

Abstract
Intratumoral hypoxia is associated with tumor progression and therapeutic resistance. The VHL tumor suppressor gene was identified in 1993, and later studies revealed that the gene product pVHL interacts with other proteins to form the VBC complex. The VBC complex functions as an E3 ubiquitin ligase and regulates the abundance of the α‐subunit of the transcription factor hypoxia‐inducible factor (HIF). Hypoxia‐inducible factor regulates thousands of genes required for cells to adapt and survive in hypoxic conditions, and thus pVHL plays a major role in oxygen‐sensing pathways. Patients with von Hippel–Lindau (VHL) disease, harboring a germline mutation of the VHL gene, develop renal cell carcinomas and a series of tumors showing hypervascular phenotypes. The extensive findings that have clarified the function of VHL have contributed to the development of novel first‐in‐human drugs, including belzutifan, a HIF‐2α inhibitor. The 2019 Nobel Prize in Physiology or Medicine was awarded to Dr. William G. Kaelin Jr., Dr. Peter J. Ratcliffe, and Dr. Gregg L. Semenza as researchers contributing to clarifying the mechanism of the oxygen‐sensing pathway of cells. The first report of VHL disease was in 1894, meaning the development of a specific drug for this disease took almost 125 years. In this article, we describe how researchers and clinician scientists successfully clarified the function of VHL and achieved a preclinical proof of concept to apply for clinical trials, key requirements for drug development.
Keywords: drug development, HIF, PHD, VHL disease
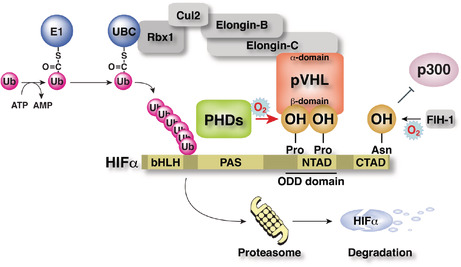
pVHL forms an E3 ubiquitin ligase complex with Elongin B/C, Cul‐2, and Rbx‐1. The beta‐domain of pVHL recognizes one or two hydroxylated proline residue(s) in the NTAD of HIF alpha, binds to HIF alpha, and targets HIF alpha for ubiquitin‐dependent protein degradation at the proteasome.
Abbreviations
- ccRCC
clear cell renal cell carcinoma
- CNS
central nervous system
- CR
complete response
- CTAD
C‐terminal transcriptional activation domain
- EF
endothelial fenestration
- EPO
erythropoietin
- FIH‐1
factor inhibiting HIF‐1
- HIF
hypoxia‐inducible factor
- mRCC
metastatic renal cell carcinoma
- PHD
prolyl hydroxylase
- pNET
pancreatic neuroendocrine tumor
- PR
partial response
- pVHL
von Hippel–Lindau gene product
- RCC
renal cell carcinoma
- SD
stable disease
- TKI
tyrosine kinase inhibitor
- VEGF
vascular endothelial growth factor
- VHL
von Hippel–Lindau
1. INTRODUCTION
Intratumoral hypoxia from severe O2 deprivation is often associated with resistance to radiation and chemotherapy as well as the poor prognosis of the patients. 1 , 2 Understanding of the molecular mechanisms of adaptation to hypoxia has progressed dramatically after the identification of the VHL tumor suppressor gene, the causative gene of VHL disease, in 1993. 3 von Hippel–Lindau disease is inherited in an autosomal dominant manner, and patients with germline mutations in VHL develop ccRCC and other types of hypervascular tumors. 4 , 5 , 6 , 7 The VHL gene product (pVHL) binds to Elongin‐C, Elongin‐B, CUL2, and RBX1 to form the VBC complex. 8 , 9 , 10 , 11 , 12 , 13 This complex functions as an E3 ubiquitin ligase and regulates the abundance of the α‐subunit of the transcription factor HIF through mediating proteasomal degradation. 14 , 15 , 16 Hypoxia‐inducible factor transcriptionally regulates most of the known genes required for cell adaptation and survival in hypoxia, and therefore VHL plays major roles in the physiological oxygen‐sensing pathway in cells. 10 , 13 The discovery of oxygen‐sensing mechanisms resulted in the development of novel first‐in‐human drugs, HIF‐PH inhibitors and HIF‐2α inhibitors. In 2019, the Nobel Prize in Physiology or Medicine was awarded to Dr. William G. Kaelin Jr., Dr. Peter J. Ratcliffe, and Dr. Gregg L. Semenza for discoveries on how cells sense and adapt to oxygen availability. In 2021, the FDA approved the HIF‐2α inhibitor belzutifan for the treatment of patients with VHL disease who require therapy for VHL‐associated ccRCC, CNS hemangioblastomas, or pNET. 17 Since the first report of VHL disease in 1894, the development of a specific drug for this disease took almost 125 years, and the extensive research clarifying the function of VHL to successfully achieve the preclinical proof of concept was pivotal for the development of belzutifan. Therefore, belzutifan could serve as a representative example of the success of the entire drug discovery process over 100 years, starting from the identification of the specific clinical characteristics of a rare disease and successfully resulting in the development of a drug for this disease.
In this review article, we discuss how researchers, including our group, successfully clarified the molecular pathways of oxygen‐sensing mechanisms and how these insights provided a basis for subsequent drug development.
2. GENERAL CHARACTERISTICS OF VHL DISEASE AND IDENTIFICATION OF THE VHL GENE
von Hippel–Lindau disease is an autosomal dominant inherited disease caused by germline mutation of the VHL gene. Patients with this disease develop multiple neoplastic lesions such as retinal hemangioma, hemangioblastoma of the CNS, pNET, adrenal pheochromocytoma/paraganglioma, ccRCC, and endolymphatic tumor in the inner ear. It is also known to cause cystadenoma of the epididymis in men and broad uterus in women (Figure 1). 4 , 5 , 6
FIGURE 1.
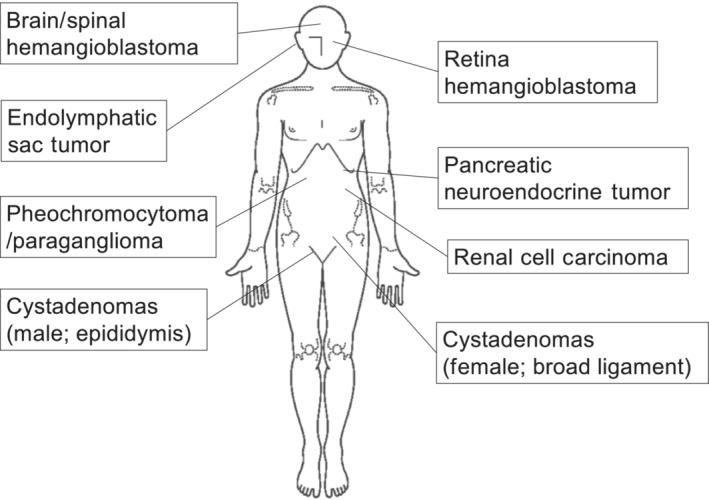
Characteristic tumors that develop in von Hippel–Lindau syndrome.
The first case of VHL disease was reported as a case of familial hemangiomas of the retina by Dr. Collins in 1894. 18 In 1904, Dr. Eugen von Hippel, a German ophthalmologist, described the familial occurrence of retinal hemangioma. 19 In 1927, Dr. Arvid Lindau, a Swedish neuropathologist, reported the pathological findings of a familial case of multiple hemangiomas not only in the retina but also in the CNS. 20
Subsequent research intensively pursued the causative gene of this disease. In 1988, Dr. Seizinger et al. 21 used pedigree linkage analysis and demonstrated that the relevant gene was located on chromosome 3p25. Finally, the VHL gene was identified by positional cloning by a group in the United States at the NCI in collaboration with other groups in England and France in 1993. 3 Then, it was revealed that the VHL gene consists of three exon regions and the protein translation region of the mRNA is 639 bases long. 3 , 10 , 12 Importantly, somatic mutation of VHL was identified in 57% (56 out of 98) of tumor specimens from patients with sporadic ccRCC, indicating that the VHL gene was also a tumor suppressor gene of ccRCC. 22
3. CLINICAL SIGNIFICANCE OF VEGF IN THE TUMOR PROGRESSION OF CCRCC AND ROLES OF PVHL IN THE REGULATION OF VEGF
In the exploration of the tumor suppressive function of pVHL, Dr. Iliopoulos et al. 23 reintroduced WT VHL into 786‐O VHL −/− RCC cells to establish WT8 cells (Table 1) and found that these cells showed reduced tumor formation ability in a mouse xenograft model compared with that of mock‐transfected pRC3 cells. These results clearly indicated that pVHL harbored tumor suppressor ability in vivo. Importantly, ccRCC tumors harbor abundant tumor blood vessels. 24 In 1994, a group from the National Cancer Center Japan reported that 26 out of 27 RCC tissues showed a markedly elevated level (3–13 fold) of VEGFA mRNA compared with adjacent normal kidney tissues. 25 Dr. Iliopoulos et al. compared the abundance of VEGFA mRNA between WT8 cells and pRC3 cells, and showed that pVHL inhibited the production of VEGFA mRNAs under normoxic conditions as the first significant therapeutic relevance of VHL in ccRCC. VEGFA is one of the genes upregulated in hypoxic conditions, and thus as early as 1996, Dr. Kaelin predicted that pVHL might play a critical role in the transduction of signals generated by changes in ambient oxygen tension. 26 In fact, this finding was quite important in the development of new treatment strategies targeting tumor vessels for mRCC.
TABLE 1.
Characteristics of clear cell renal cell carcinoma cell lines.
| Cell line | VHL status | Transactivity of HIF‐1 | Transactivity of HIF‐2 |
|---|---|---|---|
| 786‐O | Null | (−) | (+) |
| A498 | Null | (−) | (+) |
| RCC4 | Null | (+) | (+) |
| UMRC2 | Null | (+) | (+) |
| WT8 | WT | (−) | (−) |
| WT8 + HIF2αP531A | WT | (−) | (+) |
| pRC3 | Null | (−) | (+) |
Abbreviation: HIF, hypoxia inducible factor.
A randomized, placebo‐controlled phase II trial of bevacizumab, a humanized anti‐VEGF mAb, was carried out in patients with mRCC. There was a significant prolongation of the time to progression of disease in the high‐dose Ab group (10 mg/kg, given every 2 weeks) compared with the placebo group (hazard ratio, 2.55; p < 0.001). 27 In addition, multiple TKIs targeting VEGF were developed and showed significant activity in mRCC as single agents in randomized controlled trials. 28 , 29 , 30
4. POSSIBLE ROLES OF ANTI‐VEGF THERAPY IN THE MICROVASCULATURE OF CCRCC
Although previous studies on anti‐VEGF therapy in mRCC have shown promise, the actual effect of anti‐VEGF therapy on the tumor microvasculature in ccRCC patients has been largely obscure. Interestingly, previous studies validating the effect of VEGF inhibitors using mice pancreatic islet tumor models revealed that VEGF‐dependent capillaries were characterized by the presence of EFs. 31 On the basis of these findings, we characterized the tumor microvasculature of sporadic ccRCC tumor specimens using electron microscopy. 32 The microvasculature in tumors harboring mutant VHL exhibited endothelium with abundant fenestrations compared with those with WT VHL (Figure 2A). The average number of fenestrations from tumor specimens with WT and mutant VHL was 2.5 ± 0.4 and 15.1 ± 1.7/μm, 2 respectively (p < 0.001) (Figure 2B). To our knowledge, this was the first report that described the existence of EFs in the tumor microvasculature of ccRCC tumors, especially those harboring VHL mutation. Intriguingly, abundant EFs could be reproduced in a mouse subcutaneous xenograft model, and the microvasculature in tumors derived from VHL −/− pRC3 cells showed more abundant EFs than that from VHL‐reintroduced WT8 cells (Figure 2C). We next examined the effect of bevacizumab in the tumor microvasculature of these cells (Figure 2D).
FIGURE 2.
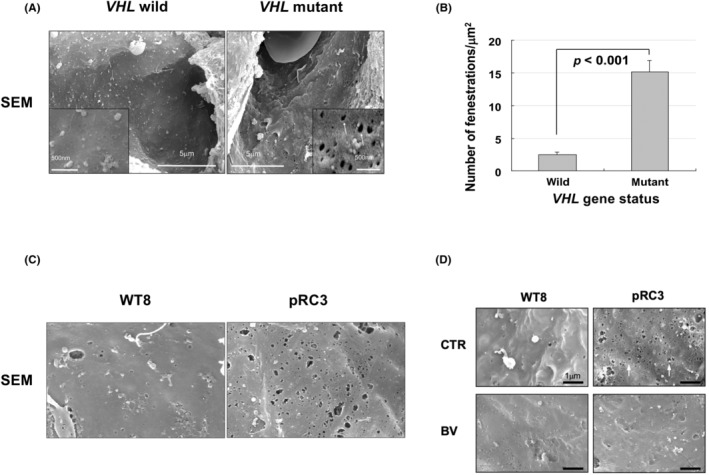
Endothelial fenestrations on microvasculature from VHL mutant clear cell renal cell carcinoma (ccRCC) might be vascular endothelial growth factor‐dependent and sensitive to bevacizumab. (A) Electron micrographs of tumor capillaries in VHL WT ccRCC and VHL mutated ccRCC. Representative data are shown. Capillaries from tumors with mutant VHL exhibit endothelium with more abundant endothelial fenestration (EFs) compared with those with WT VHL. SEM, scanning electron microscope. (B) Quantification of EFs in tumor capillaries with or without VHL mutation. The numbers of EFs were calculated per square micrometer. (C) Electron micrographs of tumor capillaries in xenografts from WT8 and pRC3 cells. Representative data are shown. Capillaries from VHL −/− pRC3 xenografts exhibited abundant EFs. (D) Electron micrographs show a reduction of EFs in VHL −/− pRC3 xenografts by the bevacizumab (BEV) treatment. CTR, control.
Importantly, bevacizumab significantly attenuated the number of EFs in VHL −/− pRC3 cells (Figure 3A). Moreover, the humanized anti‐VEGF mAb significantly attenuated both microvessel density and tumor growth in tumors derived from VHL −/− pRC3 cells (Figure 3B,C). 32 Collectively, these results indicated that microvasculature with abundant EFs on ccRCC might be VEGF‐dependent capillaries and potent targets of anti‐VEGF therapy.
FIGURE 3.
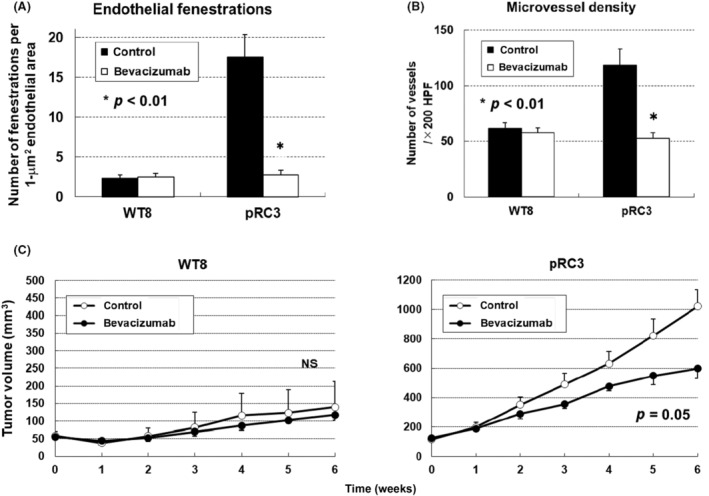
Microvasculature with abundant endothelial fenestration (EFs) on VHL null clear cell renal cell carcinoma (ccRCC) might be vascular endothelial growth factor (VEGF)‐dependent capillaries and sensitive to anti‐VEGF therapy. (A) The graph shows a significant reduction of EFs in VHL −/− pRC3 xenografts by the bevacizumab treatment. (B) Effects of bevacizumab treatment on microvessel density. Significant reduction was observed in VHL −/− pRC3 xenografts. HPF, high power filed. (C) Antitumor effects of bevacizumab in ccRCC tumors established in nude mice. Each time point represents the mean ± SE of the fold of tumor volume in each group. Statistically significant differences were observed in the tumor size of VHL −/− pRC3 xenografts between the bevacizumab treatment mice and controls. NS, not significant.
5. BIOLOGICAL ROLES OF PVHL IN SENSING AND ADAPTING TO OXYGEN AVAILABILITY
Multiple studies have explored the molecular mechanisms of pVHL in the regulation of VEGFA and the tumor suppression function of ccRCC. Gene product pVHL binds to Elongin‐C, Elongin‐B, CUL2, and RBX1 through its structural‐function α‐domain and the VBC complex functions as an E3 ubiquitin ligase complex (Figure 4A). 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15
FIGURE 4.
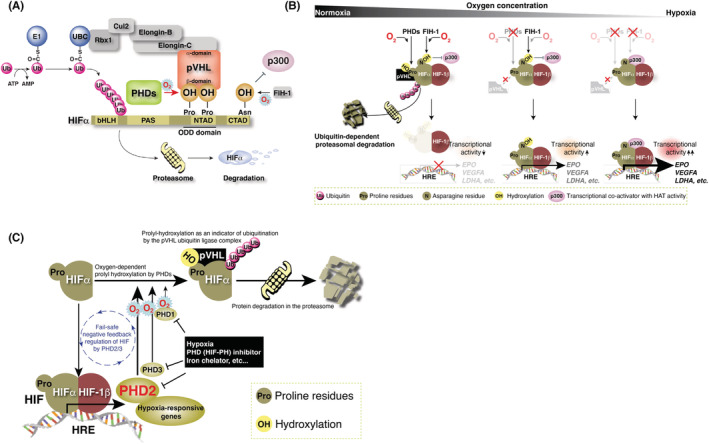
Regulation of hypoxia inducible factor (HIF) protein abundance by VHL gene product pVHL. (A) pVHL forms an E3 ubiquitin ligase complex with Elongin‐B/C, Cul2, and Rbx1. The β‐domain of pVHL recognizes one or two hydroxylated proline residue(s) in the N‐terminal transcriptional activation domain (NTAD) of HIFα, binds to HIFα, and targets HIFα for ubiquitin‐dependent protein degradation at the proteasome. (B) Molecular mechanism of oxygen sensing and adaptation. Prolyl hydroxylases (PHDs) negatively regulate the protein abundance of HIFα via the prolyl‐hydroxylation‐mediated ubiquitin‐proteasome pathway, and factor inhibiting HIF‐1 (FIH‐1) negatively regulates the transcriptional activity of HIF through asparaginyl hydroxylation. Both regulate the hypoxic response. (C) Fail‐safe negative feedback regulation of HIF by PHD2‐3. PHD1 hydroxylates HIFα independent of HIF activation. All three PHDs cooperatively hydroxylate HIFα to suppress constitutive activation of HIF.
Meanwhile, the groups of Dr. Semenza and Dr. Ratcliffe spent years searching for a transcription factor that induces erythropoietin under hypoxic conditions and the former group finally identified HIF‐1α as the master regulator of the hypoxic response. 33 , 34 , 35 Their discoveries in the early 1990s led to rapid progress in studying the hypoxic response. Then, in 1999, Dr. Ratcliffe and coworkers demonstrated that pVHL physically associated with HIFα and was necessary for its oxygen‐regulated instability. 16 , 36
Hypoxia‐inducible factor is a transcription factor formed by a heterodimer of α‐ and β‐subunits and regulates the transcription of multiple hypoxia‐responsive genes. 37 The β‐subunit of HIF (HIF‐1β/ARNT) is constitutively expressed, whereas the abundance of the α‐subunit (HIFα; HIF‐1α, ‐2α, and ‐3α) varies with oxygen concentration. Under normoxia, specific proline residues in the N‐terminal transcriptional activation domain of HIFα are hydroxylated by PHD1–3. 12 , 13 , 38 , 39 , 40 , 41 Hypoxia‐inducible factor‐α is ubiquitinated by the VBC complex, leading to proteasomal degradation, and the HIF‐mediated hypoxia response is repressed (Figure 4A). Under a hypoxic environment, the activity of PHDs decreases as the enzyme requires molecular oxygen for its enzymatic activity. Under hypoxia, HIFα escapes from ubiquitination‐dependent proteasomal degradation and binds to HIF‐1β. This heterodimer complex recognizes a specific sequence (hypoxia response element, 5′‐R(A/G)CGTG‐3′) on genomic DNA and induces the transcription of hypoxia‐related genes (Figure 4B). 2 , 42 , 43
Three PHD genes have been identified in mammals and are thought to have unique functions because their gene products are expressed in different organs and exhibit different subcellular localizations. 44 , 45 All three PHDs hydroxylate specific proline residues of HIFα in vitro. 46 In them, PHD2 is the major prolyl hydroxylase of HIFα in vivo and is an essential molecule for development. 47 , 48 Furthermore, our data revealed that PHD2 negatively regulates the HIF‐mediated hypoxia response by hydroxylating the proline residues of HIFα in cooperation with two other PHDs (Figure 4C). 49
Three HIFα isoforms have been identified that are transcribed from three distinct genes: HIF1A, EPAS1, and HIF3A. Dr. Semenza and colleagues identified FIH‐1 and revealed that specific asparagine residues in the CTADs of HIF‐1α and HIF‐2α were also hydroxylated by this enzyme in an oxygen concentration‐dependent manner. 50 Hydroxylation of this asparagine residue suppresses the transcriptional activity of the CTAD by inhibiting the binding of p300, a transcriptional coactivator with histone acetyltransferase activity. Prolyl hydroxylase regulates the expression level of HIFα, and FIH‐1 regulates the transcriptional activity of HIFα, both doubly regulated by the hydroxylation of amino acid residues (Figure 4A,B).
We and others revealed that the Km value of FIH‐1 for molecular oxygen is much lower than that of PHDs. 46 , 47 , 48 , 49 , 51 As oxygen concentration decreases, the enzymatic activity of PHD is suppressed, and the protein expression of HIFα increases, which activates HIF. A further reduction in oxygen concentration reduces the enzymatic activity of FIH‐1, which allows p300 binding to CTAD and increases the transcriptional activity of HIF to its maximum (Figure 4B). Hypoxia‐inducible factor‐3α not only lacks the CTAD and is considered less transcriptionally active than HIF‐1α and HIF‐2α but it also competitively inhibits HIF‐1α and HIF‐2α functions through several known splicing variants, 52 , 53 including IPAS and NEPAS. The splicing regulatory mechanism of HIF‐3α remains to be elucidated. Activated HIF transcribes PHD3 (and PHD2 in some cell types), negatively regulating HIF. It is as if the expression level of PHDs is trying to compensate for the weakened enzymatic activity of PHDs because of the decrease in oxygen concentration. Thus, a negative feedback mechanism exists to prevent HIF from being activated in an unregulated and constitutive manner (Figure 4C). Chronic HIF activation leads to detrimental effects, such as myocardial mitochondrial injury, resulting in severe heart failure, similar to dilated cardiomyopathy. 48 , 54 , 55 Three distinct genes encode three hydroxylases that negatively regulate HIF. Taken together, it seems that our bodies somehow avoid the constant activation of HIF. Prolyl hydroxylase inhibitors (HIF‐PH inhibitors), small compounds that inhibit PHD enzyme activities, were developed on the basis of this molecular mechanism of the hypoxic response. These inhibitors stimulate EPO production in the kidney and liver and iron absorption from the gastrointestinal tract and its transport and utilization in the body and have been used to treat renal anemia in the clinic. 56 , 57
In ccRCC cancer cells, the O2 sensing mechanism is disrupted because of VHL mutation, and HIFα escapes from proteasomal degradation. Therefore, HIF is constitutively activated and its target gene VEGFA is upregulated even under normoxic conditions. Consistent with these events, ccRCC exhibits hypervascular phenotypes, and multiple TKIs targeting VEGF have demonstrated significant activity as single agents for mRCC. 30 Therefore, HIF could represent an ideal therapeutic target for ccRCC.
6. HYPOXIA‐INDUCIBLE FACTOR‐2α PLAYS A CRITICAL ROLE AS A DRIVER IN PVHL‐DEFECTIVE RENAL CARCINOMA CELLS
When considering therapeutic strategies targeting HIF in ccRCC cells, it is critical to clarify whether HIF is the driver for the tumor. We examined whether tumor suppression by pVHL could be overridden by a HIF‐2α variant that could not be recognized by the VBC complex. We produced HIF‐2α in which the proline residue at 531 was substituted to alanine, resulting in escape from binding to pVHL. The construct was introduced to WT8 cells to establish WT8 + HIF2αP531A cells (Table 1). WT8 + HIF2αP531A cells fully restored their ability to form tumors in nude mice irrespective of their positive status of pVHL. 58 Because tumor suppression by pVHL could be overridden by a HIF‐2α variant that escapes pVHL control, our results strongly suggested that HIF‐2α might be a driver in the tumor progression of ccRCC.
In a subsequent study in 2005, Dr. Raval et al. infected 786‐O VHL −/− RCC cells with retroviruses expressing HIF‐1α, HIF‐2α, or GFP alone and implanted cells subcutaneously into nude mice. While four of the five animals with injections of 786‐O cells expressing GFP formed tumors, none with injections of 786‐O cells expressing HIF‐1α formed tumors. In contrast, those injected with cells expressing HIF‐2α formed tumors that grew at an enhanced rate compared with control animals injected with cells infected with the control virus. 59 In a later study in 2008, Dr. Gordan et al. analyzed the VHL genotype and HIFα expression among primary tumors and found that VHL −/− RCC expressed either both HIF‐1α and HIF‐2α or HIF‐2α alone. No tumors showed the HIF‐1α alone phenotype. 60 In 2011, Dr. Shen et al. reported that downregulation of HIF‐1α by shRNA promoted the growth of VHL −/− RCC4 renal carcinoma cells orthotopically implanted in the kidneys of nude mice. Downregulation of HIF‐1α in VHL −/− UMRC2 renal carcinoma cells also dramatically enhanced tumor growth in vivo. 61 As for the reasons why HIF‐1α inhibited the tumor growth of RCC cells, Dr. Gordan et al. examined the roles of both HIF‐1α and ‐2α on the transcriptional activity of c‐Myc. They clarified that HIF‐1α antagonized c‐Myc function; in contrast, HIF‐2α enhanced its transcriptional activity. 62 This result strongly suggested that HIF‐1α inhibited the tumor growth of RCC through the repression of the cell‐cycle progression regulated by c‐Myc.
Collectively, the results of our group and others clearly indicated that HIF‐2 might be a possible driver of RCC.
7. DRUG DEVELOPMENT FOR TARGETING HIF‐2α BY ALLOSTERIC BLOCKADE OF THE DIMERIZATION OF HIF‐2α AND HIF‐1β
Based on these results, an attempt was made to develop inhibitors targeting HIF‐2. Importantly, Dr. Wallace et al. succeeded in developing a series of orally active small molecules, PT2399 and PT2385, that allosterically block the dimerization of HIF‐2α and HIF‐1β. 63 , 64 , 65 Inhibition of the HIF‐2α/HIF‐1β interaction by PT2399 was confirmed by coimmunoprecipitation assay using an anti‐HIF‐1β Ab. Hypoxia inducible factor‐2α was coimmunoprecipitated from whole cell extracts from 786‐O, A498, and UMRC2 pVHL null ccRCC cells by the Ab. Hypoxia inducible factor‐1β coprecipitating with HIF‐2α protein was diminished under PT2399 treatment in a dose‐dependent manner. The effect of PT2399 was also examined in Hep3B VHL +/+ cells cultured under hypoxic conditions. The compound specifically inhibited the mRNA expression of HIF‐2‐specific genes (EPO, PAI‐1) but not those regulated by HIF‐1, indicating that PT2399 specifically inhibits HIF‐2α signals. PT2399 inhibited the in vivo tumor growth of VHL −/− RCC such as 786‐O and A498 cells as well as ccRCC patient‐derived xenografts. 65
Dr. Chen et al. evaluated the effects of PT2399 in a panel of 22 independently generated RCC patient‐derived xenografts and found that the compound decreased tumor growth by 60% across all tumor grafts (p < 0.0001). The authors also compared the antitumor effect of PT2399 and sunitinib, a representative anti‐VEGF TKI, and found that PT2399 compound was more active than sunitinib (p = 0.0126). Moreover, PT2399 inhibited tumor growth in some sunitinib‐resistant tumors. 64 Similarly, Dr. Wallace et al. revealed that the other orally active HIF‐2α inhibitor, PT2385, significantly reduced tumor weight in both 786‐O and A498 xenograft models in vivo. 63
8. DRUGS TARGETING HIF‐2 SHOWED ANTITUMOR ACTIVITY IN CLINICAL TRIALS
From the results of these preclinical studies, a phase I dose‐escalation trial of the HIF‐2α inhibitor PT2385 was undertaken in 51 patients with advanced ccRCC, 50 of whom were evaluable for the response. All patients had been previously treated with VEGF‐targeted therapy and the median number of prior systemic therapies was four, indicating that these patients were refractory to most existing drugs to ccRCC. Among the overall 50 patients, one patient (2%) had CR, six (12%) had PR, and 26 (52%) had SD. The disease control rate (CR plus PR plus SD) was as high as 66% (Table 2). 66
TABLE 2.
Efficacy of PT2385 for previously treated advanced clear cell renal cell carcinoma (NCT02293980).
| Number of patients | Dose | Primary end‐point | Treatment line | MTD | Efficacy | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CR | PR | SD | PD | NE | ORR (%) | DCR (%) | |||||
| 51 | 100–1800 mg, twice daily | MTD | 2nd line or beyond | Not identified | 1 | 6 | 26 | 17 | 1 | 14 | 66 |
Abbreviations: CR, complete response; DCR, disease control rate; MTD, maximum tolerated dose; NE, not evaluable; ORR, objective response rate; PD, progressive disease; PR, partial response; SD, stable disease.
Among the HIF‐2α inhibitors, belzutifan is the most advanced compound in clinical development, and a phase II trial has been undertaken in VHL patients in the United States and Europe. A total of 61 patients with VHL disease were enrolled. Most of them (56/61, 91.8%) showed a reduction in RCC tumor size after treatment, and 30 cases (49%) achieved a reduction in tumor size (PR) of >30%. The effect of the drug on CNS hemangioblastomas was also evaluated; the results showed that 3/50 patients (6.0%) achieved CR and 12/50 (24%) achieved PR. In addition, all patients with retinal hemangioma (n = 12) showed improvement in their condition. Belzutifan also improved pNET in VHL patients, with 3/22 (13.6%) and 17/22 (77.3%) patients achieving CR and PR, respectively (Table 3). 17 Based on these results, in 2021, FDA approved belzutifan for adult patients with VHL disease who require therapy for RCC, CNS hemangioblastoma, or pNET, not requiring immediate surgery. Thus, 127 years after the first case report of VHL disease by Dr. Collins, an effective drug for this disease was established.
TABLE 3.
Efficacy of belzutifan for renal cell carcinoma (RCC), central nervous system (CNS) hemangioblastoma, retinal hemangioblastoma, and pancreatic neuroendocrine tumor (pNET) in von Hippel–Lindau disease (NCT03401788).
| Tumor | Number of patients | Efficacy | mTTR (months) | mDOR (months) | 2 year PFS (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| CR | PR | SD | PD | NE | ORR (%) | |||||
| RCC | 61 | 0 | 30 | 30 | 0 | 1 | 49 | 8.2 | NR | 96 |
| CNS hemangioblastoma | 50 | 3 | 12 | 31 | 2 | 2 | 30 | 3.2 | NR | – |
| retinal hemangioblastoma | 12 (16 eyes evaluable) | 16 | 0 | 0 | 0 | 100 | NA | NA | – | |
| pNET | 22 | 3 | 17 | 2 | 0 | 0 | 90.9 | 5.5 | NR | – |
Abbreviations: –, no data; CR, complete response; mDOR, median duration of response; mTTR, median time to response; NA, not available; NE, not evaluable; NR, not reached; ORR, objective response rate; PD, progressive disease; PFS, progression‐free survival; PR, partial response; SD, stable disease.
Several clinical trials combining belzutifan with other drugs are ongoing or in planning. These combination therapies might be introduced into the clinic for advanced ccRCC. 30 Importantly, VHL mutation was also reported in colorectal cancer. 67 Moreover, it had been reported that HIFα was stabilized in tumor cells harboring mutations in the genes encoding a series of succinate dehydrogenase as the activity of PHDs was inhibited by the accumulation of succinate in these cells. 68 Although the exact roles of HIF‐2 in those tumors have not been elucidated, these results indicate the possibility of clinical application of HIF‐2α inhibitors against those tumors.
In addition, we, our colleagues, and other groups have successfully identified novel pVHL target proteins other than HIF. 69 , 70 , 71 , 72 , 73 , 74 , 75 , 76 Moreover, molecules exhibiting synthetic lethality against VHL −/− RCC cells have been identified. 77 , 78 , 79 Therefore, these novel molecules might be ideal candidates for new drug targets and possible new combination therapies. Together these findings indicate the potential for the successful development of additional drugs against diseases caused by mutated VHL and/or the activation of HIF‐2 with the aim of improving the prognosis of patients.
FUNDING INFORMATION
Japan Society for the Promotion of Science, Grant/Award Number: 20 K09229 and 16H04723 to YAM, 17H04328 and 24,659,712 to EN. Health, Labor and Welfare Science Research Grant for research on intractable diseases, Japan (22FC0101) to EN.
CONFLICT OF INTEREST
Yoji Andrew Minamishima receives honoraria from Astellas Pharma Inc., Kyowa Kirin Co., Ltd., and Bayer Holding Ltd. Eijiro Nakamura is an advisory board for Merck & Co., Inc. The other authors have no conflict of interest to disclose.
ETHICS STATEMENT
Approval of the research protocol by an institutional review board: The study cited as Yamasaki et al. 32 was approved by the institutional review board of Kyoto University.
Informed consent: N/A.
Registry and registration no. of the study/trial: N/A.
Animal studies: All experiments involving laboratory animals were done in accordance with the Guidelines for Animal Experiments of Kyoto University.
ACKNOWLEDGMENTS
The authors apologize to the numerous investigators whose important publications could not be cited because of space restraints.
Takamori H, Yamasaki T, Kitadai R, Minamishima YA, Nakamura E. Development of drugs targeting hypoxia‐inducible factor against tumor cells with VHL mutation: Story of 127 years. Cancer Sci. 2023;114:1208‐1217. doi: 10.1111/cas.15728
REFERENCES
- 1. Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nature Reviews. Cancer. 2008;8:967‐975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Molecular Cell. 2008;30:393‐402. [DOI] [PubMed] [Google Scholar]
- 3. Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel‐Lindau disease tumor suppressor gene. Science. 1993;260:1317‐1320. [DOI] [PubMed] [Google Scholar]
- 4. Maher ER, Kaelin WG Jr. von Hippel‐Lindau disease. Medicine (Baltimore). 1997;76:381‐391. [DOI] [PubMed] [Google Scholar]
- 5. Lonser RR, Glenn GM, Walther M, et al. von Hippel‐Lindau disease. Lancet. 2003;361:2059‐2067. [DOI] [PubMed] [Google Scholar]
- 6. Maher ER, Neumann HP, Richard S. von Hippel‐Lindau disease: a clinical and scientific review. European Journal of Human Genetics. 2011;19:617‐623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nielsen SM, Rhodes L, Blanco I, et al. Von Hippel‐Lindau disease: genetics and role of genetic counseling in a multiple neoplasia syndrome. Journal of Clinical Oncology. 2016;34:2172‐2181. [DOI] [PubMed] [Google Scholar]
- 8. Kishida T, Stackhouse TM, Chen F, Lerman MI, Zbar B. Cellular proteins that bind the von Hippel‐Lindau disease gene product: mapping of binding domains and the effect of missense mutations. Cancer Research. 1995;55:4544‐4548. [PubMed] [Google Scholar]
- 9. Kibel A, Iliopoulos O, DeCaprio JA, Kaelin WG Jr. Binding of the von Hippel‐Lindau tumor suppressor protein to Elongin B and C. Science. 1995;269:1444‐1446. [DOI] [PubMed] [Google Scholar]
- 10. Kaelin WG Jr. Molecular basis of the VHL hereditary cancer syndrome. Nature Reviews. Cancer. 2002;2:673‐682. [DOI] [PubMed] [Google Scholar]
- 11. Duan DR, Pause A, Burgess WH, et al. Inhibition of transcription elongation by the VHL tumor suppressor protein. Science. 1995;269:1402‐1406. [DOI] [PubMed] [Google Scholar]
- 12. Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. Journal of Clinical Oncology. 2004;22:4991‐5004. [DOI] [PubMed] [Google Scholar]
- 13. Kaelin WG Jr. The von Hippel‐Lindau tumour suppressor protein: O2 sensing and cancer. Na0er. 2008;8:865‐873. [DOI] [PubMed] [Google Scholar]
- 14. Lonergan KM, Iliopoulos O, Ohh M, et al. Regulation of hypoxia‐inducible mRNAs by the von Hippel‐Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Mol Cell Biol. 1998;18:732‐741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Iwai K, Yamanaka K, Kamura T, et al. Identification of the von Hippel‐Lindau tumor‐suppressor protein as part of an active E3 ubiquitin ligase complex. Proc Natl Acad Sci USA. 1999;96:12436‐12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia‐inducible factors for oxygen‐dependent proteolysis. Nature. 1999;399:271‐275. [DOI] [PubMed] [Google Scholar]
- 17. Jonasch E, Donskov F, Iliopoulos O, et al. Belzutifan for renal cell carcinoma in von Hippel‐Lindau disease. The New England Journal of Medicine. 2021;385:2036‐2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Collins ET. Intra‐ocular growths (two cases, brother and sister, with peculiar vascular new growth, probably retinal, affecting both eyes). Trans Ophthalmol Soc UK. 1894;14:141‐149. [Google Scholar]
- 19. von Hippel E. Ueber eine sehr seltene Erkrankung der Nethaut. Albrecht von Graefe's Archiv für Ophthalmologie. 1904;59:83‐106. [Google Scholar]
- 20. Lindau A. Zur Frage der Angiomatosis Retinae und Ihrer Hirncomplikation. Acta Ophthalmologica. 1927;4:193‐226. [Google Scholar]
- 21. Seizinger BR, Rouleau GA, Ozelius LJ, et al. Von Hippel‐Lindau disease maps to the region of chromosome 3 associated with renal cell carcinoma. Nature. 1988;332:268‐269. [DOI] [PubMed] [Google Scholar]
- 22. Gnarra JR, Tory K, Weng Y, et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nature Genetics. 1994;7:85‐90. [DOI] [PubMed] [Google Scholar]
- 23. Iliopoulos O, Kibel A, Gray S, Kaelin WG Jr. Tumour suppression by the human von Hippel‐Lindau gene product. Nature Medicine. 1995;1:822‐826. [DOI] [PubMed] [Google Scholar]
- 24. Cohen HT, McGovern FJ. Renal‐cell carcinoma. The New England Journal of Medicine. 2005;353:2477‐2490. [DOI] [PubMed] [Google Scholar]
- 25. Takahashi A, Sasaki H, Kim SJ, et al. Markedly increased amounts of messenger RNAs for vascular endothelial growth factor and placenta growth factor in renal cell carcinoma associated with angiogenesis. Cancer Research. 1994;54:4233‐4237. [PubMed] [Google Scholar]
- 26. Iliopoulos O, Levy AP, Jiang C, Kaelin WG Jr, Goldberg MA. Negative regulation of hypoxia‐inducible genes by the von Hippel‐Lindau protein. Proc Natl Acad Sci USA. 1996;93:10595‐10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang JC, Haworth L, Sherry RM, et al. A randomized trial of bevacizumab, an anti‐vascular endothelial growth factor antibody, for metastatic renal cancer. The New England Journal of Medicine. 2003;349:427‐434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear‐cell renal‐cell carcinoma. The New England Journal of Medicine. 2007;356:125‐134. [DOI] [PubMed] [Google Scholar]
- 29. Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal‐cell carcinoma. The New England Journal of Medicine. 2007;356:115‐124. [DOI] [PubMed] [Google Scholar]
- 30. Choueiri TK, Kaelin WG Jr. Targeting the HIF2‐VEGF axis in renal cell carcinoma. Nature Medicine. 2020;26:1519‐1530. [DOI] [PubMed] [Google Scholar]
- 31. Kamba T, Tam BY, Hashizume H, et al. VEGF‐dependent plasticity of fenestrated capillaries in the normal adult microvasculature. American Journal of Physiology. Heart and Circulatory Physiology. 2006;290:H560‐H576. [DOI] [PubMed] [Google Scholar]
- 32. Yamasaki T, Kamba T, Kanno T, et al. Tumor microvasculature with endothelial fenestrations in VHL null clear cell renal cell carcinomas as a potent target of anti‐angiogenic therapy. Cancer Science. 2012;103:2027‐2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang GL, Semenza GL. Characterization of hypoxia‐inducible factor 1 and regulation of DNA binding activity by hypoxia. The Journal of Biological Chemistry. 1993;268:21513‐21518. [PubMed] [Google Scholar]
- 34. Wang GL, Semenza GL. General involvement of hypoxia‐inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci USA. 1993;90:4304‐4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maxwell PH, Pugh CW, Ratcliffe PJ. Inducible operation of the erythropoietin 3′ enhancer in multiple cell lines: evidence for a widespread oxygen‐sensing mechanism. Proc Natl Acad Sci USA. 1993;90:2423‐2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kaelin WG Jr, Ratcliffe PJ, Semenza GL. Pathways for oxygen regulation and homeostasis: the 2016 Albert Lasker basic medical research award. JAMA. 2016;316:1252‐1253. [DOI] [PubMed] [Google Scholar]
- 37. Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nature Reviews. Cancer. 2011;12:9‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ivan M, Kondo K, Yang H, et al. HIFalpha targeted for VHL‐mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464‐468. [DOI] [PubMed] [Google Scholar]
- 39. Epstein AC, Gleadle JM, McNeill LA, et al. C. elegans EGL‐9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43‐54. [DOI] [PubMed] [Google Scholar]
- 40. Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF‐alpha to the von Hippel‐Lindau ubiquitylation complex by O2‐regulated prolyl hydroxylation. Science. 2001;292:468‐472. [DOI] [PubMed] [Google Scholar]
- 41. Ivan M, Haberberger T, Gervasi DC, et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia‐inducible factor. Proc Natl Acad Sci USA. 2002;99:13459‐13464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia‐inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc Natl Acad Sci USA. 1991;88:5680‐5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Semenza GL. Oxygen sensing, homeostasis, and disease. The New England Journal of Medicine. 2011;365:537‐547. [DOI] [PubMed] [Google Scholar]
- 44. Lieb ME, Menzies K, Moschella MC, Ni R, Taubman MB. Mammalian EGLN genes have distinct patterns of mRNA expression and regulation. Biochemistry and Cell Biology. 2002;80:421‐426. [DOI] [PubMed] [Google Scholar]
- 45. Metzen E, Berchner‐Pfannschmidt U, Stengel P, et al. Intracellular localisation of human HIF‐1 alpha hydroxylases: implications for oxygen sensing. Journal of Cell Science. 2003;116:1319‐1326. [DOI] [PubMed] [Google Scholar]
- 46. Hirsila M, Koivunen P, Gunzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4‐hydroxylases that modify the hypoxia‐inducible factor. The Journal of Biological Chemistry. 2003;278:30772‐30780. [DOI] [PubMed] [Google Scholar]
- 47. Takeda K, Ho VC, Takeda H, Duan LJ, Nagy A, Fong GH. Placental but not heart defects are associated with elevated hypoxia‐inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol. 2006;26:8336‐8346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Minamishima YA, Moslehi J, Bardeesy N, Cullen D, Bronson RT, Kaelin WG. Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood. 2008;111:3236‐3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Minamishima YA, Kaelin WG. Reactivation of hepatic EPO synthesis in mice after PHD loss. Science. 2010;329:407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mahon PC, Hirota K, Semenza GL. FIH‐1: a novel protein that interacts with HIF‐1alpha and VHL to mediate repression of HIF‐1 transcriptional activity. Genes & Development. 2001;15:2675‐2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Koivunen P, Hirsila M, Gunzler V, Kivirikko KI, Myllyharju J. Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4‐hydroxylases. The Journal of Biological Chemistry. 2004;279:9899‐9904. [DOI] [PubMed] [Google Scholar]
- 52. Makino Y, Cao R, Svensson K, et al. Inhibitory PAS domain protein is a negative regulator of hypoxia‐inducible gene expression. Nature. 2001;414:550‐554. [DOI] [PubMed] [Google Scholar]
- 53. Yamashita T, Ohneda O, Nagano M, et al. Abnormal heart development and lung remodeling in mice lacking the hypoxia‐inducible factor‐related basic helix‐loop‐helix PAS protein NEPAS. Mol Cell Biol. 2008;28:1285‐1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Minamishima YA, Moslehi J, Padera RF, Bronson RT, Liao R, Kaelin WG. A feedback loop involving the Phd3 prolyl hydroxylase tunes the mammalian hypoxic response In vivo. Mol Cell Biol. 2009;29:5729‐5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Moslehi J, Minamishima YA, Shi JR, et al. Loss of hypoxia‐inducible factor prolyl hydroxylase activity in cardiomyocytes phenocopies ischemic cardiomyopathy. Circulation. 2010;122:U1004‐U1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Maxwell PH, Eckardt KU. HIF prolyl hydroxylase inhibitors for the treatment of renal anaemia and beyond. Nature Reviews. Nephrology. 2016;12:157‐168. [DOI] [PubMed] [Google Scholar]
- 57. Suhara T, Hishiki T, Kasahara M, et al. Inhibition of the oxygen sensor PHD2 in the liver improves survival in lactic acidosis by activating the Cori cycle. Proc Natl Acad Sci USA. 2015;112:11642‐11647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG Jr. Inhibition of HIF is necessary for tumor suppression by the von Hippel‐Lindau protein. Cancer Cell. 2002;1:237‐246. [DOI] [PubMed] [Google Scholar]
- 59. Raval RR, Lau KW, Tran MG, et al. Contrasting properties of hypoxia‐inducible factor 1 (HIF‐1) and HIF‐2 in von Hippel‐Lindau‐associated renal cell carcinoma. Mol Cell Biol. 2005;25:5675‐5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gordan JD, Lal P, Dondeti VR, et al. HIF‐alpha effects on c‐Myc distinguish two subtypes of sporadic VHL‐deficient clear cell renal carcinoma. Cancer Cell. 2008;14:435‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shen C, Beroukhim R, Schumacher SE, et al. Genetic and functional studies implicate HIF1α as a 14q kidney cancer suppressor gene. Cancer Discovery. 2011;1:222‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF‐2alpha promotes hypoxic cell proliferation by enhancing c‐myc transcriptional activity. Cancer Cell. 2007;11:335‐347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wallace EM, Rizzi JP, Han G, et al. A small‐molecule antagonist of HIF2alpha is efficacious in preclinical models of renal cell carcinoma. Cancer Research. 2016;76:5491‐5500. [DOI] [PubMed] [Google Scholar]
- 64. Chen W, Hill H, Christie A, et al. Targeting renal cell carcinoma with a HIF‐2 antagonist. Nature. 2016;539:112‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cho H, Du X, Rizzi JP, et al. On‐target efficacy of a HIF‐2alpha antagonist in preclinical kidney cancer models. Nature. 2016;539:107‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Courtney KD, Infante JR, Lam ET, et al. Phase I dose‐escalation trial of PT2385, a first‐in‐class hypoxia‐inducible factor‐2alpha antagonist in patients with previously treated advanced clear cell renal cell carcinoma. Journal of Clinical Oncology. 2018;36:867‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kuwai T, Kitadai Y, Tanaka S, Hiyama T, Tanimoto K, Chayama K. Mutation of the von Hippel‐Lindau (VHL) gene in human colorectal carcinoma: association with cytoplasmic accumulation of hypoxia‐inducible factor (HIF)‐1alpha. Cancer Science. 2004;95:149‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Selak MA, Armour SM, MacKenzie ED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF‐alpha prolyl hydroxylase. Cancer Cell. 2005;7:77‐85. [DOI] [PubMed] [Google Scholar]
- 69. Okuda H, Saitoh K, Hirai S, et al. The von Hippel‐Lindau tumor suppressor protein mediates ubiquitination of activated atypical protein kinase C. The Journal of Biological Chemistry. 2001;276:43611‐43617. [DOI] [PubMed] [Google Scholar]
- 70. Lee S, Nakamura E, Yang H, et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell. 2005;8:155‐167. [DOI] [PubMed] [Google Scholar]
- 71. Nakamura E, Kaelin WG Jr. Recent insights into the molecular pathogenesis of pheochromocytoma and paraganglioma. Endocrine Pathology. 2006;17:97‐106. [DOI] [PubMed] [Google Scholar]
- 72. Nakamura E, Abreu‐e‐Lima P, Awakura Y, et al. Clusterin is a secreted marker for a hypoxia‐inducible factor‐independent function of the von Hippel‐Lindau tumor suppressor protein. The American Journal of Pathology. 2006;168:574‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yang H, Minamishima YA, Yan Q, et al. pVHL acts as an adaptor to promote the inhibitory phosphorylation of the NF‐kappaB agonist Card9 by CK2. Molecular Cell. 2007;28:15‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kanno T, Kamba T, Yamasaki T, et al. JunB promotes cell invasion and angiogenesis in VHL‐defective renal cell carcinoma. Oncogene. 2012;31:3098‐3110. [DOI] [PubMed] [Google Scholar]
- 75. Guo J, Chakraborty AA, Liu P, et al. pVHL suppresses kinase activity of Akt in a proline‐hydroxylation‐dependent manner. Science. 2016;353:929‐932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhang J, Wu T, Simon J, et al. VHL substrate transcription factor ZHX2 as an oncogenic driver in clear cell renal cell carcinoma. Science. 2018;361:290‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chakraborty AA, Nakamura E, Qi J, et al. HIF activation causes synthetic lethality between the VHL tumor suppressor and the EZH1 histone methyltransferase. Science Translational Medicine. 2017;9:eaal5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bommi‐Reddy A, Almeciga I, Sawyer J, et al. Kinase requirements in human cells: III. Altered kinase requirements in VHL−/− cancer cells detected in a pilot synthetic lethal screen. Proc Natl Acad Sci USA. 2008;105:16484‐16489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nicholson HE, Tariq Z, Housden BE, et al. HIF‐independent synthetic lethality between CDK4/6 inhibition and VHL loss across species. Science Signaling. 2019;12:eaay0482. [DOI] [PMC free article] [PubMed] [Google Scholar]