

Abstract
The identification of new effective therapeutic options for non-small-cell lung cancer (NSCLC) represents a crucial challenge in oncology. Recent studies indicate that cancer-associated fibroblasts (CAFs) participate in tumor progression by establishing a favorable microenvironment that promotes cancer progression. Therefore, the development of strategies inhibiting the interplay between CAFs and cancer cells is considered a winning approach for the development of effective anti-cancer drugs. Among other factors, the signal transducer and activator of transcription-3 (STAT3) has been reported as a key mediator of CAF oncogenic actions, representing a promising therapeutic target. Here, we applied an aptamer-based conjugate (named Gint4.T-STAT3), containing a STAT3 siRNA linked to an aptamer binding and inhibiting the platelet-derived growth factor receptor (PDGFR)β, to obtain STAT3-specific silencing and interfere with CAF pro-tumorigenic functions. We demonstrated that this molecule effectively delivers the STAT3 siRNA in NSCLC cells, and blocks CAF-induced cancer cell growth and migration and reduced spheroid dimension. In addition, we found that Gint4.T-STAT3 alters CAF phenotype, thus functioning as a double-acting molecule able to inhibit the entire tumor bulk. Our data provide a proof of principle for the targeting of CAF pro-tumor functions through an aptamer-based drug, and can open innovative horizons in NSCLC therapy.
Keywords: MT: Oligonucleotides: Therapies and Applications, aptamer, siRNA, chimeras, NSCLC, CAFs, STAT3, targeted delivery
Graphical abstract
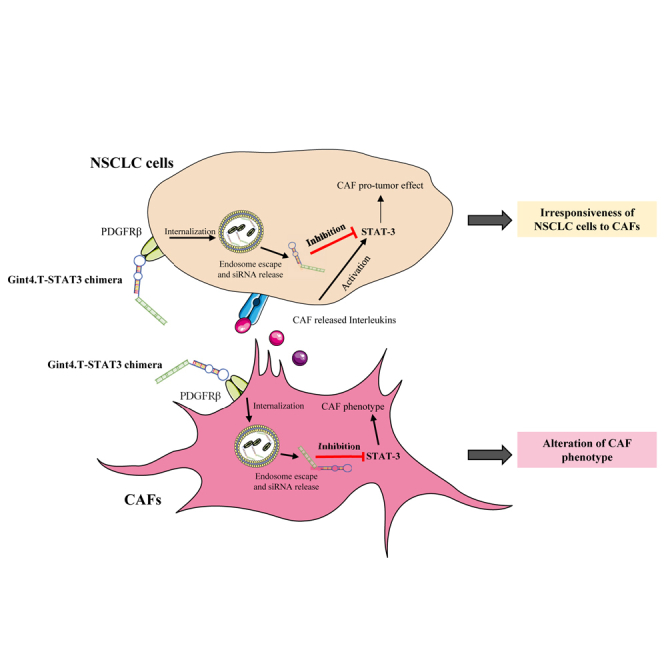
Cancer-associated fibroblasts (CAFs) are key mediators of tumor progression. This manuscript describes an innovative strategy to improve the treatment of non-small-cell lung cancer (NSCLC) patients that is a novel aptamer-based bio-drug inhibiting the interplay between cancer cells and CAFs.
Introduction
Lung cancer is the leading cause of cancer-related death in the world, with non-small-cell lung cancer (NSCLC) representing approximately 80% of all lung cancers. Patients with NSCLC have a median survival time after diagnosis generally less than 1 year, and often show tumor relapses due to drug resistance.1
An important limit of conventional anti-cancer strategies is that they do not consider the presence of a complex tumor microenvironment (TME), which is recognized to play a crucial role in tumor progression.2 Key components of the TME are cancer-associated fibroblasts (CAFs). CAFs are proliferative heterogeneous cells with a myofibroblast phenotype overexpressing specific biomarkers, such as α-smooth muscle actin (α-SMA) and fibroblast activation protein (FAP). They are able to regulate malignant progression and drug resistance through the secretion of various growth factors, cytokines, matrix remodeling proteins, and through the induction of the epithelial-mesenchymal transition in several cancer types, including NSCLC.3 Therefore, the development of therapeutic molecules hampering the interactions between tumor cells and CAFs, while preserving normal cells, offers the possibility to enhance the effectiveness of the treatment and the patient outcome.
Despite the mechanisms of tumor-CAF crosstalk are still to be uncovered, some candidate players have recently emerged. Among them, the activation of the signal transducer and activator of transcription-3 (STAT3) has been reported as a key point of convergence in CAF-tumor cell interplay. STAT3 acts as a pro-oncogenic factor in different cancers, including NSCLC, controlling the expression of specific genes involved in cell cycle progression, cell survival, angiogenesis, and immune response regulation.4 Recent studies revealed that STAT3 activation in tumor cells occurs via the CAF release of interleukins (ILs), as IL6 and IL11, and mediates CAF-induced tumor cell proliferation, migration, and drug resistance.5,6,7,8 These data suggest that STAT3 inhibition could render NSCLC cells irresponsive to CAF pro-tumor signals. STAT3 silencing in cancer cells by the use of a specific small interfering RNA (siRNA) has been addressed.9 However, a major challenge for the development of safe and reliable RNA-based therapeutics is the design of selective delivery strategies able to restrict their therapeutic action on target tumor cells, strongly reducing the occurrence of unwanted off-target effects. To address this issue, we and others have developed siRNA delivery targeted systems based on the use of nucleic acid aptamers as carriers.10,11,12,13 Aptamers are short oligonucleotides, which bind with high affinity and specificity to their target by acquiring a peculiar structured folding.14 They are very promising as antagonists of cancer-associated proteins and as selective carriers for the delivery of secondary reagents to target cells.15,16,17 Specifically, it has been shown that aptamer conjugates may internalize in a receptor-mediated manner often through the natural receptor recycling process. Despite the mechanism of conjugate processing within the cells has not been completely clarified, it has been demonstrated that once internalized aptamer-siRNA construct (AsiC) may escape from the endosome and be processed by the interference RNA machinery allowing the efficient release of the siRNA cargo.18
In the present work, we applied a previously characterized STAT3 targeting AsiC, named Gint4.T-STAT3,10,11 to alter CAF-NSCLC cell crosstalk. The AsiC contains a STAT3 siRNA linked to a cell-targeted and therapeutic aptamer (Gint4.T) binding and inhibiting the platelet-derived growth factor receptor (PDGFR)β,19 overexpressed in many tumors, including NSCLC.20
We found that NSCLC cell treatment with the conjugate effectively reduces STAT3 levels and renders the cells not responsive to the pro-tumor effects of primary CAFs on cell growth and migration. Further, we revealed a good efficacy of the conjugate to alter key CAF functions as well, and impair three-dimensional (3D) spheroids derived from NSCLC cells or CAF-NSCLC hybrid cultures, reducing their dimension.
This study represents the first proof of principle demonstrating the efficacy of an AsiC in inhibiting NSCLC-CAF interplay. It provides new insight in the therapeutic applicability and efficacy of STAT3 pathway targeting in NSCLC, offering a new therapeutic tool with the potential to target both CAFs and tumor cells.
Results
CAF primary cultures isolated from human NSCLC tissues
We isolated primary cultures of cancer fibroblasts from two NSCLC patient tissues (indicated as #16 and #17). Patient-derived CAF cultures were first characterized for the expression of epithelial (E-cadherin) and fibroblast (fibronectin and vimentin) markers, whose levels indicated that these cells show a fibroblast phenotype (Figures S1A–S1C). We then analyzed the expression of the activated myofibroblast marker α-SMA and FAP. Results were compared with normal fibroblasts (NFs) derived from the human tissues (at least 5 cm away from the tumor). As expected, we found higher mRNA levels of these markers in CAFs with respect to NFs (Figure 1A). Marker expression was confirmed by confocal microscopy comparing CAFs with primary NFs and commercial normal human lung fibroblast cells, WI-38 (Figure 1B). Further, we analyzed CAF pro-tumor effects on NSCLC cell viability and migration in vitro. To this end, Calu-1 NSCLC cells were grown in conditioned medium (CM) derived from CAFs or NFs and cell viability or migration were measured. We found that CAF CM significantly induced Calu-1 NSCLC cell survival (Figure 1C) and migration (Figure 1D). No significant effect was instead detected in the presence of CM from primary NFs. Further, in accordance with already published results,5,6,7,8 reporting that IL6 and 11 released by CAFs induce STAT3 activation, we observed that CAF CM, but not NF-CM, increases STAT3 phosphorylation levels in NSCLC cells (Figure S1D), confirming this transcription factor as a key mediator of CAF-tumor cells interplay. Accordingly, higher levels of IL6 and 11 were found in the CM derived from CAFs, as compared with that from normal human lung fibroblast cells (Figure S1E).
Figure 1.

CAF primary cultures
(A and B) Levels of CAF markers FAP and α-SMA were measured by RT-qPCR (A) or confocal microscopy (B) in the indicated cell lines. Scale bar in (B), 50 μm. (C) Calu-1 cells were grown in CAF or NF CMs and cell viability was measured following 72 h. (D) Calu-1 cells were grown in 1% FBS medium (−) or in 1% FBS CAF or NF CMs for 24 h and then migration was analyzed by Boyden chambers (8-μm pore size) using the correspondent medium as chemoattrattant. Left panels, representative photographs of migrated cells are shown; right panel, migrated cells were quantified and expressed relative to cells grown in 1% FBS. In (A), (C), and (D), error bars are standard deviation (SD) values (n = 2). Statistics by t test are reported: ∗p < 0.05; ∗∗∗p < 0.001.
Collectively, these data show that CAFs derived from primary cell cultures will retain features and properties of CAFs.
Delivery of STAT3 siRNA in NSCLC cells
In order to interfere with CAF pro-tumor effects, we addressed STAT3 inhibition in NSCLC cells by using a multifunctional complex (Gint4.T-STAT3) containing the Gint4.T aptamer, a PDGFRβ antagonist,19 linked to a STAT3 siRNA (Figure 2A). This conjugate has been previously generated by us conjugating the 34-mer Gint4.T aptamer to a 25-mer STAT3 siRNA through the extension of the 3′-ends of the aptamer and the siRNA antisense strand with complementary sticky sequences of 17 nucleotides. We previously proved that this conjugate specifically binds and delivers STAT3 siRNA to PDGFRβ-positive glioblastoma cells without affecting PDGFRβ-negative cells and producing upon treatment a silencing efficacy comparable to those obtained upon siRNA transfection.10,11 In addition, the complex is highly stable in human serum, remaining undigested up to 24 h in 85% serum.10 As a first attempt, we analyzed whether the AsiC efficiently delivered the STAT3 siRNA in NSCLC cells. To this end, Calu-1 NSCLC cells overexpressing PDGFRβ aptamer target (Figure S2), were treated with Gint4.T-STAT3 AsiC at 400 nM concentration basing on our previous report10,11 without any transfection reagents. Following treatment, the analyses of STAT3 expression level was performed by both quantitative reverse transcription polymerase chain reaction (RT-qPCR) and immunoblotting. Obtained results revealed that the conjugate treatment gives an efficient down-regulation of STAT3 both at mRNA and protein levels (Figures 2B and 2C). Notably, the levels of STAT3 were not affected in cells treated with a control complex containing an unrelated scrambled aptamer linked to siSTAT3 (CtrlApt-STAT3). Further, silencing efficacy was comparable to those obtained upon siRNA transfection and no effect was detected upon conjugate treatment of no/low PDGFRβ expressing A549 cells,19 confirming AsiC specificity (Figure 2D).
Figure 2.

Gint4.T-mediated delivery of STAT3 siRNA in NSCLLC cells
(A) Scheme of Gint4.T-STAT3 conjugate. (B and C) Calu-1 NSCLC cells (PDGFRβ+) were left untreated (NT) or treated with 400 nmol/L Gint4.T, Gint4.T-STAT3 or control chimera (CtrlApt-STAT3) or transfected with STAT3 siRNA duplex (siSTAT3) for 72 h as indicated. STAT3 mRNA (B) or protein (C) levels were analyzed by RT-qPCR or immunoblotting, respectively. Ctrl indicates the control with lipofectamine-2000 alone. In (B), error bars depict the mean ± SD (n = 2). (D) A549 NSCLC cells (PDGFRβ-) were left untreated (NT) or treated with 400 nmol/L aptamers or conjugates or transfected with STAT3 siRNA duplex (siSTAT3) for 72 h. STAT3 protein levels were analyzed by immunoblotting. In (C) and (D), Vinculin antibody was used as protein loading control. Treat: Treatment; Transf.: transfection. Statistics were calculated by t test (vs. NT): ∗p < 0.05.
These results confirm Gint4.T-STAT3 ability to effectively deliver STAT3 siRNA in PDGFRβ-positive NSCLC cells.
Gint4.T-STAT3 inhibits NSCLC cell viability and migration induced by CAF CM
Next, we investigated whether AsiC-dependent STAT3 inhibition rendered NSCLC cells unresponsive to CAF pro-tumor functions. To this end, we evaluated the effects of CM from primary CAF#16 and #17 on Calu-1 NSCLC treated with the AsiC. First, we checked the effect of CAF CM on the expression of the aptamer target that is required for the conjugate function and we found that CAF CM enhance PDGFRβ levels in Calu-1 NSCLC cells (Figure S3). Therefore, Calu-1 NSCLC cells were grown in CM from either CAF#16 or #17 in the absence or in the presence of 400 nM Gint4.T aptamer, Gint4.T-STAT3, or CtrlApt-STAT3 control complex. First, the levels of phospho-STAT3 and STAT3 were analyzed by immunoblotting (Figure 3A). As expected, both CAF-derived CM induced the activation of STAT3 as assessed by the significant increase of STAT3 tyrosine phosphorylation. Notably, Gint4.T-STAT3 AsiC treatment hampered this induction by reducing STAT3 levels. The efficacy of silencing was comparable to those obtained upon AsiC NSCLC treatment in normal medium (Figure 2C), with slight differences likely due to the different experimental protocol.
Figure 3.

Gint4.T-STAT3 inhibits NSCLC cell viability and migration induced by CAF CM
(A and B) Calu-1 cells were grown in 1% FBS medium (−) or in 1% CM from CAF#16 or #17 for 72 h in the absence (NT) or in the presence of 400 nmol/L Gint4.T, Gint4.T-STAT3, or CtrlApt-STAT3. (A) Cell lysates were analyzed by immunoblot with pSTAT3 and STAT3 antibodies. βActin or Vinculin antibodies were used as loading control. (B) Cell viability was measured and expressed relative to cells grown in 1% FBS medium (−). Error bars depict SD values (n = 2). (C–F) Calu-1 cells were grown in 1% FBS medium (−) or in 1% FBS CAF CMs for 24 h and then allowed to migrate using using Boyden chambers (8 μm pore size) uncoated (C and D) or coated with matrigel (E and F) using the correspondent medium as chemoattrattant. (C and E) Representative photographs are shown. (D and F) The percentage of migrated cells expressed relative to cells grown in 1% FBS medium (−) is reported. Error bars are SD values (n = 2). In (B), (D), and (F), statistics by t test are reported: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗∗p < 0.0001.
Then, we looked at the functional effects on NSCLC cell viability and migration. Cell viability of NSCLC Calu-1 cells grown in CM from either CAF#16 or #17 in the absence or in the presence of Gint4.T-STAT3 or controls was monitored by MTT analyses. We found that Calu-1 cells grown in CAF CM showed a significant increase in cell viability that is completely blocked by the treatment with Gint4.T-STAT3, as compared with the cells left untreated or treated with the control complex (Figure 3B). In addition, we analyzed cancer cell migration by transwell assay. To this end, Calu-1 cells were treated with either CAF#16 or #17 CM in the absence or in the presence of aptamer or conjugates for 24 h and then cell migration was monitored by using the corresponding CAF CM as chemoattractant stimulus. As shown (Figures 3C and 3D), the boosted effects of both CAF-derived CMs on NSCLC cell migration ability was significantly interfered by Gint4.T-STAT3 treatment. Indeed, Calu-1 migration rate increased with CAF#16 and #17 CM, as compared with the control medium, while remained closer to the basal when cells were treated with Gint4.T-STAT3. No changes were found, instead, in the presence of the control complex. In accordance with the already described Gint4.T functional ability,10,11,19 a reduction of about 25% was detected when cells were treated with the aptamer alone. This reduction was further enhanced up to 40% to 50% in the context of the AsiC, in which the aptamer function is combined with STAT3 siRNA effects. In addition, we found that the Gint4.T-STAT3 also strongly suppresses cell invasion ability induced by CAFs enhancing aptamer inhibitory action (Figures 3E and 3F).
Taken together, these data indicate that, by inhibiting STAT3, Gint4.T-STAT3 interferes with the enhanced viability and migration mediated by CAF-derived CM in NSCLC cells.
Gint4.T-STAT3 blocks the pro-tumor effects of CAFs in co-culture with NSCLC cells
In order to further investigate the therapeutic usefulness of Gint4.T-STAT3, we established transwell co-cultured systems of CAFs and NSCLC cells. Calu-1 cells were seeded in the lower chamber and left untreated or treated with Gint4.T aptamer or complexes before the addition of CAFs. Following 24 h, primary CAFs (#16 or #17) were added in the upper chamber by using transwell insert with pore size of 0.4 μm (Figure 4A). Cells were left to grow for 72 h. First, we confirmed that STAT3 activation was increased in Calu-1 cells when co-cultured with either CAF#16 or #17, and reduced by the treatment with Gint4.T-STAT3 (Figure 4B). STAT3 levels were significantly decreased even if with a different silencing efficacy as compared with previous data (Figures 2C and 3A), likely due to the experimental protocol and the influence of CAF presence. Then, by measuring cell viability in co-cultured systems, we showed that the survival rate of Calu-1 NSCLC cells was significantly higher when they were co-cultured with CAFs, and this increase was prevented by the Calu-1 treatment with Gint4.T-STAT3 AsiC (Figure 4C). Indeed, as compared with the controls, the complex treatment allowed the maintaining of a survival rate similar to that of Calu-1 cells grown in monoculture.
Figure 4.

Gint4.T-STAT3 inhibits viability and migration of NSCLC cells co-cultured with CAFs
(A) Scheme of Calu-1 cells and CAFs co-cultures by Boyden chambers (pore size 0.4 μm) used for immunoblot and cell viability assays. (B and C) Calu-1 were co-cultured with CAFs in 5% FBS medium in the absence or in the presence of 400 nmol/L Gint4.T, Gint4.T-STAT3 or CtrlApt-STAT3 (see materials and methods for details) for 72 h. (B) Immunoblot for pSTAT3, STAT3 was performed. βActin or Vinculin antibodies were used as loading control. (C) Cells were recovered and cell viability was measured and expressed relative to cells grown in the absence of CAFs (−). (D) Scheme of Calu-1 cells and CAFs co-cultures using Boyden chambers (pore size 8 μm) to test cell migration. (E and F) Calu-1 cells were left untreated or treated with 400 nmol/L Gint4.T, Gint4.T-STAT3, or CtrlApt-STAT3 and following 24 h were seeded in the Boyden upper chamber and exposed to 1% FBS medium (−) or to CAFs grown in 1% FBS medium (lower chamber). Left panels, representative photographs of migrated cells are shown; right panels, migrated cells were quantified and expressed relative to (−) samples. In (C), (E), and (F), error bars depict SD values (n = 2). Statistics by t test are reported: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
Next, CAF ability to promote the migration of NSCLC cancer cells was deepened by performing a transwell assay, in which Calu-1 NSCLC cells left untreated or treated with Gint4.T aptamer or AsiC, were seeded in the upper chamber (insert pore size of 8 μm) and exposed to CAF#16 or #17 (lower chamber) (Figure 4D). To analyze the consequence of STAT3 inhibition induced by the treatment with the AsiC, Calu-1 cells were left untreated or treated with Gint4.T-STAT3 for 24 h and then allowed to migrate under CAF exposure. We found that cancer cells migrated much better when co-cultured with CAFs. Importantly, CAF-mediated induction of migration was significantly reduced by the pretreatment of NSCLC cells with Gint4.T-STAT3 (Figures 4E and 4F).
These results confirm the complex ability to interfere with CAF promoting effects on cell viability and migration, because of STAT3 inhibition in NSCLC target cells.
Gint4.T-STAT3 inhibits CAF-pro-tumor effects on NSCLC primary cultures
In order to extend Gint4.T-STAT3 functional characterization, we determined its effects on the crosstalk between CAFs and PDGFRβ positive primary NSCLC cell lines (NSCLC#18 and #16) obtained from surgical specimens of lung cancer patients (Figure S2). In both cell lines, Gint4.T-STAT3 treatment reduced STAT3 levels (Figure S4). Patient-derived NSCLC cells were thus grown in either CAF#16 or #17 CM in the absence or in the presence of Gint4.T aptamer, Gint4.T-STAT3, or the control complex. The effects on cell viability and migration were then analyzed. The cell viability rate of both NSCLC #16 and #18 was strongly induced by CAF CM, as compared with cells grown in normal medium, with a fold increase of about 1.4 and 1.8, when grown in CAF#16-CM or CAF#17-CM, respectively. Gint4.T-STAT3 complex drastically opposed this effect (Figures 5A and 5B) in all the conditions tested, almost restoring the basal conditions. In addition, CM from CAF#16 and #17 significantly enhanced cell migration of both primary NSCLC cells. Despite the different extent, likely related to patient-specific responses, cancer cell treatment with Gint4.T-STAT3 greatly inhibited or completely blocked CAF CM-induced cell migration in all the cases (Figures 5C and 5D).
Figure 5.

CAF CM effects on NSCLC primary cultures
(A and B) Indicated primary NSCLC cells were grown in 5% FBS medium (−) or in 5% FBS CM from CAF#16 or #17 for 72 h in the absence (NT) or in the presence of 400 nmol/L Gint4.T, Gint4.T-STAT3, or CtrlApt-STAT3. Cell viability was measured and expressed relative to cells grown in 5% FBS medium (−). Error bars depict SD values (n = 2). (C and D) Migration of primary NSCLC cells grown in 5% FBS medium (−) or in 5% FBS CAF CMs was analyzed by Boyden chambers (8-μm pore size) containing the correspondent medium as chemoattractant. Left panels, representative photographs of migrated cells; right panels, quantization of migrated cells expressed relative to cell grown in 5% FBS medium (−). Error bars: SD values (n = 2). In (A)–(D), statistics by t test are reported: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
The results were next confirmed by co-culture systems. We demonstrated that the presence of CAFs significantly enhanced primary NSCLC#18 and #16 cell viability (Figures 6A and 6B) and migration (Figures 6C and 6D), confirming the potent CAF pro-tumor functions. Importantly, we validated Gint4.T-STAT3 ability to prevent these effects. Indeed, pretreating NSCLC cells with the STAT3 targeting complex before the addition of CAFs, results in a significant reduction of CAF-mediated pro-survival (Figures 6A and 6B) and pro-migratory (Figures 6C and 6D) consequences.
Figure 6.

CAF co-cultures with NSCLC primary cells
(A and B) NSCLC primary cells were co-cultured with CAFs in 5% FBS medium in the absence or in the presence of 400 nmol/L Gint4.T, Gint4.T-STAT3, or CtrlApt for 72 h in Boyden chambers (0.4-μm pore size). Cells were recovered and cell viability was measured and expressed relative to cells grown in the absence of CAFs. (C and D) Indicated NSCLC primary cells were left untreated or treated with 400 nmol/L Gint4.T, Gint4.T-STAT3, or CtrlApt-STAT3 and following 24 h were seeded in the Boyden upper chamber (8-μm pore size) and exposed to 5% FBS medium (−) or to CAFs grown in 5% FBS medium (lower chamber). Left panels, representative photographs of migrated cells are shown; right panels, migrated cells were quantified and expressed realtive to (−) samples. In (A)–(D) error bars depict SD values (n = 2). Statistics by t test is reported: ∗p < 0.05; ∗∗p < 0.01.
Collectively, these data strongly confirm Gint4.T-STAT3 as a promising tool to block the network of communication between CAF and tumor cells in NSCLC.
Gint4.T-STAT3 direct effects on CAFs
As previously reported,21 CAFs express high levels of PDGFRβ (Figure S5A), thus we wondered whether Gint4.T-STAT3 could have a direct therapeutic effect on this population. We first confirmed that the conjugate was able to reduce STAT3 levels in PDGFR positive primary CAFs by immunoblot and RT-qPCR (Figures S5B and S5C). Then, we analyzed the consequence of Gint4.T-STAT3 treatment on CAF viability, migration, and contraction ability. Notably, we found that upon AsiC treatment there is a significant reduction of both CAF#16 and #17 cell viability (Figure 7A) and migration (Figure 7B).
Figure 7.

Gint4.T-STAT3 direct effect on CAFs
(A) Indicated CAFs were left untreated (NT) or treated with 400 nmol/L Gint4.T, Gint4.T-STAT3, or CtrlApt for 72 h and cell viability was measured and expressed relative to NT. (B) CAFs were left untreated (NT) or treated with 400 nmol/L Gint4.T, Gint4.T-STAT3, or CtrlApt-STAT3 and following 24 h were seeded in the Boyden upper chamber (8-μm pore size) and exposed to 10% FBS medium (lower chamber). Left panels, representative photographs of migrated cells are shown; right panels, migrated cells were quantified and expressed relative to NT. In (A) and (B), error bars depict SD values (n = 2). Statistics by t test (vs. NT) are reported: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. (C and D) Collagen contraction ability of CAFs left untreated (NT) or treated as indicated. Left panels, Representative pictures of plugs at different time points; right panels, plug areas measured by ImageJ and expressed as fold over untreated samples. Means and SD of two independent experiments are reported. Statistics by two-way ANOVA (vs. NT): ∗p < 0.05; ∗∗p < 0.01. (E and F) Indicated CAF markers were analyzed by RT-qPCR in primary CAFs treated with 400 nmol/L CtrlApt or Gint4.T-STAT3. Error bars depict SD values (n = 2). Statistics by t test are reported: ∗p < 0.05; ∗∗p < 0.01.
Most importantly, since contractility is a major hallmark of CAFs, we monitored this feature by measuring fibroblast-mediated collagen contraction. CAFs were pretreated for 72 h with aptamers or Gint4.T-STAT3, and then seeded in a type 1 collagen matrix to examine their contraction ability. As shown in Figures 7C and 7D, despite a different contraction ability was detected for CAF#16 and CAF#17, STAT3 AsiC strongly impairs collagen contraction in both cell lines. Results suggest the conjugate capacity to directly alter CAF phenotype. This property was also confirmed by the reduced levels of specific CAF markers (i.e., FAP and α-SMA) detected upon Gint4.T-STAT3 treatment (Figure 7E).
Altogether, these data indicate that Gint4.T-STAT3 affects not only tumor cells, but also CAF population showing the potential to permit an improved targeting of the entire tumor bulk.
Gint4.T-STAT3 effects on 3D spheroid cultures
Three-dimensional cell in vitro models are attractive tools for investigating relevant cancer therapeutics since they recapitulate human tumor biology better than 2D cultures.22,23 Thus, we tested Gint4.T-STAT3 effects on spheroid formation. First, we analyzed homotypic cultures of Calu-1 NSCLC cells. Cells were left untreated or treated with Gint4.T-STAT3 and spheroids were formed using the hanging drop method in the absence or in the presence of the conjugate. As shown by the distribution range of spheroid diameters in Figure 8A, we found that the AsiC treatment hampers spheroid formation, reducing the median size of the spheres (>50 μm diameter) with no significant change of spheroid number (not shown). Most importantly, we then tested 3D hybrid spheroid formation, by mixing Calu-1 NSCLC cells with CAFs in the absence or in the presence of Gint4.T-STAT3. In accordance with previous reports,23 when cancer cells are mixed with CAFs, spheroids resulted in bigger round and steric aggregates (Figure 8B). Notably, in the presence of Gint4.T-STAT3, spheroid dimension resulted almost equal to that obtained from cancer cells alone. These data suggest the therapeutic potential of Gint4.T-STAT3 conjugate.
Figure 8.

Gint4.T-STAT3 effect on spheroid cultures
(A) Spheroids were derived from Calu-1 cells left untreated or treated with 400 nmol/L Gint4.T-STAT3. (B) Spheroids were derived from Calu-1 cells (−) or Calu-1 mixed with CAF#17 (ratio 1:4) in the absence or in the presence of 400 nmol/L Gint4.T-STAT3. In (A) and (B), left panels, representative photographs are shown; right panel, boxplots showing the range of spheroid diameter distribution (>50 μm) with median size. Statistics reported: (A) ∗∗∗p < 0.001 by nonparametric Mann-Whitney U test; (B) ∗p < 0.05; ∗∗∗p < 0.001 by nonparametric Kruskal-Wallis test with multiple comparison correction.
Discussion
In the present study, we provide a proof of principle of the ability of an AsiC targeting STAT3 (Gint4.T-STAT3) to impair the interplay between CAFs and NSCLC cells. By using both stable and primary NSCLC cultures, we found that cancer cell treatment with Gint4.T-STAT3, by reducing STAT3 levels, prevents primary CAF pro-tumor effects on cell viability and migration.
It is largely accepted that tumor formation and progression is a complex process involving the cooperation of cancer cells with the surrounding stroma that becomes activated and develops tumor-supporting functions.24 Key components of the tumor stroma are CAFs, for which a crucial role in triggering tumor progression, promoting drug resistance, tumor dissemination, metastasis, and recurrence has been described in several cancers, including NSCLC.25,26,27,28 It has been demonstrated that CAFs regulate the malignant progression through the secretion of various growth factors, cytokines, and matrix remodeling proteins.29 However, therapeutic approaches aimed at targeting the interactions between tumor cells and CAFs are still a challenge, and can greatly enhance the effectiveness of the treatments.
Here, to address this issue, we use an aptamer-based conjugate specifically inhibiting STAT3. In addition to its pro-oncogenic functions, STAT3 pathway has been reported to be activated by ILs (as IL6 and 11) released by CAFs,5,6,7,8 suggesting its inhibition in tumor cells can render them irresponsive to CAF-tumor-supporting functions in cancer. To this purpose, IL neutralizing antibodies or specific inhibitors of JAK2/STAT3 have been applied to block the induction of EMT, migration, and drug resistance mediated by CAFs.6,8
In our previous studies,10,11 we addressed STAT3 silencing by the use of a specific siRNA and developed a specific aptamer-based siRNA delivery system. We have generated a stable RNA conjugate (Gint4.T-STAT3), containing a STAT3 siRNA linked to the inhibitory anti-PDGFRβ aptamer as a carrier, and reported its ability in specifically inhibiting STAT3 pathway in PDGFRβ-positive cancer cells, both in vitro and in vivo. Importantly, since we used an inhibitory aptamer, Gint4.T-STAT3 can act as a multifunctional molecule combining PDGFR signaling inhibition, mediated by the aptamer, and the siRNA-dependent STAT3 suppression.
In the present study, we explored the use of this molecule to alter the interplay between CAFs and tumor cells in NSCLC. First, we demonstrated that the treatment of Calu-1 NSCLC cells with Gint4.T-STAT3, by interfering with STAT3 expression, efficiently reverts primary CAF-mediated STAT3 activation. Consequently, the increase of cancer cell viability and migration mediated by primary CAF CM or co-cultures is revoked. Results were also confirmed by using two primary cultures derived from NSCLC biopsies. Notably, the multifunctional properties of Gint4.T-STAT3 were evident on cell migration/invasion where the inhibitory function of Gint4.T was further enhanced by the presence of STAT3 siRNA. In addition, we found that Gint4.T-STAT3 directly alters CAF phenotype and impairs the formation of 3D spheroids derived from NSCLC cells or CAF-NSCLC cell hybrid cultures, reducing the median size of the spheres.
Collectively, our study provides a new AsiC as a tool to impair the interplay between CAFs and tumor cells in NSCLC, providing a first evaluation of its efficacy in vitro.
As compared with small molecules or peptide-based inhibitors, our AsiC offers several advantages as therapeutic agent, including that (1) it can be produced by chemical synthesis; (2) it is stable for several days in serum; and (3) it can provide an effective strategy for tumor-specific gene knockdown in vivo, minimizing the toxicity on normal cells.18,30 Obtained results give new insight in the therapeutic applicability of STAT3 targeting in NSCLC, and provide the rationale for a subsequent in vivo experimentation of the described conjugate.
Up to date, aptamers have been largely developed for cancer immunotherapy,31 whereas their application to alter CAF effects is poorly described. Of note, it has been shown that PDGFRβ is highly expressed also by CAFs3,21,32 and that Gint4.T aptamer inhibits the trans-differentiation of bone marrow-derived mesenchymal stem cells into CAFs and reduces their pro-tumor action in triple negative breast cancer.33 In addition, it has been demonstrated that STAT3 inhibitors have an anti-cancer activity affecting not only tumor cells, but also stromal cells.34,35,36 Accordingly, here we show that Gint.4-STAT3 has a direct effect on CAFs, suggesting its potential to revert also CAF phenotype, thus providing a double-acting molecule able to completely impair stromal component effects.
In conclusion, we present one of the first attempts in targeting the CAF-NSCLC cell interplay using aptamers and therapeutic oligonucleotides providing an AsiC-based bio-drug with the prospective to open innovative horizons in the field of NSCLC anti-cancer therapy.
Materials and methods
Cell cultures
Calu-1 and A549 NSCLC cells were obtained from American Type Culture Collection (ATCC) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) or RPMI medium, respectively, supplemented with 1% penicillin/streptomycin solution (Sigma-Aldrich) and 10% fetal bovine serum (FBS) (Sigma-Aldrich).
Primary cultures of tumor epithelial and fibroblast cells or NFs (at least 5 cm away from the tumor) were prepared from resected lung tissues of NSCLC patients provided by Prof. A. Fiorelli from the Department of Thoracic Surgery Unit, "Luigi Vanvitelli" University. Tissues were mechanically fragmented with sterile scissors and subjected to digestion with collagenase (Sigma-Aldrich). Epithelial cell pellets were obtained by centrifugation at 500 rpm for 5 min. Fibroblasts that remained in suspension were collected by centrifugation at 1,200 rpm for 5 min. Primary cells were maintained in DMEM/Nutrient F12-Ham (DMEM-F12) supplemented with 1% penicillin/streptomycin solution (Sigma-Aldrich) and 10% FBS (Sigma-Aldrich).
CAF CMs were obtained by growing cells in medium with 1% or 5% FBS for 72 h. CMs were collected and centrifuged at 1,000 × g for 10 min to remove cell debris before the use. One percent or 5% FBS CMs were used to grow Calu-1 cells or primary NSCLC cells, respectively. STAT3 siRNA duplex transfection was performed at 100 nmol/L siRNA with lipofectamine-2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Aptamers and conjugates
The following aptamer and AsiC oligos were used and purchased from TriLink Biotech or produced by the DNA/RNA Synthesis Laboratory, Beckman Research Institute of the City of Hope.
Gint4.T, 5′-UGUCGUGGGGCAUCGAGUAAAUGCAAUUCGACA-3′;
Gint4.T stick,
5′-UGUCGUGGGGCAUCGAGUAAAUGCAAUUCGACAXXXXGUACAUUCUAGAUAGCC-3′;
Control aptamer (indicated as CtrlApt),
5′-UUCGUACCGGGUAGGUUGGCUUGCACAUAGAACGUGUCA-3′;
Control aptamer stick (used in the control conjugate),
5′-GCCGCUAGAACCUUCUAAGCGAAUACAUUACCGCXXXXGUACAUUCUAGAUAGCC-3′;
Human STAT3 antisense strand (AS) stick,
5′-UUAGCCCAUGUGAUCUGACACCCUGAAGGCUAUCUAGAAUGUAC-3′;
Human STAT3 sense strand (SS), 5′-CAGGGUGUCAGAUCACAUGGGCUAA-3′.
All RNAs were modified with 2′-F-Pyrimidines (2′-F-Py). Stick sequences (underlined) were modified with 2′-F-Py and 2′-oxygen-methyl purines. The X indicates a three-carbon linker ((CH2)3) spacer. Before the use, aptamers were incubated at 85°C for 5 min, ice for 3 min, and 37°C for 5 min to allow the correct folding by denaturation-renaturation. STAT AsiC was prepared by a two-step annealing: (1) incubating in the annealing buffer (20 mM 2-[4-(2- hydroxyethyl)piperazin-1-yl] ethane sulfonic acid [HEPES; pH 7.5], 150 mM NaCl, 2 mM CaCl2) the AS and SS oligos at 95°C for 10 min, 55°C for 10 min, and 37°C for 20 min; and (2) incubating the AS-SS duplex with refolded stick aptamer at 37°C for 30 min. The correct formation of the AsiCs was coinfirmed on a 12% non-denaturing polyacrylamide gel.
Immunofluorescence
Cells were grown on glass coverslips in 24-well plates (4 × 104 cells/well) in complete medium. A day after, cells were washed in Tris-buffered saline and fixed for 10 min at −20°C with a freshly prepared ice-cold Acetone/Methanol mix (1:1). Once nonspecific sites were blocked with 1% bovine serum albumin and 0.3% Triton X-100 in Tris-buffered saline for 15 min at room temperature, cells were incubated with the primary antibody 1 h at room temperature. Cells were then washed and incubated for 1 h at room temperature in the dark with the fluorophore-conjugated secondary antibody. The coverslips were mounted with Prolong Diamond antifade reagent with DAPI (Thermo Fisher Scientific) and the cells were visualized by confocal microscopy (Zeiss LSM700). Primary antibodies used were as follows: α-smooth muscle actin (D4K9N) XP Rabbit mAb (Cell Signaling) used 1:100; FAP antibody (SS-13) mouse (Santa Cruz Biotechnology) used 1:50. Secondary antibodies used were as follows: anti-mouse Alexa fluor 488-conjugated and anti-rabbit Alexafluor 647-conjugated, used 1:500 (Thermo Fisher Scientific).
Protein extraction and western blot
Cells were grown in 3.5-cm plates (1.4 × 105 cells/plate) in normal medium or CAF CM either in the absence or in the presence of aptamers or chimeras (400 nmol/L) for 72 h.
For co-culture assays, cells were grown in a co-culture transwell apparatus with a 0.4-μm pore membrane (six-well plate; Greiner Bio-one). Calu-1 or primary NSCLC cells (1 × 105 cell/well) were seeded in the lower chamber of the transwell and treated with aptamers or conjugates (400 nmol/L). After 24 h, CAF cells (1 × 105 cell/well) were added to the upper chamber. Following an additional 48 h of incubation, co-cultured epithelial cells were harvested for western blot analysis.
Cell lysates were prepared in JS buffer (50 mM HEPES [pH 7.5], 50 mM NaCl, 1% glycerol, 1% Triton X-100, 1.5 mM MgCl2, 5 mM EGTA, 1 mM Na3VO4, protease inhibitors). To monitor IL6 and IL11 levels, CAF or NF CMs were obtained by cells grown in 5% FBS medium for 72 h and were concentrated with Amicon columns (10 kDa NMWCO; Millipore). Proteins (50 μg) from CMs were analyzed by western blot.
All samples were boiled in sodium dodecyl sulfate/β-mercaptoethanol sample buffer, and separated by electrophoresis. Gels were blotted onto polyvinylidene difluoride membranes (Millipore) and subjected to immunoblotting. The following primary antibodies were used: anti-PDGFRβ, anti-pY(705)-STAT3, anti-STAT3, anti-Vinculin (Cell Signaling Technology), anti-Actin, anti-IL6, and anti-IL11 (Santa Cruz Biotechnology).
RT-qPCR analysis
RNAs were extracted with TRiZol (Invitrogen). One microgram of total RNA was reverse transcribed with iScript cDNA Synthesis Kit. For real-time PCR, amplification was performed with IQ-SYBR Green supermix (Bio-Rad, Hercules). Relative mRNA quantization was analyzed by using the ΔΔCt method applying the equation 2−ΔΔCt. The following specific primers were used:
STAT3 Fw, 5′-ACCTGCAGCAATACCATTGAC-3′; STAT3 Rev, 5′-AAGGTGAGGGACTCAAACTGC-3′; βactin Fw, 5′-CAAGAGATGGCCACGGCTGCT-3′; βactin Rev, 5′-TCCTTCTGCATCCTGTCGGCA-3′; E-Cad Fw, 5′- GGAGGAGAGCGGTGGTCAAA-3′; E-Cad Rev, 5′-TGTGCAGCTGGCTCAAGTCA-3′; Vimentin Fw, 5′-GACAATGCGTCTCTGGCACGTCTT-3′; Vimentin Rev, 5′-TCCTCCGCCTCCTGCAGGTTCTT-3′; Fibronectin Fw, 5′-CAGTGGGAGACCTCGAGAAG-3′; Fibronectin Rev, 5′-TCCCTCGGAACATCAGAAAC-3′; FAP Fw: 5′-TACGTTTCATCACTGGCCCT-3′; FAP Rev, 5′-CATCTGCTGTTCCGTGGATG-3′; α-SMA Fw, 5′-ACCCAGCACCATGAAGATCA-3′; α-SMA Rev, 5′-AGAGACAGAGAGGAGCAGGA-3′.
Transwell migration and invasion assay
For transwell migration assay with CAF CM, cells were seeded in 3.5-cm plates (1.4 × 105 cells/plate) in medium or CAF CM either in the absence or in the presence of aptamers or chimeras (400 nmol/L).
Following 24 h, 1 × 105 cells were plated into the upper chamber of a 24-well transwell (Corning Incorporate, pre size 8 μm) in serum-free DMEM and exposed to the correspondent medium (0.6 mL, lower chamber) as stimulus of migration. Invasion assay was performed with the same protocol using upper chambers previously coated with a 20% Matrigel matrix (BD Biosciences).
For co-cultures, cells (1 × 105/point) pretreated for 24 h with 400 nmol/L aptamers or chimeras in complete DMEM, were plated into the upper chamber of a 24-well transwell (Corning Incorporate, pre size 8 μm) in serum-free DMEM and exposed to CAFs seeded in the lower chamber a day before (1 × 105 in 0.6 mL, lower chamber).
In all the experiments, migrated cells were stained with 0.1% crystal violet in 25% methanol and pictures were acquired with Leica application suite. To quantify the percentage of migrated cells, ImageJ software was used.
Cell viability
For CM assays, cells were seeded into 96-well plates (3 × 103 cells/well) in CAF CMs in the absence or presence of aptamers or chimeras (400 nmol/L). Following 72 h, cell viability was measured with CellTiter 96 Proliferation Assay (Promega).
For co-culture assays, cells were grown in co-culture transwell apparatus with a 0.4-μm pore membrane (six-well plate; Greiner Bio-one). Cells (1 × 105 cell/well) were seeded in the lower chamber of the transwell and treated with aptamers or conjugates (400 nmol/L). After 24 h, CAF cells (1 × 105 cell/well) were added to the upper chamber. Following additional 48 h of incubation, co-cultured epithelial cells were harvested and trasferred in 96-well plates to measure cell viability with CellTiter 96 Proliferation Assay (Promega).
Contraction assay
CAFs (1 × 105 cells/well in six-well plates) were left untreated or treated with 400 nmol/L control aptamer, Gint4.T, or Gint4.T-STAT3 for 72 h. Collagen mix was prepared by suspending type 1 collagen (Corning) in acetic acid (5 mM) and minimum essential Eagle’s medium (Sigma) to a final concentration of 1 mg/mL and adding NaOH (1 M) drop by drop to restore the neutral pH.
Pretreated cells were counted and 4 × 104 cells/point were suspended in 75 μL of FBS, mixed with 625 μL of collagen mix, and seeded in 24-well plates. Collagen was left to polymerize by incubating for 3 h at 37°C, collagen plugs were detached from the walls of the wells, and 5% DMEM-F12 medium (300 μL with or without treatments) was added to each well. Images of the collagen plugs were acquired at the beginning (0) and after 24 and 48 h with a scanner and plug areas were measured with ImageJ software.
Spheroid formation assay
Spheroids were formed using the hanging drop method.22,23 Calu-1 cells (1 × 105/well in six-well plates) were left untreated or treated for 24 h with 400 nmol/L Gint4.T-STAT3 in complete medium. Cells were then recovered, counted, and suspended in a minimal medium containing 20 ng/mL EGF and 10 ng/mL bFGF (Life technologies) optimized for non-adherent spheroid growth37 in the absence or in the presence of 400 nmol/L Gint4.T-STAT3. For homotypic cultures, drops (20 μL) containing 1 × 104 Calu-1 cells were seeded on the top cover lip of a 100-mm plate. For CAF-NSCLC cell hybrid cultures, pretreated Calu-1 cells were mixed with CAF#17 (ratio 1:4) and drops (20 μL) containing 2 × 103 Calu-1 cells and 8 × 103 CAF#17 were seeded on the top cover lip of a 100-mm plate. PBS was added in the plate to avoid evaporation. Spheroids were left to form for 96 h and then transferred in a 96-well plate (1 drop/well), photographed using Leica application suite, and measured (considering spheroids with a diameter >50 μm).
Statistical analysis and reproducibility
Statistics were analyzed as indicated in the figure legends. p < 0.05 was considered significant. We applied Student’s t test for general caparison between the one time effects of two groups and two-way ANOVA for the measures of gel area at different time points in the contraction assay (Figures 6C and 6D); for spheroid diameter distribution analyses, we used nonparametric Mann-Whitney U test for comparison between the two groups in Figure 8A and Kruskal-Wallis test with multiple comparison correction in Figure 8B. Experiments shown are representative of two independent replicates.
Acknowledgments
We thank F. Moscato and M. Montagna for technical assistance. This work has been supported by the Italian Ministry of Health (GR-2011-02352546; C.L.E.), H2020-MSCA-RISE-2019 cONCReTE (#872391; C.L.E.); Regione Campania PO FESR 2014-2020 SATIN (C.L.E.); Worldwide cancer research grant #22-0129 (C.L.E.).
Author contributions
M.L.I. performed the majority of the experiments and assisted with manuscript preparation. G.C., D.R., and G.C. assisted with some experiments. C.L.E. and S.C. provided intellectual input, coordinated the research, guided the experimental design and the preparation of the manuscript, and secured the funding. All authors have read and agreed to the published version of the manuscript.
Declaration of interests
The authors declared no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2023.03.003.
Contributor Information
Silvia Catuogno, Email: s.catuogno@ieos.cnr.it.
Carla L. Esposito, Email: c.esposito@ieos.cnr.it.
Supplemental information
Data availability
Data are contained within the article or in the supplemental information.
References
- 1.Viale P.H. The American cancer society's facts & figures: 2020 edition. J. Adv. Pract. Oncol. 2020;11:135–136. doi: 10.6004/jadpro.2020.11.2.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yuan Y., Jiang Y.C., Sun C.K., Chen Q.M. Role of the tumor microenvironment in tumor progression and the clinical applications (Review) Oncol. Rep. 2016;35:2499–2515. doi: 10.3892/or.2016.4660. [DOI] [PubMed] [Google Scholar]
- 3.Kalluri R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer. 2016;16:582–598. doi: 10.1038/nrc.2016.73. [DOI] [PubMed] [Google Scholar]
- 4.Harada D., Takigawa N., Kiura K. The role of STAT3 in non-small cell lung cancer. Cancers. 2014;6:708–722. doi: 10.3390/cancers6020708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ham I.H., Oh H.J., Jin H., Bae C.A., Jeon S.M., Choi K.S., Son S.Y., Han S.U., Brekken R.A., Lee D., Hur H. Targeting interleukin-6 as a strategy to overcome stroma-induced resistance to chemotherapy in gastric cancer. Mol. Cancer. 2019;18:68. doi: 10.1186/s12943-019-0972-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shien K., Papadimitrakopoulou V.A., Ruder D., Behrens C., Shen L., Kalhor N., Song J., Lee J.J., Wang J., Tang X., et al. JAK1/STAT3 activation through a proinflammatory cytokine pathway leads to resistance to molecularly targeted therapy in non-small cell lung cancer. Mol. Cancer Therapeut. 2017;16:2234–2245. doi: 10.1158/1535-7163.MCT-17-0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tao L., Huang G., Wang R., Pan Y., He Z., Chu X., Song H., Chen L. Cancer-associated fibroblasts treated with cisplatin facilitates chemoresistance of lung adenocarcinoma through IL-11/IL-11R/STAT3 signaling pathway. Sci. Rep. 2016;6:38408. doi: 10.1038/srep38408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang L., Cao L., Wang H., Liu B., Zhang Q., Meng Z., Wu X., Zhou Q., Xu K. Cancer-associated fibroblasts enhance metastatic potential of lung cancer cells through IL-6/STAT3 signaling pathway. Oncotarget. 2017;8:76116–76128. doi: 10.18632/oncotarget.18814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kortylewski M., Nechaev S. Cancer therapy using oligonucleotide-based STAT3 inhibitors: will they deliver? Ther. Deliv. 2014;5:239–242. doi: 10.4155/tde.13.152. [DOI] [PubMed] [Google Scholar]
- 10.Esposito C.L., Nuzzo S., Catuogno S., Romano S., de Nigris F., de Franciscis V. STAT3 gene silencing by aptamer-siRNA chimera as selective therapeutic for glioblastoma. Mol. Ther. Nucleic Acids. 2018;10:398–411. doi: 10.1016/j.omtn.2017.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esposito C.L., Nuzzo S., Ibba M.L., Ricci-Vitiani L., Pallini R., Condorelli G., Catuogno S., de Franciscis V. Combined targeting of glioblastoma stem-like cells by neutralizing RNA-bio-drugs for STAT3. Cancers. 2020;12:1434. doi: 10.3390/cancers12061434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Esposito C.L., Nuzzo S., Kumar S.A., Rienzo A., Lawrence C.L., Pallini R., Shaw L., Alder J.E., Ricci-Vitiani L., Catuogno S., de Franciscis V. A combined microRNA-based targeted therapeutic approach to eradicate glioblastoma stem-like cells. J. Contr. Release. 2016;238:43–57. doi: 10.1016/j.jconrel.2016.07.032. [DOI] [PubMed] [Google Scholar]
- 13.Yoon S., Wu X., Armstrong B., Habib N., Rossi J.J. An RNA aptamer targeting the receptor tyrosine kinase PDGFRalpha induces anti-tumor effects through STAT3 and p53 in glioblastoma. Mol. Ther. Nucleic Acids. 2019;14:131–141. doi: 10.1016/j.omtn.2018.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morita Y., Leslie M., Kameyama H., Volk D.E., Tanaka T. Aptamer therapeutics in cancer: current and future. Cancers. 2018;10:80. doi: 10.3390/cancers10030080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Catuogno S., Esposito C.L., Condorelli G., de Franciscis V. Nucleic acids delivering nucleic acids. Adv. Drug Deliv. Rev. 2018;134:79–93. doi: 10.1016/j.addr.2018.04.006. [DOI] [PubMed] [Google Scholar]
- 16.Esposito C.L., Catuogno S., Condorelli G., Ungaro P., de Franciscis V. Aptamer chimeras for therapeutic delivery: the challenging perspectives. Genes. 2018;9:529. doi: 10.3390/genes9110529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou G., Wilson G., Hebbard L., Duan W., Liddle C., George J., Qiao L. Aptamers: a promising chemical antibody for cancer therapy. Oncotarget. 2016;7:13446–13463. doi: 10.18632/oncotarget.7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kruspe S., Giangrande P.H. Aptamer-siRNA chimeras: discovery, progress, and future prospects. Biomedicines. 2017;5:45. doi: 10.3390/biomedicines5030045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Camorani S., Esposito C.L., Rienzo A., Catuogno S., Iaboni M., Condorelli G., de Franciscis V., Cerchia L. Inhibition of receptor signaling and of glioblastoma-derived tumor growth by a novel PDGFRbeta aptamer. Mol. Ther. 2014;22:828–841. doi: 10.1038/mt.2013.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kono S.A., Heasley L.E., Doebele R.C., Camidge D.R. Adding to the mix: fibroblast growth factor and platelet-derived growth factor receptor pathways as targets in non-small cell lung cancer. Curr. Cancer Drug Targets. 2012;12:107–123. doi: 10.2174/156800912799095144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen X., Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019;18:99–115. doi: 10.1038/s41573-018-0004-1. [DOI] [PubMed] [Google Scholar]
- 22.Jacobi N., Seeboeck R., Hofmann E., Schweiger H., Smolinska V., Mohr T., Boyer A., Sommergruber W., Lechner P., Pichler-Huebschmann C., et al. Organotypic three-dimensional cancer cell cultures mirror drug responses in vivo: lessons learned from the inhibition of EGFR signaling. Oncotarget. 2017;8:107423–107440. doi: 10.18632/oncotarget.22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakamura H., Sugano M., Miyashita T., Hashimoto H., Ochiai A., Suzuki K., Tsuboi M., Ishii G. Organoid culture containing cancer cells and stromal cells reveals that podoplanin-positive cancer-associated fibroblasts enhance proliferation of lung cancer cells. Lung Cancer. 2019;134:100–107. doi: 10.1016/j.lungcan.2019.04.007. [DOI] [PubMed] [Google Scholar]
- 24.Orimo A., Weinberg R.A. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle. 2006;5:1597–1601. doi: 10.4161/cc.5.15.3112. [DOI] [PubMed] [Google Scholar]
- 25.Bremnes R.M., Dønnem T., Al-Saad S., Al-Shibli K., Andersen S., Sirera R., Camps C., Marinez I., Busund L.T. The role of tumor stroma in cancer progression and prognosis: emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J. Thorac. Oncol. 2011;6:209–217. doi: 10.1097/JTO.0b013e3181f8a1bd. [DOI] [PubMed] [Google Scholar]
- 26.Chen C., Hou J., Yu S., Li W., Wang X., Sun H., Qin T., Claret F.X., Guo H., Liu Z. Role of cancer-associated fibroblasts in the resistance to antitumor therapy, and their potential therapeutic mechanisms in non-small cell lung cancer. Oncol. Lett. 2021;21:413. doi: 10.3892/ol.2021.12674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H., Zhang Q., Wu Q., Cui Y., Zhu H., Fang M., Zhou X., Sun Z., Yu J. Interleukin-22 secreted by cancer-associated fibroblasts regulates the proliferation and metastasis of lung cancer cells via the PI3K-Akt-mTOR signaling pathway. Am. J. Transl. Res. 2019;11:4077–4088. [PMC free article] [PubMed] [Google Scholar]
- 28.Shimoda M., Mellody K.T., Orimo A. Carcinoma-associated fibroblasts are a rate-limiting determinant for tumour progression. Semin. Cell Dev. Biol. 2010;21:19–25. doi: 10.1016/j.semcdb.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santana-Viera L., Ibba M.L., Rotoli D., Catuogno S., Esposito C.L. Emerging therapeutic RNAs for the targeting of cancer associated fibroblasts. Cancers. 2020;12:1365. doi: 10.3390/cancers12061365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou J., Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat. Rev. Drug Discov. 2017;16:181–202. doi: 10.1038/nrd.2016.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pastor F., Berraondo P., Etxeberria I., Frederick J., Sahin U., Gilboa E., Melero I. An RNA toolbox for cancer immunotherapy. Nat. Rev. Drug Discov. 2018;17:751–767. doi: 10.1038/nrd.2018.132. [DOI] [PubMed] [Google Scholar]
- 32.Aoto K., Ito K., Aoki S. Complex formation between platelet-derived growth factor receptor beta and transforming growth factor beta receptor regulates the differentiation of mesenchymal stem cells into cancer-associated fibroblasts. Oncotarget. 2018;9:34090–34102. doi: 10.18632/oncotarget.26124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Camorani S., Hill B.S., Fontanella R., Greco A., Gramanzini M., Auletta L., Gargiulo S., Albanese S., Lucarelli E., Cerchia L., Zannetti A. Inhibition of bone marrow-derived mesenchymal stem cells homing towards triple-negative breast cancer microenvironment using an anti-PDGFRbeta aptamer. Theranostics. 2017;7:3595–3607. doi: 10.7150/thno.18974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonuccelli G., Whitaker-Menezes D., Castello-Cros R., Pavlides S., Pestell R.G., Fatatis A., Witkiewicz A.K., Vander Heiden M.G., Migneco G., Chiavarina B., et al. The reverse Warburg effect: glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer associated fibroblasts. Cell Cycle. 2010;9:1960–1971. doi: 10.4161/cc.9.10.11601. [DOI] [PubMed] [Google Scholar]
- 35.Hatiboglu M.A., Kong L.Y., Wei J., Wang Y., McEnery K.A., Fuller G.N., Qiao W., Davies M.A., Priebe W., Heimberger A.B. The tumor microenvironment expression of p-STAT3 influences the efficacy of cyclophosphamide with WP1066 in murine melanoma models. Int. J. Cancer. 2012;131:8–17. doi: 10.1002/ijc.26307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xin H., Herrmann A., Reckamp K., Zhang W., Pal S., Hedvat M., Zhang C., Liang W., Scuto A., Weng S., et al. Antiangiogenic and antimetastatic activity of JAK inhibitor AZD1480. Cancer Res. 2011;71:6601–6610. doi: 10.1158/0008-5472.CAN-11-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pallini R., Ricci-Vitiani L., Banna G.L., Signore M., Lombardi D., Todaro M., Stassi G., Martini M., Maira G., Larocca L.M., De Maria R. Cancer stem cell analysis and clinical outcome in patients with glioblastoma multiforme. Clin. Cancer Res. 2008;14:8205–8212. doi: 10.1158/1078-0432.CCR-08-0644. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are contained within the article or in the supplemental information.