

Abstract
Macromolecules of various sizes induce crowding of the cellular environment. This crowding impacts on biochemical reactions by increasing solvent viscosity, decreasing the water‐accessible volume and altering protein shape, function, and interactions. Although mitochondria represent highly protein‐rich organelles, most of these proteins are somehow immobilized. Therefore, whether the mitochondrial matrix solvent exhibits macromolecular crowding is still unclear. Here, we demonstrate that fluorescent protein fusion peptides (AcGFP1 concatemers) in the mitochondrial matrix of HeLa cells display an elongated molecular structure and that their diffusion constant decreases with increasing molecular weight in a manner typical of macromolecular crowding. Chloramphenicol (CAP) treatment impaired mitochondrial function and reduced the number of cristae without triggering mitochondrial orthodox‐to‐condensed transition or a mitochondrial unfolded protein response. CAP‐treated cells displayed progressive concatemer immobilization with increasing molecular weight and an eightfold matrix viscosity increase, compatible with increased macromolecular crowding. These results establish that the matrix solvent exhibits macromolecular crowding in functional and dysfunctional mitochondria. Therefore, changes in matrix crowding likely affect matrix biochemical reactions in a manner depending on the molecular weight of the involved crowders and reactants.
Keywords: chloramphenicol, diffusion, FRAP, macromolecular crowding, mitochondria
Subject Categories: Organelles
The mitochondrial matrix solvent exhibits macromolecular crowding, which is increased under stress conditions.

Introduction
Mitochondria host a large variety of biochemical reactions, including the ATP‐generating oxidative phosphorylation (OXPHOS) system (Smeitink et al, 2001; Rath et al, 2021). Structurally, mitochondria consist of an inner (MIM) and outer membrane (MOM), which surround the mitochondrial matrix compartment. The MIM contains many folds (cristae) that protrude into the matrix thereby providing a large surface area for biochemical reactions. Alterations in metabolic state are generally paralleled by dynamic changes in mitochondrial internal and external structure, matrix volume, and physicochemical properties of the matrix solvent (Hackenbrock, 1966; Perkins & Ellisman, 2011; Willems et al, 2015; Bulthuis et al, 2019). It is to be expected that these changes are functionally relevant through affecting matrix solute diffusion and biochemical reaction dynamics (Scalettar et al, 1991; Lizana et al, 2008; Perkins & Ellisman, 2011; Dieteren et al, 2011a; Cogliati et al, 2016; Sprenger & Langer, 2019).
Diffusion‐limited bioreactions often involve “target finding” by (partially) mobile enzymes and/or substrates that need to diffuse towards a binding partner (Haggie & Verkman, 2002; Wilkens et al, 2013; Dey & Bhattacherjee, 2019). An example of such a system is the biogenesis of OXPHOS Complex I (CI), which involves matrix diffusion of an 100–200 kDa assembly intermediate in search of a MIM‐attached scaffold (Dieteren et al, 2008, 2011b; Koopman et al, 2010; Guerrero‐Castillo et al, 2017). However, our quantitative understanding of solute diffusion in the mitochondrial matrix is still limited. A key parameter in this respect is the solvent‐dependent diffusion constant of the solute (Dsolvent; Weiss, 2014). For any given solute, the value of Dsolvent is linked to the physicochemical properties of the solvent like viscosity (ηsolvent), absolute temperature (T), and solute properties like molecular structure and molecular weight (MW). The former is quantitatively described by the radius of gyration (RG) and hydrodynamic radius (RH) of the solute (Einstein, 1905; Sutherland, 1905; von Smoluchowski, 1906; Young et al, 1980; Tyn & Gusek, 1990; He & Niemeyer, 2003; Saxton, 2014). With respect to solute diffusion in the mitochondrial matrix, it has been previously demonstrated that mitochondrial cristae can act as diffusion barriers that impede the free diffusion of fluorescent proteins (FPs; Ölveczky & Verkman, 1998; Partikian et al, 1998; Dieteren et al, 2011a).
In addition to cristae, macromolecular crowding is also a potential key factor influencing solute diffusion by increasing effective solvent viscosity and reducing the water‐accessible volume via volume exclusion, the extent of which increases with crowder size (Boersma et al, 2015; Rivas & Minton, 2016). In this way, macromolecular crowding can alter the hydrodynamic volume (compaction, shape, folding, structure, and conformational stability) and/or association state (protein–protein interactions, aggregation, oligomerization, and phase separation) of proteins (Kuznetzova et al, 2014). This will affect their chemical reaction rates and/or molecular target finding (Dey & Bhattacherjee, 2019; Weilandt & Hatzimanikatis, 2019). Macromolecular crowding can also induce “molecular sieving” effects, which alter the translational mobility of biomolecules in an MW/size‐dependent manner (Papadopoulos et al, 2000; Mika et al, 2010; Junker et al, 2019; Van Tartwijk & Kaminski, 2022). In this sense, macromolecular crowding will reduce the diffusion coefficient of larger tracer molecules to a greater extent than predicted by the Stokes–Einstein equation (Mika et al, 2010; Delarue et al, 2018; Junker et al, 2019). Experimental and theoretical studies suggest that modulation of macromolecular crowding can act as a (patho)physiological control mechanism in prokaryotes and eukaryotes (Scalettar et al, 1991; Akabayov et al, 2013; Poggi & Slade, 2015; Cravens et al, 2015; Joyner et al, 2015; Hansen et al, 2016; Delarue et al, 2018; Schavemaker et al, 2018; Fuentes‐Lemus et al, 2021; Hochmair et al, 2022; Vorontsova et al, 2022). The mitochondrial matrix compartment is classically recognized as highly protein‐rich. For instance, 46% of all mitochondrial proteins were predicted to be matrix‐localized in humans (Rath et al, 2021) and 20% of the mitochondrial proteins were classified as matrix‐soluble in Saccharomyces cerevisiae (Vögtle et al, 2017). Therefore, it appears likely that the mitochondrial matrix fluid displays macromolecular crowding‐related phenomena. However, given the biological functions of these matrix proteins (e.g., metabolism, mtDNA maintenance, OXPHOS assembly; Rath et al, 2021), they might not always be expressed at the same levels and/or could be (transiently) MIM‐attached or otherwise immobilized. For example, four of the most abundant mitochondrial proteins (ATP5A1, ATP5B, ANT1, and ANT2) are membrane‐bound (Calvo & Mootha, 2010). In addition, TCA cycle enzymes and subunits of the fatty acid β‐oxidation multienzyme complex appear to be organized in (MIM‐attached) multiprotein complexes with restricted mobility (Robinson & Srere, 1985; Partikian et al, 1998; Haggie & Verkman, 2002; Fernie et al, 2018). This suggests that the protein concentration of the mitochondrial matrix solvent might be (substantially) lower than generally assumed. Moreover, quantitative live‐cell evidence demonstrating the existence of macromolecular crowding‐related phenomena in the mitochondrial matrix fluid is lacking. Here, we aimed at providing such evidence by quantifying the mobility of matrix‐targeted fluorescent protein (FP) concatemers of increasing MW (AcGFP1, AcGFP12, AcGFP13, and AcGFP14). To this end, fluorescence recovery after photobleaching (FRAP) experiments and Brownian dynamics (BD) simulations were integrated to obtain quantitative estimates of Dsolvent and ηsolvent in functionally active and dysfunctional mitochondria. The obtained results support the conclusion that the mitochondrial matrix fluid displays macromolecular crowding and that this crowding is increased during chloramphenicol (CAP)‐induced mitochondrial dysfunction.
Results
A key phenomenon associated with macromolecular crowding is that it reduces the translational mobility of solutes with a similar or larger size than the crowder to a greater extent than the mobility of smaller solutes (e.g., Mika et al, 2010; Delarue et al, 2018; Junker et al, 2019; Van Tartwijk & Kaminski, 2022). The solvent‐dependent translational mobility of a solute in the mitochondrial matrix is quantitatively described by its solvent‐dependent diffusion constant (Dsolvent). Therefore, we here quantified Dsolvent values of mitochondria‐targeted AcGFP1 concatemers of increasing MW (AcGFP1, AcGFP12, AcGFP13, and AcGFP14) in HeLa cells. To this end, fluorescence recovery after photobleaching (FRAP) analysis of these fluorescent proteins (FPs) was combined with predictive, experimentally constrained, Brownian dynamics (BD) computer simulations. This strategy aimed at delivering quantitative estimates for Dsolvent and matrix solvent viscosity (ηsolvent) in functional mitochondria, as well as in dysfunctional mitochondria treated with the mitochondrial RNA (mtRNA) translation inhibitor chloramphenicol (CAP). Interpretation of these estimates was used to gain insight into macromolecular crowding, concatemer structural conformation, and ηsolvent in the mitochondrial matrix (Fig 1).
Figure 1. Integrated strategy for quantifying the solvent‐dependent diffusion constants of mitochondrial matrix‐targeted fluorescent proteins and mitochondrial matrix solvent viscosity.
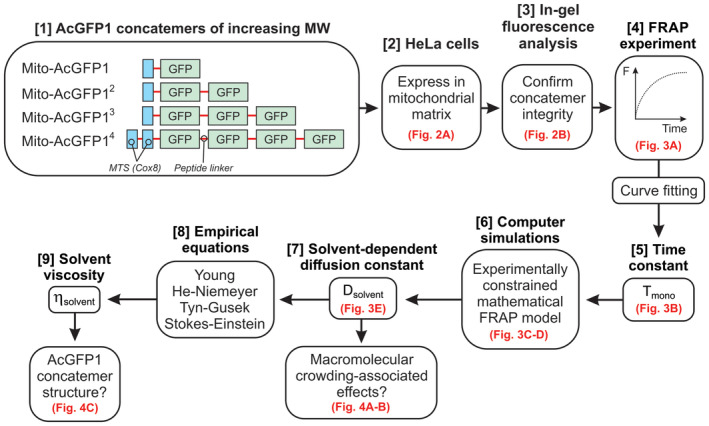
Corresponding figures are highlighted in red. Four mitochondrial matrix‐targeted concatemers of the monomeric fluorescent protein AcGFP1 [1] were stably expressed in HeLa cells [2]. The integrity and mobility of these fluorescent proteins (FPs) was investigated by in‐gel fluorescence analysis [3] and fluorescence recovery after photobleaching (FRAP) recordings [4], respectively. Curve fitting was applied to obtain a mono‐exponential FRAP time constant for each concatemer (Tmono) [5]. The latter were fed into an experimentally constrained mathematical FRAP model [6] to compute solvent‐dependent diffusion constants (Dsolvent; [7]). By inserting these constants into four empirical equations [8], the viscosity of the mitochondrial matrix solvent (ηsolvent) was determined [9]. Finally, the obtained ηsolvent and Dsolvent values were interpreted to draw conclusions on the structural conformation of the concatemers and the presence of macromolecular crowding phenomena in the mitochondrial matrix.
Expression, localization, and integrity of mitochondrial matrix‐targeted FPs
Four HeLa cell lines were created that stably expressed matrix‐targeted AcGFP1 concatemers. These proteins were targeted to the mitochondrial matrix using N‐terminal fusions with a mitochondrial targeting sequence (MTS), consisting of a 25‐residue Cox8 sequence. In case of AcGFP14, a double MTS was used to improve mitochondrial matrix delivery (Filippin et al, 2005). AcGFP1 and MTS domains were connected by flexible peptide linkers (Appendix Table S1), and FP expression was controlled by a doxycycline (DOX)‐inducible promoter. Following induction, all FPs displayed a mitochondrial localization as demonstrated by confocal microscopy and MitoTracker Red co‐staining (Fig 2A). Western blot analysis of mitochondrial fractions demonstrated that DOX treatment increased the in‐gel fluorescence and expression of the FPs (Fig 2B; marked by *). AcGFP1 and AcGFP12 displayed a single major fluorescent product, compatible with our previous findings with DOX‐inducible HEK293 cells (Dieteren et al, 2008, 2011a, 2011b). Similarly, AcGFP13‐expressing cells displayed a single fluorescence band albeit of much lower intensity. By contrast, mitochondria in AcGFP14‐expressing cells contained two fluorescence products, one of the expected size (Fig 2B; marked by *) and another of lower MW (marked by #). Fluorescence analysis by flow cytometry (Fig 2C) correlated well with in‐gel fluorescence signals (Fig 2D). This demonstrates that all inducible FP cell lines exhibit a low fluorescence signal in their noninduced state and strongly suggests that the cellular fluorescence intensities reflect mitochondria‐specific signals. The former observation is compatible with low‐level induction (“leakage”) of FP expression in the mitochondrial matrix occurring in the absence of added DOX, as observed previously in our DOX‐inducible HEK293 cell models (Dieteren et al, 2011b).
Figure 2. Expression and localization of the mitochondrial matrix‐targeted fluorescent proteins.
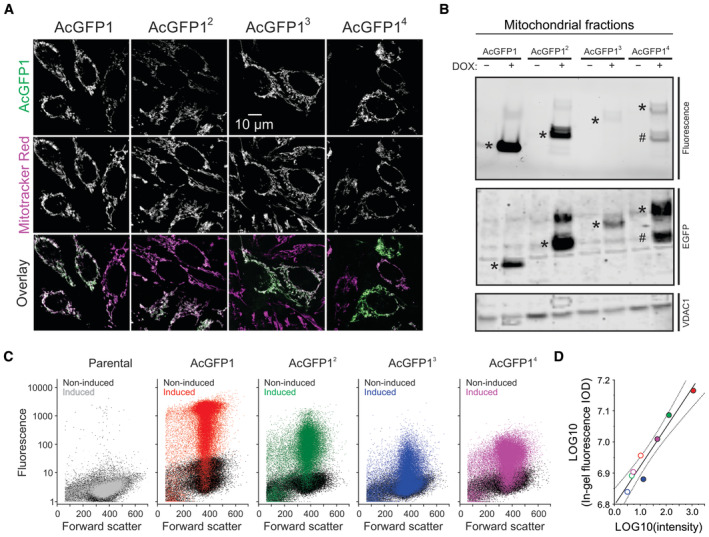
- Cellular localization of the four AcGFP1 concatemers (AcGFP1, AcGFP12, AcGFP13, AcGFP14) in HeLa cell lines cultured in the presence of the expression inducer doxycycline (DOX). Images were acquired by confocal microscopy and depict typical fluorescence signals of AcGFP1 (upper row), the mitochondrial marker Mitotracker Red CM‐H2XROS (middle row), and an overlay of these signals (lower row; AcGFP1 in green; Mitotracker Red CM‐H2XROS in magenta; colocalization in white).
- Analysis of FP expression and concatemer integrity in mitochondrial fractions isolated from HeLa cells cultured in the absence (−) and presence (+) of DOX. The panels display AcGFP1 fluorescence (SDS–PAGE fluorogram; upper panel), AcGFP1 protein signals (SDS–PAGE; anti‐EGFP immunodetection; middle panel) and VDAC1 protein signals (SDS–PAGE; loading control; lower panel). Bands for AcGFP1, AcGFP12, AcGFP3 and AcGFP14 are marked by asterisks (*). For AcGFP14, also a substantial band of lower MW was detected (marked #). Individual panels were contrast‐optimized for visualization purposes. Original blots are presented in Appendix Fig S1A.
- Flow cytometry analysis of the parental and FP‐expressing HeLa cell lines. Fluorescence intensity is plotted against forward scatter. The noninduced condition (i.e., in absence of DOX) is depicted by black symbols. The effects of DOX are marked in gray (parental cells) and various colors (FP‐expressing cells). For AcGFP1, AcGFP12 (noninduced), AcGFP13 and AcGFP14 a total of n = 40,000 cells from four technical replicates were analyzed on a single day (N = 1). For the AcGFP12‐induced condition n = 30,000 cells from three technical replicates are depicted (N = 1).
- Correlation between the average AcGFP1 fluorescence intensity in panel (C) (x‐axis) and the integrated optical density (IOD; y‐axis) for each condition (full lane) in panel (B) (in‐gel fluorescence signal; the original gel was used for quantification). Noninduced cells are marked by open symbols; induced cells are marked with colored symbols (similar to panel C). The continuous line represents a linear fit to the data (R = 0.966; P < 0.0001). Dotted lines indicate the 95% confidence limits of the fit.
Experimental FRAP analysis of mitochondrial matrix‐targeted FP mobility
FP mobility in the mitochondrial matrix was determined by confocal FRAP analysis of individual mitochondria. In this approach, mitochondrial fluorescence is rapidly bleached in a square region of interest (ROI) placed at one end of a single mitochondrial filament (Fig 3A; inset), followed by quantification of fluorescence recovery (Appendix Fig S2). As a quality control measure, this analysis included only individual mitochondria: (i) that were fully located within the focal plane (confirmed by an axial scan), and (ii) in which FRAP was paralleled by fluorescence loss in photobleaching (FLIP) in a part distal to the FRAP region (Appendix Fig S2), indicating that these mitochondria possessed a continuous matrix. Combining multiple experiments yielded an average FRAP curve for each FP (Fig 3A), which was adequately fitted by a mono‐exponential equation (Table 1). The recovery time constant of this fit (Tmono), representing the FRAP rate, increased with increasing MW (Table 1 and Fig 3B). This agrees with our previous results with mitochondrial matrix‐targeted AcGFP11 and AcGFP12 in HEK293 cells (Dieteren et al, 2011a) and demonstrates that FRAP is slower for FPs with a larger MW. The FP mobile fraction (Fm) somewhat decreased with increasing MW (Table 1 and Fig 3A), suggesting that the FPs became progressively immobilized. A proper comparison of Tmono between the different FPs requires that their MW is taken into account (Sprague & McNally, 2005). In case of AcGFP14, the fluorescent product of lower MW (Fig 2B) accelerated the average FRAP rate for this FP leading to an underestimation of Tmono. To correct for this effect, a straight line was fitted through the origin and the Tmono data points for AcGFP1, AcGFP12, and AcGFP13 (Fig 3B). Next, the measured Tmono value for AcGFP14 was projected on the fitted line yielding a corrected (“AcGFP14‐corr”) value (Fig 3B; dotted line and open symbol). This corrected Tmono value was used in the remainder of the study.
Figure 3. Solvent‐dependent mobility of the mitochondrial matrix‐targeted fluorescent proteins.
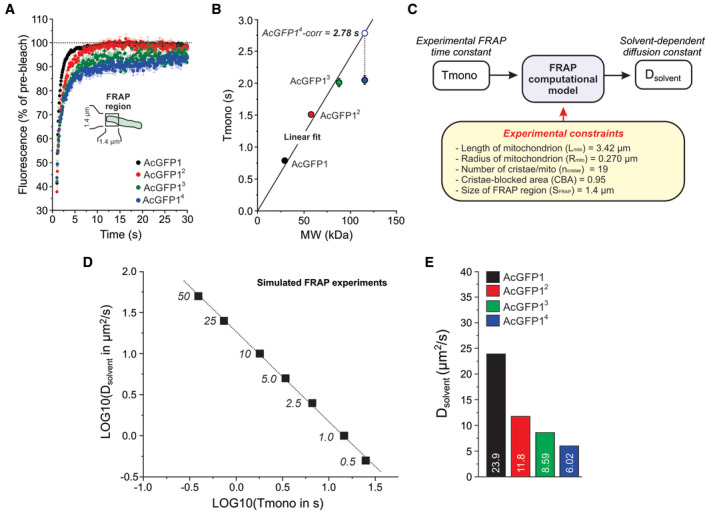
- Average mitochondrial fluorescence recovery after photobleaching (FRAP) curves for the four FPs. These curves were fitted using a mono‐exponential function to determine the FRAP recovery time constant Tmono. Symbols and error bars indicate mean ± SEM. Data was obtained in at least N = 3 independent experiments for n = 76 mitochondria (AcGFP1), n = 30 (AcGFP12), n = 37 (AcGFP13) and n = 68 (AcGFP14).
- Relationship between the molecular weight (MW) of the FPs and their FRAP time constant (Tmono; numerical data are provided in Table 1). A linear fit was calculated through the origin and the data points for AcGFP1, AcGFP12 and AcGFP13 (R = 0.993, P = 0.022; Tmono = B·MW; B (slope) = 0.0241 ± 0.00118 (SE)). The experimental Tmono value for AcGFP4 (filled blue symbol) was corrected using this linear fit (dotted line; see Results for details) yielding a new Tmono value (“AcGFP14‐corr”). Symbols and error bars reflect mean ± SE (standard error) values from the mono‐exponential fit.
- Strategy to compute Dsolvent from the experimental Tmono value for each FP using an experimentally constrained BD simulation model (see Results and Appendix Supplementary Methods for details).
- Linear relationship between LOG10 (Tmono) and LOG10 (Dsolvent) predicted by the model for a mitochondrion containing 19 cristae and with a cristae‐blocked area (CBA) of 0.95. Parameters of the linear fit (LOG10 (Dsolvent) = A + B·LOG10 (Tmono)) are: R = −0.999, P < 0.0001; A (intercept) = 1.27 ± 0.0123 (SE); B (slope) = −1.10 ± 0.0154 (SE)). Numerals indicate the Dsolvent values (μm2/s) used in the model (the corresponding FRAP data are presented in Appendix Fig S3B).
- Dsolvent values of the four FPs (numerals) obtained using the relationship in panel (D) by inserting the experimental Tmono values in the fitting equation for panel (D) (see Results for details). Numerical data are provided in Table 1.
Table 1.
Experimental data and BD modeling results.
| −CAP | +CAP | |||||||
|---|---|---|---|---|---|---|---|---|
| Cell line | AcGFP1 | AcGFP12 | AcGFP13 | AcGFP14 | AcGFP1 | AcGFP12 | AcGFP13 | AcGFP14 |
| MW (kDa) a | 29.155 | 57.586 | 87.705 | 115.337 | 29.155 | 57.586 | 87.705 | 115.337 |
| LOG10 (MW) | 1.4641 | 1.7603 | 1.9430 | 2.06197 | 1.4641 | 1.7603 | 1.9430 | 2.06197 |
| Mitochondrial length, diameter and radius determined using confocal laser scanning microscopy (CLSM) b | ||||||||
| Lmito (μm) | 3.42 (N = 55) | 3.42 (N = 30) | 3.42 (N = 37) | 3.42 (N = 68) | 3.40 (N = 31) | ND | ND | 3.42 (N = 26) |
| Dmito (μm) | 0.562 ± 0.025 (N = 26) | 0.524 ± 0.012 (N = 41) | 0.549 ± 0.020 (N = 33) | 0.525 ± 0.017 (N = 29) | 0.552 ± 0.015 (N = 24) | ND | ND | 0.546 ± 0.017 (N = 20) |
| Rmito (μm) | 0.281 | 0.262 | 0.275 | 0.263 | 0.276 | ND | ND | 0.273 |
| Vmito (μm3) | 0.848 | 0.738 | 0.813 | 0.743 | 0.819 | ND | ND | 0.801 |
| Vmito (l) | 0.848·10−15 | 0.738·10−15 | 0.813·10−15 | 0.743·10−15 | 0.819·10−15 | ND | ND | 0.801·10−15 |
| Mitochondrial diameter, radius and cristae per mitochondrion determined using electron microscopy (EM) c | ||||||||
| Dmito (μm) | 0.510 ± 0.019 (N = 52) | ND | ND | 0.504 ± 0.015 (N = 59) | 0.490 ± 0.017 (N = 51) | ND | ND | 0.494 ± 0.018 (N = 53) |
| Rmito (μm) | 0.255 | ND | ND | 0.252 | 0.245 | ND | ND | 0.247 |
| ncristae (1/μm) | 5.620 ± 0.155 (N = 244) | ND | ND | 5.462 ± 0.174 (N = 314) | 2.705 ± 0.130 (N = 295) | ND | ND | 3.50 ± 0.145 (N = 260) |
| ncristae/mito | 19 | ND | ND | 19 | 9 | ND | ND | 12 |
| Mono‐exponential fitting parameters of the fluorescence recovery after photobleaching (FRAP) curve d | ||||||||
| N | 76 | 30 | 37 | 68 | 40 | 24 | 26 | 33 |
| R 2 | 0.922 | 0.971 | 0.932 | 0.921 | 0.962 | 0.947 | 0.780 | 0.951 |
| y0 | −80.0 ± 5.85 | −0.677 ± 2.42 | 24.7 ± 2.29 | 30.7 ± 2.17 | 39.1 ± 1.20 | 30.9 ± 0.800 | 44.2 ± 0.920 | 47.5 ± 0.767 |
| Amono | 178 ± 5.84 | 98.9 ± 2.40 | 68.6 ± 2.26 | 60.5 ± 2.14 | 55.4 ± 1.17 | 45.1 ± 0.667 | 23.6 ± 0.763 | 39.0 ± 0.706 |
| Tmono (s) | 0.789 ± 0.015 | 1.51 ± 0.031 | 2.01 ± 0.065 | 2.05 ± 0.066 | 2.75 ± 0.099 | 8.76 ± 0.357 | 9.31 ± 0.883 | 5.43 ± 0.168 |
| Tmono‐corr (s) | 2.78 | |||||||
| F0 | 41.4 ± 1.61% | 37.7 ± 2.40% | 42.0 ± 1.80% | 43.6 ± 1.22% | 42.0 ± 2.60% | 22.7 ± 3.38% | 40.2 ± 4.51% | 42.6 ± 2.50% |
| F∞ | 98.8 ± 11.7% | 98.2 ± 4.82% | 93.3 ± 4.55% | 91.2 ± 4.31% | 94.5 ± 2.37% | 76.0 ± 1.47% | 67.8 ± 1.68% | 86.5 ± 1.47% |
| Fm | 0.979 | 0.972 | 0.885 | 0.844 | 0.904 | 0.689 | 0.462 | 0.765 |
| Parameters and results regarding the BD model e | ||||||||
| 1. Simulation parameters for Dsolvent prediction | ||||||||
| Identical for all simulations | Lmito = 3.42 μm; Rmito = 0.270 μm; SFRAP = 1.4 μm; CBA = 0.95 | |||||||
| ncristae | 19 | 19 | 19 | 19 | 9 | ND | ND | 12 |
| 2. Predicted Dsolvent | ||||||||
| Dsolvent (μm2/s) | 23.9 | 11.8 | 8.59 | 6.02 | 2.91 | NA | NA | NA |
| LOG10 (Dsolvent) | 1.378 | 1.072 | 0.9340 | 0.7796 | 0.464 | NA | NA | NA |
| 3. Computation of radius of gyration (RG), hydrodynamic radius (RH) and ηsolvent (cP) at 293 K | ||||||||
| MW (kDa) | 29.155 | 57.586 | 87.705 | 115.337 | 29.155 | 57.586 | 87.705 | 115.337 |
| Assuming that the FP has a compact conformation | ||||||||
| Radius of molecule R (Å) | 15 | 20 | 20 | 20 | 15 | NA | NA | NA |
| Length of molecule L (Å) | 40 | 60 | 60 | 60 | 40 | NA | NA | NA |
| RG (Å) | 15.5 | 20 | 20 | 20 | 15.5 | NA | NA | NA |
| RH (Å) | 20 | 23 | 23 | 23 | 20 | NA | NA | NA |
| ηsolvent (Young) | 3.32 | 5.36 | 6.40 | 8.34 | 27.3 | NA | NA | NA |
| ηsolvent (He–Niemeyer) | 3.69 | 5.88 | 7.53 | 10.3 | 30.3 | NA | NA | NA |
| ηsolvent (Tyn–Gusek) | 4.57 | 7.18 | 9.86 | 14.1 | 37.5 | NA | NA | NA |
| ηsolvent (Stokes–Einstein) | 4.49 | 7.91 | 10.9 | 15.5 | 36.9 | NA | NA | NA |
| Assuming that the FP has an extended conformation | ||||||||
| Radius of molecule R (Å) | 15 | 15 | 35 | 35 | 15 | NA | NA | NA |
| Length of molecule L (Å) | 40 | 130 | 247 | 349 | 40 | NA | NA | NA |
| RG (Å) | 15.5 | 38 | 73 | 102 | 15.5 | NA | NA | NA |
| RH (Å) | 20 | 30 | 61 | 75 | 20 | NA | NA | NA |
| ηsolvent (Young) | 3.32 | 5.36 | 6.40 | 8.34 | 27.3 | NA | NA | NA |
| ηsolvent (He–Niemeyer) | 3.69 | 4.27 | 3.94 | 4.55 | 30.3 | NA | NA | NA |
| ηsolvent (Tyn–Gusek) | 4.57 | 3.78 | 2.70 | 2.76 | 37.5 | NA | NA | NA |
| ηsolvent (Stokes–Einstein) | 4.49 | 6.06 | 4.10 | 4.75 | 36.9 | NA | NA | NA |
BD, Brownian dynamics; CAP, chloramphenicol; Dsolvent, solvent‐dependent diffusion constant; ηsolvent, solvent‐dependent viscosity; NA, not appropriate; ND, not determined; RG, radius of gyration; RH, hydrodynamic radius; SFRAP, size of the experimental FRAP region in the BD model; T, temperature; Tmono, experimental FRAP mono‐exponential time constant.
Molecular weight (MW) was calculated directly from the protein sequences given in Appendix Table S1 (excluding the N‐terminal mitochondrial targeting sequence) using the pI/Mw tool (web.expasy.org/compute_pi). One (1) Dalton (Da) equals 1 g/mol.
Confocal laser scanning microscopy (CLSM) analysis: The data are presented as mean ± SEM. N indicates the number of mitochondria analyzed in at least two independent experiments. Mitochondrial length (Lmito) and mitochondrial diameter (Dmito) were determined from the FRAP images by quantifying the frame width at half‐maximal height (FWHM; equaling 2∙Rmito) of a Gaussian curve fitted to a 1 pixel wide intensity profile perpendicular to the short and long axis of the mitochondrial filament (see Appendix Fig S2F). Mitochondrial volume (Vmito) was calculated using a cylindrical approximation: Vmito = π ·(Rmito)2·Lmito. One (1) μm3 equals 10−15 liter, 0.001 picoliter (1 picoliter = 10−12 liter) and 1,000 attoliter (1 attoliter = 10−18 liter). Average Vmito values equaled 0.786 ± 0.0540(SD) μm3 = 786 attoliter (−CAP) and 0.810 μm3 = 810 attoliter (+CAP).
Electron microscopy (EM) analysis: The data are presented as mean ± SEM. N indicates the number of mitochondria analyzed in two independent experiments. Dmito and Rmito indicate the mitochondrial diameter and radius, respectively.
Fitting of the fluorescence recovery after photobleaching (FRAP) curve: y = y0 + Amono[1−EXP(−t/Tmono)]. N indicates the number of mitochondria analyzed in at least three independent experiments. The coefficient of determination (R2) is used as a measure of the goodness of fit (the closer R2 is to a value of one, the closer the fit is to the data points). The parameter errors reflect the standard error (SE) value from the fit, as reported by the fitting software. F0 indicates the fluorescence signal at the start of the fluorescence recovery (expressed as % of the prebleach value). F∞ indicates the fluorescence signal to which the fluorescence signal recovers given by: y0 + Amono (expressed as % of the prebleach value). The error in F∞ was calculated by summation of the errors in y0 and Amono. Fm indicates the mobile fraction given by: [(F∞−F0+)/(Fi−F0)], with Fi being the prebleach fluorescence equaling 100%.
Mathematical modeling: One (1) Angstrom (Å) equals 1·10−10 m. CBA, cristae‐blocked area (fraction of total transecting area). See Results for further details.
Brownian dynamics simulations of FRAP experiments
To convert the experimental Tmono data into Dsolvent values, we developed a Brownian dynamics (BD) simulation model (Fig 3C). This quantitative model generated synthetic FRAP curves, used a cristae‐containing cylindrical structure to represent mitochondria, and was constrained by experimental data. The latter included mitochondrial length (Lmito), mitochondrial radius (Rmito), size of the FRAP region (SFRAP), the number of cristae per mitochondrion (ncristae), and the “cristae‐blocked area” (CBA; expressed as a fraction of the total transecting area). Validated by EM analysis and data on HeLa cristae structure from the literature (Appelhans et al, 2011; Wilkens et al, 2013; Wolf et al, 2019; Hu et al, 2020; Segawa et al, 2020; Weissert et al, 2021) we assumed in the model that (Appendix Fig S3A): (i) mitochondria contained regularly arranged cristae perpendicular to the longitudinal axis of the mitochondrion, (ii) the positions of the cristae alternated, with consecutive cristae being opposite of each other, (iii) cristae were equidistant, partially overlapping and of negligible thickness (Partikian et al, 1998), and (iv) CBA equaled 0.95. The presence of cristae increased the effective length of the “channel” that connected the two ends of the mitochondrion thereby increasing FP diffusion length. In addition, the presence of cristae reduced the diffusive flow between consecutive mitochondrial subcompartments. Therefore, as indicated previously (Ölveczky & Verkman, 1998; Partikian et al, 1998), FRAP was slower when more cristae were present (Appendix Fig S3B and C). Further details of the BD model are presented in the Appendix Supplementary Methods.
Determining the Dsolvent value of mitochondria‐targeted FPs
Confocal microscopy analysis of living FP‐expressing cells yielded values for Lmito and Rmito of 3.42 and 0.262–0.281 μm, respectively (Table 1). Similar Rmito values (0.252–0.255 μm) were obtained by electron microscopy (EM) analysis of fixed AcGFP1 and AcGFP14 cells (Table 1). EM quantification of the number of cristae yielded values of 5.620 cristae/μm (AcGFP1‐expressing cells) and 5.462 cristae/μm (AcGFP14‐expressing cells). This reflects an average ncristae value of 19 (Table 1) and an intercrista distance between 177 and 183 nm. The latter lies within a range of experimental values previously reported for HeLa cells (51–250 nm; Wilkens et al, 2013; Stephan et al, 2019; Wang et al, 2019). Applying these experimental constraints, BD simulations were carried out using the following parameters (Table 1): Lmito = 3.42 μm, Rmito = 0.270 μm, ncristae = 19 and SFRAP = 1.4 μm. Model output was determined for several values of Dsolvent (0.5–50 μm2/s) to establish the relationship between Tmono and Dsolvent. On a logarithmic scale, these parameters displayed a linear correlation (Fig 3D), allowing calculation of Dsolvent for the experimental Tmono values (Fig 3E). This yielded values of 23.9 μm2/s (AcGFP1), 11.8 μm2/s (AcGFP12), 8.59 μm2/s (AcGFP13), and 6.02 μm2/s (AcGFP14).
The mitochondrial matrix solvent reduces FP mobility in a manner compatible with macromolecular crowding
The obtained Dsolvent values (Fig 3E) reflect solvent‐dependent FP diffusion in the mitochondrial matrix. To address our primary research question on macromolecular crowding, we first compared these values with empirical data sets of free 3D diffusion of globular biomolecules in aqueous media (Fig 4A; details are provided in Appendix Tables S2 and S3). The LOG10 (Dsolvent) value of these molecules decreased linearly as a function of their LOG10 (MW) value, with a slope of −0.36 (Fig 4B). The latter is compatible with the theoretical proportionality of Dsolvent with the molecular radius, and hence MW−0.33, for globular molecules (Dross et al, 2009). Analysis of EGFP concatemer diffusion in aqueous solution suggested that these molecules display a globular conformation, that is, the open blue circles (Fig 4A) appear to be on the line fitted for the globular proteins (black symbols). However, the underlying experimental studies provided evidence that EGFP concatemers display an elongated molecular structure in aqueous solution (Pack et al, 2006; Vámosi et al, 2014). Therefore, we added the EGFP concatemers to the data set of the elongated molecules in aqueous solution (Fig 4A; open and filled blue symbols). For elongated molecules (Saxton, 2014), Dsolvent decreased faster as a function of MW than for globular molecules (Fig 4A; black vs. blue line) and a slope of −0.72 was found (Fig 4B). The latter is compatible with experimental data for elongated (i.e., supercoiled) plasmid DNA molecules in dilute aqueous solution where Dsolvent is proportional to MW−0.66 (Prazeres, 2008). Quantification of EGFP concatemer diffusion in HeLa cell nuclei and cytosol yielded steeper slopes relative to aqueous solution (Fig 4A; open red squares, filled red circles, and Fig 4B; open red bars). These slopes were relatively close to the theoretical value for elongated molecules (−0.66), suggesting that these EGFP concatemers displayed an elongated molecular structure. The latter was experimentally confirmed in the underlying studies (Pack et al, 2006; Dross et al, 2009). In case of mitochondrial matrix‐targeted FPs, a slope of −0.98 was obtained (Fig 4A; filled red squares, and Fig 4B; filled red bar). This decline was faster than for globular molecules in aqueous solution (2.7‐fold), elongated molecules in aqueous solution (1.4‐fold), elongated EGFP concatemers in the HeLa cell nucleus (1.3‐fold), and elongated EGFP concatemers in HeLa cytosol (1.5‐fold). Of note, the LOG10 (Dsolvent) value for AcGFP14 fell exactly on the fitted line (Fig 4A; filled red square for highest MW), supporting the validity of the applied Tmono correction (Fig 3B). Because the data points for the mitochondrial matrix‐targeted FPs are not parallel to the “aqueous” lines for the globular and elongated molecules (Fig 4A and B), the steeper decline in Dsolvent for the matrix‐targeted concatemers cannot be explained by differences in solvent viscosity. The steeper decline is also not due to cristae‐induced diffusion hindrance, since ncristae was similar for AcGFP1‐ and AcGFP14‐expressing cells (Table 1) and our modeling approach delivers cristae‐independent Dsolvent values. Integrating the above results, we conclude that the MW‐dependent mobility reduction of AcGFP1 concatemers in the mitochondrial matrix is not only due to their elongated structure but also involves macromolecular crowding.
Figure 4. Relationship between Dsolvent and MW for the mitochondrial matrix‐targeted fluorescent proteins, viscosity analysis and protein structure prediction.
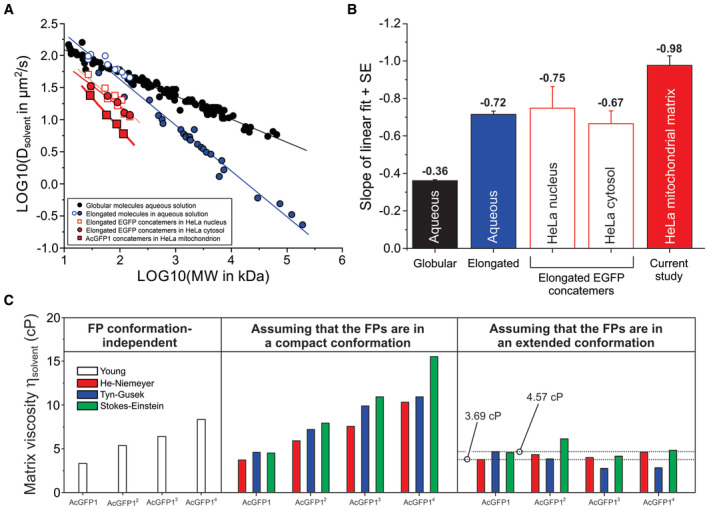
- Linear relationship between experimental Dsolvent and MW values for various biomolecules in a double logarithmic plot. Data obtained in aqueous solution is separated into globular biomolecules (filled black symbols; continuous black line), elongated EGFP concatemers (open blue symbols; continuous blue line) and elongated biomolecules (filled blue symbols; continuous blue line). Red symbols reflect intracellular diffusion measurements with EGFP concatemers in HeLa cell nuclei (open red squares; dotted red line), EGFP concatemers in HeLa cytosol (filled red circles; continuous thin red line) and AcGFP1 concatemers in the mitochondrial matrix (filled red squares; continuous thick red line; current study). Full numerical data, fitting parameters using (LOG10 (Dsolvent) = A + B·LOG10 (MW) and background information are provided in Appendix Tables S2 and S3.
- Slopes of the linear fits depicted in panel (A) for the different classes of biomolecules measured in aqueous solution and intracellular compartments. Individual bars reflect mean ± SE (standard error) values from the linear fit.
- The viscosity of the mitochondrial matrix solvent (ηsolvent) was calculated from the Dsolvent values of the FPs using four empirical equations (Young, He–Niemeyer, Tyn–Gusek and Stokes–Einstein). This allowed the prediction of ηsolvent as well as FP structural conformation (“compact” or “extended”; Fig EV1). The estimated ηsolvent ranged between 3.69 and 4.57 cP. See Results for details.
Quantifying the viscosity of the mitochondrial matrix solvent and predicting FP structural conformation
To gain further insight into the structural conformation of the mitochondrial matrix‐targeted FPs, we next determined mitochondrial matrix solvent viscosity (ηsolvent). To this end, we applied four empirical equations allowing prediction of ηsolvent from Dsolvent (Appendix Supplementary Methods): “Young” (Young et al, 1980), “He–Niemeyer” (He & Niemeyer, 2003), “Tyn–Gusek” (Tyn & Gusek, 1990), and “Stokes–Einstein” (Einstein, 1905; Sutherland, 1905; von Smoluchowski, 1906; Perrin, 1936). These equations also accounted for the effects of MW (Young, He–Niemeyer), radius of gyration (RG; He–Niemeyer, Tyn–Gusek), and hydrodynamic radius (RH; Stokes–Einstein) of the FPs. The MW of each concatemer was computed from its protein sequence (Appendix Table S1), whereas RG and RH were derived from structural information (see Table 1 and Fig EV1, and Appendix Supplementary Methods). In case of AcGFP12, AcGFP13, and AcGFP14, two extreme conformations of minimal size (“compact”) and maximal size (“extended,” i.e., elongated) were considered (Fig EV1B–G). The Young equation, which is independent of FP conformation, predicted that ηsolvent increased as a function of concatemer MW (Fig 4C; left panel). Similarly, when assuming that the FPs displayed a “compact” structural conformation also the He–Niemeyer, Tyn–Gusek and Stokes–Einstein equations predicted that ηsolvent increased as a function of MW (Fig 4C; middle panel). By contrast, the predicted ηsolvent values for AcGFP12, AcGFP13, and AcGFP14 were highly similar when assuming that these FPs displayed an “extended” structural conformation (Fig 4C; right panel). This strengthens our above conclusion that the FPs have an extended structure in the mitochondrial matrix solvent. The Dsolvent value of AcGFP1 is the most reliable and easy‐to‐interpret predictor of ηsolvent because AcGFP1‐expressing cells display a single protein band (Fig 2B), and no assumptions regarding AcGFP1 structure are required (Fig EV1A). For AcGFP1 (Fig 4C; right panel) the He–Niemeyer, Tyn–Gusek, and Stokes–Einstein equations predicted ηsolvent values between 3.69 and 4.57 cP (dotted lines). Taken together, these results support the conclusion that AcGFP12, AcGFP13, and AcGFP14 have an elongated structural conformation in the mitochondrial matrix and that the mitochondrial matrix fluid displays a three to five‐fold higher viscosity than water.
Figure EV1. Compact and extended structural conformations of AcGFP1 concatemers.
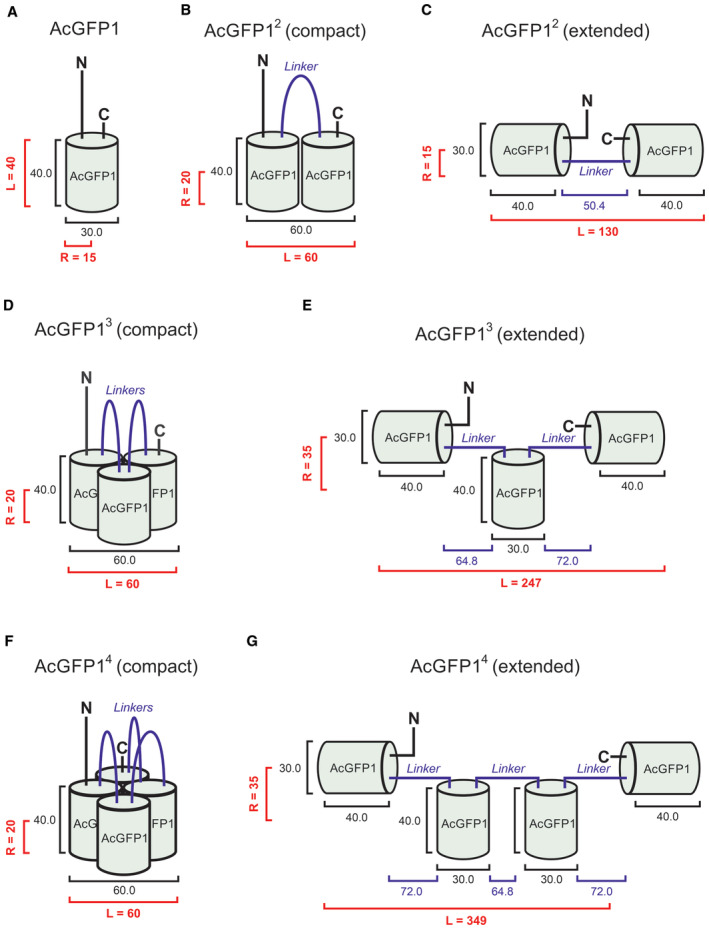
- Dimensions (numerals) of AcGFP1 in Angstrom (Å) based upon GFP crystal structure. R and L indicate the radius and length of the protein, respectively. Geometric data are summarized in Table 1.
- Same as panel (A) but now for AcGFP12 displaying a minimal structural conformation (“compact”).
- Same as panel (A) but now for AcGFP12 displaying a structural conformation of maximal size (“extended”).
- Same as panel (A) but now for AcGFP13 displaying a compact structure.
- Same as panel (A) but now for AcGFP13 displaying an extended structure.
- Same as panel (A) but now for AcGFP14 displaying a compact structure.
- Same as panel (A) but now for AcGFP14 displaying an extended structure.
Chloramphenicol decreases the number of cristae, increases matrix electron density, and partially immobilizes matrix‐targeted FPs
Although the above data provide evidence that FP diffusion in the mitochondrial matrix is reduced by macromolecular crowding, this effect appears to be relatively small. Therefore, we aimed to increase matrix macromolecular crowding levels by treating the cells with chloramphenicol (CAP), which inhibits the biosynthesis of mitochondrial DNA (mtDNA)‐encoded OXPHOS subunits. The latter normally function as MIM‐embedded binding platforms for nDNA‐encoded OXPHOS subunits and/or assembly intermediates during OXPHOS biogenesis (Guerrero‐Castillo et al, 2017; Hock et al, 2020). As such, CAP treatment induces the formation of stable subassemblies of mitochondrial CI with MWs of 170‐, 67‐, and 72‐kDa (Guerrero‐Castillo et al, 2017). Compositional analysis of these subassemblies demonstrated that the 72‐kDa subassembly consisted of the NDUFV1 and NDUFV2 subunit. Previous analysis of mitochondrial matrix protein diffusion in HEK293 cells demonstrated that CAP treatment: (i) particularly increased the Fm of AcGFP1‐tagged NDUFV1 subunits and/or NDUFV1‐containing assembly intermediates, (ii) slowed the FRAP rate of AcGFP1 and AcGFP12, and (iii) did not slow the FRAP rate of the integral MIM protein adenine nucleotide translocase 1 (ANT1; Dieteren et al, 2011b). This demonstrates that NDUFV1 subunits and/or NDUFV1‐containing subassemblies are not MIM‐attached but diffusing in the mitochondrial matrix solvent. Considering the currently known mechanisms of OXPHOS biogenesis (Hock et al, 2020; Fernández‐Vizarra & Ugalde, 2022), it is to be expected that CAP treatment also increases the amount of nDNA‐encoded subunits and/or assembly intermediates from other OXPHOS complexes (i.e., III, IV, and V) in the matrix solvent. CAP treatment did not alter mitochondrial dimensions and FP expression patterns (Table 1 and Appendix Fig S1B). EM analysis demonstrated that the number of cristae/μm was ~twofold reduced (Fig 5A and B, and Table 1), compatible with previous studies in HeLa and HEK293 cells (Lenk & Penman, 1971; Kislev et al, 1973; Dieteren et al, 2011b). Moreover, the mitochondrial matrix was more electron‐dense in CAP‐treated cells (Fig 5C). The FRAP signal recovered slower, that is, Tmono was larger for all FPs in CAP‐treated relative to untreated cells (Fig 5D and E, and Table 1). CAP treatment further induced a substantial decrease in Fm (Fig 5D and Table 1) and F∞ (i.e., the predicted FP fluorescence intensity at t = t∞; Fig 5F), in particular for AcGFP12 and AcGFP13. By contrast, CAP treatment was much less effective in reducing Tmono, Fm, and F∞ for AcGFP14, compatible with the detected fluorescent degradation product of smaller MW (Appendix Fig S1B). CAP‐treated AcGFP12‐ and AcGFP13‐expressing cells displayed a substantial decrease in Fm and F∞, which could not be quantitatively reproduced by the BD simulation model (Appendix Fig S3). In light of the above, prediction of Dsolvent values for AcGFP12, AcGFP13, and AcGFP14 using BD simulations was not possible. However, AcGFP1 displayed Tmono and Fm values that were still reasonably compatible with BD simulations, predicting a Dsolvent of 2.91 μm2/s for an experimental ncristae value of 9 (Table 1; Appendix Fig S3C and D). This predicted an ηsolvent value between 27.3 and 37.5 cP (Fig 5G and Table 1). Collectively, our FRAP analysis of CAP‐treated cells demonstrates that mitochondrial matrix‐targeted FPs become progressively immobilized as a function of MW, associated with an eightfold increase in ηsolvent. These phenomena are compatible with increased macromolecular crowding.
Figure 5. Chloramphenicol decreases the number of cristae, increases matrix electron density, reduces the mobility of mitochondrial matrix‐targeted fluorescent proteins and increases matrix viscosity.
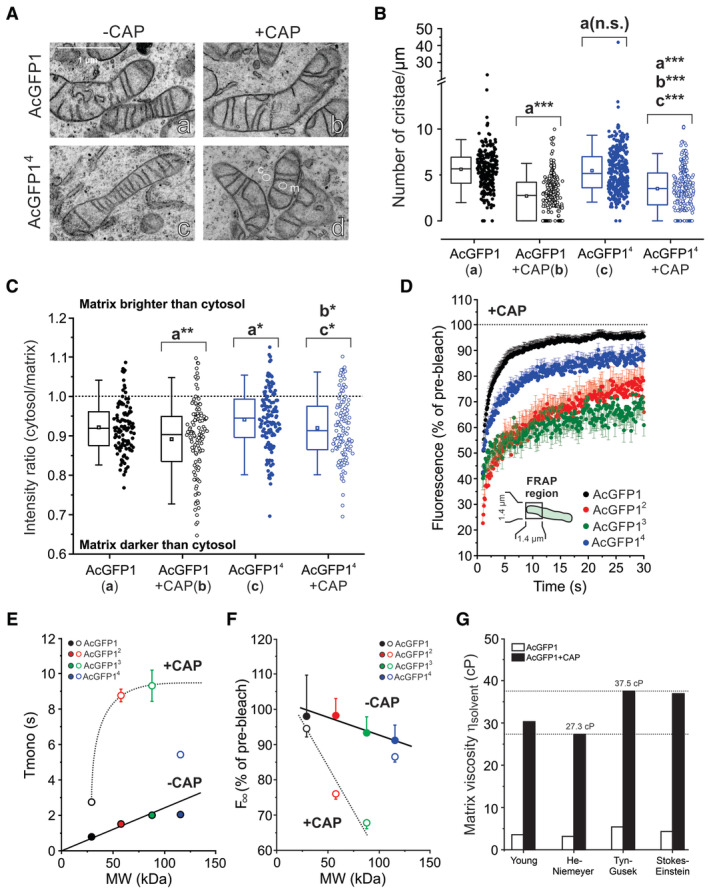
- Transmission electron microscopy (EM) images of AcGFP1 and AcGFP14‐expressing HeLa cells cultured in the absence and the presence of chloramphenicol (CAP). Images were contrast‐optimized to better visualize cristae. Cytosolic and mitochondrial regions of interest typically used for electron density analysis (panel C) are marked by “c” and “m,” respectively.
- Effect of CAP on the average number of cristae/μm calculated by dividing the number of cristae by the mitochondrial length for individual mitochondria. Data was obtained in N = 2 independent EM experiments for n = 244 mitochondria (AcGFP1), n = 295 (AcGFP1 + CAP), n = 314 (AcGFP14) and n = 259 (AcGFP14 + CAP).
- Effect of CAP on the electron density of the mitochondrial matrix quantified from EM images (original images were used). The y‐axis depicts the intensity ratio between a cytosolic region of interest (ROI; e.g., panel A‐d, marked “c”) and a close by ROI in the mitochondrial matrix (e.g., panel A‐d, marked “m”). Equal intensities of these ROIs are marked by the dotted line. Only mitochondria with clearly visible cristae (i.e., that were fully within the EM section) were included (N = 2 independent experiments) from n = 112 mitochondria (AcGFP1), n = 119 (AcGFP1 + CAP), n = 134 (AcGFP14) and n = 116 (AcGFP14 + CAP).
- Average FRAP curves (mean ± SEM) for mitochondrial matrix‐targeted FPs in the presence of CAP (Table 1). Data was obtained in at least N = 3 independent experiments for n = 40 mitochondria (AcGFP1 + CAP), n = 24 (AcGFP12 + CAP), n = 26 (AcGFP13 + CAP) and n = 33 (AcGFP14 + CAP).
- Relationship between MW and Tmono in the absence of CAP (−CAP; data taken from Fig 3B) and in the presence of CAP (+CAP; curve manually drawn). Symbols and error bars reflect mean ± SE (standard error) values from the mono‐exponential fit. For data points without error bar, the latter fell within the size of the symbol.
- Same as panel (E) but now for the relationship between MW and the fluorescence intensity (F∞) of the FPs by extrapolating the fitted exponential recovery to t = t∞ (lines manually drawn). Symbols and error bars for F∞ reflect mean ± SEM and were computed from the mono‐exponential fit parameters as described in Table 1.
- Impact of CAP on the solvent‐dependent viscosity (ηsolvent) of the mitochondrial matrix fluid determined using the Dsolvent values of AcGFP1 (see Results for details). The estimated ηsolvent ranged between 27.3 and 37.5 cP.
Data information: In panels (B and C) each symbol represents an individual mitochondrion, error bars mark the 95% (upper) and 5% (lower) percentile, the boundary boxes mark the 75% (upper) and 25% (lower) percentile, the square marks the mean value of the data, and the horizontal line within the box indicates the median value of the data. Data in panel (B and C) was compared using an independent Student's t‐test and significant differences are indicated by *P < 0.05, **P < 0.01, ***P < 0.001 between the marked conditions (a–c). Not significant is marked by n.s. The exact P‐values for panel (B) were: AcGFP1 (a) vs. AcGFP1 + CAP (b): P = 1.894·10−40; AcGFP1 (a) vs. AcGFP14 + CAP: P = 1.458·10−21; AcGFP1 + CAP (b) vs. AcGFP14 + CAP: P = 4.846·10−5; AcGFP14 (c) vs. AcGFP14 + CAP: P = 2.725·10−16. The exact P‐values for panel (C) were: AcGFP1 (a) vs. AcGFP1 + CAP (b): P = 0.00545; AcGFP1 (a) vs. AcGFP14 (c): P = 0.0348; AcGFP1 + CAP (b) vs. AcGFP14 + CAP: P = 0.0142; AcGFP14 (c) vs. AcGFP14 + CAP: P = 0.0366.
Chloramphenicol impairs mitochondrial function, but neither induces the mitochondrial unfolded protein response, not alters mtDNA copy number and mitochondrial fission/fusion protein levels
Evidence in the literature suggests that both DOX and CAP inhibit mitochondrial function, thereby potentially inducing the mitochondrial unfolded protein response (UPRmt; Houtkooper et al, 2013; Moullan et al, 2015; Shpilka & Haynes, 2018). The latter is classically linked to the accumulation of misfolded proteins in the mitochondrial matrix, which could hinder FP diffusion in our experiments by increasing macromolecular crowding. In case of DOX, cellular oxygen consumption rate (OCR) was not reduced (Appendix Supplementary Results and Fig EV2A–C) and the levels of proteinaceous UPRmt‐linked markers (LONP1, mtHSP70, mtHSP60, CLPP, and CHOP) were not affected (Figs EV2E, and EV3A and B). This makes it unlikely that DOX treatment induces mitochondrial dysfunction and UPRmt in our experiments. CAP treatment greatly reduced OCR values and increased extracellular acidification rate (ECAR), potentially suggesting induction of a glycolytic switch (Schmidt et al, 2021; Bulthuis et al, 2022; Divakaruni & Jastroch, 2022). Further analysis demonstrated that CAP‐treated cells do not display UPRmt activation (Appendix Supplementary Results and Fig EV2A–C) and exhibit lower and higher TMRM fluorescence signals in the nucleus and mitochondrion, respectively (Appendix Supplementary Results and Fig EV2D). The latter suggests that CAP induces mitochondrial membrane potential (Δψ) hyperpolarization. FP diffusion might potentially be altered by changes in mtDNA organization and the function of the mitochondrial MIM fusion protein Optic Atrophy 1 (OPA1). However, no CAP‐induced changes in mtDNA copy number and expression of mitochondrial fission/fusion proteins were detected (Appendix Supplementary Results, and Fig EV2F and G). The latter is compatible with CAP not altering mitochondrial length and diameter. Taken together, these results suggest that the progressive FP immobilization observed in CAP‐treated cells is neither caused by UPRmt induction nor by alterations in the level of mtDNA and mitochondrial fission/fusion proteins. This supports our conclusion that CAP treatment affects FP mobility by increasing macromolecular crowding.
Figure EV2. Functional consequences of chloramphenicol (CAP) treatment in HeLa parental cells.
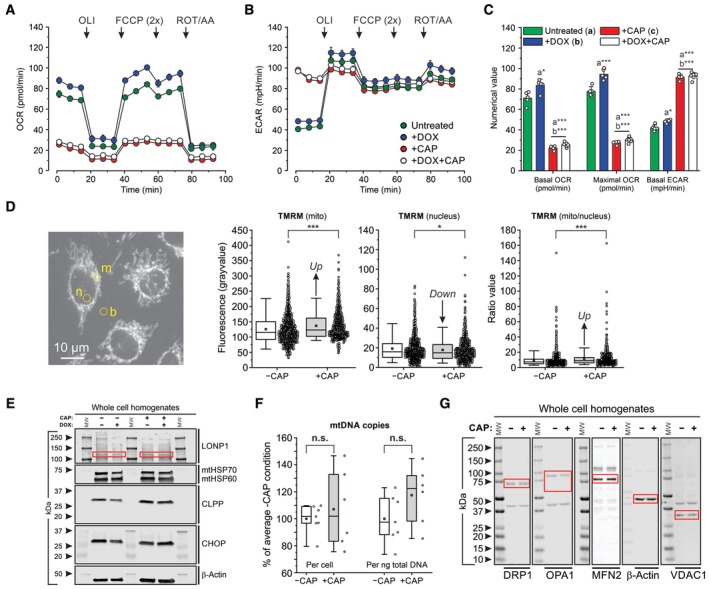
- Average oxygen consumption rate (OCR) in untreated cells, and cells treated with DOX, CAP, or DOX + CAP (for legend see panel B). Oligomycin (OLI), mitochondrial uncoupler (FCCP), and antimycin A + rotenone (AA/ROT) were added at the indicated time points.
- Same as panel (A) but now for the average extracellular acidification rate (ECAR).
- Statistical analysis of the data in panel (A and B) with respect to the basal OCR, maximal OCR and basal ECAR.
- Effect of CAP on the TMRM fluorescence signal in mitochondria and nucleus. Left panel: Typical example of fluorescence microscopy image of TMRM‐stained cells. Fluorescence signals were manually determined in two regions of interest (yellow) defined in a mitochondria‐dense (“m”) and nucleoplasmic part (“n”) of the cell and corrected for background using a close by ROI outside of the cell (“b”). Data panels: numerical values for the mitochondrial fluorescence signal (mito), nuclear fluorescence signal (nucleus) and fluorescence ratio value (mito/nucleus).
- Effect of DOX, CAP or DOX + CAP on the cellular protein levels of LONP1 (specific bands marked by red boxes), mtHSP70, mtHSP60, CLPP and CHOP. β‐actin was used as cellular loading control. Arrowheads indicate molecular weight in kDa. Individual panels were contrast‐optimized for visualization purposes. Original blots are presented in Appendix Fig S5B.
- Effect of CAP on mitochondrial DNA (mtDNA) levels expressed as number of mtDNA copies per cell and per nanogram (ng) of total DNA.
- Effect of CAP on the cellular levels of key mitochondrial fission and fusion proteins (specific bands are marked by red boxes): DRP1 (Dynamin‐related protein 1, OPA1 (Optic atrophy protein 1), MFN2 (Mitofusin 2). β‐actin and VDAC1 were used as cellular and mitochondrial loading controls, respectively. Arrowheads indicate molecular weight in kDa. Individual panels were contrast‐optimized for visualization purposes. Original blots are presented in Appendix Fig S5A.
Data information: OCR and ECAR data (panels A–C) was obtained in a single (N = 1) experiment and the following number (n) of technical replicates: Untreated (n = 5), +DOX (n = 5), +CAP (n = 4), +DOX+CAP (n = 6). TMRM data (Panel D) was obtained in N = 2 independent experiments for n = 991 cells (−CAP) and n = 668 cells (+CAP). MtDNA data (panel F) was obtained in N = 2 independent experiments in n = 7 assays (−CAP) and n = 6 assays (+CAP). Each symbol represents an individual well (panel C), cell (panel D) or assay (panel F). In panels (A and B), individual data points reflect mean ± SEM. In panel (C), bars and errors reflect mean ± SEM. In panels (D and F), error bars mark the 95% (upper) and 5% (lower) percentile, the boundary boxes mark the 75% (upper) and 25% (lower) percentile, the square marks the mean value of the data, and the horizontal line within the box indicates the median value of the data. Significant differences, obtained using an independent Student's t‐test, are indicated by *P < 0.05, **P < 0.01, ***P < 0.001 between the marked conditions (a–c) in panel (C) and between the −CAP and +CAP condition (panel D and F). Not significant is marked by n.s. The exact P‐values for panel (C) (basal OCR) were: Untreated (a) vs. +DOX (b): P = 0.0126; Untreated (a) vs. +CAP (c): P = 1.554·10−6; Untreated (a) vs. +DOX+CAP: P = 4.082·10−8; +DOX (b) vs. +CAP (c): P = 2.671·10−7; +DOX (b) vs. +DOX + CAP: P = 3.806·10−9. The exact P‐values for panel (C) (maximal OCR) were: Untreated (a) vs. +DOX (b): P = 6.509·10−4; Untreated (a) vs. +CAP (c): P = 2.019·10−7; Untreated (a) vs. +DOX+CAP: P = 4.900·10−9; +DOX (b) vs. +CAP (c): P = 4.558·10−8; +DOX (b) vs. +DOX + CAP: P = 5.713·10−10. The exact P‐values for panel (C) (basal ECAR) were: Untreated (a) vs. +DOX (b): P = 0.0023; Untreated (a) vs. +CAP (c): P = 4.571·10−8; Untreated (a) vs. +DOX+CAP: P = 7.784·10−10; +DOX (b) vs. +CAP (c): P = 2.412·10−8; +DOX (b) vs. +DOX+CAP: P = 7.039·10−10. The exact P‐value for panel (C) (mito) was: P = 7.145·10−6. The exact P‐value for panel (C) (nucleus) was: P = 0.0237. The exact P‐value for panel (C) (mito/nucleus) was: P = 5.702·10–5. The exact P‐value for panel (F) (per cell) was: P = 0.556. The exact P‐value for panel (F) (per ng total DNA) was: P = 0.139.
Figure EV3. Chloramphenicol (CAP) and doxycycline (DOX) do not increase protein markers typically associated with induction of the mitochondrial unfolded protein response (UPRmt) in parental and FP‐expressing cells.
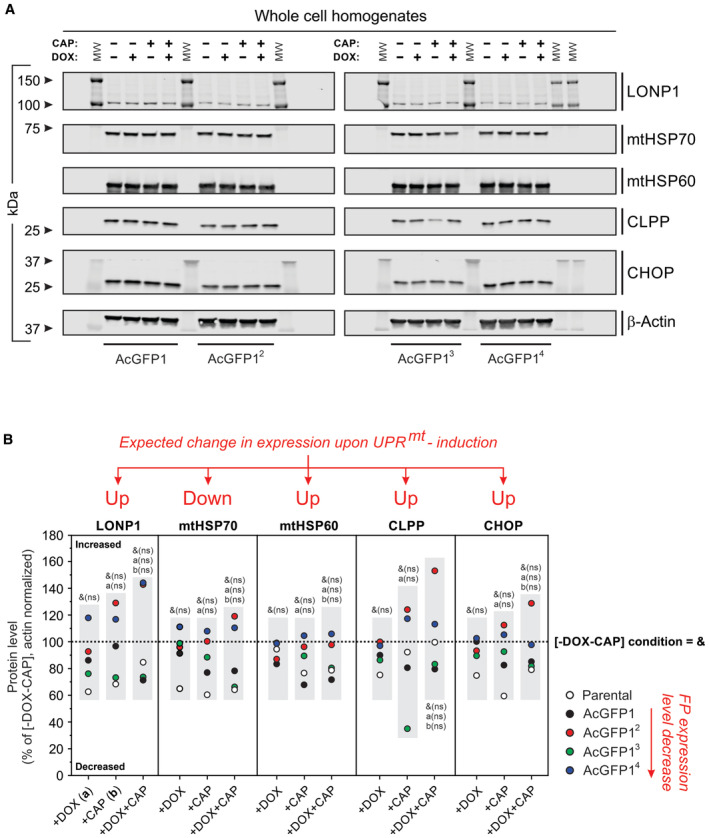
- Western blot analysis (SDS–PAGE) of whole‐cell homogenates was used to assess the levels of the following proteins: LONP1, mtHSP70, mtHSP60, CLPP, and CHOP. β‐actin was used as a cellular loading control. MW indicates molecular weight in kDa. Effect of the expression inducer DOX (1 μg/ml, 24 h) and CAP (40 μg/ml, 72 h), alone and in combination (i.e., 48‐h CAP treatment followed by 24 h CAP + DOX treatment), on protein levels in FP‐expressing HeLa cell lines (AcGFP1, AcGFP12, AcGFP13, and AcGFP14). Individual panels were contrast‐optimized for visualization purposes. Original blots are presented in Appendix Fig S4.
- Quantitative analysis of protein levels in panel A and in HeLa parental cells (the open symbols reflect data for the parental cells in Appendix Fig S5B). All signals were normalized on β‐actin and expressed as % of the condition without DOX and CAP (“−DOX−CAP”). Effects previously associated with UPRmt induction are marked: “Up” indicates proteins that are expected to be upregulated upon UPRmt induction, “Down “indicates proteins that are expected to be downregulated upon UPRmt induction.
Data information: The effects of DOX, CAP, and DOX + CAP were compared with the “−DOX−CAP” (untreated) condition (i.e., with the dotted line marked “&”) by testing whether the mean value for each protein (i.e., within each gray box in panel B) differed from 100% (using a one‐sample Student's t‐test). Comparisons with the DOX only condition (+DOX; marked “a”) and CAP‐only condition (+CAP; marked “b”) were performed using an independent two‐population t‐test (i.e., between each gray box for each protein). Not significant is marked by n.s.
Discussion
The primary aim of this study was to determine whether the mitochondrial matrix solvent displays macromolecular crowding. To this end, we studied the diffusional behavior of four inert monomeric FPs of increasing MW in the mitochondrial matrix of HeLa cells in the absence and presence of CAP. Mitochondrial FP fluorescence intensity per cell decreased as a function of concatemer MW (Fig 2C) and correlated well with in‐gel fluorescence signals (Fig 2D). This strongly suggests that the latter signals faithfully reflect the cellular situation and that AcGFP1 concatemers are not denatured during mitochondrial isolation. Similar to the current findings, the fluorescence signal and expression of nucleus‐targeted EGFP concatemers (containing up to 12 monomers) decreased as a function of MW in H1299 cells (Böhm et al, 2017). The latter study also demonstrated the presence of minor amounts of fluorescent degradation products of smaller MW. Similarly, expression of EGFP concatemers in Escherichia coli cytoplasm (containing up to eight monomers) was paralleled with the formation of degradation products of smaller MW (Nenninger et al, 2010). By contrast, FP concatemers of up to four EGFP molecules remained intact when expressed in the nucleus of HeLa cells (Dross et al, 2009), whereas concatemers of up to five EGFP molecules remained intact in the cytosol of HEK293 cells (Pack et al, 2006). Inspection of MW marker positions (Appendix Fig S1A and B) revealed that AcGFP1‐expressing cells contained a single fluorescent product with an estimated MW between 25 and 37 kDa (predicted: 29‐kDa; Table 1). The fluorescent product in AcGFP12‐expressing cells displayed an MW between 37 and 50 kDa (predicted: 58‐kDa). For AcGFP13 a product of 50–75 kDa was observed (predicted: 88‐kDa). In case of AcGFP14 (predicted: 115‐kDa), two fluorescent products with MWs between 37 and 50 kDa (smaller product) and 75–100 kDa (larger product) were detected. With the exception of AcGFP1, these data highlight a discrepancy between the marker‐reported MW of the FPs and their predicted MW. This is probably due to the fact that the structural dimensions of the FPs, which co‐determine gel migration distance, scale differently with their MW than for the MW markers. Moreover, SDS binding to the various FPs might be different, since samples were not heated (to prevent FP fluorescence loss), thereby affecting their migration behavior. In this context, our SDS–PAGE analysis suggests that the smaller fluorescent product in AcGFP14‐expressing cells is similar to AcGFP12. This might indicate that AcGFP14, once imported in the mitochondrial matrix, is cleaved by a mitochondrial protease at the position of its 2nd linker (DIPPHPAFLYKVVDDPPV; Appendix Table S1). However, an identical linker connected the 1st and 2nd AcGFP1 monomer within AcGFP13 but no fluorescent breakdown product was detected. This suggests that the size and/or MW of AcGFP14 is responsible for its partial breakdown within mitochondria, in both the presence and absence of CAP.
Viscosity of the mitochondrial matrix solvent in the absence of CAP
Analysis of AcGFP1 experiments predicted a Dsolvent of 23.9 μm2/s (Fig 3E). This is within the range of previous FP‐employing FRAP and fluorescence correlation spectroscopy (FCS) studies (Partikian et al, 1998; Verkman, 2002; Koopman et al, 2007, 2008b; Dieteren et al, 2011a). Free EGFP in aqueous solution displayed a Dsolvent value between 87 and 104 μm2/s (Appendix Table S2). This means that the Dsolvent of AcGFP1 is between 3.6‐ and 4.4‐fold lower in the mitochondrial matrix than in aqueous solution. As a consequence, computed matrix ηsolvent values were between 3.69 and 4.57 cP (Fig 4C), being three to five‐fold higher than pure water and similar to those observed previously in HEK293 cells (Dieteren et al, 2011a) and E. coli cytoplasm (Chen et al, 2018). In the literature, mitochondrial matrix viscosity was also analyzed using viscosity‐sensitive fluorescent rotor molecules (Xiao et al, 2021; Yin et al, 2021). In the case of HeLa cells, these studies reported heterogeneous and relatively high viscosity values between 20.7 and 275 cP (Yang et al, 2013; Jímenez‐Sánchez et al, 2018; Ren et al, 2018, 2020; Steinmark et al, 2019). Similarly, variable high mitochondrial viscosity values between < 100 and > 900 cP were reported in rat hepatic tissue slices (Liu et al, 2018), whereas COS7 cells displayed a viscosity value of 325 cP, measured with a BODIPY‐HaloTag rotor molecule targeted to mitochondria using the mitochondrial Cox8a targeting sequence (Chambers et al, 2018). It is currently unknown why the rotor‐based viscosity values exceed those reported in the current study and earlier FP‐based research (Partikian et al, 1998; Verkman, 2002; Koopman et al, 2007, 2008b; Dieteren et al, 2011a). Potentially, the observed discrepancy in cP values could be due to the BODIPY and/or TPP+‐containing part of the rotors displaying a (transient) physical or electrochemical interaction with the inside‐negative MIM. In this context, and to the best of our knowledge, there is currently no information regarding the translational mobility of these rotors in the mitochondrial matrix. Therefore, the rotor properties in the mitochondrial matrix might differ from those in solution, meaning that the viscosity reporting behavior of the sensor is altered (Puchkov, 2013; Chen et al, 2018). This hypothesis is supported by the observation that some of the rotor‐reported matrix viscosities are close to that of the MIM (i.e., 900–950 cP; Chazotte & Hackenbrock, 1988) and that a plasma membrane‐directed BODIPY‐based rotor‐reported viscosity values between 270 and 380 cP (Kubankova et al, 2019). This means that the rotor‐reported mitochondrial matrix viscosity values are similar to those of the plasma membrane, which is unrealistic. It was further argued that accurate viscosity analysis with BODIPY‐C10‐based probes requires calibration in the same polarity environment as the sample of interest and that the size of the surrounding molecules should not exceed that of BODIPY‐C10 (Polita et al, 2020). We here demonstrate that CAP‐induced impairment of mitochondrial function is associated with an eightfold increase in ηsolvent (Fig 5G). Therefore, differences in rotor‐ and FP‐reported matrix viscosities might also be due to interstudy differences in mitochondrial functional state.
Evidence for matrix macromolecular crowding in the absence of CAP
Applying empirical relationships, we provided evidence that AcGFP12, AcGFP13 and AcGFP14 display an elongated (structurally extended) conformation in the mitochondrial matrix solvent (Fig 4C). Similarly, EGFP concatemers displayed an elongated structure in the nucleus and cytosol of HeLa cells (Pack et al, 2006; Dross et al, 2009). This is compatible with the behavior of single DNA molecules in nanochannels, which displayed an elongated structure at low crowding levels but, when exceeding a certain volume fraction of the crowding agent, condensed into a compact form (Zhang et al, 2009). Interestingly, analyzing the dependency of Dsolvent on MW revealed a steeper linear decline for matrix‐targeted FPs, than for globular/elongated molecules in aqueous solution and elongated EGFP concatemers in the nucleus/cytoplasm of HeLa cells (Fig 4B). This means that the elongated structure of the matrix‐targeted FPs cannot fully explain the steeper decline. In addition, our BD modeling approach renders Dsolvent cristae‐independent and both Fm and F∞ increased as a function of MW (Table 1 and Fig 5F). Therefore, we conclude that matrix FP mobility is reduced by the combined impact of their elongated structure and macromolecular crowding. This latter probably is low level, since substantial crowding is associated with molecule compaction (Zhang et al, 2009; Kuznetzova et al, 2014).
CAP treatment slows solute diffusion and increases mitochondrial matrix viscosity
Triggered by our previous findings (see Results), cells were treated with CAP to increase the protein concentration of the matrix solvent. Although technical limitations prevented assessing Dsolvent for AcGFP12, AcGFP13, and AcGFP14, analysis of AcGFP1 mobility demonstrated that CAP treatment eightfold decreased and increased Dsolvent and ηsolvent, respectively (Table 1 and Fig 5G). By itself, this viscosity increase is highly relevant, since it will slow down any diffusion‐limited process, including metabolite diffusion and macromolecular association/dissociation reactions (Van den Bogaart et al, 2007; Molines et al, 2022). Mitochondria in CAP‐treated cells displayed a progressive immobilization of AcGFP12 and AcGFP13 (Fig 5D–F), contained three to five‐fold cristae and displayed a more electron‐dense matrix (Fig 5C). This strongly suggests that matrix macromolecular crowding is increased in CAP‐treated cells (Mika et al, 2010; Mittal et al, 2015; Delarue et al, 2018; Junker et al, 2019). Electron density analysis was performed on EM images of osmium tetroxide‐treated samples, which preferentially binds to unsaturated fatty acids (Belazi et al, 2009). However, it appears that osmium tetroxide also can react with proteins (e.g., Porter & Kallman, 1953) and that electron density increases when proteins with a relatively high affinity for this fixative are present (Bisht et al, 2016). In this context, it has been argued that differences in electron density of cryo‐electron tomograms can be interpreted as differences in macromolecular density and that mitochondrial matrices are expected to display a higher density than the surrounding cytoplasm (Ader et al, 2019). Therefore, we propose that the increased electron density in CAP‐treated cells (Fig 5C) represents a minor though significant increase in the concentration of soluble matrix macromolecules. This conclusion is supported by the fact that mitochondria in AcGFP14‐expressing cells displayed a lower fluorescent signal (Fig 2B–D), as well as a less electron‐dense matrix relative to AcGFP1‐expressing cells (Fig 5C). Compatible with a previous study in CAP‐treated mouse embryonic fibroblasts (Sasaki et al, 2020), CAP‐treated HeLa cells lacked a detectable increase in proteinaceous UPRmt‐linked markers (Figs EV2E and EV3). Although this suggests that the increase in matrix protein levels is relatively small, such an increase can substantially elevate macromolecular crowding levels due to the relatively small matrix volume.
CAP treatment does not induce mitochondrial orthodox‐to‐condensed transition
Classically, changes in matrix electron density are linked to alterations in mitochondrial bioenergetic state during mitochondrial “orthodox‐to‐condensed transition” (Hackenbrock, 1966, 1972). Orthodox mitochondria are characterized by a low OCR, Δψ hyperpolarization and a relatively large matrix volume, whereas condensed mitochondria display a high OCR and a relatively small matrix volume (Perkins & Ellisman, 2011). During orthodox‐to‐condensed transition, matrix volume decrease is accompanied by an increase in electron density without alterations in total mitochondrial volume (Hackenbrock, 1966). In CAP‐treated cells, mitochondria displayed functional properties typical of orthodox mitochondria (i.e., a low OCR and Δψ hyperpolarization; Fig EV2A–D) but a more electron‐dense (darker) matrix (typical of condensed mitochondria; Fig 5C). Although CAP did reduce the number of cristae, mitochondrial dimensions and volume were not detectably affected (Table 1). With respect to CAP‐induced Δψ hyperpolarization, we observed that mitochondrial TMRM fluorescence was increased, whereas nucleoplasmic TMRM fluorescence was decreased. Since TMRM accumulates in the cytoplasm and mitochondrial matrix according to the plasma membrane and MIM electrical potential, respectively, this suggests that Δψ is more negative (Koopman et al, 2008a). Similarly, CAP pretreatment (48 h) induced Δψ hyperpolarization in HepG2 cells, as reported by fluorescence analysis of mitochondrial DiOC6 accumulation (Li et al, 2005). However, when the rotational motion of fluorescent molecules is impeded their fluorescence quantum yield might increase (Dragan et al, 2014; Vyšniauskas et al, 2015). Therefore, it cannot be ruled out that the increased mitochondrial TMRM fluorescence signal in CAP‐treated cells is due to a higher viscosity/crowding environment within the mitochondrial matrix. Taken together, our results indicate that CAP treatment might hyperpolarize Δψ and does not induce mitochondrial orthodox‐to‐condensed transition.
Predicting the degree of macromolecular crowding and volume exclusion in the mitochondrial matrix
As stated above, macromolecular crowding will reduce the translational mobility of solutes with a similar or larger size than the crowder to a greater extent than the mobility of smaller solutes. Therefore, to gain some theoretical insight into how CAP might quantitatively affect matrix crowding levels, we analyzed the predicted effect of human serum albumin (HSA) as a model crowding protein (Appendix Supplementary Results). HSA was selected because its MW (≈92‐kDa) is relatively close to that of the expected NDUFV1/NDUFV2 assembly intermediates, potentially acting as (co)crowders (see Results), and similar to the MW of AcGFP13 (≈88‐kDa), which was demonstrated to be substantially immobilized in CAP‐treated cells. Using the obtained ηsolvent values, and assuming that HSA is the sole crowding entity, it was predicted that HSA would occupy between ~20% (absence of CAP) and ~33% (presence of CAP) of the total mitochondrial matrix volume (Appendix Supplementary Results and Fig 6A). Obviously, the magnitude of this volume exclusion effect will increase with the number/size of the crowding macromolecules and with decreasing matrix volume.
Figure 6. Key results and interplay between mitochondrial (ultra)structure, matrix solvent macromolecular crowding/viscosity and mitochondrial function.
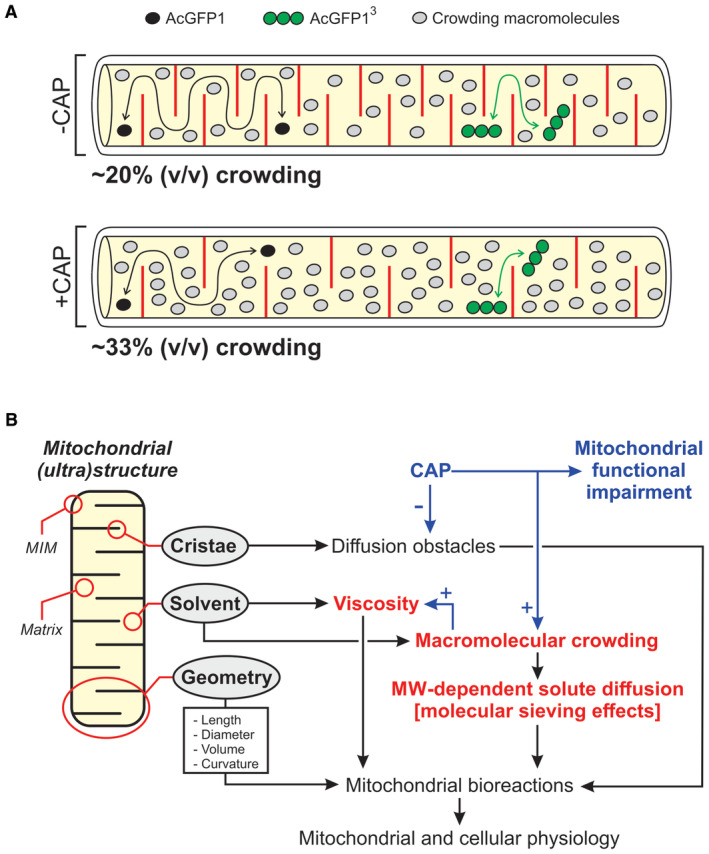
- Graphical representation (not to scale) of the key results of this study, illustrating the hindrance effect of cristae (red lines) and crowding macromolecules (gray symbols) on the mobility of AcGFP1 (black symbols) and AcGFP13 (green symbols) in the mitochondrial matrix (yellow). In the absence of chloramphenicol (−CAP), low levels of macromolecular crowding reduce the mobility of AcGFP13 to a greater extent than the mobility of AcGFP1. In the presence of CAP (+CAP), the number of cristae is reduced but crowding is increased. This impedes the mobility of AcGFP1 and AcGFP3, although the latter is affected to a greater extent. In this panel, the number of crowding macromolecules approximately reflects the predicted maximal degree of crowding determined using human serum albumin (HSA) as a model crowder (see Discussion).
- Integration of mitochondrial morphofunction. The mitochondrial matrix is surrounded by the mitochondrial inner membrane (MIM), which contains matrix‐protruding folds (cristae). Biochemical reactions in the mitochondrial matrix are affected by hindrance of solute diffusion by cristae, the physicochemical properties of the mitochondrial matrix solvent (e.g., viscosity) and the geometry of the mitochondrial compartment (e.g., volume). We here demonstrate that the mitochondrial matrix solvent can reduce the diffusion of fluorescent proteins in an MW‐dependent manner typical of macromolecular crowding. The latter appeared only minor during “normal” conditions but increased during CAP‐induced mitochondrial functional impairment. We propose that crowding‐induced molecular sieving effects, in combination with alterations in matrix solvent viscosity, impact on mitochondrial bioreactions and thereby on mitochondrial and cellular physiology.
Summary and conclusion
The results presented in this study support the conclusion that the mitochondrial matrix solvent displays macromolecular crowding phenomena, particularly during conditions of CAP‐induced mitochondrial dysfunction. In analogy to the cytosol (Delarue et al, 2018), it is therefore likely that (changes in) matrix crowding will induce molecular sieving effects that will affect matrix bioreactions dependent on the size of the involved crowding molecules and reactants. It is expected that macromolecular crowding will decrease the rate of diffusion‐limited, fast association reactions whereas it will increase this rate for slow, transition‐state‐limited association reactions (Zhou et al, 2008). Therefore, crowding impacts on complex formation, surface binding, aggregation and folding/compaction of matrix proteins and, via volume exclusion, on the diffusion and (local) concentration of metabolites, ions, and other small reactants (Minton, 1990; Yu et al, 2016; Andrews, 2020). Crowding‐induced effects on the Michaelis–Menten mechanism were predicted in silico (Weilandt & Hatzimanikatis, 2019) and demonstrated experimentally for the mitochondrial matrix enzymes malate hydrogenase (Poggi & Slade, 2015) and citrate synthase (Wilcox et al, 2020). We hypothesize that alterations in mitochondrial matrix crowding level, cristae number/architecture, and/or mitochondrial geometry exert control over mitochondrial bioreactions, which exemplifies the intricate link between mitochondrial (ultra)structure and function (Fig 6B). In this sense, the combined crowding and cristae‐induced hindrance of matrix Ca2+ diffusion might be involved in heterogeneous spreading of mitochondrial Ca2+ signals (Gerencser & Adam‐Vizi, 2005) and compartmentalization of matrix reaction networks (Kekenes‐Huskey et al, 2015).
Materials and Methods
Generation of inducible HeLa cell lines
HeLa T‐REx Flp‐in cell lines stably expressing mitochondria‐targeted concatemers of monomeric AcGFP1 (AcGFP1, AcGFP12, AcGFP13, and AcGFP14) were generated as described in the Appendix Supplementary Methods and cultured at 37°C in a humid atmosphere (95% air, 5% CO2). Expression of these fluorescent proteins (FPs) was induced by adding 1 μg/ml doxycycline (DOX) to the culture medium for 24 h. In certain experiments, cells were treated with 40 μg/ml chloramphenicol (CAP) for 72 h.
Mitochondrial staining and flow cytometry
Localization of the FPs and the mitochondrial marker MitoTracker Red CM‐H2XROS was visualized using confocal microscopy. Flow cytometry was applied to analyze the cellular fluorescence intensity of the AcGFP1 concatemers and forward scatter. Details are provided in the Appendix Supplementary Methods.
Fluorescence recovery after photobleaching (FRAP) analysis
Mitochondrial FRAP experiments were performed as described previously (Dieteren et al, 2008, 2009, 2011a, 2011b). Details are provided in the Appendix Supplementary Methods.
Western blotting
Mitochondrial fractions or whole‐cell lysates were used for SDS–PAGE, in‐gel fluorescence analysis, and Western blotting as described in the Appendix Supplementary Methods.
Electron microscopy (EM)
Electron microscopy was performed as described previously (Koopman et al, 2008a). Details are provided in the Appendix Supplementary Methods.
Functional cell analysis
Oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) were quantified using a Seahorse XFe96 flux Analyzer (Agilent, Santa Clara, CA, USA). Mitochondrial membrane potential (Δψ) was analyzed using the fluorescent cation TMRM (tetramethylrhodamine methyl ester). Mitochondrial DNA (mtDNA) copy number was quantified using real‐time quantitative PCR (qPCR) analysis. Details are provided in the Appendix Supplementary Methods.
Numerical simulations
A Brownian dynamics (BD) computer model was developed to determine the solvent‐dependent diffusion constant (Dsolvent) of the AcGFP1 concatemers. This model was constrained by experimental data and implemented in GNU Fortran (https://gcc.gnu.org) under Ubuntu 20.04 LTS (https://releases.ubuntu.com). Details are provided in the Results section and in the Appendix Supplementary Methods.
Image analysis
Image visualization, processing, and quantification were carried out using Image Pro Plus software (Media Cybernetics, Rockville, MD, USA), Zeiss LSM 510 Meta software (Carl Zeiss AG, Oberkochen, Germany) and FIJI (https://imagej.net/Fiji).
Statistical analysis
Curve fitting and statistical analyses were performed using Origin Pro software (Originlab Corp., Northampton, MA, USA). Levenberg–Marquardt and least squares algorithms were used for nonlinear and linear regression, respectively. Average data were expressed as mean ± SEM (standard error of the mean), and significance was tested using an independent Student's t‐test unless stated otherwise.
Author contributions
Elianne P Bulthuis: Formal analysis; supervision; validation; investigation; visualization; writing – original draft; writing – review and editing. Cindy E J Dieteren: Formal analysis; supervision; funding acquisition; validation; investigation; visualization; writing – original draft; writing – review and editing. Jesper Bergmans: Data curation; investigation; writing – review and editing. Job Berkhout: Formal analysis; investigation. Jori A Wagenaars: Formal analysis; investigation; visualization. Els M A van de Westerlo: Formal analysis; supervision; investigation; visualization. Emina Podhumljak: Investigation. Mark A Hink: Resources; formal analysis; supervision; investigation; writing – original draft. Laura F B Hesp: Investigation. Hannah S Rosa: Formal analysis; investigation. Afshan N Malik: Formal analysis; supervision; investigation; writing – original draft. Mariska Kea‐te Lindert: Formal analysis; investigation; writing – original draft. Peter H G M Willems: Funding acquisition; writing – original draft. Han J G E Gardeniers: Writing – original draft; writing – review and editing. Wouter K den Otter: Conceptualization; resources; software; validation; visualization; methodology; writing – review and editing. Merel J W Adjobo‐Hermans: Conceptualization; supervision; validation; investigation; visualization; writing – original draft; writing – review and editing. Werner J H Koopman: Conceptualization; data curation; formal analysis; supervision; funding acquisition; validation; visualization; methodology; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
WJHK is an ad hoc scientific advisor of Khondrion B.V. (Nijmegen, The Netherlands). This SME had no involvement in the data collection, analysis and interpretation, writing of the manuscript, and in the decision to submit the manuscript for publication.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Appendix
PDF+
Acknowledgements
We thank Stephen S. Taylor (Division of Cancer Sciences, University of Manchester, UK) for providing the parental HeLa T‐REx Flp‐in cells, Michael J. Saxton (Department of Biochemistry and Molecular Medicine, University of California, Davis, USA) for providing numerical data (Saxton, 2014), Laszlo Groh (Department of Internal Medicine, Radboudumc, The Netherlands) for assistance with OCR and ECAR measurements, Hanka Venselaar (Centre for Molecular and Biomolecular Informatics, Radboudumc, the Netherlands) for discussions on protein conformation, Riekelt H. Houtkooper (Amsterdam UMC, University of Amsterdam, Amsterdam, The Netherlands) for discussions on UPRmt and Anna O. Chertkova (Section of Molecular Cytology and van Leeuwenhoek Centre for Advanced Microscopy, University of Amsterdam, The Netherlands) for cell culture. We thank the Radboudumc Microscopy Technology Center for equipment maintenance, training, and assistance with the FRAP and EM recordings. This work was supported by an equipment grant from NWO (Netherlands Organization for Scientific Research, No: 911‐02‐008). CEJD was supported by an NWO‐VENI grant (#863.13.019). JB was supported by an EJP‐RD grant (“CureMILS”) through ZonMW (Netherlands Organisation for Health Research and Development; #463003001). WJHK was supported by a junior researcher grant (EPB; Radboudmc, Nijmegen, The Netherlands), the Next Level Animal Sciences (NLAS) initiative (“Data and Models”) of the Wageningen University (Wageningen, The Netherlands) and by Principal Investigator (PI) support funding (Radboudumc).
The EMBO Journal (2023) 42: e108533
Data availability
Source data, materials, and computer code are available from the corresponding authors at reasonable request. All other data are contained in the main manuscript, Expanded View and Appendix. This study includes no data deposited in external repositories.
References
- Ader NR, Hoffmann PC, Ganeva I, Borgeaud AC, Wang C, Youle RJ, Kukulski W (2019) Molecular and topological reorganizations in mitochondrial architecture interplay during Bax‐mediated steps of apoptosis. Elife 8: e40712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akabayov B, Akabayov SR, Lee SJ, Wagner G, Richardson CC (2013) Impact of macromolecular crowding on DNA replication. Nat Commun 4: 1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews SS (2020) Effects of surfaces and macromolecular crowding on bimolecular reaction rates. Phys Biol 17: 045001 [DOI] [PubMed] [Google Scholar]
- Appelhans T, Richter CP, Wilkens V, Hess ST, Piehler J, Busch KB (2011) Nanoscale organisation of mitochondrial microcompartments revealed by combining tracking and localization microscopy. Nano Lett 12: 610–616 [DOI] [PubMed] [Google Scholar]
- Belazi D, Sole‐Domenech S, Johansson B, Schalling M, Sjovall P (2009) Chemical analysis of osmium tetroxide staining in adipose tissue using imaging ToF‐SIMS. Histochem Cell Biol 132: 105–115 [DOI] [PubMed] [Google Scholar]
- Bisht K, Sharma K, Lacoste B, Tremblay ME (2016) Dark microglia: why are they dark? Commun Integr Biol 9: e1230575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersma AJ, Zuhorn IS, Poolman B (2015) A sensor for quantification of macromolecular crowding in living cells. Nat Methods 12: 227–229 [DOI] [PubMed] [Google Scholar]
- Böhm J, Thavaraja R, Giehler S, Nalaskowski MM (2017) A set of enhanced green fluorescent protein concatemers for quantitative determination of nuclear localization signal strength. Anal Biochem 533: 48–55 [DOI] [PubMed] [Google Scholar]
- Bulthuis EP, Adjobo‐Hermans M, Willems PHGM, Koopman WJH (2019) Mitochondrial morphofunction in mammalian cells. Antioxid Redox Signal 30: 2066–2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulthuis EP, Einer C, Distelmaier F, Groh L, van Emst‐de Vries SE, van de Westerlo E, van de Wal M, Wagenaars J, Rodenburg RJ, Smeitink JAM et al (2022) The decylTPP mitochondria‐targeting moiety lowers electron transport chain supercomplex levels in primary human skin fibroblasts. Free Radic Biol Med 188: 434–446 [DOI] [PubMed] [Google Scholar]
- Calvo SE, Mootha VK (2010) The mitochondrial proteome and human disease. Annu Rev Genomics Hum Genet 11: 25–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers JE, Kubánková M, Huber RG, López‐Duarte I, Avezov E, Bond PJ, Marciniak SJ, Kuimova MK (2018) An optical technique for mapping microviscosity dynamics in cellular organelles. ACS Nano 12: 4398–4407 [DOI] [PubMed] [Google Scholar]
- Chazotte B, Hackenbrock CR (1988) The multicollisional, obstructed, long‐range diffusional nature of mitochondrial electron transport. J Biol Chem 263: 14359–14367 [PubMed] [Google Scholar]
- Chen E, Esquerra RM, Meléndez PA, Chandrasekaran SS, Kliger DS (2018) Microviscosity in E coli cells from time‐resolved linear dichroism measurements. J Phys Chem B 122: 11381–11389 [DOI] [PubMed] [Google Scholar]
- Cogliati S, Enriquez JA, Scorrano L (2016) Mitochondrial cristae: where beauty meets functionality. Trends Biochem Sci 41: 261–273 [DOI] [PubMed] [Google Scholar]
- Cravens SL, Schonhoft JD, Rowland MM, Rodriguez AA, Anderson BG, Stivers JT (2015) Molecular crowding enhances facilitated diffusion of two human DNA glycosylases. Nucleic Acids Res 43: 4087–4097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delarue M, Brittingham GP, Pfeffer S, Surovtsev IV, Pinglay S, Kennedy KJ, Schaffer M, Gutierrez JI, Sang D, Poterewicz G et al (2018) mTORC1 controls phase separation and the biophysical properties of the cytoplasm by tuning crowding. Cell 174: 338–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey P, Bhattacherjee A (2019) Disparity in anomalous diffusion of proteins searching for their target DNA sites in a crowded medium is controlled by the size, shape and mobility of macromolecular crowders. Soft Matter 15: 1960–1969 [DOI] [PubMed] [Google Scholar]
- Dieteren CEJ, Willems PHGM, Vogel RO, Swarts HG, Fransen J, Roepman R, Crienen G, Smeitink JAM, Nijtmans LGJ, Koopman WJH (2008) Subunits of mitochondrial complex I exist as part of matrix‐ and membrane‐associated subcomplexes in living cells. J Biol Chem 283: 34753–34761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieteren CEJ, Koopman WJH, Nijtmans LGJ (2009) Tracing human mitochondrial complex I assembly by use of GFP‐tagged subunits. Methods Enzymol 456: 133–151 [DOI] [PubMed] [Google Scholar]
- Dieteren CEJ, Gielen SCAM, Nijtmans LGJ, Smeitink JAM, Swarts HG, Brock R, Willems PHGM, Koopman WJH (2011a) Solute diffusion is hindered in the mitochondrial matrix. Proc Natl Acad Sci USA 108: 8657–8662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieteren CEJ, Willems PHGM, Swarts HG, Fransen J, Smeitink JAM, Koopman WJH, Nijtmans LGJ (2011b) Defective mitochondrial translation differentially affects the live cell dynamics of complex I subunits. Biochim Biophys Acta 1807: 1624–1633 [DOI] [PubMed] [Google Scholar]
- Divakaruni AS, Jastroch M (2022) A practical guide for the analysis, standardization and interpretation of oxygen consumption measurements. Nat Metab 4: 978–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragan A, Graham AE, Geddes CD (2014) Fluorescence‐based broad dynamic range viscosity probes. J Fluoresc 24: 397–402 [DOI] [PubMed] [Google Scholar]
- Dross N, Spriet C, Zwerger M, Müller G, Waldeck W, Langowski J (2009) Mapping eGFP oligomer mobility in living cell nuclei. PLoS ONE 4: e5041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einstein A (1905) Über die von der molekularkinetischen Theorie der Wärme geforderte Bewegung von in ruhenden Flüssigkeiten suspendierten Teilchen. AdP 322: 549–560 [Google Scholar]
- Fernández‐Vizarra E, Ugalde C (2022) Cooperative assembly of the mitochondrial respiratory chain. Trends Biochem Sci 47: 999–1008 [DOI] [PubMed] [Google Scholar]
- Fernie AR, Zhang Y, Sweetlove LJ (2018) Passing the baton: substrate channelling in respiratory metabolism. Research 2018: 1539325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippin L, Abad MC, Gastaldello S, Magelhães PJ, Sandonà D, Pozzan T (2005) Improved strategies for the delivery of GFP‐based Ca2+ sensors into the mitochondrial matrix. Cell Calcium 37: 129–136 [DOI] [PubMed] [Google Scholar]
- Fuentes‐Lemus E, Reyes JS, Gamon LF, López‐Alarcón C, Davies MJ (2021) Effect of macromolecular crowding on protein oxidation: consequences on the rate, extent and oxidation pathways. Redox Biol 48: 102202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerencser AA, Adam‐Vizi V (2005) Mitochondrial Ca2+ dynamics reveals limited intramitochondrial Ca2+ diffusion. Biophys J 88: 698–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero‐Castillo S, Baertling F, Kownatzki D, Wessels HJ, Arnold S, Brandt U, Nijtmans LGJ (2017) The assembly pathway of mitochondrial respiratory chain complex I. Cell Metab 25: 128–139 [DOI] [PubMed] [Google Scholar]
- Hackenbrock CR (1966) Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J Cell Biol 30: 269–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackenbrock CR (1972) Energy‐linked ultrastructural transformations in isolated liver mitochondria and mitoplasts. J Cell Biol 53: 450–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggie PM, Verkman AS (2002) Diffusion of tricarboxylic acid cycle enzymes in the mitochondrial matrix in vivo. J Biol Chem 277: 40782–40788 [DOI] [PubMed] [Google Scholar]
- Hansen MMK, Mijer LHH, Spruijt E, Maas RJM, Ventosa Roquelles M, Groen J, Heus HA, Huck WTS (2016) Macromolecular crowding develops heterogeneous environments of gene expression in picoliter droplets. Nat Nanotechnol 11: 191–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Niemeyer B (2003) A novel correlation for protein diffusion coefficients based on molecular weight and radius of gyration. Biotechnol Prog 19: 544–548 [DOI] [PubMed] [Google Scholar]
- Hochmair J, Exner C, Franck M, Dominguez‐Baquero A, Diez L, Brognaro H, Kraushar ML, Mielke T, Radbruch H, Kaniyappan S et al (2022) Molecular crowding and RNA synergize to promote phase separation, microtubule interaction, and seeding of tau condensates. EMBO J 17: e108882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock DH, Robinson DRL, Stroud DA (2020) Blackout in the powerhouse: clinical phenotypes associated with defects in the assembly of OXPHOS complexes and the mitoribosome. Biochem J 477: 4085–4132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J (2013) Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497: 451–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Shu L, Huang X, Yu J, Li L, Gong L, Yang M, Wu Z, Gao Z, Zhao Y et al (2020) OPA1 and MICOS regulate mitochondrial crista dynamics and formation. Cell Death Dis 11: 940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jímenez‐Sánchez A, Lei EK, Kelley SO (2018) A multifunctional chemical probe for the measurement of local micropolarity and microviscosity in mitochondria. Angew Chem Int Ed Engl 57: 8891–8895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyner RP, Tang JH, Helenius J, Dultz E, Brune C, Holt LJ, Huet S, Müller DJ, Weis K (2015) A glucose‐starvation response regulates the diffusion of macromolecules. Elife 5: e09376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junker NO, Vaghefikia F, Albarghash A, Höfig H, Kempe D, Walter J, Otten J, Pohl M, Katranidid A, Wiegand S et al (2019) Impact of molecular crowding on translational mobility and conformational properties of macromolecules. J Phys Chem B 123: 447–4486 [DOI] [PubMed] [Google Scholar]
- Kekenes‐Huskey PM, Eun C, McCammon JA (2015) Enzyme localization, crowding, and buffers collectively modulate diffusion‐influenced signal transduction: insights from continuum diffusion modelling. J Chem Phys 143: 094103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kislev N, Spolsky CM, Eisenstadt JM (1973) Effect of chloramphenicol on the ultrastructure of mitochondria in sensitive and resistant strains of HeLa. J Cell Biol 57: 571–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman WJH, Hink MA, Verkaart S, Visch HJ, Smeitink JAM, Willems PHGM (2007) Partial complex I inhibition decreases mitochondrial motility and increases matrix protein diffusion as revealed by fluorescence correlation spectroscopy. Biochim Biophys Acta 1767: 940–947 [DOI] [PubMed] [Google Scholar]
- Koopman WJH, Distelmaier F, Esseling JJ, Smeitink JAM, Willems PHGM (2008a) Computer‐assisted live cell analysis of mitochondrial membrane potential, morphology and calcium handling. Methods 46: 304–311 [DOI] [PubMed] [Google Scholar]
- Koopman WJH, Distelmaier F, Hink MA, Verkaart S, Wijers M, Fransen J, Smeitink JAM, Willems PHGM (2008b) Inherited complex I deficiency is associated with faster protein diffusion in the matrix of moving mitochondria. Am J Physiol Cell Physiol 294: C124–C1132 [DOI] [PubMed] [Google Scholar]
- Koopman WJH, Nijtmans LGJ, Dieteren CEJ, Roestenberg P, Valsecchi F, Smeitink JAM, Willems PHGM (2010) Mammalian mitochondrial complex I: biogenesis, regulation, and reactive oxygen species generation. Antioxid Redox Signal 12: 1431–1470 [DOI] [PubMed] [Google Scholar]
- Kubankova M, Summers PA, Ljpez‐Duarte I, Kiryushko D, Kuimova KM (2019) Microscopic viscosity of neuronal plasma membranes measured using fluorescent molecular rotors: effects of oxidative stress and neuroprotection. ACS Appl Mater Interfaces 11: 36307–36315 [DOI] [PubMed] [Google Scholar]
- Kuznetzova IM, Turoverov KK, Uversky VN (2014) What macromolecular crowding can do to a protein. Int J Mol Sci 15: 23090–23140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenk R, Penman S (1971) Morphological studies of cells grown in the absence of mitochondrial‐specific protein synthesis. J Cell Biol 49: 541–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CH, Tzeng SL, Cheng YW, Kang JJ (2005) Chloramphenicol‐induced mitochondrial stress increases p21 expression and prevents cell apoptosis through a p21‐dependent pathway. J Biol Chem 280: 26193–26199 [DOI] [PubMed] [Google Scholar]
- Liu F, Luo Y, Xu M (2018) Viscosity measurements using a two-photon ratiometric fluorescent sensor with two rotors. Tetrahedron Lett 59: 4540–4544 [Google Scholar]
- Lizana L, Konkoli Z, Bauer B, Jesorka A, Orwar O (2008) Controlling chemistry by geometry in nanoscale systems. Annu Rev Phys Chem 60: 449–468 [DOI] [PubMed] [Google Scholar]
- Mika JT, van den Bogaart G, Veenhoff L, Krasnikov V, Poolman B (2010) Molecular sieving properties of the cytoplasm of Escherichia coli and consequences of osmotic stress. Mol Microbiol 77: 200–207 [DOI] [PubMed] [Google Scholar]
- Minton AP (1990) Holobiochemistry: the effect of local environment upon the equilibria and rates of biochemical reactions. Int J Biochem 22: 1063–1067 [DOI] [PubMed] [Google Scholar]
- Mittal S, Chowhan RK, Singh LR (2015) Macromolecular crowding: macromolecules friend or foe. Biochim Biophys Acta 1850: 1822–1831 [DOI] [PubMed] [Google Scholar]
- Molines AT, Lemière J, Gazzola M, Steinmark IE, Edrington CH, Hsu CT, Real‐Calderon P, Suhling K, Goshima G, Holt LJ et al (2022) Physical properties of the cytoplasm modulate the rates of microtubule polymerization and depolymerization. Dev Cell 57: 466–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moullan N, Mouchiroud L, Wang X, Ryu D, Williams EG, Mottis A, Jovaisaite V, Frochaux MV, Quiros PM, Deplancke B et al (2015) Tetracyclines disturb mitochondrial function across eukaryotic models: a call for caution in biomedical research. Cell Rep 10: 1681–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nenninger A, Mastroianni G, Mullineaux CW (2010) Size dependence of protein diffusion in the cytoplasm of Escherichia coli . J Bacteriol 192: 4535–4540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ölveczky BP, Verkman AS (1998) Monte Carlo analysis of obstructed diffusion in three dimensions: application to molecular diffusion in organelles. Biophys J 74: 2722–2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pack C, Saito K, Tamura M, Kinjo M (2006) Microenvironment and effect of energy depletion in the nucleus analyzed by mobility of multiple oligomeric EGFPs. Biophys J 91: 3921–3936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos S, Jürgens KD, Gros G (2000) Protein diffusion in living skeletal muscle fibers: dependence on protein size, fiber type, and contraction. Biophys J 79: 2084–2094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partikian A, Olveczky B, Swaminathan R, Li Y, Verkman AS (1998) Rapid diffusion of green fluorescent protein in the mitochondrial matrix. J Cell Biol 140: 821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins GA, Ellisman MH (2011) Mitochondrial configurations in peripheral nerve suggest differential ATP production. J Struct Biol 173: 117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin F (1936) Mouvement Brownien d'un ellipsoide (II). Rotation libre et dépolarisation des fluorescences. Translation et diffusion de molécules ellipsoidales. J Phys Radium 7: 1–11 [Google Scholar]
- Poggi CG, Slade KM (2015) Macromolecular crowding and the steady‐state kinetics of malate dehydrogenase. Biochemistry 54: 260–267 [DOI] [PubMed] [Google Scholar]
- Polita A, Toliautas S, Žvirblis R, Vyšniauskas A (2020) The effect of solvent polarity and macromolecular crowding on the viscosity sensitivity of a molecular rotor BODIPY‐C10 . Phys Chem Chem Phys 16: 8296–8303 [DOI] [PubMed] [Google Scholar]
- Porter KR, Kallman F (1953) The properties and effects of osmium tetroxide as a tissue fixative with special reference to its use for electron microscopy. Exp Cell Res 4: 127–141 [Google Scholar]
- Prazeres DMF (2008) Prediction of diffusion coefficients of plasmids. Biotechnol Bioeng 99: 1040–1044 [DOI] [PubMed] [Google Scholar]
- Puchkov EO (2013) Intracellular viscosity: methods of measurement and role in metabolism. Biochem (Mosc) Suppl A Membr Cell Biol 7: 279 [Google Scholar]
- Rath S, Sharma R, Gupta R, Ast T, Chan C, Durham JH, Goodman RP, Grabarek Z, Haas ME, Hung WHW et al (2021) MitoCarta 3.0: an updated mitochondrial proteome now with sub‐organelle localization and pathway annotations. Nucl Acids Res 49: D1541–D1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren M, Zhou K, Wang L, Liu K, Lin W (2018) Construction of a ratiometric two‐photon fluorescent probe to monitor the changes of mitochondrial viscosity. Sens Actuators B 262: 452–459 [Google Scholar]
- Ren M, Xu Q, Wang S, Liu L, Kong F (2020) A biotin‐guided fluorescent probe for dual‐mode imaging of viscosity in cancerous cells and tumor tissues. Chem Commun 56: 13351–13354 [DOI] [PubMed] [Google Scholar]
- Rivas G, Minton AP (2016) Macromolecular crowding in vitro, in vivo and in between. Trends Biochem Sci 41: 970–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JB, Srere PA (1985) Organization of Krebs Tricarboxylic acid cycle enzymes in mitochondria. J Biol Chem 260: 10800–10805 [PubMed] [Google Scholar]
- Sasaki K, Uchiumi T, Toshima T, Yagi M, Do Y, Hirai H, Igami K, Gotoh K, Kang D (2020) Mitochondrial translation inhibition triggers ATF4 activation, leading to integrated stress response but not to mitochondrial unfolded protein response. Biosci Rep 40: BSR20201289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton MJ (2014) Wanted: scalable tracers for diffusion measurements. J Phys Chem B 118: 12805–12817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalettar BA, Abney JR, Hackenbrock CR (1991) Dynamics, structure and function are coupled in the mitochondrial matrix. Proc Natl Acad Sci USA 88: 8057–8061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schavemaker PE, Boersma AJ, Poolman B (2018) How important is protein diffusion in prokaryotes? Front Mol Biosci 5: 93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt CA, Fisher‐Wellman KH, Neufer PD (2021) From OCR and ECAR to energy: perspectives on the design and interpretation of bioenergetics studies. J Biol Chem 4: 101140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segawa M, Wolf DM, Hultgren NW, Williams DS, van der Bliek AM, Schackelford DB, Liesa M, Shirihai OS (2020) Quantification of cristae architecture reveals time‐dependent characteristics of individual mitochondria. Life Sci Alliance 3: e201900620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shpilka T, Haynes CM (2018) The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat Rev Mol Cell Biol 19: 109–120 [DOI] [PubMed] [Google Scholar]
- Smeitink JAM, van den Heuvel L, DiMauro S (2001) The genetics and pathology of oxidative phosphorylation. Nat Rev Genet 2: 342–352 [DOI] [PubMed] [Google Scholar]
- Sprague BL, McNally JG (2005) FRAP analysis of binding: proper and fitting. Trends Cell Biol 15: 84–91 [DOI] [PubMed] [Google Scholar]
- Sprenger HG, Langer T (2019) The good and the bad of mitochondrial breakups. Trends Cell Biol 29: 888–900 [DOI] [PubMed] [Google Scholar]
- Steinmark IE, James AL, Chung PH, Morton PE, Parsons M, Dreiss CA, Lorentz CD, Yahioglu G, Suhling K (2019) Targeted fluorescence lifetime probes reveal responsive organelle viscosity and membrane fluidity. PLoS ONE 14: e0211165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan T, Roesch A, Riedel D, Jakobs S (2019) Live‐cell STED nanoscopy of mitochondrial cristae. Sci Rep 9: 12419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland W (1905) A dynamical theory of diffusion for non‐electrolytes and the molecular mass of albumin. Philos Mag 9: 781–785 [Google Scholar]
- Tyn MT, Gusek TD (1990) Prediction of diffusion coefficients of proteins. Biotechnol Bioeng 35: 327–338 [DOI] [PubMed] [Google Scholar]
- Vámosi G, Mücke N, Müller G, Krieger JW, Curth U, Langowski J, Tóth K (2014) EGFP oligomers as natural fluorescence and hydrodynamic standards. Sci Rep 6: 33022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bogaart G, Hermans N, Krasnikov V, Poolman B (2007) Protein mobility and diffusive barriers in Escherichia coli: consequences of osmotic stress. Mol Microbiol 64: 858–871 [DOI] [PubMed] [Google Scholar]
- Van Tartwijk FW, Kaminski CF (2022) Protein condensation, cellular organization and spatiotemporal regulation of cytoplasmic properties. Adv Biol 6: e2101328 [DOI] [PubMed] [Google Scholar]
- Verkman AS (2002) Solute and macromolecular diffusion in cellular aqueous compartments. Trends Biochem Sci 27: 27–33 [DOI] [PubMed] [Google Scholar]
- Vögtle FN, Burkhart JM, Gonczarowska‐Jorge H, Kücükköse C, Taskin AA, Kopczynski D, Ahrends R, Mossmann D, Sickmann A, Zahedi RP et al (2017) Landscape of submitochondrial protein distribution. Nat Commun 8: 290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Smoluchowski M (1906) Zur kinetischen theorie der Brownschen Molekularbewegung und der suspensionen. Ann Phys 326: 756–780 [Google Scholar]
- Vorontsova I, Vallmitjana A, Torrado B, Schilling TF, Hall JE, Gratton E, Malacrida L (2022) In vivo macromolecular crowding is differentially modulated by aquaporin 0 in zebrafish lens: insights from a nanoenvironment sensor and spectral imaging. Sci Adv 8: eabj4833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyšniauskas A, Qurashi M, Gallop N, Balaz M, Anderson HL, Kuimova MK (2015) Unravelling the effects of temperature on viscosity‐sensitive fluorescence molecular rotors. Chem Sci 6: 5773–5778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Taki M, Sato Y, Tamura Y, Yaginuma H, Okada Y, Yamaguchi S (2019) A photostable fluorescent marker for the superresolution live imaging of the dynamic structure of the mitochondrial cristae. Proc Natl Acad Sci USA 116: 15817–15822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weilandt DR, Hatzimanikatis V (2019) Particle‐based simulation reveals macromolecular crowding effects on the Michaelis‐Menten mechanism. Biophys J 117: 355–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss M (2014) Crowding, diffusion, and biochemical reactions. Chapter 11 in: new models of the cell nucleus: crowding, entropic forces, phase separation, and fractals. Int Rev Cell Mol Biol 307: 383–417 [DOI] [PubMed] [Google Scholar]
- Weissert V, Rieger B, Morris S, Arroum T, Psathaki OE, Zobel T, Perkins G, Busch KB (2021) Inhibition of the mitochondrial ATPase function by IF1 changes the spatiotemporal organisation of ATP synthase. Biochim Biophys Acta Bioenerg 1862: 148322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox XE, Ariola A, Jackson JR, Slade KM (2020) Overlap concentration and the effect of macromolecular crowding on citrate synthase activity. Biochemistry 59: 1737–1746 [DOI] [PubMed] [Google Scholar]
- Wilkens V, Kohl W, Busch K (2013) Restricted diffusion of OXPHOS complexes in dynamic mitochondrial delays their exchange between cristae and engenders a transitory mosaic distribution. J Cell Sci 126: 103–116 [DOI] [PubMed] [Google Scholar]
- Willems PHGM, Rossignol R, Dieteren CEJ, Murphy MP, Koopman WJH (2015) Redox homeostasis and mitochondrial dynamics. Cell Metab 22: 207–218 [DOI] [PubMed] [Google Scholar]
- Wolf DM, Segawa M, Kondadi AK, Anand R, Bailey ST, Reichert AS, van der Bliek AM, Schackelford DB, Liesa M, Shirihai O (2019) Individual cristae within the same mitochondrion display different membrane potentials and are functionally independent. EMBO J 38: e101056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Li P, Tang B (2021) Small molecular fluorescent probes for imaging of viscosity in living biosystems. Chemistry 27: 6880–6898 [DOI] [PubMed] [Google Scholar]
- Yang Z, He Y, Lee JH, Park N, Suh M, Chae WS, Cao J, Peng X, Jung H, Kang C et al (2013) A self‐calibrating bipartite viscosity sensor for mitochondria. J Am Chem Soc 135: 9181–9185 [DOI] [PubMed] [Google Scholar]
- Yin J, Huang L, Wu L, Li J, James TD, Lin W (2021) Small molecule based fluorescent chemosensors for imaging the microenvironment within specific cellular regions. Chem Soc Rev 50: 12098–12150 [DOI] [PubMed] [Google Scholar]
- Young ME, Carroad PA, Bell RL (1980) Estimation of diffusion coefficients of proteins. Biotechnol Bioeng 22: 947–955 [Google Scholar]
- Yu I, Mori T, Ando T, Harada R, Jung J, Sugita Y, Feig M (2016) Biomolecular interactions modulate macromolecular structure and dynamics in atomistic model of a bacterial cytoplasm. Elife 5: e19274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Shao PG, van Kan JA, van der Maarel JRC (2009) Macromolecular crowding induced elongation and compaction of single DNA molecules confined in a nanochannel. Proc Natl Acad Sci USA 106: 16651–16656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HX, Rivas G, Minton AP (2008) Macromolecular crowding and confinement: biochemical, biophysical and potential physiological consequences. Annu Rev Biophys 37: 375–397 [DOI] [PMC free article] [PubMed] [Google Scholar]