

Abstract
Rationale:
Sildenafil, a well-known vasodilator known to interfere with purinergic signaling through effects on cGMP, is a mainstay in the treatment of pulmonary hypertension (PH). However, little is known regarding its effects on the metabolic reprogramming of vascular cells, which is a hallmark of pulmonary hypertension (PH). Purine metabolism, especially intracellular de novo purine biosynthesis is essential for vascular cell proliferation. Since adventitial fibroblasts are critical contributors to proliferative vascular remodeling in pulmonary hypertension, in this study we aimed to investigate if sildenafil, beyond its well-known vasodilator role in smooth muscle cells, impacts intracellular purine metabolism and proliferation of fibroblasts derived from human PH patients
Methods:
Integrated omics approaches (plasma and cell metabolomics) and pharmacological inhibitor approaches were employed in plasma samples and cultured pulmonary artery fibroblasts from PH patients.
Measurements and main results:
Plasma metabolome analysis of 27 PH patients before and after treatment with sildenafil, demonstrated a partial, but specific effect of sildenafil on purine metabolites, especially adenosine, adenine, and xanthine. However, circulating markers of cell stress, including lactate, succinate, and hypoxanthine were only decreased in a small subset of sildenafil-treated patients. To better understand potential effects of sildenafil on pathological changes in purine metabolism (especially purine synthesis) in PH, we performed studies on pulmonary fibroblasts from PAH patients (PH-Fibs) and corresponding controls (CO-Fibs), since these cells have previously been shown to demonstrate stable and marked PH associated phenotypic and metabolic changes. We found that PH-Fibs exhibited significantly increased purine synthesis. Treatment of PH-Fibs with sildenafil was insufficient to normalize cellular metabolic phenotype and only modestly attenuated the proliferation. However, we observed that treatments which have been shown to normalize glycolysis and mitochondrial abnormalities including a PKM2 activator (TEPP-46), and the histone deacetylase inhibitors (HDACi), SAHA and apicidin, had significant inhibitory effects on purine synthesis. Importantly, combined treatment with HDACi and sildenafil exhibited synergistic inhibitory effects on proliferation and metabolic reprogramming in PH-Fibs.
Conclusions:
While sildenafil alone partially rescues metabolic alterations associated with PH, treatment with HDACi, in combination with sildenafil, represent a promising and potentially more effective strategy for targeting vasoconstriction, metabolic derangement and pathological vascular remodeling in PH.
Keywords: Pulmonary hypertension, pulmonary artery fibroblasts, purine synthesis, metabolomics, sildenafil, HDAC inhibitors
INTRODUCTION
Chronic Pulmonary Hypertension (PH) is an incurable disease with poor outcomes and a median survival of 5–7 years [1, 2]. Pulmonary artery (PA) vasoconstriction and pathological vascular remodeling characterized by dysregulated proliferation, persistent inflammatory signaling and extracellular matrix synthesis and remodeling collectively contributes to pathogenic disease outcomes. Targeting both aspects would appear to be required for optimal treatment effect. Purinergic signaling plays a pivotal role in the control of vascular tone and remodeling [3–5] and thus has been proposed as a potential contributor to the pathogenesis of PH [6] Adenosine triphosphate (ATP), released from endothelial cells in response to changes in blood flow and hypoxia, acts on ATP and ADP P2 purinergic receptors (particularly A2AR) on endothelial cells leading to the production of endothelium-derived hyperpolarizing factors including nitric oxide (NO) and prostaglandins to cause vasodilation [4]. On the other hand, A2BR activation can induce vascular remodeling through P2X7 as well as P2Y1 and P2Y12 receptors. Further, intracellular purines serve as building blocks for nucleic acids, cofactors and energy store molecules, the key molecules controlling bioenergetics and anabolism to sustain proliferation of smooth muscle cells (SMCs), endothelial cells (ECs) in the pulmonary vasculature. It has been shown that intracellular purine synthesis is increased in proliferative vascular SMCs and plays an important role in vascular proliferative remodeling [7]. However, whether intracellular purine metabolism, especially de novo purine synthesis is a druggable target in PH and other cardiovascular diseases remains unexplored.
Several therapeutic interventions in PH rely on the use of vasodilators interfering with purinergic signaling, such as the phosphodiesterase type 5 (PDE5) inhibitors Sildenafil and Tadalafil, which block the degradation of cyclic guanylate monophosphate (cGMP), providing the pharmacological basis for modulation of the NO/cGMP pathway in vascular ECs and SMCs [8–11]. It is known that Sildenafil improves exercise capacity and quality of life in patients with symptomatic PAH [12] and is effective for cardiovascular and oxygenation endpoints in pediatric PH [13]. However, it is increasingly recognized that only some patients respond to vasodilator therapy with improved pulmonary hemodynamics and symptomatic status [14, 15]. Reasons for lack of success include the presence of chronic vascular remodeling and inflammation, rather than simply active vasoconstriction [16–18] and the involvement of multiple cell types including adventitial fibroblasts and recruited monocytes, macrophages, and lymphocytes where the effects of vasodilating drugs such as sildenafil are less clear [19–23]. Thus, the observed proliferative vascular remodeling in PH patients raises questions about the potential metabolic changes, including purine metabolism in PH vascular cells, especially in response to sildenafil treatment.
In contrast to normal vascular cells, the cells of hypertensive subjects and particularly adventitial fibroblasts, exhibit complex metabolic reprogramming characterized by an increase in glucose uptake, glycolysis, pentose phosphate pathway (PPP) and biosynthetic processes to sustain increased cell growth and proliferation [19–21, 23, 24]. Purines are basic components of the nucleic acids DNA and RNA and also provide the necessary energy (purine nucleotides such as ATP and guanosine triphosphate (GTP)) and cofactors (such as nicotinamide adenine dinucleotide (NAD) and its phosphate, reduced counterpart (NADPH) – essential for almost all anabolic reactions - and coenzyme A) to promote cell growth [25, 26]. Thus, alterations in purine metabolism can be associated with the progression of proliferative vascular responses in PH. Notably, altered purine metabolism is common in tumor cells. This discovery led to the development of the earliest antitumor drugs (purine antimetabolites) to treat cancers by blocking DNA and RNA synthesis and halting metastatic cell growth [27–30]. Importantly, de novo biosynthesis of purines requires the generation of 5-phosphoribose-1-pyrophosphate (PRPP) produced through the PPP parallel to glycolysis, which was recently observed in PH-Fibs [20, 23].
In the current study, we investigated purine metabolism in plasma and cells derived from PH patients and controls and investigated the changes in response to sildenafil treatment. Given the role of glycolysis in fueling de novo purine synthesis, we also determined the effect of previously validated glycolysis inhibiting interventions (TEPP-46 and HDACi) on purine metabolism in PH-Fibs. Finally, we investigated the combined effect of sildenafil and HDACi on PH-Fibs’ purine metabolism and proliferation. The results of our study provide evidence of the feasibility and promising efficacy of a novel treatment strategy for PH that combines the vasodilatory and anti-inflammatory effects of sildenafil, with treatments inhibiting metabolic reprogramming and de novo purine synthesis, such as HDACi.
MATERIALS and METHODS
Plasma Metabolomics from PH patients treated with sildenafil
Metabolomics analyses were performed on 20 ul of plasma from 27 PH patients (Age: 53.5 + 14.0; Sex: 24 female, 3 male; Ethnicity: 20 non-Hispanic white; 4 African American, 3 Hispanic Latino), prior to and after treatment with sildenafil. Detailed information in Supplemental Table 1 and supplemental Figure 1.
Cell Isolation and Culture
Human pulmonary artery fibroblasts were derived from patients with idiopathic PH (n = 4) undergoing lung transplantation at Papworth Hospital, Cambridge, UK or from control non-PH donors (n = 4) as previously described [22, 23, 31]. Cells were cultured under normoxic conditions. Cell phenotype was thoroughly characterized, and all experiments were performed on cells at passages 5–10. All patients provided written informed consent.
Proliferation assay
Cell proliferation was evaluated using CyQUANT proliferation analysis Kit (Thermo Fisher Scientific, REF# C7026) as previously described [20, 23]. After various days in culture, plates were gently inverted to remove medium by blotting it onto paper towels and washed once with PBS. Cells in the plates were frozen and stored at −80 °C until samples to be assayed. Measurement was done following manufacture’s instruction. Briefly, plates were thawed at room temperature, then 250 μl of the CyQUANT GR dye/cell-lysis buffer was added to each sample well. Mixed gently, incubated at room temperature for 5 min, protected from light, then fluorescence was measured using a fluorescence plate reader with filters at 480 nm excitation and 520 nm emission maxima.
Real-time RT-PCR
As described previously [22, 23], extraction of total RNA was performed using RNeasy Mini Kit (Qiagen, Inc). Total RNA was reverse-transcribed to cDNA, using an iScript cDNA Synthesis Kit (Bio-Rad, Inc) for mRNA detection. The levels of mRNA were determined quantitatively using Quantitative-Reverse Transcriptase PCR (qRT-PCR). The iTaq Universal SYBR Green Supermix (Bio-Rad, Inc) and unique primer pair (HPRT forward primer: 5’- ACGTCTTGCTCGAGATGTGA −3’ and reverse primer: 5’- AATCCAGCAGGTCAGCAAAG −3’; ADA forward primer: 5’- CCTGGCCAAGTTTGACTACTAC–3’ and reverse primer: 5’- CCTCTTTGGCCTTCATCTCTAC −3’) were used to detect mRNAs. qRT-PCR product analysis was performed on CFX96 Real-Time System (Bio-Rad, Inc, Hercules, CA, USA). The data were normalized using the endogenous HPRT as control. Gene expression was calculated after normalization to control group using the delta-delta CT method.
Sildenafil, TEEP-46 and HDAC inhibitor treatment
The cultured cells (under normoxic conditions) were treated with sildenafil (50nM, Pfizer) or TEPP-46 (100 μM, EMD Millipore Corp, Billerica, MA) or pan-HDAC inhibitor, suberoylanilide hydroxamic acid (SAHA; 10 μM, ChemieTek, Indianapolis, IN) or class I HDAC inhibitor apicidin (3 μM) (Enzo, New York, NY) as previously described [23]. The cells were harvested for Ultra-high Pressure Liquid Chromatography-Mass Spectrometry-Based Metabolomics Analyses after 48h treatment.
Ultra-high Pressure Liquid Chromatography-Mass Spectrometry-Based Metabolomics Analyses
Steady state and U13C-glucose tracing metabolomics analyses were performed as previously reported [20, 32]. Briefly, CO-Fibs and PH-Fibs (n=3–4, each group) were either untreated or treated for 48h with (i) sildenafil (50nM) ; (ii) TEPP-46 (100 μM); (iii) HDACi (SAHA, 10 μM or apicidin, 3 μM); (iv) sildenafil (50 nM) + apicidin (3 μM). Cells were extracted (2×106 cells/ml ice-cold buffer methanol:acetonitrile:water 5:3:2). For tracing experiments, cells were incubated with U-13C-glucose (SIGMA) replaced DMEM media for 24h. Extracts run on a C18 reversed phase column (Phenomenex, Torrance, CA) through an ultra-high performance chromatographic system (UHPLC – Vanquish, Themo Fisher) coupled online with a high resolution quadrupole orbitrap instrument (Qexactive – Thermo Fisher) operated in both polarity modes at 70,000 resolution. Metabolite assignment, peak integration for relative quantitation and isotopologue distributions in tracing experiments were calculated through the software Maven (Princeton), against the KEGG pathway database and an in-house validated standard library (>800 compounds; SIGMA Aldrich; IROATech). Integrated peak areas were exported into Excel (Microsoft, Redmond, CA) for statistical analysis (t-test, ANOVA through the software GraphPad Prism, GENE E (Broad Institute) or Metaboanalyst 3.0 [33], for hierarchical clustering and partial-least square discriminant analysis (PLS-DA).
Statistical analysis.
Unpaired Student t test was used to compare two groups. For more than 2 groups with 1 variable, 1-way ANOVA was performed and for more than 2 groups with 2 independent variables, 2-way ANOVA followed by Tukey’s multiple comparisons test was performed. The Kolmogorov-Smirnov, Shapiro-Wilk, and D’Agostino tests were used to assess for normality before applying parametric statistical tests. Nonparametric test was performed if data did not pass the parametric assumption. Statistical analysis for metabolomics analyses, including hierarchical clustering and partial-least square discriminant analysis (PLS-DA), were performed using GraphPad Prism, GENE E (Broad Institute), or Metaboanalyst 5.0. Data are presented as mean ± SEM. Differences with p values <0.05 were considered statistically significant.
RESULTS
Sildenafil treatment in PH patients impacts plasma levels of purines with mixed effects on other metabolic pathways
Sildenafil was developed to mimic the purine ring of cGMP and thus inhibit the main enzyme that catalyzes its degradation, phosphodiesterase E5 (PDE5) [34]. While metabolic reprogramming is a hallmark of PH and sildenafil treatment is a mainstay in PH treatment, little is known about the impact of sildenafil on the plasma metabolome of PH patients in vivo. To bridge this knowledge gap, we evaluated the plasma metabolome of 27 patients with PH (including the patients with idiopathic pulmonary arterial hypertension (IPAH) and sickle cell PH), before and after treatment with sildenafil using UHPLC-MS (Figure 1, Supplementary Figure 1 and Supplementary Table 1).
Figure 1. Metabolomics analysis of plasma from 27 PH patients, before and after treatment with sildenafil.
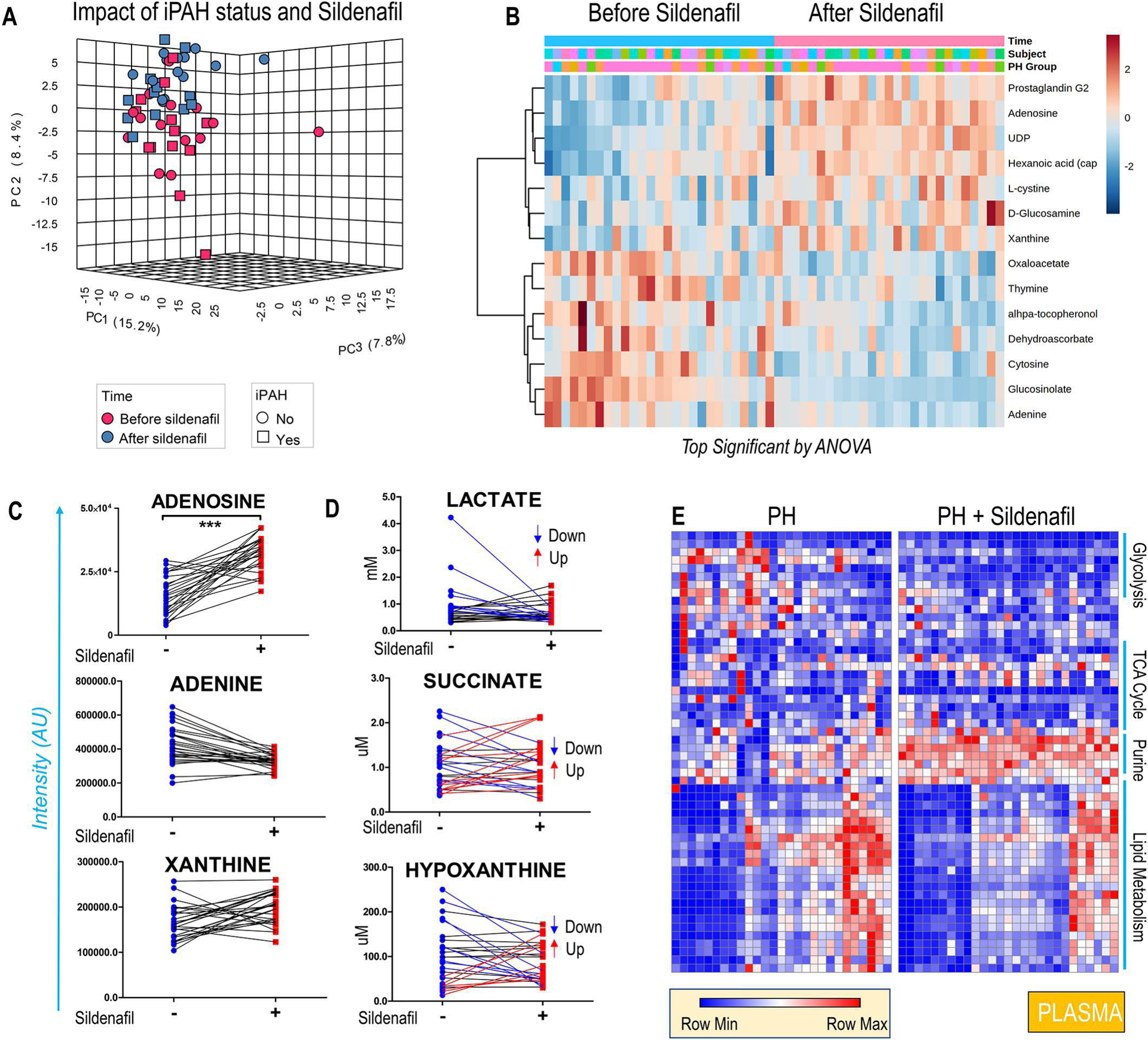
Sildenafil partially reverses plasma metabolic changes altered by PH as shown by the principal component analysis in A. Hierarchical clustering of the most significant metabolites by repeated measures ANOVA (B) shows a significant impact of sildenafil on the plasma levels of adenosine and several purines (C). Of note, mixed effects were observed in the population with respect to circulating markers of hypoxia, lactate, succinate and hypoxanthine (D). The heat map in E provides a more comprehensive of the impact (or lack thereof) of sildenafil on specific pathways, in particular lipid metabolism.
Treatment with sildenafil had a significant effect on the plasma metabolome, as shown by the principal component analysis in Figure 1. A. Significant effects were noted in the circulating levels of purines – including adenosine, adenine, and xanthine (Figure 1. C) as well as several oxidant stress-related metabolites (prostaglandins, cystine, alpha-tocopherol, dehydroascorbate, glucosinolate – Figure 1. B). However, circulating markers related to glycolysis/hypoxia including lactate, succinate and hypoxanthine were decreased only in a subset of patients following treatment with sildenafil (Figure 1. D). Importantly, neither the carboxylic acid (TCA cycle) nor lipid metabolism was affected by sildenafil treatment (heat map in Figure 1. E).
Clinical variables like 6-minute walk distance (6MWD), body mass index (BMI), and age were recorded before and after sildenafil treatment and used to identify poor vs good responders to the treatment (Figure 2. A). No correlation was observed between changes in 6MWD and circulating levels of sildenafil and its metabolite desmethylsildenafil, or the ratios of desmethylsildenafil/sildenafil (data not shown). BMI exhibited a significant negative correlation to 6MWD independently of sildenafil treatment and purine levels, especially adenosine and urate (Figure 2. B). Improvements in 6MWD were positively associated with decreases in carboxylic acids (alpha-ketoglutarate), sulfur metabolites (taurine) and increases in arginine metabolism (arginine, guanidinoacetate - Figure 2. A) and fatty acyl-carnitines. Patients’ age had a significant negative correlation with 6MWD (Figure 2. A) and plasma adenosine levels (Figure 2. C).
Fig 2. Functional clinical measurements including 6-minute walk test to determine the efficacy of the treatment with sildenafil were performed in 27 PH patients, before and after treatment with sildenafil.
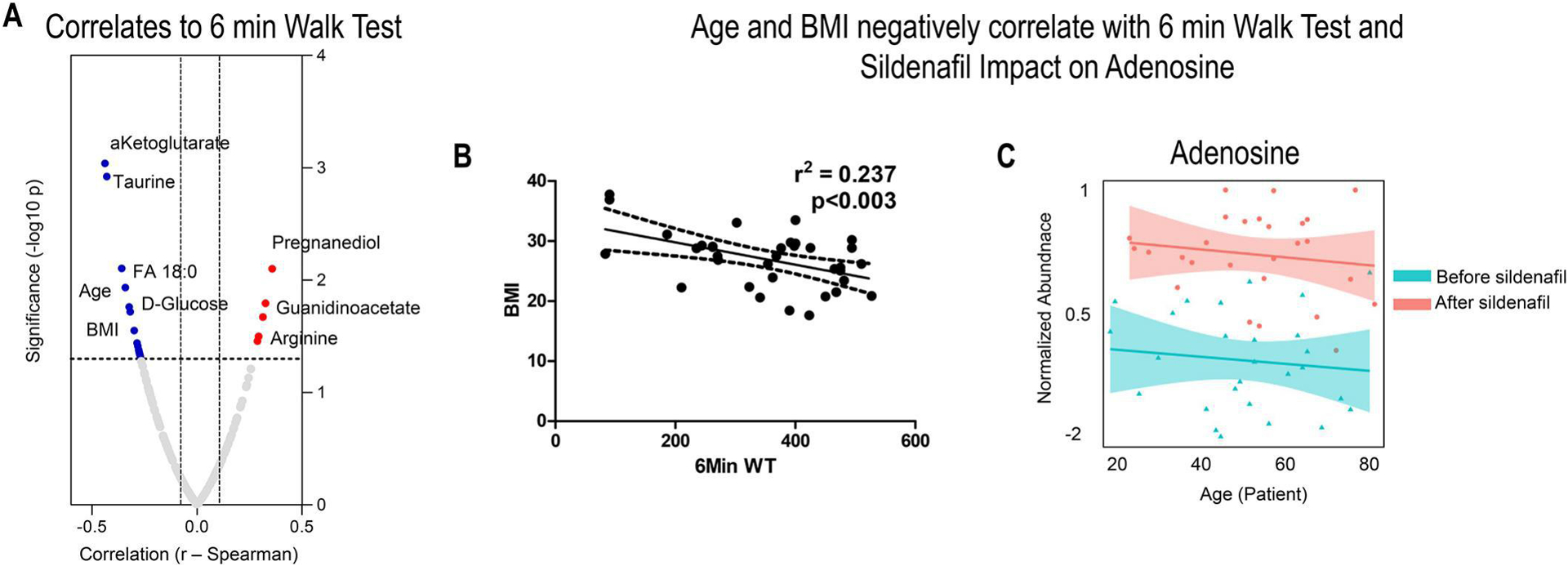
Top metabolic correlates to 6MWD included multiple metabolites in fatty acid and arginine metabolism, lipid oxidation products (A). However, patients’ BMI and age were identified as critical covariates impacting such measurements and factors. Patients’ BMI exhibited a significant negative correlation to 6MWD independently of sildenafil treatment and purine levels in B. Patients’ age at least in part impacting the effect of sildenafil on purine metabolism (e.g., adenosine as a function of sildenafil treatment) in C. Adenosine/urate ranked amongst the top correlates to BMI independently from sildenafil treatment in Supplementary Table 1).
Of note, we noticed divergent responses across the subjects enrolled in this study, with Sildenafil either decreasing or increasing the circulating levels of metabolites, which may explain the diversity of responses of patients to Sildenafil.
Metabolomics analyses reveal the up-regulation of purines and purine de novo synthesis in PH-Fibs cultured under normoxic conditions
To explore the role of purine metabolisms in the hyperproliferative state of vascular cells, human pulmonary artery fibroblasts derived from patients with IPAH (PH-Fibs) or from control donors (CO-Fibs) were used as in vitro model system. We have previously reported that these PH-Fibs, cultured under normoxic conditions, exhibit an imprinted proliferative and reprogrammed metabolic phenotype [20, 23, 35–37]. Herein we sought to examine the state of intracellular purine metabolism, especially de novo purine synthesis in CO- and PH-Fibs.
UHPLC-MS metabolomics revealed that PH-Fibs exhibited significant up-regulation of purines in comparison to corresponding controls as shown by the heatmap (Figure 3. A). Purine levels successfully informed clustering of CO-Fibs vs PH-Fibs through PLS-DA analysis with PC1 explaining 71.8% of the total variance across samples (Figure 3. B). An overview of purine metabolism and intermediates showing the most statistically significant changes between the two groups is provided in Figure 3. C and D, respectively. We observed that PH-Fibs exhibited the elevated levels of nucleotides (e.g., AMP) and nucleosides (e.g., adenosine). Of note, significant decreases in the transcripts for adenosine deaminase (ADA) in PH-Fibs in comparison to CO-Fibs was accompanied by significant decreases in the levels of inosine (the byproduct of the ADA enzyme) and inosine-derived hypoxanthine in PH-Fibs. On the other hand, guanosine and downstream purine catabolism/oxidation byproducts xanthine and allantoate were increased significantly in PH-Fibs compared to CO-Fibs as illustrated by purine metabolism and oxidation cascades and purine metabolism intermediates (Figure 3. C, D).
Figure 3. PH-Fibs exhibit upregulation of purine metabolism.
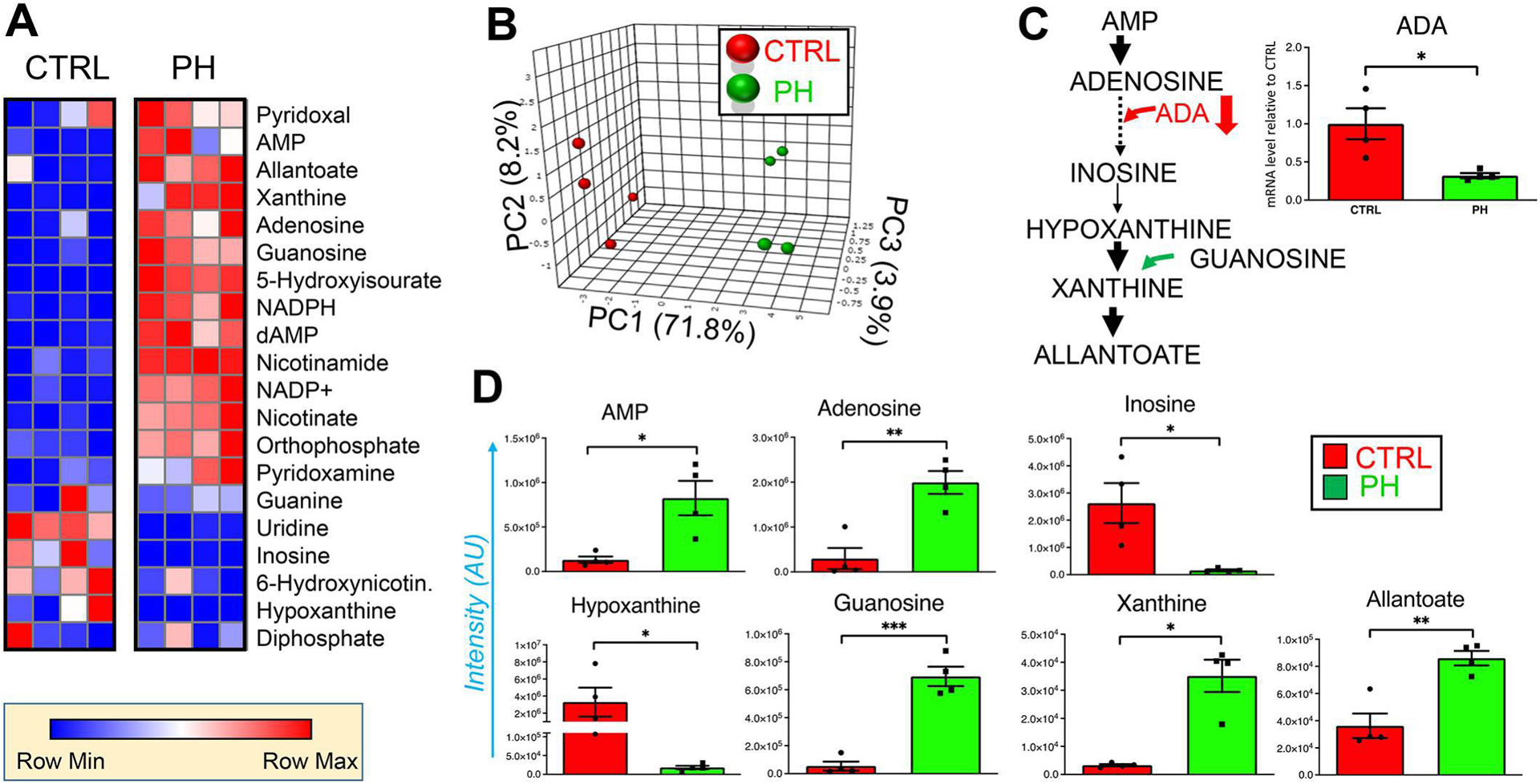
Deranged purine metabolism in fibroblasts derived from the pulmonary artery of health donors (CTRL) and PH patients (PH), as represented by heat maps (blue to red = low to high levels of Z score normalized metabolite abundances, n = 4) in (A). PLS-DA of PH-Fibs is shown in B, based on purine metabolite levels. In C, an overview of purine metabolism and oxidation cascades. Metabolites from this pathway are highlighted in D. (*, **, *** p <0.05, 0.01, 0.001, respectively; n = 4).
Significant increases in the steady state levels of purine nucleotides and nucleosides are suggestive of either decreased catabolism (e.g., by ADA) and/or increased anabolism, a necessary requirement for the biosynthesis of DNA and RNA in highly proliferating cells. Metabolic tracing experiments upon incubation of cells with U-13C-glucose for 24h were performed to test whether human PH-Fibs engaged in increased nucleoside biosynthesis. An overview of the expected isotopologue distributions is presented in Figure 4. A. Increased serine biosynthesis was observed, along with increased activation of the pentose phosphate pathway (either at the oxidative or non-oxidative branch level) which resulted in the generation of 13C5-ribose phosphate (Figure 4. B). Since both pathways fuel de novo nucleoside synthesis (according to the scheme in Figure 4. A), we identified significantly higher adenosine synthesis from both ribose and serine in PH-Fibs at 24h from incubation with U-13C-glucose (Figure 4. B).
Figure 4. Metabolic tracing of U-13C-glucose for 24h reveals incorporation of heavy carbon atoms in de novo synthesized purines through activation of the pentose phosphate pathway and serine metabolism in PH-Fibs.
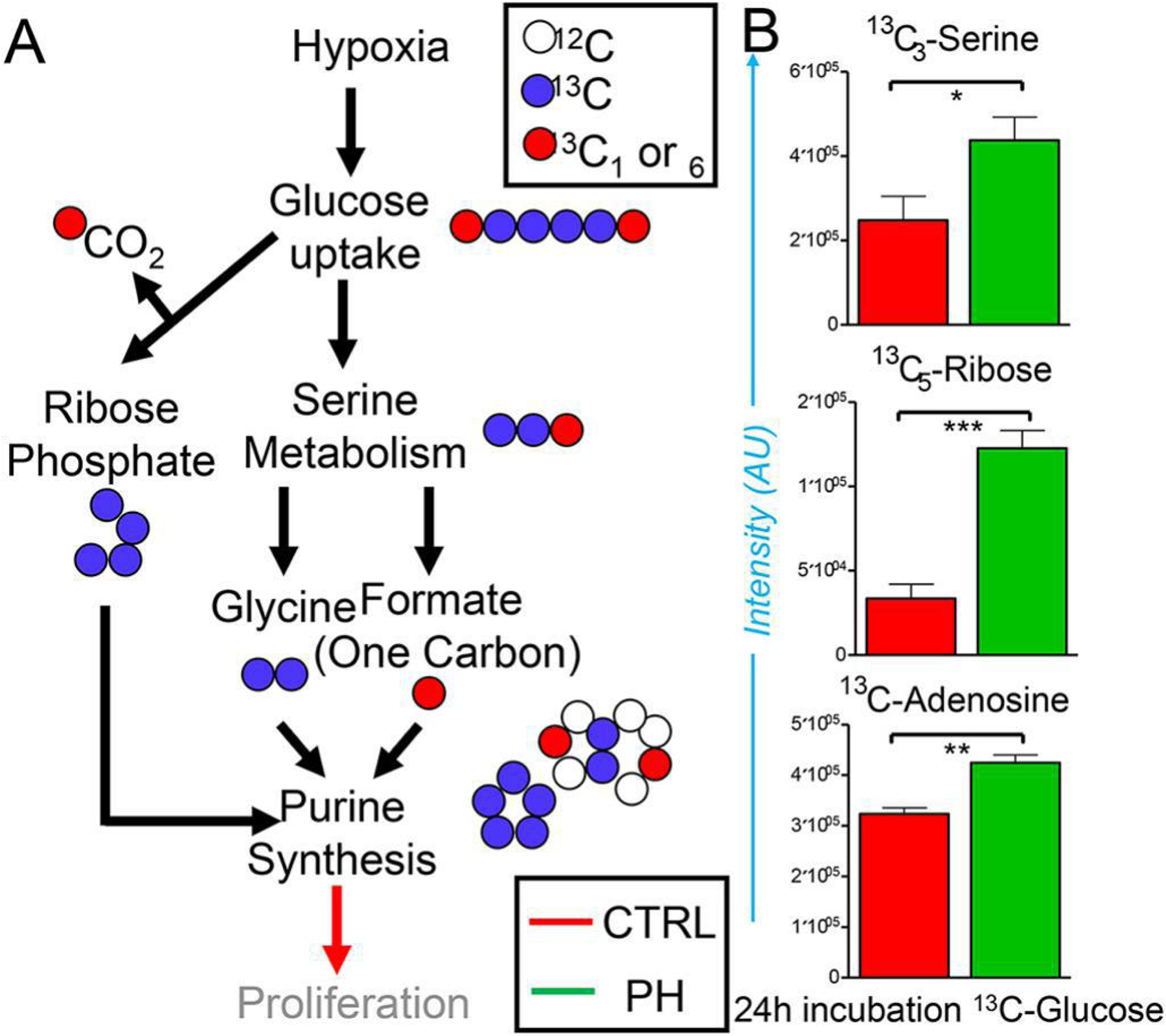
An overview of the expected isotopologue distributions is presented in A. Increased serine biosynthesis was observed, along with increased generation of 13C5-ribose phosphate and higher adenosine synthesis in PH-Fibs at 24h from incubation with U-13C-glucose in B. (*, **, *** T-test p <0.05, 0.01, 0.001, respectively; n = 3).
Effects of sildenafil on metabolism and phenotype of PH-Fibs
Next, we sought to determine the effect of sildenafil on the metabolic and proliferative state of human PH-Fibs. We treated PH-Fibs with Sildenafil (50 nM) and performed metabolomics analyses and proliferation assay. UHPLC-MS metabolomics revealed that sildenafil was insufficient to induce significant changes in the cellular levels of lactate and carboxylates including alpha-ketoglutarate, fumarate, and malate (Figure 5. A, B), which are the metabolites that have been proven significantly increased in PH-Fibs [20, 23, 36]. Further, we observed that sildenafil didn’t significantly impact the levels the purine metabolites such as AMP, adenosine and inosine in PH-Fibs (Figure 5. C). Importantly, we found sildenafil exhibited modest effect on inhibiting the proliferation of PH-Fibs (Figure 5. D).
Figure 5. Sildenafil was insufficient to correct metabolic and proliferative state in PH-Fibs.
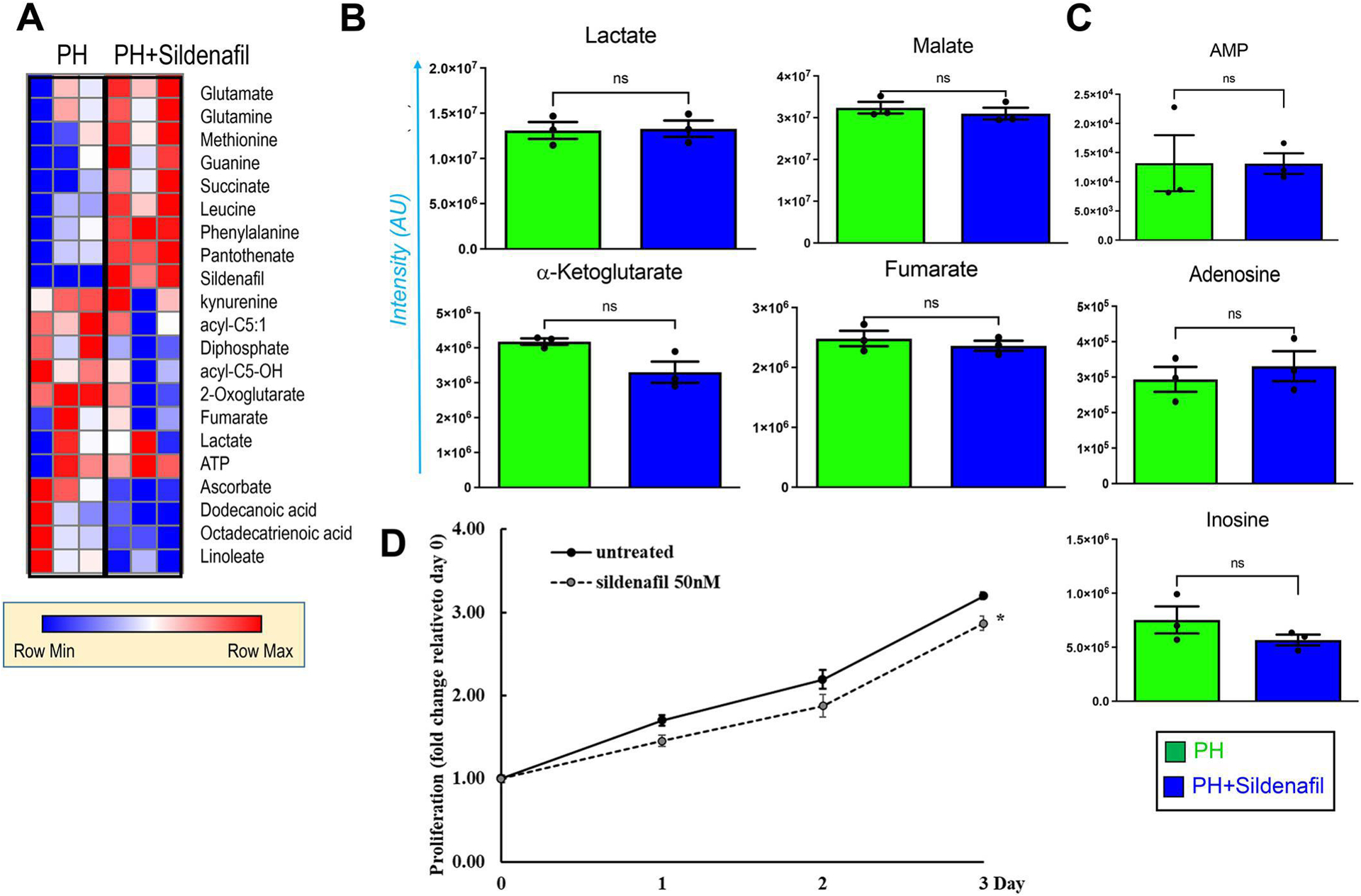
Metabolic analysis revealed that treatment with sildenafil did not decrease lactate generation and Krebs cycle aberrations as represented by heat maps (A) and plots (B), as well as purine metabolites (C) in PH-Fibs. (n = 3). (D) Proliferation assay was performed with CyQuant Kit in PH-Fibs with or without treatment of sildenafil (50nM) up to 3 days (n=3). Results were calculated as fold changes relative to day 0. *p <0.05 compared to untreated.
Promoting PKM activity through pharmacological treatment with TEPP-46 or HDACi decreases the subtracts of purine de novo synthesis (serine and ribose) and purines in PH-Fibs
Given the non-significant effect of sildenafil on the metabolism of PH-Fibs, then we have a reason for looking at other pathways. Next, we sought to examine whether suppression of serine metabolism and the pentose phosphate pathway is sufficient to decrease purine synthesis. In other pathologies characterized by highly proliferative states such as cancer, increased serine biosynthesis is sustained by isoform switching of PKM and the up-regulation of the PKM2/PKM1 ratio [38]. Increases in this ratio result in total decreases in PKM activity and promote a metabolic bottleneck in late glycolysis fueling serine anabolism[39, 40], which is required for purine synthesis as shown in Figure 4.A. Recently, we noted that PH-Fibs undergo a similar transition towards up-regulation of the PKM2/PKM1 ratio by decreasing PKM1 expression, paralleled with glycolytic reprogramming, mitochondrial abnormalities and activation of serine metabolism and pentose phosphate pathway [23, 41]. Furthermore, we have shown that normalization of PKM activity by both PKM activator (TEPP-46) and HDAC inhibitors (HDACi) reverses the PH-Fibs phenotype by limiting glycolysis and promoting the normalization of mitochondrial metabolism [23]. Since purine synthesis requires ribose-5-phosphate from the pentose phosphate pathway and formate from the serine synthesis pathway, here we hypothesized that normalization of PKM activity in PH-Fibs would limit purine biosynthesis by normalizing serine anabolism and pentose phosphate pathway (Figure 6. A), a prediction that we sought to validate through UHPLC-MS. We observed that both TEPP-46 (Figure 6. B) and the HDACi SAHA and Apicidin (Figure 6. C) decreased serine, ribose phosphate, and thus AMP significantly in PH-Fibs.
Figure 6. TEEP-46 and HDACi decreased upstream to purine synthesis via inhibiting serine metabolism and pentose phosphate pathway in PH-Fibs.
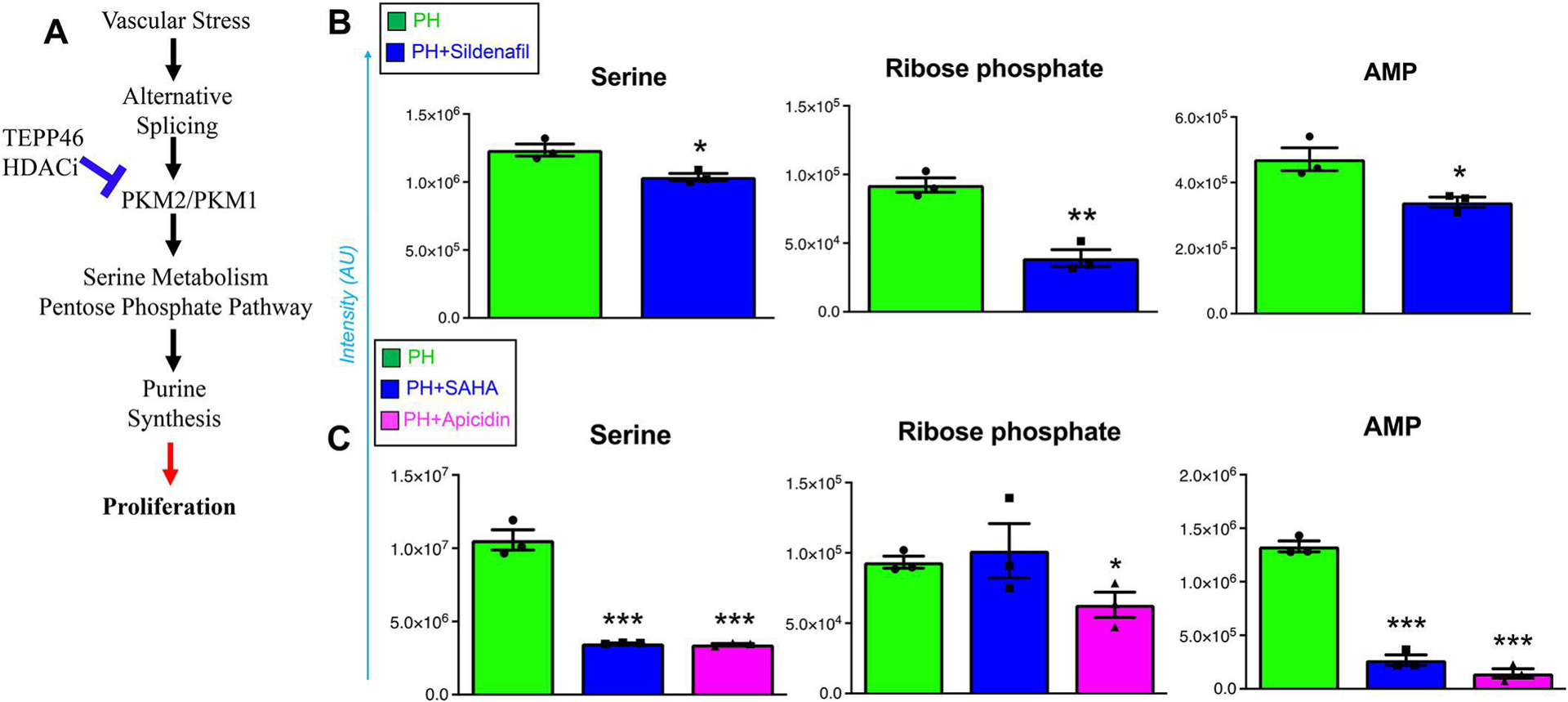
The impaired PKM activity resulting from increased PKM2/PKM1 ratio can be rescued by TEPP46 and HDACi treatment (A). TEPP-46 (B) and HDACi (C) decreased the levels of serine, ribose phosphate and AMP in PH-Fibs. *, **, *** p <0.05, p <0.01, 0.001, compared to untreated PH-Fibs, n = 3).
HDACi and sildenafil synergize to correct metabolic reprogramming and inhibit PH-Fibs proliferation.
To further explore the potential treatment strategy of using an HDACi in combination with sildenafil to target both vasoconstriction and vascular remodeling, we treated human PH-Fibs with Sildenafil alone (50 nM), HDACi alone (Apicidin, 3 uM) or in combination. Metabolomics analyses revealed that the combination of Apicidin and sildenafil was more effective than either treatment alone in correcting the metabolic derangements of PH-Fibs (Figure 7. A), especially with respect to hypoxic markers of carboxylates (two critical metabolites that contribute to the stabilization and regulate the degradation of Hypoxia-inducible factor 1alpha – alpha-ketoglutarate and 2-hydroxyglutarate [42, 43], the substrates of purine de novo synthesis (serine (p=0.069) and ribose) and purine nucleotides (the building blocks of RNA and DNA, ATP and ADP) (Figure 7. B). More importantly, we found that the combination of Apicidin and sildenafil suppressed the proliferation of human PH-Fibs more effectively than either treatment alone (Figure 7. C). It should be noted that Apicidin had a stronger effect on the inhibition of proliferation than sildenafil.
Figure 7. Combined treatment with sildenafil and Apicidin is more effective than either treatment alone in correcting metabolism and arresting PH-Fibs proliferation.
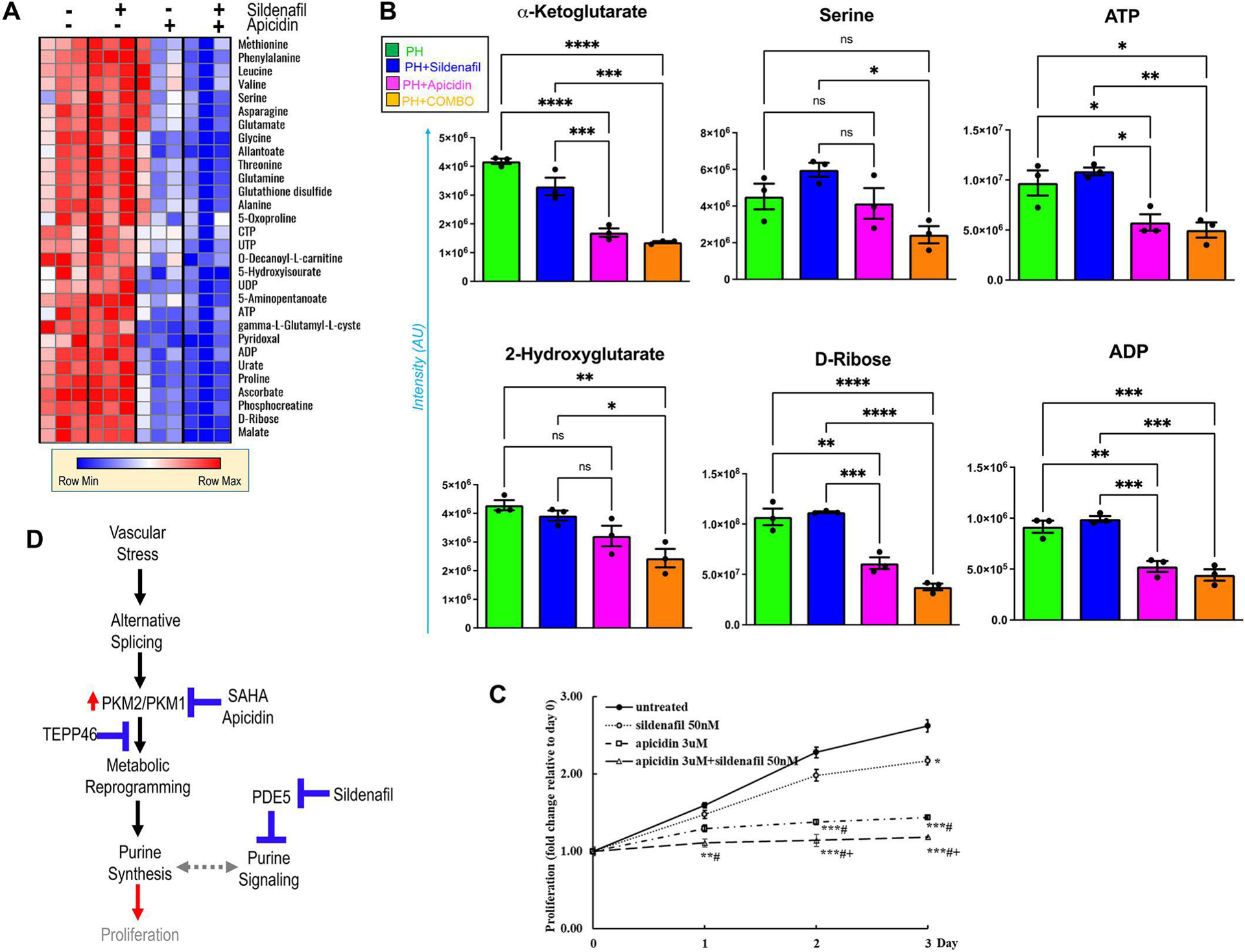
This effect was revealed by metabolic analysis in heatmap (A) and representative plots for metabolites associated with hypoxic metabolic reprogramming in the Krebs cycle (alpha-ketoglutarate and 2-hydroxyglutarate), the substrates of purine de novo synthesis (serine and ribose) and purine nucleotides (ATP and ADP) in (B), *p<0.05, **p< 0.01, ***p< 0.001, ****p< 0.0001 as indicated. (C), Proliferation assay was performed with CyQuant Kit in human PH-Fibs with different treatment (sildenafil 50nM or Apicidin 3uM, alone or the in combination) or left untreated up to 3 days (n=3). Results were calculated as fold changes relative to day 0, *p<0.05, **p< 0.01, ***p< 0.001 compared to untreated; #p<0.05, compared to sildenafil; +p< 0.05, compared to Apicidin. (D), overview of the proposed strategies to target dysregulated purine metabolism and signaling in PH.
DISCUSSION
Advancements in our understanding of the etiological factors contributing to PH have fostered the introduction of drugs that specifically target PA vasoconstriction and potentially pathological vascular remodeling. Among these treatments, sildenafil showed significant promise as a strategy to inhibit PDE5 and thus prevent the degradation of cGMP, which in turn prolongs the smooth muscle relaxation mediated by NO production [34] and improves symptoms and hemodynamics in patients with symptomatic PAH [12]. However, conflicting data from currently available clinical trials showed general safety but overall inconsistent efficacy across patients, with some patients responding better than others [9]. These inconsistencies may be in part explained by considerations of sildenafil metabolism, which occurs primarily in the liver, where cytochrome P450 enzymes yield one active metabolite with a potency of approximately 50% of the parent drug. Sildenafil metabolism exerts its efficacy in some categories of patients, such as in patients older than 65, with creatinine clearance less than 30, and with hepatic cirrhosis, which are all characterized by reduced clearance of sildenafil [12, 44]. Here, we performed metabolomics analyses on a clinical cohort of PH patients undergoing treatment with sildenafil. Results indicate that sildenafil alone only partially reversed the plasma metabolic derangements, which are commonly observed in PH patients [45, 46]. Clinical (6MWD) and metabolic responses to sildenafil were found to be impacted by patients’ age and BMI, suggesting that these patient characteristics may be leveraged in the future for targeted therapy in combination with sildenafil. On the other hand, circulating levels of sildenafil or desmethylsildenafil/sildenafil ratio – a marker of sildenafil metabolism by cytochrome P450 – were not predictive of in vivo responses in our clinical cohort.
Metabolic reprogramming of vascular cells is a critical hallmark of PH and is recognized as a potential target for PH treatment [19]. Hyperproliferative phenotypes of endothelial cells, smooth muscle cells, and fibroblasts have been associated with a Warburg-like metabolic reprogramming towards aerobic glycolysis in animal models or patients with PH [20, 36]. Pulmonary artery fibroblasts play a critical role in PA pathological remodeling, however little is known about the effects of sildenafil on these cells. In the present study, we used human PA fibroblasts as an in vitro model to explore the effect of sildenafil on the metabolic reprogramming and the effect of combined treatment of sildenafil with HDACi. Using UHPLC-MS metabolomics and 13C tracing experiments, we documented that incomplete glucose oxidation in PH-Fibs feeds into purine nucleoside synthesis via serine synthesis and activation of the pentose phosphate pathway (PPP), which is similar to cancer cells [39]. Supportively, it was reported that the genetic abnormalities of the rate-limiting enzyme of the PPP, glucose 6-phosphate dehydrogenase, are protective against PH in sickle cell disease patients [47]. On the other hand, sildenafil is insufficient to reverse the incomplete glucose oxidation and the subsequently elevated purine synthesis which may partially be responsible for the inconsistencies of sildenafil’s effect on PH patients.
The molecular structure of sildenafil is similar to that of cGMP, suggesting that as a purine compound, it may, through yet unexplored mechanisms, directly interact and/or be a cofactor for intracellular purine-converting enzymes involved in intracellular purine metabolism. In addition, intracellular cGMP is a second messenger, involved in the inhibition of proliferation in several cell types including SMC, endothelial, and tumor cells, suggesting its potential anti-proliferative effects in vascular cells [48, 49]. Of note, we observed an increased guanine level in PH-Fibs after sildenafil treatment. The physiological role of increased intracellular guanine in vascular cells, except its function as a canonical nucleotide base in DNA and RNA, remains largely unknown. However, its ribosylated product, guanosine, has been shown to exhibit a protective role against neuroinflammation and oxidative stress and to exert trophic effects in neuronal and glial cells [50]. Moreover, guanine nucleotides, as GDP and GTP contribute to protein biosynthesis and signal transduction, also playing a role in cancer cell energy metabolism [51–53]. It cannot be excluded that in adventitial fibroblasts, de novo-produced GTP may serve as a substrate for guanylate cyclase-mediated cGMP production and thereby contribute to the suppression of cell proliferation.
Serine biosynthesis in cancer has been linked to increased purine synthesis, a phenomenon promoted by decreased PKM activity owing to increases in the ratios of PKM isoforms 2 to 1, the former being constitutively less active than the latter [38]. PKM activity is also allosterically regulated by early glycolytic intermediates (e.g., fructose 1,6-bisphosphate) and serine levels [40]. We previously proved that decreases in PKM1 expression in PH-Fibs underlie the increases in PKM2/PKM1 ratios and thus decreased PKM activity and contribute to the Warburg-like glycolytic phenotype of PH-Fibs. The pharmacological intervention with TEPP-46 treatment resulting in PKM tetramerization and increasing its activity and HDACi treatment resulting in decreasing the PKM2/PKM1 ratio (through miR-124/PTBP1/PKM axis by regulating PKM isoform alternative splicing), can reverse glycolytic reprogramming and highly proliferative phenotypes in these cells [23]. Here we show that serine, PPP, and purine levels are normalized in PH-Fibs treated with TEPP-46 and HDACi which both promote PKM activity. Furthermore, we found that combined treatment with HDACi (Apicidin) and sildenafil showed the synergistic effects on normalization of metabolism and inhibition of proliferation of PH-Fibs, paving the way for future studies on animal models and clinical trials exploiting combination therapeutic approaches.
In conclusion, HDAC inhibitors, alone or in combination with sildenafil, may represent a viable therapeutic strategy to target deranged (purine) metabolism in PH by influencing PKM activity (Figure 7. D). The combined inhibitory effect of HDACi and sildenafil on pulmonary artery fibroblast proliferation reflect the existence of complex regulatory mechanisms that include coordinated action of epigenetic, signaling, and metabolic inputs. Further studies are necessary to evaluate the exact molecular mechanisms underlying the beneficial effect of sildenafil in combination with HDACi for the treatment of PAH.
Supplementary Material
Supplemental Figure 1. Metabolomics analysis of 27 PH patients before and after sildenafil treatment
Supplemental Table 1. Detailed information of PH patients
ACKNOWLEDGEMENTS
This work has been supported by NIH Program Project Grant HL152961, and Department of Defense Grant W81XWH1910259, W81XWH2010249 to KRS, ML, HZ. AD was supported by funds from the National Institute of General and Medical Sciences RM1GM131968 and R01HL146442, R01HL149714, R01HL148151, R01HL161004 from the National Heart, Lung, and Blood Institute.
The authors would like to thank Marcia McGowan and Andy Poczobutt for helping with manuscript preparation, Nick Morrell (University of Cambridge, UK) for providing human pulmonary artery fibroblasts.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: All the authors disclose no conflicts of interest relevant to this study.
REFERENCES
- [1].Humbert M, Sitbon O, Yaici A, Montani D, O’Callaghan DS, Jais X, Parent F, Savale L, Natali D, Gunther S, Chaouat A, Chabot F, Cordier JF, Habib G, Gressin V, Jing ZC, Souza R, Simonneau G, French N Pulmonary Arterial Hypertension, Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension, Eur Respir J 36(3) (2010) 549–55. [DOI] [PubMed] [Google Scholar]
- [2].Wijeratne DT, Lajkosz K, Brogly SB, Lougheed MD, Jiang L, Housin A, Barber D, Johnson A, Doliszny KM, Archer SL, Increasing Incidence and Prevalence of World Health Organization Groups 1 to 4 Pulmonary Hypertension: A Population-Based Cohort Study in Ontario, Canada, Circ Cardiovasc Qual Outcomes 11(2) (2018) e003973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Burnstock G, Purinergic signalling and the autonomic nervous system in health and disease, Auton Neurosci 191 (2015) 1. [DOI] [PubMed] [Google Scholar]
- [4].Burnstock G, Purinergic Signaling in the Cardiovascular System, Circ Res 120(1) (2017) 207–228. [DOI] [PubMed] [Google Scholar]
- [5].Headrick JP, Ashton KJ, Rose’meyer RB, Peart JN, Cardiovascular adenosine receptors: expression, actions and interactions, Pharmacol Ther 140(1) (2013) 92–111. [DOI] [PubMed] [Google Scholar]
- [6].Cai Z, Tu L, Guignabert C, Merkus D, Zhou Z, Purinergic Dysfunction in Pulmonary Arterial Hypertension, J Am Heart Assoc 9(18) (2020) e017404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ma Q, Yang Q, Xu J, Zhang X, Kim D, Liu Z, Da Q, Mao X, Zhou Y, Cai Y, Pareek V, Kim HW, Wu G, Dong Z, Song WL, Gan L, Zhang C, Hong M, Benkovic SJ, Weintraub NL, Fulton D Jr., Asara JM, Ben-Sahra I, Huo Y, ATIC-Associated De Novo Purine Synthesis Is Critically Involved in Proliferative Arterial Disease, Circulation 146(19) (2022) 1444–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Andersson KE, PDE5 inhibitors - pharmacology and clinical applications 20 years after sildenafil discovery, Br J Pharmacol 175(13) (2018) 2554–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Barnett CF, Machado RF, Sildenafil in the treatment of pulmonary hypertension, Vasc Health Risk Manag 2(4) (2006) 411–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tsai H, Sung YK, de Jesus Perez V, Recent advances in the management of pulmonary arterial hypertension, F1000Res 5 (2016) 2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wang H, Ye M, Robinson H, Francis SH, Ke H, Conformational variations of both phosphodiesterase-5 and inhibitors provide the structural basis for the physiological effects of vardenafil and sildenafil, Mol Pharmacol 73(1) (2008) 104–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Galie N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, Fleming T, Parpia T, Burgess G, Branzi A, Grimminger F, Kurzyna M, Simonneau G, Sildenafil G Use in Pulmonary Arterial Hypertension Study, Sildenafil citrate therapy for pulmonary arterial hypertension, N Engl J Med 353(20) (2005) 2148–57. [DOI] [PubMed] [Google Scholar]
- [13].Unegbu C, Noje C, Coulson JD, Segal JB, Romer L, Pulmonary Hypertension Therapy and a Systematic Review of Efficacy and Safety of PDE-5 Inhibitors, Pediatrics 139(3) (2017). [DOI] [PubMed] [Google Scholar]
- [14].McGoon MD, Vlietstra RE, Vasodilator therapy for primary pulmonary hypertension, Mayo Clin Proc 59(10) (1984) 672–7. [DOI] [PubMed] [Google Scholar]
- [15].Sitbon O, Humbert M, Jais X, Ioos V, Hamid AM, Provencher S, Garcia G, Parent F, Herve P, Simonneau G, Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension, Circulation 111(23) (2005) 3105–11. [DOI] [PubMed] [Google Scholar]
- [16].Humbert M, Guignabert C, Bonnet S, Dorfmuller P, Klinger JR, Nicolls MR, Olschewski AJ, Pullamsetti SS, Schermuly RT, Stenmark KR, Rabinovitch M, Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives, Eur Respir J 53(1) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Prins KW, Thenappan T, Weir EK, Kalra R, Pritzker M, Archer SL, Repurposing Medications for Treatment of Pulmonary Arterial Hypertension: What’s Old Is New Again, J Am Heart Assoc 8(1) (2019) e011343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tuder RM, Pulmonary vascular remodeling in pulmonary hypertension, Cell Tissue Res 367(3) (2017) 643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].D’Alessandro A, El Kasmi KC, Plecita-Hlavata L, Jezek P, Li M, Zhang H, Gupte SA, Stenmark KR, Hallmarks of Pulmonary Hypertension: Mesenchymal and Inflammatory Cell Metabolic Reprogramming, Antioxid Redox Signal 28(3) (2018) 230–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li M, Riddle S, Zhang H, D’Alessandro A, Flockton A, Serkova NJ, Hansen KC, Moldovan R, McKeon BA, Frid M, Kumar S, Li H, Liu H, Caanovas A, Medrano JF, Thomas MG, Iloska D, Plecita-Hlavata L, Jezek P, Pullamsetti S, Fini MA, El Kasmi KC, Zhang Q, Stenmark KR, Metabolic Reprogramming Regulates the Proliferative and Inflammatory Phenotype of Adventitial Fibroblasts in Pulmonary Hypertension Through the Transcriptional Corepressor C-Terminal Binding Protein-1, Circulation 134(15) (2016) 1105–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Plecita-Hlavata L, Tauber J, Li M, Zhang H, Flockton AR, Pullamsetti SS, Chelladurai P, D’Alessandro A, El Kasmi KC, Jezek P, Stenmark KR, Constitutive Reprogramming of Fibroblast Mitochondrial Metabolism in Pulmonary Hypertension, Am J Respir Cell Mol Biol 55(1) (2016) 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang H, Laux A, Stenmark KR, Hu CJ, Mechanisms Contributing to the Dysregulation of miRNA-124 in Pulmonary Hypertension, Int J Mol Sci 22(8) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang H, Wang D, Li M, Plecita-Hlavata L, D’Alessandro A, Tauber J, Riddle S, Kumar S, Flockton A, McKeon BA, Frid MG, Reisz JA, Caruso P, El Kasmi KC, Jezek P, Morrell NW, Hu CJ, Stenmark KR, Metabolic and Proliferative State of Vascular Adventitial Fibroblasts in Pulmonary Hypertension Is Regulated Through a MicroRNA-124/PTBP1 (Polypyrimidine Tract Binding Protein 1)/Pyruvate Kinase Muscle Axis, Circulation 136(25) (2017) 2468–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Joshi SR, Kitagawa A, Jacob C, Hashimoto R, Dhagia V, Ramesh A, Zheng C, Zhang H, Jordan A, Waddell I, Leopold J, Hu CJ, McMurtry IF, D’Alessandro A, Stenmark KR, Gupte SA, Hypoxic activation of glucose-6-phosphate dehydrogenase controls the expression of genes involved in the pathogenesis of pulmonary hypertension through the regulation of DNA methylation, Am J Physiol Lung Cell Mol Physiol 318(4) (2020) L773–L786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pedley AM, Benkovic SJ, A New View into the Regulation of Purine Metabolism: The Purinosome, Trends Biochem Sci 42(2) (2017) 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Huang Z, Xie N, Illes P, Di Virgilio F, Ulrich H, Semyanov A, Verkhratsky A, Sperlagh B, Yu SG, Huang C, Tang Y, From purines to purinergic signalling: molecular functions and human diseases, Signal Transduct Target Ther 6(1) (2021) 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Parker WB, Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer, Chem Rev 109(7) (2009) 2880–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Robak P, Robak T, Older and new purine nucleoside analogs for patients with acute leukemias, Cancer Treat Rev 39(8) (2013) 851–61. [DOI] [PubMed] [Google Scholar]
- [29].Yin J, Ren W, Huang X, Deng J, Li T, Yin Y, Potential Mechanisms Connecting Purine Metabolism and Cancer Therapy, Front Immunol 9 (2018) 1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Pettitt AR, Mechanism of action of purine analogues in chronic lymphocytic leukaemia, Br J Haematol 121(5) (2003) 692–702. [DOI] [PubMed] [Google Scholar]
- [31].Wang D, Zhang H, Li M, Frid MG, Flockton AR, McKeon BA, Yeager ME, Fini MA, Morrell NW, Pullamsetti SS, Velegala S, Seeger W, McKinsey TA, Sucharov CC, Stenmark KR, MicroRNA-124 controls the proliferative, migratory, and inflammatory phenotype of pulmonary vascular fibroblasts, Circ Res 114(1) (2014) 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Nemkov T, Hansen KC, D’Alessandro A, A three-minute method for high-throughput quantitative metabolomics and quantitative tracing experiments of central carbon and nitrogen pathways, Rapid Commun Mass Spectrom 31(8) (2017) 663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Xia J, Sinelnikov IV, Han B, Wishart DS, MetaboAnalyst 3.0--making metabolomics more meaningful, Nucleic Acids Res 43(W1) (2015) W251–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Corbin JD, Mechanisms of action of PDE5 inhibition in erectile dysfunction, Int J Impot Res 16 Suppl 1 (2004) S4–7. [DOI] [PubMed] [Google Scholar]
- [35].Kumar S, Frid MG, Zhang H, Li M, Riddle S, Brown RD, Yadav SC, Roy MK, Dzieciatkowska ME, D’Alessandro A, Hansen KC, Stenmark KR, Complement-containing small extracellular vesicles from adventitial fibroblasts induce pro-inflammatory and metabolic reprogramming in macrophages, JCI Insight (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Li M, Riddle S, Kumar S, Poczobutt J, McKeon BA, Frid MG, Ostaff M, Reisz JA, Nemkov T, Fini MA, Laux A, Hu CJ, El Kasmi KC, D’Alessandro A, Brown RD, Zhang H, Stenmark KR, Microenvironmental Regulation of Macrophage Transcriptomic and Metabolomic Profiles in Pulmonary Hypertension, Front Immunol 12 (2021) 640718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Plecitá-Hlavatá L, D’Alessandro A, El Kasmi K, Li M, Zhang H, Ježek P, Stenmark KR, Metabolic Reprogramming and Redox Signaling in Pulmonary Hypertension, Adv Exp Med Biol 967 (2017) 241–260. [DOI] [PubMed] [Google Scholar]
- [38].Dong G, Mao Q, Xia W, Xu Y, Wang J, Xu L, Jiang F, PKM2 and cancer: The function of PKM2 beyond glycolysis, Oncol Lett 11(3) (2016) 1980–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Maddocks OD, Labuschagne CF, Adams PD, Vousden KH, Serine Metabolism Supports the Methionine Cycle and DNA/RNA Methylation through De Novo ATP Synthesis in Cancer Cells, Mol Cell 61(2) (2016) 210–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chaneton B, Hillmann P, Zheng L, Martin ACL, Maddocks ODK, Chokkathukalam A, Coyle JE, Jankevics A, Holding FP, Vousden KH, Frezza C, O’Reilly M, Gottlieb E, Serine is a natural ligand and allosteric activator of pyruvate kinase M2, Nature 491(7424) (2012) 458–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Caruso P, Dunmore BJ, Schlosser K, Schoors S, Dos Santos C, Perez-Iratxeta C, Lavoie JR, Zhang H, Long L, Flockton AR, Frid MG, Upton PD, D’Alessandro A, Hadinnapola C, Kiskin FN, Taha M, Hurst LA, Ormiston ML, Hata A, Stenmark KR, Carmeliet P, Stewart DJ, Morrell NW, Identification of MicroRNA-124 as a Major Regulator of Enhanced Endothelial Cell Glycolysis in Pulmonary Arterial Hypertension via PTBP1 (Polypyrimidine Tract Binding Protein) and Pyruvate Kinase M2, Circulation 136(25) (2017) 2451–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E, Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase, Cancer Cell 7(1) (2005) 77–85. [DOI] [PubMed] [Google Scholar]
- [43].Ryan DG, Murphy MP, Frezza C, Prag HA, Chouchani ET, O’Neill LA, Mills EL, Coupling Krebs cycle metabolites to signalling in immunity and cancer, Nat Metab 1 (2019) 16–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Morales A, Gingell C, Collins M, Wicker PA, Osterloh IH, Clinical safety of oral sildenafil citrate (VIAGRA) in the treatment of erectile dysfunction, Int J Impot Res 10(2) (1998) 69–73; discussion 73–4. [DOI] [PubMed] [Google Scholar]
- [45].Bujak R, Mateo J, Blanco I, Izquierdo-García JL, Dudzik D, Markuszewski MJ, Peinado VI, Laclaustra M, Barberá JA, Barbas C, Ruiz-Cabello J, New Biochemical Insights into the Mechanisms of Pulmonary Arterial Hypertension in Humans, PLoS One 11(8) (2016) e0160505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rhodes CJ, Ghataorhe P, Wharton J, Rue-Albrecht KC, Hadinnapola C, Watson G, Bleda M, Haimel M, Coghlan G, Corris PA, Howard LS, Kiely DG, Peacock AJ, Pepke-Zaba J, Toshner MR, Wort SJ, Gibbs JS, Lawrie A, Gräf S, Morrell NW, Wilkins MR, Plasma Metabolomics Implicates Modified Transfer RNAs and Altered Bioenergetics in the Outcomes of Pulmonary Arterial Hypertension, Circulation 135(5) (2017) 460–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kitagawa A, Kizub I, Jacob C, Michael K, D’Alessandro A, Reisz JA, Grzybowski M, Geurts AM, Rocic P, Gupte R, Miano JM, Gupte SA, CRISPR-Mediated Single Nucleotide Polymorphism Modeling in Rats Reveals Insight Into Reduced Cardiovascular Risk Associated With Mediterranean G6PD Variant, Hypertension 76(2) (2020) 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Heller R, Polack T, Gräbner R, Till U, Nitric oxide inhibits proliferation of human endothelial cells via a mechanism independent of cGMP, Atherosclerosis 144(1) (1999) 49–57. [DOI] [PubMed] [Google Scholar]
- [49].Yu SM, Hung LM, Lin CC, cGMP-elevating agents suppress proliferation of vascular smooth muscle cells by inhibiting the activation of epidermal growth factor signaling pathway, Circulation 95(5) (1997) 1269–77. [DOI] [PubMed] [Google Scholar]
- [50].Bettio LE, Gil-Mohapel J, Rodrigues AL, Guanosine and its role in neuropathologies, Purinergic Signal 12(3) (2016) 411–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hamel E, Cashel M, Role of guanine nucleotides in protein synthesis. Elongation factor G and guanosine 5’-triphosphate,3’-diphosphate, Proc Natl Acad Sci U S A 70(11) (1973) 3250–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Rodbell M, The role of GTP-binding proteins in signal transduction: from the sublimely simple to the conceptually complex, Curr Top Cell Regul 32 (1992) 1–47. [DOI] [PubMed] [Google Scholar]
- [53].Sasaki AT, Dynamic Role of the GTP Energy Metabolism in Cancers, Keio J Med 65(1) (2016) 21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Metabolomics analysis of 27 PH patients before and after sildenafil treatment
Supplemental Table 1. Detailed information of PH patients