

Abstract
Purpose:
ALK-activating mutations are identified in approximately 10% of newly diagnosed neuroblastomas and ALK amplifications in a further 1%–2% of cases. Lorlatinib, a third-generation anaplastic lymphoma kinase (ALK) inhibitor, will soon be given alongside induction chemotherapy for children with ALK-aberrant neuroblastoma. However, resistance to single-agent treatment has been reported and therapies that improve the response duration are urgently required. We studied the preclinical combination of lorlatinib with chemotherapy, or with the MDM2 inhibitor, idasanutlin, as recent data have suggested that ALK inhibitor resistance can be overcome through activation of the p53-MDM2 pathway.
Experimental Design:
We compared different ALK inhibitors in preclinical models prior to evaluating lorlatinib in combination with chemotherapy or idasanutlin. We developed a triple chemotherapy (CAV: cyclophosphamide, doxorubicin, and vincristine) in vivo dosing schedule and applied this to both neuroblastoma genetically engineered mouse models (GEMM) and patient-derived xenografts (PDX).
Results:
Lorlatinib in combination with chemotherapy was synergistic in immunocompetent neuroblastoma GEMM. Significant growth inhibition in response to lorlatinib was only observed in the ALK-amplified PDX model with high ALK expression. In this PDX, lorlatinib combined with idasanutlin resulted in complete tumor regression and significantly delayed tumor regrowth.
Conclusions:
In our preclinical neuroblastoma models, high ALK expression was associated with lorlatinib response alone or in combination with either chemotherapy or idasanutlin. The synergy between MDM2 and ALK inhibition warrants further evaluation of this combination as a potential clinical approach for children with neuroblastoma.
Translational Relevance.
Neuroblastoma is a pediatric tumor of the developing sympathetic nervous system. Around 50% of patients with high-risk neuroblastoma are curable. Mutations or amplification of anaplastic lymphoma kinase (ALK) have emerged as a marker with which to further risk-stratify patients. The ALK inhibitor lorlatinib will soon be used alongside chemotherapy in upfront treatment of high-risk patients with ALK-aberrant disease. In this preclinical study, we used a panel of ALK-aberrant neuroblastoma models to evaluate ALK inhibitors focusing on lorlatinib in combination with conventional chemotherapy and the small-molecule MDM2 inhibitor idasanutlin. In both approaches, we found synergy in models with high basal ALK expression without MAPK pathway alterations. We conclude that in neuroblastoma the level of ALK expression could be an additional biomarker predictive of clinical response to ALK inhibitors.
Introduction
Neuroblastoma is an embryonal tumor that arises in the developing sympathetic nervous system. Neuroblastoma is dramatically heterogeneous, ranging from spontaneous and complete regression (1) to very aggressive high-risk tumors. High-risk neuroblastoma defined as metastatic disease over 1 year of age or MYCN-amplified disease, has a 5-year overall survival of 50% despite advances in treatment over the last 30 years and accounts for approximately 15% of all childhood cancer deaths (2). New treatments and a better understanding of drug resistance are required to improve disease survival.
The heterogeneity of neuroblastoma is highly dependent on tumor biology. Whereas large-scale copy-number alterations occur in nearly all neuroblastoma as either numerical and/or segmental chromosome alterations, few recurrent molecular alterations targeting single genes have been described in neuroblastoma, of which some are correlated with poor outcome. MYCN amplification (3), TERT rearrangements (4–6), loss-of-function (LoF) ATRX mutations (7, 8), and ALK-activating mutations and amplifications (9) are the most recognized. Both MYCN amplification and TERT rearrangements lead to telomere maintenance by induction of telomerase, whereas ATRX LoF mutations induce telomere maintenance by activation of the alternative lengthening of telomeres pathway. These alterations identify three almost nonoverlapping groups of high-risk neuroblastomas, each associated with very poor prognosis, in particular when associated with mutations in the RAS/MAPK pathway (6–8). As driver oncogenic events, these alterations could represent potential therapeutic targets for neuroblastoma.
ALK-activating mutations are identified in approximately 10% of newly diagnosed neuroblastomas (10, 11) and ALK amplifications (12) in 1%–2% of cases. Furthermore, the incidence of ALK mutations increases in relapsed neuroblastomas, occurring in around 20% of cases (13–15). Three mutation hotspots in the kinase domain (F1174, R1275, and F1245) represent 85% of all forms of ALK mutations (16). Together with ALK amplifications, these gain-of-function alterations lead to phosphorylation of anaplastic lymphoma kinase (ALK) and constitutive increased kinase activity, as well as activation of downstream signaling molecules (such as PI3K-AKT, JAK-STAT, and MAPK pathways), which results in enhanced cellular survival, migration, and proliferation of neuroblastoma (10, 11). In addition, germline ALK mutations are the major cause of hereditary neuroblastoma, which account for 1%–2% of all neuroblastoma cases (10, 11). The high incidence of ALK activation justifies the study of ALK inhibition as a therapeutic target in neuroblastoma.
The most extensively studied ALK inhibitor in neuroblastoma, crizotinib, is a small-molecule competitive inhibitor of ALK and MET kinase activity and was the first FDA-approved drug for use in adult patients with ALK-translocated non–small cell lung cancer. A phase II trial involving crizotinib has been completed by the Children's Oncology Group (COG) in pediatric patients with relapsed/refractory ALK-driven neuroblastoma (NCT00939770). Only 3 of 20 patients showed objective responses (17), mainly explained by the intrinsic resistance of F1174 and F1245 hotspot mutations to crizotinib (16). Preclinical studies showed that crizotinib resistance can be overcome when combined with chemotherapy (18), and was the rationale for the COG phase I trial combining crizotinib with topotecan and cyclophosphamide in children with relapsed and refractory solid tumors (NCT01606878; ref. 19). The second-generation ALK inhibitor ceritinib resulted in objective responses in patients with neuroblastoma harboring ALK F1275 mutations whereas patients with F1174 mutations did not benefit (20).
Later generation ALK inhibitors have been developed to overcome both intrinsic and de novo resistance observed with crizotinib and ceritinib. Lorlatinib, a third-generation ROS1 and ALK inhibitor has been shown to exert preclinical activity against cell line xenografts, patient-derived xenografts (PDX), and genetically engineered mouse models (GEMM) harboring all hotspot ALK mutations (21, 22). These findings led to a phase I trial of lorlatinib for patients with ALK-driven neuroblastoma that is currently ongoing through the New Approaches to Neuroblastoma Therapy (NANT) consortium (NCT03107988), but even following initial response, relapse can occur. Resistance to ALK inhibition is thought to occur through several different mechanisms, including changes in the epigenetic landscape and reactivation of the RAS-MAPK pathway (23, 24).
Furthermore, it has been shown that it is also possible to overcome resistance to ALK inhibition by activating the p53-MDM2 pathway (25, 26). In contrast to other malignancies, neuroblastomas rarely harbor TP53 mutations. Nevertheless, there is evidence of p53 pathway inactivation in 15% of neuroblastomas at the time of relapse, that may contribute to chemotherapy resistance (27). Several mechanisms for p53 inactivation have been proposed, including MYCN-mediated MDM2 overexpression or MDM2 gene amplification or homozygous p14 deletion (28–30). Thus, enhancing or reactivating the functional activity of p53 by targeting the p53-MDM2 pathway via MDM2 inhibition may represent a plausible approach for neuroblastoma treatment.
We hypothesized that combining lorlatinib with chemotherapy or MDM2 inhibitors will lead to enhanced efficacy in neuroblastoma and, ultimately in a clinical setting, improved patient survival. In this work, the in vitro and in vivo activity of ALK inhibitors has been explored alone and in combination with chemotherapy or MDM2 inhibitors in various ALK-mutated or -amplified preclinical models of neuroblastoma, including in vitro (cell lines), ex vivo (PDX-derived tumor cells, PDTC), and in vivo GEMM and PDX models. These models were also used to study the activity of the combination of ALK and MDM2 inhibition for the treatment of ALK-aberrant neuroblastomas. We found that in the context of ALK-aberrant neuroblastoma, treatment efficacy with lorlatinib, either alone or as part of combination therapy, was associated with high basal ALK expression, and we suggest this might be evaluated as an additional biomarker in addition to genetic status in future neuroblastoma clinical trials of ALK inhibitors in neuroblastoma.
Materials and Methods
Treatment of ALK-aberrant neuroblastoma cell lines
Human neuroblastoma cell lines were cultured in RPMI1640 (Sigma) supplemented with 10% v/v FCS (Gibco/Life Technologies Ltd; Supplementary Table S1; ref. 31). Cell lines were authenticated using short tandem repeat genotyping and/or cytogenetic analyses. Cell lines were sequenced for ALK by Sanger sequencing (exons 20–29) and targeted next-generation sequencing (NGS) for ALK and RAS-MAPK pathway genes (31) and ALK amplification was assessed by FISH and/or SNP array, the latter analyzed using Nexus Biodiscovery software (Supplementary Fig. S1A).
The MDM2 antagonist idasanutlin was provided by Hoffman-La Roche/Genentech. All other compounds were purchased from Selleck Chemicals (Stratech Scientific Ltd). All compounds for in vitro studies were dissolved in DMSO (Sigma-Aldrich). A total of 72-hour XTT growth inhibition assays with determination of the concentration required to inhibit growth by 50% (GI50) and median effect analysis were performed as previously described in cells growing exponentially (32). Synergy and combination index (CI) values with the ALK inhibitors TAE684 and alectinib (as a prelude to in vivo studies with the clinical inhibitor lorlatinib) in combination with idasanutlin, were determined using CalcuSyn v2 (Biosoft). Flow cytometry and caspase 3/7 assays were performed as previously described and according to manufacturer's protocols (32).
Ex vivo drug screenings in PDTC models
Obtaining PDTCs from PDX tumors
For viable PDTC generation, we used a dissociation protocol adapted from Stewart and colleagues (33), which included a first step of PDX tumor mechanical dissociation with sterile scalpels and then enzymatic dissociation by trypsin (10 mg/mL) and type II collagenase (275 U/mg, Worthington Biochemical). The tube was then placed in a warm 37°C water bath for 60 minutes. Dissociation was stopped by adding Soybean Trypsin Inhibitor (10 mg/mL, Sigma). Deoxyribonuclease I (2 mg/mL, Sigma) and magnesium chloride (1 mol/L) were added in equal amounts. Tumor suspension was filtered with a 40 μm cell strainer and then centrifuged at 500 × g for 5 minutes. Supernatant was discarded and the cell pellet was resuspended in PBS-minus/10%FBS for cell counting. The suspension was then recentrifuged and resuspended in serum-free stem-cell (SC) medium, which contained DMEM/Nutrient Mixture F-12 (DMEM/F12, Gibco) supplemented with 40 ng/mL basic FGF, 20 ng/mL EGF, 1× B27 supplement (Gibco), and 500 U/mL of penicillin/streptomycin.
Monotherapy drug screening of PDTCs
The same experimental pipeline was used for all drug screens in PDTCs. ALK inhibitors crizotinib, alectinib, ceritinib, and lorlatinib were purchased from Selleck Chemicals (Stratech Scientific Ltd) as 10 mmol/L stock solution in 100% DMSO. PDTCs in SC medium were seeded in 384-well plates (ViewPlate-384 Black Perkin Elmer, ref. 6007460) using a MultiDrop combi (Thermo Fisher Scientific) at a concentration of 20,000 cells in 40 μL per well. Twenty-four hours after cell seeding, drugs were robotically added to each well and titrated by 3-fold dilutions covering 12 concentrations from 10,000 to 0.056 nmol/L, across three 384-well plates. Control wells received the same distributed volumes with either DMSO (DMSO control) or cell media alone (empty). Plates were then incubated at 37°C in 5% CO2 for 72 hours, then cell viability was evaluated in culture based on quantification of the ATP present using CellTiter Glo Luminescent cell viability assay (Promega). Raw luminescence was recorded using a CLARIOStar (BMG Labtech) and obtained values were normalized on a per-plate basis by dividing the raw luminescence value in each well by the median value obtained for the DMSO-treated wells condition (100% viability). Dose–response curves were fitted with a 4-parameter log-logistic model with a fixed viability of 100% at 0 nmol/L using R package drc. Drug responses were represented by the quantitative drug sensitivity score (DSS), as described by Yadav and colleagues (34). Each PDX model was screened in two independent biological replicates (two different experiments for each PDX model, at a different PDX passage number, with high correlation across the results; Supplementary Fig. S2A).
In vivo experiments with GEMMs
All experiments, including the breeding of transgenic animals, were performed in accordance with the local ethical review panel, the UK Home Office Animals (Scientific procedures) Act 1986, the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines (35) and the UK NCRI guidelines (36).
For GEMM experiments, crizotinib and lorlatinib were provided by Pfizer, alectinib was provided by Hoffman-La Roche/Genentech, and ceritinib was purchased from Selleck Chemicals (Stratech Scientific Ltd).
Th-ALKF1174L/MYCN tumor-bearing animals were enrolled into therapeutic trials when their abdominal tumors reached 5 mm in diameter according to palpation. Changes in tumor volume in the TH-ALKF1174L/TH-MYCN mice were quantified using MRI on a 7T horizontal bore MicroImaging system (Bruker Instruments) using a 3 cm birdcage coil. Anatomic T2-weighted coronal images were acquired through the mouse abdomen, from which tumor volumes were determined using segmentation from regions of interest drawn on each tumor-containing slice (37).
Short-term studies
For in vivo oral dosing, crizotinib was dissolved in sterile water with 10% Tween 20. Ceritinib was dissolved into 0.5% methylcellulose, 0.5% Tween 80 with sterile water. Alectinib was dissolved in 10% DMSO, 10% cremophor, 15% PEG400, 15% HPCD, 0.02N HCl in sterile water. Lorlatinib was dissolved into 0.5% methylcellulose, 0.5% Tween 80 with sterile water. The following doses were used for CAV chemotherapy, given as two intraperitoneal injections: (C) cyclophosphamide: 28 mg/kg; (A) doxorubicin: 0.7 mg/kg; (V) vincristine: 0.015 mg/kg (38). Two hours following the final dose of compound, tumor tissue was excised and snap frozen prior to analysis.
Longer-term survival studies
For longer-term survival studies, tumor volume was monitored by daily palpation, and the animal was sacrificed when UK Home Office License limits were reached to record survival. Daily dosing of compounds was continuous for maximum of 56 doses. Mice receiving the CAV chemotherapy regimen were treated with an intraperitoneal injection on day 1 with doses as described above. In the combination regimen, lorlatinib was commenced from day 2 at 10 mg/kg orally, once daily. In the single-drug regimen, lorlatinib was commenced on day 1 at 10 mg/kg orally, twice daily. Mice were sacrificed 2 hours following the final dose of lorlatinib and any remaining tumor tissue was collected for pharmacodynamic studies.
Pharmacodynamic studies on GEMM tumors
Snap frozen tumors were lysed in 5% CHAPS buffer and quantitation of ALK and pY1586 ALK was performed using immunoassay as described previously (37).
PDX in vivo experiments
All in vivo experiments on PDX were performed according to the European and National Regulation in force for the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes (Directive 86/609). This study was approved by the Ministère de l'Education Nationale, de l'Enseignement Supérieur et de la Recherche (authorization number APAFIS # 13980-2018030813227748 v2).
PDX treatments
Swiss Nude mice (Charles River) were engrafted in the interscapular fat pad. Twelve mice were engrafted per experimental treatment arm to ensure at least 5 treated mice for analysis of tumor growth inhibition and 3 for pharmacodynamic studies (IHC) were available. For PDX experiments, all drugs were purchased from Selleck Chemicals (Stratech Scientific Ltd), and dissolved as per the GEMM experiments. Treatment started when tumor reached 60–180 mm3. All mice received the CAV chemotherapy regimen intraperitoneally on day 1 with the same doses as used in the GEMM. For efficacy studies, mice received idasanutlin and lorlatinib orally at 75 mg/kg/dose from day 1 and 10 mg/kg/dose from day 2, respectively, 5 days per week. Targeted therapies were continued until tumor ethical size at 2,000 mm3. For IHC pharmacodynamic studies, mice received CAV chemotherapy on day 1, idasanutlin on day 1 and day 2, and lorlatinib on day 2 and day 3 (all drugs at the doses described above). Mice were sacrificed on day 3, 4 hours after the second dose of lorlatinib.
Antitumor efficacy assessment on PDX tumors
Tumor volumes were calculated by measuring two perpendicular diameters with calipers. Each tumor volume (V) was calculated according to the following formula: V = a × b2/2, where a and b are the largest and smallest perpendicular tumor diameters. Relative tumor volumes (RTV) were calculated from the following formula: RTV = (Vx/V1), where Vx is the tumor volume on day x and V1 is the tumor volume at initiation of therapy (day 1). Growth curves were obtained by plotting the mean values of RTV on the Y axis against time (X axis, expressed as days after the start of treatment). Antitumor activity was evaluated according to tumor growth inhibition (TGI), calculated according to the following formula: percent GI = (1 − RTVt/RTVc) × 100, where RTVt is the median RTV of treated mice and RTVc is the median RTV of controls, both at a given timepoint when the antitumor effect was optimal. A total of 50% TGI was considered to be the limit for a meaningful biological effect. Statistical significance of differences observed between the individual RTVs corresponding to the treated mice and control groups was calculated by the two-tailed Student t test.
Pharmacodynamic studies on PDX tumors
Tumors for IHC were extracted on day 3 of treatment, 4 hours after the second dose of lorlatinib. All tissue blocks were cut to provide sections of 3 μm. IHC was performed for Ki67 (Abcam ab15580), cleaved caspase-3 (Cell Signaling Technology #9661), and ALK to assess for cell proliferation, apoptosis, and the level of ALK expression, respectively. The evaluation of immunostaining was conducted by two independent pathologists. The intensity of the immunostaining in the cell cytoplasm and the percentage of neoplastic cells with positive immunostaining were estimated. The score is the result of the multiplication of color intensity and percentage of cells with positive immunostaining, giving scores in the range of 0–3. For tumors harvested at ethical size limit, Western blotting was carried out for ALK (Cell Signaling Technology #3633), pY1586 ALK (Cell Signaling Technology #3348), ERK1/2 (Cell Signaling Technology #4695), pERK1/2 (Cell Signaling Technology #9101), AKT (Cell Signaling Technology #9272), pAKT (Cell Signaling Technology #4060), and GAPDH (Cell Signaling Technology #2118) as described previously (37). Densitometry was performed using ImageJ.
Data availability statement
The data generated in this study are publicly available at BioProject, ID PRJNA848127.
Results
Lorlatinib is the most potent ALK inhibitor in both in vitro and in vivo models of neuroblastoma
Given the poorer prognostic impact of ALK amplifications and clonal ALK mutations in patients with high-risk neuroblastoma (9), we sought to further study the preclinical efficacy of ALK inhibitors in neuroblastoma models. In a first step, GI50 values for the ALK inhibitors TAE-684 (a preclinical ALK inhibitor included to provide comparison with previous studies), crizotinib, alectinib, ceritinib, and lorlatinib were determined in a panel of neuroblastoma cell lines of varying ALK status using 72-hour XTT assays. Sensitivity to the tested ALK inhibitors except for lorlatinib correlated with the presence and type of ALK alteration (i.e., mutation or amplification; Fig. 1Ai–v). Cell lines with ALK alterations were generally less sensitive to lorlatinib than crizotinib, with a much wider range of reported GI50 values also seen for lorlatinib. A recent article reported that the coexistence of RAS/MAPK pathway mutation following ALK inhibitor treatment confers resistance to ALK inhibitors, particularly the most selective ALK inhibitor lorlatinib, due to by-pass ALK signaling (24). Using targeted NGS, we found that ALK-aberrant cell lines most resistant to lorlatinib were those with additional RAS/MAPK pathway mutations, for example, NBLW, LAN6, SH-SY5Y (Supplementary Table S1).
Figure 1.

Lorlatinib is the most potent ALK inhibitor tested across ALK-mutant or ALK-amplified neuroblastoma cell lines and PDTC ex vivo models. 72-hour GI50 values for ALK inhibitors. A, (i) TAE-684, (ii) crizotinib, (iii) ceritinib, (iv) alectinib, and (v) lorlatinib in a panel of neuroblastoma cell lines (M & A: mutant and amplified ALK; Mut.: mutant ALK; Amp.: amplified ALK; WT: wild-type ALK). Cell lines were grouped on the basis of the type of ALK alteration. Statistically significant differences were determined by one-way ANOVA with Bonferroni post hoc tests and paired testing versus WT. *, P ≤ 0.05; **, 0.01; ***, 0.001; ****, 0.0001. B, Neuroblastoma cell lines treated with 20 nmol/L of indicated inhibitor for 3 hours, and lysates subjected to ALK immunoassay for total ALK and pY1586 ALK. Statistically significant differences determined by paired, two-tailed t test. *, P ≤ 0.05; **, 0.01; ***, 0.001. C, Study of cell viability by analysis of the DSS after treatment with (i) crizotinib, (ii) ceritinib, (iii) alectinib, or (iv) lorlatinib in ALK-mutant and -amplified PDTC models. R1: replicate 1; R2 replicate 2. Statistically significant differences determined by unpaired, two-tailed t test. **, P ≤ 0.01.
We quantified in vitro inhibition of ALK phosphorylation by immunoassay, comparing the different ALK inhibitors in a subset of our panel of neuroblastoma cell lines and found that lorlatinib was the most potent inhibitor of ALK phosphorylation at this dose and timepoint (Fig. 1B; ref. 37), which would not be affected by concomitant RAS pathway mutation.
To extend this work, we screened a selection of five neuroblastoma PDX-derived ex vivo tumor cell models (PDTC) carrying ALK alterations: GR-NB4 carried ALK amplification and IC-pPDX-112 carried an intron 3 ALK amplification (Supplementary Fig. S1B), and IC-pPDX-75, HSJD-NB-011, and HSJD-NB-012 carried ALKF1174L, ALKI1171N, and ALKF1174C mutations, respectively (Supplementary Table S2). None of these models had concomitant RAS-MAPK pathway alterations. Notably, the patient from whom the IC-pPDX-75 model was derived had received crizotinib treatment for 12 months before establishment of the PDX model.
PDTCs were treated with a panel of four ALK inhibitors (Fig. 1Ci–iv). Drug responses were represented by the quantitative DSS (33), which integrates a multiparameter analysis, including the potency (EC50), slope of the dose–response curve, the AUC, and the maximum effect of the drug. In PDTC models, while DSS values of lorlatinib were higher than those of ceritinib or similar to those of crizotinib, only lorlatinib showed significantly higher cytotoxicity (lower DSS values) in ALK-aberrant PDTC models compared with wild-type models. The correlation between two experimental repeats (R1 and R2) was good across all four models, with good distribution of viability for each PDTC in the control wells, and evidence that lorlatinib treatment increased the percentage of apoptotic cells compared with DMSO control (Supplementary Fig. S2A–S2D). Altogether our data support previous work that lorlatinib is the most potent ALK inhibitor (21, 22). However, its efficacy in in vitro neuroblastoma models is not superior to other ALK inhibitors when a concomitant RAS/MAPK pathway mutation is present.
To evaluate the in vivo efficacy of ALK inhibitors, we performed a short-term response assessment study in the Th-ALKF1174L/MYCN GEMM of ALK mutant neuroblastoma. Th-ALKF1174L/MYCN double transgenic animals develop spontaneous abdominal neuroblastoma at a shorter latency and 100% penetrance, compared with their Th-MYCN littermates, indicating that the addition of ALKF1174L plays a key role in tumorigenesis in this model (39). Tumor-bearing Th-ALKF1174L/MYCN mice, which had been treated with lorlatinib showed the greatest reduction in tumor volume compared with other clinical ALK inhibitors, over a 3-day interventional dosing schedule (Fig. 2A). When tumors were harvested for immunoassay measurement of total and pY1586 ALK, it was found that alectinib had an equivalent effect to lorlatinib on reduction of pY1586/total ALK in the tumor cells, suggesting that the tumor volume response observed in these animals following lorlatinib treatment, may be due to a secondary off-target effect of lorlatinib, not shared with alectinib (Fig. 2B). Treatment of Th-MYCN heterozygous mice with crizotinib and alectinib also resulted in a highly variable tumor volume response over 3 days of therapy, confirming the activity of these compounds against targets other than mutant ALK (Supplementary Fig. S3A). Subsequently, survival of the Th-ALKF1174L/MYCN GEMM treated continuously with lorlatinib twice daily, a schedule suggested by previous pharmacokinetic studies in mice (40), was assessed against vehicle control (Fig. 2C). Tumor volume measurements using MRI indicated that lorlatinib only slowed tumor progression in this model, with no tumor-free survivors observed (Fig. 2D and E). Analysis of tumor tissue taken at the end of the study revealed sustained ALK dephosphorylation and inhibition of expression of endogenous murine Mycn as a downstream marker of ALK activity (39), suggesting that lorlatinib was still active against ALK signaling in these tumors (Supplementary Fig. S3B and S3C). Furthermore, RNA sequencing of tumor tissue taken at the end of the experiment, comparing vehicle-treated controls against lorlatinib-treated Th-ALKF1174L/Mycn, revealed reduced expression of noradrenergic genes, which was associated with lower ALK expression (Supplementary Fig S3E; ref. 42). The adrenergic phenotype has been associated with ALK expression in neuroblastoma, suggesting that lorlatinib treatment either allows expansion of non–ALK-expressing cells or that ALK inhibition can drive a cell plasticity response (43). In addition, there was an enhanced reduction of ALK expression in tumors classified as “poor responders” compared with “early responders,” based upon evidence of tumor regression at day 7 of the study (Supplementary Fig. S3D and S3E).
Figure 2.

Lorlatinib treatment of Th-ALKF1174L/MYCN tumor-bearing animals gives a survival advantage over vehicle control. A,In vivo analysis of a panel of ALK inhibitors including crizotinib, ceritinib, alectinib, and lorlatinib was carried out using the Th-ALKF1174L/MYCN model. Tumor-bearing Th-ALKF1174L/MYCN mice, were treated with the indicated inhibitor or its corresponding vehicle (veh.) over a 3-day interventional dosing schedule, and tumor volume change was monitored by MRI on day 0 and day 3. Each bar represents tumor volume change in an individual animal. Crizotinib versus vehicle ****, P < 0.0001. Alectinib versus vehicle **, P = 0.0021. Lorlatinib versus vehicle ***, P = 0.0002. B, Tumors from A were harvested for immunoassay testing of ALK and pY1586 ALK status. Alectinib versus vehicle ***, P = 0.0005. Lorlatinib versus vehicle **, P = 0.0037. C, Th-ALKF1174L/MYCN animals were treated with lorlatinib twice daily versus vehicle control to assess survival. ****, P < 0.0001 according to log-rank (Mantel–Cox) test. D, Tumor volume was monitored by MRI. E, Representative abdominal MRI of an animal from the lorlatinib survival study versus vehicle control. Tumor outlined by white line.
Lorlatinib combined with chemotherapy improves tumor growth control in immunocompetent GEMM
We next evaluated whether concurrent ALK inhibition could increase the efficacy of conventional multi-agent chemotherapy regimens representative of those used in clinical practice. To this end, we developed a multi-agent chemotherapy regime, consisting of CAV, given as a single dose, prior to commencement of once-daily lorlatinib (38). We employed this schedule in the Th-ALKF1174L/MYCN and Th-MYCN GEMM and found that the addition of lorlatinib to CAV chemotherapy significantly increased survival in this model, but not in the heterozygous Th-MYCN GEMM (Fig. 3A). Notably, animals treated with lorlatinib alone exhibited no survival advantage against vehicle controls with the once daily dosing schedule. Animals receiving the combination therapy underwent an early tumor response to treatment as measured by MRI (Fig. 3B), but ultimately no tumor-free survivors were observed.
Figure 3.

CAV chemotherapy in combination with lorlatinib leads to significantly enhanced survival in preclinical Th-ALKF1174L/MYCN GEMM neuroblastoma. A, Survival study of lorlatinib, with and without CAV chemotherapy in Th-ALKF1174L/MYCN and Th-MYCN (dashed line) GEMMs. Vehicle versus CAV and lorlatinib ***, P = 0.0006; CAV versus CAV and lorlatinib ***, P = 0.0005; lorlatinib versus CAV and lorlatinib ***, P = 0.0005 in the Th-ALKF1174L/MYCN model according to log-rank (Mantel–Cox) test. B, MRI growth monitoring of Th-ALKF1174L/MYCN tumors from the survival study in A. No statistical significance between groups. C, Heatmap of the expression of 34 selected genes across all the treatment groups. Noradrenergic (ADRN) and MES-like (mesenchymal cell–like). D, Mean of log2 TMM (trimmed mean of M values) score for genes in ADRN (i) or MES (ii) panel across treatment groups.
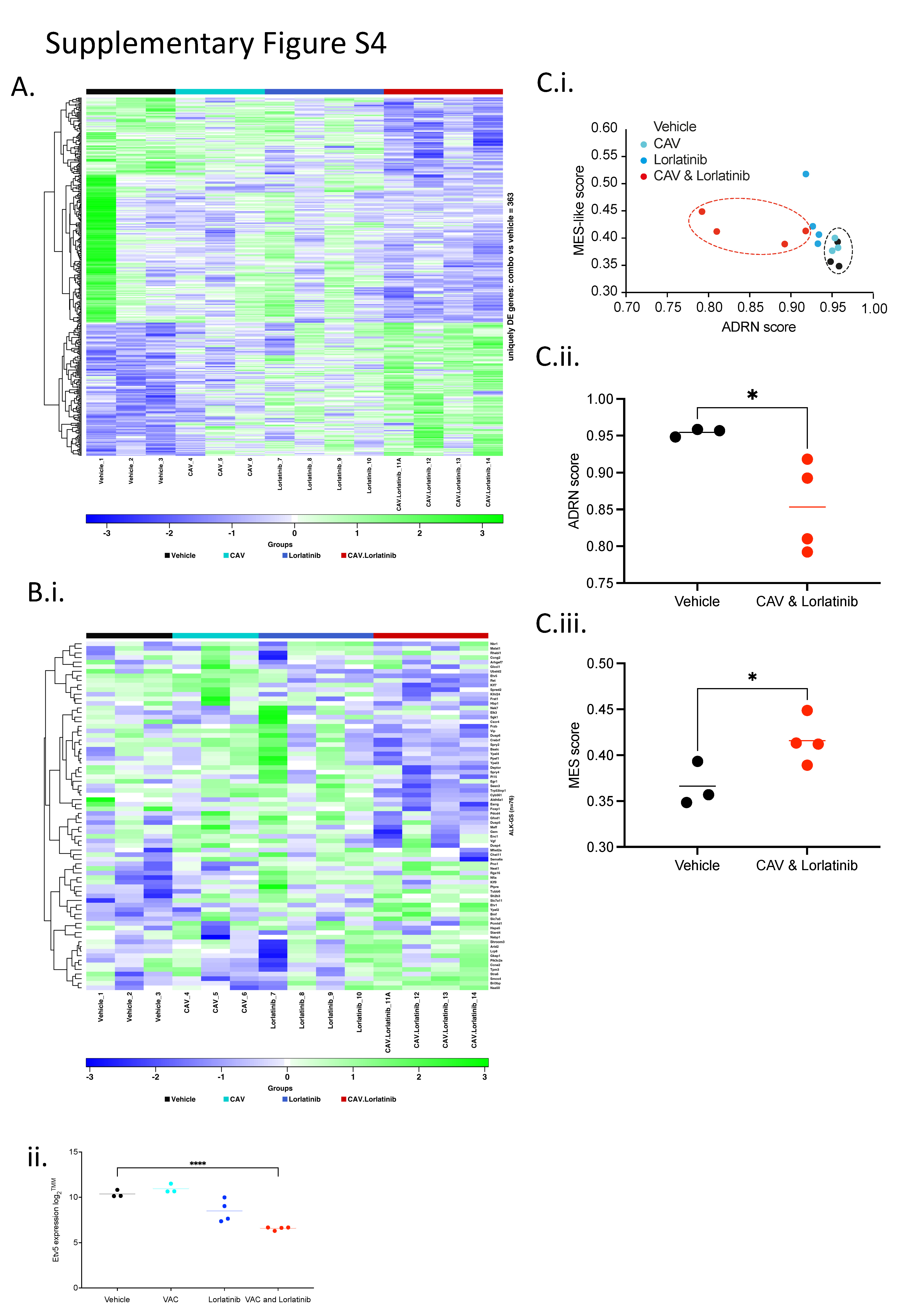
We carried out RNA sequencing of tumors from the endpoint of this CAV-lorlatinib survival study to further investigate the reason for development of resistance on continuous daily dosing of lorlatinib. The greatest difference in overall gene expression was found between the vehicle-treated and combination-treated arms, within which we found upregulation of 135 genes and downregulation of 228 genes in tumors treated with CAV-lorlatinib (Supplementary Fig S4A). Furthermore, we considered ALK-signature genes in this experimental cohort, and found that while there was no significant change in the mean ALK gene set expression, individually the expression of ETV5, known to be a robust marker of ALK activity, was significantly lowered between vehicle- and combination-treated tumors (Supplementary Fig. S4Bi and ii; refs. 43, 44). We formally tested the published signature score (42, 45) for mesenchymal and adrenergic phenotypic expression in neuroblastoma cells, and found a trend toward upregulation of the mesenchymal-like score (*, P = 0.0476) and downregulation of the adrenergic score (*, P = 0.0452) in combination-treated tumors versus vehicle-treated controls; however, this was not necessarily of biological significance (Fig. 3C and D; Supplementary Fig. S4C). In addition, in both the lorlatinib and lorlatinib-chemotherapy arms, we found activation of the Tp53 pathway according to analysis of gene expression (Fig. 3C), consistent with previous studies showing activation of the Tp53 pathway after ALK inhibition and chemotherapy (18). However, there was no apparent upregulation of RAS/MAPK-associated genes in the combination-treated group, activation of which is also associated with neuroblastoma relapse and resistance to ALK inhibition (24).
To expand our study of CAV in combination with lorlatinib in vivo, we used the identical dosing schedule as developed for Th-ALKF1174L/MYCN tumor-bearing animals in our neuroblastoma PDX panel. We employed this dosing schedule in an ALK-amplified and intron 3 ALK-amplified and one ALK-mutant (ALKF1174C) PDX models (Supplementary Table S2). Pharmacodynamic studies were performed by treating a cohort of PDX up until day 3. We analyzed levels of total ALK, P53, cleaved caspase 3, and P21 expression by IHC in PDX samples in treated and vehicle-treated tumors at day 3 (Supplementary Table S3). The GR-NB4 model exhibited the highest levels of ALK mRNA and protein expression in vehicle-treated tumors among the three models (Fig. 4A; Supplementary Fig. S5A and S5B). However overall, the level of ALK protein expression was otherwise stable at this day 3 timepoint in all PDX models independently of the treatment received. No differences in cleaved caspase-3 and Ki67 protein expression were observed in ALK-amplified and intron 3 ALK-amplified PDX models in untreated and lorlatinib or lorlatinib-CAV treated PDX at this early timepoint (Supplementary Table S3). Of note, GR-NB4 had very low CDKN2A expression (Supplementary Fig. S5A) consistent with CDKN2A homozygous deletion (Supplementary Table S2).
Figure 4.

High baseline in vivo expression of ALK determines sensitivity to lorlatinib in an ALK-amplified PDX neuroblastoma model. A, Hematoxylin and Eosin (H&E) and ALK IHC staining of vehicle-treated tumors from indicated PDX models, treated for 3 days. GR-NB4 (B), IC-pPDX-112 (C), and HSJD-NB-012 (D) PDX models were treated with vehicle control, one dose of chemotherapy (CAV: cyclophosphamide, doxorubicin, and vincristine), continuous lorlatinib, or chemotherapy and continuous lorlatinib combination. (i) Tumor volumes were monitored during treatment. (ii) pY1586/total ALK measured by immunoassay in tumor lysates taken at the end of the experiment (mean of two technical replicates; Veh.: vehicle; Lorla.: lorlatinib; C&L: CAV and lorlatinib). One-way ANOVA: B.ii: P < 0.0001; C.ii: not significant; D.ii: P = 0.0010 (iii). Immunoblots of signaling pathways downstream of ALK from tumor lysates as per (ii).
A longer-term tumor volume study was undertaken in a second cohort of mice. Total ALK expression was compared across the three models by immunoblot and immunoassay in vehicle-treated tumors, confirming the highest level of ALK expression in the ALK-amplified GR-NB4 model (Supplementary Fig. S5Bi and ii). In this ALK-amplified GR-NB4 model, lorlatinib alone showed significant activity (TGI 99% at 24 days; Fig. 4Bi). The addition of CAV did not significantly increase the efficacy of lorlatinib alone. Conversely, there was no effect on tumor growth in the intron 3 ALK-amplified model (Fig.4Ci). The ALK-mutant PDX model showed a modest response to lorlatinib, both alone, and combined with CAV (Fig. 4Di). ALK immunoassay analysis of tumor lysates taken at the end of these experiments (Fig. 4Bii, Cii, and Dii) revealed robust dephosphorylation of pY1586 ALK in GR-NB4 and HSJD-NB-012 but not IC-pPDX-112. There was good evidence of dephosphorylation of both AKT and ERK1/2 on immunoblotting following lorlatinib treatment in the GR-NB4 model, and dephosphorylation limited to ERK1/2 in the IC-pPDX-112 model (Fig. 4Biii, Ciii, and Diii; Supplementary Fig. S5C). Furthermore, studying the ALK expression intensity evaluated by RNA sequencing in 44 patients with neuroblastoma from the MAPPYACTS trial (46), indicated that mutation or amplification of ALK is associated with increased RNA expression of ALK, in comparison with non–ALK-aberrant neuroblastoma (Supplementary Fig. S5D).
Idasanutlin synergizes with ALK inhibitors in TP53 wild-type and ALK-aberrant neuroblastoma cell lines
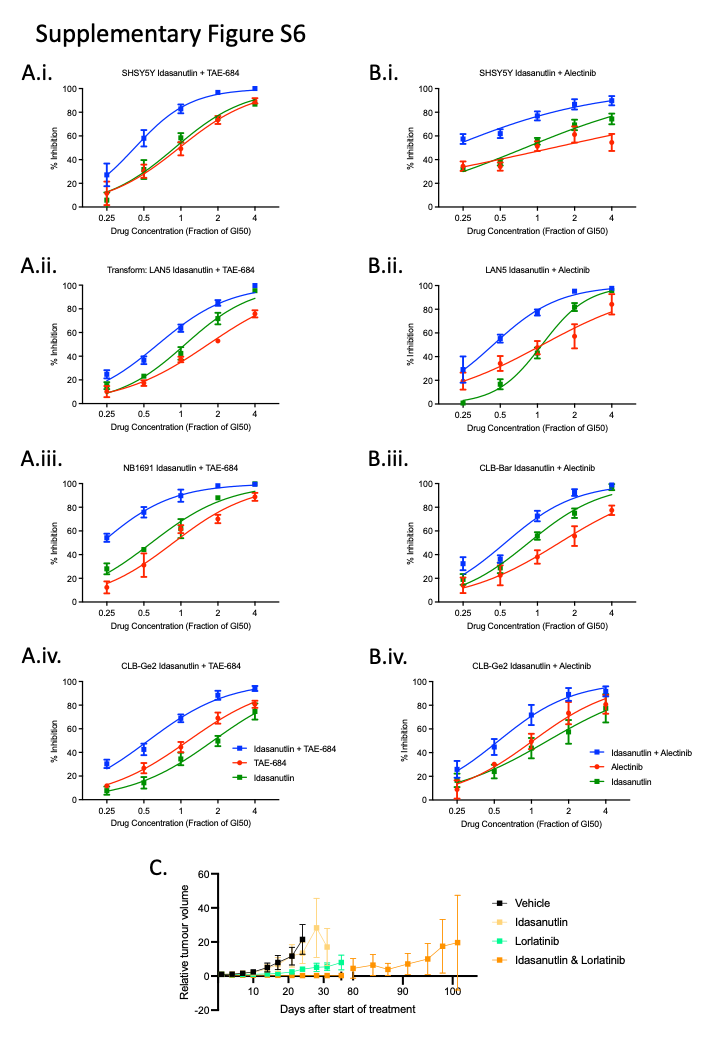
While the combination of lorlatinib with chemotherapy will soon be introduced into upfront therapy for high-risk ALK-aberrant neuroblastoma, other combinations that might be suitable for this subgroup of patients following relapse and chemotherapy resistance are still warranted. To this end, we also investigated combinations of ALK inhibitors with MDM2 inhibitors, which have previously been suggested as potential strategies to overcome resistance to ALK inhibitors (25, 26). Because none of the PDXs had RAS-MAPK mutations, this was not a mechanism of resistance to ALK inhibitors in these models and all of them were TP53 wild-type (Supplementary Table S2). The MDM2 antagonist, idasanutlin, is being evaluated in combination with venetoclax or chemotherapy for patients with relapsed and/or refractory neuroblastoma in a phase I clinical trial (NCT04029688). In TP53 wild-type and ALK-aberrant neuroblastoma cell lines including a previously unreported ALK-amplified cell line, NB1691 with MDM2 amplification (Supplementary Fig. S1A), 2 ALK-mutant and 2 ALK-mutant and amplified cell lines (Supplementary Table S1), median effect analysis demonstrated a synergistic interaction between idasanutlin and ALK inhibitors, TAE-684 and alectinib in vitro (Fig. 5A and B). Further assessment showed that the combination treatment led to enhanced levels of apoptosis as evident by the greater proportion of sub-G1 events and caspase 3/7 activity determined using FACS and caspase 3/7 assays (Fig. 5C–F; Supplementary Fig. S6A and S6B).
Figure 5.

Idasanutlin synergizes with ALK inhibitors in TP53 wild-type and ALK-aberrant neuroblastoma cell lines. CI values at each constant 1:1 ratio combination and average of CI values at ED50, ED75, and ED90 of idasanutlin in combination with TAE-684 (A) and alectinib (B). Functional analysis of idasanutlin in combination with TAE-684 and alectinib using sub-G1 and cell-cycle phase distribution (C and D), and caspase 3/7 activity (E and F). IDA, idasanutlin; TAE, TAE-684; I&T, idasanutlin and TAE-684; ALE, alectinib; I&A, idasanutlin and alectinib. One-way ANOVA: (C): SHSY5Y *, P = 0.0441; NB1691 **, P = 0.0070; (D): SHSY5Y *, P = 0.0273; CLB-Ge2 *, P = 0.0230; (E): SHSY5Y **, P = 0.0043; NB1691 *, P = 0.0322; (F): SHSY5Y *, P = 0.0150; CLB-Ge2 ***, P = 0.0008.
Idasanutlin enhances antitumor activity of lorlatinib in an ALK-amplified neuroblastoma PDX model
In vivo we studied the efficacy of lorlatinib in combination with idasanutlin in two ALK-amplified and one ALK-mutant PDX model. In the high-ALK expressing ALK-amplified GR-NB4 model, whereas idasanutlin alone had a modest effect on TGI (38% at 24 days), the combination of lorlatinib and idasanutlin showed impressive additive effect with complete remission (TGI 50% and 0% at 24 days for combination and inhibitors alone, respectively; Fig. 6Ai and ii; Supplementary Table S4). The animals in this combination arm were treated until day 84 and then left until tumor relapse occurred and the tumors reached the ethical size limit (Supplementary Fig. S6C). Conversely, the ALK mutant model was completely resistant to both drugs as monotherapy as well as the combination (Fig. 6Ci). Immunoassay analysis for pY1586ALK expression from the samples at the end of the experiment on days 35–40 revealed robust dephosphorylation of ALK in most tumors receiving either lorlatinib alone or in combination with idasanutlin (Fig. 6Aiii, Bii, and Cii). The exception was from the lorlatinib-idasanutlin arm of the GR-NB4 model, as all animals had survived several weeks following termination of treatment (Fig. 6Ai and ii; Supplementary Fig. S6C). Pharmacodynamics were performed on day 3 on a separate cohort of animals. No differences in cleaved caspase-3 and Ki67 protein expression were observed in ALK-amplified and ALK mutant PDX models in samples treated with lorlatinib alone and in combination with idasanutlin (Supplementary Table S3). The level of ALK, TP53, and P21 expression by IHC in ALK -amplified model was unchanged in treated and untreated tumors.
Figure 6.
![Figure 6. Combination effects of lorlatinib and idasanutlin in ALK-altered neuroblastoma PDX models. A, GR-NB4 (B) IC-pPDX-112 (C) HSJD-NB-012 PDX models were treated with vehicle control, idasanutlin, lorlatinib, or idasanutlin and lorlatinib combination. I, Tumor volumes were monitored during treatment [with (A.ii) inset of GR-NB4 PDX up to day 10]. (A.iii, B.ii, and C.ii) pY1586/total ALK measured by immunoassay in tumor lysates taken at the end of the experiment (mean of two technical replicates). One-way ANOVA: A.iii: P = <0.0001; B.ii: P = 0.0331; C.ii: P = 0.0020.](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=10068437_1317fig6.jpg)
Combination effects of lorlatinib and idasanutlin in ALK-altered neuroblastoma PDX models. GR-NB4 (A), IC-pPDX-112 (B), and HSJD-NB-012 (C) PDX models were treated with vehicle control, idasanutlin, lorlatinib, or idasanutlin and lorlatinib combination. (i) Tumor volumes were monitored during treatment [with (A.ii) inset of GR-NB4 PDX up to day 10]. (A.iii, B.ii, and C.ii) pY1586/total ALK measured by immunoassay in tumor lysates taken at the end of the experiment (mean of two technical replicates). One-way ANOVA: A.iii: P = <0.0001; B.ii: P = 0.0331; C.ii: P = 0.0020.
Bulk RNA sequencing of representative PDX tumors from the experiments shown in Fig. 4B–D and Fig. 6A–C was undertaken, illustrating differences in gene expression across the three models (Supplementary Fig. S5A). This showed increased TP53 and MDM2 expression following combination treatment in IC-pPDX-112 consistent with functional TP53, but not in the GR-NB4 or HSJD-NB-012 PDX models.
Discussion
The goal of this study was to provide robust preclinical insights into the activity of ALK inhibitors in combination with chemotherapy or targeted MDM2 inhibition in neuroblastomas carrying mutations or amplifications of the ALK gene.
The ongoing International Society of Paediatric Oncology European Neuroblastoma trial HRNBL2 (NCT04221035) for first-line treatment of high-risk neuroblastoma will soon be amended to introduce lorlatinib in upfront treatment of patients with ALK-aberrant high-risk neuroblastoma. Concurrently, the COG is amending the ANBL1531 phase III trial to replace crizotinib with lorlatinib. Since 2008, when the role of ALK in neuroblastoma was first described (10, 11, 47), the study of ALK inhibitors in neuroblastoma has grown as a result of progress in the development of these drugs for adult malignancies and advances made by the scientific neuroblastoma community.
Preclinical studies in ALK-aberrant neuroblastomas have shown intrinsic resistance of ALKF1174 and ALKF1245, two of the three main mutational hotspots, to crizotinib (16). The second-generation molecule ceritinib inhibits the kinases of ALK F1245 and ALK F1275 but also exhibits intrinsic resistance to the F1174 variant (20). Lorlatinib, a third-generation ALK inhibitor overcomes resistance to first- and second-generation ALK inhibitors by its macrocyclic structure with optimized physicochemical properties, which are associated with improved metabolic stability, blood–brain barrier permeability, and low propensity for multi-drug resistance efflux (48).
Cell lines with ALK alterations were in general less sensitive to lorlatinib than to ceritinib and other ALK inhibitors based on growth inhibition assays (Fig. 1Ai–v) which we believe is due to the presence of a concomitant RAS-MAPK pathway mutation in cell lines which were hyperresistant to lorlatinib (except for cell line NB1691, which has a highly complex genotype; Supplementary Fig. S1A). Despite these GI50 data in cell lines, albeit with an assay measuring metabolic activity as a surrogate for cell viability, lorlatinib showed the greatest inhibition of ALK phosphorylation in cell lines (Fig. 1B), which reflects its selectivity for the ALK signaling pathway upstream of the RAS-MAPK pathway compared with other ALK inhibitors, but inhibition of ALK signaling by lorlatinib can be bypassed in the presence of a downstream RAS-MAPK pathway mutation. Indeed, lorlatinib showed the greatest in vivo reduction in tumor volume over a 3-day interventional dosing schedule in ALKF1174L/MYCN GEMMs which do not have concomitant RAS-MAPK pathway mutations. However, alectinib had a similar effect to lorlatinib on ALK dephosphorylation in tumor cells, suggesting other off-target effects of lorlatinib which could lead to enhanced efficacy compared with alectinib (Fig. 2A and B). Furthermore, whereas CAV chemotherapy or lorlatinib alone did not show any survival advantage, the association of both was synergistic and induced significantly increased survival in the Th-ALKF1174L/MYCN neuroblastoma model against continuous once-daily lorlatinib or one dose of CAV alone (Fig. 3A). In addition, our observation that GEMM tumors relapsing during treatment with lorlatinib alone (Fig. 2C) or the CAV-lorlatinib combination (Fig. 3A) exhibit a decrease in their adrenergic gene expression (Fig. 3D), is in keeping with recently published data describing ALK inhibitor resistance of mesenchymal-type neuroblastoma xenografts which express low levels of ALK (41). We can therefore speculate that either there was a subpopulation of tumor cells inherently resistant to therapy or that phenotypic plasticity might have played a role in the acquisition of resistance to ALK inhibition and chemotherapy in our GEMM.
Interest in PDXs has increased in recent years due to the development of personalized cancer medicines based on genomic profiling and they have improved the predictive power of preclinical therapeutic studies (49). From ex vivo screening of PDTC models, whereas ceritinib was more active and crizotinib showed similar activity to lorlatinib in ALK-aberrant models, lorlatinib was the only ALK inhibitor to show statistically significant enhanced cytotoxicity in ALK-aberrant models compared with wild-type, reinforcing the hypothesis of off-target effects of ceritinib or crizotinib (Fig. 1Ci–iv). In vivo studies showed high efficacy of lorlatinib alone in the GR-NB4 ALK-amplified model, where the addition of chemotherapy did not increase the tumor growth inhibition and chemotherapy alone was ineffective (Fig. 4Bi). This may be due to the low doses of chemotherapy chosen for these experiments, as CAV doses were low to help reveal a potential synergy between chemotherapy and lorlatinib. It is also possible that an immunocompetent mouse model might be required to demonstrate preclinical efficacy of chemotherapy via immunostimulation, therefore the study of the microenvironment in our GEMM is ongoing. Unexpectedly, the ALKF1174C mutated model (HSJD-NB-012) exhibited only a modest response to lorlatinib alone and in combination with chemotherapy (Fig. 4Di), the reasons for which are currently unclear. The lack of response by the PDX with a “silent” intron 3 ALK amplification (IC-pPDX-112), as opposed to amplification across the entire coding sequence, fits with the relatively low expression of ALK RNA and protein. The high mutational burden in this model, which is unusual for a diagnostic neuroblastoma, could give rise to especially aggressive tumor growth.
In view of the future amended HRNBL2 protocol combining upfront lorlatinib with chemotherapy in ALK-aberrant tumors from patients with high-risk neuroblastoma, we investigated the possibility of combined ALK and MDM2 inhibition for ALK-aberrant relapsed neuroblastoma, as ALK and MDM2 inhibitors have already been explored as potential strategies to overcome resistance to ALK inhibitors (25, 26), and TP53 mutations are rare in relapsed neuroblastoma. In several TP53 wild-type and ALK-aberrant (mutated, amplified or both) cell lines, the combination of idasanutlin and ALK inhibitors was found to be synergistic and induced enhanced levels of apoptosis (Fig. 5). In the ALK-amplified GR-NB4 PDX model, the combination of lorlatinib and idasanutlin showed impressive additive effect with complete remission (Fig. 6Ai), which may be partly explained by the additional presence of a homozygous CDKN2A/B deletion in this model. Although it was not possible to test idasanutlin, which is specific for human MDM2 (50) in the Th-ALKF1174L/MYCN GEMM, our previous studies have shown that ALKF1174L promotes antiapoptotic activity in this model of neuroblastoma (39). Therefore, future work to investigate whether a mouse reactive MDM2 inhibitor is synergistic with ALK inhibition via enhanced apoptosis is warranted.
Importantly, our comprehensive study of ALK inhibitor combinations included a pharmacodynamic assessment of ALK expression, ALK phosphorylation, and downstream activity of ALK across our models. We found a high baseline level of tumoral ALK expression was associated with sensitivity to ALK inhibition. This finding supports measurement of total ALK expression alongside ALK aberrations in future clinical studies with ALK inhibitors to determine whether there is a threshold of ALK expression that might predict a response to ALK inhibitor combinations in ALK-aberrant neuroblastoma.
Supplementary Material
Cell line and PDX genetics
{kind=link}
PDTC experimental data
{kind=link}
GEMM ALK inhibitor data
{kind=link}
GEMM sequencing data
{kind=link}
PDX data and clinical data
{kind=link}
Cell line growth curves and PDX data for idasanutlin combination
{kind=link}
Supplementary methods, supplementary tables, supplementary figure legends
Acknowledgments
In the United Kingdom, E.R. Tucker, E. Poon, H. Che, B. Martins Da Costa, K. Barker, S. Shrestha, and L. Chesler were supported by a Cancer Research UK (CRUK) Programme Grant (A28278). E. Calton was supported by a Children with Cancer Clinical PhD Fellowship (CWL022X). Q. Gao was supported by a Cancer Research UK (A24566). Y. Jamin was supported by a Children with Cancer UK Research Fellowship (2014/176). S.L. George was supported by Royal Marsden NIHR Biomedical Research Centre funding, a CRUK Postdoctoral Research Bursary, and a CRUK Clinical Scientist Fellowship. D.A. Tweddle, L. Chen, S. Dhariwal, and A. Goodman received funding from Neuroblastoma UK, Niamh's Next Step, Children's Cancer North, the Newcastle National Institute for Health Research Biomedical Research Centre, and the Children's Cancer & Leukaemia Group/Little Princess Trust. In France, this work was supported by the Annenberg Foundation, the Association Hubert Gouin Enfance et Cancer, the Fédération Enfants Cancers Santé, the Société Française de lutte contre les Cancers et les leucémies de l'Enfant et l'adolescent (SFCE), Les Bagouz à Manon, Les amis de Claire, and the Fondation ARC pour la Recherche contre le Cancer (ARC). Funding was also obtained from SiRIC/INCa (Grant INCa-DGOS-4654), and PHRC IC2007-09 grant. High-throughput sequencing was performed by the ICGex NGS platform of the Institut Curie supported by the grants ANR-10-EQPX-03 (Equipex) and ANR-10-INBS-09-08 (France Génomique Consortium) from the Agence Nationale de la Recherche (“Investissements d'Avenir” program), by the Canceropole Ile-de-France, and by the SiRIC-Curie program—SiRIC Grant “INCa-DGOS—4654.” Tumor sequencing data and clinical data have been provided under the MAPPYACTS protocol (clinicaltrial.gov: NCT02613962). The drug screening was supported in the framework of ERA PerMed (ERAPERMED2018-121, COMPASS). Molecular characterization of the PDX models was supported within the framework of the IMI2 ITCC P4 program funded by IMI2 grant agreement no. 116064 ITCC-P4. B. Geoerger is supported by the Parrainage médecin-chercheur of Gustave Roussy.
The authors would like to thank Pfizer for providing lorlatinib for the in vivo GEMM work, and Roche-Genentech for providing both idasanutlin for the in vitro work and alectinib for the in vivo GEMM work. We wish to thank the Biological Services Unit at The Institute of Cancer Research (London), and the Animal Platform of the Institut Curie-Orsay (Paris), as well as Adnan Naguez and Marine Michou for their technical assistance.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Disclosures
D.A. Tweddle reports non-financial support from Hoffman-La Roche during the conduct of the study; D.A. Tweddle also reports personal fees from Eusa Pharma outside the submitted work. G. Schleiermacher reports grants from BMS, Roche, Pfizer, and MSDavenir outside the submitted work. No disclosures were reported by the other authors.
Authors' Contributions
E.R. Tucker: Conceptualization, formal analysis, validation, investigation, methodology, writing–original draft, writing–review and editing. I. Jiménez: Conceptualization, investigation, methodology, writing–original draft. L. Chen: Conceptualization, investigation, methodology, writing–review and editing. A. Bellini: Investigation, writing–review and editing. C. Gorrini: Investigation, methodology, writing–review and editing. E. Calton: Investigation, methodology. Q. Gao: Formal analysis, methodology, writing–review and editing. H. Che: Formal analysis. E. Poon: Supervision. Y. Jamin: Formal analysis, investigation, methodology. B. Martins Da Costa: Investigation. K. Barker: Investigation. S. Shrestha: Investigation. J.C. Hutchinson: Formal analysis. S. Dhariwal: Investigation. A. Goodman: Investigation. E. Del Nery: Supervision, investigation. P. Gestraud: Investigation. J. Bhalshankar: Investigation. Y. Iddir: Investigation. E. Saberi-Ansari: Formal analysis. A. Saint-Charles: Investigation. B. Geoerger: Investigation, writing–review and editing. M.E. Marques Da Costa: Investigation. C. Pierre-Eugène: Investigation, methodology. I. Janoueix-Lerosey: Investigation, writing–review and editing. D. Decaudin: Investigation. F. Nemati: Investigation. A.M. Carcaboso: Investigation. D. Surdez: Investigation. O. Delattre: Supervision. S.L. George: Conceptualization, supervision, writing–review and editing. L. Chesler: Conceptualization, resources, supervision, funding acquisition, writing–review and editing. D.A. Tweddle: Conceptualization, resources, supervision, funding acquisition, methodology, writing–review and editing. G. Schleiermacher: Conceptualization, resources, formal analysis, supervision, funding acquisition, visualization, writing–review and editing.
References
- 1. Brodeur GM. Spontaneous regression of neuroblastoma. Cell Tissue Res 2018;372:277–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, et al. Neuroblastoma. Nat Rev Dis Primers 2016;2:16078. [DOI] [PubMed] [Google Scholar]
- 3. Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984;224:1121–4. [DOI] [PubMed] [Google Scholar]
- 4. Ackermann S, Cartolano M, Hero B, Welte A, Kahlert Y, Roderwieser A, et al. A mechanistic classification of clinical phenotypes in neuroblastoma. Science 2018;362:1165–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Peifer M, Hertwig F, Roels F, Dreidax D, Gartlgruber M, Menon R, et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 2015;526:700–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Valentijn LJ, Koster J, Zwijnenburg DA, Hasselt NE, van Sluis P, Volckmann R, et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat Genet 2015;47:1411–4. [DOI] [PubMed] [Google Scholar]
- 7. Cheung NK, Zhang J, Lu C, Parker M, Bahrami A, Tickoo SK, et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA 2012;307:1062–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hartlieb SA, Sieverling L, Nadler-Holly M, Ziehm M, Toprak UH, Herrmann C, et al. Alternative lengthening of telomeres in childhood neuroblastoma from genome to proteome. Nat Commun 2021;12:1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bellini A, Potschger U, Bernard V, Lapouble E, Baulande S, Ambros PF, et al. Frequency and prognostic impact of ALK amplifications and mutations in the European Neuroblastoma Study Group (SIOPEN) High-Risk Neuroblastoma Trial (HR-NBL1). J Clin Oncol 2021;39:3377–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008;455:967–70. [DOI] [PubMed] [Google Scholar]
- 11. Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008;455:930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. De Brouwer S, De Preter K, Kumps C, Zabrocki P, Porcu M, Westerhout EM, et al. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin Cancer Res 2010;16:4353–62. [DOI] [PubMed] [Google Scholar]
- 13. Eleveld TF, Oldridge DA, Bernard V, Koster J, Colmet Daage L, Diskin SJ, et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet 2015;47:864–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Padovan-Merhar OM, Raman P, Ostrovnaya I, Kalletla K, Rubnitz KR, Sanford EM, et al. Enrichment of targetable mutations in the relapsed neuroblastoma genome. PLoS Genet 2016;12:e1006501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schleiermacher G, Javanmardi N, Bernard V, Leroy Q, Cappo J, Rio Frio T, et al. Emergence of new ALK mutations at relapse of neuroblastoma. J Clin Oncol 2014;32:2727–34. [DOI] [PubMed] [Google Scholar]
- 16. Bresler SC, Wood AC, Haglund EA, Courtright J, Belcastro LT, Plegaria JS, et al. Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma. Sci Transl Med 2011;3:108ra14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Foster JH, Voss SD, Hall DC, Minard CG, Balis FM, Wilner K, et al. Activity of crizotinib in patients with ALK-aberrant relapsed/refractory neuroblastoma: a Children's Oncology Group study (ADVL0912). Clin Cancer Res 2021;27:3543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Krytska K, Ryles HT, Sano R, Raman P, Infarinato NR, Hansel TD, et al. Crizotinib synergizes with chemotherapy in preclinical models of neuroblastoma. Clin Cancer Res 2016;22:948–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Greengard E, Mosse YP, Liu X, Minard CG, Reid JM, Voss S, et al. Safety, tolerability and pharmacokinetics of crizotinib in combination with cytotoxic chemotherapy for pediatric patients with refractory solid tumors or anaplastic large cell lymphoma (ALCL): a Children's Oncology Group phase 1 consortium study (ADVL1212). Cancer Chemother Pharmacol 2020;86:829–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fischer M, Moreno L, Ziegler DS, Marshall LV, Zwaan CM, Irwin MS, et al. Ceritinib in paediatric patients with anaplastic lymphoma kinase-positive malignancies: an open-label, multicentre, phase 1, dose-escalation and dose-expansion study. Lancet Oncol 2021;22:1764–76. [DOI] [PubMed] [Google Scholar]
- 21. Infarinato NR, Park JH, Krytska K, Ryles HT, Sano R, Szigety KM, et al. The ALK/ROS1 inhibitor PF-06463922 overcomes primary resistance to crizotinib in ALK-driven neuroblastoma. Cancer Discov 2016;6:96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guan J, Tucker ER, Wan H, Chand D, Danielson LS, Ruuth K, et al. The ALK inhibitor PF-06463922 is effective as a single agent in neuroblastoma driven by expression of ALK and MYCN. Dis Model Mech 2016;9:941–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Debruyne DN, Dries R, Sengupta S, Seruggia D, Gao Y, Sharma B, et al. BORIS promotes chromatin regulatory interactions in treatment-resistant cancer cells. Nature 2019;572:676–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Berlak M, Tucker E, Dorel M, Winkler A, McGearey A, Rodriguez-Fos E, et al. Mutations in ALK signaling pathways conferring resistance to ALK inhibitor treatment lead to collateral vulnerabilities in neuroblastoma cells. Mol Cancer 2022;21:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miyazaki M, Otomo R, Matsushima-Hibiya Y, Suzuki H, Nakajima A, Abe N, et al. The p53 activator overcomes resistance to ALK inhibitors by regulating p53-target selectivity in ALK-driven neuroblastomas. Cell Death Discov 2018;4:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang HQ, Halilovic E, Li X, Liang J, Cao Y, Rakiec DP, et al. Combined ALK and MDM2 inhibition increases antitumor activity and overcomes resistance in human ALK mutant neuroblastoma cell lines and xenograft models. Elife 2017;6:e17137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Carr-Wilkinson J, O'Toole K, Wood KM, Challen CC, Baker AG, Board JR, et al. High frequency of p53/MDM2/p14ARF pathway abnormalities in relapsed neuroblastoma. Clin Cancer Res 2010;16:1108–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen Z, Lin Y, Barbieri E, Burlingame S, Hicks J, Ludwig A, et al. Mdm2 deficiency suppresses MYCN-driven neuroblastoma tumorigenesis in vivo. Neoplasia 2009;11:753–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Swarbrick A, Woods SL, Shaw A, Balakrishnan A, Phua Y, Nguyen A, et al. miR-380–5p represses p53 to control cellular survival and is associated with poor outcome in MYCN-amplified neuroblastoma. Nat Med 2010;16:1134–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Veschi V, Thiele CJ. High-SETD8 inactivates p53 in neuroblastoma. Oncoscience 2017;4:21–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen L, Humphreys A, Turnbull L, Bellini A, Schleiermacher G, Salwen H, et al. Identification of different ALK mutations in a pair of neuroblastoma cell lines established at diagnosis and relapse. Oncotarget 2016;7:87301–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen L, Rousseau RF, Middleton SA, Nichols GL, Newell DR, Lunec J, et al. Pre-clinical evaluation of the MDM2-p53 antagonist RG7388 alone and in combination with chemotherapy in neuroblastoma. Oncotarget 2015;6:10207–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stewart E, Federico SM, Chen X, Shelat AA, Bradley C, Gordon B, et al. Orthotopic patient-derived xenografts of paediatric solid tumours. Nature 2017;549:96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yadav B, Pemovska T, Szwajda A, Kulesskiy E, Kontro M, Karjalainen R, et al. Quantitative scoring of differential drug sensitivity for individually optimized anticancer therapies. Sci Rep 2014;4:5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. J Pharmacol Pharmacother 2010;1:94–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, et al. Guidelines for the welfare and use of animals in cancer research. Br J Cancer 2010;102:1555–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tucker ER, Tall JR, Danielson LS, Gowan S, Jamin Y, Robinson SP, et al. Immunoassays for the quantification of ALK and phosphorylated ALK support the evaluation of on-target ALK inhibitors in neuroblastoma. Mol Oncol 2017;11:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Garaventa A, Poetschger U, Valteau-Couanet D, Luksch R, Castel V, Elliott M, et al. Randomized trial of two induction therapy regimens for high-risk neuroblastoma: HR-NBL1.5 International Society of Pediatric Oncology European Neuroblastoma Group study. J Clin Oncol 2021;39:2552–63. [DOI] [PubMed] [Google Scholar]
- 39. Berry T, Luther W, Bhatnagar N, Jamin Y, Poon E, Sanda T, et al. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell 2012;22:117–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yamazaki S, Lam JL, Zou HY, Wang H, Smeal T, Vicini P. Translational pharmacokinetic-pharmacodynamic modeling for an orally available novel inhibitor of anaplastic lymphoma kinase and c-Ros oncogene 1. J Pharmacol Exp Ther 2014;351:67–76. [DOI] [PubMed] [Google Scholar]
- 41. Westerhout EM, Hamdi M, Stroeken P, Nowakowska NE, Lakeman A, van Arkel J, et al. Mesenchymal-type neuroblastoma cells escape ALK inhibitors. Cancer Res 2022;82:484–96. [DOI] [PubMed] [Google Scholar]
- 42. Boeva V, Louis-Brennetot C, Peltier A, Durand S, Pierre-Eugene C, Raynal V, et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat Genet 2017;49:1408–13. [DOI] [PubMed] [Google Scholar]
- 43. Lambertz I, Kumps C, Claeys S, Lindner S, Beckers A, Janssens E, et al. Upregulation of MAPK negative feedback regulators and RET in mutant ALK neuroblastoma: implications for targeted treatment. Clin Cancer Res 2015;21:3327–39. [DOI] [PubMed] [Google Scholar]
- 44. Mus LM, Lambertz I, Claeys S, Kumps C, Van Loocke W, Van Neste C, et al. The ETS transcription factor ETV5 is a target of activated ALK in neuroblastoma contributing to increased tumour aggressiveness. Sci Rep 2020;10:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. van Groningen T, Koster J, Valentijn LJ, Zwijnenburg DA, Akogul N, Hasselt NE, et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat Genet 2017;49:1261–6. [DOI] [PubMed] [Google Scholar]
- 46. Berlanga P, Pierron G, Lacroix L, Chicard M, Adam de Beaumais T, Marchais A, et al. The European MAPPYACTS trial: precision medicine program in pediatric and adolescent patients with recurrent malignancies. Cancer Discov 2022;12:1266–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. George RE, Sanda T, Hanna M, Frohling S, Luther W 2nd, Zhang J, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008;455:975–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zou HY, Friboulet L, Kodack DP, Engstrom LD, Li Q, West M, et al. PF-06463922, an ALK/ROS1 inhibitor, overcomes resistance to first and second generation ALK inhibitors in preclinical models. Cancer Cell 2015;28:70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tucker ER, George S, Angelini P, Bruna A, Chesler L. The promise of patient-derived preclinical models to accelerate the implementation of personalised medicine for children with neuroblastoma. J Pers Med 2021;11:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen L, Esfandiari A, Reaves W, Vu A, Hogarty MD, Lunec J, et al. Characterisation of the p53 pathway in cell lines established from TH-MYCN transgenic mouse tumours. Int J Oncol 2018;52:967–77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cell line and PDX genetics
PDTC experimental data
GEMM ALK inhibitor data
GEMM sequencing data
PDX data and clinical data
Cell line growth curves and PDX data for idasanutlin combination
Supplementary methods, supplementary tables, supplementary figure legends
Data Availability Statement
The data generated in this study are publicly available at BioProject, ID PRJNA848127.