

RNA editing has long been known to play a critical role in curtailing the immune’s early response to foreign RNA viruses, which is also referred to as innate immunity. This process of editing cellular premRNAs as endogenous (self) RNAs is instrumental for self-RNA to be distinguished from foreign viral (nonself) RNAs. Double-stranded RNA I editors, such as ADAR1 (adenosine deaminase acting on RNA 1), are at the heart of this process as they catalyze unwinding and adenosine-to-inosine (A-to-I) conversion of the RNA sequence.1 It is ADAR enzymatic/editing functions that are thought to confer innate immunity as the conversion of A to I is interpreted as guanosine (G) upon translation.1 This functional A-to-G substitution in RNA is what marks the RNA as self RNA and prevents the upregulation of cytoplasmic immune-related RNA sensors such as MDA5 (melanoma differentiation-associated protein 5) and downstream inflammatory (interferon-based) pathways to trigger an immune response (Figure). Studies using global ADAR1-deficient mice have highlighted the functional importance of ADAR1 in mouse embryonic viability and cell survival in numerous organs, including the heart.1,2 ADAR1-deficient mice targeting the embryonic (Nkx2.5-Cre) and adult (α-MHCMerCreMer) heart have more specifically shown the importance of ADAR1 in cardiomyocyte survival as cardiomyocyte apoptosis was observed in these settings.3,4 Loss of ADAR1 in embryonic cardiomyocytes resulted in reduction of trabeculation in the embryonic heart (due to loss of cardiomyocyte proliferation and presence of apoptosis), while loss in adult cardiomyocytes resulted in lack of endoplasmic reticulum stress response and survival (presence of apoptosis) resulting in cardiomyopathy and rapid premature death in mice, respectively.3,4 However, the mechanisms driving cardiomyocyte cell survival deficits in adult ADAR1 deficient mice and whether innate immunity is integral in these settings remained unclear. In this issue of Circulation Research, Garcia-Gonzalez et al5 provide new insight to the field as they showcase editor dependent and independent functions of ADAR1, which target cardiac innate immunity to drive cardiomyopathy and premature death in adult ADAR1-deficient mice.
Figure. Canonical and noncanonical role of ADAR1 (adenosine deaminase acting on RNA 1), in mediating autoinflammatory cardiomyopathy.
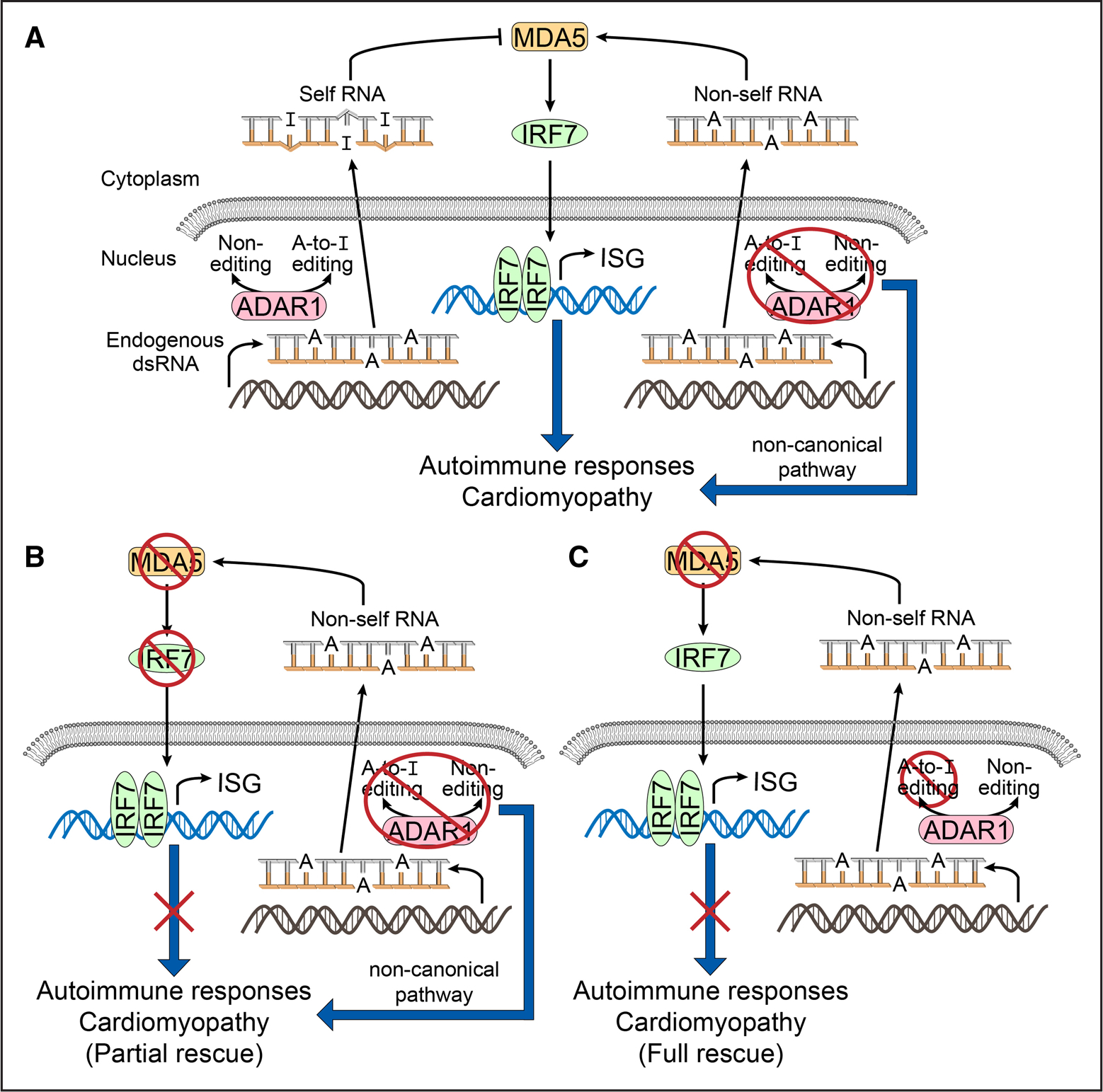
A, left, In the normal heart, endogenous RNA is recognized as “self” by ADAR1 mediated A-to-I editing. Thus, “self” dsRNA is not “sensed” by canonical pathways associated with the RNA sensor, MDA5 (melanoma differentiation-associated protein 5) to trigger immune activation via IRF7 (interferon regulatory factor 7) and interferon stress responsive (ISF) gene activation. A, right, ADAR1-deficient cardiomyocytes activate both canonical (via MDA5) and noncanonical pathways (undetermined) to trigger autoimmune cardiomyopathy/myocarditis and premature death in mice. B, Noncanonical pathways of ADAR1 were identified when loss of Ifih1 (gene coding for MDA5) and loss of Irf7 (gene coding for IRF7) could only partially rescue/attenuate the autoimmune cardiomyopathy/myocarditis and premature death in cardiomyocyte-specific ADAR1-deficient mice, resulting in late-onset cardiomyopathy. C, Canonical pathways of ADAR1 were identified when mice expressing a catalytically inactive ADAR1 (disrupted RNA editing functions) and loss of Ifih1 (gene coding for MDA5) resulted in the absence (full rescue) of autoimmune cardiomyopathy/myocarditis and premature death.
Through an elegant series of compound mutant mouse models targeting cardiac-specific loss of ADAR1 or enzymatically deficient ADAR1, Garcia-Gonzalez et al5 highlight a critical role for editor and RNA sensor (MDA5-interferon 7 (Irf7)) dependent (canonical) and independent (noncanonical) pathways driving ADAR1-based cardiac innate immunity and disease in adult mice in vivo. To circumvent the embryonic lethality using Nkx2.5-Cre as well as rapid premature death in adult heart using α-MHCMerCreMer,3,4 the authors have used a cardiomyocyte Cre driver (α-MHC-Cre) that focuses on the early postnatal period to drive ADAR1 loss in the postnatal heart.5 Their studies revealed that cardiomyocyte ADAR1 was dispensable for early postnatal mouse survival, however, was essential for late onset cardiomyocyte innate immunity protection, function and survival of adult mice.5
A critical difference between the Garcia-Gonzalez et al study and previous studies focused on cardiac ablation of ADAR1 is the timing of ADAR-1 ablation in mice. The authors utilized the α-MHC-Cre driver to constitutively ablate ADAR1 primarily within early postnatal cardiomyocytes (with transient effects in the embryo), which resulted in postnatal viability of mice.5,6 These findings were distinct from a previous study utilizing the Nkx2.5-Cre to constitutive deplete ADAR1 as it resulted in embryonic lethality.3 Given that Nkx2.5 may target other cell types in the heart and embryo,7 Garcia-Gonzalez et al5 exploited the cardiomyocyte restricted, Xenopus laevis myosin light chain-2-Cre (XMLC2-Cre) to drive ADAR1 loss in the embryonic heart, which also resulted in postnatal viability of mice, suggesting that noncardiomyocyte effects of the Nkx2.5 promoter may be responsible for driving embryonic lethality. Key findings bridging this study to a previous mouse model focused on postnatal deletion of ADAR1,4 are that Garcia-Gonzalez et al5 also demonstrate that postnatal loss of ADAR1 drives cardiomyopathy and premature death of mice. These data highlight the importance of RNA editing machinery in postnatal cardiomyocyte survival and function. Interestingly, the mechanisms likely differ between studies as the authors show that postnatal cardiomyocyte ADAR1 loss of function effects were independent of effects on cardiomyocyte cytotoxicity (apoptosis), cytokine storm (upregulation of NFkB signaling) and engagement of endoplasmic reticulum-stress responsive pathways.5 This could likely relate to the timing of ADAR1 ablation (12 weeks old mice) and severity of the previous mouse model, where mice exhibited premature death < 3 weeks after tamoxifen injection/ADAR1 gene ablation.4 Instead, Garcia-Gonzalez et al5 have mechanistically reinforced the sufficiency of ADAR1 in the cardiac innate immunity response during the postnatal period as the expression of inflammatory markers, including IRF7 (interferon regulatory 7) and interferon-stress genes were upregulated, and found to precede late onset of cardiac dysfunction, failure, and premature death. These studies altogether highlight the autoinflammatory response of the postnatal cardiomyocyte to even low level alterations in RNA editing, which appears to be dynamically regulated during postnatal cardiac development.
To determine if the RNA editing functions of ADAR1 drive cardiac inflammatory pathways and disease, Garcia-Gonzalez et al5 studied outcomes from a compound mutant mouse model that harbored germline expression of a catalytically inactive ADAR1 in the setting of germline loss of the RNA sensor, MDA5 (Adar1ki/ki;Ifih1−/−). This would essentially prevent the cell from detecting foreign nonedited/self RNA and thus, no downstream inflammatory responses would be triggered. True to the hypothesis, Garcia-Gonzalez et al5 showed that hearts from this model did not display an autoinflammatory response and did not exhibit cardiomyopathy resulting in a normal lifespan (>50 weeks), suggesting that RNA editing (canonical) functions of ADAR1 likely drive the autoinflammatory response in the heart. Only when determining whether the RNA sensor, MDA5 could rescue the phenotypes driven by postnatal cardiomyocyte specific-loss of ADAR1 did the authors reveal that ADAR1 harbored noncanonical functions. Two compound mutant mouse models that combined postnatal cardiomyocyte-specific loss of ADAR1 (ADAR1 cm-KO) with loss of MDA5 (Adar1 cm-cKO; Ifih1−/−) or loss of IRF7, downstream of the MDA5 pathway (Adar1 cm-cKO; Irf7−/−) were instrumental to these findings.5 Both models demonstrated that they could not completely rescue but significantly attenuated cardiac inflammation and cardiomyopathy as well as prolonged median lifespan of the ADAR1 cm-KO mouse by 6 weeks (Adar1 cm-cKO; Ifih1−/−) and 10 weeks (Adar1 cm-cKO; Irf1−/−), respectively.5 In contrast to classic inflammatory signaling pathways (eg., NFkB, TNF-α, IL-6, etc) found immediately upregulated in ADAR1 deficient cells and associated with cytotoxic effects,8,9 the auto-inflammatory actions of ADAR1 in this study seem to depend on downstream IRF7 signaling, as Adar1 cm-cKO; Irf7−/− mouse showed little to no signs of cardiac inflammation.5 This pathway may also help explain the late-onset autoinflammation (lack of apoptosis) observed in hearts of Adar1 cm-KO mice.
In conclusion, work by Garcia-Gonzales et al provide new insights on editor versus noneditor functions of ADAR1 and the importance of IRF7 in driving the graded intolerance to inflammation and cardiomyopathy in the postnatal heart over time (Figure). These pathways have important implications in better understanding viral-induced myocarditis, where ADAR1 appears to be dynamically regulated in the postnatal heart.10,11 ADAR1’s known association with Dicer via its Z-DNA and dsRNA binding domains12 may also provide important clues on defining other RNA processing (noncanonical) functions of ADAR1 in cardiac inflammation and disease in the future. Since low level of editing is observed in ADAR1 deficient cardiomyocytes in this study5 and ADAR family members are also found in the developing human heart,13 future studies directed at targeting other ADAR members in cardiomyocytes, will provide further insight into the distinct roles of ADAR family members in cardiac inflammation and function.
Sources of Funding
F. Sheikh is supported by NIH/NHLBI (HL142251;HL145534) and Department of Defense Grant (W81XWH-1810380) grants.
Footnotes
Disclosures
F. Sheikh is a co-founder and shareholder of Papillon Therapeutics, Inc as well as consultant and shareholder of LEXEO Therapeutics, Inc.
REFERENCES
- 1.Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science. 2015;349:1115–1120. doi: 10.1126/science.aac7049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, Nishikura K. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J Biol Chem. 2004;279:4952–4961. doi: 10.1074/jbc.M310162200 [DOI] [PubMed] [Google Scholar]
- 3.Moore JBt, Sadri G, Fischer AG, Weirick T, Militello G, Wysoczynski M, Gumpert AM, Braun T, Uchida S. The A-to-I RNA Editing enzyme adar1 is essential for normal embryonic cardiac growth and development. Circ Res. 2020;127:550–552. doi: 10.1161/CIRCRESAHA.120.316932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.El Azzouzi H, Vilaça AP, Feyen DAM, Gommans WM, de Weger RA, Doevendans PAF, Sluijter JPG. Cardiomyocyte specific deletion of adar1 causes severe cardiac dysfunction and increased lethality. Front Cardiovasc Med. 2020;7:30. doi: 10.3389/fcvm.2020.00030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia-Gonzalez C DC, Maroli G, Wiesnet M, Wietelmann A, Li X, Graumann J, Stellos K, Kubin T, Schneider A, Braun T. ADAR1 prevents autoinflammatory processes in the heart mediated by IRF7. Circ Res. 2022;131:580–597. doi: 10.1161/CIRCRESAHA.122.320839 [DOI] [PubMed] [Google Scholar]
- 6.Ng WA, Grupp IL, Subramaniam A, Robbins J. Cardiac myosin heavy chain mRNA expression and myocardial function in the mouse heart. Circ Res. 1991;68:1742–1750. doi: 10.1161/01.res.68.6.1742 [DOI] [PubMed] [Google Scholar]
- 7.Stanley EG, Biben C, Elefanty A, Barnett L, Koentgen F, Robb L, Harvey RP. Efficient Cre-mediated deletion in cardiac progenitor cells conferred by a 3’UTR-ires-Cre allele of the homeobox gene Nkx2–5. Int J Dev Biol. 2002;46:431–439. [PubMed] [Google Scholar]
- 8.Ben-Shoshan SO, Kagan P, Sultan M, Barabash Z, Dor C, Jacob-Hirsch J, Harmelin A, Pappo O, Marcu-Malina V, Ben-Ari Z, et al. ADAR1 deletion induces NFκB and interferon signaling dependent liver inflammation and fibrosis. RNA Biol. 2017;14:587–602. doi: 10.1080/15476286.2016.1203501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y, Peng H, Zhou S, Zhuang Y. ADAR1 is targeted by miR-143 to regulate IL-1β-induced endothelial activation through the NFκB pathway. Int J Biochem Cell Biol. 2017;89:25–33. doi: 10.1016/j.biocel.2017.05.021 [DOI] [PubMed] [Google Scholar]
- 10.Dong N, Dong C, Xiong S. Janus effects of ADAR1 on CVB3-induced viral myocarditis at different infection stages. Int J Cardiol. 2016;223:898–905. doi: 10.1016/j.ijcard.2016.08.315 [DOI] [PubMed] [Google Scholar]
- 11.Zhang X, Gao X, Hu J, Xie Y, Zuo Y, Xu H Zhu S. ADAR1p150 forms a complex with dicer to promote mirna-222 activity and regulate pten expression in cvb3-induced viral myocarditis. Int J Mol Sci. 2019;20:407 doi: 10.3390/ijms20020407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ota H, Sakurai M, Gupta R, Valente L, Wulff BE, Ariyoshi K, Iizasa H, Davuluri RV, Nishikura K. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell. 2013;153:575–589. doi: 10.1016/j.cell.2013.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Altaf F, Vesely C, Sheikh AM, Munir R, Shah STA, Tariq A. Modulation of ADAR mRNA expression in patients with congenital heart defects. PLoS One. 2019;14:e0200968. doi: 10.1371/journal.pone.0200968 [DOI] [PMC free article] [PubMed] [Google Scholar]