

Abstract
Lenvatinib is the favorable treatment for advanced hepatocellular carcinoma (HCC), and it is currently undergoing phase III clinical trials. However, the specific effects of lenvatinib on PD1+ CD8+ T cells in HCC microenvironment have not been systematically studied. Here, we established an orthotopic hepa1‐6 mouse model treated with lenvatinib to investigate CD8+ T cells’ role in the tumor and spleen. We found an increasing proportion of TCF‐1+ in PD1+ CD8+ T cells and proliferation of PD1+ CD8+ T cells after lenvatinib treatment. Meanwhile, lenvatinib treatment upregulated the expression of granzyme B on PD1+ CD8+ T cells both in vitro and in vivo. Lenvatinib activated the endogenous mTOR pathway of exhausted CD8+ T cells, and mTOR pathway blockade eliminated the antitumor effect of lenvatinib and function of PD1+ CD8+ T cells. The effects of the mTOR pathway on PD1+ CD8+ T cells after lenvatinib treatment were mediated by VEGFR2 inhibition. Overall, our work provides insight into the mechanism of lenvatinib's antitumor efficacy through exhausted CD8+ T cells in HCC treatment.
Keywords: HCC, lenvatinib, mTOR, PD1, TCF1
In this work, we observed TCF1+ PD1+ CD8+ T cells gradually decreased with tumor progression. We discovered that lenvatinib promoted the production of pTex and increased the proportion of the pTex subset in all exhausted CD8+ T cells both in tumor and spleen. At the same time, the proliferation activity of PD1+ CD8+ T cells was significantly increased, and the PD1+ CD8+ T cells secreted more granzyme B, which means greater tumor‐killing function under the influence of lenvatinib. Furthermore, lenvatinib affects PD1+ CD8+ T cell differentiation by affecting the PI3K/Akt/mTOR signaling pathway. After the combination of lenvatinib and rapamycin, the effect of lenvatinib on pTex and granzyme B expression was significantly inhibited. Finally, we found lenvatinib affects the PI3K/Akt/mTOR signaling pathway by blocking VEGFR2. Overall, our work provides insight into the mechanism of lenvatinib's antitumor efficacy through exhausted CD8+ T cells in HCC treatment.
Abbreviations
- FGFR
fibroblast growth factor receptor
- HCC
hepatocellular carcinoma
- PD‐1
programmed cell death‐1
- PDGFR
platelet‐derived growth factor receptor
- pTex
progenitor exhausted CD8+ T cells
- TCF1
transcription factor T cell factor 1
- TIM‐3
T cell immunoglobulin and mucin domain–containing protein 3
- tTex
terminal exhausted CD8+ T cells
- VEGFR2
vascular endothelial growth factor receptor 2
1. INTRODUCTION
TCF1 is an effector transcription factor downstream of the WNT signaling pathway. Recent research has found that TCF1 plays a role in regulating the multifunctional, environment‐dependent functions of mature T cell responses. In the exhausted CD8+ T cells, TCF1 is required for the self‐renewal of stem‐like exhausted CD8+ T cells generated in response to tumor antigens and for preserving responses to checkpoint blockade immunotherapy. 1 Through ectopic expression of TCF1, a higher fraction of tumor infiltrating lymphocyte (TILs) adopts a stem‐like exhausted CD8+ T cell phenotype, enhances polyfunctionality, and has lower expression of coinhibitory receptors including PD1 and TIM3, leading to improved tumor control. 2 The PI3K/Akt/mTOR signaling pathway also plays an important role in regulating T cell self‐proliferation and differentiation. It was found that mTORC1 activity was higher in the “effector‐like” daughter cell and lower in the “memory‐like” daughter cell due to asymmetric partitioning of amino acid transporters during the division of CD8+ T cells. 3
In many tumors, the mTOR pathway has been found to be abnormally activated, so several inhibitors of the mTOR pathway have been investigated in clinical studies. 4 However, the effects of the mTOR pathway on the phenotype and function of PD1+ CD8+ T cells are poorly understood.
Tumor‐specific CD8+ T cells are exposed to persistent antigenic stimulation, causing a dysfunctional state known as “exhaustion.” This pathway is a dynamic process from activation to “progenitor exhaustion” through to “terminally exhaustion” with different phenotype and properties. 5 In the tumor microenvironment, after receiving continuous tumor antigen stimulation, CD8+ T cells are exhausted, and a part of them differentiate into TCF1+PD1+ Ki‐67+ TIM3− T progenitor cells (pTex), which have strong self‐proliferation and differentiation ability, but weak cytotoxicity, and these cells further differentiate into TCF1− PD1+ Ki‐67− TIM3− T cells (intermediate Tex), which have no self‐proliferative ability but have tumor‐killing function and play a major role in limiting tumor growth. Then, intermediate exhausted CD8+ T cells eventually become TCF1− PD1+ KI‐67− TIM3+ terminal exhausted CD8+ T (tTex) cells with no tumor‐killing function. 6 , 7 , 8 A large number of studies have found that immune checkpoint blockade therapy achieved efficacy through promoting the expansion of progenitor exhausted CD8+ T subsets and their differentiation into intermediate Tex instead of the reversal of the CD8+ T exhaustion status. 1 , 9 , 10 , 11 , 12
Lenvatinib is a multi‐target inhibitor that inhibits vascular endothelial growth factor receptor (VEGFR) 1‐3, fibroblast growth factor receptor (FGFR) 1‐4, platelet‐derived growth factor receptor (PDGFR) α, and proto‐oncogenes RET and KIT. 13 At present, lenvatinib has been demonstrated to hamper tumor progress in numerous ways: (i) it inhibits tumor angiogenesis via inhibiting the VEGF/VEGFR signaling pathway 14 ; (ii) it inhibits tumor cell proliferation and shows antitumor activity by inhibiting downstream RET signaling in tumor models of RET gene fusion 15 ; and (iii) it reverses the immunosuppressive tumor microenvironment caused by VEGF‐A, reduces the infiltration of tumor associated macrophage (TAMs), and increases the activity of CD8+ T cells by inhibiting VEGFR. 16 , 17 Existing studies mainly focused on lenvatinib's anti‐immunosuppressive and anti‐angiogenic effects. However, the effect of lenvatinib on tumor‐infiltrating progenitor exhausted CD8+ T cells has been little studied.
In this work, we observed TCF1+ PD1+ CD8+ T cells gradually decreased with tumor progression. We discovered that lenvatinib promoted the production of pTex and increased the proportion of the pTex subset in all exhausted CD8+ T cells both in tumor and spleen. At the same time, the proliferation activity of PD1+ CD8+ T cells was significantly increased and the PD1+ CD8+ T cells secreted more granzyme B, which means greater tumor‐killing function under the influence of lenvatinib. Furthermore, lenvatinib affects PD1+ CD8+ T cell differentiation by affecting the PI3K/Akt/mTOR signaling pathway. After the combination of lenvatinib and rapamycin, the effect of lenvatinib on pTex and granzyme B expression was significantly inhibited. Finally, we found lenvatinib affects the PI3K/Akt/mTOR signaling pathway by blocking VEGFR2. Taken together, these findings supplement understanding of the mechanism of lenvatinib on exhausted CD8+ T cells in tumor microenvironment (TME).
2. MATERIALS AND METHODS
2.1. Establishment of a mouse tumor model and treatment
The mice were 6‐8 weeks of age. All the mice were fed in the specific pathogen free (SPF) experimental animal center. After the establishment of the mouse tumor model, lenvatinib and rapamycin were administered daily by gavage. In the recovery experiment, mice were randomly divided into four groups: control group, lenvatinib group (100 mg/kg, daily), rapamycin group (4 mg/kg, daily), and combined group. The mice in the four groups were given drugs on day 9 of tumor bearing, and the tumors in situ were removed for analysis on day 17 of tumor bearing.
2.2. Cell culture and induced differentiation assay
For CD8+ T cell differentiation experiment, naive CD8+ T cells were sorted from fresh spleen tissue by magnetic beads (BD, 551516) and stimulated with DynabeadsTM Mouse T‐activator CD3/CD28 (Thermo Fisher, 11542D; at a ratio of 1:1) for 6 days for further experiment in vitro. The induction conditions of CD8+ T cells were RMPI 1640 containing IL‐2 (25 ng/ml). The concentration of lenvatinib (Selleck S1164) and ISCK03 were 10 μM (Selleck S2070). The concentration of rapamycin was 20 nM (Selleck S1039). The concentration of BIBF1120 was 2 μM (Selleck S1010). The concentration of BLU667 was 5 μM (Selleck S8716).
2.3. Single‐cell suspension preparation and flow cytometry
The mice were sacrificed on the indicated day, and the tumor in the liver was collected. The tumor tissues were treated with a mouse tumor dissociation kit (Miltenyi, 130‐096‐730) according to the manufacturer's instruction. The CD8+ T cells in tumor were enriched by the CD8 (TIL) mouse MicroBead Kit (Miltenyi, 130‐116‐478), and the CD8+ T cells in spleen were enriched by Magnetic beads (BD, 551516) and then detected by multicolor flow cytometry after antibody incubation. CD8+ T cells were stimulated by Leukocyte Activation Cocktail (550583, BD Pharmingen) for 5 hours for the analysis of cytotoxic function. Specific staining steps and corresponding antibodies are shown in the supplementary materials.
2.4. Immunohistochemistry
Tissue sections with a thickness of 3 μm were used for immunohistochemical assay, and the images were analyzed by two independent pathologists. Specific immunohistochemistry method is provided in the supplementary materials.
2.5. RNA‐seq
CD8+ T cells were treated with lenvatinib in a concentration of 10 μM for 24 hours. Total RNA of normal control and treated cells was isolated by TRIzol (Life Technologies). The mRNA was sequenced by Illumina HiSeq 2500. Clean data were deposited in the NCBI Gene Expression Omnibus (PRJNA852361) The sequencing reads were aligned to the mouse reference sequence (UCSC/GRCm38/mm10) by HISAT2. The feature Counts function was performed for each gene count from trimmed reads against the GENCODE (release 30) transcript models. Differential gene expression analysis was quantitated by edgeR. The top 200 significantly differentially expressed genes were used for gene ontology (GO) and kyoto encyclopedia of genes and genomes (KEGG) enrichment. The analysis of differentially expressed genes and gene set enrichment analysis was implemented by the R package of Cluster Profiler.
2.6. Statistical approach
Flow cytometry data were analyzed by FlowJo.10 (TreeStar). Statistical analysis was performed using GraphPad Prism 8 software. Student's two‐tailed t test was used for comparison of quantitative data from different groups. p < 0.05 was considered statistically significant (*p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001).
3. RESULTS
3.1. The variation of expression of TCF1 on exhausted CD8 + T cells in tumor and spleen with HCC progression
Given that exhaustion of CD8+ T cells occurred when consistently receiving antigen stimulation, we constructed an orthotopic hepa1‐6 mouse model 18 and dynamically observed the changes of PD1+ CD8+ T cells with the prolongation of tumor‐bearing time. Then, we collected CD8+ T cells from liver and spleen of tumor‐bearing mice at day 13 and day 17 (Figure 1A). Flow cytometry was used to analyze the phenotypic and functional changes of CD8+ T cells. We found that the stem‐like exhausted CD8+ T cells showed decreased proliferative activity (Figure 1B). Furthermore, the proportion of the pTex subset (PD1+ TCF1+ TIM3− CD8+ T cells) decreased significantly in spleen and tumor, and the proportion of the tTex subgroup (PD1+ TCF1− TIM3+ CD8+ T cells) increased with HCC progression in tumor but not spleen (Figure 1C,D). To assess the expression levels of TCF1 in tumor‐infiltrating lymphocytes of HCC patients, we performed an immunofluorescence (IF) assay (Figure 1E). As shown in Table 1, the high proportion of TCF1+ tumor‐infiltrating lymphocytes was significantly correlated with TNM stage (p = 0.037). Taken together, the proportion of pTex was decreased with tumor bearing.
FIGURE 1.
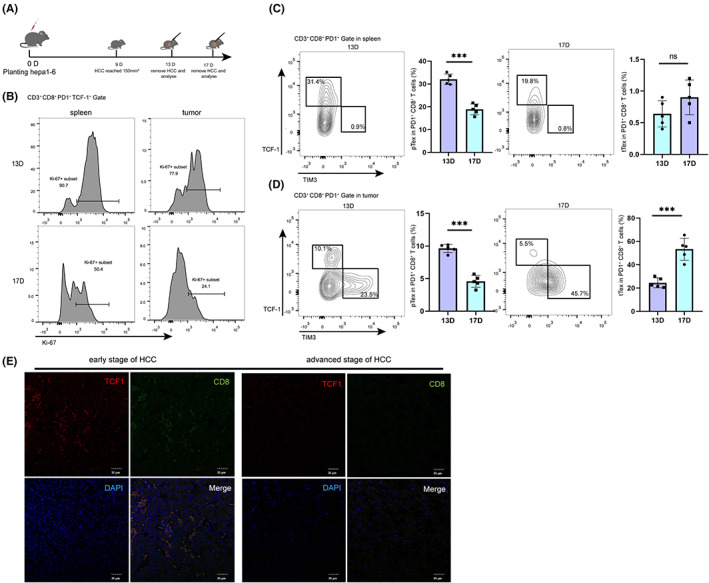
The variation of expression of TCF1 on exhausted CD8+ T cells in tumor and spleen with hepatocellular carcinoma (HCC) progression. A, Schematic diagram of tumor‐bearing mice. Hepa1‐6 cells were in situ inoculated into C57BL/6 mice on day 0, and the tumors were removed for analysis on days 13 and 17. B‐D, Phenotype and proliferative activity of PD1+ CD8+ T cells from spleen and tumor were analyzed at 13 and 17 d, respectively, including proliferative markers (marked by expression of Ki‐67) and exhausted PD1+ CD8+ T cell phenotype markers (marked by expression of TCF1 and TIM3). Data were expressed as mean ± SEM (n = 5). E, Under the field of view of 10 × 50, we randomly selected five fields in a sample, and the average number of cells expressing both CD8 and TCF1 in each field was recorded as the high group if the number was greater than 27, and the low group if the number was <27 (27 was the average number of double‐positive cells in each field of 40 samples)
TABLE 1.
Association of TCF1 expression in tumor‐infiltrating lymphocytes with clinicopathological features of 40 hepatocellular carcinoma (HCC) patients
| Characteristics | TCF1 expression | P value | |
|---|---|---|---|
| Low | High | ||
| (n = 20) | (n = 20) | ||
| Age y ≥ 55 y old | 12 | 14 | 0.715 |
| Gender female | 4 | 5 | >0.999 |
| Serum AFP ≥ 20 | 10 | 12 | 0.735 |
| Tumor size ≥ 5.0 cm | 16 | 14 | >0.999 |
| TNM stage III/IV | 17 | 6 | 0.037 |
| Survival time ≥ 48 mo | 9 | 15 | 0.158 |
3.2. Effects of lenvatinib on infiltration of TCF1 + PD1 + CD8 + T cells in TME and spleen
Next, we explored whether lenvatinib could halt or reverse the decline of pTex. Lenvatinib treatment was started on day 9 of tumor bearing, and the samples were harvested on day 17. The tumor volume of lenvatinib group was significantly smaller than that of the control group (Figure 2A).
FIGURE 2.
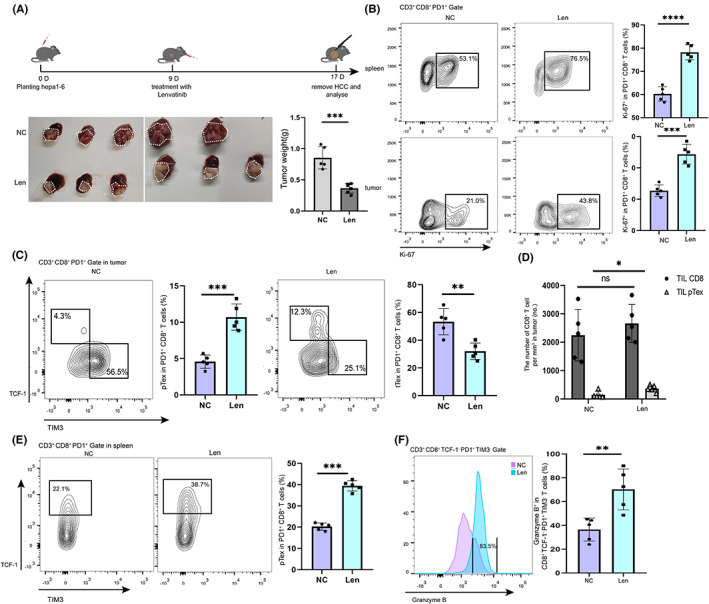
Effects of lenvatinib on infiltration of tcf1+ PD1+ CD8+ T cells in the TME and spleen in mice. A, Schematic of lenvatinib therapy. Hepa1‐6 cells were in situ inoculated into C57BL/6 mice on day 0, lenvatinib was intragastrically administered daily when the tumor volume reached 150 mm3 on day 9, and tumor samples were collected for analysis on days 13 and 17. B, Proliferative activities (marked by expression of Ki‐67) of PD1+ CD8+ T cells from spleen and tumor in the negative control (NC) group and lenvatinib group were analyzed. Data were expressed as mean ± SEM (n = 5). C, Phenotypes of exhausted PD1+ CD8+ T cells (marked by expression of TCF1 and TIM3) from tumor in the NC group and the lenvatinib group were analyzed. Data were expressed as mean ± SEM (n = 5). D, The total number of tumor‐infiltrating CD8+ T cells and the number of pTex cells were detected. Data were expressed as mean ± SEM (n = 5). E, Phenotypes of exhausted PD1+ CD8+ T cells (marked by expression of TCF1 and Ki‐67) from spleen in the NC group and the lenvatinib group were analyzed. Data were expressed as mean ± SEM (n = 5). F, Tumor‐killing functions of intermediate Tex cells (marked by expression of granzyme B) from tumor in the NC group and the lenvatinib group were analyzed. Data were expressed as mean ± SEM (n = 5). *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001
In order to explore the specific effect of lenvatinib on PD1+ CD8+ T cells, we examined the overall expression of Ki‐67 in PD1+ CD8+ T cells in the lenvatinib group and found it was significantly higher than that in the control group (Figure 2B) both in the spleen and tumor. In the tumor, the proportion of pTex (PD1+ TCF1+ TIM3− CD8+ T cells) in the lenvatinib group increased significantly compared with the control group. Meanwhile, the proportion of terminal exhausted CD8+ T cells (PD1+ TCF1− TIM3+ CD8+ T cells) decreased (Figure 2C). Notably, there was no significant difference in the total number of tumor‐infiltrating CD8+ T cells between the two groups, while there was a significant difference in the number of pTex cells (Figure 2D). Similarly, the proportion of pTex in the lenvatinib group was increased compared with that in the control group in the spleens (Figure 2E).
To further explore the tumor‐killing function of the two groups of intermediate exhausted CD8+ T cells, we examined the expression of granzyme B and IFN‐γ in PD1+ TCF1− TIM3− CD8+ intermediate Tex in the lenvatinib group and found it was significantly higher than that in the control group (Figure 2F and Figure S1). Taken, together, it can be concluded that lenvatinib promotes the self‐proliferation and cytokine secretion of progenitor exhausted CD8+ T cells in the TME and spleen.
Given that lenvatinib has a direct impact on tumor cell growth and angiogenesis, we next aimed to explore whether lenvatinib has a direct effect on the proliferation and differentiation of progenitor exhausted CD8+ T cells. Considering the dose‐dependent effect of lenvatinib, we first studied the effects of different concentrations (0‐10 μM) of lenvatinib on CD8+ T cells and found that the maximum effect was achieved at the concentration of 5 μM, which was not significantly different from that at the concentration of 10 μM (Figure S4). Naive CD8+ T cells were isolated from the spleen and stimulated with anti‐CD3 and anti‐CD28 for 5‐7 days; then, they were treated with lenvatinib 24 hours (Figure 3A). In vitro, 5,6‐carboxyfluorescein diacetate, succinimidyl ester (CFSE) assay showed lenvatinib promoted the proliferation of CD8+ PD1+ T cells (Figure 3B). Furthermore, we observed that lenvatinib increased the proportion of pTex but decreased the proportion of tTex compared with the control group in the 5‐ and 7‐day groups (Figure 3C). At the same time, the expression of granzyme B in CD8+ TCF1− PD1+ TIM3− T cells (intermediate Tex in the lenvatinib group was significantly higher than that in the negative control (NC) group (Figure 3D), indicating lenvatinib directly increased the cell‐killing function of intermediate Tex and the proliferation and of CD8+ PD1+ T cells.
FIGURE 3.

Lenvatinib increased the proportion of progenitor exhausted CD8+ T cells in vitro. A, Schematic diagram of logical door frame selection of exhausted CD8+ T cells was listed. Naïve CD8+ T cells were continuously stimulated by CD3 and CD28 in vitro for 6 d and cocultured with lenvatinib for 48 h. B, Proliferative activities (marked by CFSE) of PD1+ CD8+ T cells from the NC group and the lenvatinib group were analyzed. Data were expressed as mean ± SEM (n = 5). C, Five‐day and seven‐day phenotypes of exhausted PD1+ CD8+ T cells (marked by expression of TCF1 and TIM3) from the NC group and the lenvatinib group were analyzed. Data were expressed as mean ± SEM (n = 5). D, Tumor‐killing functions of intermediate Tex cells (marked by expression of granzyme B) from the NC group and the lenvatinib group were analyzed. Data were expressed as mean ± SEM (n = 5). *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001
3.3. Lenvatinib affects PD1 + CD8 + T cell differentiation by affecting the PI3K/Akt/mTOR signaling pathway
MTOR is a major regulator of T cell metabolism. 19 To understand whether the proliferation differences in exhausted CD8+ T cells observed in the lenvatinib group and control group are related to changes in the mTOR signaling pathway, we used our RNA‐seq data to perform gene set enrichment analysis (GSEA) on Tex cells. Notably, there was a negative enrichment of genes involved in mTOR signaling in the control group (Figure 4A). Although both the Lenvatinib and NC groups showed expression of marker genes associated with the mTOR complex 1 (mTORC1) signaling pathway, the expression was significantly lower in the NC group (Figure 4B), suggesting that the mTORC1 pathway was expressed differently in the two groups. Some immune‐related genes (including tcf7) and pathways were also activated. Meanwhile, the expressions of TGF‐β1 and TGF‐β3 were decreased (Figure S2). In line with these data, pTex and tTex in the lenvatinib group displayed increased expression of p‐S6, the direct target of mTORC1 activity (Figure 4C). Ki‐67 was highly expressed in both groups in vitro, suggesting that the reduced mTORC1 activity was not the result of reduced Tex cell activation status (Figure 4D).
FIGURE 4.
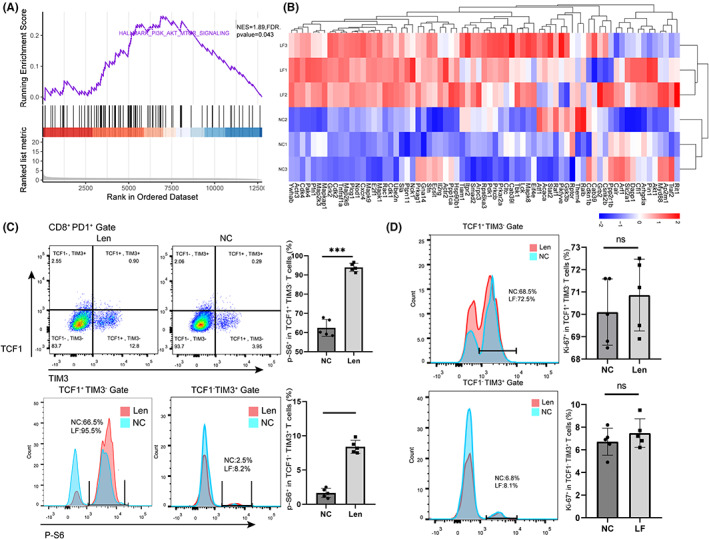
Lenvatinib affects CD8+ T cell differentiation by affecting the PI3K/Akt/mTOR signaling pathway. A, GSEA barcode plot of the mTORC1 pathway in the NC group and the lenvatinib group derived from cultured cells in vitro. B, Heatmap of relative expression (Z score) of the top 50 leading‐edge genes from GSEA of the mTORC1 signaling pathway. C, Phosphorylations of the mTOR pathway (marked by expression of p‐S6) of pTex and tTex cells in the NC group and the lenvatinib group were analyzed. Data were expressed as mean ± SEM (n = 5). D, pTex and tTex cells’ activation status (marked by expression of Ki‐67), in the NC group and lenvatinib group and derived from cultured cells in vitro, was analyzed. Data were expressed as mean ± SEM (n = 5). *p < 0.05; **p < 0.01
3.4. Blockade of the mTOR pathway undermined the antitumor effect of lenvatinib and the function of PD1 + CD8 + T cells
Mice were divided into NC group, lenvatinib group, rapamycin group, and combination group according to the treatment of the four groups. We found that lenvatinib significantly inhibited tumor growth and prolonged survival in mice when compared with other groups. However, compared with the lenvatinib group, the combination group was not better at controlling tumor size or extending survival (Figure 5A,B). Rapamycin, which blocks the mTOR pathway in tumor cells, also blocks the mTOR pathway in T cells. In fact, in vivo, we did not find a significant difference in tumor size between the combination and lenvatinib groups. At the same time, we conducted the same experiment in CD8+ T cell–deleted mice and RAG1 mice (Figure S7). The results showed that lenvatinib and rapamycin could still reduce tumor weight in RAG1 or CD8+ T cell–deleted mice, and unlike in WT group, the tumor weight in the combined group was smaller than that in the lenvatinib group, especially in CD8+ T cell–deleted mice. This suggests that rapamycin can act on both tumor cells and CD8+ T cells, by inhibiting the proliferation of both tumor cells and Tex, and explains why there was no significant difference in tumor weight between the combined treatment group and the lenvatinib group in WT mice.
FIGURE 5.

Blockade of the mTOR pathway undermined the antitumor effect of Len and function of PD1+ CD8+ T cells. A, All tumors were harvested, and the tumor appearance and weight were compared between the groups. Data are shown as mean ± SEM (n = 3‐5). B, Survival curve analysis of four groups of mice. C, Phenotypes of exhausted PD1+ CD8+ T cells (marked by expression of TCF1 and TIM3) from tumor in four groups were analyzed. Data were expressed as mean ± SEM (n = 5). D, Proliferative activities (marked by expression of Ki‐67) of pTex and tTex cells from tumor in four groups were analyzed. Data were expressed as mean ± SEM (n = 5). E, Phenotypes of exhausted PD1+ CD8+ T cells (marked by expression of TCF1 and TIM3) from four groups cultured in vitro were analyzed. Data were expressed as mean ± SEM (n = 5). F, Proliferative activity (marked by CFSE) of PD1+ CD8+ T cells from four groups cultured in vitro were analyzed. Data were expressed as mean ± SEM (n = 5). G, Tumor‐killing function of intermediate Tex cells (marked by expression of granzyme B) from four groups cultured in vitro were analyzed. Data were expressed as mean ± SEM (n = 5). *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001
To further confirm our idea, we measured the proportions of progenitor exhausted CD8+ T cells in the four groups and found that combination of lenvatinib and rapamycin weakened the effect of lenvatinib on PD1+ CD8+ T cells (Figure 5C). At the same time, compared with pTex in the lenvatinib group, the expression of Ki‐67 was significantly decreased in the combined treatment group (Figure 5D).
In vitro, we evaluated the proliferation and proportion of progenitor exhausted CD8+ T cells upon anti‐CD3 and anti‐CD28 stimulation in the four groups, and the phosphorylations of the mTOR pathway were compared. We found that the phosphorylation of the mTOR pathway was activated in the lenvatinib group and weakened by rapamycin (Figure S5). CFSE assay showed that the proliferation was reduced by rapamycin processing and the effect of lenvatinib on exhausted CD8+ T cells was also weakened by rapamycin (Figure 5E,F). Meanwhile, lenvatinib‐induced promotion of granzyme B expression was inhibited when treated with rapamycin (Figure 5G). Through in vitro and in vivo experiments, we confirmed that the inhibition of the mTOR pathway attenuates the effect of lenvatinib on PD1+ CD8+ T cells.
3.5. Lenvatinib affects the PI3K/Akt/mTOR signaling pathway by blocking VEGFR2
Considering that lenvatinib is a multitarget inhibitor, we conducted in vitro experiments with antibodies targeted at each target to determine through which target lenvatinib affects the mTOR pathway (Figure 6A). We found that there was no significant diversity in exhausted CD8+ T cell differentiation and p‐S6 expression between BIBF1120 and lenvatinib, and other blockers had no obvious effect on PD1+ CD8+ T cells (Figure 6B). According to previous reports, continuous antigenic stimulation induces VEGFR1 and VEGFR2 expression in CD8+ T cells. To further identify which target affects the mTOR pathway, we investigated the VEGFR1 and VEGFR2 in CD8+ T cells at different stages in vitro. With the exhaustion of CD8+ T cells, the expression of VEGFR2 increased, while VEGFR1 expression was not significantly altered (Figure 6C). We first performed small interfering RNA–mediated knockdown to deplete VEGFR2 in CD8+ T cells (Figure S6). Then, VEGFR2‐specific blocker SU5408 was applied to further validate the results. The expression level of p‐S6 in pTex and tTex cells was increased in the si‐VEGFR2 group and SU5408 group, while it was decreased in the VEGF‐A group (Figure 6D). Through these experiments, we found that lenvatinib affects the mTOR pathway through VEGFR2.
FIGURE 6.
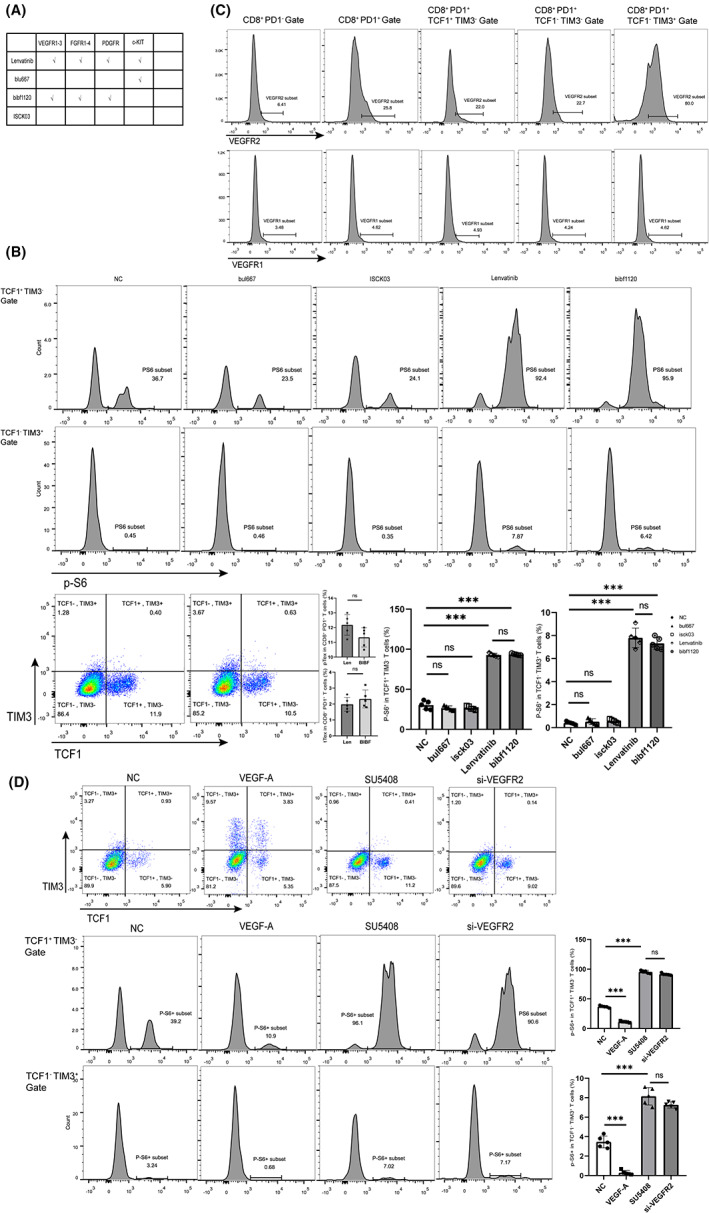
Lenvatinib affects the PI3K/Akt/mTOR signaling pathway by blocking VEGFR2. A, Schematic diagram of the target of each drug. B, Phosphorylations of the mTOR pathway (marked by expression of p‐S6) of pTex and tTex cells in each group were analyzed. Data were expressed as mean ± SEM (n = 5). C, In the different stages of CD8+ T cells, the expression of VEGFR2 was analyzed. Data were expressed as mean ± SEM (n = 5). D, Phosphorylations of the mTOR pathway (marked by expression of p‐S6) of pTex and tTex T cells in each group were analyzed. Data were expressed as mean ± SEM (n = 5). *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001
4. DISCUSSION
Immunotherapy that affects tumor‐specific T cells by blocking the PD1/PD‐L1 axis has emerged as a treatment for HCC. However, only 15%‐20% response rate was observed in HCC patients treated with immune checkpoint blockade, and most patients were drug resistant. So, we are considering how we can improve patients’ response to immunotherapy. A large number of studies have found that the reactivity of targeted immunotherapies was mainly related to tumor‐infiltrating Tcf1+ PD1+ CD8+ T cells with progenitor properties. Among the reported effects of lenvatinib, we wondered whether lenvatinib might also affect Tcf1+ PD1+ CD8+ T cells, thereby improving the efficacy of targeted immunotherapy. In this study, we explored the proportion and functional changes of progenitor exhausted and terminal exhausted cell subsets in spleen and tumor‐infiltrating CD8+ T cells during the progression of liver cancer and the effect of lenvatinib on tumor‐infiltrating exhausted CD8+ T cells and the differentiation of naive CD8+ T cells in the spleen. We demonstrated that with the progression of HCC, the proportion and proliferation capacity of progenitor exhausted CD8+ T cells will gradually decrease. However, lenvatinib reversed this trend and promoted the expansion of Tcf1+ PD1+ CD8+ T cells in tumors and spleens by exciting the PI3K/Akt/mTOR signaling pathway. The Ki‐67‐positive rate of exhausted CD8+ T cells in spleen was also significantly increased. Moreover, we supported our findings in vitro and in vivo: Blockade of the PI3K/Akt/mTOR signaling pathway inhibited progenitor exhausted CD8+ T cell proliferation, and combined lenvatinib and rapamycin attenuated the effect of lenvatinib on promoting the proliferation of progenitor exhausted CD8+ T cells. Collectively, our data suggest that lenvatinib's ability to activate mTOR in exhausted CD8+ T cells provides the rationale for improving the reactivity of immunotherapy, in addition to its inhibitory activity against the VEGFR2 and FGFR signaling pathways.
Cell proliferation–related signal transduction pathway PI3K/Akt/mTOR is involved in regulating various cell proliferation and apoptosis functions. Abnormally elevated mTOR activity is often observed in human malignancies. Therefore, in the clinical treatment of liver cancer, mTOR blockade is considered to be an effective treatment method and has achieved significant clinical effects. 20 However, PI3K/Akt/mTOR also plays a critical role in the adaptive immune system. Phosphorylation and activation of PI3K/Akt/mTOR affects cell cycle, cell function, cell differentiation, and apoptosis by mediating downstream signals and has a significant impact on immune cell subgroup differentiation. Previous studies have shown that the PI3K/Akt/mTOR pathway can affect IFN‐γ secretion and memory CD8+ T cell production. 21 , 22
In our study, we found that the PI3K/Akt/mTOR pathway also affected the proliferation and differentiation of exhausted CD8+ T cells. By stimulating the mTOR pathway, lenvatinib promoted the proliferation of TCF1+ PD1+ CD8+ T cells and eventually differentiated into more TCF1− PD1+ CD8+–exhausted T cells with tumor‐killing function. In particular, after the effect of lenvatinib on exhausted CD8+ T cells was blocked by rapamycin, the proportion of progenitor exhausted CD8+ T cells recovered to a similar level to that of the control group.
We also compared the function of exhausted CD8+ T cells and found that lenvatinib influenced both phenotype and function of exhausted CD8+ T cells. The expression level of granzyme B in the lenvatinib group was significantly higher than that in the control group. A recent study found that CD101− CD8+ PD1+ TCF1− T cells are a transient state in stem‐like exhausted CD8+ T cells that eventually differentiate into terminal exhausted CD8+ T cells, and these newly generated TIM3− CD101− CD8+ PD1+ TCF1− T cells were more functional than terminal exhausted CD8+ T cells (TIM3+ CD101+ CD8+ PD1+ TCF1− T cells). To further understand the effects of lenvatinib on exhausted CD8+ T cells, we also investigated its phenotypic changes through CD101 (Figure S3). We confirmed that lenvatinib also decreased the proportion of CD101+ TIM3+ tTex cells, which was also associated with greater tumor control.
Lenvatinib has previously been implicated in the immunosuppressive and angiogenic effects on tumor microenvironment by inhibiting the VEGFA/VEGFR pathway. However, it has remained unknown whether lenvatinib affects exhausted CD8+ T cell differentiation and function. Here, we found that lenvatinib promotes the development and differentiation of exhausted CD8+ T cells by activating the PI3K/Akt/mTOR signaling pathway via inhibiting the VEGFA/VEGFR pathway. However, mTOR activity is regulated by a multitude of factors, including nutrients, oxygen, and growth factors, and whether lenvatinib activates the mTOR pathway merely through VEGFR2 deserves further investigation. 23 Confusingly, as opposed to it is function in PD1+ CD8+ T cells, VEGFR2 promotes the mTOR pathway in the tumor. We suspect that VEGFR2 inhibits the mTOR pathway by stimulating TGF‐β expression. In line with this hypothesis, researchers found that TGF‐β could suppress the activation of the mTOR signaling pathway, 24 and we found the expression of TGF‐β in exhausted CD8+ T cells was also inhibited in the lenvatinib group (Figure S2). In addition, lenvatinib's function in other immune cells has not been fully researched. The phenomenon observed in animal studies may not be caused by CD8+ T cells alone but also by other types of cells, including Treg and antigen‐presenting cells, and it will be important to identify subset‐specific regulators of the mTOR activity.
Finally, immune checkpoint blockade therapy is a promising approach that can increase the number of exhausted T cells and improve their function; it may also lead to a decreased durability of the T cell response. Excessive immunotherapy can overdraw the potential of CD8+ T cells. By accelerating the differentiation of pTex cells into intermediate exhausted CD8+ T cells, the long‐term control mechanism of the immune system on the tumor will be broken. Blindly applying immunotherapy in the late stage of tumor progression will undoubtedly promote tumor progression. In advanced live cancer, using drugs to promote pTex rather than promote its differentiation is a smarter strategy. Recently phase III clinical trials in HCC have demonstrated that lenvatinib in combination with pembrolizumab is not significantly different to lenvatinib monotherapy. Our study revealed the dysfunction of tTex in the tumor microenvironment uncovered the mechanism of inefficacy of combination strategy, and provided a potential solution with the mTOR pathway.
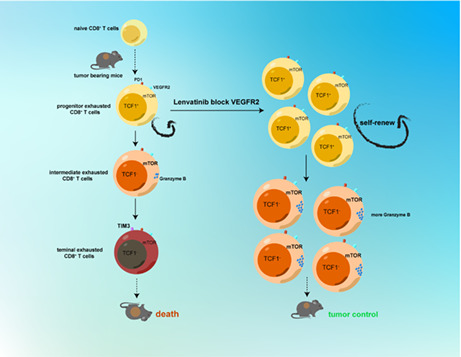
Overall, this study allows us to propose the concept that lenvatinib can activate the mTOR pathway in target cells, including exhausted CD8+ T cells. 25 Lenvatinib has been found to inhibit the TGF‐β pathway, 24 , 26 which may be the mechanism of indirect activation of the MTOR pathway. Or, as in NK cells, it directly activates the mTOR pathway by inhibiting the C‐kit pathway. 27 Our findings have potential implications for combinatory immunotherapy and target agents in HCC and other anti‐PD1–resistant cancers and reveal that lenvatinib suppresses tumor progression not only by anti‐immunosuppressive and anti‐angiogenic effects but also by affecting the mTOR pathway of exhausted T cells via VEGFR2 (Figure 7).
FIGURE 7.
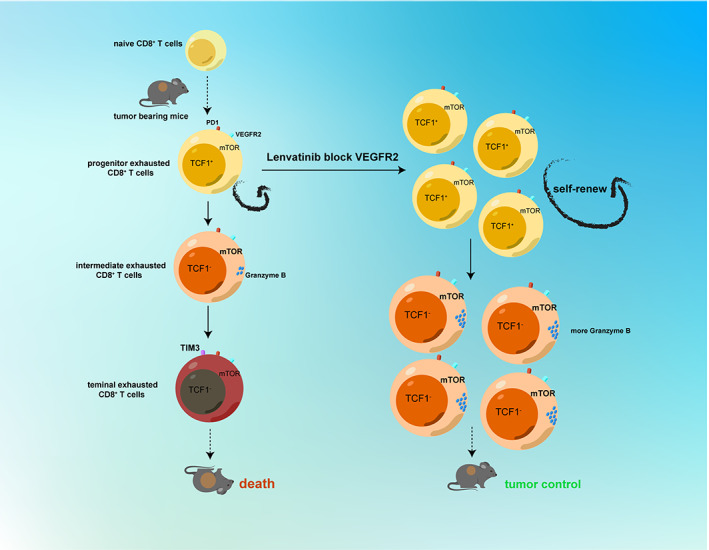
Lenvatinib promotes the phosphorylation of the mTOR pathway by blocking VEGFR2 of PD1+CD8+ T cells and enhances the tumor‐killing function of intermediate exhausted CD8+ T cells and amplification of pTex subsets. Schematic diagram showing that lenvatinib enhances production of progenitor exhausted T cells and then pTex produce more intermediate exhausted T cells with tumor‐killing function by promoting the phosphorylation of the mTOR pathway via blocking VEGFR2
FUNDING INFORMATION
This work was supported by the Health Commission of Zhejiang Province under Grant [JBZX‐202004]; Research Unit Project of Chinese Academy of Medical Science under Grant [2019‐I2M‐5‐030]; and Research Project of Jinan Microecological Biomedicine Shandong Laboratory under Grant [JNL‐2022002A, JNL‐2022008B].
DISCLOSURE
The authors have no conflict of interest to disclose.
ETHICS STATEMENT
Approval of the research protocol by an Institutional Review Board: N/A.
Informed consent: The present study was approved by the Ethical Committee of the First Affiliated Hospital of Zhejiang University School of Medicine (20191421), and written informed consent was obtained from all patients.
Registry of the study: N/A.
Animal Studies: The present study was approved by the Ethical Committee of the First Affiliated Hospital of Zhejiang University School of Medicine (20221145).
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
Data S1
ACKNOWLEDGMENTS
We thank xingxing gao for the data collation and the First Affiliated Hospital of Zhejiang University for the tumor samples.
Mei Z, Gao X, Pan C, et al. Lenvatinib enhances antitumor immunity by promoting the infiltration of TCF1 + CD8 + T cells in HCC via blocking VEGFR2 . Cancer Sci. 2023;114:1284‐1296. doi: 10.1111/cas.15719
Contributor Information
Lin Zhou, Email: zhoulin99@zju.edu.cn.
Shushen Zhen, Email: shusenzheng@zju.edu.cn.
DATA AVAILABILITY STATEMENT
All data needed to evaluate the conclusions in the paper are presented in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
REFERENCES
- 1. Siddiqui I, Schaeuble K, Chennupati V. Intratumoral Tcf1+PD‐1+CD8+ T cells with stem‐like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity. 2019;50(1):195‐211.e10. [DOI] [PubMed] [Google Scholar]
- 2. Shan Q, Sheng'en H, Chen X. Ectopic Tcf1 expression instills a stem‐like program in exhausted CD8+ T cells to enhance viral and tumor immunity. Cell Mol Immunol. 2021;18(5):1262‐1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pollizzi KN, Sun IH, Patel CH. Asymmetric inheritance of mTORC1 kinase activity during division dictates CD8(+) T cell differentiation. Nat Immunol. 2016;17:704‐711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960‐976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jiang W, Yinjun W. Exhausted CD8+ T cells in the tumor immune microenvironment: new pathways to therapy. Front Immunol. 2020;11:622509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Miller BC, Sen DR, al Abosy R, et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. 2019;20:326‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pauken KE, Sammons MA, Odorizzi PM. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD‐1 blockade. Science. 2016;354(6316):1160‐1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kallies A, Zehn D, Utzschneider DT. Precursor exhausted T cells: key to successful immunotherapy? Nat Rev Immunol. 2020;20(2):128‐136. [DOI] [PubMed] [Google Scholar]
- 9. Im SJ, Hashimoto M, Gerner MY, et al. Defining CD8+ T cells that provide the proliferative burst after PD‐1 therapy. Nature. 2016;537:417‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8+ T cells by αPD‐L1 blockade. Proc Natl Acad Sci. 2008;105:15016‐15021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Utzschneider DT, Charmoy M, Chennupati V, et al. T cell factor 1‐expressing memory‐like CD8+ T cells sustain the immune response to chronic viral infections. Immunity. 2016;45:415‐427. [DOI] [PubMed] [Google Scholar]
- 12. Li H, van der Leun AM, Yofe I, et al. Dysfunctional CD8+ T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell. 2019;7(4):775‐789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tohyama O, Matsui J, Kodama K. Antitumor activity of lenvatinib (e7080): an angiogenesis inhibitor that targets multiple receptor tyrosine kinases in preclinical human thyroid cancer models. J Thyroid Res. 2014;2014:638747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ogasawara S, Mihara Y, Kondo R. Antiproliferative effect of lenvatinib on human liver cancer cell lines in vitro and in vivo. Anticancer Res. 2019;39:5973‐5982. [DOI] [PubMed] [Google Scholar]
- 15. Okamoto K, Kodama K, Takase K. Antitumor activities of the targeted multi‐tyrosine kinase inhibitor lenvatinib (E7080) against RET gene fusion‐driven tumor models. Cancer Lett. 2013;340:97‐103. [DOI] [PubMed] [Google Scholar]
- 16. Zhao Y, Zhang YN, Wang KT. Lenvatinib for hepatocellular carcinoma: from preclinical mechanisms to anti‐cancer therapy. Biochim Biophys Acta rev. Cancer. 2020;1874(1):188391. [DOI] [PubMed] [Google Scholar]
- 17. Voron T, Marcheteau E, Pernot S, Colussi O. Control of the immune response by pro‐angiogenic factors. Front Oncol. 2014;4:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qinchuan W, Zhou W, Yin S. Blocking triggering receptor expressed on myeloid Cells‐1‐positive tumor‐associated macrophages induced by hypoxia reverses immunosuppression and anti‐programmed cell death ligand 1 resistance in liver cancer. Hepatology. 2019;70(1):198‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rostamzadeh D. mTOR signaling pathway as a master regulator of memory CD8+ T‐cells, Th17, and NK cells development and their functional properties. J Cell Physiol. 2019;234(8):12353‐12368. [DOI] [PubMed] [Google Scholar]
- 20. Schnitzbauer AA, Filmann N, Adam R, et al. mTOR inhibition is most beneficial after liver transplantation for hepatocellular carcinoma in patients with active tumors. Randomized controlled trial. Ann Surg. 2020;272(5):855‐862. [DOI] [PubMed] [Google Scholar]
- 21. Araki K. mTOR regulates memory CD8+ T‐cell differentiation. Nature. 2009;460(7251):108‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Macintyre AN, Finlay D. Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is dispensable for T cell metabolism. Immunity. 2011;34:224‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang H, Long L, Zhou P. mTOR signaling at the crossroads of environmental signals and T‐cell fate decisions. Immunol Rev. 2020;295(1):15‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gabriel SS, Tsui C. Transforming growth factor‐b‐regulated mTOR activity preserves cellular metabolism to maintain long‐term T cell responses in chronic infection. Immunity. 2021;54:1698‐1714. [DOI] [PubMed] [Google Scholar]
- 25. Hutson TE, Michaelson MD, Kuzel TM, et al. A single‐arm, multicenter, phase 2 study of Lenvatinib plus Everolimus in patients with advanced non‐clear cell renal cell carcinoma. Eur Urol. 2021;80(2):162‐170. [DOI] [PubMed] [Google Scholar]
- 26. Torrens L. Immunomodulatory effects of Lenvatinib plus anti–programmed cell death protein 1 in mice and rationale for patient enrichment in hepatocellular carcinoma. Hepatology. 2021;74(5):2652‐2669. [DOI] [PubMed] [Google Scholar]
- 27. Bösken B, Hepner‐Schefczyk M. An inverse relationship between c‐kit/CD117 and mTOR confers NK cell dysregulation late after severe injury. Front Immunol. 2020;11:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
Data S1
Data Availability Statement
All data needed to evaluate the conclusions in the paper are presented in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.