palade bodies of endothelial cells [51].
Our study has several notable strengths. The biospecimens and linked clinical data were collected in a prospective protocolized fashion at major centers of excellence for vasculitis. The comprehensive set of assays measured were using established techniques in a moderately large number of samples given the rarity of each disease under study.
Our study also has a few limitations to consider. Patients in the study were on a variety of immunosuppressive therapeutic agents and the effects of these drugs on the levels of NETs is not fully known, especially considering that NETs are associated with inflammation. Additionally, these results need to be confirmed in a separate validation study.
Our findings highlight both excessive NET formation and defective NET clearance as common mechanisms in vasculitis, supporting the development of novel preventative treatment strategies against NET formation for these diseases. Specific inhibitors of protein kinase C (PKC), c-rapidly accelerated fibrosarcoma (c-Raf) kinase and mitogen-activated protein kinase (MEK) can prevent NET formation, by blocking the downregulation of the anti-apoptotic protein Mcl-1, highlighting the seminal role of the Raf-MEK-ERK pathway in NET formation [52]. Alvelestat, a neutrophil elastase inhibitor was shown to inhibit tissue damage and inflammation in a mouse model of acute lung injury/acute respiratory distress syndrome by inhibiting NETs [53]. BAY 85-8501 is another novel selective and reversible inhibitor of NE that inhibits NET formation in vitro following stimulation with PMA, that reduced pulmonary inflammation in a protease-induced acute lung injury mouse model [54].
In conclusion, neutrophil activation and NET formation is a potential common disease process in AAV and LVV. Impaired NET degradation, possibly due to presence of anti-NET antibodies, may promote NET accumulation in the peripheral blood of patients with vasculitis. Although more frequent in AAV, also some patients with LVV have anti-NET IgG antibodies. In patients with TAK, the association of anti-histone antibodies with NETs suggests that NETs could be a source of autoantigens in that subgroup of vasculitis. Finally, the association of the platelet-derived TSP-1 with NET formation, indicates a link between markers of neutrophil and platelet activation in these conditions.
Highlights.
Neutrophils and neutrophil extracellular traps (NETs) not only possess antimicrobial properties, but are also key players in the pathophysiology of many autoimmune diseases.
Although there is extensive literature about NET formation in ANCA-associated vasculitis, studies about neutrophil cell death in large-vessel vasculitis are extremely limited.
Increased NET levels associated with platelet activation, along with impaired NET degradation are common features in both ANCA-associated and large-vessel vasculitis.
Whilst more frequent in ANCA-associated vasculitis, some patients with large-vessel vasculitis also have antibodies targeting NETs.
Targeting both NET formation and degradation could be beneficial therapeutic approaches in the management of both ANCA-associated and large-vessel vasculitis.
Funding:
DM is supported by the NIH training grant, award #5T32HL007028-44 and Pfizer US Pharmaceuticals Group grant (sponsor award number 53857367). PG is funded by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) intramural program. CL is supported by the NIH grants 1R21EY029391, 1R01HL158606, and R21AR075129.
The VCRC is part of the Rare Diseases Clinical Research Network, an initiative of the Office of Rare Diseases Research, National Center for Advancing Translational Science (NCATS). The VCRC has received funding from NCATS, the NIAMS (U54 AR057319), the National Center for Research Resources (U54 RR019497), and GlaxoSmithKline.
Footnotes
Declarations
Conflict of Interest: Dr. Michailidou received Advisory Board fees from ChemoCentryx. Dr. Khalidi received clinical trial support from BMS, Sanofi and Abbvie, travel support from Astra Zeneca, and Advisory Board fee from Roche. Dr. Koening served on the advisory board for Chemocentryx and Genentech. Dr. Specks reports receiving funds for the following activities: Consulting: AstraZeneca, ChemoCentryx. Research Support: AstraZeneca, GlaxoSmithKline, Bristol-Myers Squibb, Genentech/Roche, InflaRx. Dr. Sreih works at Bristol-Myers Squibb and owns Astra Zeneca and Alexion Stocks. Dr. Warrington received clinical trial support from Eli Lilly and Kiniksa. Dr. Monach received consulting fees from Kiniksa, Celgene/BMC, and ChemoCentryx. Dr. Merkel reports receiving funds for the following activities: Consulting and Research Support: AbbVie, AstraZeneca, Boeringher-Ingelheim, Bristol-Myers Squibb, ChemoCentryx, Forbius, Genentech/Roche, Genzyme/Sanofi, GlaxoSmithKline, InflaRx, Takeda. Consulting only: CSL Behring, Dynacure, EMDSerono, Janssen, Kiniksa, Kyverna, Magenta, MiroBio, Neutrolis, Novartis, Pfizer, Sparrow, Talaris. Royalties: UpToDate. Dr. Lood received research funding from Pfizer, Gilead Sciences, Boehringer Ingelheim, Redd Pharma, Amytryx, and Eli Lilly.
Patient consent for publication: Informed written consent was obtained from all participants in accordance with the Helsinki Declaration
Ethics approval: The study was approved by the regional ethics board at University of Washington, Seattle, WA, (#3100).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data availability statement:
All data relevant to this study are included in the article
References
- 1.Grayson PC, Kaplan MJ. At the Bench: Neutrophil extracellular traps (NETs) highlight novel aspects of innate immune system involvement in autoimmune diseases. J Leukoc Biol, 99 (2016), pp.253–64, 10.1189/jlb.5BT0615-247R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sørensen OE, Borregaard N. Neutrophil extracellular traps-the dark side of neutrophils. J Clin Invest, 126 (2016), pp. 1612–20, 10.1172/JCI84538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Bont CM, Eerden N, Boelens WC, Pruijn GJM. Neutrophil proteases degrade autoepitopes of NET-associated proteins. Clin Exp Immunol, 199 (2020), pp. 1–8, 10.1111/cei.13392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Michailidou D, Mustelin T, Lood C. Role of Neutrophils in Systemic Vasculitides. Front Immunol, 11 (2020), 10.3389/fimmu.2020.619705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum, 65 (2013), pp. 1–11, 10.1002/art.37715. [DOI] [PubMed] [Google Scholar]
- 6.McKinney EF, Willcocks LC, Broecker V, S Smith KG. The immunopathology of ANCA-associated vasculitis. Semin Immunopathol., 36 (2014), pp. 461–78, 10.1007/s00281-014-0436-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jennette JC, Falk RJ. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat Rev Rheumatol., 10 (2014), pp. 463–73, 10.1038/nrrheum.2014.103. [DOI] [PubMed] [Google Scholar]
- 8.Michailidou DM, Rosenblum JS, Rimland CA, Marko J, Ahlman MA, Grayson PC. Clinical symptoms and associated vascular imaging findings in Takayasu’s arteritis compared to giant cell arteritis. Ann Rheum Dis, 79 (2020), pp. 262–267, 10.1136/annrheumdis-2019-216145. [DOI] [PubMed] [Google Scholar]
- 9.Yuan J, Gou SJ, Huang J, Hao J, Chen M, Zhao MH. C5a and its receptors in human anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Arthritis Res Ther., 14 (2012), p.140, 10.1186/ar3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Porges AJ, Redecha PB, Kimberly WT, Csernok E, Gross WL, Kimberly RP. Anti-neutrophil cytoplasmic antibodies engage and activate human neutrophils via Fc gamma RIIa. J Immunol, 153 (1994), pp.1271–80. [PubMed] [Google Scholar]
- 11.Matsumoto K, Yasuoka H, Yoshimoto K, Suzuki K, Takeuchi T. Platelet CXCL4 mediates neutrophil extracellular traps formation in ANCA-associated vasculitis. Sci Rep, 11 (2021), p. 222, 10.1038/s41598-020-80685-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakazawa D, Tomaru U, Yamamoto C, Jodo S, Ishizu A Abundant neutrophil extracellular traps in thrombus of patient with microscopic polyangiitis. Front Immunol, 3 (2012), p. 333, 10.3389/fimmu.2012.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knight JS, Carmona-Rivera C C, Kaplan MJ. Proteins derived from neutrophil extracellular traps may serve as self-antigens and mediate organ damage in autoimmune diseases. Front Immunol, 3 (2012), p. 380, 10.3389/fimmu.2012.00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Bavel CC, Dieker JW, Kroeze Y, Tamboer WP, Voll R, Muller S, et al. Apoptosis-induced histone H3 methylation is targeted by autoantibodies in systemic lupus erythematosus. Ann Rheum Dis, 70 (2011), pp. 201–7, 10.1136/ard.2010.129320. [DOI] [PubMed] [Google Scholar]
- 15.Sato S, Ihn H, Kikuchi K, Takehara K. Anti-histone antibodies in systemic sclerosis: association with pulmonary fibrosis. Arthritis Rheum, 37 (1994), pp. 391–394. 10.1002/art.1780370313. [DOI] [PubMed] [Google Scholar]
- 16.Kubo M, Ihn H, Yazawa N, Sato S, Kikuchi K, Tamaki K. Prevalence and antigen specificity of anti-histone antibodies in patients with polymyositis/dermatomyositis. J Invest Dermatol, 112 (1999), pp. 711–5, 10.1046/j.1523-1747.1999.00580.x. [DOI] [PubMed] [Google Scholar]
- 17.de Bont CM, Stokman MEM, Faas P, Thurlings RM, Boelens WC, Wright HL, et al. Autoantibodies to neutrophil extracellular traps represent a potential serological biomarker in rheumatoid arthritis. J Autoimmun, 113 (2020), Article102484, 10.1016/j.jaut.2020.102484. [DOI] [PubMed] [Google Scholar]
- 18.Leffler J, Martin M, Gullstrand B, Tydén H, Lood C, Truedsson L, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol, 188 (2012), pp. 3522–31, 10.4049/jimmunol.1102404. [DOI] [PubMed] [Google Scholar]
- 19.Bach M, Moon J, Moore R, Pan T, Nelson JL, Lood C. A neutrophil activation bomarker panel in prognosis and monitoring of patients with rheumatoid arthritis. Arthritis Rheum, 72 (2020), pp. 47–56, 10.1002/art.41062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA. 107 (2010), pp. 9813–8, 10.1073/pnas.0909927107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hattanda F, Nakazawa D, Watanabe-Kusunoki K, Kusunoki Y, Shida H, Masuda S, et al. The presence of anti-neutrophil extracellular trap antibody in patients with microscopic polyangiitis. Rheumatology (Oxford). 58 (2019), pp. 1293–1298, 10.1093/rheumatology/kez089. [DOI] [PubMed] [Google Scholar]
- 22.Mahr AD, Neogi T, Lavalley MP, Davis JC, Hoffman GS, McCune WJ, et al. Assessment of the item selection and weighting in the Birmingham vasculitis activity score for Wegener’s granulomatosis. Arthritis Rheum, 59 (2008), pp. 884–91, 10.1002/art.23707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med, 22 (2016), pp. 146–53, 10.1038/nm.4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med, 5 (2013), 178ra40, 10.1126/scitranslmed.3005580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR et al. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol, 184 (2010), pp. 3284–97, 10.4049/jimmunol.0902199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodriguez-Nuevo A, Zorzano A. The sensing of mitochondrial DAMPs by non-immune cells. Cell Stress. 3 (2019), pp. 195–207, 10.15698/cst2019.06.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leffler J, Gullstrand B, Jönsen A, Nilsson JÅ, Martin M, Blom AM, et al. Degradation of neutrophil extracellular traps co-varies with disease activity in patients with systemic lupus erythematosus. Arthritis Res Ther, 15 (2013) R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michailidou D, Duvvuri B, Kuley R, Cuthbertson D, Grayson PC, Khalidi NA, et al. Markers of neutrophil activation in patients with anti-neutrophil cytoplasmic autoantibody-associated vasculitis and large-vessel vasculitis. Arthritis Res Ther. 24 (2022), p. 160, 10.1186/s13075-022-02849-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kessengrock K, Krumbholz M, Schonermark U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 15 (2009), pp. 623–5. doi: 10.1038/nm.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palamidas DA, Argyropoulou OD, Georgantzoglou N, Karatza E, Xingi E, Kapsogeorgou EK et al. Neutrophil extracellular traps in giant cell arteritis biopsies: presentation, localization and co-expression with inflammatory cytokines. Rheumatology (Oxford). 61 (2022), pp. 1639–44, 10.1093/rheumatology/keab505. [DOI] [PubMed] [Google Scholar]
- 31.Wang H, Sha LL, Ma TT, Zhang LX, Chen M M, Zhao MH. Circulating level of neutrophil extracellular traps is not a useful biomarker for assessing disease activity in antineutrophil cytoplasmic antibody-associated vasculitis. PLoS ONE. 11 (2016), e0148197, 10.1371/journal.pone.0148197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soderberg D, Kurz T, Motamedi A, Hellmark T, Eriksson P, Segelmark M. Increased levels of neutrophil extracellular trap remnants in the circulation of patients with small vessel vasculitis, but an inverse correlation to anti-neutrophil cytoplasmic antibodies during remission. Rheumatology (Oxford). 54 (2015), pp. 2085–94, 10.1093/rheumatology/kev217. [DOI] [PubMed] [Google Scholar]
- 33.Moore S, Juo HH, Nielsen CT, Tyden H, A Bengtsson A, Lood C. Role of Neutrophil Extracellular Traps Regarding Patients at Risk of Increased Disease Activity and Cardiovascular Comorbidity in Systemic Lupus Erythematosus. J Rheumatol, 47 (2020), pp. 1652–60, 10.3899/jrheum.190875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakazawa D, Shida H, Tomaru U, Yoshida M, Nishio S, Atsumi T, et al. Enhanced formation and disordered regulation of NETs in myeloperoxidase-ANCA-associated microscopic polyangiitis. J Am Soc Nephrol, 25 (2014), pp. 990–7, 10.1681/ASN.2013060606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kelley JM, Monach PM, Ji C, Zhou Y, Wu J, Tanaka S, et al. IgA and IgG antineutrophil cytoplasmic antibody engangement of Fc receptor genetic variants influences granulomatosis with polyangiitis. Proc Natl Acad Sci USA. 108 (2011), pp. 20736–41, 10.1073/pnas.1109227109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kraaij T, Kamerling SWA, van Dam LS, Bakker JA, Bajema IM, Page T, et al. Excessive neutrophil trap formation in ANCA-associated vasculitis is independent of ANCA. Kidney Int, 94 (2018), pp. 139–49, 10.1016/j.kint.2018.01.013. [DOI] [PubMed] [Google Scholar]
- 37.Duvvuri B, Pachman LM, Morgan G, Khojah AM, Klein-Gitelman M, Curran ML, et al. Neutrophil extracellular traps in Tissue and Periphery in Juvenile Dermatomyositis. Arthritis Rheum, 72 (2019), pp. 348–358, 10.1002/art.41078. [DOI] [PubMed] [Google Scholar]
- 38.Frangou E, Vassilopoulos D, Boletis J, Boumpas DT. An emerging role of neutrophils and NETosis in chronic inflammation and fibrosis in systemic lupus erythematosus (SLE) and ANCA-associated vasculitides (AAV): implications for the pathogenesis and treatment. Autoimmun Rev., 18 (2019), pp. 751–60, 10.1016/j.autrev.2019.06.011. [DOI] [PubMed] [Google Scholar]
- 39.Wiesner O, Russell KA, Lee AS, Jenne DE, Trimarchi M, Gregorini G, et al. Antineutrophil cytoplasmic antibodies reacting with human neutrophil elastase as a diagnostic marker for cocaine-induced midline destructive lesions but not autoimmune vasculitis. Arthritis Rheum, 50 (2004), pp. 2954–65. doi: 10.1002/art/20479. [DOI] [PubMed] [Google Scholar]
- 40.Lood C, Hughes GC. Neutrophil extracellular traps as a potential source of autoantigen in cocaine-associated autoimmunity. Rheumatology (Oxford). 56 (2017), pp. 638–43, 10.1093/rheumatology/kew256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carmona-Rivera C, Purmalek MM, Moore E, Waldman M, Walter PJ, Garraffo HM, et al. A role for muscarinic receptors in neutrophil extracellular trap formation and levamisole-induced autoimmunity. JCI Insight. 2 (2017). e89780, 10.1172/jci.insight.89780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Allam R, Scherbaum CR, Darisipudi MN, Mulay SR, Hägele H, Lichtnekert J, et al. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol, 23 (2012), pp. 1375–88, 10.1681/ASN.2011111077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Corsiero E, Pratesi F, Prediletto E, Bombardieri M, Migliorini P. NETosis as source of autoantigens in rheumatoid arthritis. Front Immunol, 7 (2016), p. 485, doi: 10.3389/fimmu.2016.00485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maugeri N, Baldini M, Rovere-Querini P, Maseri A, Sabbadini MG. Leukocyte and platelet activation in patients with giant cell arteritis and polymyalgia rheumatica: a clue to thromboembolic risks? Autoimmunity. 42 (2009), pp. 386–8, 10.1080/08916930902832629. [DOI] [PubMed] [Google Scholar]
- 45.Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood. 123 (2014), pp. 2768–2776, 10.1182/blood-2013-10-463646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patsouras M, Tsiki E, Karagianni P, Vlachoyiannopoulos PG. The role of thrombospondin-1 in the pathogenesis of antiphospholipid syndrome. J Autoimmun, 115 (2020), Article 102527, 10.1016/j.jaut.2020.102527. [DOI] [PubMed] [Google Scholar]
- 47.Everts-Graber J, Martin KR, Thieblemont N, Mocek J, Roccabianca A, Chafey P, et al. Proteomic analysis of neutrophils in ANCA-associated vasculitis reveals a dysregulation in proteinase 3-associated proteins such as annexin-A1 involved in apoptotic cell clearance. Kidney Int, 96 (2019), pp. 397–408, 10.1016/j.kint.2019.02.017. [DOI] [PubMed] [Google Scholar]
- 48.Merkel PA, Lo GH, Holbrook JT, Tibbs AK, Allen NB, Davis JC Jr, et al. High incidence of venous thrombotic events among patients with Wegener granulomatosis: the Wegener’s Clinical Occurrence of Thrombosis (WeCLOT) Study. Ann Intern Med, 142 (2005), pp. 620–6, 10.7326/0003-4819-142-8-200505030-00011. [DOI] [PubMed] [Google Scholar]
- 49.Unizony S, Lu N, Tomasson G, Zhang Y, Merkel PA, Stone JH, et al. Temporal trends of venous thromboembolism risk before and after diagnosis of giant cell arteritis. Arthritis Rheumatol, 69 (2017), pp. 176–184, 10.1002/art.39847. [DOI] [PubMed] [Google Scholar]
- 50.Michailidou DD, Zhang T, Stamatis P, Ng B. Risk of venous and arterial thromboembolic evens in giant cell arteritis: A US veterans health administration population-based study. J Intern Med, 291 (2022), pp. 665–675. doi: 10.1111/joim.13446. [DOI] [PubMed] [Google Scholar]
- 51.Mosher DF, Doyle MJ, Jaffe EA. Synthesis and secretion of thrombospondin by cultured human endothelial cells. J Cell Biol, 93 (1982), pp. 343–348, 10.1083/jcb.93.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, et al. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol, 75 (2011), pp. 75–7, 10.1038/nchembio.496. [DOI] [PubMed] [Google Scholar]
- 53.Li H, Zhou X, Tan H, Hu Y, Zhang L, Liu S, et al. Neutrophil extracellular traps contribute to the pathogenesis of acid-aspiration-induced ALI/ARDS. Oncotarget. 9 (2018), pp. 1772–84, 10.18632/oncotarget.22744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nagelschmitz JKD, Von Nussbaum F, Delbeck M, Lusting K, Bandel T, Watz H. The novel inhibitor BAY 85-8501 provides a new approach in the treatment of pulmonary diseases. Eur Respirat J., 44 (2014), p. 1510. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data relevant to this study are included in the article