

Abstract
Dietary interventions are promising approaches to treat pain associated with metabolic changes because they impact both metabolic and neural components contributing to painful neuropathy. Here, we tested whether consumption of a ketogenic diet could affect sensation, pain, and epidermal innervation loss in type 1 diabetic mice. C57Bl/6 mice were rendered diabetic using streptozotocin and administered a ketogenic diet at either three weeks (prevention) or nine weeks (reversal) of uncontrolled diabetes. We quantified changes in metabolic biomarkers, sensory thresholds, and epidermal innervation to assess impact on neuropathy parameters. Diabetic mice consuming ketogenic diet had normalized weight gain, reduced blood glucose, elevated blood ketones, and reduced hemoglobin-A1C levels. These metabolic biomarkers were also improved following nine weeks of diabetes followed by four weeks of a ketogenic diet. Diabetic mice fed a control chow diet developed rapid mechanical allodynia of the hind paw that was reversed within a week of consumption of a ketogenic diet in both prevention and reversal studies. Loss of thermal sensation was also improved by consumption of a ketogenic diet via normalized thermal thresholds. Finally, diabetic mice consuming a ketogenic diet had normalized epidermal innervation, including after nine weeks of uncontrolled diabetes and four weeks of consumption of the ketogenic diet. These results suggest that, in mice, a ketogenic diet can prevent and reverse changes in key metabolic biomarkers, altered sensation, pain and axon innervation of the skin. These results identify a ketogenic diet as a potential therapeutic intervention for patients with painful diabetic neuropathy and/or epidermal axon loss.
Introduction
Painful small-fiber neuropathy is a common complication of diabetes, as patients with diabetes have a 3.6-fold increased risk of developing this chronic pain condition [4]. Diabetic peripheral neuropathy (DPN) affects 10- to 50-percent of all patients with diabetes [22], and small fiber DPN commonly presents as both reduced sensation and pain in the lower extremities and hands. During disease progression, diabetes patients experience a progressive loss of peripheral innervation in the skin [13; 23; 34], which is represented clinically as a reduction in intraepidermal nerve fiber (IENF) density. Hyperglycemia and hemoglobin-A1C (Hb-A1C), a glycosylated hemoglobin adduct, are strong predictors for the incidence and severity of DPN in patients with diabetes [21; 37]. Glucotoxicity and toxicity from glycolysis-derived metabolites are also strongly associated with DPN [1; 6; 42]. These processes lead to hyperexcitability of sensory neurons in the dorsal root ganglia and can directly activate pronociceptive signaling circuits [1; 6], leading to pain. Despite the prevalence of DPN and its negative effect on quality of life, there are few therapeutic options available to patients with painful DPN and no clinically-available treatments that can improve lost sensation or restore peripheral innervation.
Prior to the discovery and mass production of insulin, very-low carbohydrate, ketogenic diets were used by Allen and others to manage type 1 diabetes. In recent years, ketogenic diets have experienced a resurgence of interest as therapeutic modalities in a range of neurologic diseases, including epilepsy [7; 18; 20], Alzheimer’s disease [8; 48], and various chronic pain conditions [16; 17; 40; 41]. While the mechanism(s) by which a ketogenic diet ameliorates chronic pain remains loosely defined, the low-carbohydrate nature of this diet has been demonstrated to reduce fasting blood glucose in models of diabetes [32] and diabetic nephropathy [39].
Previously, our lab has published that a ketogenic diet can reverse mechanical allodynia associated with consumption of a high fat diet [16]. Additionally, β-hydroxybutyrate (β-HB), the primary circulating ketone body in mice and humans on a ketogenic diet, stimulated neurite outgrowth in vitro. We therefore hypothesized that a ketogenic diet might improve DPN in a model of experimental diabetes. We used streptozotocin (STZ) to induce uncontrolled hyperglycemia in mice and measured whether a ketogenic diet altered biomarkers for diabetes progression, nociceptive behavior, and peripheral innervation of the hind paw. Here, we report that a ketogenic diet 1) is well tolerated in a type I-like diabetes mouse model, 2) rescues both mechanical allodynia and loss of thermal sensation, and 3) stimulates regeneration of intraepidermal nerve fibers that are lost in DPN. These findings have important implications for dietary interventions associated with painful neuropathy related to metabolic dysfunction.
Materials and Methods
Animals and Diet
All animal work was performed following review and approval by the Institutional Animal Care and Use Committee of Kansas University Medical Center. Eight-week-old C57/Bl6 mice #027 were purchased from Charles River Laboratories (Wilmington, MA) and maintained on a 12:12 light:dark cycle in the animal research facility at the University of Kansas Medical Center. Mice were given ad libitum access to water and a control rodent chow (TD.8604; Envigo, Madison, WI; 14% fat, 32% protein, and 54% carbohydrate by kcal) or a ketogenic diet (TD.96355; Envigo, 90.5% fat, 9.2% protein, and 0.3% carbohydrate by kcal). Mice fed a ketogenic diet were given fresh diet every 3–4 days.
Experimental Induction of Type 1 Diabetes
Diabetes was experimentally induced by intraperitoneal (i.p.) injection of streptozotocin (STZ; Sigma, Lot #WXBC8740V; St. Louis, MO). Briefly, mice were fasted for 3 hours before receiving an i.p. injection of either control sodium citrate buffer (pH 4.5) or 180 mg/kg STZ dissolved in sodium citrate buffer (pH 4.5). We measured fasting blood glucose 72 hours later as described below to confirm induction of diabetes (fasting blood glucose ≥ 250 mg/dL). Mice that did not develop hyperglycemia were given subsequent injections of 30 mg/kg STZ and reevaluated by fasting blood glucose every 72 hours until diabetic or the mice had received a cumulative dose of 240 mg/kg STZ.
Blood Measurements
Fasting blood glucose was determined biweekly as described by Groover, et. al. [27]. Briefly, mice were fasted for 3 hours before drawing 20 μL blood from the tail vein by clipping off the tip of the tail or the resulting scab in subsequent blood draws. We used Vaseline during blood draw to avoid skin abrasion related to the blood draw. All standards and samples were mixed with molecular H2O, ZnSO4 (Sigma), and Ba(OH)2 (Sigma) and centrifuged at 13000 rpm and 4 °C for 5 minutes. The resultant supernatant was incubated for 30 minutes at 37 °C with color reagent (PGO capsule, Sigma; o-Dianisidine Dihydrochloride, Sigma; in molecular water). The absorbance of standards and samples was then determined by plate reader (SpectraMax M5) at 450 nm.
Blood ketones were measured using a hand-held blood monitor and β-hydroxybutyrate blood ketone strips (β-Ketone blood test strips, Abbott Laboratories, Chicago, IL; Precision Xtra, Abbott Laboratories). Blood ketones were determined biweekly after a 3 hour fast in STZ- and sodium citrate-injected mice. Saline- and MGO-injected mice were not fasted prior to blood ketone measures.
During the early ketogenic diet intervention, insulin levels were determined following a 3 hour fast biweekly. Tail vein blood was collected and allowed to coagulate on ice. Samples were then centrifuged for 30 minutes at 4 °C and 3000 rpm, and serum was drawn off for storage at −80 °C until analysis by ELISA (Alpco; Salem, NH). A1CNow+ Test Kits (PTSDiagnostics) were used to determine Hb-A1c measures from tail blood immediately before sacrifice. Mice were not fasted prior to Hb-A1c measurement.
Sensory Behavioral Testing
Sensory behavioral testing was performed at baseline and biweekly for STZ-injected mice (Figure 1). Prior to collection of baseline data, mice were acclimated to testing areas for 30 minutes and either the mesh table or thermal testing apparatus for 30 minutes on at least two occasions separated by 24 hours. Prior to collection of all sensory behavioral data, mice were again acclimated to the testing area, mesh table, and thermal testing apparatus for 30 minutes each.
Figure 1.
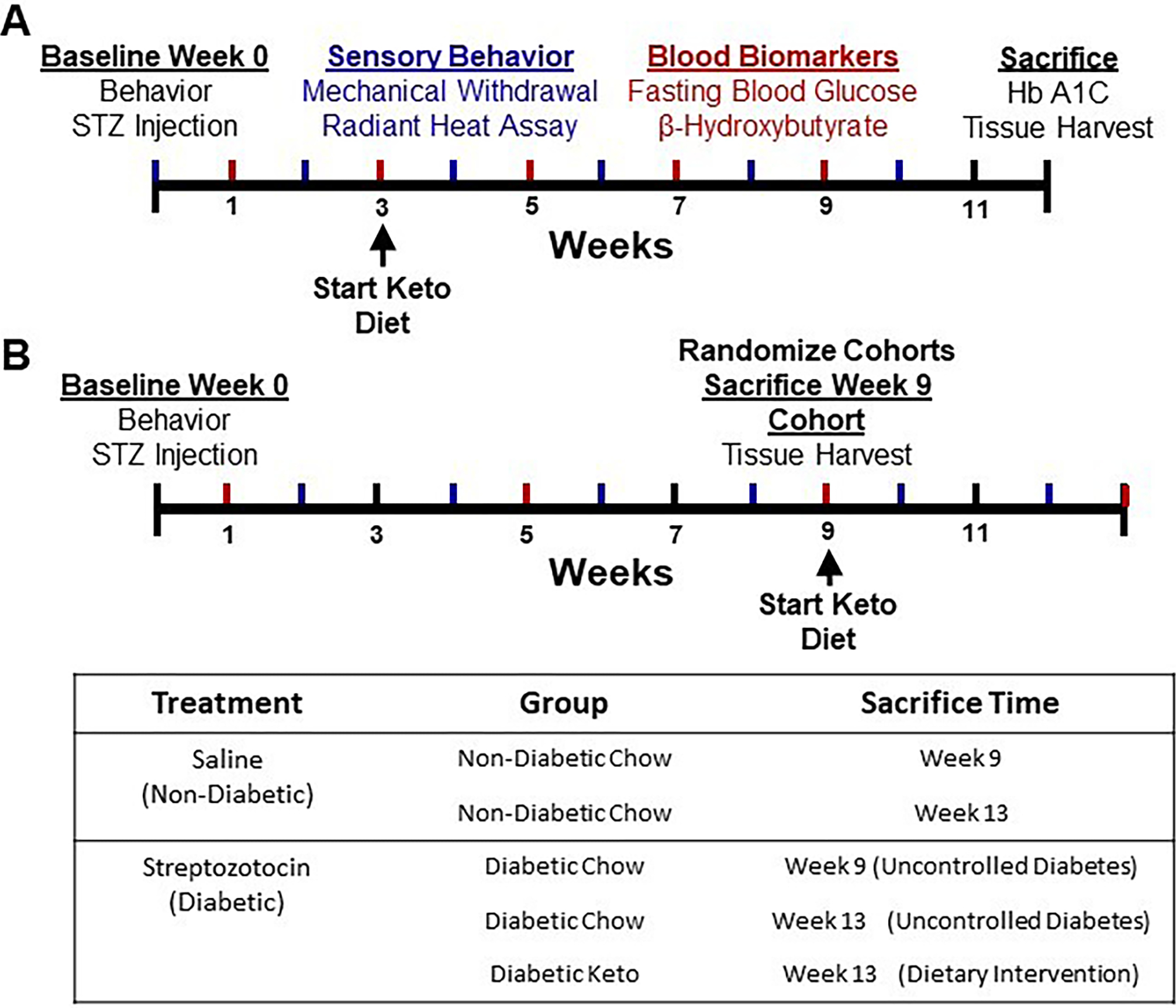
Graphical timelines of experimental design for early-intervention and rescue paradigms. Diabetes was induced by intraperitoneal injection of streptozotocin (180 mg/kg) and confirmed three days later by blood glucose measurement. Sensory behavioral testing was performed at baseline and biweekly following diabetes induction. (A) Mice in the early-intervention paradigm were administered a ketogenic diet three weeks post-injection or maintained on a chow diet. Blood biomarkers were assessed biweekly and at sacrifice. (B) Mice in the rescue paradigm had uncontrolled diabetes for nine weeks before randomization into three groups: immediate sacrifice for peripheral innervation assessment, maintenance on chow diet, or administration of a ketogenic diet for four weeks. Blood biomarkers were assessed every four weeks.
Different Von Frey monofilaments were applied to the plantar surface of the hind paw following the “up-down” method for one second [10]. Mice were observed for either a negative or a positive response, and mechanical withdrawal threshold was calculated following five positive responses. Thermal sensitivity was determined by radiant heat assay using a thermal testing apparatus. A 4.2 V radiant heat source was applied to the plantar surface of the hind paw and latency to withdrawal was recorded three times. Cold-sensitivity was determined using a cold-plate assay. Each animal was subject to two trials each, the results of which were averaged. During each trial, the temperature of the cold plate ramped down from 27 °C to 0 °C at a rate of 10 °C/minute. After observing a nocifensive behavior, the assay was stopped and the final temperature and time in seconds to reach it were recorded.
Intraepidermal Nerve Fiber Density
Footpads were collected from all mice and post-fixed in Zamboni’s fixative for 18 hours before overnight incubation in 30% sucrose and cryopreservation in Optimal Cutting Temperature Compound (Sekura Tissue-Tek). Thirty-micron sections of footpad were blocked for two hours in Superblock (ThermoFisher; Grand Island, NY), 1.5% Normal Donkey Serum, 0.5% Porcine Gelatin, and 0.5% Triton X-100 (Sigma) at room temperature. Slides were incubated overnight with rabbit α-PGP9.5 (1:1500, UCHL1; ProteinTech; Rosemont, IL). Slides were incubated with AlexaFluor-555 tagged donkey-α-rabbit secondary antibody (1:1000; Molecular Probes) for one hour, then imaged with a Nikon Eclipse 90i microscope using a 20X objective. Nikon Elements software was used to measure length of the dermal-epidermal junction. IENF was quantified as the number of fibers crossing that junction and expressed as number of fibers per millimeter. Nine images were taken per slide, and the average IENF density for each mouse was used for statistical analyses.
Experimental Design
Following STZ injection, mice were assigned to either dietary prevention (Figure 1A) or dietary rescue paradigm (Figure 1B). In both paradigms, mice were randomized by baseline mechanical withdrawal threshold into diabetic and non-diabetic cohorts. In the early intervention paradigm, diabetic and healthy mice were randomized by mechanical withdrawal threshold again three weeks following induction of diabetes into groups fed a ketogenic diet or maintained on chow diet for nine weeks, for a total length of twelve weeks. In the rescue paradigm, mice were randomized nine weeks following induction of diabetes by mechanical withdrawal threshold (Figure 1B). Non-diabetic mice in this cohort were randomized to either immediate sacrifice for tissue harvest or maintenance on chow diet for four weeks. Diabetic mice were randomized to either immediate sacrifice for tissue harvest, maintenance on chow diet for four weeks, or administration of a ketogenic diet for four weeks, for a total of thirteen weeks.
Statistics and Data Analysis
All statistical analyses were performed using R version 3.6.2 and packages “Rmisc”, “car”, “nlme”, “multcomp”, and “ggpubr”. All analyses for which data were collected over time were performed using a mixed-model analysis of variance (ANOVA) with repeated measures. All other analyses were performed using a two-way ANOVA or Student’s t-test, as appropriate. Data were analyzed posthoc by pairwise t-test or Tukey’s Honest Significant Difference (HSD), as indicated. Assumptions of normal distribution and homogeneity of variance were confirmed by Shapiro-Wilks and Levene tests, respectively. All data are presented as mean +/− standard error of the mean.
Results
Type 1 Diabetic Mice Tolerate a Ketogenic Diet
We used an intraperitoneal streptozotocin (STZ) injection model to induce diabetes in 8-week-old mice. Diabetic mice fed a chow diet developed a significant decrease in bodyweight compare to chow-fed non-diabetic mice fed (Figure 2A; mixed-model ANOVA with repeated measures, time: p < 0.001, diabetes: p < 0.05, diet: p < 0.78, diabetes-diet interaction: p < 0.001; Figure 3A–B; mixed-model ANOVA with repeated measures, time: p < 0.0001, group: p < 0.0001). Notably, diabetic ketogenic diet-fed mice had increased bodyweight relative to chow-fed diabetic mice in the early intervention paradigm, which was mirrored to a lesser degree in the rescue paradigm (Figure 2A, 3A, B).
Figure 2.
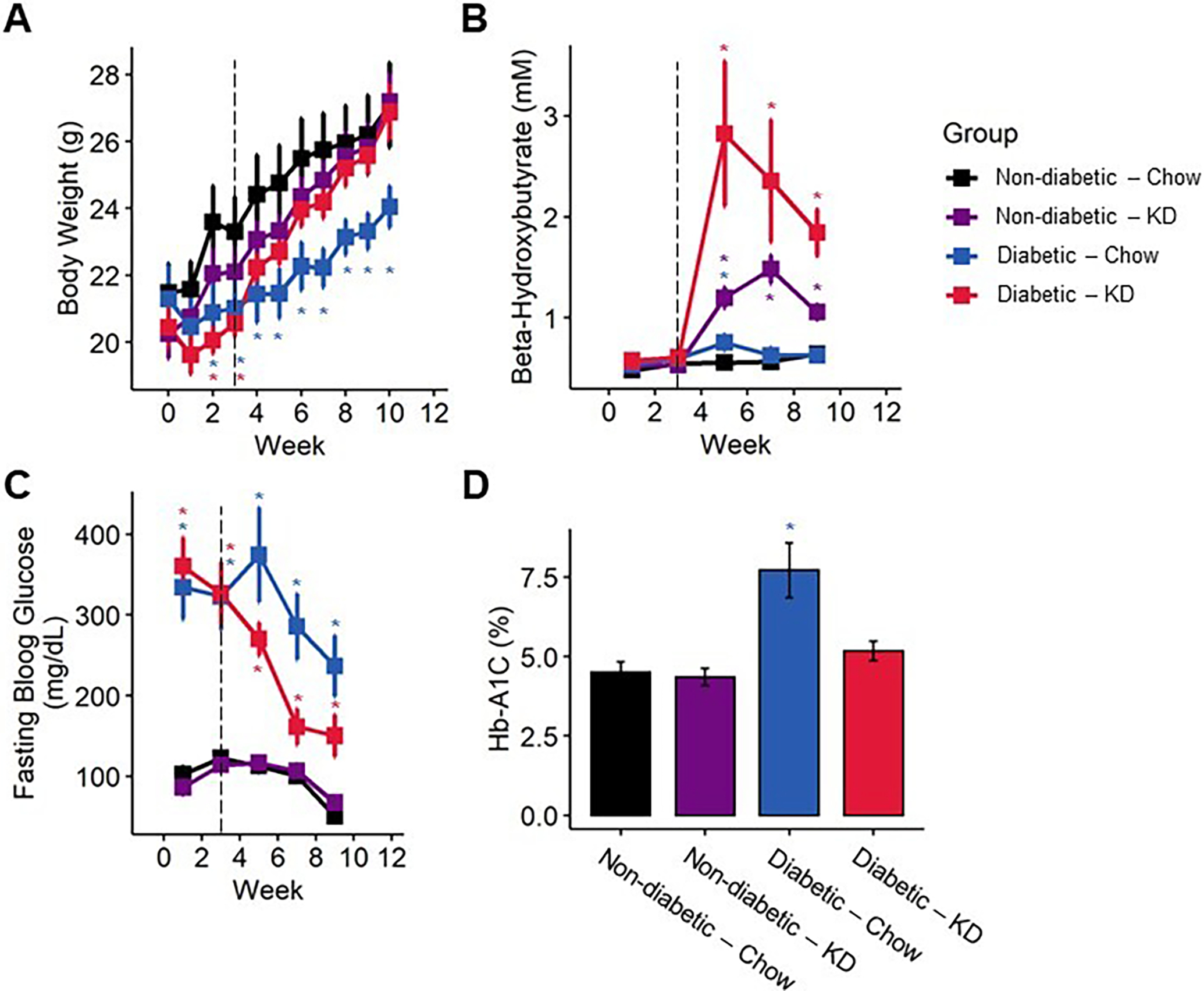
Bodyweight and blood biomarkers in diabetic mice following administration of a ketogenic diet (KD) as a preventative intervention. (A) Diabetic mice exhibited reduced gain in bodyweight compared to non-diabetic mice, which was ameliorated by a ketogenic diet (n=7–12). (B) Mice fed a ketogenic diet displayed elevated ketone body levels. Though elevated ketones are much more pronounced in diabetic mice fed a ketogenic diet, the blood β-HB levels remain below the threshold for ketoacidosis (n=7–12). (C) Diabetic mice exhibit significantly elevated fasting blood glucose compared to non-diabetic controls, which is partially ameliorated following early administration of a ketogenic diet (n=7–12). (D) Diabetic chow-fed, but not ketogenic diet-fed, mice exhibit elevated % Hb-A1C (n=7–10). (A-C) Mixed-models ANOVA with repeated measures and Tukey’s post hoc test; * p < 0.05 compared to chow-fed non-diabetic mice. (D) 2-way ANOVA and Tukey’s post hoc test; * p < 0.05 compared to chow-fed non-diabetic mice.
Figure 3.
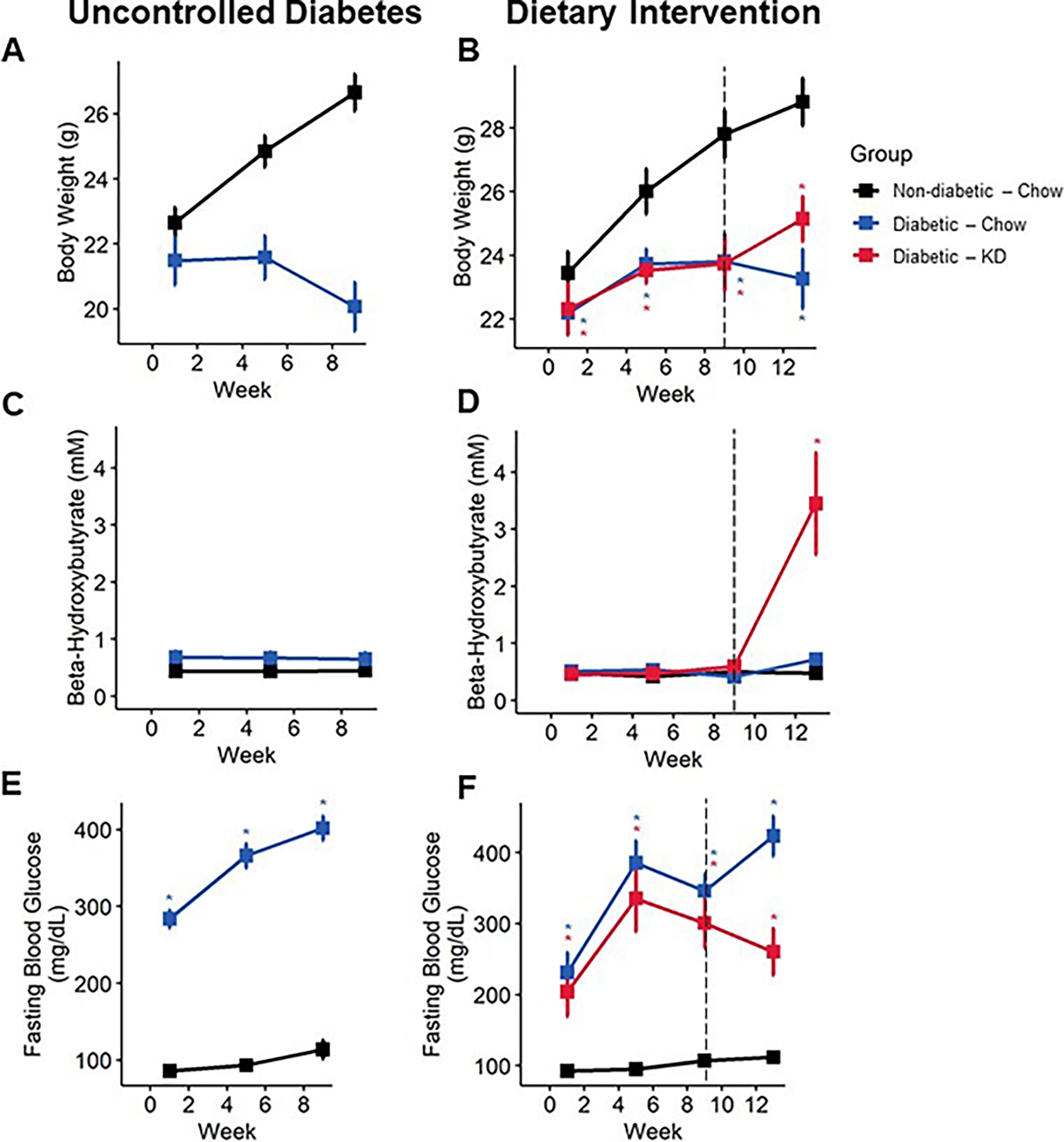
Bodyweight and blood biomarkers in diabetic mice following an uncontrolled diabetes (A, C, E) or dietary intervention (B, D, E) paradigm. Diabetic mice fail to gain bodyweight (A), which is modestly rescued following administration of a ketogenic diet (B) (n=6–10). Diabetic mice fed a ketogenic diet develop nutritional ketosis yet remain below the threshold of life-threatening ketoacidosis (C and D) (8 mM β-HB; n=6–10). Diabetic mice exhibit robust increase in fasting blood glucose (E), which is significantly reduced following administration of a ketogenic diet (F) (n=6–10). (A-E) Mixed-models ANOVA with repeated measures and Tukey’s post hoc test; * p < 0.05 compared to chow-fed non-diabetic mice.
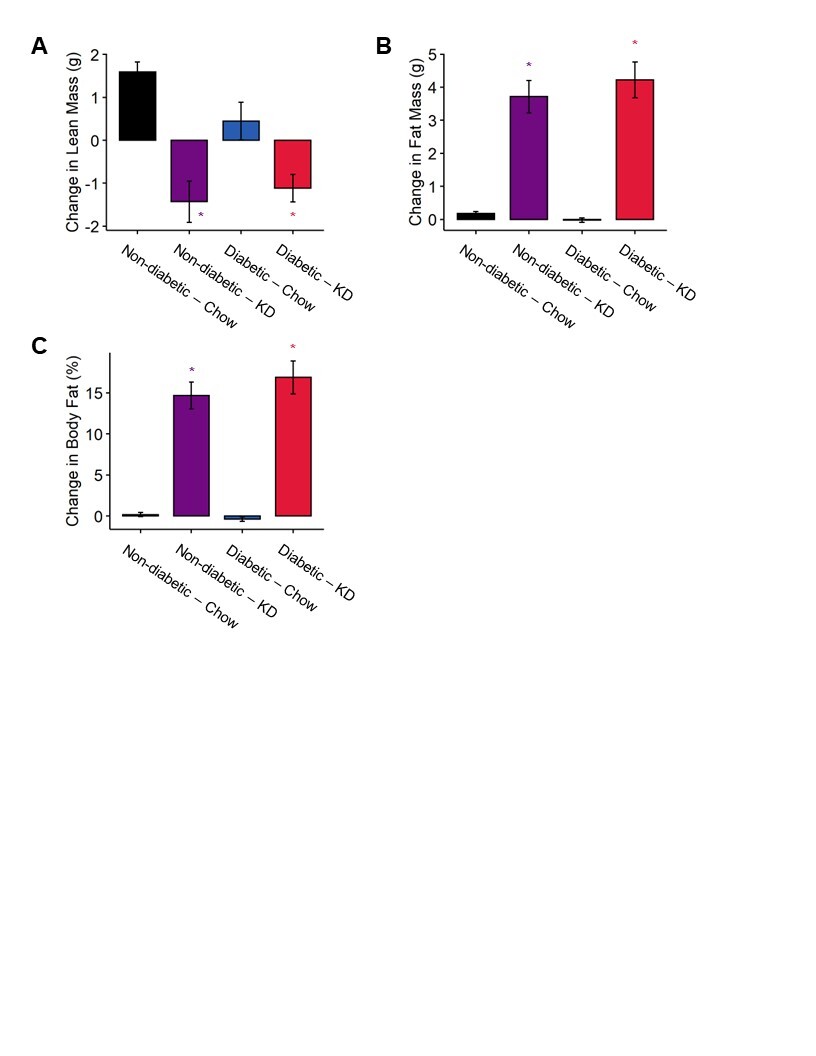
We tested whether the impact of a ketogenic diet on bodyweight in diabetic mice was related to changes in lean and fat mass. We quantified changes in lean mass, fat mass, and percent bodyfat over a four-week period between week 3 and week 7. We detected no significant effect of diabetes or a diabetes-diet interaction on lean mass, fat mass, or percent bodyfat (Figure S1). Diet alone, however, accounted for significant decreases in lean mass (2-way ANOVA, diet: p < 0.0001) and increases in fat mass (2-way ANOVA, diet: p < 0.0001) and percent bodyfat (2-way ANOVA, diet: p < 0.0001).
We examined whether consumption of a ketogenic diet significantly increased concentrations of β-hydroxy-butyrate (β-HB), the primary ketone body detected in blood (Figure 2B; mixed-models ANOVA with repeated measures, diabetes: p < 0.003, diet: p < 0.0001, diabetes-diet interaction: p < 0.036; Figure 3C, D, mixed-models ANOVA with repeated measures, group: p < 0.0001). Mice fed a ketogenic diet had increased circulating β-HB in both diabetic and non-diabetic groups compared to chow-fed mice following administration of a ketogenic diet (Tukey’s HSD, p = 0.00295). However, ketogenic diet-fed diabetic mice did not exhibit blood β-HB concentrations above 8 mM at any timepoint, indicating that a ketogenic diet does not cause onset of diabetic ketoacidosis in this diabetic mouse model.
A Ketogenic Diet Improves Type 1 Diabetes Blood Biomarkers
Ketogenic diets are defined by their very-low carbohydrate components, which results in the production and consumption of ketone bodies as an alternative fuel source. To test whether reduced carbohydrate intake from a ketogenic diet altered fasting blood glucose levels, we quantified blood glucose on a bi-weekly basis. Both chow- and ketogenic diet-fed diabetic mice initially developed high fasting blood glucose typical of this model. However, following early intervention with a ketogenic diet, diabetic ketogenic diet-fed mice displayed lower fasting blood glucose than chow-fed diabetic mice (Figure 2C; mixed-models ANOVA with repeated measures, time: p < 0.0001, diabetes: p 0.0001, diet: p < 0.168, diabetes-diet interaction: p < 0.085; Tukey’s HSD, p = 0.12). Notably, these levels dipped below the generally accepted level of fasting blood glucose for diabetes in mice. While ketogenic diet-fed diabetic mice in rescue experiments did not dip below the threshold of being considered diabetic, these ketogenic-diet fed mice did exhibit significantly reduced fasting blood glucose compared to chow-fed diabetic mice after four weeks (Figure 3E, F; mixed-models ANOVA with repeated measures, time: p < 0.0001, group: p < 0.0001; Tukey’s HSD, p < 0.01).
We measured percent glycosylated hemoglobin HbA1C in all mice 12 weeks post-STZ injection (Figure 2D). Chow-fed diabetic mice exhibited significantly higher HbA1C than all other groups, while ketogenic diet-fed diabetic mice were not significantly different from either chow- or ketogenic diet-fed non-diabetics (2-way ANOVA, diabetes: p < 0.00012, diet: p < 0.011, diabetes-diet interaction: p < 0.0094; Tukey’s HSD, STZ-Chow and STZ-Keto adjusted p < 0.0027, STZ-Keto and Ctrl-Chow adjusted p < 0.676).
STZ induces diabetes by causing DNA damage to pancreatic β-islets and preventing insulin production. We measured fasting insulin levels across all four groups for 8 weeks. As expected, STZ-injection led to a significant reduction in blood insulin levels (Figure S2; mixed-models ANOVA with repeated measure, time: p < 0.0001, diabetes: p < 0.0001, diet: p < 0.0729, diabetes-diet interaction: p < 0.65). Insulin levels in ketogenic diet-fed diabetic mice were not different than chow-fed diabetic mice (Tukey’s HSD, p = 0.45).
Consumption of a Ketogenic Diet Reverses Diabetes-Induced Mechanical Allodynia
Mice developed significant lasting hind paw mechanical allodynia within two weeks of diabetes induction in both early intervention (Figure 4A) and rescue paradigms (Figure 5A, B). However, following early intervention with a ketogenic diet, diabetic mice regained and maintained mechanical thresholds similar to non-diabetic, chow-fed mice throughout the duration of the experiment (Figure 4A; mixed-model ANOVA with repeated measures, time: p < 0.0001, diabetes: p < 0.0001, diet: p < 0.0001, diabetes-diet interaction: p < 0.0001; Tukey’s HSD, diabetic and non-diabetic chow-fed p < 0.001, diabetic chow- and ketogenic diet-fed p < 0.001). The rescue paradigm showed similar results, as within one week of administering a ketogenic diet, mechanical withdrawal thresholds of diabetic mice returned to baseline (Figure 5B; mixed-model ANOVA with repeated measures, time: p = 0.6225, group: p <0.0001; Tukey’s HSD, diabetic and non-diabetic chow-fed p < 0.001, diabetic chow- and ketogenic diet-fed p < 0.00123).
Figure 4.
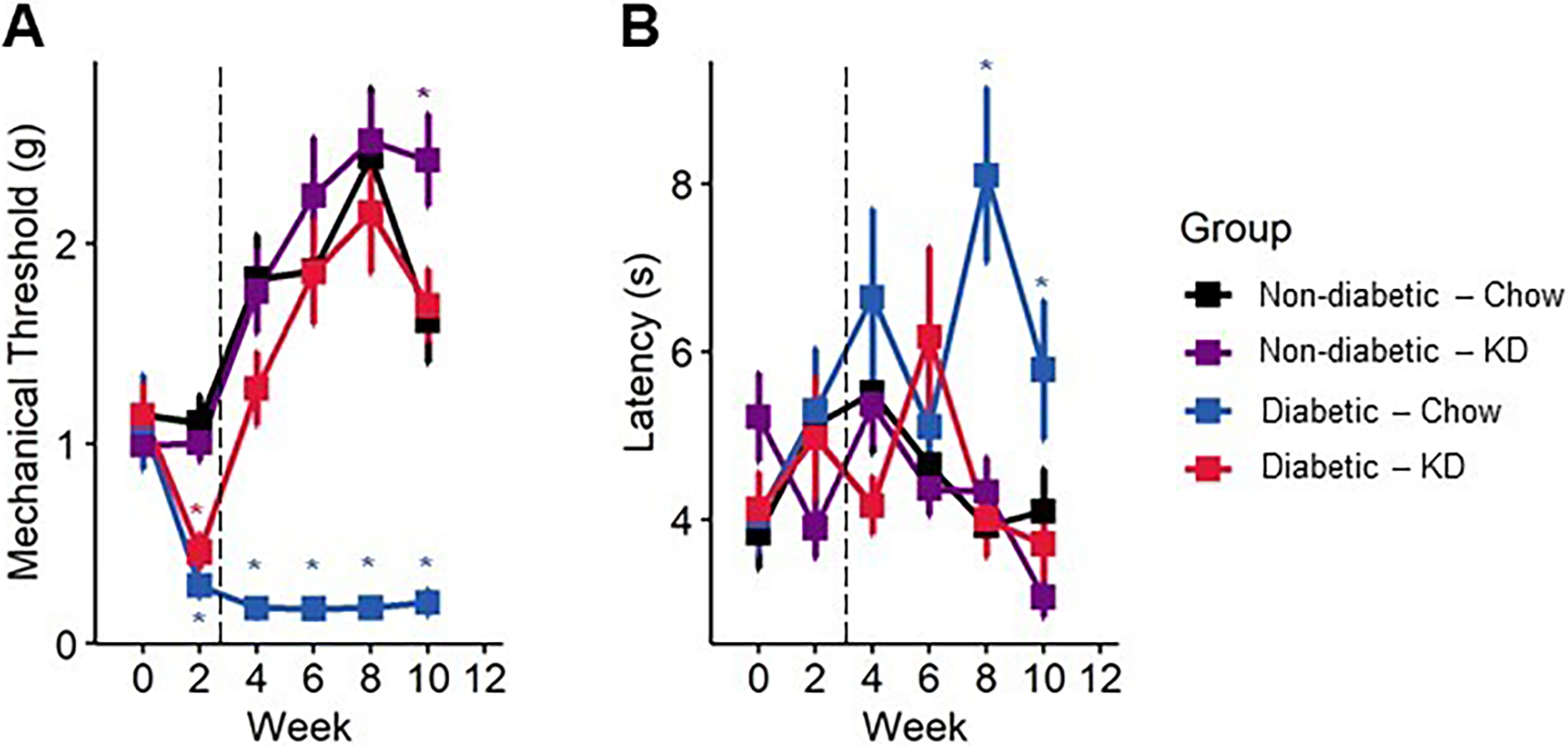
Sensory behavior in diabetic mice following administration of a ketogenic diet as a preventive intervention. (A) Diabetes induces a sharp, lasting decline in mechanical withdrawal thresholds as assessed by Von Frey filaments, indicative of mechanical allodynia. Within a week of administration of a ketogenic diet, diabetic mice returned to normal levels of mechanical sensitivity (n=7–12). (B) Diabetic mice slowly develop a delayed response to noxious heat stimuli, as assayed by radiant heat assay. The thermal hyporesponsiveness was prevented in diabetic mice fed a ketogenic diet (n=7–12). (A-B) Mixed-models ANOVA with repeated measures and Tukey’s post hoc test; * p < 0.05 compared to chow-fed non-diabetic mice.
Figure 5.
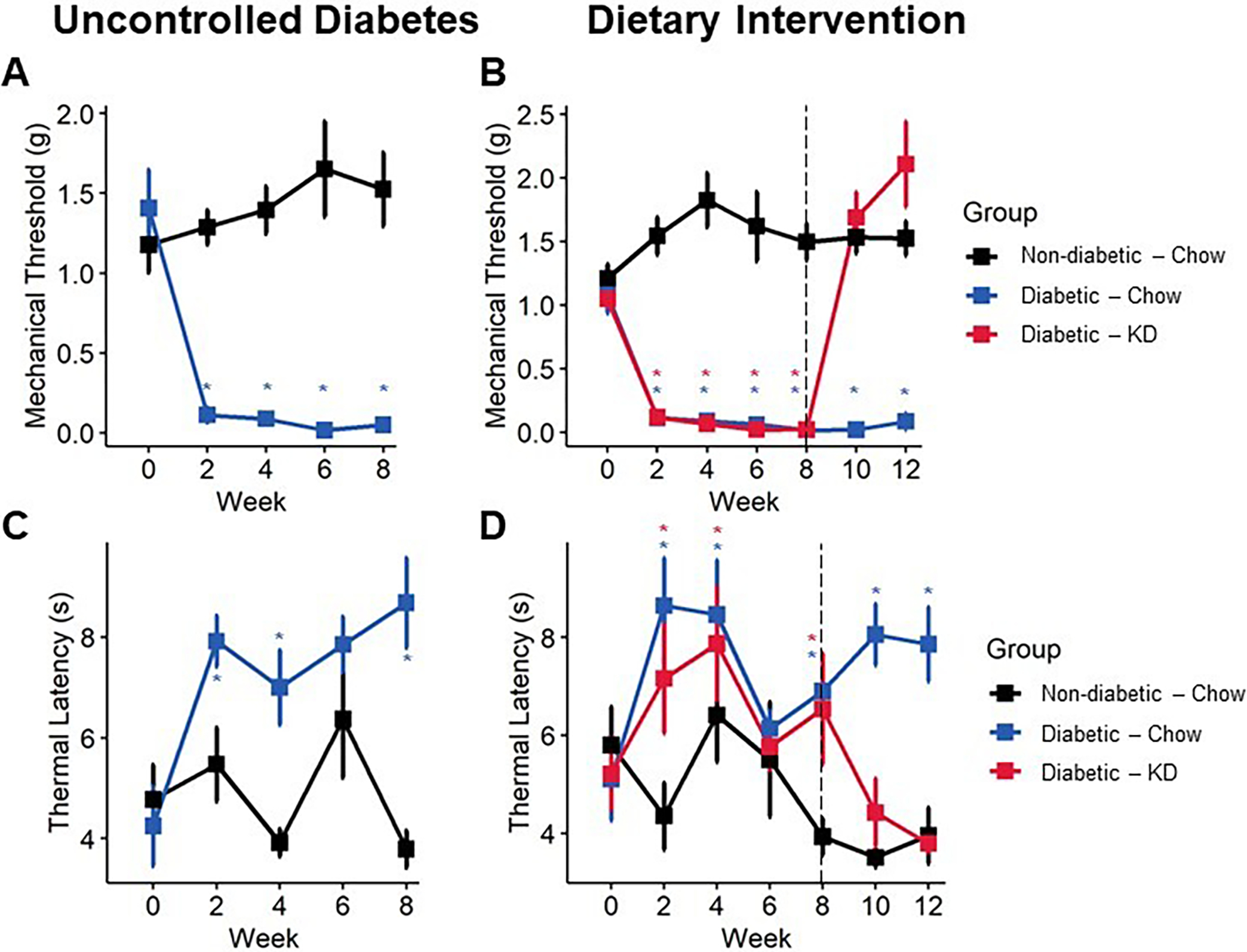
Sensory behavior in diabetic mice following an uncontrolled diabetes (A, C) or dietary intervention (B, D) paradigm. Diabetes induces a sharp and lasting onset of mechanical allodynia (A), which is reversed within one week of administering a ketogenic diet (B) (n=6–10). Diabetic mice develop delayed responsiveness to radiant heat following 9 weeks of uncontrolled diabetes (C). Following administration of a ketogenic diet, thermal latency returns to normal (D) (n=6–10). (A-B) Mixed-models ANOVA with repeated measures and Tukey’s post hoc test; * p < 0.05 compared to chow-fed non-diabetic mice.
We also observed a modest but significant improvement in thermal sensitivity with ketogenic diet-fed mice using a radiant heat assay. In both experiments, diabetic mice exhibited an increase in paw withdrawal latency by week four (Figure 4B; mixed-model ANOVA with repeated measures, time: p < 0.38, diabetes: p < 0.065, diet: p < 0.077, diabetes-diet interaction: p < 0.098; Figure 5C, D; mixed-model ANOVA with repeated measures, time: p = 0.39, group: p <0.0001), indicating diminished thermal sensation. Administration of a ketogenic diet occurred before onset of thermal hyposensitivity and prevented increased latency in diabetic mice (Tukey’s HSD, STZ-diabetic chow- and ketogenic diet-fed p = 0.08). In rescue experiments, a ketogenic diet was provided after the onset of thermal hyposensitivity. Diabetic mice exhibiting diminished thermal sensation showed improvements in thermal latency following administration of a ketogenic diet and were not different from healthy controls by the end of the experiment (Tukey’s HSD, non-diabetic and diabetic ketogenic diet-fed p = 1.00).
We tested whether a ketogenic diet modified cold sensation. Using a cold-plate assay, we observed no significant effects of either diabetes or diet on cold thresholds (Figure S3; 2-way ANOVA, p > 0.5).
Consumption of a Ketogenic Diet Prevents Reductions in Diabetes-Induced Intraepidermal Nerve Fiber Density
IENF density is significantly reduced in patients with diabetic neuropathy [13; 23] and in preclinical models of diabetic neuropathy [34]. Therefore, we assessed the impact of a ketogenic diet on IENF density in diabetic mice (Figure 6). Consistent with previous findings, diabetic chow-fed mice exhibited an approximately 20% reduction in IENF density (2-way ANOVA, diabetes: p < 0.0055, diet: p < 0.015, diabetes-diet interaction: p < 0.21). The diabetic ketogenic diet-fed group, however, was not significantly different from chow-fed non-diabetic mice (Tukey’s HSD, adjusted p < 0.98) and displayed a statistical trend of higher IENF density than diabetic chow-fed mice (Tukey’s HSD, adjusted p < 0.052).
Figure 6.
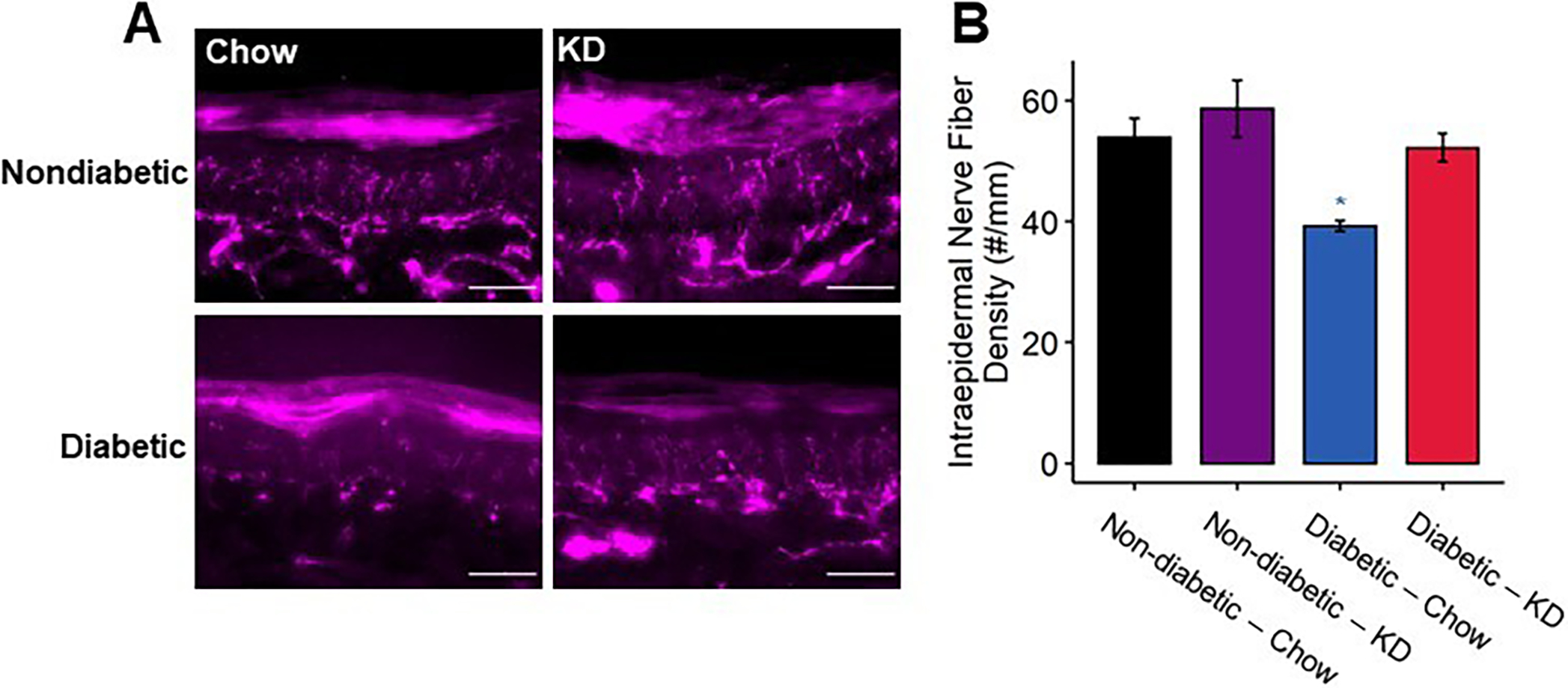
Intraepidermal innervation of diabetic footpads following using a ketogenic diet as a preventative intervention. (A) Representative images of fixed footpads fluorescently labeled with anti-protein gene product 9.5 (PGP9.5) to stain all nerve fibers. Scale bar set to 50 μm. (B) Quantification of PGP9.5+ fibers/mm. Diabetic mice exhibit approximately 30% fewer IENFs compared to non-diabetic controls, whereas diabetic mice fed a ketogenic diet exhibit normal IENFD (n=4). 2-Way ANOVA and Tukey’s post hoc test; * p < 0.05 compared to chow-fed non-diabetic mice.
Consumption of a Ketogenic Diet Stimulates Recovery of Intraepidermal Nerve Innervation in Diabetic Mice
Once intraepidermal axons degenerate in diabetic neuropathy, there are few experimental interventions shown to improve transient reinnervation of the epidermis [2; 14; 29]. The above results suggest that consumption of a ketogenic diet prevents IENF density reductions associated with the progression of diabetes. Here, we tested whether initiation of a ketogenic diet following a timepoint in which mice had demonstrable reductions in IENF density could reverse this neurodegenerative change. We sacrificed chow-fed non-diabetic and diabetic mice nine weeks after STZ-injection to confirm that diabetic mice had reduced IENF density. Consistent with this diabetes model and mouse strain, chow-fed diabetic mice displayed a significant reduction in IENF density after nine weeks of diabetes compared to chow-fed non-diabetic mice (Figure 7; Tukey’s HSD, non-diabetic and diabetic chow-fed at 9 weeks p < 0.001). The remaining nondiabetic mice were maintained on chow diet, while diabetic mice were either provided a ketogenic diet or maintained on chow. After four additional weeks, diabetic chow-fed mice continued to exhibit significantly reduced IENF density compared to non-diabetic mice (Tukey’s HSD, non-diabetic compared to diabetic chow-fed at 13 weeks p < 0.0002). Diabetic mice fed a ketogenic diet, however, displayed significantly increased levels of IENF density that were comparable to levels in chow-fed non-diabetic mice (Tukey’s HSD, diabetic chow- and ketogenic diet-fed at 13 weeks p < 0.0005, non-diabetic and diabetic ketogenic diet-fed p = 0.98).
Figure 7.
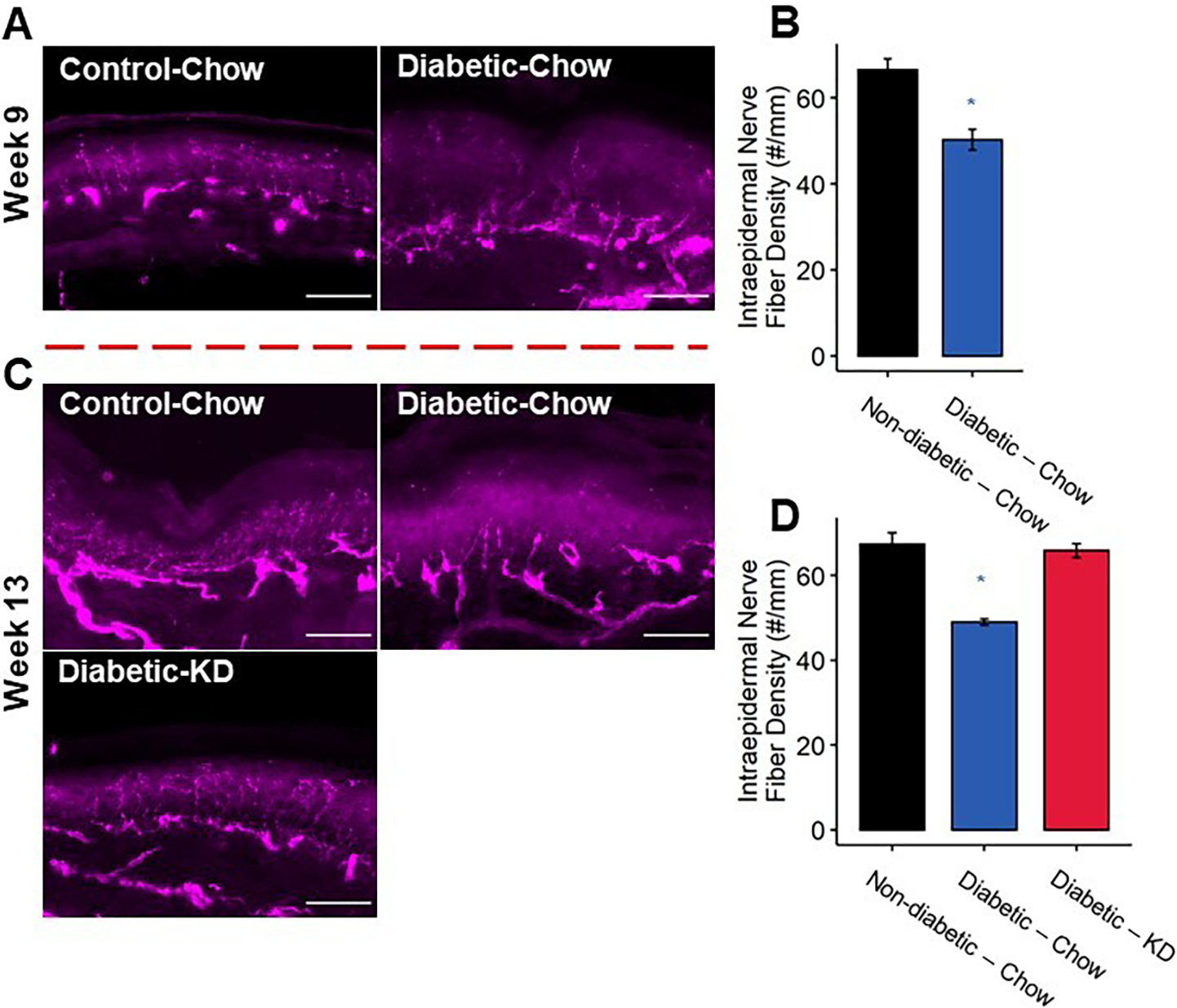
Intraepidermal innervation of diabetic footpads following a rescue paradigm of ketogenic diet administration. (A) Representative images of epidermal axons. Scale bar set to 50 μm. (B) Quantification of IENFD. Diabetic mice exhibited approximately 25% reduction in IENFD compared to non-diabetic controls at 9 weeks. (C) Representative images of epidermal axons. Axon loss was also apparent in chow-fed diabetic mice at 13 weeks. (D) Diabetic mice fed a ketogenic diet as a rescue paradigm for 4 weeks exhibit normal IENFD (n=3–5). 2-Way ANOVA and Tukey’s post hoc test; * p < 0.05 compared to chow-fed non-diabetic mice.
Discussion
In this study, we demonstrated that administration of a ketogenic diet is effective at rescuing positive and negative sensory abnormalities indicative of DPN and stimulating regeneration of peripheral nerve fibers in the distal limb. We tested a ketogenic diet (prevention) and ketogenic diet rescue experimental paradigms in an experimental diabetes model in mice with known sensory deficits and axonal loss. From a safety perspective, consumption of a ketogenic diet in diabetic mice was well tolerated and improved body weight, fasting blood glucose, and Hb-A1C (Figures 2 and 3), congruent with previous literature [9; 18; 39; 50]. Diabetic mice developed mechanical allodynia and thermal hyposensitivity (Figures 4 and 5) and displayed reduced IENF density (Figures 6 and 7), all of which are well recapitulated in models of DPN [28; 34] and serve as clinical indicators of the condition. Importantly, administration of a ketogenic diet reversed mechanical allodynia and lost thermal sensation and stimulated regeneration of epidermal nerve fibers in diabetic mice exhibiting symptoms of DPN.
Improvements in fasting blood glucose and reduced Hb-A1c suggest that a ketogenic diet may ameliorate aberrant nociception and sensory loss in part by improving metabolic parameters in diabetes. The peripheral nervous system is generally thought to be insulin-independent related to glucose uptake and utilization [38], rendering these tissues more susceptible to glucotoxicity and subsequent generation of reactive oxygen species (ROS) [46; 54] and toxic secondary metabolites like dicarbonyls. For instance, increased glycolytic flux is known to increase accumulation of methylglyoxal, which has been shown to contribute to nociception in DPN and other neuropathies [6; 19; 52] via activation of the integrated stress response [3] and transient receptor potential ankyrin 1 (TRPA1)[1; 26]. These complications can lead to problems in the redox balance of neurons and are known to be strongly associated with pain [1; 6; 46].
Ketogenic diets feature a drastic reduction in carbohydrate intake, presumably allowing insulin-independent tissues to use available circulating glucose without replenishing and overburdening these tissues with glucose. While we observed a reduction in fasting blood glucose in diabetic mice fed a ketogenic diet (Figures 2C and 3F), it is important to note that these effects occurred temporally after improvements in mechanical allodynia and thermal sensation. While consuming a ketogenic diet, glucose is replaced as the primary circulating fuel source by ketone bodies such as β-HB and acetoacetate. We observed elevated ketosis within two weeks of administering a ketogenic diet (Figure 2B). Importantly, the concentration of circulating ketones remained below the threshold of ketoacidosis, a common life-threatening complication of diabetes (Figure 2B, Figure 3D). Ketone bodies have been further implicated in reducing glycolytic flux [51] and scavenging ROS [15; 25; 35]. Thus, peripheral neurons in the somatosensory system of diabetic mice may experience reduced glycolytic flux due to a concomitant decrease in carbohydrate intake and ketone-mediated regulation of glycolysis.
In addition to changes in metabolism, additional mechanisms associated with elevated ketone bodies must also be considered. Increasing evidence now reveals that ketone bodies can function as signaling molecules that are relevant to sensation and pain. For example, β-HB signals through hydroxy-carboxylic acid receptor (HCAR) −1 and −2 [12; 30] and inhibit the NLRP3 inflammasome [36; 43]. NLRP3 and its product interleukin-1β are heavily implicated in both diabetes [11; 45] and chronic pain [24; 33; 47], making them attractive potential mechanisms for the anti-nociceptive effects of a ketogenic diet.
Ketogenic diet may also improve peripheral sensation in DPN by stimulating growth of distal axons innervating peripheral tissues. Human patients with DPN exhibit greatly reduced IENF density compared to those without neuropathy [13; 34]. The relationship between reduced IENF density and painful DPN is complex, and causality of axon loss and pain is not clearly delineated. In mice, the, appearance of mechanical allodynia occurs before reductions in IENF density (Figures 6 and 7)[34], yet that may be due in part to increased regrowth and degradation cycles of IENF terminals in diabetics with pain. For example, Feldman and colleagues reported reduced IENF density and an increased proportion of IENFs positive for growth-associated protein 43 (GAP43), a marker for axonal growth and regeneration, in patients with painful DPN compared to those with neuropathy and no pain [13]. Truini and colleagues likewise reported increased GAP43 staining and reduced total IENFD in diabetic patients with either mechanical allodynia or burning sensation [23]. These studies suggest for the possibility of IENFD reduction with concomitant cycles of regrowth and degeneration as contributors to pain in DPN. A much clearer relationship exists between reduced IENF density and loss of sensation. Epidermal axon subtypes include polymodal C-fibers exhibiting responsiveness to heat and noxious stimuli [31] and are known to degenerate in DPN [34]. Loss of the distal tips of these fibers are associated with reduced sensitivity to heat stimuli. Indeed, latency to nocifensive response in the hot plate assay is negatively correlated with IENF density [31]. Here, our studies suggest that a ketogenic diet prevents IENF loss in DPN, but importantly, even after axons have been lost, can lead to the regeneration of distal axons.
Mitigating hyperglycemia may also contribute to improved IENF density in diabetic mice fed a ketogenic diet. Sensory axon growth is positively impacted by neurotrophin release and transport from nonneuronal cell types that include keratinocytes and Schwann cells. Schwann cells reduce nerve growth factor (NGF) expression and release in high glucose conditions, leading to reduced neurite outgrowth from cultured sensory neurons [49]. Future studies should examine whether high glucose conditions similarly reduce neurotrophin expression in target tissues, such as keratinocytes. Prolonged exposure to high glucose can lead to receptor glycation, and glycation of the receptor for NGF, TrkA, reduces NGF-binding affinity three-fold [5]. Further, treatment with 2-deoxyglucose, a glucose analog that inhibits glycolysis, upregulated NGF and brain-derived neurotrophic factor (BDNF) in a mouse model of Alzheimer’s disease [53]. Reducing hyperglycemia may therefore be sufficient to rescue diabetes associated IENF loss, yet it is also possible that elevated ketone bodies may contribute to axon regrowth. β-HB is known to upregulate the neurotrophin brain-derived neurotrophic factor (BDNF) through histone deacetylase inhibition [44]. It is also important to note the increase in NGF and BDNF following 2-deoxyglucose treatment was concomitant to both decreased glycolysis and increased circulating β-HB availability in the brain. Our results include the possibility for improved metabolic changes and noncanonical actions of ketone bodies to both contribute to positive changes we observed in mice with DPN.
DPN is a devastating condition with a multifaceted etiology, resulting in chronic pain and loss of sensation in patients that leads to reduced quality of life. Our results are consistent with the findings that, as an intervention, a ketogenic diet is well tolerated in diabetes [9; 18; 39; 50] and may offer significant benefits to prevent and reverse diabetes-associated abnormalities in mechanical and thermal sensitivity. Moreover, our results suggest the promising possibility that consumption of a ketogenic diet can stimulate the regeneration of peripheral axons.
Supplementary Material
Figure S1. Changes in body composition in diabetic and ketogenic diet-fed mice. (A) Diabetic chow-fed mice have a decreased gain in lean mass compared to non-diabetic controls. Both non-diabetic and diabetic mice fed a ketogenic diet experience a loss in lean mass relative to chow-fed mice (n=7–12). Mice fed a ketogenic diet experience a robust increase in fat mass (B) and percent body fat (C) regardless of diabetes status (n=7–12). (A-C) 2-Way ANOVA and Tukey’s post hoc test; * p < 0.05 compared to chow-fed non-diabetic mice.
{kind=link}
Figure S2. Insulin in non-diabetic and type 1 diabetic mice fed a ketogenic diet. Diabetic mice exhibited reduced serum insulin compared to non-diabetic controls, as expected. Diabetic mice fed a ketogenic diet do not exhibit elevated serum insulin relative to chow-fed diabetics (n=7–12). Mixed-models ANOVA with repeated measures and Tukey’s post hoc test; * p < 0.05 compared to chow-fed non-diabetic mice.
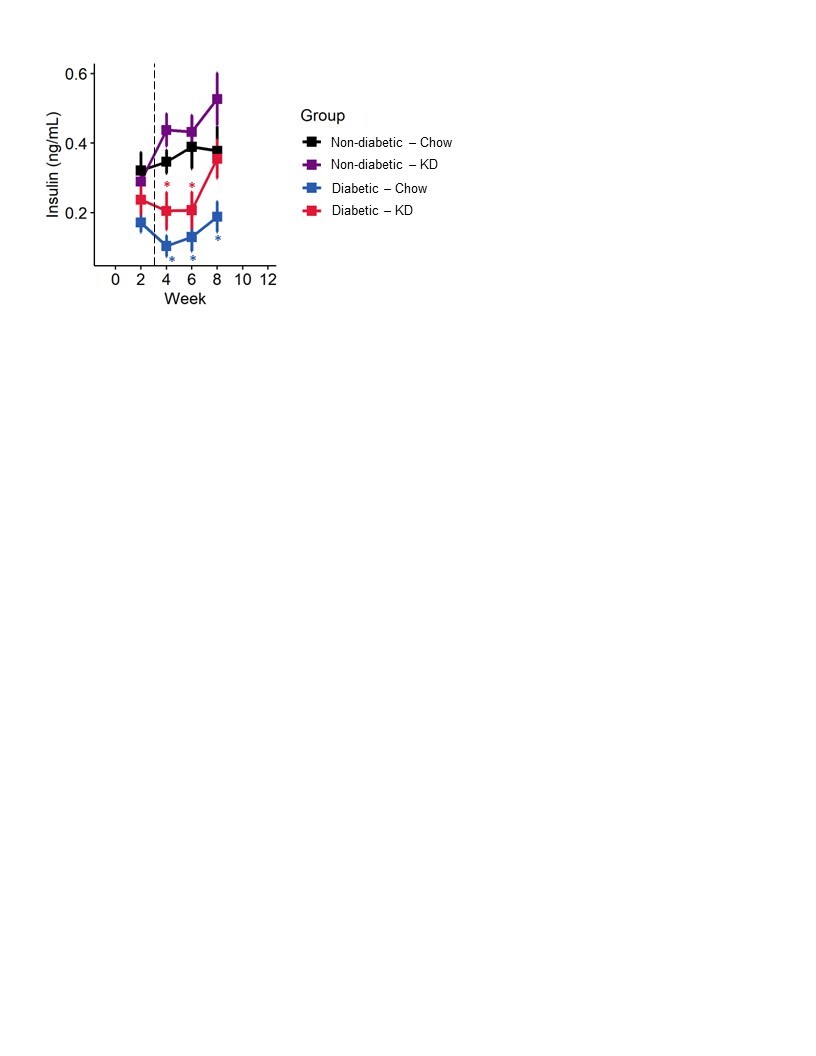{kind=link}
Figure S3. Cold plate assay in diabetic and non-diabetic mice fed a ketogenic diet. Mice displayed no changes in thermal thresholds to elicit nocifensive behavior regardless of diet or diabetic status (n=7–12). 2-Way ANOVA and Tukey’s post hoc test.
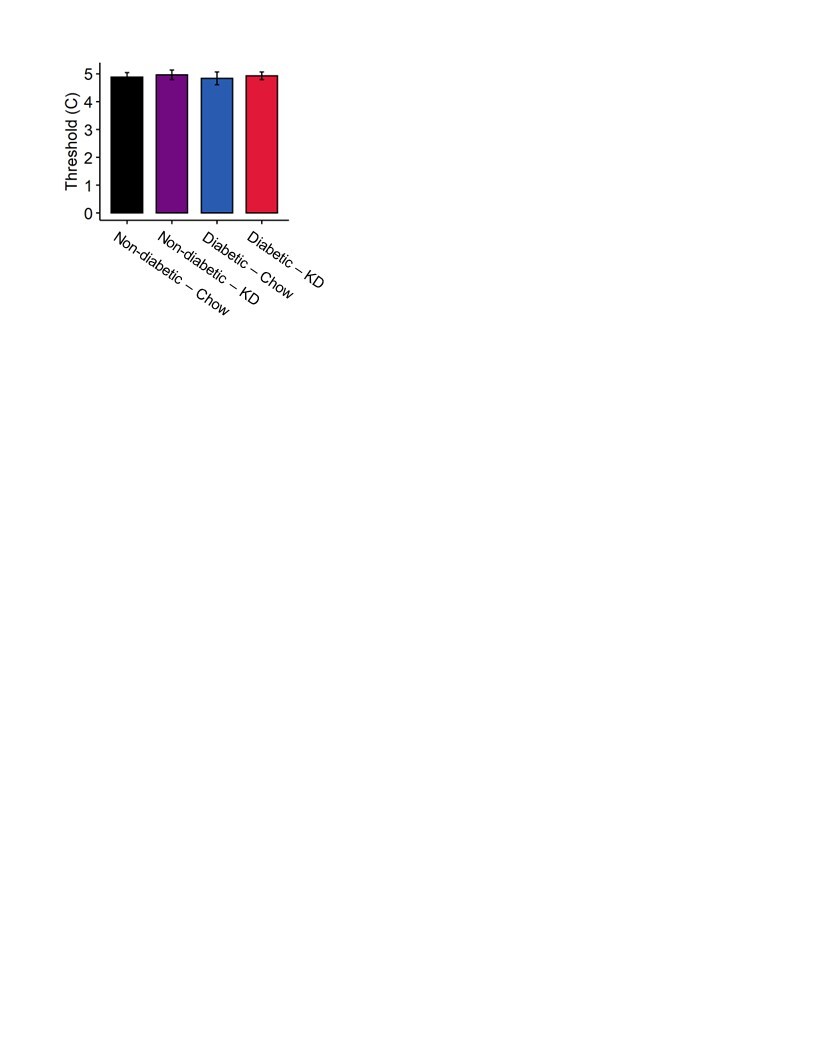{kind=link}
Acknowledgments
This work was supported by NIH grants RO1 NS043314 (DEW), the Kansas Institutional Development Award (IDeA) P20 GM103418, and core support from the Kansas IDDRC P30 HD00228.
Footnotes
Conflict of interest
The authors declare no competing financial interests.
Bibliography
- [1].Andersson DA, Gentry C, Light E, Vastani N, Vallortigara J, Bierhaus A, Fleming T, Bevan S. Methylglyoxal evokes pain by stimulating TRPA1. PloS one 2013;8(10):e77986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Apfel SC. Nerve growth factor for the treatment of diabetic neuropathy: What went wrong, what went right, and what does the future hold? International Review of Neurobiology, Vol. 50: Academic Press, 2002. pp. 393–413. [DOI] [PubMed] [Google Scholar]
- [3].Barragán-Iglesias P, Kuhn J, Vidal-Cantú GC, Salinas-Abarca AB, Granados-Soto V, Dussor GO, Campbell ZT, Price TJ. Activation of the integrated stress response in nociceptors drives methylglyoxal-induced pain. Pain 2019;160(1):160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bednarik J, Vlckova‐Moravcova E, Bursova S, Belobradkova J, Dusek L, Sommer C. Etiology of small‐fiber neuropathy. Journal of the Peripheral Nervous System 2009;14(3):177–183. [DOI] [PubMed] [Google Scholar]
- [5].Bennmann D, Kannicht C, Fisseau C, Jacobs K, Navarette-Santos A, Hofmann B, Horstkorte R. Glycation of the high affinity NGF-receptor and RAGE leads to reduced ligand affinity. Mechanisms of Ageing and Development 2015;150:1–11. [DOI] [PubMed] [Google Scholar]
- [6].Bierhaus A, Fleming T, Stoyanov S, Leffler A, Babes A, Neacsu C, Sauer SK, Eberhardt M, Schnölzer M, Lasitschka F. Methylglyoxal modification of Na v 1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nature medicine 2012;18(6):926–933. [DOI] [PubMed] [Google Scholar]
- [7].Bough KJ, Schwartzkroin PA, Rho JM. Calorie restriction and ketogenic diet diminish neuronal excitability in rat dentate gyrus in vivo. Epilepsia 2003;44(6):752–760. [DOI] [PubMed] [Google Scholar]
- [8].Brownlow ML, Benner L, D’Agostino D, Gordon MN, Morgan D. Ketogenic diet improves motor performance but not cognition in two mouse models of Alzheimer’s pathology. PloS one 2013;8(9):e75713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chandrasekaran P, Rani PK. Reversal of diabetic tractional retinal detachment attributed to keto diet. BMJ Case Reports 2020;13(10):e235873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. Journal of neuroscience methods 1994;53(1):55–63. [DOI] [PubMed] [Google Scholar]
- [11].Chen K, Zhang J, Zhang W, Zhang J, Yang J, Li K, He Y. ATP-P2X4 signaling mediates NLRP3 inflammasome activation: a novel pathway of diabetic nephropathy. The international journal of biochemistry & cell biology 2013;45(5):932–943. [DOI] [PubMed] [Google Scholar]
- [12].Chen Y, Ouyang X, Hoque R, Garcia-Martinez I, Yousaf MN, Tonack S, Offermanns S, Dubuquoy L, Louvet A, Mathurin P. β-Hydroxybutyrate protects from alcohol-induced liver injury via a Hcar2-cAMP dependent pathway. Journal of hepatology 2018;69(3):687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cheng HT, Dauch JR, Porzio MT, Yanik BM, Hsieh W, Smith AG, Singleton JR, Feldman EL. Increased axonal regeneration and swellings in intraepidermal nerve fibers characterize painful phenotypes of diabetic neuropathy. J Pain 2013;14(9):941–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Christianson JA, Ryals JM, Johnson MS, Dobrowsky RT, Wright DE. Neurotrophic modulation of myelinated cutaneous innervation and mechanical sensory loss in diabetic mice. Neuroscience 2007;145(1):303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cooper MA, McCoin C, Pei D, Thyfault JP, Koestler D, Wright DE. Reduced mitochondrial reactive oxygen species production in peripheral nerves of mice fed a ketogenic diet. Experimental physiology 2018;103(9):1206–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cooper MA, Menta BW, Perez-Sanchez C, Jack MM, Khan ZW, Ryals JM, Winter M, Wright DE. A ketogenic diet reduces metabolic syndrome-induced allodynia and promotes peripheral nerve growth in mice. Experimental Neurology 2018;306:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Di Lorenzo C, Coppola G, Bracaglia M, Di Lenola D, Sirianni G, Rossi P, Di Lorenzo G, Parisi V, Serrao M, Cervenka MC. A ketogenic diet normalizes interictal cortical but not subcortical responsivity in migraineurs. BMC neurology 2019;19(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dressler AR E; Trimmel-Schwahofer P; Klbermaxz K; Prayer D; Kaspiran G; Rami B; Schober E; Feucht M Type 1 diabetes and epilepsy: efficacy and safety of the ketogenic diet. Epilepsia 2010;51(6):1806–1809. [DOI] [PubMed] [Google Scholar]
- [19].Düll MM, Riegel K, Tappenbeck J, Ries V, Strupf M, Fleming T, Sauer SK, Namer B. Methylglyoxal causes pain and hyperalgesia in human through C-fiber activation. Pain 2019;160(11):2497–2507. [DOI] [PubMed] [Google Scholar]
- [20].Dupuis N, Curatolo N, Benoist JF, Auvin S. Ketogenic diet exhibits anti-inflammatory properties. Epilepsia 2015;56(7):e95–98. [DOI] [PubMed] [Google Scholar]
- [21].Dyck PJ, Davies JL, Wilson DM, Melton LJ, O’Brien PC. Risk factors for severity of diabetic polyneuropathy: intensive longitudinal assessment of the Rochester Diabetic Neuropathy Study cohort. Diabetes care 1999;22(9):1479–1486. [DOI] [PubMed] [Google Scholar]
- [22].Feldman EL, Callaghan BC, Pop-Busui R, Zochodne DW, Wright DE, Bennett DL, Bril V, Russell JW, Viswanathan V. Diabetic neuropathy. Nat Rev Dis Primers 2019;5(1):41. [DOI] [PubMed] [Google Scholar]
- [23].Galosi E, La Cesa S, Di Stefano G, Karlsson P, Fasolino A, Leone C, Biasiotta A, Cruccu G, Truini A. A pain in the skin. Regenerating nerve sprouts are distinctly associated with ongoing burning pain in patients with diabetes. European Journal of Pain 2018;22(10):1727–1734. [DOI] [PubMed] [Google Scholar]
- [24].Grace PM, Strand KA, Galer EL, Urban DJ, Wang X, Baratta MV, Fabisiak TJ, Anderson ND, Cheng K, Greene LI. Morphine paradoxically prolongs neuropathic pain in rats by amplifying spinal NLRP3 inflammasome activation. Proceedings of the National Academy of Sciences 2016;113(24):E3441–E3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Greco T, Glenn TC, Hovda DA, Prins ML. Ketogenic diet decreases oxidative stress and improves mitochondrial respiratory complex activity. Journal of Cerebral Blood Flow & Metabolism 2016;36(9):1603–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Griggs RB, Laird DE, Donahue RR, Fu W, Taylor BK. Methylglyoxal requires AC1 and TRPA1 to produce pain and spinal neuron activation. Frontiers in neuroscience 2017;11:679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Groover AL, Ryals JM, Guilford BL, Wilson NM, Christianson JA, Wright DE. Exercise-mediated improvements in painful neuropathy associated with prediabetes in mice. PAIN® 2013;154(12):2658–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Guilford BL, Ryals JM, Wright DE. Phenotypic Changes in Diabetic Neuropathy Induced by a High-Fat Diet in Diabetic C57Bl/6 Mice. Experimental Diabetes Research 2011;2011:848307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Guo G, Kan M, Martinez JA, Zochodne DW. Local insulin and the rapid regrowth of diabetic epidermal axons. Neurobiology of disease 2011;43(2):414–421. [DOI] [PubMed] [Google Scholar]
- [30].Harun-Or-Rashid M, Inman DM. Reduced AMPK activation and increased HCAR activation drive anti-inflammatory response and neuroprotection in glaucoma. Journal of neuroinflammation 2018;15(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hsieh Y-L, Lin C-L, Chiang H, Fu Y-S, Lue J-H, Hsieh S-T. Role of Peptidergic Nerve Terminals in the Skin: Reversal of Thermal Sensation by Calcitonin Gene-Related Peptide in TRPV1-Depleted Neuropathy. PLOS ONE 2012;7(11):e50805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hussain TA, Mathew TC, Dashti AA, Asfar S, Al-Zaid N, Dashti HM. Effect of low-calorie versus low-carbohydrate ketogenic diet in type 2 diabetes. Nutrition 2012;28(10):1016–1021. [DOI] [PubMed] [Google Scholar]
- [33].Jia M, Wu C, Gao F, Xiang H, Sun N, Peng P, Li J, Yuan X, Li H, Meng X. Activation of NLRP3 inflammasome in peripheral nerve contributes to paclitaxel-induced neuropathic pain. Molecular pain 2017;13:1744806917719804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Johnson MS, Ryals JM, Wright DE. Early loss of peptidergic intraepidermal nerve fibers in an STZ-induced mouse model of insensate diabetic neuropathy. Pain 2008;140(1):35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Julio-Amilpas A, Montiel T, Soto-Tinoco E, Gerónimo-Olvera C, Massieu L. Protection of hypoglycemia-induced neuronal death by β-hydroxybutyrate involves the preservation of energy levels and decreased production of reactive oxygen species. Journal of Cerebral Blood Flow & Metabolism 2015;35(5):851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kajitani N, Iwata M, Miura A, Tsunetomi K, Yamanashi T, Matsuo R, Nishiguchi T, Fukuda S, Nagata M, Shibushita M. Prefrontal cortex infusion of beta‐hydroxybutyrate, an endogenous NLRP3 inflammasome inhibitor, produces antidepressant‐like effects in a rodent model of depression. Neuropsychopharmacology reports 2020;40(2):157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Papanas N, Ziegler D. Risk Factors and Comorbidities in Diabetic Neuropathy: An Update 2015. Rev Diabet Stud 2015;12(1–2):48–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Patel N, Llewelyn J, Wright D, Thomas P. Glucose and leucine uptake by rat dorsal root ganglia is not insulin sensitive. Journal of the neurological sciences 1994;121(2):159–162. [DOI] [PubMed] [Google Scholar]
- [39].Poplawski MM, Mastaitis JW, Isoda F, Grosjean F, Zheng F, Mobbs CV. Reversal of Diabetic Nephropathy by a Ketogenic Diet. PLOS ONE 2011;6(4):e18604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ruskin DN, Kawamura M Jr, Masino SA. Reduced pain and inflammation in juvenile and adult rats fed a ketogenic diet. PloS one 2009;4(12):e8349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ruskin DN, Sturdevant IC, Wyss LS, Masino SA. Ketogenic diet effects on inflammatory allodynia and ongoing pain in rodents. Scientific Reports 2021;11(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Russell JW, Sullivan KA, Windebank AJ, Herrmann DN, Feldman EL. Neurons Undergo Apoptosis in Animal and Cell Culture Models of Diabetes. Neurobiology of Disease 1999;6(5):347–363. [DOI] [PubMed] [Google Scholar]
- [43].Shang S, Wang L, Zhang Y, Lu H, Lu X. The beta-hydroxybutyrate suppresses the migration of glioma cells by inhibition of NLRP3 inflammasome. Cellular and molecular neurobiology 2018;38(8):1479–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sleiman SF, Henry J, Al-Haddad R, El Hayek L, Abou Haidar E, Stringer T, Ulja D, Karuppagounder SS, Holson EB, Ratan RR, Ninan I, Chao MV. Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body β-hydroxybutyrate. eLife 2016;5:e15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Song S, Qiu D, Luo F, Wei J, Wu M, Wu H, Du C, Du Y, Ren Y, Chen N. Knockdown of NLRP3 alleviates high glucose or TGFB1-induced EMT in human renal tubular cells. Journal of molecular endocrinology 2018;61(3):101–113. [DOI] [PubMed] [Google Scholar]
- [46].Sözbir E, Nazıroğlu M. Diabetes enhances oxidative stress-induced TRPM2 channel activity and its control by N-acetylcysteine in rat dorsal root ganglion and brain. Metabolic brain disease 2016;31(2):385–393. [DOI] [PubMed] [Google Scholar]
- [47].Sweitzer S, Colburn R, Rutkowski M, DeLeo J. Acute peripheral inflammation induces moderate glial activation and spinal IL-1β expression that correlates with pain behavior in the rat. Brain research 1999;829(1–2):209–221. [DOI] [PubMed] [Google Scholar]
- [48].Taylor MK, Sullivan DK, Mahnken JD, Burns JM, Swerdlow RH. Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimer’s & Dementia: Translational Research & Clinical Interventions 2018;4:28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tosaki T, Kamiya H, Yasuda Y, Naruse K, Kato K, Kozakae M, Nakamura N, Shibata T, Hamada Y, Nakashima E, Oiso Y, Nakamura J. Reduced NGF secretion by Schwann cells under the high glucose condition decreases neurite outgrowth of DRG neurons. Experimental Neurology 2008;213(2):381–387. [DOI] [PubMed] [Google Scholar]
- [50].Tóth CC Z Type 1 diabetes mellitus successfully managed with the paleolithic ketogenic diet. Int J Case Rep Images 2014;5(10):699–703. [Google Scholar]
- [51].Valdebenito R, Ruminot I, Garrido-Gerter P, Fernández-Moncada I, Forero-Quintero L, Alegría K, Becker HM, Deitmer JW, Barros LF. Targeting of astrocytic glucose metabolism by beta-hydroxybutyrate. Journal of Cerebral Blood Flow & Metabolism 2015;36(10):1813–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wei J-Y, Liu C-C, Ouyang H-D, Ma C, Xie M-X, Liu M, Lei W-L, Ding H-H, Wu S-L, Xin W-J. Activation of RAGE/STAT3 pathway by methylglyoxal contributes to spinal central sensitization and persistent pain induced by bortezomib. Experimental neurology 2017;296:74–82. [DOI] [PubMed] [Google Scholar]
- [53].Yao J, Chen S, Mao Z, Cadenas E, Brinton RD. 2-Deoxy-D-Glucose Treatment Induces Ketogenesis, Sustains Mitochondrial Function, and Reduces Pathology in Female Mouse Model of Alzheimer’s Disease. PLOS ONE 2011;6(7):e21788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zherebitskaya E, Akude E, Smith DR, Fernyhough P. Development of selective axonopathy in adult sensory neurons isolated from diabetic rats: role of glucose-induced oxidative stress. Diabetes 2009;58(6):1356–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Changes in body composition in diabetic and ketogenic diet-fed mice. (A) Diabetic chow-fed mice have a decreased gain in lean mass compared to non-diabetic controls. Both non-diabetic and diabetic mice fed a ketogenic diet experience a loss in lean mass relative to chow-fed mice (n=7–12). Mice fed a ketogenic diet experience a robust increase in fat mass (B) and percent body fat (C) regardless of diabetes status (n=7–12). (A-C) 2-Way ANOVA and Tukey’s post hoc test; * p < 0.05 compared to chow-fed non-diabetic mice.
Figure S2. Insulin in non-diabetic and type 1 diabetic mice fed a ketogenic diet. Diabetic mice exhibited reduced serum insulin compared to non-diabetic controls, as expected. Diabetic mice fed a ketogenic diet do not exhibit elevated serum insulin relative to chow-fed diabetics (n=7–12). Mixed-models ANOVA with repeated measures and Tukey’s post hoc test; * p < 0.05 compared to chow-fed non-diabetic mice.
Figure S3. Cold plate assay in diabetic and non-diabetic mice fed a ketogenic diet. Mice displayed no changes in thermal thresholds to elicit nocifensive behavior regardless of diet or diabetic status (n=7–12). 2-Way ANOVA and Tukey’s post hoc test.