

SUMMARY
How environmental nutrient availability impacts T cell metabolism and function remains poorly understood. Here, we report that the presence of physiologic carbon sources (PCSs) in cell culture medium broadly impacts glucose utilization by CD8+ T cells, independent of transcriptional changes in metabolic reprogramming. The presence of PCSs reduced glucose contribution to the TCA cycle and increased effector function of CD8+ T cells, with lactate directly fueling the TCA cycle. In fact, CD8+ T cells responding to Listeria infection preferentially consumed lactate over glucose as a TCA cycle substrate in vitro, with lactate enhancing T cell bioenergetic and biosynthetic capacity. Inhibiting lactate-dependent metabolism in CD8+ T cells by silencing lactate dehydrogenase A (Ldha) impaired both T cell metabolic homeostasis and proliferative expansion in vivo. Together, our data indicate that carbon source availability shapes T cell glucose metabolism and identifies lactate as a bioenergetic and biosynthetic fuel for CD8+ effector T cells.
In brief
Kaymak and Luda et al. report that environmental metabolite availability directly impacts glucose utilization and effector function of CD8+ T effector (Teff) cells, independent of transcriptional changes in metabolic programming. Lactate is a physiologic fuel for CD8+ Teff cells, serving as a preferred TCA cycle and biosynthetic substrate.
Graphical Abstract
INTRODUCTION
CD8+ T cells are critical for type I adaptive immune responses against pathogens (i.e., bacteria and viruses) and tumors (Tscharke et al., 2015; Williams and Bevan, 2007). In response to antigenic stimulation and appropriate costimulation, CD8+ T cells undergo sequential phases of activation starting with clonal expansion and differentiation into T effector (Teff) cells that control the threat, ultimately transitioning to memory (Tmem) cells that provide long-term protective immunity (Kaech and Cui, 2012). Differentiation into CD8+ Teff cells is marked by acquisition of effector functions, such as cytolytic activity and cytokine production (Badovinac et al., 2004; Obar et al., 2011; Teixeiro et al., 2009). One of the fundamental adaptable biological programs supporting T cells—from cellular homeostasis to effector function—is cellular metabolism (Buck et al., 2017; O’Neill et al., 2016; Olenchock et al., 2017). T cells display flexibility in metabolic pathway utilization and bioenergetic capacity to function in different environments in vivo (Chapman and Chi, 2022), but the metabolites that fuel the TCA cycle and biosynthetic metabolism in CD8+ T cells, particularly in vivo, have yet to be fully defined.
Our understanding of cellular metabolism has been aided by advances in metabolomics and 13C-based stable isotope labeling (SIL) techniques that facilitate tracking of metabolite usage in cells and tissues (Jang et al., 2018). Glucose has traditionally been held as the primary fuel for T cell metabolic needs and effector function, as glucose is rapidly taken up by activated T cells and is a major substrate fueling central carbon metabolism (i.e., glycolysis; the pentose phosphate pathway [PPP]; serine, glycine, and one-carbon [SGOC] metabolism; the TCA cycle; and nucleotide metabolism) in T cells (Frauwirth et al., 2002; Wang et al., 2011). In addition to supporting Teff cell expansion and survival (Macintyre et al., 2014), glucose has been linked to regulation of effector cytokine production in CD8+ Teff cells (Blagih et al., 2015; Cham and Gajewski, 2005; Chang et al., 2013). However, infusion of 13C-labeled metabolites into mice and humans has revealed differences in nutrient utilization by cells in vivo when compared with in vitro culture (Davidson et al., 2016; Hensley et al., 2016), including the use of lactate as a TCA cycle fuel by some tumors and non-transformed tissues in vivo (Faubert et al., 2017; Hui et al., 2017; Tasdogan et al., 2020). Using 13C-based metabolite infusion and SIL methods to quantify immune cell metabolism in live mice (Sheldon et al., 2021), we recently established that glucose is a prominent biosynthetic substrate in CD8+ Teff cells but is used less prominently for TCA cycle metabolism in vivo (Ma et al., 2019). These data suggest the possibility that undefined substrates in immune microenvironments may influence glucose utilization by T cells or themselves serve as mitochondrial substrates to fuel optimal T cell function in vivo.
Recognition that nutrient availability can impact cellular metabolic phenotypes has spurred efforts to quantify metabolite levels in tissues and serum (Cantor et al., 2017; Sullivan et al., 2019) and design synthetic media and cell culture systems to better recapitulate physiologic environments (Birsoy et al., 2014; Cantor et al., 2017; Vande Voorde et al., 2019). Here, we applied 13C-based SIL techniques to cell culture medium augmented with physiologic carbon sources (PCSs, nutrients found at >100 μM in mouse serum) to determine the impact of nutrient availability on CD8+ T cell metabolism and function. Our data highlight how environmental nutrient availability can influence T cell metabolic phenotypes to shape immune outcomes.
RESULTS
Physiologic carbon sources influence glucose utilization by T cells
To assess the impact of nutrient availability on T cell glucose utilization, we cultured in vitro-activated CD8+ T cells with standard T cell culture medium (IMDM, Iscove’s modified Delbecco’s medium) or medium modified to approximate polar metabolite concentrations in mouse serum (denoted VIM, Van Andel Institute-modified Iscove’s medium). The major features of VIM include adjusting glucose and glutamine levels to lower concentrations (5 and 0.5 mM, respectively), as well as several glucogenic amino acids (i.e., glycine, alanine, aspartate, methionine, threonine, and serine) (Figure 1A; Table S1). Mouse serum glucose concentrations can vary based on feeding behavior (4.4 mM blood glucose for fasted mice versus 8 mM for fed mice), with our plasma profiling indicating an average blood glucose of 7.0 ± 0.7 mM in mice fed ad libitum (data not shown).We settled on an intermediate glucose concentration (5 mM) used previously for physiologic culture of T cells(Leney-Greene et al., 2019; Ma et al., 2019). In addition, IMDM or VIM was augmented with nutrients highly abundant (>100 μM) in mouse serum but absent from most standard culture media (acetate, β-hydroxybutyrate [βOHB], citrate, lactate, and pyruvate; referred hereafter as physiologic carbon sources [PCSs]) (Hui et al., 2017; Sugimoto et al., 2012). Each of these carbon sources has been previously verified as an oxidizable substrate in mice, in some cases displaying circulatory turnover fluxes approaching or exceeding that of glucose and glutamine (Hui et al., 2017).
Figure 1. Physiologic carbon sources influence glucose utilization by T cells.
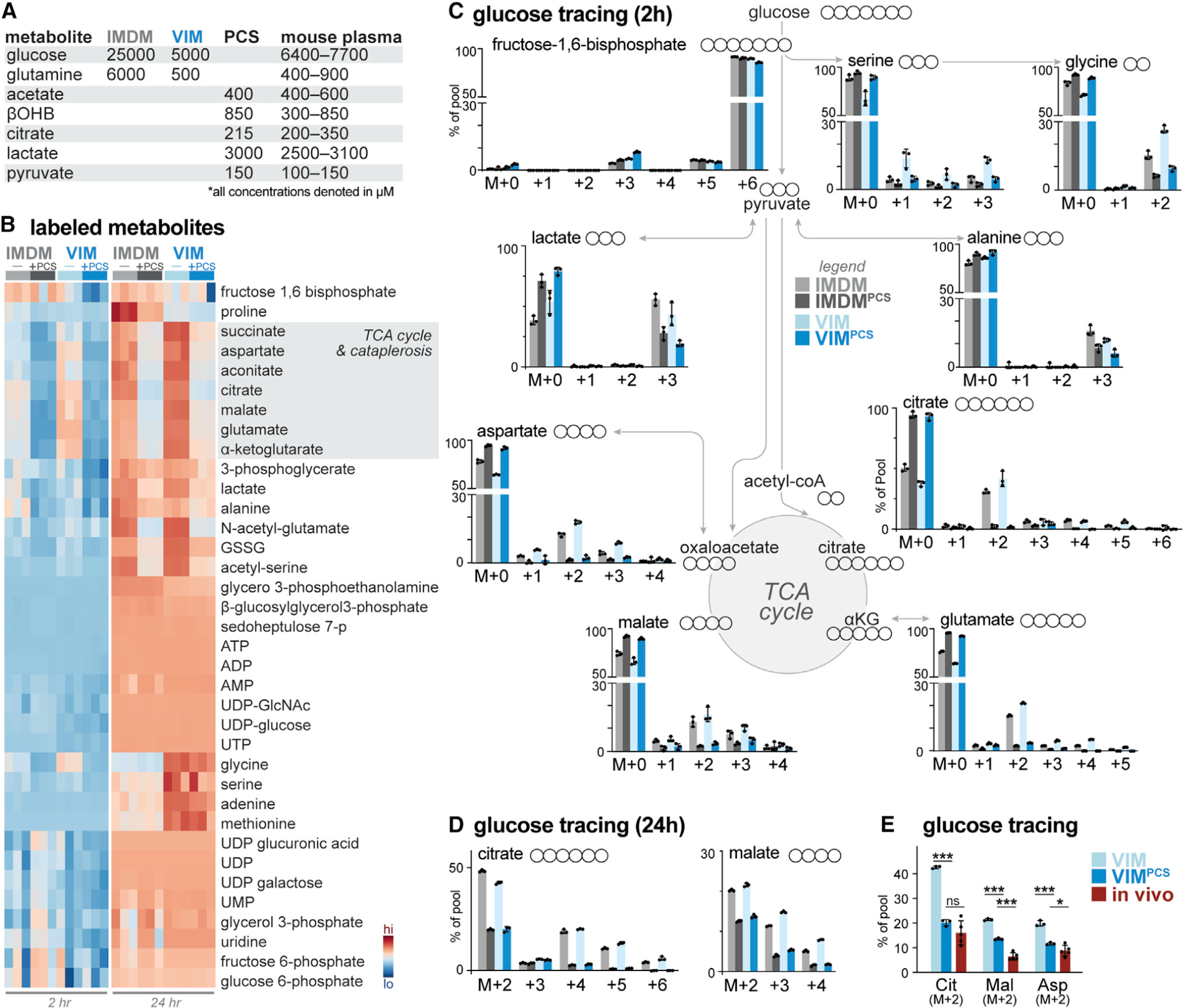
(A) Table highlighting major medium components of IMDM and VIM and physiologic carbon sources (PCSs) used in this study. Concentration ranges for metabolites in mouse serum are provided for comparison (Table S1).
(B) Heatmap depicting relative incorporation of 13C from [U-13C]-glucose into the indicated intracellular metabolites from activated CD8+ T cells cultured in IMDM or VIM with (+) or without (−) PCSs. Activated (CD44+) T cells were cultured in the indicated medium containing [U-13C]-glucose for 2 or 24 h (n = 3/group).
(C) Mass isotopologue distribution (MID) for [U-13C]-glucose-derived metabolites for activated CD8+ T cells cultured in IMDM (gray) or VIM (blue) with or without PCSs. Shown are MID labeling patterns for select metabolites after 2 h of culture as in (B) (mean ± SEM, n = 3/group).
(D) MID labeling patterns for [U-13C]-glucose-derived citrate and malate for CD8+ T cells cultured in IMDM (gray) or VIM (blue) with or without PCSs for 24 h as in (B) (mean ± SEM, n = 3/group).
(E) Fractional enrichment of [U-13C]-glucose-derived carbon in TCA cycle intermediates in activated CD8+ T cells. T cells activated as in (B) were cultured with [U-13C]-glucose in VIM or VIM plus PCS (VIMPCS) for 24 h. For infusion samples (in vivo), fractional enrichment of [U-13C]-glucose into TCA cycle intermediates was determined relative to [U-13C]-glucose abundance in spleen (mean ± SEM, n = 3–6/sample) (data from Ma et al., 2019).
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, not significant.
Naive CD8+ T cells were stimulated for 2 days in standard IMDM using plate-bound anti-CD3 and -CD28 antibodies, after which activated (CD44+) T cells were cultured in IMDM or VIM containing [U-13C]-glucose up to 24 h. T cell extracts were analyzed by mass spectrometry (MS) to identify 13C-glucose-derived metabolites generated over short-term (2 h) or long-term (24 h) culture (Figures 1B and S1A). We observed steady-state labeling from 13C-glucose into intracellular TCA cycle intermediates and lactate within 2–6 h of labeling (Figure S1B), consistent with previous results(Blagih et al., 2015; Chang et al., 2013).The intracellular metabolic fate of 13C-glucose into major metabolite classes was similar, regardless of base medium (IMDM or VIM), with 13C-glucose extensively labeling intermediates of glycolysis (i.e., fructose-1,6-bisphosphate; lactate), the PPP (i.e., D-sedoheptulose), nucleotides (i.e., ATP and UTP), and the TCA cycle (i.e., citrate, malate, and α-ketoglutarate) in T cells cultured in IMDM or VIM (Figure 1B). One notable exception was enrichment of 13C-glucose-derived serine and glycine—intermediates of one-carbon metabolism—in T cells cultured with VIM (Figure 1C), which may reflect increased intracellular biosynthesis to compensate for 4-fold lower levels of these metabolites in VIM relative to IMDM (Table S1). In contrast, the relative contribution of 13C from glucose into TCA cycle intermediates was significantly reduced upon addition of PCSs at their physiologic concentration(s) found in mouse serum (Figures 1B and 1C). However, total metabolite pool sizes segregated based on base medium (IMDM versus VIM) and were largely unaffected by the presence of additional PCSs (Figure S1A).
Analysis of metabolite levels in culture medium over time revealed several trends (Figures S1C–S1E). First, glucose and glutamine levels decreased over the 24 h culture period, consistent with active consumption of these carbon sources, but their consumption was not significantly altered by the presence of PCSs (Figure S1C). Levels of other carbon sources, including βOHB and pyruvate levels, displayed slower rates of decrease than glucose in PCS-supplemented medium (Figure S1C). Interestingly, we observed accumulation of several TCA cycle intermediates (i.e., α-ketoglutarate, fumarate, and malate; Figure S1D) and TCA cycle-derived amino acids (i.e., glutamate, aspartate; Figure 1E) in extracellular medium over time, suggesting net export of these metabolites.
Further examination of SIL patterns confirmed that the presence of PCSs drives differential partitioning of 13C-glucose-derived carbon in T cells (Figure 1C). Under standard culture conditions, citrate is readily labeled from 13C-glucose, with M+2 labeling reflecting the initial entry of 13C-glucose-derived pyruvate into the TCA cycle (via pyruvate dehydrogenase [PDH]) and higher-order labeling (M+4–5) reflecting 13C-glucose labeling through additional turns of the TCA cycle (Figures 1C and 1D). In the presence of PCSs, glucose contribution to citrate synthesis was significantly decreased (from 62.6% ± 2.2% to 7.0% ± 3.0% in VIM after 2 h of labeling) with minimal labeling beyond citrate M+2 (Figure 1C), despite high abundance of glucose (>5 mM) in the extracellular medium at 2 h (Figure S1C). Similarly, 13C-glucose labeling into downstream TCA cycle intermediates (i.e., malate M+2–3) and metabolites arising from TCA cataplerosis (i.e., glutamate M+2, +4; aspartate M+2–3) were reduced upon culture with PCSs (Figure 1C), and these 13C labeling patterns were retained over the 24 h time course (Figure 1D). It is unlikely that the decrease in 13C-glucose-derived citrate observed upon addition of PCSs was due to unlabeled citrate in the medium, as the intracellular citrate pool was not significantly altered by the addition of PCSs (Figure S1A) and extracellular citrate levels remained constant over the culture period (Figure S1C). These data suggest that PCSs reduced contribution of 13C-glucose to the mitochondrial acetyl-CoA pool and subsequent entry into the TCA cycle.
We have previously demonstrated that T cells activated by pathogen in vivo display distinct patterns of glucose utilization—marked by lower rates of lactate production—compared with in vitro-activated T cells (Ma et al., 2019). The addition of PCSs to VIM resulted in similar fractional enrichment of 13C-glucose-derived carbon into the TCA cycle intermediates citrate, malate, and aspartate compared with OT-I CD8+ T cells isolated from mice infected with Listeria monocytogenes expressing ovalbumin (LmOVA) that had received 13C-glucose through intravenous infusion 3 days post infection (dpi) (Figure 1E).
Physiologic carbon sources influence T cell metabolism independent of transcriptional metabolic programming
Upon activation, CD8+ T cells undergo extensive metabolic reprogramming characterized by transcriptional changes in metabolic gene expression (Wang et al., 2011). To assess whether differences in metabolic programming could account for the changes in metabolic glucose partitioning triggered by PCSs, we conducted parallel metabolic and transcriptional analysis of CD8+ T cells. Principal component analysis (PCA) of mRNA expression for activated CD8+ T cells cultured in different nutrient environments revealed significant differences in gene expression between different media (i.e., IMDM versus VIM) but minimal impact of PCSs on gene expression even after 24 h of culture (Figure 2A). PCA of KEGG pathway genes that encompass central carbon metabolism similarly indicated a high degree of similarity between CD8+ T cells cultured with or without PCSs (Figure 2B). Strikingly, for CD8+ T cells cultured in VIM or IMDM, we observed no significant difference in mRNA expression for genes encoding KEGG metabolic pathway enzymes upon addition of PCSs (Figure S2). In contrast, the presence of PCSs strongly influenced the metabolic fate of 13C-glucose, regardless of medium, with the variance driven primarily by changes in glucose partitioning in the TCA cycle (Figure 2C). Collectively, these data indicate that (1) the presence of PCSs repartitions glucose carbons away from the TCA cycle and into other glucose sinks, and (2) the shift in glucose utilization driven by PCSs occurs independent of global transcriptional changes in metabolic programming.
Figure 2. Physiologic carbon sources influence T cell metabolism independent of metabolic programming.
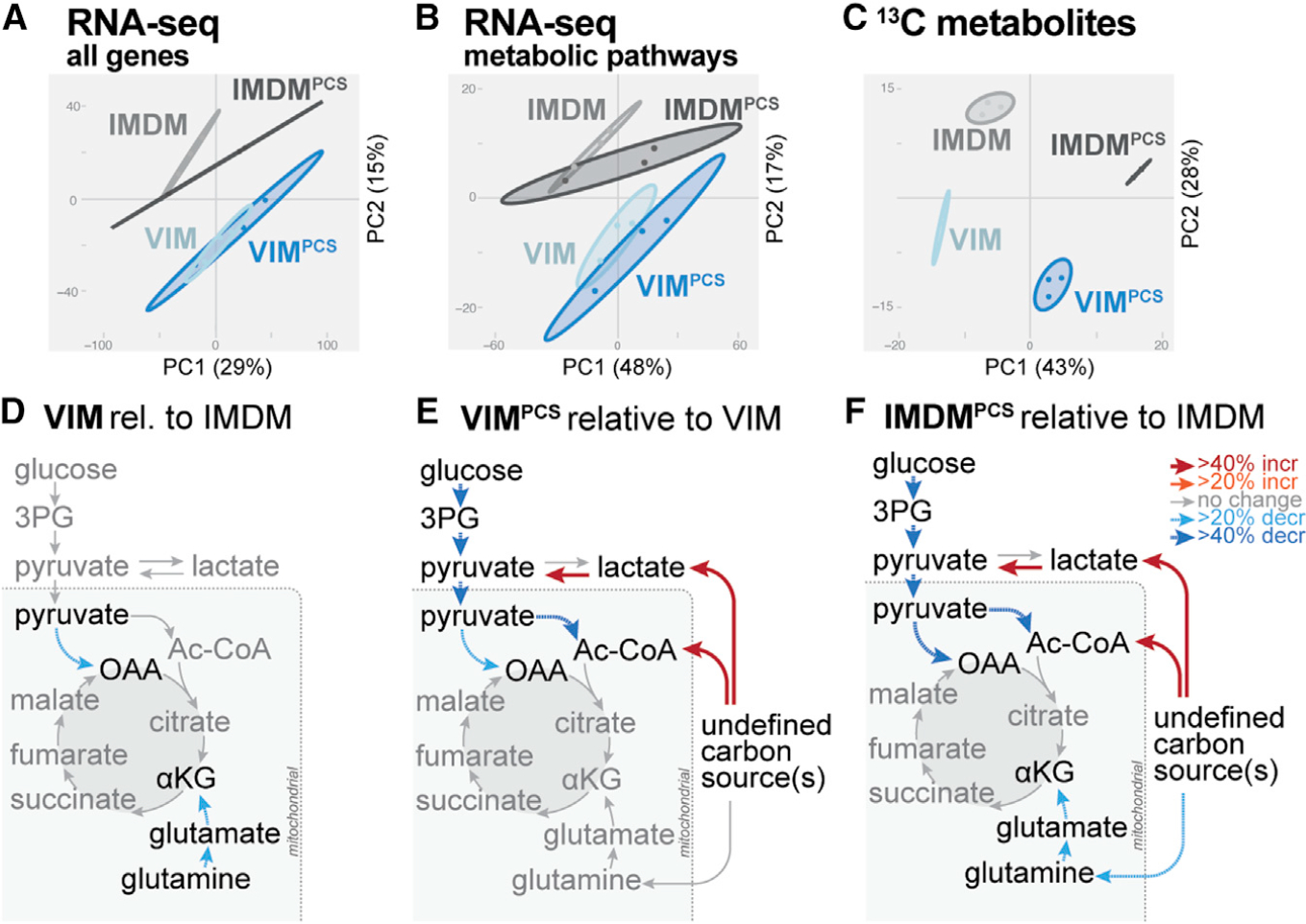
A–C) PCA for CD8+ T cells cultured for 24 h in IMDM or VIM with or without PCSs as in Figure 1B. Shown are data for total mRNA expression (A), mRNA expression for KEGG central carbon metabolism genes (B), and [U-13C]-glucose-derived metabolites (C) (mean ± SEM, n = 3/group).
(D–F) Metabolic flux modeling of central carbon metabolism in CD8+ T cells as determined using 13C-glucose tracing data from Figure 1B. Significant (p < 0.05) differences in net flux are indicated by color and bar weight (red, increased; blue, decreased).
(D) Net fluxes for CD8+ T cells cultured for 24 h in VIM relative to IMDM.
(E and F) Net fluxes for CD8+ T cells cultured for 24 h in VIM (E) or IMDM (F) containing or lacking PCSs (i.e., PCSs versus unsupplemented medium). Refer to Table S2 for details of flux analysis.
To better understand the impact of PCSs on T cell central carbon metabolism, we utilized [U-13C]-glucose-derived mass isotopologue distributions (MIDs) from the steady-state (24 h) labeling experiments to perform metabolic flux analysis (MFA). Using the isotopomer network compartmental analysis (INCA) package (Young, 2014), we created a stoichiometry reaction network tracking carbon trafficking through glycolysis and the TCA cycle, starting from canonical carbon inputs (e.g., glucose) as well as each PCS. Flux ratios between media conditions are presented in Figures 2D–2F. Despite observing significant differences in gene expression between CD8+ T cells cultured in IMDM versus VIM (Figures 2A and S2), changing medium from IMDM to VIM resulted in few changes in metabolic flux (Figure 2D). In contrast, addition of PCSs resulted in broader metabolic changes relative to the base medium (VIM or IMDM). Culturing cells in VIM or IMDM plus PCSs decreased relative flux through glycolytic enzymes and impacted pyruvate entry into the TCA cycle through both pyruvate dehydrogenase (PDH) and pyruvate carboxylase (PC) (Figures 2E and 2F). However, flux through the TCA cycle enzymes was not significantly impacted when base medium (VIM or IMDM) was supplemented with PCSs. Rather, the predicted source of acetyl-CoA (which fuels the TCA cycle) was affected by PCSs, suggesting overall TCA cycle activity was being fueled (>40%) by carbon flux from non-glucose sources.
Physiologic carbon sources contribute to TCA cycle metabolism and biosynthesis
To further understand the contribution of non-glucose carbon sources to T cell metabolism, we cultured CD8+ T cells in VIM supplemented with 13C-labeled PCSs at their physiologic concentrations (Figure 1A) and evaluated labeling patterns in metabolites involved in central carbon metabolism (schematic in Figure 3A). TCA cycle intermediates were labeled from 13C-glucose and 13C-glutamine, as previously reported (Blagih et al., 2015; Chang et al., 2013); however, lactate, acetate, and βOHB—which enter the TCA cycle as acetyl-CoA at the same point as glucose (Figure 3A)—also contributed carbon to the TCA cycle (Figure 3B). Examination of individual 13C labeling patterns revealed that lactate, acetate, and βOHB contribute carbon to the TCA cycle by 2-carbon entry via citrate synthase (M+2 citrate) and carry through the entire cycle (M+2 malate, M+2 aspartate, and M+4 citrate) (Figure 3C). The strong preference for these PCSs over glucose for TCA cycle use is especially striking when considering the lower concentration of lactate and βOHB (3 and 0.85 mM, respectively) compared with glucose (5 mM) (Figure 3C). TCA cycle intermediates were not readily labeled from 13C-alanine (Figures 3B and S3A), as previously reported (Ron-Harel et al., 2019). Although pyruvate can enter the TCA cycle at the same point as glucose (Figure 3A), we observed minimal labeling of citrate (Figure S3A), downstream TCA cycle intermediates (Figure S3B) and alanine (Figure S3C) from 13C-pyruvate under physiologic conditions (150 μM pyruvate). Under physiologic culture conditions (VIMPCS), approximately 70% of intracellular pyruvate is derived from glucose at steady state (Figure S3D). 13C-pyruvate was capable of contributing carbon to the TCA cycle when provided in excess (1.5 mM, 10-fold higher than physiologic levels) but was still outcompeted for the TCA cycle by other PCSs (Figure S3B), suggesting that extracellular pyruvate is a minor fuel for T cell oxidative metabolism under the physiologic culture conditions described here.
Figure 3. Physiologic carbon sources contribute to TCA cycle metabolism and biosynthesis.
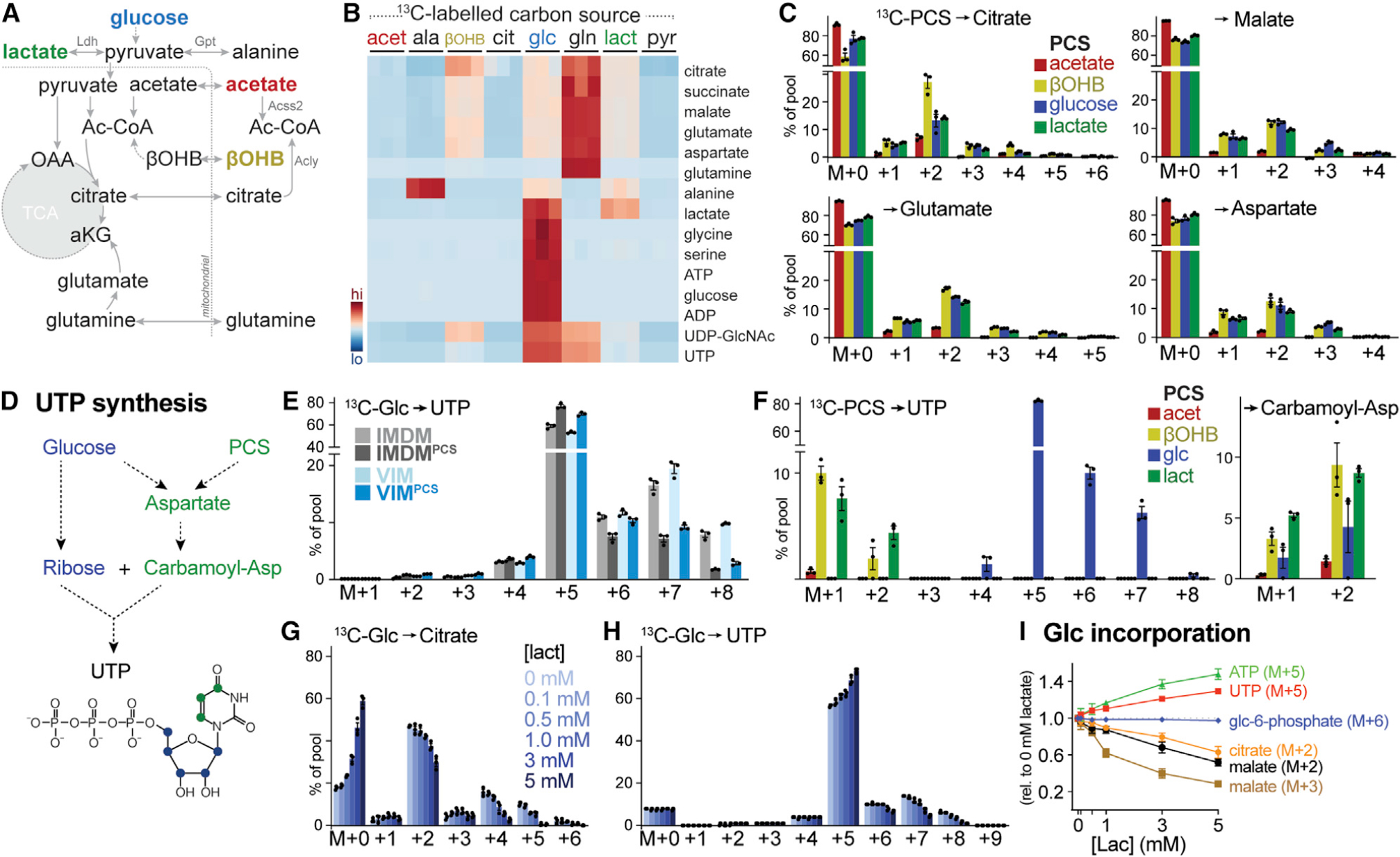
(A) Schematic depicting potential contribution of carbon sources to TCA cycle metabolism. Enzyme reactions localized to the cytosol and mitochondrion are indicated, with metabolic enzymes listed in gray.
(B) Heatmap depicting relative contribution of 13C from indicated [U-13C]-labeled substrates into metabolites of central carbon metabolism in activated CD8+ T cells after 24 h of culture (n = 3/group).
(C) MIDs for [U-13C]-PCS-derived carbon into citrate, malate, glutamate, and aspartate for CD8+ T cells cultured as in Figure 1B. Shown are individual isotopologues derived from acetate, βOHB, glucose, and lactate after 24 h of culture (mean ± SEM, n = 3/group).
(D) Schematic showing pathways for UTP synthesis from glucose and PCSs.
(E) MIDs for [U-13C]-glucose-derived UTP for activated CD8+ T cells cultured in IMDM or VIM with or without PCSs. Shown are MID labeling patterns from [U-13C]-glucose into UTP after 24 h of culture (mean ± SEM, n = 3/group).
(F) MID labeling pattens in UTP and carbamoyl-aspartate for in vitro-activated CD8+ T cells cultured in VIMPCS for 24 h. Shown are individual isotopologues derived from specific metabolites as in (C) (mean ± SEM, n = 3/group).
(G and H) MIDs for [U-13C]-glucose-derived citrate (G) and UTP (H) for activated CD8+ T cells cultured for 24 h in VIM supplemented with indicated concentrations of lactate (mean ± SEM, n = 3/group).
(I) Change in MID from [U-13C]-glucose into indicated metabolite isotopologues in activated CD8+ T cells cultured for 24 h in VIM containing increasing concentrations of lactate. Changes in MID were determined relative to cells cultured in VIM containing no lactate (mean ± SEM, n = 3/group).
TCA cycle metabolism in proliferating T cells is used to fuel both bioenergetics and cellular biosynthesis (Olenchock et al., 2017). Pyrimidine nucleotides such as UTP are synthesized from both glycolytic and TCA cycle intermediates, with ribose and aspartate functioning as essential components for biosynthesis (Figure 3D). Similar to that observed with TCA cycle intermediates (Figure 1C), the presence of PCSs altered 13C-glucose labeling patterns in UTP, maintaining M+5 ribose labeling in UTP from [U-13C]-glucose but reducing glucose contributions to pyrimidine ring synthesis via carbamoyl-aspartate synthesis (i.e., UTP M+6–8) (Figure 3E). Using parallel 13C-labeling, we observed significant labeling of UTP (M+1–2) from both 13C-lactate and 13C-βOHB, corresponding to labeling of carbamoyl-aspartate from these carbon sources (Figure 3F).
The addition of PCSs to culture medium had minimal impact on the overall abundance of TCA cycle and downstream metabolites in CD8+ T cells (Figure S1A), suggesting that PCSs have a distinct metabolic impact on glucose partitioning in T cells. To test this hypothesis directly, we examined 13C-glucose labeling patterns in response to increasing extracellular abundance of lactate, a PCS found at millimolar concentrations in both mouse and human serum (Figure 1A; Table S1) (Cantor et al., 2017; Hui et al., 2017). Lactate promoted a dose-dependent decrease in 13C-glucose incorporation into the TCA cycle, as noted by increasing levels of unlabeled citrate (M+0) coupled with decreasing abundance of 13C-glucose-labeled citrate (M+2, +4, +5) (Figure 3G). Increasing extracellular lactate levels also reduced glucose contribution to pyrimidine ring biosynthesis (UTP M+7–8) (Figure 3H), corresponding to reduced glucose-dependent aspartate production. Notably, increasing extracellular lactate levels had no inhibitory effect on the commitment of 13C-glucose to upper glycolysis (glucose-6-phosphate M+6) and the PPP (via ribose M+5 of ATP, UTP) (Figure 3I). Overall, these data establish that non-glucose carbon sources, including the glycolytic product lactate, redistribute cellular glucose fates from oxidative to biosynthetic pathways and can themselves directly participate in TCA cycle-dependent biosynthesis.
Physiologic carbon sources influence T cell survival and effector function
Reduced glucose availability has been shown to impact effector cytokine expression (Blagih et al., 2015; Cham and Gajewski, 2005; Chang et al., 2013); however, it is unclear whether other metabolites influence effector cytokine production under physiologic glucose concentrations. To address this, naive CD8+ T cells were activated with anti-CD3 and -CD28 antibodies in VIM culture medium with or without PCS supplementation, and 3 days later, intracellular and surface marker expression, proliferation, and cytokine production were measured. Activation in the presence of PCSs did not alter expression of CD8+ T cell activation/effector markers (i.e., CD69, KLRG1, and CX3CR1) or the transcription factor (TF) T-bet (Figures 4A and S4A). Interestingly, expressions of the TFs T cell factor 1 (Tcf1) and Tox were elevated in PCS-activated CD8+ T cells (Figure 4A). Co-expression of Tcf1 and Tox is a feature of polyfunctional “progenitorlike” CD8+ T cells, a differentiation state observed in chronic infection and associated with self-renewal properties (Beltra et al., 2020; Sekine et al., 2020; Yao et al., 2019). CD8+ T cells cultured with or without PCSs displayed similar levels of proliferation, as determined by fluorescent dye dilution and Ki67 expression, when cultured in medium containing glucose (Figure 4B). However, addition of PCSs specifically enhanced CD8+ T cell viability under conditions of glucose deprivation (Figures 4C and S4B).
Figure 4. Physiologic carbon sources influence T cell survival and effector function.
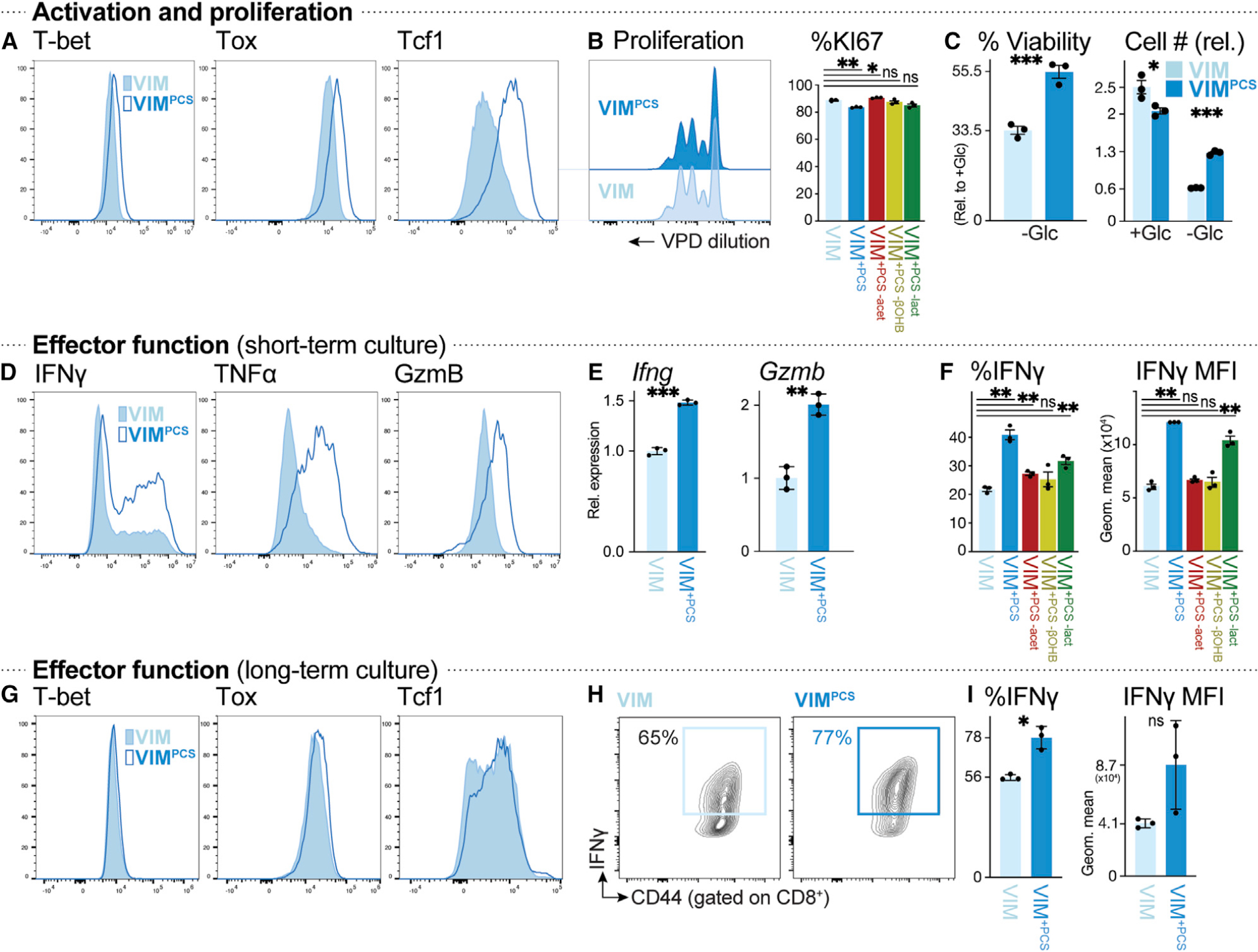
(A) Histograms of T-bet, Tox, and Tcf1 expression in CD8+ T cells activated for 3 days with plate-bound anti-CD3 and -CD28 antibodies in VIM (closed) or VIMPCS (open).
(B) Proliferation of anti-CD3- and anti-CD28-stimulated CD8+ T cells cultured in VIM or VIMPCS for 3 days (n = 3/group). Left: violet proliferation dye (VPD) dilution. Right: percentage of Ki67+ CD8+ T cells after activation in VIM, VIMPCS, or VIM containing PCSs but lacking acetate (red), βOHB (yellow), or lactate (green).
(C) Cell viability (left) or cell number (right) of activated CD8+ T cells after 48 h of culture in VIM plus IL-2 and containing (+Glc) or lacking (−Glc) glucose. Percent viability was calculated relative to cell viability in glucose-replete conditions (5 mM) (mean ± SEM, n = 3/group). Cell number was expressed relative to initial cell number at day 0 (mean ± SEM, n = 3/group).
(D) Intracellular IFN-γ, TNF-α, and granzyme B levels in CD8+ T cells activated as in (A) (n = 3/group).
(E) Relative expression of Ifng and Gzmb mRNA in CD8+ T cells cultured as in (D) (mean ± SD, n = 3/group).
(F) Percentage of IFN-γ+ CD8+ T cells (left) and MFI for IFN-γ expression (right) for CD8+ T cells cultured as in (D) in VIM, VIMPCS, or VIM containing PCSs but lacking acetate, βOHB, or lactate (n = 3/group).
(G–I) CD8+ T cells were activated for 3 days with plate-bound anti-CD3 and -CD28 antibodies, followed by culture for an additional 4 days with IL-2, in VIM or VIMPCS. Analysis of CD8+ T cells was conducted on day 7.
(G) T-bet, Tox, and Tcf1 expression.
(H) Plot of CD44 versus intracellular IFN-γ expression.
(I) Percentage of IFN-γ+ CD8+ T cells and MFI for IFN-γ expression (mean ± SEM, n = 3/group).
We next examined the impact of PCSs on CD8+ T cell function by assessing expression of intracellular effector molecules by flow cytometry. Supplementing T cell medium with PCSs boosted intracellular levels of IFN-γ, TNF-α, and granzyme B production by CD8+ T cells, promoting both an increase in the number of cytokine-producing cells and protein production per cell (as determined by increased mean fluorescence intensity [MFI] of these effector molecules) (Figures 4D and S4C). Mechanistically, PCS-activated CD8+ T cells displayed increased expression of Ifng and Gzmb mRNA, which encode IFN-γ and granzyme B proteins, respectively, relative to controls (Figure 4E). Levels of mechanistic target of rapamycin complex 1 (mTORC1) activity, as determined by intracellular staining for phosphorylation of the mTORC1 target ribosomal S6 protein, were unaffected by PCSs (Figure S4D), suggesting that PCSs influence effector molecule expression at the level of mRNA transcription rather than translation. Metabolite drop-out experiments revealed that removing any one of lactate, acetate, or βOHB from PCSs could partially reverse the enhanced cytokine production triggered by PCSs (Figure 4F) but had minimal impact on T cell proliferation (Figure 4B). Long-term culture (7 day) in PCS-containing medium did not dramatically alter the immunologic phenotype of CD8+ T cells (Figures 4G and S4E), except for increased Tcf1 expression (Figure 4G) and a higher percentage of IFN-γ+ CD8+ T cells in long-term cultures supplemented with PCSs (Figures 4H and 4I). These data identify oxidizable carbon sources (i.e., lactate, acetate, and βOHB) that specifically influence T cell effector function and survival without impacting T cell proliferative capacity.
Lactate is a physiologic fuel for CD8+ T cells
Lactate is a prominent metabolic endpoint of glucose metabolism, particularly in tumor cells undergoing the Warburg effect, and is often considered a metabolic waste product of glycolysis (DeBerardinis and Chandel, 2020; Vander Heiden et al., 2009). High concentrations (>5 mM) of lactic acid, such as found in the tumor microenvironment, can dampen T cell proliferation and cytokine production, which has been attributed in part to lowering of extracellular pH (Brand et al., 2016; Fischer et al., 2007; Quinn et al., 2020; Watson et al., 2021). However, recent work has identified lactate as a bona fide TCA cycle fuel for some tumor cells and non-transformed tissues in vivo (Faubert et al., 2017; Hui et al., 2017; Tasdogan et al., 2020). Functional analysis of in vitro-activated CD8+ T cells revealed suppressive effects of exogenous lactate supplementation on T cell proliferation (Figure S5A) and IFN-γ production (Figure S5B) at high concentrations (>5 mM), whereas exposure to physiologic lactate concentrations (0.5–2 mM) invoked a slight increase in IFN-γ production compared with controls (Figures S5B and S5C).
Although CD4+ regulatory T (Treg) and effector T cells have been shown to metabolize lactate under high lactate (~5–20 mM) and low glucose conditions (Angelin et al., 2017; Quinn et al., 2020; Watson et al., 2021), we examined whether lactate is a fuel for CD8+ T cells at concentrations found in mouse serum (1–3 mM) (schematic in Figure 5A). Activated CD8+ T cells readily took up 13C-lactate even in the presence of physiologic glucose concentrations (5 mM) (Figure 5B), and under these conditions, 13C-lactate contributed to the intracellular pyruvate pool of CD8+ T cells to equal levels as 13C-glucose (Figure 5C). Interestingly, the presence of lactate did not decrease the overall abundance 13C-glucose-derived pyruvate; rather, lactate exposure promoted an overall increase in intracellular pyruvate abundance, with pyruvate being generated in equal abundance from both 13C-glucose and 13C-lactate (Figure 5C). We observed similar labeling patterns in TCA cycle intermediates, with both 13C-glucose and 13C-lactate contributing carbon to the first turn of the TCA cycle (citrate M+2) (Figure S5D) as well as aspartate synthesis (Figure S5E).
Figure 5. Lactate is a physiologic fuel for CD8+ T cells.
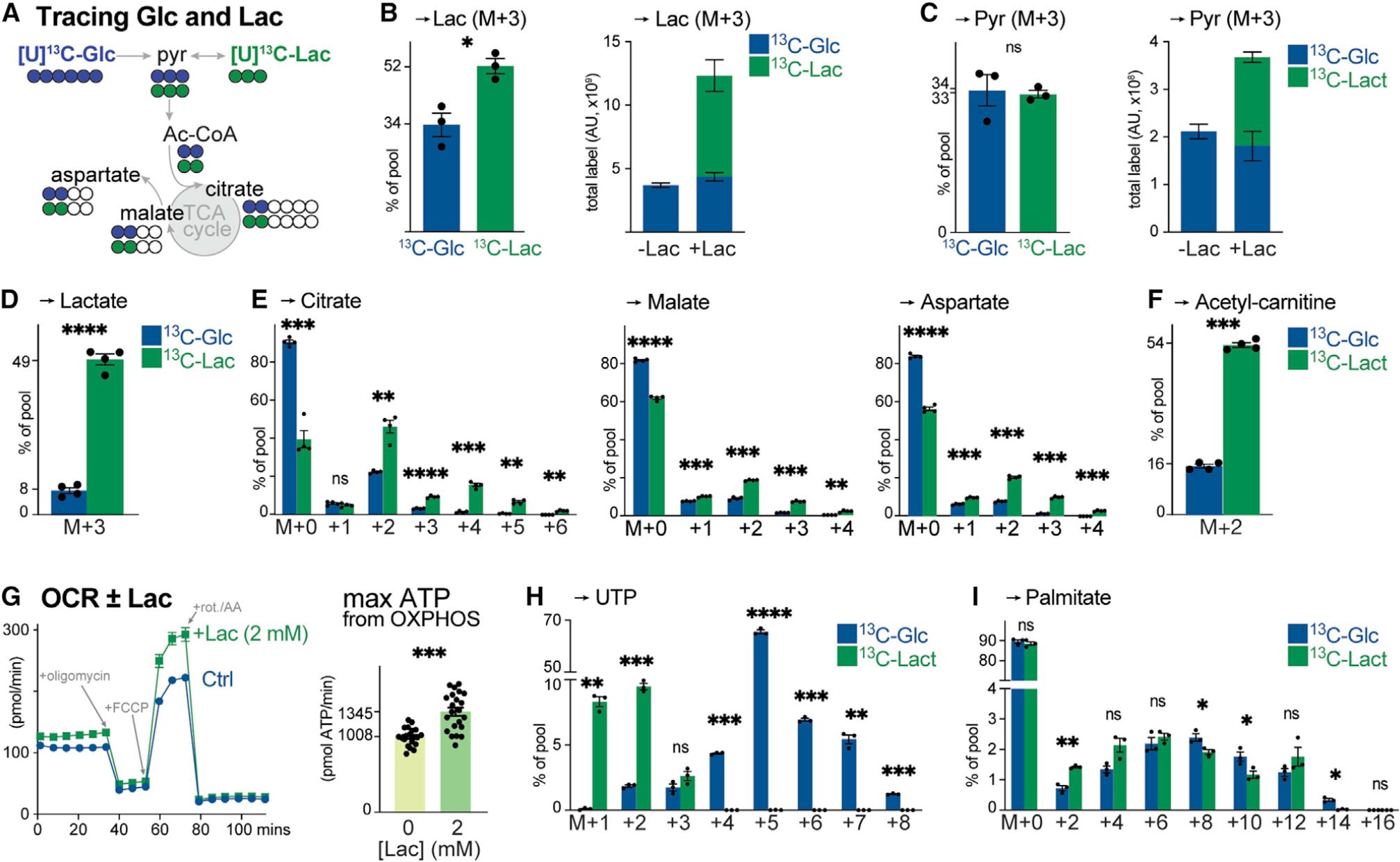
(A) Schematic of [U-13C]-glucose (blue) and [U-13C]-lactate (green) tracing into TCA cycle intermediates and TCA cycle-derived metabolites (i.e., aspartate).
(B) [U-13C]-glucose (5 mM) and [U-13C]-lactate (2 mM) labeling into lactate in activated CD8+ T cells after 4 h of culture (mean ± SEM, n = 3/group). Left: fractional enrichment of [U-13C]-glucose and [U-13C]-lactate carbon into the lactate pool. Right: total abundance of [U-13C]-glucose- or [U-13C]-lactate-derived lactate in activated CD8+ T cells cultured in VIM lacking (−) or containing (+) 2 mM lactate.
(C) [U-13C]-glucose and [U-13C]-lactate labeling into pyruvate for T cells cultured as in (B) (mean ± SEM, n = 3/group). Left: fractional enrichment of [U-13C]-glucose and [U-13C]-lactate carbon into the pyruvate pool. Right: total abundance of [U-13C]-glucose- or [U-13C]-lactate-derived pyruvate in T cells cultured without (−) or with (+) 2 mM lactate.
(D–F) Fractional enrichment of [U-13C]-glucose and [U-13C]-lactate in intracellular (D) lactate, (E) TCA cycle intermediates (citrate, malate, and aspartate), and (F) acetyl-carnitine in OT-I CD8+ T cells isolated from LmOVA-infected mice at 3 dpi. Isolated T cells were cultured ex vivo in VIM containing 5 mM glucose and 2 mM lactate (either 12C or 13C) for 4 h (mean ± SEM, n = 4/group).
(G) Bioenergetic profile of in vitro-activated CD8+ T cells cultured with no (Ctrl) or 2 mM lactate (+Lac) (mean ± SD, n = 20–22/group). Left: OCR plot for activated CD8+ T cells over time. Time of addition of oligomycin, fluoro-carbonyl cyanide phenylhydrazone (FCCP), and rotenone and antimycin A (rot./AA) are indicated. Right: maximal ATP production rates from OXPHOS.
(H and I) MID of [U-13C]-glucose and [U-13C]-lactate carbon in (H) UTP or (I) palmitate for in vitro-activated CD8+ T cells cultured in VIM for 4 h (H) or 24 h (I) (mean ± SEM, n = 3/group).
To confirm lactate as a physiologic fuel for proliferating antigen-specific CD8+ T cells, we conducted ex vivo 13C tracing experiments in which CD8+ OT-I T cells isolated from LmOVA-infected mice (3 dpi) were subjected to short-term culture in VIM containing 13C-glucose or 13C-lactate. LmOVA-specific CD8+ T cells readily consumed lactate, with ~50% of the intracellular lactate pool derived from 13C-lactate (Figure 5D). Less than 10% of the intracellular lactate pool was derived from 13C-glucose (Figure 5D), consistent with previous observations of low glucose-to-lactate turnover by antigen-specific T cells in vivo (Ma et al., 2019). LmOVA-specific CD8+ T cells displayed preference for lactate over glucose as a TCA cycle fuel, as evidenced by the enrichment of M+2 and higher order isotopologues of citrate (M+3–6), malate (M+3–4), and aspartate (M+3–4) from 13C-lactate (Figure 5E). Moreover, 13C-lactate carbon contributed almost 4-fold more than 13C-glucose to intracellular acetyl-CoA production (measured as surrogate by M+2 enrichment in acetyl-carnitine) in LmOVA-specific CD8+ T cells (Figure 5F). Together, these data indicate that lactate is a physiologic TCA cycle substrate for CD8+ Teff cells.
We next assessed the impact of exogenous lactate on T cell bioenergetics using a Seahorse extracellular flux analyzer. Exposure to exogenous lactate (2 mM) promoted a slight but significant increase in the basal oxygen consumption rate (OCR) of in vitro-activated CD8+ T cells (Figure 5G), corresponding to a 15%–20% increase in basal ATP production (Figure S5F) and 30% increase in maximal ATP production from oxidative phosphorylation (OXPHOS) in response to lactate versus glucose alone (Figure 5G). Consistent with its contribution to TCA cycle-dependent aspartate and acetyl-CoA biosynthesis, we also detected prominent 13C-lactate labeling in UTP (M+1–3; Figure 5H) and palmitate (Figure 5I). We conclude from these data that antigen-specific CD8+ T cells concomitantly use lactate and glucose for TCA cycle-dependent bioenergetics and biosynthesis, with lactate contributing directly as a carbon source for the TCA cycle under physiologic conditions.
Ldha regulates T cell metabolism and effector T cell expansion in vivo
Lactate metabolism is regulated by the tetrameric enzyme complex lactate dehydrogenase (LDH), which interconverts lactate and pyruvate using NAD+/NADH as a co-factor. T cells express both M and H subunits of LDH (encoded by the Ldha and Ldhb genes, respectively) (Rambotti and Davis, 1981; Ringoir and Plum, 1975); thus, we performed shRNA-mediated knockdown of Ldha to determine the metabolic and functional roles of this enzyme complex in CD8+ T cells. Expression of shRNA targeting Ldha resulted in depletion of Ldha protein expression (Figure 6A) and reduced LDH enzyme activity (Figure S6A) in CD8+ T cells. Although 13C-lactate uptake in Ldha-depleted cells remained intact (Figure 6B), both the conversion of 13C-lactate to 13C-pyruvate (Figure 6B), as well as OXPHOS-dependent ATP production from lactate (Figure 6C), were reduced in CD8+ T cells expressing Ldha-targeting shRNA. 13C-glucose-to-lactate conversion was also reduced in shLdha-expressing CD8+ T cells (Figure 6D), consistent with the function of the LDH enzyme complex mediating pyruvate and lactate interconversion.
Figure 6. Ldha regulates T cell metabolism and effector cell expansion in vivo.
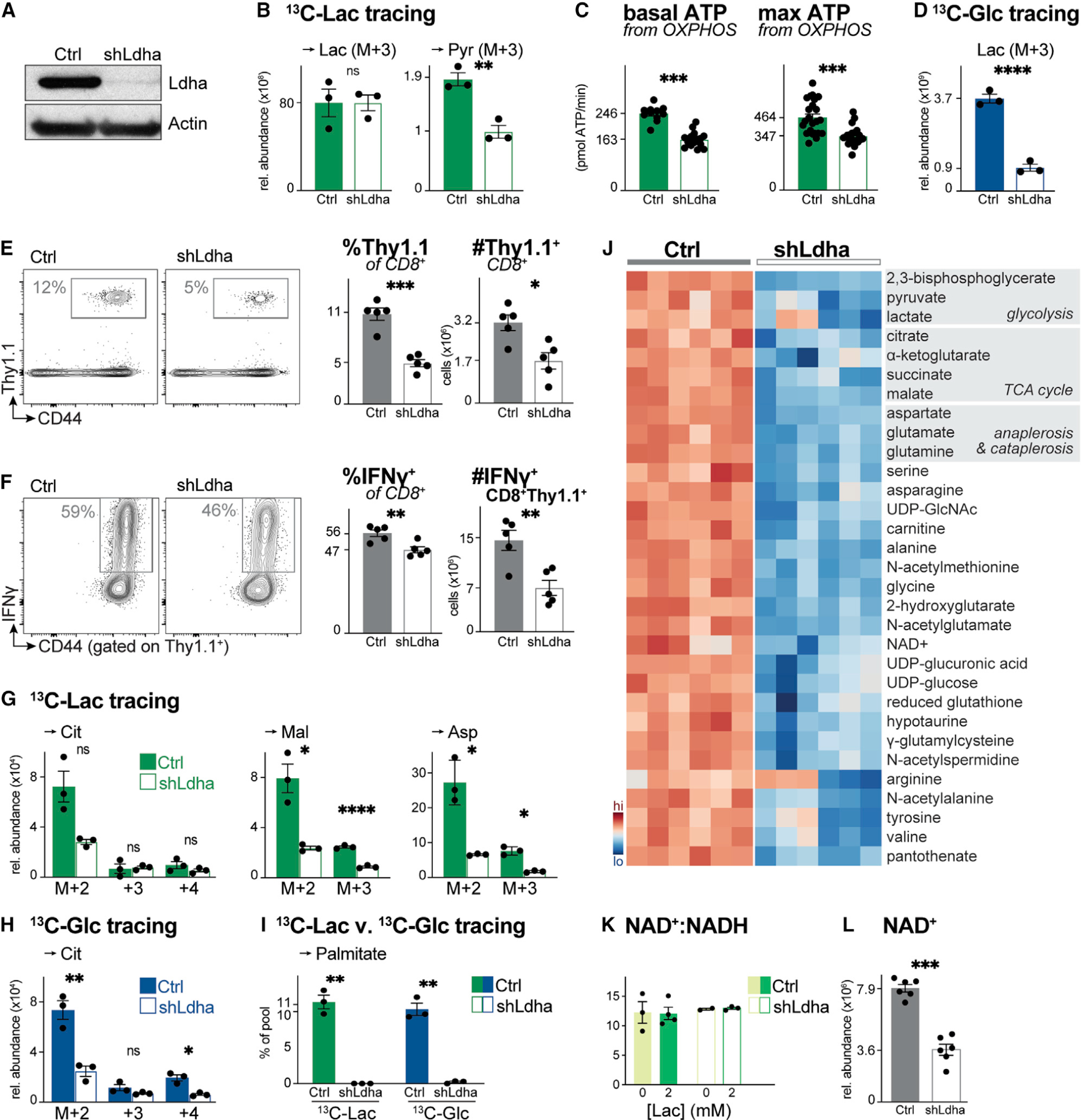
(A) Immunoblot of Ldha and actin protein levels in whole cell lysates from activated CD8+ T cells expressing a control (Ctrl) or Ldha-targeting (shLdha) shRNA.
(B) Relative abundance of intracellular [U-13C]-lactate-derived lactate (Lac) and pyruvate (Pyr) in activated CD8+ T cells expressing a control (Ctrl) or Ldha-targeting (shLdha) shRNA. T cells were cultured in VIM containing 5 mM glucose and 2 mM [U-13C]-lactate for 4 h (mean ± SEM, n = 3/group).
(C) Basal (left) and maximal (right) ATP production rates from OXPHOS for CD8+ T cells expressing a control (Ctrl) or Ldha-targeting (shLdha) shRNA cultured as in (B) (mean ± SD, n = 10–24/group).
(D) Relative abundance of intracellular [U-13C]-glucose-derived lactate (Lac) in control (Ctrl) or shLdha-expressing CD8+ T cells cultured with 5 mM [U-13C]-glucose and 2 mM lactate for 4 h (mean ± SEM, n = 3/group).
(E) Flow cytometry plots showing abundance of OVA-specific Thy1.1+ OT-I T cells in the spleen of LmOVA-infected mice 7 dpi. Right: percentage and total number of Thy1.1+ CD8+ OT-I T cells (mean ± SEM, n = 5 mice/shRNA).
(F) Flow cytometry plots showing the percentage of IFN-γ+ control (Ctrl) or shLdha-expressing Thy1.1+ CD8+ OT-I T cells in the spleen of LmOVA-infected mice 7 dpi. Right: percentage and total number of IFN-γ+ Thy1.1+ CD8+ T cells (mean ± SEM, n = 5 mice/shRNA).
(G and H) MIDs for indicated intracellular metabolites in activated control (Ctrl) or shLdha-expressing CD8+ T cells. Cells were cultured for 4 h in VIM containing (G) 2 mM [U-13C]-lactate and unlabeled 5 mM glucose or (H) 5 mM [U-13C]-glucose and unlabeled 2 mM lactate (mean ± SEM, n = 3/group).
(I) [U-13C]-lactate and [U-13C]-glucose labeling into palmitate (in VIM for 24 h) in activated CD8+ T cells expressing a control (Ctrl) or Ldha-targeting (shLdha) shRNA (mean ± SEM, n = 3/group).
(J) Heatmap depicting relative abundance of intracellular metabolites in activated control (Ctrl) or shLdha-expressing CD8+ T cells following 4 h culture in VIM. Shown are the top 45 significant metabolites (p < 0.05) based on differential metabolite levels between groups (n = 6/group).
(K) NAD+:NADH ratio for control (Ctrl) or shLdha-expressing CD8+ T cells cultured for 2 h in VIM containing 0 or 2 mM sodium lactate (n = 2–4/group).
(L) NAD+ abundance in activated CD8+ T cells expressing a control (Ctrl) or Ldha-targeting (shLdha) shRNA following 4 h culture in VIM (mean ± SEM, n = 6/group).
We next examined the role of Ldha in CD8+ T cell responses to LmOVA infection, which we demonstrated stimulates responding T cells to take up and oxidize lactate (Figures 5D and 5E). Thy1.1-expressing CD8+ OT-I T cells were transduced with a control or Ldha-targeting shRNA, adoptively transferred into naive Thy1.2+ hosts, then infected with LmOVA 1 day later (Figure S6B). In animals that received shLdha-expressing OT-I T cells, we observed a significant reduction in antigen-specific T cell expansion, as measured by the percentage and number of OVA-specific CD8+ Teff cells in the spleen of mice 7 dpi (Figure 6E). These data are consistent with our observations that IL-2-dependent proliferation is reduced in shLdha-expressing CD8+ T cells in vitro (Figure S6C). Silencing Ldha reduced both the proportion and total number of IFN-γ-producing CD8+ T cells (Figure 6F), the latter attributed to the reduced expansion of OVA-specific CD8+ Thy1.1+ T cells due to Ldha knockdown. Our results independently confirm similar observations using mice with T cell-specific deletion of Ldha (Xu et al., 2021) that Ldha expression is critical for CD8+ T cell expansion in vivo.
To define the mechanisms that underpin Ldha-dependent regulation of CD8+ T cell expansion and effector function, we conducted SIL on control and shLdha-expressing CD8+ T cells using 13C-lactate and 13C-glucose. We observed a decrease in the overall abundance of 13C-lactate-derived citrate, malate, and aspartate in shLdha-expressing CD8+ T cells, consistent with reduced metabolic flux of lactate into the TCA cycle (Figure 6G). Notably, except for citrate M+2, the fractional enrichment of 13C-lactate in TCA cycle intermediates and aspartate was not significantly altered by Ldha knockdown (Figure S6D), suggesting that flux from 13C-lactate to the TCA cycle was altered. Based on results using LDH inhibitors (Hermans et al., 2020), we hypothesized that T cells may promote metabolic rewiring to compensate for decreased Ldha expression. However, reduced abundance of 13C-glucose-derived carbon in citrate and downstream metabolites (i.e., malate, aspartate) was also observed in shLdha-expressing CD8+ T cells (Figures 6H and S6E), indicating no compensation from glucose. Other metabolic features of Ldha silencing included reduced glucose-dependent serine biosynthesis (which supports T cell proliferation; Ma et al., 2017, 2019) (Figure S6E) and reduced lactate- and glucose-dependent lipid synthesis (Figure 6I).
Silencing Ldha led to an overall decrease in intracellular metabolite abundance that was not restricted to intermediates of the TCA cycle; other prominent metabolite pools affected by Ldha silencing included PPP intermediates, nucleotides, and components of SGOC metabolism (Figure 6J). We hypothesized that the LDH complex may influence metabolite levels in T cells through effects on NAD+/NADH balance, given previous reports that Ldha-deficient T cells display a decrease in their intracellular NAD+:NADH ratio (Xu et al., 2021). However, we did not observe a difference in the NAD+:NADH ratio between control and shLdha-expressing CD8+ T cells either at baseline or physiologic concentrations of lactate (2 mM) (Figure 6K). Rather, we observed a significant (~50%) reduction in overall NAD+ abundance in shLdha-expressing CD8+ T cells compared with controls (Figure 6L). Collectively, these data establish the processing of lactate through Ldha as a critical metabolic node in CD8+ T cells that regulates both lactate-dependent bioenergetics and T cell metabolic homeostasis.
DISCUSSION
Much of the recent work on metabolic pathway utilization in immune cells has focused on cell-intrinsic factors, such as metabolic enzyme expression. However, environmental nutrient availability can strongly influence metabolic pathway utilization (Muir et al., 2018). For example, cancer cells display changes in cell growth and stress resistance when cultured with physiologic medium due to differences in substrate utilization (Cantor et al., 2017; Rossiter et al., 2021). Quantifying metabolic fluxes in yeast grown under defined nutrient conditions, Hackett and colleagues identified substrate concentrations as the major driver of metabolic pathway utilization, exerting twice the impact on metabolic output than enzyme expression (Hackett et al., 2016). In addition, homeostasis of circulating metabolites at the whole organism level is driven in large part by mass action-driven oxidation via the TCA cycle (Li et al., 2022). Our present work expands on these studies by using SIL metabolomics approaches to investigate how nutrient availability impacts CD8+ T cell metabolism and function.
Our data reveal four important conclusions about the impact of environmental nutrient availability on T cell metabolism. First, CD8+ T cells display a high degree of plasticity in mitochondrial fuel choice, consuming conventional fuels such as glucose and glutamine but also other PCSs when available. Exploring this in further detail, we identified lactate as a prominent mitochondrial fuel in CD8+ Teff cells that contributes both to TCA cycle metabolism and biomass generation even when glucose is available. Second, T cells are capable of oxidizing non-glucose fuels independent of transcriptional changes in metabolic enzyme expression, indicating that flexibility in metabolic pathway choice is inherent to activated T cells rather than reliant on transcriptional changes in response to changes in the metabolic environment. Third, when PCSs such as lactate are readily available, they are preferentially oxidized by T cells. Metabolism of PCSs occurs concomitantly with glucose metabolism and reduces the fraction of TCA cycle carbon that comes from glucose. These observations are consistent with the predominant oxidative phenotype of proliferating CD8+ T cells in vivo (Klarquist et al., 2018; Ma et al., 2019) and may explain the differences in glucose metabolism often observed between in vivo and cell culture systems (Davidson et al., 2016; Hensley et al., 2016; Hui et al., 2020). Finally, we found that T cell viability and the production of effector molecules (i.e., IFN-γ) by CD8+ Teff cells is enhanced in the presence of PCSs, providing evidence that both fuel availability and fuel choice can impact T cell function. This last observation suggests that T cell function and survival in diverse immune microenvironments, such as solid tumors or inflamed tissues, may be shaped by environmental nutrient availability and impact immune outcomes.
Cell culture has allowed us to map out major metabolic pathways that underpin immune cell metabolism. However, one limitation of current cell culture medium formulations is that they were designed to promote cell growth and survival ex vivo and as such often represent supraphysiological concentrations of some nutrients, while lacking other nutrients relevant to cellular metabolism (Cantor, 2019). An important implication of this work is that glucose utilization by T cells is highly influenced by the presence of other non-glucose substrates, specifically some of the nutrients left out of most current medium formulations. Adding PCSs to cell culture media largely did not affect glucose utilization in the upper phase of glycolysis—shunting of glucose carbon into the PPP and SGOC metabolism; rather, the presence of PCSs reduced glucose utilization for TCA cycle-dependent metabolism and biosynthesis. Our flux modeling (Figure 2) indicates that the presence of PCSs favors the partitioning of glucose away from the TCA cycle. One interpretation is that the primary fate of glucose is not oxidative energy production but rather to fuel key biosynthetic pathways that specifically rely on glucose (i.e., nucleotide synthesis). One may speculate that glucose actually plays a backup role for bioenergetic metabolism in T cells, providing oxidizable fuel to mitochondria when more favorable oxidative fuels such as lactate are in low supply.
Both our transcriptional profiling and flux modeling indicate that environmental nutrient availability can impact TCA cycle flux and fuel utilization independent of acute changes in metabolic gene expression. Thus, we put forth the concept that activation-related metabolic programming lays out a network of possibilities for T cell metabolism but that metabolic pathway usage ultimately depends on substrate availability. We speculate that environmental nutrient availability influences metabolic pathway usage largely through the balance of substrates and products and that pathway flux is further regulated through allosteric regulation and/or posttranslational modifications of metabolic enzymes (Kaymak et al., 2021). In this regard, regulation of systemic metabolite availability may be a strategy for the organism to modulate immune responses. Using single-cell methods for bioenergetic (Argüello et al., 2020) and metabolic enzyme (Hartmann et al., 2020; Levine et al., 2021) profiling may help define the metabolic potential of T cells within distinct immune microenvironments.
Using SIL, we have identified three major substrates—acetate, βOHB, and lactate—not found in conventional medium but readily oxidized in the TCA cycle, while demonstrating that exogenous pyruvate is a minor oxidative fuel for CD8+ T cells. Notably, CD8+ T cells responding to Listeria infection in vivo display high rates of lactate oxidation (Figure 5E), highlighting lactate as a fuel for CD8+ Teff cells in vivo. When present, lactate contributes equally with glucose to the intracellular pyruvate pool and is favored ~4-fold over glucose as a TCA cycle substrate by pathogen-specific CD8+ T cells. The presence of 13C-lactate carbon in pyrimidine nucleotides and palmitate indicate that lactate can serve as a biosynthetic substrate through its contribution to TCA cycle metabolism. Thus, although lactate has traditionally been viewed as a metabolic waste product of glycolysis, the data presented here indicate that, at its physiologic concentration, lactate can contribute significantly to both T cell bioenergetics (fueling the TCA cycle and OXPHOS) and cellular biosynthesis, particularly during functional T cell responses in vivo. We speculate that lactate may function as a prominent fuel during T cell priming in secondary lymphoid organs, where local lactate concentrations may be significantly higher than circulating levels. Glucose-dependent lactate production is a key early event in dendritic cell (DC) activation (Everts et al., 2014; Krawczyk et al., 2010), and lactate produced by DCs may provide bioenergetic support to T cells while facilitating glucose partitioning to nucleotide biosynthesis for T cell proliferation. Similarly, local lactate production by innate immune cells (Mehta et al., 2017; O’Neill et al., 2016) or tumor cells may feed Teff cells in inflamed tissues. Lactate appears to function within a tight dynamic range, with lower concentrations (0.5–3 mM) facilitating T cell metabolism and function but high lactate levels (>5 mM) suppressing T cell function.
Mechanistically, we show that Ldha is required for lactate metabolism in CD8+ T cells, regulating 13C-lactate-to-pyruvate conversion, subsequent fueling of the TCA cycle and ultimately CD8+ T cell proliferative responses in vivo. Based on our data, we hypothesize that the LDH complex is critical for regulating T cell metabolic homeostasis by maintaining NAD+:NADH balance, fueled in part through the bidirectional interconversion of pyruvate and lactate but also via maintenance of overall NAD+ levels. Without a compensatory increase in NAD+ production, we expect T cells with low LDH activity to decrease both glycolysis- and NAD+-dependent metabolic pathways due to decreased NAD+ abundance, which we observed. Our data suggest that LDH functions as a key metabolic node in CD8+ T cells, effectively coupling the cellular redox state with substrate availability to make critical decisions regarding fuel choice for T cell bioenergetics and biosynthesis.
In summary, our study highlights a previously underappreciated role for physiologic carbon substrates (including lactate) in fueling T cell metabolism and function. Our data highlight the ability of the TCA cycle to integrate multiple fuels to support bioenergetics, biosynthesis, and effector function in T cells. One implication is that specialized media approximating physiologic conditions, such as VIM (this study) or HPLM (Cantor et al., 2017), may be useful in identifying metabolic nodes relevant to T cell metabolism and function in vivo (MacPherson et al., 2022). A key observation of our study is that PCSs can directly enhance CD8+ T cell effector responses. Although the mechanism(s) underlying this phenomenon is the focus of future studies, the data provide evidence that non-glucose carbon sources can influence inflammatory CD8+ T cell function. This emphasizes the importance of better defining nutrient microenvironments to understand and tailor metabolic interventions, such as diet, to modulate T cell responses and improve protective immunity.
Limitations of study
While the results of our study agree with recent reports of fuel utilization in CD8+ T cells in vivo (Ma et al., 2019), there are several important caveats. First, we restricted our focus to major PCSs (>100 μM) that display high turnover fluxes in mice in vivo (Hui et al., 2017). It is possible that additional nutrients found at low micromolar concentrations in vivo not considered in our study may impact glucose utilization and/or serve as bioenergetic fuels, particularly when nutrients are limiting (Wang et al., 2020). In addition, oxygen availability, which has recently been shown to impact effector T cell dysfunction (Scharping et al., 2021), is an environmental factor that may influence glucose partitioning. Nutrient availability is highly influenced by inflammation (Lercher et al., 2019), and local availability of nutrients such as acetate (Balmer et al., 2016) may change over the course of infection or differ at sites of T cell priming (i.e., spleen and lymph nodes). Finally, we cannot rule out the effect of residual Ldha activity or compensation from Ldhb contributing to residual lactate metabolism in shLdha-expressing T cells.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Additional information and request for resources and reagents should be directed to and will be made available by the corresponding author, Russell G. Jones (russell.jones@vai.org).
Materials availability
All unique/stable reagents generated in this study will be made available from the Lead Contact with a completed Materials Transfer Agreement. Plasmids generated in this study will be deposited to Addgene.
Data and code availability
Unprocessed data underlying the display items in the manuscript, related to Figures 1, 2, 3, 4, 5, 6, and S1–S6, are provided in Data S1. Data tables for flux modeling in Figure 2 are shown in Table S2 (Flux equations used in INCA modeling). RNA sequencing data from Figure 2 has been deposited at NCBI GEO (GEO: GSE205002). Bioenergetics data analysis was based on protocols developed by Mookerjee and Brand (Mookerjee et al., 2017) which is available for download at https://russelljoneslab.vai.org/tools.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6, CD90.1 (Thy1.1+), and OT-I transgenic mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Mice were bred and maintained under specific pathogen-free conditions at VAI under approved protocols. Experiments were performed using mice between 8 and 12 weeks of age.
METHOD DETAILS
T cell purification and culture
For mouse T cell isolation, naive CD8+ T cells were purified from spleen and peripheral lymph nodes by negative selection (StemCell Technologies, Vancouver, BC), as previously described (Ma et al., 2019). Cells were cultured in Iscove’s Modified Delbecco’s Medium lacking sodium pyruvate (IMDM) or Van Andel Institute-modified Iscove’s Medium (VIM) supplemented with 10% dialyzed FBS (Wisent, St. Bruno, QC), penicillin-streptomycin (Invitrogen), and 2-ME (Sigma-Aldrich, St. Louis, MO). Glucose, L-glutamine, and PCS were added to cell culture media at concentrations indicated in Table S1. In vitro-activated CD8+ T cells were generated by stimulating naive CD8+ T cells (1 × 106 cells/mL) with plate-bound anti-CD3ε (clone 2C11) and anti-CD28 (clone 37.51) antibodies (eBioscience, San Diego, CA) and 50 U/mL IL2 (PeproTech, Rocky Hill, NJ) for 3 days. For ex vivo 13C tracing experiments, activated Thy1.1+ OT-I CD8+ T cells were isolated from the spleen of LmOVA-infected mice 3 days post infection (dpi) by magnetic bead isolation for Thy1.1 as described (Ma et al., 2019; Sheldon et al., 2021), followed by short-term (up to 4 h) cell culture in vitro. For retroviral transduction experiments, CD8+ Thy1.1+ OT-I T cells were transduced with retrovirus 24 h post activation and expanded for 2 additional days in IMDM containing IL-2. Transduced T cells were sorted by FACS Aria and cultured overnight prior to adoptive transfer into naive Thy1.2+ C57BL/6 hosts or cultured for 2 additional days in vitro prior to incubation in medium containing 13C tracers as described above.
Adoptive transfer and infection with L. monocytogenes
Mice were immunized intravenously with a sublethal dose of recombinant attenuated Listeria monocytogenes expressing OVA (LmOVA, 2 × 106 CFU), as previously described (Blagih et al., 2015; Ma et al., 2017). For adoptive transfer experiments using non-transduced or transduced cells, 5 × 103 CD8+ OT-I T cells (Thy1.1+) were injected intravenously into C57BL/6 mice, followed by LmOVA infection 1 day later. Splenocytes were isolated from mice 7 dpi and analyzed for the presence of OVA-specific CD8+ T cells by Thy1.1 (for adoptive transfer experiments of transduced T cells). Cytokine production by CD8+ T cells was analyzed by intracellular cytokine staining (ICS) following peptide re-stimulation (OVA257) as previously described (Blagih et al., 2015; Ma et al., 2017).
Stable isotope labeling (SIL) and metabolomics
For SIL experiments with in vitro-activated T cells, naive CD8+ T cells were activated as above, washed in IMDM or VIM containing 10% dialyzed FBS, and re-cultured (2.5 × 106 cells/well in 24-well plates) for indicated times (up to 24 h) in IMDM or VIM containing 13C-labeled metabolites (Cambridge Isotope Laboratories) added back to concentrations indicated in Table S1. For SIL experiments ex vivo, activated Thy1.1+ OT-I CD8+ T cells isolated from the spleen of LmOVA-infected mice 3 dpi were cultured (2.5 × 106 cells/well in 24-well plates) in VIM containing 5 mM [U-13C]-glucose or 2 mM [U-13C]-lactate for up to 4 h. Cells were transferred from tissue culture plates to falcon tubes and centrifuged at 500g 4°C for 3 min. The cell pellet was washed with ice-cold saline and centrifuged before being snap frozen on dry-ice and stored at −80°C. Metabolites were extracted by modified Bligh-Dyer extraction (Bligh and Dyer, 1959) by the addition of ice-cold methanol (A456, Fisher Scientific) directly to frozen cells, to which one volume of chloroform (A456, Fisher Scientific) was added. The sample was vortexed for 10 sec, incubated on ice for 30 min, and then 0.9 parts of LC/MS grade water (W6–4, Thermo Fisher Scientific) was added. The samples were vortexed vigorously and centrifuged at maximum speed to achieve phase separation. The top layer containing polar metabolites was aliquoted into a fresh tube and dried in a speedvac for LC/MS analysis. The bottom layer was retained for fatty acid methyl-ester measurement.
For LC-MS analysis, metabolite extracts were resuspended in 50 μL of 60% acetonitrile (A955, Fisher Scientific) and analyzed by high resolution accurate mass spectrometry using an ID-X Orbitrap mass spectrometer (Thermo Fisher Scientific) coupled to a Thermo Vanquish Horizon liquid chromatography system. 2 μL of sample volume was injected on column. Chromatographic separations were accomplished with Acquity BEH Amide (1.7 μm, 2.1 mm × 150 mm) analytical columns (#176001909, Waters, Eschborn, Germany) fitted with a pre-guard column (1.7 μm, 2.1mm × 5 mm; #186004799, Waters) using an elution gradient with a binary solvent system. Solvent A consisted of LC/MS grade water (W6–4, Fisher), and Solvent B was 90% LC/MS grade acetonitrile (A955, Fisher). For negative mode analysis, both mobile phases contained 10 mM ammonium acetate (A11450, Fisher Scientific), 0.1% (v/v) ammonium hydroxide, and 5 μM medronic acid (5191–4506, Agilent Technologies). For positive mode analysis, both mobile phases contained 10 mM ammonium formate (A11550, Fisher), and 0.1% (v/v) formic acid (A11710X1, Fisher). For both negative and positive mode analyses the 20-min analytical gradient at a flow rate of 400 μL/min was: 0–1.0 min ramp from 100% B to 90% B, 1.0–12.5 min from 90% B to 75% B, 12.5–19 min from 75% B to 60% B, and 19–20 min hold at 60% B. Following every analytical separation, the column was re-equilibrated for 20 min as follows: 0–1 min hold at 65%B at 400 μL/min, 1–3 min hold at 65% B and ramp from 400 μL/min to 800 μL/min, 3–14 min hold at 65% B and 800 μL/min, 14–14.5 min ramp from 65% B to 100% B at 800 μL/min, 14.5–16 min hold at 100% B and increase flow from 800 μL/min to 1200 μL/min, 16–18.4 min hold at 100% B at 1200 μL/min, 18.4–19.5 min hold at 100% B and decrease flow from 1200 μL to 400 μL/min, 19.5–20 min hold at 100% B and 400 μL/min. The column temperature was maintained at 40°C. The H-ESI source was operated at spray voltage of 2500 V(negative mode)/3500 V(positive mode), sheath gas: 60 a.u., aux gas: 19 a.u., sweep gas: 1 a.u., ion transfer tube: 300°C, vaporizer: 300°C. For isotopically labelled experimental replicates, high resolution MS1 data was collected with a 20-min full-scan method with m/z scan range using quadrupole isolation from 70 to 1000 m/z, mass resolution of 120,000 FWHM, RF lens at 35%, and standard automatic gain control (AGC). Unlabelled control samples were used for data dependent MS2 (ddMS2) fragmentation for compound identification and annotation via the AquireX workflow (Thermo Scientific). In this workflow, first blank and experimental samples are injected to generate exclusion and inclusion lists, respectively, followed by iterative sample injections for ddMS2 fragmentation where triggered ions are added to the exclusion list for subsequent injections. ddMS2 data was collected using MS1 resolution at 60,000, MS2 resolution at 30,000, intensity threshold at 2.0 × 104, and dynamic exclusion after one trigger for 10 s. MS2 fragmentation was completed first with HCD using stepped collision energies at 20, 35, and 50% and was followed on the next scan by CID fragmentation in assisted collision energy mode at 15, 30, and 45% with an activation Q of 0.25. Both MS2 scans used standard AGC and a maximum injection time of 54 ms. The total cycle time of the MS1 and ddMS2 scans was 0.6 s.
For metabolite profiling in culture medium, 40 μL of conditioned media were extracted with ice-cold 40% acetonitrile, 40% methanol, and 20% water and the supernatant was dried in a speedvac. Samples were reconstituted in 50 μL of water and analyzed by LC/MS using an ion-paired liquid chromatography as described previously (Ma et al., 2019) coupled to an Orbitrap Exploris 240 (Thermo Scientific) operated in ESI negative mode. The H-ESI source was operated at spray voltage of −2500 V, sheath gas: 60 a.u., aux gas: 19 a.u., sweep gas: 1 a.u., ion transfer tube: 300°C, vaporizer: 250°C. The mass spectrometer was operated in full scan mode from 70 to 800 m/z, mass resolution of 240,000 FWHM, RF lens at 35%, and standard automatic gain control (AGC).
Full scan LC-MS data were analyzed in Compound Discoverer (v 3.2, Thermo Scientific). Compounds were identified by chromatography specific retention time of external standards and MS2 spectral matching using the mzCloud database (Thermo Scientific).
TCA cycle intermediates were measured via GC-MS following LC-MS analysis. Briefly, following LC/MS extracts were dried and derivatized with 30 μL of methoxyamine (11.4 mg/mL) in pyridine and 70 μL of MTBSFA+1%TMCS as described previously (Ma et al., 2019). In addition, GC-MS was used to evaluate incorporation of 13C into de novo synthesized fatty acids from metabolic precursors using fatty acid methyl-esters (FAMEs). The bottom organic fraction from the Bligh-Dyer extraction (above) was aliquoted, dried in a speedvac, and FAMEs were generated as described previously (Griss et al., 2015). GC-MS analysis of both TBDMS derivatives and FAMEs were conducted on an Agilent 7890/5977b GC/MSD equipped with a DB-5MS+DG (30 m × 250 μm × 0.25 μm) capillary column (Agilent J&W, Santa Clara, CA, USA) was used. Data were collected by electron impact set at 70 eV. A total of 1 μL of the derivatized sample was injected in the GC in split mode (1:2 or 1:4) with inlet temperature set to 280°C, using helium as a carrier gas with a column flow rate of 1.2 mL/min. The oven program for all metabolite analyses started at 95°C for 1 min, increased at a rate of 40°C/min until 118°C and held for 2 min, then increased to 250°C at a rate of 12°C/min, then increased to 320°C at a rate of 40°C/min and finally held at 320°C for 7 min. The source temperature was 230°C, the quadrupole temperature was 150°C, and the GC/MS interface at 285°C. Data were acquired both in scan mode (50–800 m/z) and 2 Hz.
MassHunter software (v10, Agilent Technologies) was used for peak picking and integration of GC/MS data. Peak areas of all isotopologues for a molecular ion of each compound in both labeled experimental and unlabeled control samples were used for mass isotopologue distribution analysis via a custom algorithm developed at VAI. This algorithm uses matrices correcting for natural contribution of isotopologue enrichment and were generated for each metabolite as described previously (Fernandez et al., 1996; Trefely et al., 2016).
RNA sequencing
Total RNA was isolated from murine T cells following metabolite extraction using QIAzol (QIAGEN, Germantown, MD, USA). RNA preparation and library construction for RNA sequencing was conducted by the VAI Genomics Core as previously described (Roy et al., 2020). Libraries were sequenced using a HiSeq 2500 (Illumina) using 40 bp by 10 bp pair-end sequencing. Gene-set enrichment analysis (GSEA) on RNA-seq data was conducted using the gage function and non-parametric Kolmogorov–Smirnov test from the GAGE (version 2.22.0) R Bioconductor package (Luo et al., 2009). RNA-seq data has been deposited in NCBI GEO: GSE205002.
Metabolic flux analysis
Metabolic fluxes were calculated using mass isotopologue distributions (MIDs) of glycolytic (glucose, glucose-6-phosphate, fructose 6-phosphate, 3-phosphoglycerate, phosphoenolpyruvate, lactate) and TCA cycle (citrate, succinate, fumarate, malate, aspartate, glutamine and glutamate) metabolites with the INCA software package (Young, 2014) as previously described (Faubert et al., 2017). INCA applies an elementary metabolite unit framework to simulate MIDs. We developed reaction networks for central carbon metabolism (e.g., glycolysis and TCA cycle) to describe carbon stoichiometry and transitions, with the assumptions of the model summarized below. Fluxes through individual reactions were inferred by minimizing the sum of squared residuals to experimentally measured MIDs. To constrain the model, ratiometric flux values were made relative to citrate synthase, setting the flux value for CS as 1. Parameter sensitivity and 95% confidence intervals were then assessed for each flux value (Table S2).
Steady state (24 h, [U-13C]-glucose) MIDs were used to fit the model and estimate flux values. Flux estimations were initiated from random values and repeated a minimum of 50 times to ensure the identification of a global minimum of fluxes. MFA procedures and assumptions were as follows: 1) Cells are at metabolic and isotopic steady state; 2) 13CO2 is not reincorporated via carboxylation reactions; 3) Succinate and fumarate are symmetrical, and neither orientation is favored in the TCA cycle; 4) Metabolite extraction mixes the cytosolic and mitochondrial pools, and this is reflected in the isotopic analysis; and 5) All flux values are made relative to citrate synthase (CS), with CS set at an arbitrary value of 100. Goodness-of-fit was assessed by a chi-square test and 95% confidence intervals were calculated by assessing the sensitivity of the sum of squared residuals (SRR) to variations in the flux parameters. Table S2 contains the degrees of freedom and sum-of-squared residuals (SSR) for the best fit model and the lower and upper bounds of 95% confidence intervals for all fluxes.
Metabolic assays
T cell oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using a Seahorse XF96 Extracellular Flux Analyzer following established protocols (Ma et al., 2019). Activated T cells (2 × 105) were cultured in XF medium following centrifugation onto poly-D-lysine-coated XF96 plates, and cellular bioenergetics assessed at 5-min intervals following the sequential addition of oligomycin (2.0 μM), fluoro-carbonyl cyanide phenylhydrazone (FCCP, 2 μM), rotenone/antimycin A (2 μM), and monensin (10 mM). Data were normalized to cell number.
NAD+:NADH ratios were determined using the NAD/NADH-Glo Assay (Promega Corporation, Madison, WI) following previously described protocols (Luengo et al., 2021). In brief, 2×106 shRNA-transduced CD8+ T cells were cultured for 2 h in VIM containing 0 or 2 mM lactate, followed by centrifugation at 600g for 1 min and a quick wash with cold PBS. Cell pellets were resuspended in 100 μL of DTAB solution [0.5% dodecyltrimethylammonium bromide (DTAB) and 0.1 N NaOH in PBS], snap frozen on dry ice, and stored at −80°C. For the assay, thawed samples were diluted 1:10 in DTAB solution (20 μL final volume). To measure NADH, samples were incubated at 75°C for 30 min, allowed to cool to room temperature, and then quenched with 20 μL of a 1:1 mixture of 0.5 M Tris base and 0.4 M HCl. To measure NAD+, 20 μL of DTAB solution and 20 μL of 0.4 M HCl were added to each sample. The NAD+ samples were then incubated at 60°C for 15 min, allowed to cool to room temperature, and quenched with 20 μL of 0.5 M Tris base. A master mix of luciferin detection reagent (prepared according to the manufacturer’s protocol) was added 1:1 with each NAD+ or NADH sample in a white-walled 96-well plate and incubated at room temperature for 45 min. Luminescence was detected using a BioTek Synergy Neo2 plate reader. NAD+:NADH ratios were calculated by the following: (NAD-blank)/(NADH-blank)*2 (the multiplication by 2 is to correct for dilution differences between the NAD+ and NADH samples).
Flow cytometry
Single-cell suspensions were surface stained with fluorescently conjugated antibodies listed in key resources table. Cell viability was assessed by using Fixable Viability Dye eFluor 780 (eBioscience) according to manufacturer’s protocols. Cell proliferation was assessed by CellTrace Violet dye dilution or Ki67 staining following manufacturer’s protocols (Thermo Fisher). For p-S6 staining, cells were incubated in 1×Lyse/Fix Buffer (BD Biosciences) at 37°C for 10min, permeabilized with cold Perm Buffer III (BD Biosciences)on ice for 30 min, and then stained with anti-p-S6 antibody for 1 hr at room temperature. For intracellular cytokine staining(ICS)for IFN-γ and TNF-α, T cells were stimulated in vitro with PMA (50 ng/mL) and ionomycin (500 ng/mL) for 2 h, followed by treatment with Brefeldin A for 2 h. After restimulation, cells were surface stained, fixed, and permeabilized using Foxp3/Transcription Factor Staining Buffer Set (eBioscience), stained for intracellular proteins, and analyzed by flow cytometry using an Aurora Cytek cytometer. Data analysis was performed using FlowJo (Tree Star). Fluorescence-activated cell sorting (FACS) was performed on an BD FACSAria Fusion cell sorter (BD Biosciences).
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Hamster monoclonal anti-mouse CD3 | eBioscience | Clone 145-2C11 |
| Hamster monoclonal anti-mouse CD28 | eBioscience | Clone 37.51 |
| Rat monoclonal anti-mouse IFN-gamma | eBioscience | Clone AN-18 |
| Rat monoclonal anti-mouse CD4 | eBioscience | Clone GK1.5 |
| Rat monoclonal anti-mouse IFN-gamma | eBioscience | Clone XMG1.2 |
| Rat monoclonal anti-mouse CD8 | eBioscience | Clone 53-6.7 |
| Rat monoclonal anti-mouse TNF-α | eBioscience | Clone MP6-XT22 |
| Rat monoclonal anti-mouse Granzyme B | eBioscience | Clone NGZB |
| Rat monoclonal anti-mouse CD44 | eBioscience | Clone IM7 |
| Hamster monoclonal anti-mouse CD69 | eBioscience | Clone H1.2F3 |
| Hamster monoclonal anti-mouse KLRG1 | eBioscience | Clone 2F1 |
| Anti-mouse CX3CR1 | BioLegend | Clone SA011F11 |
| Anti-mouse CD127 (IL-7R) | BioLegend | Clone A7R34 |
| Anti-mouse CD279 (PD-1) | BD Biosciences | Clone RMP1-30 |
| Anti-mouse Ly108 (Slamf6) | BioLegend | Clone 330-AJ |
| Anti-mouse Tim-3 | BioLegend | Clone RMT3-23 |
| Anti-human/mouse TOX | Miltenyi Biotec | Clone REA473 |
| Anti-human/mouse T-bet | BioLegend | Clone 4B10 |
| Rabbit monoclonal anti-mouse TCF1 | Cell Signaling | Clone C63D9 |
| Mouse monoclonal anti-mouse CD90.1 (Thy-1.1) | eBioscience | Clone HIS51 |
| Rabbit polyclonal anti-human/mouse beta-actin | Cell Signaling | #4967 |
| Rabbit polyclonal anti-Ldha | Cell Signaling | #2012 |
| Rat monoclonal anti-mouse Ki67 | eBioscience | Clone SolA15 |
| Rabbit monoclonal anti-phospho-S6 ribosomal protein (Ser235/236) | Cell Signaling | #8520 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Luciferase shRNA (LMPd-Amt backbone): AGCTCCCGTGAATTGGAATCCTAGTGAAGCCACAGAT GTAGGATTCCAATTCAGCGGGAGCC | vector backbone (Chen et al., 2014) | custom |
| Ldha shRNA1 (LMPd-Amt backbone): TGCTGTTGACAGTGAGCGAACTCAATTTGGTCCAGCGAAATAGTGAAGCCACAGATGTATTTCGCTGGACCAAATTGAGTCTGCCTACTGCCTCGGA | This paper | custom |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| D-glucose [13C6] | Cambridge Isotopes | CLM-1396 |
| L-glutamine [13C5] | Cambridge Isotopes | CLM-1822 |
| sodium acetate [13C2] | Cambridge Isotopes | CLM-440 |
| sodium D-3-hydroxybutyrate [13C4] | Cambridge Isotopes | CLM-3853 |
| citric acid [13C6] | Sigma Aldrich | 606081 |
| sodium lactate [13C3] | Cambridge Isotopes | CLM-10768 |
| sodium pyruvate [13C3] | Cambridge Isotopes | CLM-2440 |
| L-alanine [13C3] | Cambridge Isotopes | CLM-2184 |
| IMDM GlutaMAX | Thermo Fisher | 3198-022 |
| Heat-inactivated FCS | Biochrom | S0115– Lot# 1289W |
| Penicillin-streptomycin | Millipore | A2213 |
| 2-mercaptoethanol | Gibco | #21985-023 |
| Recombinant murine IL-2 | Peprotech | 212-12 |
| Phorbol 12-myristate 13-acetate (PMA) | Sigma-Aldrich | P-8139 |
| Ionomycin | Sigma-Aldrich | 10634 |
| Glucose ≥99,5 % D(+) | Roth | HN06.1 |
| L-Glutamine | Biochrom AG | M11-004 |
| Sodium Pyruvate | Gibco | #11360-070 |
| Fixable Viability Dye eFluor 780 | eBioscience | #65-0865-14 |
| CellTrace Violet Cell Proliferation Kit | Thermo Fisher | C34557 |
| BD GolgiStop | BD Biosciences | #51-2092KZ |
| Foxp3/transcription factor staining buffer set | eBioscience | #00-5523-00 |
| Lyse/Fix Buffer, 5X | BD Biosciences | 558049 |
| Perm Buffer III | BD Biosciences | 558050 |
| 16% Formaldehyde | Polysciences | 18814-20 |
| Hexadimethrine bromide (Polybrene) | Sigma-Aldrich | 107689 |
| OVA(257-264) SIINFEKL peptide | Bio-Synthesis | custom |
|
| ||
| Critical commercial assays | ||
|
| ||
| EasySep Mouse Naïve CD8+ T cell isolation kit | StemCell technologies | #19858 |
| EasySep Mouse CD90.1 positive selection kit | StemCell technologies | #18958 |
| Qiagen RNAeasy Kit | Qiagen | #74106 |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Roche | 11873580001 |
| Seahorse XFe96 FluxPak | Agilent technologies | #102416-100 |
| NAD:NADH-Glo Assay | Promega | G9071 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| 293T | ATCC | CRL-3216 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6J mice | Charles River Laboratories | CR:027 |
| OT-I mice | Jackson Laboratories | JAX:003831 |
| CD90.1 (Thy.1.1) mice | Jackson Laboratories | JAX:000406 |
|
| ||
| Software and algorithms | ||
|
| ||
| FlowJo 9.9.5 | FlowJo LLC | https://www.flowjo.com/ |
| GraphPad Prism V6 or V7 | GraphPad Software | https://www.graphpad.com/ |
| El-Maven | Elucidata | https://elucidatainc.github.io/ElMaven/ |
| Compound Discoverer V3.2 | Thermo Scientific | https://www.thermofisher.com/ |
| Mass Hunter V10 | Agilent Technologies | https://www.agilent.com |
Immunoblotting
Cells were lysed in modified Laemmli lysis buffer (240 mM Tris/HCl pH 6.8, 40% glycerol, 8% SDS, 5% 2-ME) supplemented with protease and phosphatase inhibitors (Roche/Sigma-Aldrich). Protein from whole cell lysates was quantified using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). Lysates were resolved by SDS-PAGE, transferred to nitrocellulose, and incubated with primary antibodies to Ldha or actin and HRP-conjugated secondary antibodies (Cell Signaling Technology, Danvers, MA).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as mean ± SD for technical replicates or mean ± SEM for biological replicates and were analyzed using unpaired Student’s t test (assuming unequal variance) or One-Way ANOVA. Statistical significance is indicated in all figures by the following annotations: *, p < 0.05; **, p < 0.001; ***, p < 0.0001.
Supplementary Material
Highlights.
Environmental physiologic carbon sources (PCSs) alter glucose usage by CD8+ T cells
PCSs enhance CD8+ T cell bioenergetics, survival, and cytokine production
Lactate is a physiologic TCA cycle fuel for CD8+ T cells
Ldha regulates CD8+ T cell metabolism and effector responses in vivo
ACKNOWLEDGMENTS
We acknowledge Jason Cantor, Connie Krawczyk, and Julian Lum for scientific discussions contributing to this manuscript. We thank Sarah Chang, Hongbo Chi, Peter Crawford, Evan Lien, Patrycja Puchalska, and Matthew Vander Heiden for technical assistance. We thank members of the VAI Core Facilities (Metabolomics and Bioenergetics, Genomics, Bioinformatics and Biostatistics, and Flow Cytometry) for technical assistance, and Jeanie Wedberg and Michelle Minard for administrative assistance. M.D. is supported by a Postdoctoral Fellowship Award from the Fonds de la Recherche du Québec—Santé (FRQS). J.L. is supported by a VAI Metabolism & Nutrition (MeNu) Program Pathway-to-Independence Award. M.J.W. is supported by a National Cancer Institute (NCI) T32 training grant (T32CA251066-01A1). R.J.D. is supported by the HHMI Investigator Program and the NCI (R35CA22044901). R.G.J. is supported by the Paul G. Allen Frontiers Group Distinguished Investigator Program, the National Institute of Allergy and Infectious Diseases (NIAID, R01AI165722), and the Van Andel Institute (VAI).
Footnotes
DECLARATION OF INTERESTS
R.J.D. is a founder and consultant for Atavistik Biosciences and an advisor for Agios Pharmaceuticals, Nirogy Therapeutics, and Vida Ventures. R.G.J. is a scientific advisor for Agios Pharmaceuticals and Servier Pharmaceuticals and is a member of the Scientific Advisory Board of Immunomet Therapeutics.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cmet.2022.07.012.
REFERENCES
- Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, Wang Z, Quinn WJ, Kopinski PK, Wang L, et al. (2017). Foxp3 reprograms T cell metabolism to function in low-glucose, high lactate environments. Cell Metab. 25, 1282–1293.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argüello RJ, Combes AJ, Char R, Gigan JP, Baaziz AI, Bousiquot E, Camosseto V, Samad B, Tsui J, Yan P, et al. (2020). SCENITH: a flow cytometry-based method to functionally profile energy metabolism with single-cell resolution. Cell Metab. 32, 1063–1075.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badovinac VP, Porter BB, and Harty JT (2004). CD8+ T cell contraction is controlled by early inflammation. Nat. Immunol. 5, 809–817. [DOI] [PubMed] [Google Scholar]
- Balmer ML, Ma EH, Bantug GR, Gärhlert J, Pfister S, Glatter T, Jauch A, Dimeloe S, Slack E, Dehio P, et al. (2016). Memory CD8+ T cells require increased concentrations of acetate induced by stress for optimal function. Immunity 44, 1312–1324. [DOI] [PubMed] [Google Scholar]
- Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, Casella V, Ngiow SF, Khan O, Huang YJ, et al. (2020). Developmental relationships of four exhausted CD8 + T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity 52, 825–841.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, and Sabatini DM (2014). Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 508, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vázquez G, Yurchenko E, Raissi TC, van der Windt GJW, Viollet B, Pearce EL, et al. (2015). The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 42, 41–54. [DOI] [PubMed] [Google Scholar]
- Bligh EG, and Dyer WJ (1959). A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917. [DOI] [PubMed] [Google Scholar]
- Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, Matos C, Bruss C, Klobuch S, Peter K, et al. (2016). LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 24, 657–671. [DOI] [PubMed] [Google Scholar]
- Buck MD, Sowell RT, Kaech SM, and Pearce EL (2017). Metabolic instruction of immunity. Cell 169, 570–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor JR (2019). The rise of physiologic media. Trends Cell Biol. 29, 854–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor JR, Abu-Remaileh M, Kanarek N, Freinkman E, Gao X, Louissaint A, Lewis CA, and Sabatini DM (2017). Physiologic medium rewires cellular metabolism and reveals uric acid as an endogenous inhibitor of UMP synthase. Cell 169, 258–272.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cham CM, and Gajewski TF (2005). Glucose availability regulates IFN-gamma production and p70S6 kinase activation in CD8+ effector T cells. J. Immunol. 174, 4670–4677. [DOI] [PubMed] [Google Scholar]
- Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, et al. (2013). Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman NM, and Chi H (2022). Metabolic adaptation of lymphocytes in immunity and disease. Immunity 55, 14–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Bélanger S, Frederick MA, Li B, Johnston RJ, Xiao N, Liu YC, Sharma S, Peters B, Rao A, Crotty S, and Pipkin ME (2014). In vivo RNA interference screens identify regulators of antiviral CD4(+) and CD8(+) T cell differentiation. Immunity 41, 325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O’Brien JP, Pierce KA, et al. (2016). Environment impacts the metabolic dependencies of Ras-driven non-small cell lung cancer. Cell Metab. 23, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, and Chandel NS (2020). We need to talk about the Warburg effect. Nat. Metab. 2, 127–129. [DOI] [PubMed] [Google Scholar]
- Everts B, Amiel E, Huang SCC, Smith AM, Chang CH, Lam WY, Redmann V, Freitas TC, Blagih J, Van Der Windt GJW, et al. (2014). TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nat. Immunol. 15, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, et al. (2017). Lactate metabolism in human lung tumors. Cell 171, 358–371.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez CA, Des Rosiers C, Previs SF, David F, and Brunengraber H (1996). Correction of 13C mass isotopomer distributions for natural stable isotope abundance. J. Mass Spectrom. 31, 255–262. [DOI] [PubMed] [Google Scholar]
- Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, et al. (2007). Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 109, 3812–3819. [DOI] [PubMed] [Google Scholar]
- Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, Elstrom RL, June CH, and Thompson CB (2002). The CD28 signaling pathway regulates glucose metabolism. Immunity 16, 769–777. [DOI] [PubMed] [Google Scholar]
- Griss T, Vincent EE, Egnatchik R, Chen J, Ma EH, Faubert B, Viollet B, DeBerardinis RJ, and Jones RG (2015). Metformin antagonizes cancer cell proliferation by suppressing mitochondrial-dependent biosynthesis. PLoS Biol. 13, e1002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett SR, Zanotelli VRT, Xu W, Goya J, Park JO, Perlman DH, Gibney PA, Botstein D, Storey JD, and Rabinowitz JD (2016). Systems-level analysis of mechanisms regulating yeast metabolic flux. Science 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann FJ, Mrdjen D, McCaffrey E, Glass DR, Greenwald NF, Bharadwaj A, Khair Z, Verberk SGS, Baranski A, Baskar R, et al. (2020). Single-cell metabolic profiling of human cytotoxic T cells. Nat. Biotechnol. 39, 186–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, et al. (2016). Metabolic heterogeneity in human lung tumors. Cell 164, 681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans D, Gautam S, García-Cañaveras JC, Gromer D, Mitra S, Spolski R, Li P, Christensen S, Nguyen R, Lin J-X, et al. (2020). Lactate dehydrogenase inhibition synergizes with IL-21 to promote CD8+ T cell stemness and antitumor immunity. Proc. Natl. Acad. Sci. USA 117, 6047–6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Le Zhan L, Yanxiang Guo J, et al. (2017). Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui S, Cowan AJ, Zeng X, Yang L, TeSlaa T, Li X, Bartman C, Zhang Z, Jang C, Wang L, et al. (2020). Quantitative fluxomics of circulating metabolites. Cell Metab. 32, 676–688.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C, Chen L, and Rabinowitz JD (2018). Metabolomics and isotope tracing. Cell 173, 822–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, and Cui W (2012). Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 12, 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaymak I, Williams KS, Cantor JR, and Jones RG (2021). Immunometabolic interplay in the tumor microenvironment. Cancer Cell 39, 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klarquist J, Chitrakar A, Pennock ND, Kilgore AM, Blain T, Zheng C, Danhorn T, Walton K, Jiang L, Sun J, et al. (2018). Clonal expansion of vaccine-elicited T cells is independent of aerobic glycolysis. Sci. Immunol. 3, 9822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, and Pearce EJ (2010). Toll-like receptor–induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115, 4742–4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leney-Greene MA, Boddapati AK, Su HC, Cantor J, and Lenardo MJ (2019). Human plasma-like medium improves T lymphocyte activation. Preprint at bioRxiv. 10.1101/740845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lercher A, Bhattacharya A, Popa AM, Caldera M, Schlapansky MF, Baazim H, Agerer B, Gu€rtl B, Kosack L, Má jek P, et al. (2019). Type I interferon signaling disrupts the hepatic urea cycle and alters systemic metabolism to suppress T cell function. Immunity 51, 1074–1087.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine LS, Hiam-Galvez KJ, Marquez DM, Tenvooren I, Madden MZ, Contreras DC, Dahunsi DO, Irish JM, Oluwole OO, Rathmell JC, and Spitzer MH (2021). Single-cell analysis by mass cytometry reveals metabolic states of early-activated CD8+ T cells during the primary immune response. Immunity 54, 829–844.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Hui S, Mirek ET, Jonsson WO, Anthony TG, Lee WD, Zeng X, Jang C, and Rabinowitz JD (2022). Circulating metabolite homeostasis achieved through mass action. Nat. Metab. 4, 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luengo A, Li Z, Gui DY, Sullivan LB, Zagorulya M, Do BT, Ferreira R, Naamati A, Ali A, Lewis CA, et al. (2021). Increased demand for NAD + relative to ATP drives aerobic glycolysis. Mol. Cell 81, 691–707.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Friedman MS, Shedden K, Hankenson KD, and Woolf PJ (2009). GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics 10, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, Mainolfi N, Suri V, Guak H, Balmer ML, et al. (2017). Serine is an essential metabolite for effector T cell expansion. Cell Metab. 25, 345–357. [DOI] [PubMed] [Google Scholar]
- Ma EH, Verway MJ, Johnson RM, Roy DG, Steadman M, Hayes S, Williams KS, Sheldon RD, Samborska B, Kosinski PA, et al. (2019). Metabolic profiling using stable isotope tracing reveals distinct patterns of glucose utilization by physiologically activated CD8+ T cells. Immunity 51, 856–870.e5. [DOI] [PubMed] [Google Scholar]
- Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, and Rathmell JC (2014). The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 20, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson S, Keyes S, Kilgour MK, Smazynski J, Chan V, Sudderth J, Turcotte T, Devlieger A, Yu J, Huggler KS, et al. (2022). Clinically relevant T cell expansion media activate distinct metabolic programs uncoupled from cellular function. Mol. Ther. Methods Clin. Dev. 24, 380–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta MM, Weinberg SE, and Chandel NS (2017). Mitochondrial control of immunity: beyond ATP. Nat. Rev. Immunol. 17, 608–620. [DOI] [PubMed] [Google Scholar]
- Mookerjee SA, Gerencser AA, Nicholls DG, and Brand MD (2017). Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem. 292, 7189–7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir A, Danai LV, and Vander Heiden MG (2018). Microenvironmental regulation of cancer cell metabolism: implications for experimental design and translational studies. Dis. Model. Mech 11, dmm035758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LAJ, Kishton RJ, and Rathmell J (2016). A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 16, 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obar JJ, Jellison ER, Sheridan BS, Blair DA, Pham Q-M, Zickovich JM, and Lefranç ois L (2011). Pathogen-induced inflammatory environment controls effector and memory CD8 + T cell differentiation. J. Immunol. 187, 4967–4978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olenchock BA, Rathmell JC, and Vander Heiden MG (2017). Biochemical underpinnings of immune cell metabolic phenotypes. Immunity 46, 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn WJ, Jiao J, TeSlaa T, Stadanlick J, Wang Z, Wang L, Akimova T, Angelin A, Schäfer PM, Cully MD, et al. (2020). Lactate limits T cell proliferation via the NAD(H) redox state. Cell Rep. 33, 108500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambotti P, and Davis S (1981). Lactic dehydrogenase in normal and leukemia lymphocyte subpopulations: evidence for the presence of abnormal T cells and B cells in chronic lymphocytic leukemia. Blood 57, 324–327. [PubMed] [Google Scholar]
- Ringoir S, and Plum J (1975). LDH isozymes of human T and B lymphocytes. Clin. Chim. Acta 60, 379–383. [DOI] [PubMed] [Google Scholar]
- Ron-Harel N, Ghergurovich JM, Notarangelo G, LaFleur MW, Tsubosaka Y, Sharpe AH, Rabinowitz JD, and Haigis MC (2019). T cell activation depends on extracellular alanine. Cell Rep. 28, 3011–3021.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossiter NJ, Huggler KS, Adelmann CH, Keys HR, Soens RW, Sabatini DM, and Cantor JR (2021). CRISPR screens in physiologic medium reveal conditionally essential genes in human cells. Cell Metab. 33, 1248–1263.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy DG, Chen J, Mamane V, Ma EH, Muhire BM, Sheldon RD, Shorstova T, Koning R, Johnson RM, Esaulova E, et al. (2020). Methionine metabolism shapes T helper cell responses through regulation of epigenetic reprogramming. Cell Metab. 31, 250–266.e9. [DOI] [PubMed] [Google Scholar]
- Scharping NE, Rivadeneira DB, Menk AV, Vignali PDA, Ford BR, Rittenhouse NL, Peralta R, Wang Y, Wang Y, DePeaux K, and Delgoffe GM (2021). Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 22, 205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine T, Perez-Potti A, Nguyen S, Gorin JB, Wu VH, Gostick E, Llewellyn-Lacey S, Hammer Q, Falck-Jones S, Vangeti S, et al. (2020). TOX is expressed by exhausted and polyfunctional human effector memory CD8 + T cells. Sci. Immunol 5, eaba7918. [DOI] [PubMed] [Google Scholar]
- Sheldon RD, Ma EH, DeCamp LM, Williams KS, and Jones RG (2021). Interrogating in vivo T-cell metabolism in mice using stable isotope labeling metabolomics and rapid cell sorting. Nat. Protoc. 16, 4494–4521. [DOI] [PubMed] [Google Scholar]
- Sugimoto M, Ikeda S, Niigata K, Tomita M, Sato H, and Soga T (2012). MMMDB: mouse multiple tissue metabolome database. Nucleic Acids Res. 40, D809–D814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, Dennstedt EA, Vander Heiden MG, and Muir A (2019). Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife 8, e44235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdogan A, Faubert B, Ramesh V, Ubellacker JM, Shen B, Solmonson A, Murphy MM, Gu Z, Gu W, Martin M, et al. (2020). Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 577, 115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeiro E, Daniels MA, Hamilton SE, Schrum AG, Bragado R, Jameson SC, and Palmer E (2009). Different T cell receptor signals determine CD8+ memory versus effector development. Science 323, 502–505. [DOI] [PubMed] [Google Scholar]
- Trefely S, Ashwell P, and Snyder NW (2016). FluxFix: automatic isotopologue normalization for metabolic tracer analysis. BMC Bioinformatics 17, 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tscharke DC, Croft NP, Doherty PC, and La Gruta NL (2015). Sizing up the key determinants of the CD8+ T cell response. Nat. Rev. Immunol. 15, 705–716. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, and Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Voorde J, Ackermann T, Pfetzer N, Sumpton D, Mackay G, Kalna G, Nixon C, Blyth K, Gottlieb E, and Tardito S (2019). Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci. Adv. 5, eaau7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Dillon CPP, Shi LZZ, Milasta S, Carter R, Finkelstein D, McCormick LLL, Fitzgerald P, Chi H, Munger J, and Green DR (2011). The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Gnanaprakasam JNR, Chen X, Kang S, Xu X, Sun H, Liu L, Rodgers H, Miller E, Cassel TA, et al. (2020). Inosine is an alternative carbon source for CD8+-T-cell function under glucose restriction. Nat. Metab. 2, 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson MJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, Menk AV, Rittenhouse NL, DePeaux K, Whetstone RD, et al. (2021). Metabolic support of tumor-infiltrating regulatory T cells by lactic acid. Nature 591, 645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MA, and Bevan MJ (2007). Effector and memory CTL differentiation. Annu. Rev. Immunol. 25, 171–192. [DOI] [PubMed] [Google Scholar]
- Xu K, Yin N, Peng M, Stamatiades EG, Shyu A, Li P, Zhang X, Do MH, Wang Z, Capistrano KJ, et al. (2021). Glycolysis fuels phosphoinositide 3-kinase signaling to bolster T cell immunity. Science 371, 405–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao C, Sun HW, Lacey NE, Ji Y, Moseman EA, Shih HY, Heuston EF, Kirby M, Anderson S, Cheng J, et al. (2019). Single-cell RNA-seq reveals TOX as a key regulator of CD8+ T cell persistence in chronic infection. Nat. Immunol. 20, 890–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JD (2014). INCA: a computational platform for isotopically non-stationary metabolic flux analysis. Bioinformatics 30, 1333–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Unprocessed data underlying the display items in the manuscript, related to Figures 1, 2, 3, 4, 5, 6, and S1–S6, are provided in Data S1. Data tables for flux modeling in Figure 2 are shown in Table S2 (Flux equations used in INCA modeling). RNA sequencing data from Figure 2 has been deposited at NCBI GEO (GEO: GSE205002). Bioenergetics data analysis was based on protocols developed by Mookerjee and Brand (Mookerjee et al., 2017) which is available for download at https://russelljoneslab.vai.org/tools.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.