

Abstract
Purpose:
Recurrent 16p11.2 duplications produce a wide range of clinical outcomes with varying impacts on cognition and social functioning. Family-based studies of copy number variants (CNV) have revealed significant contributions of genomic background on variable expressivity. In this study, we measured the phenotypic impact of 16p11.2 duplications and quantified the modulating effect of familial background on cognitive and social outcomes.
Methods:
Genomic and clinical data were ascertained from 41 probands with a 16p11.2 duplication and their first-degree relatives. Paired comparisons were completed to determine the duplication’s impact on expected versus actual performance on standardized tests of intelligence (IQ) and social functioning (SRS-2). Intraclass correlations (ICCs) between relatives and probands were also calculated.
Results:
Cognitive and social functioning were significantly lower among individuals with 16p11.2 duplications compared to their CNV-negative relatives, while ICCs between the groups remained high for full-scale IQ and SRS-2 scores.
Conclusion:
16p11.2 duplications confer deleterious effects on cognition and social functioning, while familial background significantly influences phenotypic expression of these traits. Understanding variable expressivity in CNV disorders has implications for anticipatory clinical care, particularly for individuals who receive a genetic diagnosis at an early age, long before the full scope of manifestations becomes evident.
Introduction
Genomic copy number variants (CNVs), including recurrent deletions and duplications of 16p11.2 (OMIM #614671), are implicated in the etiology of autism spectrum disorder (ASD), schizophrenia, and other neurodevelopmental psychiatric disorders (NPD). 1,2 Rare CNVs confer deleterious impacts on brain function which manifest as clinical NPD that can vary widely in both presentation and severity. A major contributor to this variable expressivity is inherited genomic background which can be assessed through measurement of parental phenotypes (e.g., neurocognitive functioning), and more recently, polygenic scores.3,4 While recognizing that the term “familial background” encompasses both environmental and genomic influences on phenotypic outcomes, here we primarily define it as a proxy for genomic background, given the high heritability of the traits examined in this study.
We previously examined the impact of familial background on expression of inherited quantitative traits in individuals with 16p11.2 deletion syndrome, a rare genetic disorder characterized by obesity, relative macrocephaly, and variable NPD.3 In our family-based study of 56 probands with a de novo recurrent 16p11.2 deletion, the continuous distributions of these quantitative traits were preserved, but their means were “shifted” in a deleterious direction compared to relatives without the CNV. Notably, intraclass correlations (ICCs) between probands and family controls were maintained, suggesting that an individual’s baseline functioning related to family background influences his/her phenotypic expression in the presence of a 16p11.2 deletion.
Here, we expand on our original study of 16p11.2 deletions to examine recurrent 16p11.2 duplications which result in a different rare disorder due to overexpression of genes in the same chromosomal region. Like its reciprocal deletion, the 16p11.2 duplication presents with variable NPD but is distinguished by contrasting “mirror” phenotypes related to body mass index (BMI) and head circumference, with both being increased in the deletion and decreased in the duplication.5 The current family-based study of quantitative traits in individuals with 16p11.2 duplications was carried out to determine if “shift” is a generalizable phenomenon across different NPD-related CNVs, that contributes to phenotypic variability.6
Materials and Methods
Data were ascertained from the Simons VIP study7 which has extensively phenotyped families with rare genetic conditions, including recurrent 16p11.2 duplications (GRCh37/hg19 chr16:29649997-30199852). Simons VIP data includes in-person phenotypic evaluations and online questionnaires. 16p11.2 duplication probands included in the current study completed at least one phenotypic measure of interest and had a CNV-negative family member available for comparison. Individuals with additional known pathogenic variants (“second hits”) other than 16p11.2 duplication were excluded from analysis.
Full scale IQ (FSIQ) scores (M=100, SD=15) from standardized measures, as well as verbal (VIQ) and nonverbal performance (PIQ) scores were used to assess cognitive functioning. Social functioning was evaluated using the Social Responsiveness Scale (SRS)8, a 65-item questionnaire that assesses multiple domains of reciprocal social interaction. SRS raw scores were used to provide greater variability in the sample, with higher scores indicating increased impairment. For BMI analysis, probands were matched to a closest-in-age CNV-negative sibling, and BMI was normalized to z-scores for comparisons.
As 16p11.2 duplications are frequently inherited, we included both de novo and inherited cases in our study; inheritance status was unknown in three cases. Since the effect of parental background functioning could potentially be obscured in inherited cases, we employed a novel best-estimate strategy to identify appropriate relatives from the Simons project to compare to each proband. When available, the biparental mean of CNV-negative biological parent pairs was used as the best-estimate comparison. When a biparental mean was not available, a full biological CNV-negative sibling closest in age to the proband was used. In the absence of a biparental mean or CNV-negative sibling, the assessment scores of a single confirmed CNV-negative parent were used. One limitation of this best-estimate approach is that we could not fully exclude the impact of unrelated genetic influences, such as assortative mating, on our findings.
We used Statistical Product and Service Solutions8 software to perform paired t-test comparisons to quantify differences in performance on quantitative measures between 16p11.2 duplication probands and their best-estimate relatives. Further, we calculated ICCs between the groups on all domains assessed.
Results
IQ and SRS scores were available on 41 and 39 probands, respectively (Table 1). Among those in our IQ analysis, the 16p11.2 duplication was de novo in ten, inherited in 28, and of unknown inheritance in three individuals. CNV-negative controls for IQ included nine parent pairs (biparental means), 13 siblings, and 19 single parents. For SRS analysis, the duplication was de novo in 11, inherited in 25, and of unknown inheritance in three probands. CNV-negative SRS controls included eight parent pairs (biparental means), 15 siblings, and 16 single parents.
Table 1.
Descriptive statistics of probands and CNV-negative relatives across domains
| Domain | Proband Mean (SD) |
CNV-Negative Relative Mean (SD) |
ICC |
|---|---|---|---|
| Cognition | |||
| FSIQ | 75.05 (21.26) | 105.65 (15.75) | 0.411 |
| Verbal IQ | 77.10 (25.72) | 102.99 (15.01) | 0.447 |
| Performance IQ | 75.46 (20.34) | 107.27 (16.51) | 0.241 |
| Social responsiveness | 80.08 (26.99) | 40.17 (27.55) | 0.218 |
| BMI z-score | 0.72 (0.99) | 0.36 (0.89) | 0.222 |
BMI, body mass index; CNV, copy number variation; FSIQ, full-scale IQ; ICC, intraclass correlation.
Comparative IQ and SRS results are summarized in Figure 1. Probands with 16p11.2 duplications had lower FSIQs (M=75.05, SD=21.26) than their CNV-negative familial controls (M=105.65, SD=15.75), (t(40)=8.600, p<.001). A significant shift was also noted in both VIQ and PIQ domains, with mean score differences ranging from −26.71 (deleterious shift of 1.78 SD) to −32.63 (deleterious shift of 2.2 SD), respectively. This ~2SD shift is similar to that reported in 16p11.2 deletions3 and 22q11.2 deletions.9 ICCs between probands and family controls were significantly positive for VIQ (ICC=.447, p=.032) and FSIQ (ICC=.411, p=.049); correlations were not significant for PIQ (ICC=.241, p=.194).
Figure 1:
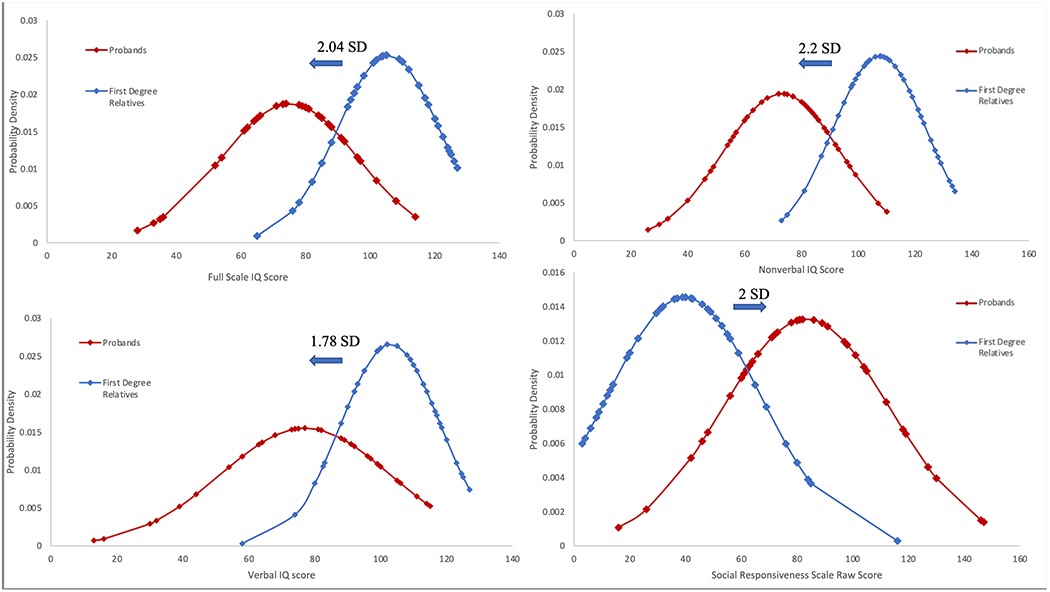
Normative Distributions of Quantitative Traits in 16p11.2 Duplications and CNV-negative Relatives
Raw SRS scores were elevated in 16p11.2 duplication probands (M=80.08, SD=30.04) compared to their familial controls (M=40.17, SD=27.55), (t(40)=7.814, p<.001), consistent with observed higher rates of ASD in this CNV population. We recognized one extreme outlier among the controls, with an SRS raw score of 120 which fell well outside 1.5x the interquartile range of the dataset. While overall raw SRS scores were not significantly correlated between probands and relatives (ICC=.218, p=0.17), the correlation approached significance (ICC=.403, p=.056) when the outlier and matched proband were excluded from the analysis. No significant difference was found in BMI Z-scores between probands and their biological CNV-negative siblings (p=.91).
Discussion
Recurrent CNVs confer large and variable effects on cognition and behavior, leading to a wide range of NPD outcomes. Understanding variable expressivity in CNV disorders has important implications for anticipatory clinical care, particularly for children who receive a genetic diagnosis at an early age, long before the full scope of manifestations becomes evident5. The modulating effect of parental performance on cognitive and neuropsychiatric measures has been reported in several rare genetic conditions, including fragile X, Prader-Willi, Klinefelter, and 22q11.2 deletion syndromes.9–11 We previously expanded this line of inquiry to other quantitative traits, including social functioning and BMI, in individuals with 16p11.2 deletions, and our current study of 16p11.2 duplications further confirms shift as a generalizable phenomenon across genetic disorders (Figure 1). The magnitude of the observed shift was similar in both our current and previous 16p11.2 studies, with an FSIQ shift of 1.78 SD observed in deletion probands3 and 2.04 SD in duplication probands. Similarly, SRS scores were increased by 2.2 SD in deletion probands3, and by 2.0 SD in duplication probands. One limitation to both our previous work on the 16p11.2 deletion and the current study is the use of the Simons VIP cohort. The Simons VIP cohort is a group of clinically ascertained probands which represent the more significant phenotypic range of the deletion and duplication. Additionally, many of the individuals in Simons VIP were ascertained due to diagnosis of ASD and may represent a cohort skewed towards those with more autism-related symptoms.
Conceptually, the deleterious shift from expected to observed NPD-related outcomes provides useful clinical insights into the phenotypic variability seen in many CNVs. Depending on the familial “starting point” for a particular trait, a shift in functioning due to a CNV may or may not cause an individual to reach the diagnostic threshold for a clinical NPD. A child with a 16p11.2 duplication, for example, may have an average IQ score compared to the general population; however, compared to her/his parents with above-average scores, this may represent a significant deviation from the expected outcome (i.e., an IQ score similar to that of the parents). Viewed out of family context, her/his average cognition gives the illusion of non-penetrance, until familial studies reveal a significant deleterious impact on the expected IQ.
While functional assessment of family members is a useful research tool, it is an impractical proxy measurement of genetic background. Recently, polygenic scores have more directly investigated the familial background contribution to variable expressivity in rare genetic disorders.4,10,11 Future refinement of polygenic scores for NPD and related traits may prove useful for narrowing prognoses in children with CNVs that have widely variable developmental and psychiatric outcomes. By better quantifying the type and magnitude of NPD risks in these children, clinicians could take a more proactive approach to targeted interventions, rather than waiting for symptoms to emerge.
Acknowledgments
This study was supported by grants U01MH119705 from the National Institute of Mental Health and the National Institute of Child Health and Human Development to Drs. Christa Martin & David Ledbetter and R01MH107431 from the National Institute of Mental Health to Dr. Christa Martin.
Footnotes
Ethics Declaration
The Geisinger Institutional Review Board provided oversight for this work. Individuals in this study were obtained from a deidentified database where consent was previously collected. Obtaining additional consent for this analysis was not required.
Conflicts of Interest
D.H.L. has received compensation as a consultant to Natera, Inc. and owns stock in the company, and is a consultant to MyOme, Inc. and has received stock in the company. All other authors declare no conflicts of interest.
Data Availability
Data used in this study was ascertained from the Simons Foundation funded SFARI Base research database that is freely available to any qualified researcher.
References
- 1.Chung WK, Roberts TP, Sherr EH, Snyder LG, Spiro JE. 16p11.2 deletion syndrome. Curr Opin Genet Dev. 2021;68:49–56. doi: 10.1016/j.gde.2021.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.D’Angelo D, Lebon S, Chen Q, et al. Defining the Effect of the 16p11.2 Duplication on Cognition, Behavior, and Medical Comorbidities. JAMA Psychiatry. 2016;73(1):20–30. doi: 10.1001/jamapsychiatry.2015.2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moreno-De-Luca A, Evans DW, Boomer KB, et al. The role of parental cognitive, behavioral, and motor profiles in clinical variability in individuals with chromosome 16p11.2 deletions. JAMA Psychiatry. 2015;72(2):119–126. doi: 10.1001/jamapsychiatry.2014.2147 [DOI] [PubMed] [Google Scholar]
- 4.Oetjens MT, Kelly MA, Sturm AC, Martin CL, Ledbetter DH. Quantifying the polygenic contribution to variable expressivity in eleven rare genetic disorders. Nat Commun. 2019;10(1):4897. doi: 10.1038/s41467-019-12869-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacquemont S, Reymond A, Zufferey F, et al. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature. 2011;478(7367):97–102. doi: 10.1038/nature10406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finucane B, Challman TD, Martin CL, Ledbetter DH. Shift happens: family background influences clinical variability in genetic neurodevelopmental disorders. Genet Med Off J Am Coll Med Genet. 2016;18(4):302–304. doi: 10.1038/gim.2015.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simons VIP Consortium. Simons Variation in Individuals Project (Simons VIP): a genetics-first approach to studying autism spectrum and related neurodevelopmental disorders. Neuron. 2012;73(6):1063–1067. doi: 10.1016/j.neuron.2012.02.014 [DOI] [PubMed] [Google Scholar]
- 8.IBM Corp. Released 2013. IBM SPSS Statistics for Mac, Version 22.0. Armonk, NY: IBM Corp. [Google Scholar]
- 9.Olszewski AK, Radoeva PD, Fremont W, Kates WR, Antshel KM. Is child intelligence associated with parent and sibling intelligence in individuals with developmental disorders? An investigation in youth with 22q11.2 deletion (velo-cardio-facial) syndrome. Res Dev Disabil. 2014;35(12):3582–3590. doi: 10.1016/j.ridd.2014.08.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davies RW, Fiksinski AM, Breetvelt EJ, et al. Using common genetic variation to examine phenotypic expression and risk prediction in 22q11.2 deletion syndrome. Nat Med. 2020;26(12):1912–1918. doi: 10.1038/s41591-020-1103-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cleynen I, Engchuan W, Hestand MS, et al. Genetic contributors to risk of schizophrenia in the presence of a 22q11.2 deletion. Mol Psychiatry. 2021;26(8):4496–4510. doi: 10.1038/s41380-020-0654-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data used in this study was ascertained from the Simons Foundation funded SFARI Base research database that is freely available to any qualified researcher.