

Abstract
The year 2022 will be remembered as the turning point for accurate long-read sequencing, which now establishes the gold standard for speed and accuracy at competitive costs. We discuss the key bioinformatics techniques needed to power long reads across application areas and close with our vision for long-read sequencing over the coming years.
While originally slow, error-prone and expensive, long-read sequencing has dramatically improved in the past decade. The foremost example of this transformation is the publication of the first complete human telomere-to-telomere (T2T) genome based entirely on long reads1. In addition to revealing ~200 Mbp of missing sequence and 99 missing genes in the standard reference genome (GRCh38), the T2T consortium showed that using the T2T genome as a reference improves the analysis of globally diverse samples, plus offers up to 12-fold better accuracy within challenging, clinically relevant genes2. In terms of speed, long-read nanopore sequencing set a new clinical record by identifying a likely pathogenic variant in an infant in critical condition less than 9 h after enrollment3. Long reads have substantially improved the genome, transcriptome and epigenome sequences in many other humans and non-human species4–7.
Long reads have an intrinsic appeal for genomics. Whereas short-read sequencing is analogous to completing a jigsaw puzzle made of a huge number of tiny pieces, long reads are analogous to completing that same puzzle with much larger and fewer pieces. While a 30× coverage of the human genome requires 450 million Illumina reads (averaging 200 bp), only 4.5 million Pacific Biosciences (PacBio) HiFi reads (averaging 20 kbp) or 900,000 Oxford Nanopore (ONT) ultra-long reads (averaging 100 kbp) are needed8. Moreover, long reads are intrinsically more suited to resolving repetitive sequences like long terminal repeat (LTR) retrotransposons or long interspersed nuclear elements (LINEs) that have stymied short reads. Incredibly, the human T2T genome and the Arabidopsis Col-CEN genome6 show that even centromeres can be correctly and completely resolved using long reads by bridging the sparse variation in them.
While long reads have had this tantalizing potential since their introduction, this revolution has had a rocky progression. Early PacBio data were limited in read length, accuracy and throughput, so it was only practical to sequence small microbial genomes on the platform’s commercial release in 2010. Eukaryotic genomes came years later and required hybrid analysis with short reads to reach usable quality. The earliest ONT data in 2015 were even more problematic, with a ~40% error rate and highly variable yields, sometimes with only a few usable reads per flow cell. Some researchers at this time questioned whether long reads would ever be useful for anything. Fortunately, these early metrics are a distant memory, and both PacBio9 and ONT10 sequencing now average over 99% accuracy (Q30) per read in their highest accuracy modes, with substantially improved throughput and costs.
We emphasize that this revolution has required equal parts improvements to biotechnology and bioinformatics. For example, simply reprocessing electrical data from earlier ONT PromethION runs with the newest base callers can yield dramatic read quality improvements so that average contig lengths can jump from below 1 Mb to nearly 10 Mb. And while early attempts to assemble human genomes from long reads required over 1 million CPU core hours using thousands of computers, excellent long-read human genomes can now be assembled on your laptop in a few minutes11. Here, we highlight the key bioinformatics techniques that made long-read sequencing the gold standard it is today, the remaining challenges, and the long term future for the genomics field as a whole.
Signal-level analysis, epigenomics and epitranscriptomics
ONT and PacBio are sequencing technologies that operate on individual DNA or RNA molecules (Fig. 1)8. Their read lengths range from kilobases to a few megabases (Fig. 2a,b), without the drop-off in quality that limits short-read platforms. Nanopore sequencing works by passing DNA or RNA strands through a small pore via an electric potential, which produces varying currents depending on which bases are in the pore. PacBio sequencing works by sequentially incorporating fluorescent nucleotides into a template DNA molecule, often using a circularized molecule to allow multiple passes for accurate consensus — that is, HiFi sequencing9. The highest quality long-read sequencing approaches (HiFi sequencing for PacBio9 and duplex sequencing for ONT10) effectively reread the same molecule two or more times, which helps to cancel out most sequencing noise. The electrical and fluorescence signals can be base-called using algorithms like hidden Markov models and neural networks to obtain nucleotide sequences. Modern nanopore base callers predominantly use neural networks for higher accuracy, especially around low-complexity sequences, and the first PacBio neural network base caller was recently introduced12. A potential downside of these machine learning approaches is overtraining, in which accuracy may be inflated on species used to train the model (for example, humans).
Fig. 1 |. Long-read sequencing methods and applications.
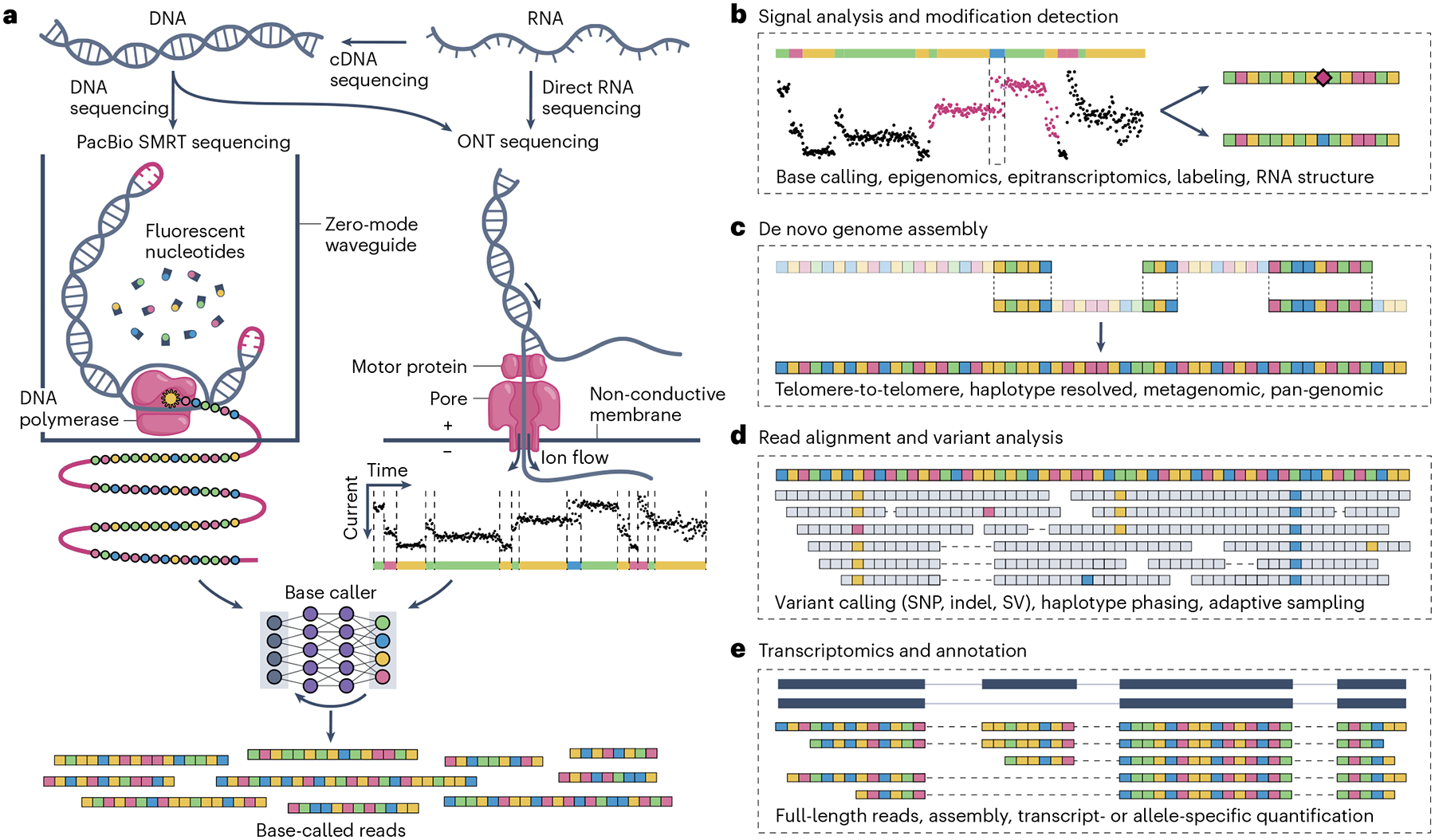
a, Long strands of unamplified DNA can be sequenced by PacBio and ONT sequencers. ONT can also directly sequence RNA molecules while PacBio requires synthesis of cDNA. PacBio Single Molecule, Real-Time (SMRT) sequencing observes fluorescent nucleotides within zero mode waveguides as they are incorporated into a circular molecule, generating a series of forward and reverse complement fluorescence signals. ONT sequencing generates a time series of electric current for different nucleotides. These signals are input into a technology-specific base caller using neural nets or related techniques, which output reads represented as a series of adenines (green), cytidines (blue), guanines (yellow) and thymines (red). b, Signal-level analyses include base calling and DNA or RNA modification detection (purple diamond). c, Long reads improve de novo genome assembly by spanning more repetitive DNA and haplotype-specific variants. d, Long reads can span complex variants with unique alignments, allowing more accurate structural variant detection and phasing. SNP, single-nucleotide polymorphism; SV, structural variant. e, Long-read RNA sequencing can often span full transcripts in single reads, improving annotation and transcript-level quantification.
Fig. 2 |. Improvement of sequencing technologies.
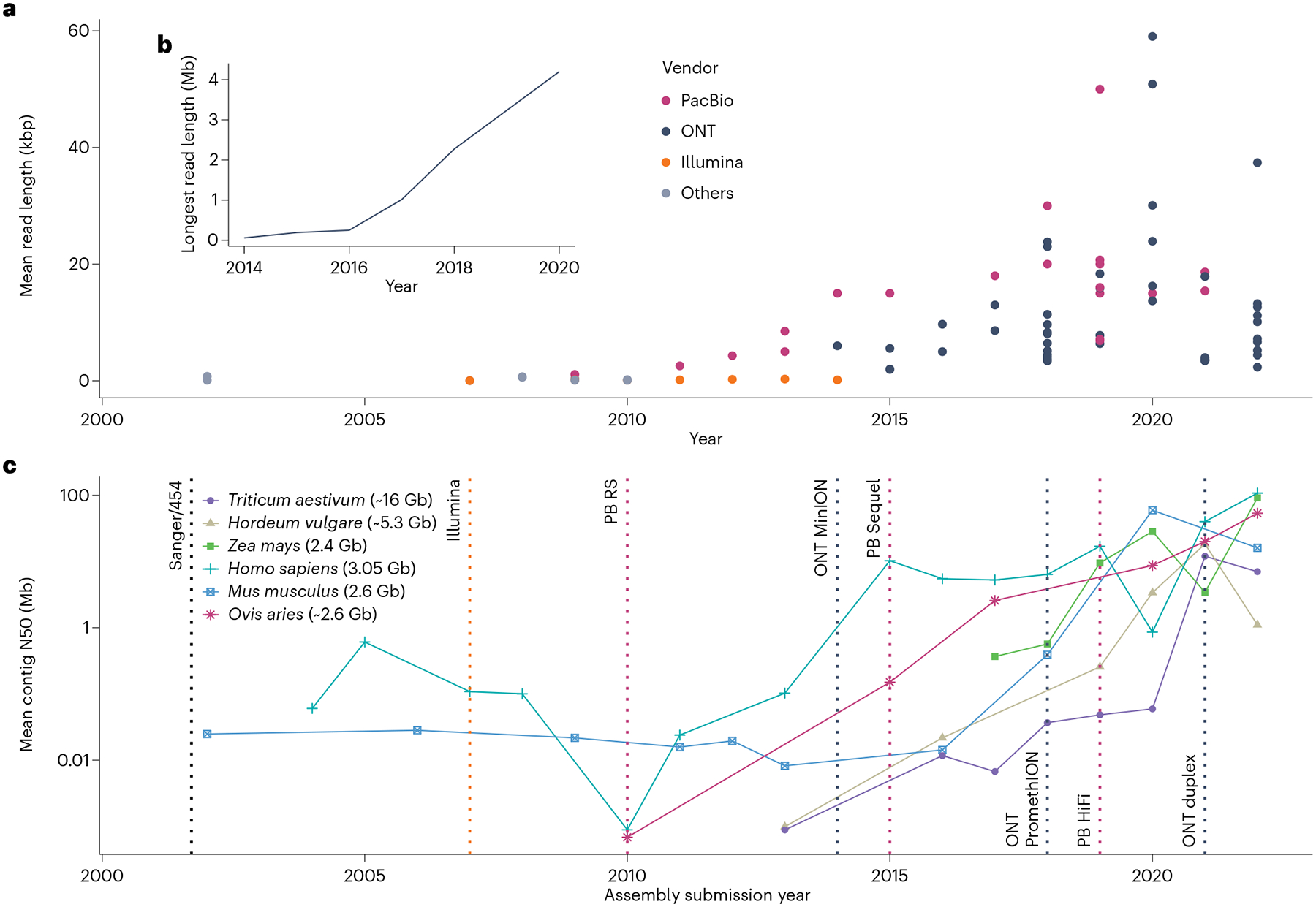
a, Mean read length reported by a selection of published genome sequencing studies. Each dot represents a report, colored by sequencing platform. b, Longest read length of ONT sequencing studies per year. c, Average contig N50 (meaning that 50% of the genome is assembled into contigs of at least the indicated size) of genomes submitted to the US National Center for Biotechnology Information for a selection of six model species. The six species were selected as having the most assembly submissions for genomes at least 2 Gb in size. Vertical dotted lines denote the release of new sequencing technologies. PB, PacBio; ONT, Oxford Nanopore Technologies. Data used in this figure may be accessed through https://github.com/schatzlab/long-read-commentary.
Besides sequencing the four canonical bases (adenine, cytosine, guanine, and thymine or uracil), raw ONT and PacBio data can also signal chemical modifications such as methylation. Unlike conventional epigenetic profiling such as short-read bisulfite sequencing, long-read sequencing preserves the underlying molecular configuration, allowing simultaneous genetic and epigenetic variant calling without specialized preparation. This was underscored in the human T2T project, where methylation patterns in centromeric repeats could be accurately profiled, a task that was challenging with short-read methods7. ONT can also directly sequence RNA molecules, with the potential to identify over 150 known epitranscriptomic modifications, most of which cannot be detected by other methods. However, only a few RNA modifications have been detected thus far, with varying levels of accuracy due to limited training data and weak statistical signals13. Beyond natural modifications, artificial modifications have been used to study DNA and RNA secondary structure and replication or transcription dynamics13.
Adaptive sampling is a unique capability of nanopore sequencing, whereby reads can be selectively ejected from the pore before sequencing finishes14,15. Reads of interest can be identified in real time, which enables software-based targeted enrichment or depletion — that is, in silico exome-capture-style sequencing. Applications include metagenomics, where certain species may be over-represented in a sample, or gene panel enrichment, where genes can be targeted without amplification bias and while preserving read lengths and epigenetics. In the longer term this could even enable lower coverage sequencing assays by undercutting Poisson coverage requirements, which currently dictate the need for oversampling a genome many fold.
Long-read sequencing has necessitated the development of new file formats to represent these unconventional types. PacBio has converged on storing their signal data within SAM/BAM files with specialized tags to encode molecular kinetics. Raw nanopore electrical data have historically been stored in FAST5 files, an HDF5-based format that suffers from large file sizes and slow read/write speeds. The recent SLOW5 format demonstrated that performance can be improved using a lightweight SAM-inspired implementation16, and similar results were later achieved with the POD5 Apache Arrow format that ONT is developing. Long-read modification detection has necessitated simultaneous encoding of genetic and epigenetic information, leading to official support for modification tags in the widely used SAM/BAM format for both PacBio and ONT, which allows convenient visualization and analysis of epigenetics.
Genome assembly and annotation
Accurate genome assemblies provide the foundation of most genomics research. In short-read assemblies, centromeres, ribosomal DNA and other repeats are largely absent, but long reads have the potential to span them and determine their exact sequences. Both model and non-model species have benefited from long-read sequencing, and assembly contiguity has steadily improved since 2010, when long reads were first introduced (Fig. 2c). In earlier long-read assemblies, the reads themselves were plagued with errors, which reduced consensus quality despite this higher contiguity17. Assembling telomere-to-telomere, error-free genomes has only recently become possible with highly accurate long-read sequencing.
While short reads are commonly assembled using de Bruijn graphs of short k-mers, long reads are more commonly assembled using string graphs whereby entire reads are compared to each other using minimizers and MinHash techniques, or through specialized de Bruijn graphs with very long k-mers11. Notably, the T2T human genome was constructed from a string graph using PacBio HiFi reads, augmented by ONT ultra-long reads and reaching a consensus accuracy over Q70 (<1 error per 10 million nucleotides) after polishing1. Fully phased, haplotype-resolved assemblies that leverage trio family pedigrees or Hi-C integration are now also possible, further improving accuracy and contiguity for diploid genomes. With enough long-read data, telomere-to-telomere assemblies of diploid genomes are on the horizon, although this currently requires high coverage with ultra-long reads (>50× ONT with >100-kbp reads and >50× HiFi with >20-kbp reads). The new Verkko assembler was designed specifically for these sequencing data to produce nearly telomere-to-telomere assemblies with minimal manual intervention18. This brings the exciting possibility for telomere-to-telomere genomes for all model and non-model organisms and, eventually, telomere-to-telomere pan-genomes to explore previously challenging variants at a population scale.
The rapid influx of new genomes is spurring parallel developments for genome annotation, which was once considered relatively easy compared to the moonshot task of whole-genome sequencing. However, with new genomes being assembled daily, properly annotating and aligning genomes is becoming the new bottleneck. Genomes assembled from long reads were once prone to large numbers of small-indel errors, which created frequent frame shifts and incomplete gene annotations17. Fortunately, long-read consensus accuracy has improved, and the resulting gene assemblies can now achieve similar or even higher qualities compared to those generated by previous technologies.
In addition to revealing more genes, long-read assemblies afford a more complete view of transposable elements, satellites, and other repetitive sequences. Transposable element annotation algorithms, such as the Pan-genome Extensive De novo TE Annotator (panEDTA)19, are shifting focus from repeat masking of single genomes to detailed annotation of transposable element structure and homology in pan-genomes. Newly resolved satellites, centromeres and other complex repeats have spurred development for specialized analysis algorithms and higher order visualizations. For example, the StainedGlass20 package can visualize massive tandem repeats with ‘identity heatmaps’, revealing features never before seen. Moving forward, many widely used annotation methods would benefit from deeper integration of long-read sequencing advances, specifically long-read transcriptome and methylome data.
Genome variation analysis
One of the primary uses of genomics is to discover and analyze genetic variation, with some studies, such as the US National Institutes of Health’s All Of Us project, considering as many as 1 million genomes sequenced with short reads in a single project. This work brings profound insights into human genomic variation, and similar work in non-humans has broadly transformed our understanding of variation in agriculture, evolution and beyond. However, we have come to appreciate that these short-read studies have systematically missed certain classes of genomic variation, especially complex structural variants (SVs), as a result of the fundamental limitations of short-read sequencing.
SVs are variants at least 50 bp in size, including insertions, deletions, inversions, duplications and transversions21. Our ability to detect SVs is intrinsically linked to the sequencing technology: variants longer than the read length are harder to fully span and resolve. Furthermore, SVs are often flanked by repetitive sequences so that certain parts of the genome were inaccessible using short reads. However, it is now possible to fully capture all types of genomic variation with long reads, leading to tens of thousands more variants spanning tens of megabases, effectively doubling the identifiable variation per person and across populations22,23. This includes variants associated with neurodegenerative diseases in humans24 and a tandem duplication leading to larger fruit sizes in plants25, among many others.
Achieving these results has required the development of several specialized algorithms. The earliest algorithms required very sensitive methods for aligning noisy long reads. However, current data typically has at most a few percent error, shifting the challenges to alignment speed and accuracy. One of the key advances has been adopting minimizers and other sketching techniques to enable rapid and space-efficient mapping algorithms with little to no loss in sensitivity21,26. In addition, optimized dynamic programming methods were introduced to compute the base-by-base alignment of long reads to a genome, accommodating their distinct error models along with any true biological variation21,26,27. Many of the informatics challenges are now focused on population-scale analysis, starting with methods to robustly compare variation from one sample to many others and, crucially, how to interpret the functional consequences of the newly resolved variation28. Understanding common structural variation has advanced by adopting techniques first developed for single-nucleotide variants, especially association or expression quantitative trait loci (eQTL) studies28, although understanding rare SVs remains more difficult.
Transcriptomics
While short-read high-throughput RNA sequencing (RNA-seq) enabled extensive measurements of gene expression across many biological samples, it has struggled to accurately capture the full complexity of the eukaryotic transcriptome, which features widespread alternative splicing. Like genomic repeats, exons shared between gene isoforms cause ambiguity in reconstructing or aligning transcripts from short reads. This makes detecting all isoforms challenging, which stunts applications such as gene annotation or sequencing in individuals with diseases such as cancer, where genomic rearrangements and break-down of splicing machinery often generate new transcripts.
Long reads more completely represent the transcriptome, as many transcripts can be fully spanned by individual long reads. The major remaining obstacles to long-read RNA-seq are error rate, read fragmentation and throughput. Indel errors challenge the accurate placement of splice sites, and deletion errors can be mistaken for introns in low-coverage transcripts. Consensus calling methods reduce many of these errors29, except read fragmentation, which occurs during library preparation or premature ending of sequencing. These can be overcome using transcriptome assembly methods, as in short-read sequencing30. Finally, lower read counts limit expression quantification and make assembly of low-abundance transcripts inaccurate and incomplete.
Improvements to throughput have enabled gene-level quantification with similar accuracy to that of short reads and transcript-level quantification surpassing that of short reads31. The ability to capture full RNA molecules in single reads has been used to study long-range transcript dynamics, such as intron splicing order in nascent RNA transcripts32. Long reads have been extended to single-cell RNA sequencing33, revealing full-length cell-specific transcripts; these historically could be sequenced only on the 3′ end. The benefits of long-read RNA-seq were recently demonstrated in a large-scale project wherein samples from the Genotype–Tissue Expression (GTEx) project were sequenced with ONT, enabling the study of allele-specific expression in several phased genomes5.
While ONT and PacBio cDNA sequencing are similar in read lengths and error characteristics, ONT can directly sequence RNA molecules. This avoids limitations of cDNA synthesis such as transcript fragmentation and erasure of epitranscriptomic modifications. PacBio announced a prototype of direct RNA sequencing in 2013; however, it was never commercialized. ONT direct RNA sequencing remains more error-prone than cDNA sequencing, despite operating at a slower sequencing speed for a higher sampling rate. This slower speed also reduces yield, in our view making direct RNA sequencing currently a somewhat niche method for studying modifications or long transcripts that cannot be fully captured by cDNA.
The future of long-read genomics
We have entered the era of high-throughput, highly accurate long-read sequencing. This presents many exciting opportunities for discovery, starting with an explosion of references and telomere-to-telomere genomes. For example, the Vertebrate Genomes Project and the Earth BioGenome Project aim to establish reference genomes for all vertebrates and all eukaryotic species, respectively, as resources for studying diversity and evolution. Long-read translational and clinical sequencing are also accelerating, with several studies in progress to observe the genetic, transcriptomic and epigenetic components of disease, especially neurological diseases and cancer, along with a variety of other traits and phenotypes. We anticipate major discoveries to be made; previous short-read sequencing systematically missed certain classes of variation across the genome, but long-read telomere-to-telomere sequencing leaves no place for variation to hide.
Will long-read sequencing entirely displace short-read sequencing? In our opinion, the long-term prospects are high, especially if a few key challenges are addressed.
At the molecular level, sample preparation for long-read sequencing is more demanding than for short reads, especially to extract high-molecular-weight DNA to enable the longest read lengths. However, the protocols have substantially improved, and for many applications the requirements are manageable: for example, genome resequencing requires a few micrograms of modest read length (≤50 kbp) to capture the majority of variation. For short-molecule and count-based assays (for example, cell-free DNA, copy number variation analysis, RNA-seq or metagenomics), short-molecule nanopore sequencing is emerging as a competitive alternative to short-read sequencing.
Computationally, further advances in base calling are needed to robustly identify variations, especially in a clinical context. The high-accuracy modes of long-read platforms are promising, but the error characteristics are not as well understood and the platforms are currently throughput limited: HiFi by the relatively limited number of DNA molecules that can be read at once per SMRT cell, ONT by the relatively low yield of high-accuracy duplex versus lower-quality single-pass reads. Throughput can be offset by pricing, although short-read sequencing is cheaper, especially considering that the introduction of the Illumina NovaSeq X in fall 2022 has reduced the reagent cost for a 30× human genome to ~$200 while the best pricing for ONT is ~$600 (PromethION) and that of PacBio is ~$995 (Revio).
Finally, and perhaps most importantly, researchers need to continually demonstrate the biological and medical advantages of long reads over short reads. The human T2T project and ultra-fast clinical diagnostics are universally praised, but they are just the beginning of potential applications. We still need faster, more accurate genome assemblies and annotations, along with more accurate and complete detection of genomic variation, isoform diversity and epigenomic modifications. Moreover, we need new assays that exploit the unique powers of long reads, such as the simultaneous detection of genomic variation, methylation and chromatin status in a single read.
Ultimately, genomics is advancing beyond the analysis of individual genomes or individual samples to population-wide analyses whereby many genomes and many datasets are compared to each other. This era of pan-genomics will be anchored in graph genomes, where similarities and differences between genomes can be concisely represented and mined to explore distant homology. Methods that can jointly analyze long-read and short-read datasets are very important to exploit the huge number of short-read datasets available. In parallel, we anticipate a rise in machine learning and deep learning, building on the breakthrough algorithms recently published for base calling, variation identification, splicing prediction, chromatin analysis, repeat annotation and related techniques. These approaches require large, harmonized training data and extensive computational resources, such as cloud computing systems like Galaxy and AnVIL. Our collective goal is to make reliable predictions about a sample’s phenotype directly from sequence data. We are confident that major strides will be made in the coming years powered by long reads.
Acknowledgements
We would like to thank all past and current members of the Schatz lab, as well as our long-read collaborators, especially Timour Baslan, Andrew Carroll, Jason Chin, Megan Dennis, Evan Eichler, Tom Gingeras, Mark Gerstein, Sara Goodwin, Ian Henderson, Candice Hirsch, Matthew Hufford, Alison Klein, Ben Langmead, Zach Lippman, Erich Jarvis, W. Richard McCombie, Rajiv McCoy, Karen Miga, Rachel O’Neill, Mihaela Pertea, Adam Phillippy, Fritz Sedlazeck, Steven Salzberg, Winston Timp, Eli Van Allen, Justin Zook, and many others. Finally, we would also like to thank the researchers at PacBio and Oxford Nanopore for their developments and collaborations. This work was supported in part by the US National Science Foundation (IOS-2216612, IOS-1758800), the US National Institutes of Health (U24HG010263, U41HG006620, U01CA253481), and the Human Frontier Science Program (RGP0025/2021).
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Nurk S et al. Science 376, 44–53 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aganezov S et al. Science 376, eabl3533 (2022).35357935 [Google Scholar]
- 3.Gorzynski JE et al. N. Engl. J. Med 386, 700–702 (2022). [DOI] [PubMed] [Google Scholar]
- 4.Hufford MB et al. Science 373, 655–662 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glinos DA et al. Nature 608, 353–359 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Naish M et al. Science 374, eabi7489 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gershman A et al. Science 376, eabj5089 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goodwin S, McPherson JD & McCombie WR Nat. Rev. Genet 17, 333–351 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wenger AM et al. Nat. Biotechnol 37, 1155–1162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Silvestre-Ryan J & Holmes I Genome Biol. 22, 38 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ekim B, Berger B & Chikhi R Cell Syst. 12, 958–968.e6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baid G et al. Nat. Biotechnol 10.1038/s41587-022-01435-7 (2022). [DOI] [Google Scholar]
- 13.Furlan M et al. RNA Biol. 18 (Suppl. 1), 31–40 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kovaka S, Fan Y, Ni B, Timp W & Schatz MC Nat. Biotechnol 39, 431–441 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Payne A et al. Nat. Biotechnol 39, 442–450 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gamaarachchi H et al. Nat. Biotechnol 40, 1026–1029 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watson M & Warr A Nat. Biotechnol 37, 124–126 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Rautiainen M et al. Preprint at bioRxiv 10.1101/2022.06.24.497523 (2022). [DOI] [Google Scholar]
- 19.Ou S et al. Preprint at bioRxiv 10.1101/2022.10.09.511471 (2022). [DOI] [Google Scholar]
- 20.Vollger MR, Kerpedjiev P, Phillippy AM & Eichler EE Bioinformatics 10.1093/bioinformatics/btac018 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sedlazeck FJ et al. Nat. Methods 15, 461–468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Audano PA et al. Cell 176, 663–675.e19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alonge M et al. Cell 182, 145–161.e23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sone J et al. Nat. Genet 51, 1215–1221 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Della Coletta R, Qiu Y, Ou S, Hufford MB & Hirsch CN Genome Biol. 22, 3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H Bioinformatics 34, 3094–3100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marco-Sola S, Moure JC, Moreto M & Espinosa A Bioinformatics 37, 456–463 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirsche M et al. Preprint at bioRxiv 10.1101/2021.05.27.445886 (2021). [DOI] [Google Scholar]
- 29.Wyman D & Mortazavi A Bioinformatics 35, 340–342 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kovaka S et al. Genome Biol. 20, 278 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Y et al. Preprint at bioRxiv 10.1101/2021.04.21.440736 (2021). [DOI] [Google Scholar]
- 32.Drexler HL et al. Nat. Protoc 16, 1343–1375 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lebrigand K, Magnone V, Barbry P & Waldmann R Nat. Commun 11, 4025 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]