

Summary
Class IB phosphoinositide 3-kinase (PI3Kγ) is activated in immune cells and can form two distinct complexes (p110γ-p84 and p110γ-p101), which are differentially activated by G protein-coupled receptors (GPCRs) and Ras. Using a combination of X-ray crystallography, hydrogen deuterium exchange mass spectrometry (HDX-MS), electron microscopy, molecular modeling, single-molecule imaging, and activity assays, we identify molecular differences between p110γ-p84 and p110γ-p101 that explain their differential membrane recruitment and activation by Ras and GPCRs. The p110γ-p84 complex is dynamic compared with p110γ-p101. While p110γ-p101 is robustly recruited by Gβγ subunits, p110γ-p84 is weakly recruited to membranes by Gβγ subunits alone and requires recruitment by Ras to allow for Gβγ activation. We mapped two distinct Gβγ interfaces on p101 and the p110γ helical domain, with differences in the C-terminal domain of p84 and p101 conferring sensitivity of p110γ-p101 to Gβγ activation. Overall, our work provides key insight into the molecular basis for how PI3Kγ complexes are activated.
Keywords: PI3K, PIK3CG, p101, p84, PIK3R5, PIK3R6, phosphoinositide 3-kinase, HDX-MS, TIRF, GPCR
Graphical abstract
Highlights
-
•
Structure of p110γ-p84 reveals mechanism of differential stability compared with p110γ-p101
-
•
p110γ-p84 requires Ras for robust membrane activation by Gβγ subunits
-
•
Identification of Gβγ binding site in both p101 and p110γ
-
•
A unique p101 extension mediates differential GPCR sensitivity compared with p84
PI3Kγ is a master regulator of PI(3,4,5)P3 synthesis during immune cell signaling and consists of two complexes (p110γ-p101 and p110γ-p84) that are differentially activated. Here, Rathinaswamy et al., using structural and biophysical approaches, reveal how Ras and GPCRs distinctly activate PI3Kγ complexes. This has relevance for developing future anti-PI3Kγ therapeutics.
Introduction
The class IB phosphoinositide 3-kinase PI3Kγ is a lipid kinase that generates the lipid signaling molecule phosphatidylinositol 3,4,5 trisphosphate (PIP3) downstream of diverse cell surface receptors.2 PI3Kγ can form two distinct complexes composed of a single catalytic subunit (p110γ, encoded by PIK3CG) binding to one of two regulatory subunits (p101 and p84, encoded by PIK3R5 and PIK3R6, respectively).3,4,5,6 PI3Kγ is highly expressed in immune cells and is a master regulator of the adaptive and innate immune systems,2 with key roles in chemotaxis,7 reactive oxide production,8 and cytokine production.9 It also plays important roles in endothelial cells, neurons, cardiomyocytes, and pulmonary cells.10 Studies on catalytically dead PI3Kγ or the use of selective ATP-competitive inhibitors have defined important roles for PI3Kγ in the inflammatory response, and it shows promise as a therapeutic target for inflammatory disease including lupus,11 arthritis,12 atherosclerosis,13 asthma,14 and obesity-related changes in metabolism.15,16 PI3Kγ inhibition has also shown promise for cancer treatment, with PIK3CG being frequently overexpressed in numerous cancers,17,18 and inhibiting PI3Kγ in the tumor microenvironment can promote anti-tumor immune responses.19,20 This has led to PI3Kγ selective inhibitors being in phase II clinical trials to treat triple-negative breast cancer, renal cell carcinoma, and urothelial carcinoma.21 However, the discovery of primary immunodeficiency patients harboring loss-of-function mutations in PI3Kγ22,23 highlights potential challenges that may exist for long-term inhibition of PI3Kγ as a therapeutic approach.
The two complexes of PI3Kγ (p110γ-p101 and p110γ-p84) play distinct roles in cell signaling, with these putatively mediated by their differential ability to be activated by stimuli, including G protein-coupled receptors (GPCRs),24 the immunoglobulin E (IgE)/antigen receptor,8 receptor tyrosine kinases,25 and Toll-like receptors (TLRs).26 Experiments examining immune cells with selective knockout of the p101 or p84 regulatory subunits show that p101 is required for PI3Kγ’s role in chemotaxis, while the p84 subunit is required for reactive oxide generation,27,28,29 with knockout of both regulatory subunits leading to complete loss of PI3Kγ activity.28 Biochemical reconstitution studies have defined two major signaling proteins that mediate PI3Kγ activation downstream of cell surface receptors, lipidated Gβγ subunits released by activated GPCRs, and GTP-loaded lipidated Ras. The presence of p101 and p84 regulatory subunits dramatically alter the activation by each of these stimuli, with, in vitro, the p110γ-p101 complex activated ∼100 fold by Gβγ, while p110γ-p84 is activated ∼5-fold.30,31,32,33,34 In cells, the p110γ-p84 complex is poorly recruited to cell membranes by Gβγ subunits, with it requiring Ras for membrane localization.34 The p101 subunit forms an obligate heterodimer with p110γ, while p84 forms a weaker transient interaction with p110γ,33 but the molecular basis for this is currently not understood.
Extensive biophysical experiments on the free p110γ catalytic subunit and the p110γ-p101 complex have revealed insight into the architecture and regulation of p110γ.30,32,35,36,37 The p110γ catalytic subunit is composed of an adaptor-binding subunit (ABD), a Ras-binding domain (RBD) that mediates activation downstream of Ras, a C2 domain, a helical domain, and a bilobal kinase domain. The cryoelectron microscopy (cryo-EM) structure of the p110γ-p101 complex revealed that p110γ binds to the p101 regulatory subunit through the C2 domain and the RBD-C2 and C2-helical linkers.32 We previously mapped a putative Gβγ binding interface in the helical domain of p110γ,30 with an additional binding site in the C-terminal domain of p101.30,32 Mutations in Gβγ have differential effects on either p110γ or p110γ-p101 activation,38 but the full molecular details of how Gβγ binds to either p110γ or p101 are still unclear.
To decipher the molecular mechanism for why p101 and p84 subunits differentially regulate p110γ activation, we determined the structure of the p110γ-p84 complex using a combined X-ray crystallography, EM, and computational modeling approach. Hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments revealed that the p110γ-p84 is dynamic relative to the p110γ-p101 complex. Membrane reconstitution experiments using HDX-MS and single-molecule total internal reflection fluorescence (TIRF) microscopy to study membrane recruitment of p110γ-p84 and p110γ-p101 by lipidated Gβγ and Ras shows that p110γ-p84 requires Ras for membrane localization. The p110γ-p84 complex can only be potently activated and membrane recruited by Gβγ when Ras is present, where the p110γ-p101 complex can be robustly activated by Gβγ subunits alone. Finally, computational modeling, HDX-MS, and mutagenesis were used to define the Gβγ binding interfaces with both the C-terminal domain of p101 and the helical domain of p110γ. Overall, this work provides insight into the molecular mechanisms mediating differential PI3Kγ activation by Ras and GPCR signaling.
Results
Structure of the p110γ-p84 complex
To understand differences in the regulation of p110γ-p84 versus p110γ-p101 required molecular details of the p110γ-p84 complex. We purified full-length human p110γ in complex with either mouse p84 or porcine p101, with gel filtration profiles consistent with the formation of heterodimers. The domain architectures of p110γ, p84, and p101 are shown in Figure 1A, with SDS-PAGE gels of all proteins and protein complexes purified in this article shown in the source data. To determine the structure of the p110γ-p84 complex, we utilized a combination of X-ray crystallography, EM, and AlphaFold2 computational modeling. Initial negative-stain EM data revealed that purified p110γ-p84 was homogeneous and formed a similar-shaped complex to our recently determined p110γ-p101 cryo-EM complex. However, even with extensive optimization, we could not generate high-quality vitrified specimens for cryo-EM, as the p110γ-p84 complex always dissociated into free p110γ and p84 particles. Extensive screening of precipitant conditions allowed us to obtain crystals of p110γ-p84 that diffracted to ∼8.5 Å, with initial attempts to phase this using molecular replacement with the p110γ-p101 cryo-EM structure being unsuccessful. To provide additional molecular details on this complex, we utilized an AlphaFold239 model specifically trained for multimeric complexes.40 Extensive computational modeling of different sequences of p110γ and p84 resulted in a consensus solution for the interface of p110γ with p84. These models had low predicted alignment error (PAE) between the p110γ and p84 subunits, which is a measure of the confidence of protein-protein interfaces (Figure S1). This model was then used as a search model for the low-resolution X-ray diffraction data, with only rigid body refinement resulting in a solution with high confidence (Rwork = 0.28, Rfree = 0.34; Table S1), despite the low resolution of the X-ray diffraction (Figures 1C and S2). While the positioning of side chains is impossible at this resolution, analysis of the 2mfo-Dfc density revealed the orientation of the helices in p84 at the p110γ interface, validating the inter-subunit orientation (Figure S2B), with this solution fitting well in the low-resolution negative-stain EM density (Figure S2A).
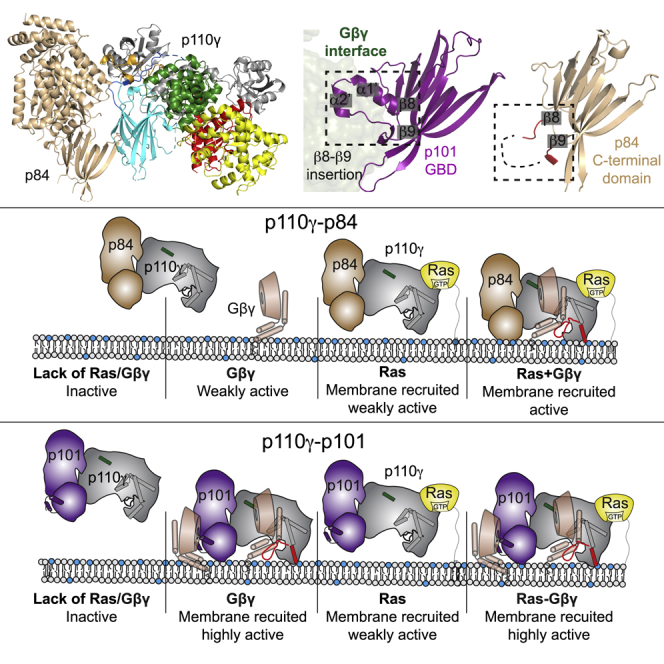
Figure 1.

The structure of the p110γ-p84 complex and comparison with p110γ-p101
(A) Cartoon schematic of the PI3Kγ catalytic (p110γ) and regulatory subunits (p101 and p84) with domain boundaries indicated.
(B) Cartoon of differences in activation between p110γ-p84 and p110γ-p101 complexes downstream of GPCRs and RTKs.
(C) Structure of the p110γ-p84 complex based on X-ray crystallography, negative-stain EM, and AlphaFold modeling (PDB: 8AJ8, supporting data in Figures S1 and S2). Domains are indicated from (A), with a cartoon schematic shown in the bottom left.
(D) Structure of the p110γ-p101 complex (PDB: 7MEZ).32 Domains are indicated from (A), with a cartoon schematic shown in the bottom left.
(E and F) Differences in the C-terminal domain of p84 (E) and p101 (F) are shown with this domain shown as an electrostatic surface.
The overall architecture of the p84 subunit is conserved compared with p101, with it containing an N-terminal helical domain, a central α/β barrel domain, and a C-terminal β-sandwich domain. The orientation of the regulatory subunit in the p110γ-p84 complex versus the p110γ-p101 complex32 was strikingly similar (Figures 1C and 1D), with p84 binding to the C2 domain and the RBD-C2 and C2-helical linkers of p110γ. The primary interface for p110γ in p84 was located at the N-terminal helical region of p84, with additional interactions involving the C-terminal domain and the C terminus. One of the primary differences between p101 and p84 regulatory subunits is their differential ability to be recruited by lipidated Gβγ subunits. We have previously identified that this binding occurs at the C-terminal domain of p101 in a region we defined as the Gβγ binding domain (GBD).30,32 The AlphaFold2 model of the p110γ-p84 structure allowed us to examine differences in this domain. The C-terminal domain of p84 contains the same β-sandwich fold; however, there are distinct differences compared with p101 at the face of this domain distal from p110γ. This can be clearly highlighted by visualizing the electrostatics of the GBD between p101 and p84, showing a strikingly different interfacial surface for Gβγ binding (Figures 1E and1F). One of the primary differences that was immediately apparent was the presence of a helical extension in the loop between the β8-β9 strands of the C-terminal domain of p101 that is part of the Gβγ binding face,30,32 which is not present in p84.
Differences in the interface of p84 with p110γ compared with p101
Previous in vitro assays testing subunit exchange of p110γ-p101 and p110γ-p84 complexes suggest that the p101 complex forms a constitutive complex with p110γ, with the p84 complex forming a weaker dynamic interaction with p110γ.33 To explore the dynamics of the two complexes, we used HDX-MS, which measures dynamic differences in protein secondary structure. These experiments were carried out with human p110γ bound to porcine p101 and human p110γ bound to mouse p84. We compared the HDX rates in p110γ between the two complexes at four different time points (3, 30, 300, and 3,000 s). The full details of HDX-MS data processing are in Table S2, with all raw HDX-MS data for all time points available in the source data. We observed statistically significant decreased exchange (defined as differences at any time point >5%, >0.4 Da, and a p value less than 0.01) in the p110γ-p101 complex versus the p110γ-p84 complex in the helical domain, C2 domain, the RBD-C2 linker, and the C2-helical linker (Figures 2A and 2B). These changes were all localized to either the interface with regulatory subunits or the helical domain adjacent to the interface, which is consistent with p101 forming a more stable complex with p110γ compared with p84.
Figure 2.

Differences in the p110γ interface in p84 versus p101
(A) HDX-MS differences in the p110γ subunit between the p110γ-p101 and p110γ-p84 complexes. Significant differences in deuterium exchange (defined as greater than 5%, 0.4 Da, and a two-tailed t test p < 0.01 at any time point) are mapped on to the structure of p110γ-p84 and cartoon of p110γ according to the legend.
(B) Sum of the number of deuteron differences between the p110γ-p101 and p110γ-p84 complexes over the entire deuterium exchange time course. Positive difference is indicative of enhanced exchange in p110γ-p84. Each point is representative of the center residue of an individual peptide. Peptides that met the significance criteria described in (C) are colored red. Error is shown as the sum of the standard deviation across all time points (n = 3 for each time point). All HDX-MS data are provided in the source data.
(C) Selected deuterium exchange at 30 s for peptides in p110γ for p110γ-p101 and p110γ-p84 complexes at either a high concentration (1,500 nM) or a low concentration (175 nM). Error is shown as standard deviation (n = 3) with two-tailed p values as indicated: ∗∗p < 0.01; not significant (ns) > 0.05. Full HDX-MS data for all peptides in this experiment are shown in the source data.
(D) Cartoon schematic of the p110γ interface for p101 (top) and p84 (bottom), with a zoom in on the residue’s located at the p110γ interface for both p84 and p101. Dotted lines indicate cation-pi or electrostatic interactions.
(E) Sequence alignment of both p101 and p84 residues in the α3 to α6 helices located at the p110γ interface. The residues annotated in panel are indicated on the alignment. A full alignment of p101 and p84 is shown in Figure S3.
These differences in exchange could possibly arise from differential dynamics of the intact complexes, an increased propensity for the p110γ-p84 complex to fall apart compared with p110γ-p101, or some combination of both. To further explore this point, we carried out HDX-MS experiments with varying concentrations of p110γ-p84 or p110γ-p101. In addition, for these experiments, we purified p110γ bound to either the human p84 or p101 regulatory subunits to validate that the changes observed were not due to minor differences in evolutionary conservation between mouse, pig, and human sequences (Figure S3). HDX-MS experiments were carried out at two time points (30 and 300 s) with a final concentration of 1,500 nM in high-concentration experiments and 175 nM in low-concentration experiments for both p110γ-p84 and p110γ-p101. Comparing p110γ-p84 with p110γ-p101 in the high-concentration experiment showed similar differences to what we observed with the mouse p84 or pig p101 complexes, showing that the difference between regulatory subunits is conserved for the human proteins (source data). For the p110γ-p101 complex, there was no significant difference in exchange between the high- and low-concentration samples, signifying that the complex remains intact in both conditions (Figure 2C). However, in the p110γ-p84 complex, there was significant increases in exchange at p84 interfacial regions in the low concentration compared with the high concentration (Figure 2C). This is consistent with the p84 complex being more likely to fall apart compared with p110γ-p101 (Figure 2D).
To possibly understand the molecular basis for this difference in complex stability, we compared the structures of p110γ-p84 and p110γ-101 at the regulatory subunit interfaces. Both p101 and p84 bind the same interface with p110γ. The most extensive binding interface for both p84 and p101 with p110γ is comprised of a set of N-terminal helices, specifically the loops between α3-α4 and α5-α6 (Figure 2E). In both p84 and p101, the interfacial residues found in these helices are strongly evolutionarily conserved (Figure 2F); however, there are distinct differences between p101 and p84 subunits. The main difference is that in the p110γ-101 complex, there are a set of cation-pi interactions between charged residues in p110γ and aromatic residues in p101,32 with these cation-pi interactions being lost in p84. Further mutational analysis will be required to define the molecular basis for the dynamic differences in stability between p84 and p101 complexes with p110γ.
Activation of the p110γ-p84/p110γ-p101 complexes by lipidated Gβγ and Ras
To provide additional insight into functional differences between p110γ-p84 and p110γ-p101, we characterized their lipid kinase activities as well as the activity of the free p110γ catalytic subunit using membrane reconstitution assays with lipidated Gβγ- and lipidated GTPγS-loaded G12V HRas (Figure 3A). We characterized the lipid kinase activities using saturating concentrations of lipidated Gβγ and lipidated HRas on membranes roughly mimicking the composition of the plasma membrane (5% PIP2, 20% phosphatidylserine, 50% phosphatidylethanolamine, 10% cholesterol, 10% phosphatidylcholine, and 5% sphingomyelin). The presence of HRas alone led to roughly similar 3-fold activation for p110γ, p110γ-p84, and p110γ-p101 (Figure 3B). The presence of Gβγ alone led to robust activation of p110γ-p101 (>100-fold activation), with weak activation of p110γ-p84 (∼3-fold), and no detectable activation of p110γ (Figure 3B). The additional presence of HRas for p110γ-p101 with Gβγ caused an approximately similar 3-fold Ras activation as was seen in the absence of Gβγ. However, for both free p110γ and p110γ-p84, there was synergistic activation when both HRas and Gβγ were present, which is not seen for p110γ-p101. This was consistent with previous observations of Gβγ- and HRas-mediated activation of PI3Kγ on other membrane systems.30,31,32,34 Because the p110γ-p84 complex is more reliant on activation by Ras, we wanted to ensure that there was no major affinity difference toward HRas for p110γ-p84 and p110γ-p101. We carried out activation assays with varying levels of HRas both in the presence and absence of saturating lipidated Gβγ subunits. Both p110γ-p84 and p110γ-p101 in the presence and absence of Gβγ showed very similar EC50 values (Figures 3C and 3D).
Figure 3.

Activation of p110γ-p84 and p110γ-p101 by lipidated HRas and Gβγ
(A) Cartoon schematic describing PI3Kγ variants tested and the lipidated activators, GTPγS-loaded HRas and Gβγ.
(B) Lipid kinase activity assays of different p110γ complexes (concentration, 100–2,000 nM) with and without lipidated Gβγ (1.5 μM) and lipidated HRas (1.5 μM) using 5% phosphatidylinositol 4,5-bisphosphate (PIP2) vesicles mimicking the plasma membrane (20% phosphatidylserine, 50% phosphatidylethanolamine, 10% cholesterol, 10% phosphatidylcholine, 5% sphingomyelin). Error bars represent standard deviation (n = 3). Two-tailed p values represented by the symbols are as follows: ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; ns > 0.05.
(C) Lipid kinase activity assays of p110γ-p84 and p110γ-p101 with varying concentrations of lipidated HRas (n = 3).
(D) Lipid kinase activity assays of p110γ-p84 and p110γ-p101 in the presence of lipidated Gβγ (1.5 μM) with varying concentrations of lipidated HRas (n = 3).
Experiments in (C) and (D) were performed using the same vesicles as in (B). The dotted red line in the graph for the p110γ-p101 complex shows the peak activity for p110γ-p84 with both activators. The EC50 and 95 confidence intervals (CIs) are indicated for both (C) and (D).
HDX-MS analysis of Gβγ and HRas activation of p110γ-p84
To define the molecular mechanism underlying the difference between p110γ-p84 and p110γ-p101 activation by lipidated Gβγ and HRas, we carried out HDX-MS experiments on membrane-reconstituted complexes. HDX experiments were measured at four time points (3, 30, 300, and 3,000 s) and five conditions: p110γ-p84 alone, p110γ-p84 with plasma membrane (PM) mimic vesicles, p110γ-p84 with HRas on PM mimic vesicles, p110γ-p84 with Gβγ on PM mimic vesicles, and p110γ-p84 with Gβγ and HRas on PM mimic vesicles (Figures 4 and S4). The full details of HDX-MS data processing are in Table S2, with all raw HDX-MS data for all time points available in the source data.
Figure 4.

HDX-MS analysis of p110γ-p84 activation by membrane-localized HRas and Gβγ and comparison with p110γ-p101
(A–C) Significant HDX-MS differences in the p110γ and p84 subunits between (A) plasma membrane mimic vesicles and plasma membrane mimic vesicles with 3 μM GTPγS-loaded lipidated HRas, (B) plasma membrane mimic vesicles and plasma membrane mimic vesicles with 3 μM Gβγ, and (C) plasma membrane mimic vesicles with 3 μM GTPγS-loaded lipidated HRas and plasma membrane mimic vesicles with both HRas and Gβγ (3 μM) are mapped on the structure of p110γ-p84 according to the legend in (A). A cartoon model is shown to the right with differences annotated. The sum of the number of deuteron difference is shown for p110γ, with red dots representing peptides showing statistically significant differences. Error is shown as the sum of the standard deviations across all time points (n = 3 for each time point).
(D) Significant HDX-MS differences in the p110γ and p101 subunits between plasma membrane mimic vesicles and plasma membrane mimic vesicles with Gβγ mapped on the structure of p110γ-p101 (PDB: 7MEZ) according to the legend. A cartoon model is shown to the right with differences annotated.
The HDX-MS data in (D) are from our previous study30 and are provided as a comparison with (B).
There were no significant differences in HDX between free p110γ-p84 and p110γ-p84 in the presence of PM mimic vesicles without lipidated activators. This is consistent with the p110γ-p84 complex being primarily in solution in the absence of either HRas or Gβγ. When HRas was present on membrane surfaces, there were multiple regions that showed significant differences compared with membranes alone (Figures 4A and S4). This included increased exchange in the helical domain and multiple regions of the regulatory motif in the kinase domain, as well as decreases in exchange in the kα12 membrane-binding C-terminal helix and the HRas interface of the RBD. The changes in the helical and kinase domains are consistent with previously observed conformational changes that accompany membrane binding in p110γ.30,36,41 Intriguingly, there were almost no significant changes in either p110γ or p84 between Gβγ membranes compared with membranes alone, with only one peptide in the kinase domain showing increased exchange (Figure 4B).
Consistent with the synergistic activation observed in the lipid kinase assays, there were significant differences in exchange observed for p110γ-p84 between HRas membranes and HRas/Gβγ membranes, including decreased exchange at the helical domain and increased exchange in the regulatory motif of the kinase domain (Figure 4C). These changes in the helical domain were similar, although of a lesser magnitude than those we have observed when examining binding of p110γ-p101 to Gβγ membranes (Figure 4D),30 indicating that the binding site for Gβγ on p110γ is conserved between the two complexes. There were no significant decreases in exchange in p84 with Gβγ membranes (Figures 4C and S4), in contrast to the protection observed in the C-terminal domain of p101 (Figure 4D), which is consistent with p84 lacking a binding site for Gβγ subunits.
Single-molecule analysis of Gβγ/Ras-mediated membrane recruitment of p110γ-p101/p110γ-p84
To compare how p110γ-p101 and p110γ-p84 associate with membrane-tethered HRas and Gβγ, we performed single-molecule TIRF microscopy experiments on supported lipid bilayers (SLBs). Membranes containing cysteine-reactive maleimide lipids were used to covalently attach HRas in a biologically relevant orientation (Figures 5A and S5A–S5C). Farnesylated Gβγ was then equilibrated into supported membranes to reach a surface density that facilitated membrane binding of PI3K as previously demonstrated.32 Using this assay, we compared the bulk membrane recruitment, single-molecule dwell times, and diffusion coefficients of fluorescently tagged PI3K complexes, DY647-p101-p110γ and DY647-p84-p110γ, in the absence and presence of HRas and/or Gβγ.
Figure 5.

p110γ-p84 and p110γ-p101 exhibit distinct membrane-binding dynamics in the presence of HRas and Gβγ
(A) Experimental setup for visualizing DY647-p101-p110γ and DY647-p84-p110γ interactions with a supported lipid bilayer containing membrane-anchored HRas(GDP or GTP) and farnesylated Gβγ. Experiments show a representative experiment, with all quantification generated from 2 to 4 technical replicates (see Table S3).
(B and C) Kinetics of PI3K complex membrane recruitment measured in the presence of 10 nM DY647-p101-p110γ or DY647-p84-p110γ using TIRF-M.
(D) Representative smTIRF-M images visualizing PI3K complex localization in the presence of 10 pM DY647-p101-p110γ or 100 pM DY647-p84-p110γ. Localization was measured on SLBs containing Ras(GDP), Ras(GTP), Gβγ, or Ras(GTP)/Gβγ.
(E and F) Single-molecule dwell time distributions for DY647-p101-p110γ or DY647-p84-p110γ measured in the presence of the indicated binding partners. Plots showing log10(1-cumulative distribution frequency [CDF]) versus dwell time (s). Data are fit to either a single or double exponential decay curve (black dashed lines). Single-molecule imaging of DY647-p84-p110γ yielded the following mean dwell times: 39 (+RasGTP), 74 (+Gβγ), and 188 ms (+RasGTP/Gβγ). Single-molecule imaging of DY647-p101-p110γ yielded the following mean dwell times: 0.146 (+RasGTP), 0.73 (+Gβγ), and 3.09 s (+RasGTP/Gβγ).
(G and H) Step size (or displacement) distributions of DY647-p101-p110γ or DY647-p84-p110γ. PI3K complex formation with Ras(GTP), Gβγ, or Ras(GTP)/Gβγ modulates the single-molecule displacement (i.e., step size, μm). Dashed black line represents the curve fit used to calculate the diffusion coefficient (see STAR Methods).
The membrane composition for membranes in (B)–(H) was 96% DOPC, 2% PI(4,5)P2, 2% MCC-PE. See Table S3 for time constants (τ1 and τ2), diffusion coefficients (μm2/s), and statistics (n = 2–4).
Visualization of the bulk membrane recruitment dynamics of fluorescently labeled PI3K complexes revealed that p101-p110γ interacts more strongly with HRas(GTP) and Gβγ compared with p84-p110γ (Figures 5B and 5C). In the presence of HRas(GTP), both DY647-p101-p110γ and DY647-p84-p110γ displayed rapid equilibration kinetics on supported membranes. In contrast, recruitment of DY647-p101-p110γ to membranes containing lipid-anchored Gβγ was more robust compared with DY647-p84-p110γ (Figures 5B–5C). The time required for DY647-p110γ-p101 to equilibrate with Gβγ-containing membranes was also longer, reflecting the ability of p101-p110γ to interact with two Gβγ molecules.32 A synergistic enhancement in membrane binding was observed for both DY647-p101-p110γ and DY647-p84-p110γ in the combined presence of HRas(GTP) and Gβγ. However, the total membrane recruitment of DY647-p101-p110γ was ∼30-fold higher compared with that of DY647-p84-p110γ.
We established conditions to directly visualize membrane association of PI3K complexes with single-molecule resolution (Figure 5D; Videos S1 and S2). In the presence of HRas(GDP), no detectable membrane-binding events were observed for either DY647-p101-p110γ or DY647-p84-p110γ (Figure 5D). Consistent with our bulk membrane recruitment measurements, the timescales of the single-molecule dwell times for DY647-p84-p110γ under all conditions were an order of magnitude shorter compared with DY647-p101-p110γ (Figures 5E, 5F, and S5K–S5M; Table S3). In addition, a 10-fold higher concentration of DY647-p84-p110γ was required to observe a similar number of single-molecule binding events compared with DY647-p101-p110γ (Figure 5D). To infer whether membrane-bound DY647-p101-p110γ simultaneously engages HRas(GTP) and Gβγ, we measured changes in the diffusion coefficients calculated from the step size distribution. In the presence of either HRas(GTP) or Gβγ, DY647-p101-p110γ displayed a range of diffusion coefficients between 0.08 and 0.64 μm2/s (Figures 5G, 5H, and S5H–S5J; Table S3). The median displacement (or step size) per frame decreased 3-fold when comparing DY647-p101-p110γ bound to membranes containing either HRas(GTP) or Gβγ (0.2 and 0.15 μm/frame, respectively) with the combination of HRas(GTP)/Gβγ (0.06 μm/frame). A similar trend was observed for DY647-p84-p110γ; however, the single-molecule trajectories measured in the presence of Gβγ were too transient to calculate a diffusion coefficient.
Membrane composition: 96% DOPC, 2% PI(4,5)P2, 2% MCC-PE. Video associated with Figure 5E and 5G. Video plays at 25 frames per second. smTIRF-M data was collected with 52 ms time intervals (i.e. 19 fps). Scale bar is 2 μm.
Membrane composition: 96% DOPC, 2% PI(4,5)P2, 2% MCC-PE. Video associated with Figure 5F and 5H. Video plays at 25 frames per second. smTIRF-M data was collected with 22 ms time intervals (i.e. 45 fps). Scale bar is 2 μm.
These data are consistent with both complexes being able to simultaneously engage Ras and Gβγ, with the p110γ-p101 complex able to bind to two Gβγ molecules and one Ras, and p110γ-p84 binding to one Gβγ molecule and one Ras. When each activator is presented alone, p110γ-p84 is more robustly recruited by Ras compared with Gβγ, and p110γ-p101 is more robustly recruited by Gβγ compared with Ras.
Analysis of Gβγ binding to p110γ and p101
We previously characterized Gβγ binding to both the p101 and p110γ subunits using HDX-MS and identified mutations in either the helical domain of p110γ or the C-terminal domain of p101 that prevent Gβγ activation.30 To provide additional insight into the molecular basis for how p110γ and p101 interact with Gβγ and why p84 lacks this ability, we carried out AlphaFold-multimer40 modeling of both interfaces (Figures S6 and S7). The search models converged on a consensus orientation of Gβγ interaction with the p101 C-terminal domain (Figure S6) and a different consensus orientation of Gβγ interaction with the helical domain (Figure S7), both with predicted alignment scores and per-residue estimate of confidence (predicted local distance difference test [pLDDT]) scores39 consistent with excellent model accuracy (Figures S6A, S6B, S7A, and S7B).
These models of Gβγ binding allowed us to make a schematic of how the p110γ-p101 complex associates with two Gβγ subunits (Figure 6A). Critically, the Gγ subunit of Gβγ is geranylated at its C terminus, and in our models, the Gγ C terminus is oriented in a direction pointed toward the membrane when p110γ is oriented toward its putative membrane interface. Examining these models compared with other Gβγ complexes showed that the same face of the Gβ subunit that binds to the pleckstrin homology (PH) domain of GPCR kinase 242 binds to the C-terminal domain of p101 and the helical domain of p110γ (Figures 6B–6D). The p101 interface with Gβγ is primarily composed of two evolutionarily conserved helices positioned in a loop between β8 and β9 of the C-terminal domain (Figure 6E), along with an extensive interface at residues 816–830. The loop between β8 and β9 is not conserved in p84 (Figure 6E). In the helical domain, the interface is entirely composed of the N-terminal helix (annotated as hα1, 548–560). These sites are also where we previously designed complex disrupting mutations for both p101 (DQDE817AAA and RKIL821AAA) and p110γ (RK552DD),30 providing further validation of the putative interface.
Figure 6.

Model of Gβγ activation of PI3Kγ complexes
(A) Model of the activation of p110γ-p101 complex by two different Gβγ subunits. The location of the Gβγ subunits bound to the C-terminal domain of p101 (Figure S6) and the helical domain of p110γ (Figure S7) was based on AlphaFold2-multimer modeling aligned to the structure of the p110γ-p101 complex (PDB: 7MEZ). The domains of p110γ-p101 are annotated, with the Gβ subunit shown as a transparent surface, and the Gγ subunit shown as cartoon, with the C terminus colored in blue. Both Gβγ subunits are positioned in an orientation compatible with membrane binding of p110γ.
(B) Model of the C-terminal domain of p101 bound to Gβγ (full details on AlphaFold2-multimer modeling is Figure S6). The unique helical extension between β8-β9 in p101 is annotated.
(C) Model of the helical domain of p110γ bound to Gβγ (full details on AlphaFold2-multimer modeling is Figure S7). The N-terminal helix of the helical domain in contact with Gβγ is annotated.
(D) Structure of the PH domain of GPCR kinase 2 (GRK2) bound to Gβγ (PDB: 1OMW).42
(E) Evolutionary conservation of the Gβγ binding site in p101. Alignment of the Gβγ binding extension in p101 between p101 and p84 (top), between different orthologs of p101 (center), and between different orthologs of p84 (bottom).
(F) Comparison of the C-terminal domain between p101 and p84. The evolutionarily conserved helical extension that occurs between β8 and β9 in p101 is annotated, with the Gβγ subunit from (B) shown as a transparent surface. The end and start of β8 and β9, respectively, are labeled, highlighting the corresponding loop between p84 and p101, with the loop colored red in p84. In p84, the majority of this loop was disordered in both the X-ray and AlphaFold2-multimer modeling and is indicated as a dotted line.
(G) Lipid kinase activity assays of different p110γ complexes (p110γ-p101, p110γ-p101 β8-β9 loop swap mutant, and p110γ-p84) with and without lipidated Gβγ (100 nM) using 5% PIP2 vesicles (5% PIP2, 30% phosphatidylserine, 65% phosphatidylethanolamine). Error bars represent standard deviation (n = 3–6). Two-tailed p values represented by the symbols are as follows: ∗∗∗p < 0.001.
There are differences in the orientation and residues mediating Gβγ binding between the p101 and p110γ sites (Figures 6B and 6C). The p101 site forms a more extensive interface with Gβγ, with multiple Gβγ contact sites that are unique compared with the helical domain. These differences in interaction are consistent with unique mutations in Gβγ having differential effects between p110γ and p110γ-p101 activation.38 Examining the structures of the C-terminal domains showed differences between p101 and p84 at the site where p101 binds Gβγ. Overall, the C-terminal domains are mainly structurally conserved, but the two helices at the interface with Gβγ in p101 between β8 and β9 are absent in p84 (Figure 6F). We generated a loop swap mutation where we cloned the p84 β8-β9 loop into p101 and measured the lipid kinase activity of p110γ-p84, p110γ-p101, and the p110γ-p101 β8-β9 loop swap mutant in the presence and absence of a low concentration of Gβγ (100 nM). This concentration of Gβγ was used as p110γ-p84 is 10-fold less sensitive to Gβγ compared with p110γ-p101,34 with 100 nM Gβγ able to robustly activate p110γ-p101 while only minimally activating p110γ-p84. Under these conditions, we observed robust activation of p110γ-p101 by Gβγ, with minimal Gβγ activation for both p110γ-p84 and the p110γ-p101 β8-β9 loop swap mutant (Figure 6G). Critically p110γ-p84 and the p110γ-p101 β8-β9 loop swap mutant had equivalent activation, clearly identifying the β8-β9 loop as a critical feature in p101 that confers sensitivity to Gβγ activation. This reveals the structural basis for the absence of Gβγ binding in p84 and provides a molecular underpinning for p110γ-p101 sensitivity toward GPCR activation. Overall, this model is consistent with our previous biochemical and TIRF microscopy data supporting the engagement of two Gβγ molecules by the p110γ-p101 complex.32
Discussion
The class IB PI3Kγ is a key regulator of the immune system2,10,43 and is a therapeutic target for multiple human diseases including cancer and inflammatory diseases.12,19,20 Selective p110γ inhibitors are currently in phase II clinical trials, so fully understanding the regulation of PI3Kγ is essential for continued therapeutic development. The activity of p110γ is fundamentally regulated by its association with regulatory subunits, p84 or p101, as neutrophils lacking both regulatory subunits have similar PIP3 responses to a p110γ kinase-dead knockin mutant.28 Here, we report clear molecular insight into how Ras and GPCRs differentially regulate the p110γ-p84 and p110γ-p101 complexes.
The structure of p110γ-p84 reveals that the p84 subunit shares a similar architecture to the p101 subunit.32 In vivo, the p101 and p84 regulatory subunits are expressed in a tissue-specific manner alongside p110γ. Biochemical evidence suggests that the p110γ-p84 complex is more dynamic compared with p110γ-p101, with p101 capable of replacing the p84 subunit, but not vice versa.33 While the overall secondary structure at the interface with p110γ is conserved, there are also numerous evolutionarily conserved differences between p101 and p84 in amino acids at the p110γ interface. We identified two specific cation-pi interactions in p110γ-p101 that are absent in p110γ-p84. Although further mutational analysis is required, we hypothesize that these interactions are responsible for the strong association between p101 and p110γ compared with p84.
The dynamic nature of p110γ-p84 has important implications for PI3Kγ signaling and inhibition, as this suggests that any stimuli that may depend on binding or modulating free p110γ will only occur in p110γ-p84. An antibody that bound the p84/p101 interface on the C2 domain of p110γ selectively inhibited only p110γ-p84 and not p110γ-p101. This is likely mediated by p110γ-p84 dissociating and the antibody sterically preventing regulatory subunit binding, with the antibody binding surface being inaccessible in p110γ-p101.44 The p110γ subunit can be activated by protein kinase C phosphorylation of the helical domain downstream of the IgE antigen receptor in mast cells, with this putatively only occurring for p110γ-p84 and not p110γ-p101.45 This phosphorylation site is in a location that may be inaccessible to p110γ when bound to either p101 or p84; this may provide a unique mechanism for why only p110γ-p84 complexes can be activated by protein kinase C (PKC). Further biochemical and structural studies will be required to examine if dynamic differences in p110γ-p84 and p110γ-p101 control regulation by post-translational modifications. Biochemical assays of HRas activation showed that in the absence of Gβγ, both p110γ-p101 and p110γ-p84 are similarly weakly activated by saturating HRas, consistent with previous observations.30,31,33,34 Dose-response experiments clearly showed that the affinity for HRas activation was equivalent between the two complexes, which is consistent with the Ras interface being distant from the p101-p84 interface.37
HDX-MS and single-molecule TIRF microscopy (smTIRF-M) experiments showed the membrane anchored HRas was able to recruit p110γ-p84 to the membrane; however, this interaction was not sufficient to fully activate kinase activity. This suggests that HRas by itself acts as a critical regulator of the membrane binding but that both PI3Kγ complexes require Gβγ for robust activation. In p110γ-p84, the presence of HRas led to large synergistic activation by Gβγ. This was supported by HDX-MS and smTIRF-M experiments showing limited Gβγ-mediated membrane recruitment of p110γ-p84. Clear differences in HDX-MS at the Gβγ interface of p110γ-p84 were only observed in the presence of both HRas and Gβγ. For p110γ-p84 with both HRas and Gβγ present at saturating concentrations, the kinase activity was still much lower than Gβγ activation of p110γ-p101. Based on the smTIRF-M experiments, the membrane avidity of p110γ-p101 under “saturating” conditions was an order of magnitude stronger compared with p110γ-p84. This is consistent with the Gβγ interfaces in both the helical domain of p110γ and the GBD of p101 being critical in orienting the p110γ catalytic subunit for maximal kinase activity. These biochemical and biophysical data provide a molecular underpinning for the observation in cells that Ras is required for p110γ-p84 activation34 and for why full activation requires an intact Gβγ binding site in p110γ.28 This also explains why in mast cells, which only express p84, inhibitors of Ras lipidation abrogate PI3Kγ signaling, while upon treatment in immune cells expressing p101, PI3Kγ signaling is maintained.46
The p110γ catalytic subunit being almost completely inactive in the absence of a regulatory subunit is unique among class I PI3K isoforms, as in the other class I PI3K isoforms (p110α, p110β, p110δ), the catalytic subunit alone is highly active,47,48,49 and the regulatory subunit acts to inhibit kinase activity and stabilize the catalytic subunit. The p110γ subunit is inhibited through the presence of an internal tryptophan lock in the regulatory motif of the kinase domain,36,41 with this putatively opened when the p110γ subunit is properly oriented on a membrane surface.30 The opening of this lock putatively reorients the C-terminal helix of the kinase domain, allowing it to interact with membrane surfaces and allowing the activation loop to bind to lipid substrate. The requirement of Gβγ for robust activation of p110γ possibly implies that it orients the catalytic subunit in a manner that disrupts this inhibitory interface. This is supported by cellular experiments that show constitutively membrane-localized p110γ is activated by GPCRs.50 Additional computational and biophysical studies of p110γ bound to membrane in inactive and active conformations will be required to fully define the molecular basis for conformational changes required for the fully active state.
Modeling of Gβγ binding to both p101 and p110γ revealed insight into how Gβγ can activate PI3Kγ complexes. This p110γ-p101 can bind two Gβγ subunits, with p110γ-p84 able to bind only a single Gβγ subunit. These models agreed well with our previous HDX-MS and mutational analysis of p101 and p110γ30 as well as smTIRF-M experiments examining membrane recruitment using varying Gβγ concentrations, which implied that the p110γ-p101 complex bound two Gβγ subunits.32 By contrast, the p110γ-p84 complex can only bind one Gβγ, with robust membrane recruitment by Gβγ requiring Ras. The interface in p110γ is located in the N-terminal helix of the helical domain. HDX-MS experiments found that this same region mediates Gβγ binding in the class IA PI3K isoform p110β.51 Like p110γ-p84, p110β requires additional activation and membrane recruitment by either RTKs or Rho GTPases to be robustly activated by Gβγ subunits, suggesting this is either a relatively weak interface or that binding is dependent on conformational changes induced by membrane binding. Intriguingly, in both p110β and p110γ, there is a conformational change in this helix upon membrane recruitment.30,51 The interface in the C-terminal domain of p101 is primarily composed of two helices between β8 and β9, which is evolutionarily conserved in p101 and is not conserved in p84. This provides a molecular underpinning for why p84 shows greatly reduced sensitivity and activation by Gβγ subunits even in the presence of Ras. The Gβγ interface in p101 is more extensive than that found in p110γ, which may explain why Gβγ alone can so potently activate p110γ-p101 and does not require additional membrane-localized activators.
The development of therapeutics targeting PI3Kγ are clinically advanced, with ATP-competitive small-molecule inhibitors currently in phase II clinical trials in cancer21 and in preclinical investigation in chronic obstructive pulmonary disease and inflammatory disease.14 There are potential challenges for even highly selective p110γ inhibitors, as immune side effects may be difficult to avoid, highlighted by patients with inactivating primary immunodeficiency clinical p110γ mutations.22,23 The molecular insight into the difference in how p110γ-p101 and p110γ-p84 are regulated could lead to inhibitors specific for either p110γ-p101 or p110γ-p84, which may maintain therapeutic benefit but with decreased side effects. This fits with our observation of nanobodies that block Ras activation strongly inhibit p110γ-p84 activation, while those blocking the p101 interface with Gβγ selectively target p110γ-p101.52 Further medicinal chemistry efforts may reveal opportunities to target these sites by small-molecule inhibitors.
Our detailed biochemical, biophysical, and structural analysis of p110γ-p84 and p110γ-p101 provides insight into how PI3Kγ complexes are assembled and activated by membrane-localized Ras and Gβγ. Our work has defined the molecular basis for how these two distinct complexes can differentially integrate upstream signals, similarly to how different regulatory subunits can alter the activation of mTOR complexes.53 A summary of the molecular differences between the p110γ-p84 and p110γ-p101 and their activation by Ras and Gβγ are shown in Figure 7. This work provides a framework for the design of allosteric modulators for both p110γ-p84 and p110γ-p101, which may inform PI3Kγ complex-specific therapeutic development in inflammatory diseases and cancer.
Figure 7.

Model of differential activation of PI3Kγ complexes by Gβγ and Ras
Schematic of how Ras and Gβγ subunits can activate p110γ-p84 (top) and p110γ-p101 (bottom). Ras in the absence of Gβγ leads to membrane recruitment for both complexes but only weakly activates kinase activity. The Gβγ binding helices in the GBD of p101 and the helical domain interface with Gβγ are shown, with the helical domain hα1 highlighted in green. The C-terminal helix in the kinase domain that reorients upon membrane binding is highlighted in red in the membrane-bound complex.
Limitations of the study
The molecular insight we present here concerning the association of p84 with p110γ and how both p110γ and p101 interact with Gβγ subunits relies on an extensive set of medium- to low-resolution data supported by AlphaFold modeling and mutagenesis analysis. Continued structural investigations of the Gβγ binding site on both p101 and p110γ will be required to unambiguously define these interfaces beyond what is possible with modeling, HDX-MS, and mutagenesis analysis. Currently, our HDX-MS data have limited resolution in that they simultaneously detect all protein conformational changes induced by Gβγ binding. To gain new insight about transitions between PI3Kγ conformations, molecular dynamic simulations could be performed on Gβγ-containing membranes. Solving higher-resolution cryo-EM structures of PI3Kγ complexes docked on membranes in the absence and presence of Ras and Gβγ could also help define the ensemble of PI3Kγ conformations and membrane orientations. In the case of the p110γ-p84 complex, our structural analysis was challenging due to the transient nature of this complex compared with p110γ-p101. It remains to be seen whether unidentified p84 specific binders or activators (post-translational modifications [PTMs], etc.) can strengthen the association between p110γ-p84. Understanding these mechanisms could facilitate solving higher-resolution structures of p110γ-p84 in complex with Ras and Gβγ.
Using smTIRF-M, we observed changes in PI3Kγ membrane-binding behavior that were consistent with distinct higher-order complexes forming between Gβγ and Ras. Our current study, however, lacks the computational tools and resolution needed to quantify the probability distribution and transition rates between membrane-tethered PI3Kγ complexes that are in dynamic equilibrium. In the future, single-molecule fluorescence resonance energy transfer (FRET) measurements could help quantify intramolecular conformational dynamics of membrane-tethered PI3Kγ and quantify the dynamics of interactions with Ras and Gβγ. Future single-molecule studies could also help decipher how allosteric regulation versus membrane orientation control PI3Kγ activity. This would help explain why there is such a large increase in kinase activity for the p110γ-p101 complex upon binding Gβγ and Ras beyond what can be explained simply by membrane association. Continued investigations on membranes that more closely mimic native biological membrane will also be necessary to understand the full biophysical details of PI3Kγ activation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E.coli XL10-GOLD KanR Ultracompetent Cells | Agilent | 200317 |
| E.coli DH10EMBacY Competent Cells | Geneva Biotech | DH10EMBacY |
| Chemicals, peptides, and recombinant proteins | ||
| Deuterium Oxide 99.9% | Sigma | 151882 |
| GTPγS | Sigma | 10220647001 |
| Guanosine 5′-diphosphate (GDP) sodium salt hydrate | Sigma | G7127-100MG |
| Guanosine 5′-triphosphate (GTP) sodium salt hydrate | Sigma | G8877-250MG |
| Sodium deoxycholate | Sigma | D6750 |
| Polyoxyethylene (10) lauryl ether | Sigma | P9769 |
| CHAPS, Molecular Biology Grade | EMD Millipore | 220201 |
| Phosphatidylserine (Porcine Brain) | Avanti | 840032C |
| Phosphatidylethanolamine (Egg yolk) | Sigma | P6386 |
| Cholesterol | Sigma | 47127-U |
| Phosphatidylcholine (Egg yolk) | Avanti | 840051C |
| Phosphatidylinositol-4,5-bisphosphate (Porcine Brain) | Avanti | 840046 |
| Sphingomyelin (Egg yolk) | Sigma | S0756 |
| 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) | Avanti | 850375C |
| 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (18:1, DOPS) | Avanti | 840035C |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[4-(p-maleimidomethyl)cyclohexane-carboxamide] (18:1 MCC-PE) | Avanti | 780201C |
| 10 mg/mL beta casein solution | ThermoFisher | 37528 |
| glucose oxidase from Aspergillus niger (225 U/mg) | Biophoretics | B01357.02 |
| catalase | Sigma | C40-100MG Bovine Liver |
| Trolox | Cayman Chemicals | 10011659 |
| Dyomics 647 maleimide dye | Dyomics | 647P1-03 |
| Coenzyme A | Sigma | C3019 |
| Sulfuric acid | Sigma | 58105–2.5L-PC |
| Critical commercial assays | ||
| Transcreener ADP2 FI Assay (1,000 Assay, 384 Well) | BellBrook Labs | 3013-1K |
| Deposited data | ||
| PDB coordinate file for p110γ-p84 structure | PDB | 8AJ8 |
| EM density file for p110γ-p84 complex | EMD | 27738 |
| HDX-MS proteomics data for all experiments | PRIDE | PXD035723 |
| Recombinant DNA | ||
| pMultiBac-Gβ1/Gγ2 | Rathinaswamy et al.32 | pOP737 |
| pACEBac1-hsp110γ | Rathinaswamy et al.32 | MR30 |
| pMultiBac-hsp110γ-ssp101 | Rathinaswamy et al.32 | MR22 |
| pMultiBac-hsp110γ-mmp84 | Rathinaswamy et al.32 | MR24 |
| pFastBac HRas G12V | Siempelkamp et al.54 | BS9 |
| biGBac hsp110γ/ybbr-hsp84 | This paper | HP28 |
| biGBac hsp110γ/ybbr-hsp101 | This paper | HP29 |
| biGBac hsp110γ/ybbr-hsp101 (β8-β9 loop swap from p84) (Δ699-727, ins p84 resi 566–587) | This paper | MJ301 |
| his6-GST-PrescissionProtease-SNAP-RBD(K65E) | This paper | pSH936 |
| his6TEV-HRas(1–184aa) C118S, C181S | This paper | pSH414 |
| his6-Gγ2, SNAP-Gβ1 (DUAL FastBac) | Rathinaswamy et al.32 | pSH651 |
| Software and algorithms | ||
| COOT-0.9.4.1 | CCP4 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Phenix-1.19.1 | Open source | https://www.phenix-online.org/ |
| PDBePISA (Proteins, Interfaces, Structures and Assemblies) | EMBL-EBI | https://www.ebi.ac.uk/pdbe/pisa/pistart.html |
| ESPript 3.0 | Robert et al.55 | https://espript.ibcp.fr |
| HDExaminer | Sierra Analytics | http://massspec.com/hdexaminer |
| GraphPad Prism 7 | GraphPad | https://www.graphpad.com |
| PyMOL | Schroedinger | http://pymol.org |
| Compass Data Analysis | Bruker | https://www.bruker.com |
| ChimeraX | UCSF | https://www.rbvi.ucsf.edu/chimerax/ |
| AlphaFold2- Multimer | DeepMind | https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb |
| ImageJ/Fiji | ImageJ | https://imagej.net/software/fiji/ |
| Nikon NIS elements | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| Other | ||
| Sf9 insect cells for expression | Expression Systems | 94–001S |
| Insect cell media | Expression Systems | 96-001-01 |
| Hellmanex III cleaning solution | Fisher | 14-385-864 |
| 6-well sticky-side chamber | IBIDI | 80608 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, John E Burke (jeburke@uvic.ca).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
All p110γ, p101, and p84 samples were grown in Spodoptera frugiperda (Sf9) cells. Sf9 cells (94–001S) were obtained from Expression Systems (CA, USA) and were cultured in ESF 921 media (96-001-01, Expression Systems, CA, USA) at 27°C.
Method details
Plasmid generation
Plasmids encoding Homo sapiens p110γ (human), Mus musculus p84 (mouse), Sus scrofa p101 (porcine), and Gβγ were used as previously described.32 The full-length human PIK3R5 (p101) gene was purchased from Addgene (70464), and the full-length human PIK3R6 (p84) gene was purchased from DanaFarber (HsCD00462228). Plasmids encoding HRas was used as previously described.54 PI3K genes were subcloned into pLIB vectors for expression with no engineered tags, while in the case of p110γ a TEV cleavable C-terminal 10x histidine and 2x strep tag was added. Genes were subsequently amplified following the biGBac protocol to generate plasmids containing hsP110γ/hsP101 and hsP110γ/hsP84.
For purification, a 10× histidine tag, a 2× strep tag, and a tobacco etch virus protease cleavage site were cloned to the N terminus of the regulatory subunits for the complex and to p110γ for constructs without regulatory subunits.
Virus generation and amplification
The plasmids encoding genes for insect cell expression were transformed into DH10MultiBac cells (MultiBac, Geneva Biotech) to generate baculovirus plasmid (bacmid) containing the genes of interest. Successful generation was identified by blue-white colony screening and the bacmid was purified using a standard isopropanol-ethanol extraction method. Bacteria were grown overnight (16 h) in 3–5 mL 2xYT (BioBasic #SD7019). Cells were spun down and the pellet was resuspended in 300 μL of 50 mM Tris-HCl, pH 8.0, 10 mM EDTA, 100 mg/mL RNase A. The pellet was lysed by the addition of 300 μL of 1% sodium dodecyl sulfate (SDS) (W/V), 200 mM NaOH, and the reaction was neutralized by addition of 400 μL of 3.0 M potassium acetate, pH 5.5. Following centrifugation at 21130 RCF and 4°C (Rotor #5424 R), the supernatant was mixed with 800 μL isopropanol to precipitate bacmid DNA. Following centrifugation, the pelleted bacmid DNA was washed with 500 μL 70% Ethanol three times. The pellet was then air dried for 1 min and re-suspended in 50 μL Buffer EB (10 mM Tris-Cl, pH 8.5; All buffers from QIAprep Spin Miniprep Kit, Qiagen #27104). Purified bacmid was then transfected into Sf9 cells. 2 mL of Sf9 cells at 0.6X106 cells/mL were aliquoted into a 6-well plate and allowed to attach to form a confluent layer. Transfection reactions were prepared mixing 8–12 μg of bacmid DNA in 100 μL 1xPBS and 12 μg polyethyleneimine (Polyethyleneimine ‘‘Max’’ MW 40.000, Polysciences #24765, USA) in 100 μL 1xPBS and the reaction was allowed to proceed for 20–30 min before addition to an Sf9 monolayer containing well. Transfections were allowed to proceed for 5–6 days before harvesting virus containing supernatant as a P1 viral stock.
Viral stocks were further amplified by adding P1 to Sf9 cells at ∼2 × 106 cells/mL (2/100 volume ratio). This amplification was allowed to proceed for 4–5 days and resulted in a P2 stage viral stock that was used in final protein expression. Harvesting of P2 viral stocks was carried out by centrifuging cell suspensions in 50 mL Falcon tubes at 2281 RCF (Beckman GS-15). To the supernatant containing virus, 5–10% inactivated fetal bovine serum (FBS; VWR Canada #97068-085) was added and the stock was stored at 4°C.
Expression and purification of PI3Kγ constructs
PI3Kγ constructs were expressed in Sf9 insect cells using the baculovirus expression system. Following 55 h of expression, cells were harvested by centrifuging at 1680 RCF (Eppendorf Centrifuge 5810 R) and the pellets were snap-frozen in liquid nitrogen. The complex was purified through a combination of nickel affinity, streptavidin affinity and size exclusion chromatographic techniques.
Frozen insect cell pellets were resuspended in lysis buffer (20 mM Tris pH 8.0, 100 mM NaCl, 10 mM imidazole pH 8.0, 5% glycerol (v/v), 2 mM βME), protease inhibitor (Protease Inhibitor Cocktail Set III, Sigma)) and sonicated for 2 min (15s on, 15s off, level 4.0, Misonix sonicator 3000). Triton X- was added to the lysate to a final concentration of 0.1% and clarified by spinning at 15,000 RCF at 4°C for 45 min (Beckman Coulter JA-20 rotor). The supernatant was loaded onto a 5 mL HisTrap™ FF crude column (GE Healthcare) equilibrated in NiNTA A buffer (20 mM Tris pH 8.0, 100 mM NaCl, 20 mM imidazole pH 8.0, 5% (v/v) glycerol, 2 mM βME). The column was washed with high salt NiNTA A buffer (20 mM Tris pH 8.0, 1 M NaCl, 20 mM imidazole pH 8.0, 5% (v/v) glycerol, 2 mM βME), NiNTA A buffer, 6% NiNTA B buffer (20 mM Tris pH 8.0, 100 mM NaCl, 250 mM imidazole pH 8.0, 5% (v/v) glycerol, 2 mM βME) and the protein was eluted with 100% NiNTA B. The eluent was loaded onto a 5 mL StrepTrap™ HP column (GE Healthcare) equilibrated in gel filtration buffer (20mM Tris pH 8.5, 100 mM NaCl, 50 mM Ammonium Sulfate and 0.5 mM TCEP). The column was washed with the same buffer and loaded with tobacco etch virus protease. After cleavage on the column overnight, the protein was eluted in gel filtration buffer. The protein was concentrated in a 50,000 MWCO Amicon Concentrator (Millipore) to <1 mL and injected onto a Superdex™ 200 10/300 GL Increase size-exclusion column (GE Healthcare) equilibrated in gel filtration buffer. After size exclusion, the protein was concentrated, aliquoted, frozen, and stored at −80°C.
Expression and purification of lipidated Gβγ for kinase activity assays
Full length, lipidated human Gβγ (Gβ1γ2) was expressed in Sf9 insect cells and purified as described previously.56 After 65 h of expression, cells were harvested, and the pellets were frozen as described above. Pellets were resuspended in lysis buffer (20 mM HEPES pH 7.7, 100 mM NaCl, 10 mM βME, protease inhibitor (Protease Inhibitor Cocktail Set III, Sigma)) and sonicated for 2 min (15s on, 15s off, level 4.0, Misonix sonicator 3000). The lysate was spun at 500 RCF (Eppendorf Centrifuge 5810 R) to remove intact cells and the supernatant was centrifuged again at 25,000 RCF for 1 h (Beckman Coulter JA-20 rotor). The pellet was resuspended in lysis buffer and sodium cholate was added to a final concentration of 1% and stirred at 4°C for 1 h. The membrane extract was clarified by spinning at 10,000 RCF for 30 min (Beckman Coulter JA-20 rotor). The supernatant was diluted 3 times with NiNTA A buffer (20 mM HEPES pH 7.7, 100 mM NaCl, 10 mM Imidazole, 0.1% C12E10, 10mM βME) and loaded onto a 5 mL HisTrap™ FF crude column (GE Healthcare) equilibrated in the same buffer. The column was washed with NiNTA A, 6% NiNTA B buffer (20 mM HEPES pH 7.7, 25 mM NaCl, 250 mM imidazole pH 8.0, 0.1% C12E10, 10 mM βME) and the protein was eluted with 100% NiNTA B. The eluent was loaded onto HiTrap™ Q HP anion exchange column equilibrated in Hep A buffer (20 mM Tris pH 8.0, 8 mM CHAPS, 2 mM Dithiothreitol (DTT)). A gradient was started with Hep B buffer (20 mM Tris pH 8.0, 500 mM NaCl, 8 mM CHAPS, 2 mM DTT) and the protein was eluted in ∼50% Hep B buffer. The eluent was concentrated in a 30,000 MWCO Amicon Concentrator (Millipore) to <1 mL and injected onto a Superdex™ 75 10/300 GL size exclusion column (GE Healthcare) equilibrated in Gel Filtration buffer (20 mM HEPES pH 7.7, 100 mM NaCl, 10 mM CHAPS, 2 mM TCEP). Fractions containing protein were pooled, concentrated, aliquoted, frozen and stored at −80°C.
Expression and purification of lipidated HRas G12V for ATPase and HDX-MS experiments
Full-length HRas G12V was expressed by infecting 500 mL of Sf9 cells with 5 mL of baculovirus. Cells were harvested after 55 h of infection and frozen as described above. The frozen cell pellet was resuspended in lysis buffer (50 mM HEPES pH 7.5, 100 mM NaCl, 10 mM βME and protease inhibitor (Protease Inhibitor Cocktail Set III, Sigma)) and sonicated on ice for 1 min 30 s (15s ON, 15s OFF, power level 4.0) on a Misonix sonicator 3000. Triton X-114 was added to the lysate to a final concentration of 1%, mixed for 10 min at 4°C and centrifuged at 25,000 rpm for 45 min (Beckman Ti-45 rotor). The supernatant was warmed to 37°C for few minutes until it turned cloudy following which it was centrifuged at 11,000 rpm at room temperature for 10 min (Beckman JA-20 rotor) to separate the soluble and detergent-enriched phases. The soluble phase was removed, and Triton X-114 was added to the detergent-enriched phase to a final concentration of 1%. Phase separation was performed 3 times. Imidazole pH 8.0 was added to the detergent phase to a final concentration of 15 mM and the mixture was incubated with Ni-NTA agarose beads (Qiagen) for 1 h at 4°C. The beads were washed with 5 column volumes of Ras-NiNTA buffer A (20mM Tris pH 8.0, 100mM NaCl, 15mM imidazole pH 8.0, 10mM βME and 0.5% Sodium Cholate) and the protein was eluted with 2 column volumes of Ras-NiNTA buffer B (20mM Tris pH 8.0, 100mM NaCl, 250mM imidazole pH 8.0, 10mM βME and 0.5% Sodium Cholate). The protein was buffer exchanged to Ras-NiNTA buffer A using a 10,000 kDa MWCO Amicon concentrator, where protein was concentrated to ∼1mL and topped up to 15 mL with Ras-NiNTA buffer A and this was repeated a total of 3 times. GTPγS was added in 2-fold molar excess relative to HRas along with 25 mM EDTA. After incubating for an hour at room temperature, the protein was buffer exchanged with phosphatase buffer (32 mM Tris pH 8.0, 200 mM Ammonium Sulfate, 0.1 mM ZnCl2, 10 mM βME and 0.5% Sodium Cholate). 1 unit of immobilized calf alkaline phosphatase (Sigma) was added per milligram of HRas along with 2-fold excess nucleotide and the mixture was incubated for 1 h on ice. MgCl2 was added to a final concentration of 30 mM to lock the bound nucleotide. The immobilized phosphatase was removed using a 0.22-micron spin filter (EMD Millipore). The protein was concentrated to less than 1 mL and was injected onto a Superdex 75 10/300 GL size exclusion column (GE Healthcare) equilibrated in gel filtration buffer (20 mM HEPES pH 7.7, 100 mM NaCl, 10 mM CHAPS, 1 mM MgCl2 and 2 mM TCEP). The protein was concentrated to 1 mg/mL using a 10,000 kDa MWCO Amicon concentrator, aliquoted, snap-frozen in liquid nitrogen and stored at −80°C.
Expression and purification of complex of porcine p110γ with mouse p84 (X-Ray crystallography)
Constructs of full-length porcine p110γ were cloned into pVL1393 (Invitrogen). The plasmid for EE-tagged mouse p84 was a gift from Len Stephens (The Babraham Institute, UK). The constructs were transfected into Spodoptera frugiperda 9 (Sf9) insect cells with ExGen500 (Fermentas) and incubated at 27°C for 5 days to make baculoviruses. The heterodimeric p110γ-p84 complexes were obtained by co-infection of 3 L of SF9 cells with p110γ-expressing and p84-expressing viruses. Cells were inoculated at a density of 1 × 107 cells/mL and grown in 2L roller bottles standing vertically, with 500 mL of Sf9 cells per bottle. After 62 h incubation at 27°C, cells were harvested, washed in PBS, pelleted, snap-frozen in liquid nitrogen and stored at −80°C.
Purification of complex of porcine p110γ with mouse p84 (X-Ray crystallography)
Frozen cells were resuspended in sonication buffer (50mM TrisHCl pH 8, 100 mM NaCl, 1 mM PEFA, 25 mM imidazole) and lysed by sonication on ice at power 8 for 10 min (Sf9 cells). The lysates were ultracentrifuged at 35,000 rpm for 45 min at 4°C in Ti45 rotor. The soluble cell lysate was filtered through a 0.45 μm filter. Subsequently, the lysate was passed over a 5 mL Ni-NTA Fast Flow column (GE Healthcare) that had been equilibrated with Ni wash buffer (20 mM Tris pH8, 1% Betaine, 0.1 M NaCl, 50 mM potassium phosphate pH 7, 0.05% Tween), washed with 15 mL of Ni wash buffer then eluted in a gradient from Ni A buffer (20 mM Tris pH 8, 300 mM NaCl, 25 mM imidazole) to Ni B (20 mM Tris pH 8, 300 mM NaCl, 500 mM imidazole). Fractions containing the p110-p84 complex were pooled and diluted 1:2 with QA buffer (50 mM Tris pH 8, 2 mM DTT). The diluted sample was loaded onto tandem HiTrap Q (5mL, GE Healthcare) and HiTrap Heparin (5 mL, GE Healthcare) columns that had been equilibrated in tandem with QA buffer. The protein was eluted from the tandem columns with a gradient of QA buffer to QB buffer (50 mM Tris pH 8, 1 M NaCl, 2 mM DTT). The eluted fractions containing the heterodimer were pooled and concentrated to 2 mL in a 50 kD MWCO Amicon Ultra concentrator (Millipore). The concentrated sample was then purified using a Superdex 200 (16/60) gel-filtration column with gel filtration buffer (20 mM Tris pH 7.5, 100 mM NaCl, 2 mM DTT). The fractions containing the heterodimer were pooled and concentrated to 10 mg/mL. One preparation from 3L of Sf9 cells yielded about 11 mg of purified, concentrated heterodimer.
Purification of proteins for TIRF microscopy
Purification of recombinant farnesyl Gβ1Gγ2 and SNAP-Gβ1Gγ2
Proteins were expressed, purified, and labeled with Alexa 488 SNAP dye as previously described.32
Purification of HRas
The coding sequence for human HRas (1–189aa, Uniprot #P01112) was cloned into a pProEX vector to generate a his6-TEV-HRas fusion protein that lacked the last 5 amino acids (i.e. ΔKCVLS∗). In addition, 2 solvent exposed cysteines (C118S and C181S) were mutated leaving a single reactive cysteine (C184), which was used for site-specific conjugation to MCC-PE lipids incorporated in supported lipid bilayers. This form of HRas has been used extensively to characterize GEF-mediated activation of Ras on supported membrane (Iversen et al. 2014). The his6-TEV-HRas fusion protein was expressed in BL21 (DE3) E. coli. Bacteria were initially grown at 37°C in Terrific Broth to an OD600 of 0.8. Cultures were then shifted to 18°C for 1 h, induced with 0.1 mM IPTG, and allowed to express overnight for 20 h at 18°C before being harvested. Cells were lysed into 50 mM Na2HPO4 [pH 8.0], 300 mM NaCl, 0.4 mM BME, 1 mM PMSF, 100 μg/mL DNase using microtip sonication. Lysate was clarified by centrifugation at 16,000 rpm (35,172 x g) for 60 min in a Beckman JA-17 rotor chilled to 4°C. Lysate was circulated over 5 mL HiTrap Chelating column (GE Healthcare, Cat# 17-0409-01) charged with 100 mM CoCl2. Bound protein was eluted with a linear gradient of lysis buffer containing up to 500 mM imidazole [pH 8.0] (8 CV, 40 mL total, 2 mL/min flow rate). Peak fractions were pooled, combined with TEV protease (75 μg/mL final concentration), and dialyzed against 4 L of buffer containing 50 mM Na2HPO4 [pH 8.0], 300 mM NaCl, and 0.4 mM BME for 16–18 h at 4°C. Dialysate containing TEV protease cleaved his6-TEV-HRas was recirculated for 2 h over a 5 mL HiTrap Chelating column. Flow-through containing cleaved HRas was concentrated in a 10 kDa MWCO Amicon spin concentrator before being loaded on a 120 mL Superdex 75 (10/300 GL) gel filtration column equilibrated in 20 mM Tris [pH 8.0], 200 mM NaCl, 10% glycerol, 1 mM TCEP. Peak fractions were pooled and concentrated 400–500 μM before snap freezing in liquid nitrogen. Note that absence of 10% glycerol in the storage buffer causes purified HRas to crystalize during spin concentration.
Purification and labeling of SNAP-RBD (Ras binding domain)
The Ras binding domain (RBD, 56–131aa) derived from human C-Raf kinase (Uniprot, #P04049) was cloned into a pETM-33 vector containing a gene sequence encoding his6-GST-3C-SNAP. Using site-directed mutagenesis, a K58E mutation was introduced into the RBD gene sequence to create a fast-cycling and high specificity Ras(GTP) binding protein as previously reported.57,58 The his6-GST-3C-SNAP-RBD(K65E) fusion protein was expressed in BL21 (DE3) E. coli. Bacteria were initially grown at 37°C in Terrific Broth to an OD600 of 0.8. Cultures were then shifted to 18°C for 1 h, induced with 0.1 mM IPTG, and allowed to express overnight for 20 h at 18°C before being harvested. Cells were lysed into 50 mM Na2HPO4 [pH 8.0], 400 mM NaCl, 0.4 mM BME, 1 mM PMSF, 100 μg/mL DNase using microtip sonication. Lysate was clarified by centrifugation at 16,000 rpm (35,172 x g) for 60 min in a Beckman JA-17 rotor chilled to 4°C. Lysate was circulated over 5 mL HiTrap Chelating column (GE Healthcare, Cat# 17-0409-01) charged with 100 mM CoCl2. Bound protein was eluted with a linear gradient of lysis buffer containing up to 500 mM imidazole [pH 8.0] (8 CV, 40 mL total, 2 mL/min flow rate). Peak fractions were pooled, combined with 0.1 mg/mL prescission protease, and dialyzed against 4 L of buffer containing 20 mM HEPES [pH 7], 400 mM NaCl, 5% glycerol, and 0.4 mM BME for 16–18 h at 4°C. Dialysate containing prescission protease cleaved his6-GST-3C-SNAP-RBD(K65E) was recirculated for 2 h over a 5 mL HiTrap Chelating column. Flow-through containing cleaved SNAP-RBD(K65E) was concentrated in a 10 kDa MWCO Amicon spin concentrator before being loaded on a 120 mL Superdex 75 (10/300 GL) gel filtration column equilibrated in 20 mM HEPES [pH 7], 200 mM NaCl, 10% glycerol, 1 mM TCEP. Peak fractions were pooled and concentrated ∼100 μM before snap freezing in liquid nitrogen and storing in the −80°C.
To fluorescently label SNAP-RBD(K65E), we combined 1 mL of 20 μM protein with 25 μM Alexa 546-SNAP surface dye (New England Biolabs, Cat# S9132S). SNAP dye labeling was performed in buffer containing 20 mM HEPES [pH 7], 200 mM NaCl, 10% glycerol, 1 mM TCEP overnight at 4°C. Labeled protein was then separated from free Alexa 546-SNAP dye using a 5 kDa MWCO Amicon spin concentrator followed by size exclusion chromatography (i.e. Superdex 75 10/300 GL). Peak SEC fractions containing Alexa 546-SNAP-RBD(K65E) were pooled and centrifuged in a 5 kDa MWCO Amicon spin concentrator to reach a final concentration of 20 μM before snap freezing in liquid nitrogen and storing in the −80°C. To calculate the SNAP dye labeling efficiency, we determined that Alexa 546 contributes 10% of the peak A550 signal to the measured A280. We calculate the final concentration of Alexa 546-SNAP-RBD(K65E) using an adjusted A280 (i.e. A280(protein) = A280(observed) – A550(dye)∗0.10) and the following extinction coefficients: ε280(SNAP-RBD) = 26,470 M−1 cm−1, ε650(Alexa 546) = 104,000 M−1 cm−1.
Fluorescent labeling of ybbr-p84-p110γ and ybbr-p101-p110γ
We generated a Dyomics647-CoA conjugate in-house by combining 15 mM Dyomics647 maleimide (Dyomics, Cat #647P1-03) in DMSO with 10 mM CoA (Sigma, #C3019, MW = 785.33 g/mol) dissolved in 1x PBS. This mixture was incubated overnight at 23°C. Unreacted Dyomics647 maleimide was quenched by the addition of 5 mM DTT. We labeled recombinant ybbr-p84-p110γ and ybbr-p101-p110γ containing an N-terminal ybbR13 motif (DSLEFIASKLA) using Sfp transferase and DY647-CoA.59,60 Chemical labeling was achieved by combining 5 μM ybbr-tagged PI3K complexes, 4 μM Sfp-his6, and 10 μM DY647-CoA, in 2 mL of buffer containing 20 mM Tris [pH 8], 150 mM NaCl, 10 mM MgCl2, 10% Glycerol, 1 mM TCEP, 0.05% CHAPS. Following a 4-h labeling reaction on ice, excess DY647-CoA was removed using a PD-10 desalting column. The DY647-ybbr-p84-p110γ and DY647-ybbr-p101-p110γ was concentrate in a 50 kDa MWCO Amicon centrifuge tube and loaded on a Superdex 200 gel filtration column equilibrated in 20 mM Tris [pH 8], 150 mM NaCl, 10% glycerol, 1 mM TCEP, 0.05% CHAPS. Peak fractions were pooled and concentrated to 2–5 μM before flash freezing in liquid nitrogen and storing in the −80°C. We calculated the final concentration of DY647-labeled PI3K complexes using an adjusted A280 (i.e. A280(protein) = A280(observed) – A650(dye)∗0.06) and the following extinction coefficients: ε280(p101-p110γ) = 250,730 M−1 cm−1, ε280(p84-p110γ) = 233,730 M−1 cm−1, ε650(DY647) = 220,000 M−1 cm−1.
Preparation of supported lipid bilayers
The following lipids were used to generated small unilamellar vesicles (SUVs) and subsequently supported lipid bilayers: 1,2-dioleoyl-sn-glycero-3-phosphocholine (18:1 DOPC, Avanti #850375C), L-α-phosphatidylinositol-4,5-bisphosphate (Brain PI(4,5)P2, Avanti #840046X), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[4-(p-maleimidomethyl)cyclohexane-carboxamide] (18:1 MCC-PE, Avanti #780201C). To make liposomes, 2 μmol total lipids are combined in a 35 mL glass round bottom flask containing 2 mL of chloroform. Lipids are dried to a thin film using rotary evaporation with the glass round-bottom flask submerged in a 42°C water bath. After evaporating all the chloroform, the round bottom flask was flushed with nitrogen gas for at least 30 min. Resuspend lipid film in 2 mL of PBS [pH 7.2], making a final concentration of 1 mM total lipids. All lipid mixtures expressed as percentages are equivalent to molar fractions. To generate 50 nm SUVs, 1 mM total lipid mixtures were extruded through a 0.05 μm pore size 19 mm polycarbonate membrane (Sigma, #WHA800308) with filter supports (Avanti #610014) on both sides of the PC membrane.
Supported lipid bilayers are formed on 25 × 75 mm coverglass (IBIDI, #10812). Coverglass was first cleaned with 2% Hellmanex III (Fisher, Cat#14-385-864) heated to 60–70°C in a glass coplin jar. Incubate for at least 30 min. Wash coverglass extensively with MilliQ water and then etched with Pirahna solution (1:3, hydrogen peroxide:sulfuric acid) for 10–15 min the same day SLBs were formed. Etched coverglass, in water, is rapidly dried with nitrogen gas before adhering to a 6-well sticky-side chamber (IBIDI, Cat# 80608). Form SLBs by flowing 30 nm SUVs diluted in PBS [pH 7.2] to a total lipid concentration of 0.25 mM (i.e. 1:4 dilution of extruded lipids). After 30 min, IBIDI chambers are washed with 5 mL of PBS [pH 7.2] to remove non-absorbed SUVs. Membrane defects are blocked for 5–10 min with a 1 mg/mL beta casein (Thermo FisherSci, Cat# 37528) diluted in 1x PBS [pH 7.4]. Before use as a blocking protein, frozen 10 mg/mL beta casein stocks were thawed, centrifuged for 30 min at 21370 x g, and 0.22 μm syringe filtered. After blocking SLBs with beta casein, membranes were washed with 2 mL of PBS, followed by 1 mL of TIRF-M imaging buffer.
Supported membrane containing with MCC-PE lipids were used to covalently couple HRas(GDP). For these SLBs, 100 μL of 30 μM HRas diluted in a 1x PBS [pH 7.4] and 0.1 mM TCEP buffer was added to the IBIDI chamber and incubated for 2 h at 23°C. The addition of TCEP significantly increases the coupling efficiency. SLBs with MCC-PE lipids were then washed with 2 mL of 1x PBS [pH 7.4] containing 5 mM beta-mercaptoethanol (BME) and incubated for 15 min to neutralize the unreacted maleimide headgroups. SLBs were washed with 1mL of 1x PBS, followed by 1 mL of kinase buffer before starting smTIRF-M experiments.
Activation of HRas on supported lipid bilayers
Membrane conjugated HRas(GDP) was converted to HRas (GTP) using either chemical activation (i.e. EDTA/GTP/MgCl2) or with a guanine nucleotide exchange factor (GEF). Chemical activation was achieved by first washing supported membranes containing maleimide conjugated HRas (GDP) with buffer containing 1x PBS, 1 mM EDTA, 1 mM GTP. This was followed by a 15-min incubation to exchange GDP for GTP. To stably associate the newly loaded GTP with HRas, chambers containing the SLBs were subsequently washed with 1x PBS, 1 mM MgCl2, 50 μM GTP. A complementary approach that utilizes GEF-mediated activation of H-Ras was achieved by flowing 50 nM Sos1 catalytic domain over HRas (GDP) conjugated membranes. The mechanism of activation was carried out in buffer containing 1x PBS, 1 mM MgCl2, 50 μM GTP. Both methods of activation yielded the same density of HRas (GTP) and have been validated in previous studies. Efficient activation of membrane tethered HRas was assessed by visualizing the nucleotide dependent localization of SNAP Alexa 546 labeled Ras binding domain (Ax546-RBD) derived from c-Raf kinase using TIRF microscopy (Figures S5A-S5C). The experimental data shown in Figure 5 was collected under conditions that utilized chemical activation of HRas.
Quantification of supported membrane localization
In the presence HRas(GDP), we detected no membrane binding events for either DY647-p101-p110γ and DY647-p84-p110γ. This condition served as a negative control for determining the background level of fluorescence for bulk membrane absorption experiments shown in Figures 5B-5C. The average membrane fluorescence intensity measured by TIRF-M in the presence of HRas(GDP) was subtracted from data collected in the presence of membrane tethered HRas(GTP) and/or Gβγ to yield the plots in Figures 5B-5C.
Single molecule TIRF microscopy experiments
All smTIRF-M experiments were performed on an inverted Nikon Ti2 microscope using a 100× Nikon objective (1.49 NA) oil immersion TIRF objective. The x axis and y axis positions were controlled using a Nikon motorized stage. Fluorescently labeled proteins were excited with either a 488, 561, or 637 nm diode lasers (OBIS laser diode, Coherent Inc. Santa Clara, CA) controlled by a Vortran laser drive with acousto-optic tunable filters (AOTF). The power output measured through the objective for single particle imaging was 1–3 mW. Excitation light passing through quad multi-pass dichroic filter cube (Semrock). Fluorescence emission passed through Nikon emission filter wheel containing the following 25 mm emission filters: ET525/50M, ET600/50M, ET700/75M (Semrock) and then detected using an iXion Life 897 EMCCD camera (Andor Technology Ltd., UK). All experiments smTIRF-M were performed at room temperature (23°C). Microscope hardware was controlled using Nikon NIS elements.
All smTIRF-M experiments were performed on supported membrane used the following imaging buffer: 20 mM HEPES [pH 7.0], 150 mM NaCl, 50 μM GTP, 1 mM ATP, 5 mM MgCl2, 0.5 mM EGTA, 20 mM glucose, 200 μg/mL beta casein (ThermoScientific, Cat# 37528), 20 mM BME, 320 μg/mL glucose oxidase (Biophoretics, Cat# B01357.02), 50 μg/mL catalase (Sigma, #C40-100MG Bovine Liver), and 2 mM Trolox (Cayman Chemical, Cat#10011659).61 Perishable reagents (i.e. glucose oxidase, catalase, and Trolox) were added 5–10 min before image acquisition.
Alphafold2 modeling
We utilized the AlphaFold2 using Mmseqs2 notebook of ColabFold at colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb62 to make structural predictions of p110γ bound to p84, p101 bound to Gβγ, and p110γ bound to Gβγ. The pLDDT confidence values consistently scored above 90% for all models, with the predicted aligned error and pLDDT scores for all models are shown in Figures S1, S6, S7. The best models for Gβγ bound to the helical domain of p110γ and the C-terminal domain of p101 are included as PDB files in the source data.
Crystallization of porcine p110γ/mouse p84
For initial screens, 100 nL drops of purified, concentrated heterodimer at 10 mg/mL were dispensed into LMB 96-well plates with 100 nL of reservoir solution. The initial screen was the 2000 condition LMB screen,63,64 containing a wide range of crystallisation solutions, using an Innovadyne crystallisation robot. The plates were stored at 17°C. To improve initial crystals, 1μL drops of protein and 1μL drops of well solution were manually pipetted into 24 well plates (either sitting drop or hanging drop). Seeding from the existing crystals into the fresh drop was performed using a Hampton seeding tool. The plates were then stored at 17°C or 4°C. Crystals were initially obtained from a Morpheus screen.65 Optimized crystals were grown from a crystallization solution containing 16% EDO_P8K (20% w/v PEG 8000, 40% v/v ethylene glycol), 0.06 M amino acids (0.2 M sodium L-glutamate, 0.2 M DL-alanine, 0.2 M glycine, 0.2 M DL-lysine, 0.2 M DL-serine), 0.08 M buffer 2 pH7.5 (0.5 M HEPES, 0.5 M MOPS), 0.4 M Na/K phosphate pH 6.3. Crystals were 120 μm × 50 μm x 10μm plates that diffracted to 8 Å resolution (Table S1).
X-Ray data collection/refinement for complex of porcine p110γ with mouse p84
Diffraction data collected with remote control at ESRF beamline ID29, using a wavelength of 0.9762. Images were integrated with MOSFLM66 and scaled with SCALA.67 Molecular replacement and refinement were carried out using PHASER68 and Phenix.refine.69 For molecular replacement a model of the p110γ-p84 complex was generated in COOT70 from a composite of the alphafold2 model of the p110γ C2 domain and RBD-C2 and C2-helical linkers bound to full length p84 with the rest of the sus scrofus p110γ subunit assembled from an alphafold generated model templated on the human p110γ from the PDB entry 7MEZ.32 There were four heterodimers per asymmetric unit. The entire assembly was then subjected to rigid-body, xyz reciprocal space, and group B-factor refinement in phenix-refine69 using NCS and secondary structure restraints. Due to the low resolution, no manual adjustments were made in the model. Statistics for the final model are shown in Table S1.
Negative stain electron microscopy
Purified human p110γ-mouse p84 was adsorbed to glow discharged carbon coated grids at a concentration of 0.02 mg/mL for 5s and stained with uranyl formate. The stained specimen was examined using a Tecnai Spirit transmission electron microscope (ThermoFisher Scientific) operated at an accelerating voltage of 120 kV and equipped with an FEI Eagle 4K charged-coupled-device (CCD) camera. 50 micrographs were acquired at a nominal magnification of 49,000x at a defocus of −1.2mm and binned twice to obtain a final pixel size of 4.67 Å/pixel. The contrast transfer function (CTF) of each micrograph was estimated using CTFFind4.1 within Relion 3.0.8. 200 particles were manually picked then aligned to generate 2D class averages for template-based autopicking. These templates were then used to autopick 20,610 particles which were extracted with a box size of 336 Å. Particles were then exported to cryoSPARC v2.14.2 for 2D classification and 10,344 particles which classified to “good” classes were selected and subjected to ab initio reconstruction with a max alignment resolution of 12 Å. The same particles were then used for homogeneous refinement of the ab initio model, yielding the final map at 19 Å resolution, as calculated by the gold standard Fourier Shell Correlation (FSC) at 0.143 cutoff.
Lipid vesicle preparation for kinase activity assays
Lipid vesicles for HDX-MS and bulk kinase assays were prepared by mixing the lipids solutions in organic solvent. The solvent was evaporated in a stream of argon following which the lipid film was desiccated in a vacuum for 45 min. The lipids were resuspended in lipid buffer (20 mM HEPES pH 7.0, 100 mM NaCl and 10% glycerol) at a concentration of 5 mg/mL and the solution was bath sonicated for 15 min. The vesicles were subjected to five freeze thaw cycles and extruded 11 times through a 100-nm filter (T&T Scientific: TT-002-0010). The extruded vesicles were aliquoted and stored at −80°C.
Kinase assays
All kinase assays were done using Transcreener ADP2 Fluorescence Intensity (FI) assays (Bellbrook labs) which measures ADP production. PM-mimic vesicles [5% phosphatidylinositol 4,5-bisphosphate (PIP2), 20% phosphatidylserine (PS), 10% phosphatidylcholine (PC), 35% phosphatidylethanolamine (PE), 25% cholesterol, 5% sphingomyelin (SM)] were at a final concentration of 0.5 mg/mL, ATP at a final concentration of 100 μM and HRas at final concentrations ranging from 10 nM to 1.5 μM were used. For assays measuring co-stimulation with Gβγ, 1.5 μM of the activator was used in the reaction. Final concentration of kinase ranged from 400 nM to 2000 nM for both p110γ-p101 and p110γ-p84. For conditions with Gβγ, final kinase concentrations of kinase ranged from 100 nM to 400 nM for p110γ-p84 and from 3 nM to 10 nM for p110γ-p101. Assays comparing the activation of p110γ-p101, the loop swap mutant, and p110γ-p84 by Gβγ (Figure 6G) used lipid vesicles containing 5% PIP2, 65% phosphatidylethanolamine (PE), and 30% phosphatidylserine (PS) at a final concentration of 0.75 mg/mL, ATP at a final concentration of 100 μM, and 100 nM Gβγ. Kinase was present at concentrations ranging from 17 to 400 nM.
2 μL of 2X substrate solution containing vesicles, the appropriate concentration of Ras and Gβγ (for conditions assaying co-stimulation) was mixed with 2 μL of 2X kinase solution and the reaction was allowed to proceed for 60 min. The reactions were stopped with 4 μL of 2X stop and detect solution containing Stop and Detect buffer, 8 nM ADP Alexa Fluor 594 Tracer and 93.7 μg/mL ADP2 Antibody IRDye QC-1 and incubated for 50 min. The fluorescence intensity was measured using a SpectraMax M5 plate reader at excitation 590 nm and emission 620 nm. The % ATP turnover was interpolated from a standard curve (0.1–100 μM ADP) using Graphpad prism, with these values converted into specific activity based on the concentration of protein.
Hydrogen deuterium eXchange mass spectrometry- activators
Exchange reactions were carried out at 18°C in 12 μL volumes with final concentrations of 1.5 μM, 3 μM, 3 μM for p110γ-p84, HRas (G12V) and Gβγ respectively. A total of five conditions were assessed: p110γ-p84, p110γ-p84 + HRas (G12V), p110γ-p84 + Gβγ, and p110γ-p84 + HRas(G12V) + Gβγ. All conditions were in the presence of PM mimic membranes [5%PIP2, 20% phosphatidylserine (PS), 10% phosphatidylcholine (PC), 35% phosphatidylethanolamine (PE), 25% cholesterol, 5% sphingomyelin (SM)] at a final concentration of 0.42 mg/mL. Mixtures of lipid vesicles and activators (HRas(G12V)/Gβγ) were prepared by combining 1 μL of lipid vesicles or vesicle buffer (25mM HEPES 7.0, 100mM NaCl, 10% glycerol) with 0.85 μL of HRas(G12V) or HRas buffer (20mM HEPES pH 7.7, 100mM NaCl, 10mM CHAPS, 2mM TCEP), and 0.63 μL of Gβγ or Gβγ buffer (20mM HEPES pH 7.7, 100mM NaCl, 8mM CHAPS, 2mM TCEP). Prior to the addition of D2O, 1.2 μL of p110γ-p84 was added to the lipid-activator mixture, and the solution was left to incubate at 18°C for 2 min. The hydrogen-deuterium exchange reaction was initiated by the addition of 8.32 μL D2O buffer (94.3% D2O, 100 mM NaCl, 20 mM HEPES pH 7.5) to the 3.68 μL protein or protein-lipid solutions for a final D2O concentration of 65.5%. Exchange was carried out over four time points (3s, 30s, 300s, 3000s) and terminated by the addition of 60 μL ice-cold acidic quench buffer (0.6 M guanidine-HCl, 0.9% formic acid final).
Hydrogen deuterium eXchange mass spectrometry- regulators
Exchange reactions were carried out at 18°C in 50 μL volumes with final concentrations of 0.4 μM, human p110γ/mouse p84 or human p110γ/porcine p101. The hydrogen-deuterium exchange reaction was initiated by the addition of 48.6 μL D2O buffer (94.3% D2O, 100 mM NaCl, 20 mM HEPES pH 7.5) to the 1.4 μL protein solutions for a final D2O concentration of 91.7% Exchange was carried out over five time points (3s, 30s, 300s, 3000 s at 18°C and 3s at 4°C) and terminated by the addition of 20 μL ice-cold acidic quench buffer (0.6 M guanidine-HCl, 0.9% formic acid final).
Hydrogen deuterium eXchange mass spectrometry-human regulators
Exchange reactions were carried out at 18°C in either 6 μl (high concentration) or 50 μL(low concentration) volumes with final concentrations of 1.5 μM(high) or 0.175 μM (low) human p110γ/mouse p84 or human p110γ/porcine p101. The hydrogen-deuterium exchange reaction was initiated by the addition of 3 μL or 25 μL D2O buffer (94.3% D2O, 100 mM NaCl, 20 mM HEPES pH 7.5) to the 3 μL or 25 μL protein solutions for a final D2O concentration of 47.2% Exchange was carried out over two time points (30s, 300 s at 18°C) and terminated by the addition of 64 μL or 20 μL ice-cold acidic quench buffer (0.6 M guanidine-HCl, 0.9% formic acid final).
Protein digestion and MS/MS data collection
Protein samples were rapidly thawed and injected onto an integrated fluidics system containing a HDx-3 PAL liquid handling robot and climate-controlled chromatography system (LEAP Technologies), a Dionex Ultimate 3000 UHPLC system, as well as an Impact HD QTOF Mass spectrometer (Bruker). The protein was run over two immobilized pepsin columns (Applied Biosystems; Poroszyme™ Immobilized Pepsin Cartridge, 2.1 mm × 30 mm; Thermo-Fisher 2-3131-00; at 10°C and 2°C respectively), or for the high low human regulator HDX over one immobilized Nepenthesin-2 column from Affipro (AP-PC-004), at 200 μL/min for 3 min. The resulting peptides were collected and desalted on a C18 trap column [Acquity UPLC BEH C18 1.7 mm column (2.1 × 5 mm); Waters 186003975]. The trap was subsequently eluted in line with an ACQUITY 1.7 μm particle, 100 × 1 mm2 C18 UPLC column (Waters 186002352), using a gradient of 3–35% B (buffer A, 0.1% formic acid; buffer B, 100% acetonitrile) over 11 min immediately followed by a gradient of 35–80% B over 5 min. MS experiments acquired over a mass range from 150 to 2200 mass/charge ratio (m/z) using an electrospray ionization source operated at a temperature of 200°C and a spray voltage of 4.5 kV.
Peptide identification
Peptides were identified using data-dependent acquisition following tandem MS/MS experiments (0.5 s precursor scan from 150 to 2000 m/z; twelve 0.25 s fragment scans from 150 to 2000 m/z). MS/MS datasets were analyzed using PEAKS7 (PEAKS), and a false discovery rate was set at 0.1% using a database of purified proteins and known contaminants.71 The search parameters were set with a precursor tolerance of 20 parts per million, fragment mass error 0.02 Da, and charge states 1–8.
Quantification and statistical analysis
Single particle tracking
Particle detection and tracking of DY647-p101-p110γ and DY647-p84-p110γ was performed using the ImageJ/Fiji TrackMate plugin.72 Image stacks were loaded into ImageJ/Fiji as .nd2 files and cropped to 400 × 400 pixels to minimize differences in field illumination caused by TIRF illumination. Using the LoG detector option, particles were identified based on brightness and their signal-to-noise ratio. Working with dimension in pixels, particles with a “blob” diameter of 6 pixels were identified. After identifying the position of all fluorescent particles, we used the LAP tracker to generate particle trajectories that monitor molecular displacement as a function of time. Particle trajectories were filtered based on the following parameters: Track Start (removed trajectories that began in first frame), Track End (removed trajectories present in last frame), Duration ( ≤2 frames), Track displacement (removed immobilized particles), and X – Y location (removed particles on the edge of the images). Our filtering parameters removed 1–5% of particle trajectories that were either immobilized or displayed fluorescence brightness that was either too bright or dim compared to the mean particle intensity. The TrackMate output files were analyzed using Prism 9 to calculate the single molecule dwell times and diffusion coefficients.
For all analysis presented in this manuscript, the bin size for the step size distribution equals 0.01 μm. For curving fitting, the step-size distributions were plotted as probability density versus step size (μm). Probability density was calculated by dividing the frequency by a bin size of 0.01 μm. The probability density versus step size plots were fit to the following one- or two-species distribution models:
Single species model
Two species model
Variables are defined as the D1 = diffusion coefficient species 1 (μm2/s), D2 = diffusion coefficient species 2 (μm2/s), alpha (α = % of species 1), r = step size (μm), τ = time interval between steps in seconds. The final step size distribution plots were fit in Prism graphing software using the following equations: f(r) = x/(2∗D1∗t)∗exp(-(xˆ2/(4∗D1∗t))) for a 1 species model, f(r) = alpha∗(x/(2∗D1∗t)∗exp(-(xˆ2/(4∗D1∗t))))+(1-alpha)∗(x/(2∗D2∗t)∗exp(-(xˆ2/(4∗D2∗t)))) for a 2 species model.
To calculate the single molecule dwell times for DY647-p101-p110γ and DY647-p84-p110γ we generated a cumulative distribution frequency (CDF) plot using the frame interval as the bin size (e.g. 50 ms). After plotting the log10(1-CDF) as a function of dwell time the data was fit to either a single or double exponential decay curve. See Figures S5D-S5G for an example of our data processing.
Single exponential model
Two exponential model
Fitting procedure initiated with a single exponential. In cases of a low-quality single exponential fit, a maximum of two species model was used. For double exponential fit, alpha (α) represents the fraction of fast dissociating molecules characterized by the exponential decay time constant, τ1.
Mass analysis of peptide centroids and measurement of deuterium incorporation for HDX-MS experiments
HD-Examiner Software (Sierra Analytics) was used to automatically calculate the level of deuterium incorporation into each peptide. All peptides were manually inspected for correct charge state, correct retention time, and appropriate selection of isotopic distribution. Deuteration levels were calculated using the centroid of the experimental isotope clusters. HDX-MS results are presented with no correction for back exchange shown in the Source data, with the only correction being applied correcting for the deuterium oxide percentage of the buffer used in the exchange. Changes in any peptide at any time point greater than specified cut-offs (5% and 0.3 Da, or 7% and 0.5Da for human regulator HDX) and with an unpaired, two-tailed t test value of p < 0.01 was considered significant.
The raw peptide deuterium incorporation graphs for a selection of peptides with significant differences are shown, with the raw data for all analyzed peptides in the source data. To allow for visualization of differences across all peptides, we utilized number of deuteron difference (#D) plots. These plots show the total difference in deuterium incorporation over the entire H/D exchange time course, with each point indicating a single peptide. These graphs are calculated by summing the differences at every time point for each peptide and propagating the error (example Figures 2E, 4A-4C). For a selection of peptides we are showing the %D incorporation over a time course, which allows for comparison of multiple conditions at the same time for a given region (Figure S4). Samples were only compared within a single experiment and were never compared to experiments completed at a different time with a different final D2O level. The data analysis statistics for all HDX-MS experiments are in Table S2 according to the guidelines of.73 The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with identifier PXD035723.1
Acknowledgments
We thank Alex Berndt and ESRF ID29 beamline scientist Christoph Müller-Dieckmann for assistance with X-ray diffraction data collection. J.E.B. is supported by an operating grant from the Canadian Institutes of Health Research (CIHR, 168998), with salary support from the Michael Smith Foundation for Health Research Scholar award (MSFHR-17686). S.D.H. was supported by an NSF CAREER Award (MCB-2048060). B.R.D. was supported by the Molecular Biology and Biophysics Training Program (NIH T32 GM007759). C.K.Y. is supported by CIHR (FDN-143228) and the Natural Sciences and Engineering Research Council of Canada (RGPIN-2018-03951). R.L.W. is supported by the Medical Research Council (MC_U105184308) and Cancer Research UK (grant DRCPGM\100014). X.Z. was supported by an MRC-LMB Cambridge Scholarship and by the Cambridge Overseas Trust.
Author contributions
Conceptualization, M.K.R., S.D.H., and J.E.B.; validation, N.J.H.; formal analysis, M.K.R., M.L.J., N.J.H., X.Z., S.D.H., and J.E.B.; investigation, M.K.R., B.R.D., M.L.J., X.Z., N.J.H., U.D., J.T.E., J.T.B.S., and K.D.F.; writing – original draft, M.L.J., S.D.H., and J.E.B.; writing – review & editing, M.K.R., M.L.J., R.L.W., C.K.Y., S.D.H., and J.E.B.; supervision, R.L.W., C.K.Y., S.D.H., and J.E.B.; project administration, S.D.H. and J.E.B.; funding acquisition, R.L.W., S.D.H., and J.E.B.
Declaration of interests
J.E.B. reports personal fees from Scorpion Therapeutics and Olema Oncology and research grants from Novartis.
Published: February 26, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112172.
Contributor Information
Scott D. Hansen, Email: shansen5@uoregon.edu.
John E. Burke, Email: jeburke@uvic.ca.
Supplemental information
The highest ranked alphafold models underlying the Gβγ interfaces in both p110γ and p101 are included in a zip file, with the b-factor column in the PDB file indicating the pLDDT score.
Data and code availability
-
•
The data underlying this manuscript are available at the following databases: The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository1 with the dataset identifier PXD035723, the X-ray crystallography data and model for the p110γ-p84 structure was deposited in the Protein DataBank (PDB), with the identifier PDB: 8AJ8, the EM density for the p110γ-p84 complex was deposited in the Electron Microscopy DataBank with the identifier 27738, and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Perez-Riverol Y., Bai J., Bandla C., García-Seisdedos D., Hewapathirana S., Kamatchinathan S., Kundu D.J., Prakash A., Frericks-Zipper A., Eisenacher M., et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022;50:D543–D552. doi: 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lanahan S.M., Wymann M.P., Lucas C.L. The role of PI3Kγ in the immune system: new insights and translational implications. Nat. Rev. Immunol. 2022;22:687–700. doi: 10.1038/s41577-022-00701-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stephens L.R., Eguinoa A., Erdjument-Bromage H., Lui M., Cooke F., Coadwell J., Smrcka A.S., Thelen M., Cadwallader K., Tempst P., Hawkins P.T. The G beta gamma sensitivity of a PI3K is dependent upon a tightly associated adaptor, p101. Cell. 1997;89:105–114. doi: 10.1016/s0092-8674(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 4.Suire S., Coadwell J., Ferguson G.J., Davidson K., Hawkins P., Stephens L. p84, a new Gbetagamma-activated regulatory subunit of the type IB phosphoinositide 3-kinase p110gamma. Curr. Biol. 2005;15:566–570. doi: 10.1016/j.cub.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 5.Voigt P., Dorner M.B., Schaefer M. Characterization of p87PIKAP, a novel regulatory subunit of phosphoinositide 3-kinase γ that is highly expressed in heart and interacts with PDE3B. J. Biol. Chem. 2006;281:9977–9986. doi: 10.1074/jbc.M512502200. [DOI] [PubMed] [Google Scholar]
- 6.Rathinaswamy M.K., Burke J.E. Class I phosphoinositide 3-kinase (PI3K) regulatory subunits and their roles in signaling and disease. Adv. Biol. Regul. 2020;75:100657. doi: 10.1016/j.jbior.2019.100657. [DOI] [PubMed] [Google Scholar]
- 7.Li Z., Jiang H., Xie W., Zhang Z., Smrcka A.V., Wu D. Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science. 2000;287:1046–1049. doi: 10.1126/science.287.5455.1046. [DOI] [PubMed] [Google Scholar]
- 8.Laffargue M., Calvez R., Finan P., Trifilieff A., Barbier M., Altruda F., Hirsch E., Wymann M.P. Phosphoinositide 3-kinase gamma is an essential amplifier of mast cell function. Immunity. 2002;16:441–451. doi: 10.1016/s1074-7613(02)00282-0. [DOI] [PubMed] [Google Scholar]
- 9.Hirsch E., Katanaev V.L., Garlanda C., Azzolino O., Pirola L., Silengo L., Sozzani S., Mantovani A., Altruda F., Wymann M.P. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science. 2000;287:1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- 10.Nürnberg B., Beer-Hammer S. Function, regulation and biological roles of PI3Kγ variants. Biomolecules. 2019;9:E427. doi: 10.3390/biom9090427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barber D.F., Bartolomé A., Hernandez C., Flores J.M., Redondo C., Fernandez-Arias C., Camps M., Rückle T., Schwarz M.K., Rodríguez S., et al. PI3Kgamma inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat. Med. 2005;11:933–935. doi: 10.1038/nm1291. [DOI] [PubMed] [Google Scholar]
- 12.Camps M., Rückle T., Ji H., Ardissone V., Rintelen F., Shaw J., Ferrandi C., Chabert C., Gillieron C., Françon B., et al. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat. Med. 2005;11:936–943. doi: 10.1038/nm1284. [DOI] [PubMed] [Google Scholar]
- 13.Fougerat A., Gayral S., Gourdy P., Schambourg A., Rückle T., Schwarz M.K., Rommel C., Hirsch E., Arnal J.-F., Salles J.-P., et al. Genetic and pharmacological targeting of phosphoinositide 3-kinase-gamma reduces atherosclerosis and favors plaque stability by modulating inflammatory processes. Circulation. 2008;117:1310–1317. doi: 10.1161/CIRCULATIONAHA.107.720466. [DOI] [PubMed] [Google Scholar]
- 14.Campa C.C., Silva R.L., Margaria J.P., Pirali T., Mattos M.S., Kraemer L.R., Reis D.C., Grosa G., Copperi F., Dalmarco E.M., et al. Inhalation of the prodrug PI3K inhibitor CL27c improves lung function in asthma and fibrosis. Nat. Commun. 2018;9:5232. doi: 10.1038/s41467-018-07698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Becattini B., Marone R., Zani F., Arsenijevic D., Seydoux J., Montani J.P., Dulloo A.G., Thorens B., Preitner F., Wymann M.P., Solinas G. PI3Kgamma within a nonhematopoietic cell type negatively regulates diet-induced thermogenesis and promotes obesity and insulin resistance. Proc. Natl. Acad. Sci. USA. 2011;108:E854–E863. doi: 10.1073/pnas.1106698108/-/DCSupplemental. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Breasson L., Becattini B., Sardi C., Molinaro A., Zani F., Marone R., Botindari F., Bousquenaud M., Ruegg C., Wymann M.P., Solinas G. PI3Kγ activity in leukocytes promotes adipose tissue inflammation and early-onset insulin resistance during obesity. Sci. Signal. 2017;10:eaaf2969. doi: 10.1126/scisignal.aaf2969. [DOI] [PubMed] [Google Scholar]
- 17.Torres C., Mancinelli G., Cordoba-Chacon J., Viswakarma N., Castellanos K., Grimaldo S., Kumar S., Principe D., Dorman M.J., McKinney R., et al. p110γ deficiency protects against pancreatic carcinogenesis yet predisposes to diet-induced hepatotoxicity. Proc. Natl. Acad. Sci. USA. 2019;116:14724–14733. doi: 10.1073/pnas.1813012116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang S., Chung W.-C., Wu G., Egan S.E., Miele L., Xu K. Manic fringe promotes a claudin-low breast cancer phenotype through notch-mediated PIK3CG induction. Cancer Res. 2015;75:1936–1943. doi: 10.1158/0008-5472.CAN-14-3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaneda M.M., Messer K.S., Ralainirina N., Li H., Leem C.J., Gorjestani S., Woo G., Nguyen A.V., Figueiredo C.C., Foubert P., et al. PI3Kγ is a molecular switch that controls immune suppression. Nature. 2016;539:437–442. doi: 10.1038/nature19834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Henau O., Rausch M., Winkler D., Campesato L.F., Liu C., Cymerman D.H., Budhu S., Ghosh A., Pink M., Tchaicha J., et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature. 2016;539:443–447. doi: 10.1038/nature20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H., Prever L., Hirsch E., Gulluni F. Targeting PI3K/AKT/mTOR signaling pathway in breast cancer. Cancers. 2021;13:3517. doi: 10.3390/cancers13143517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takeda A.J., Maher T.J., Zhang Y., Lanahan S.M., Bucklin M.L., Compton S.R., Tyler P.M., Comrie W.A., Matsuda M., Olivier K.N., et al. Human PI3Kγ deficiency and its microbiota-dependent mouse model reveal immunodeficiency and tissue immunopathology. Nat. Commun. 2019;10:4364. doi: 10.1038/s41467-019-12311-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thian M., Hoeger B., Kamnev A., Poyer F., Köstel Bal S., Caldera M., Jiménez-Heredia R., Huemer J., Pickl W.F., Groß M., et al. Germline biallelic PIK3CG mutations in a multifaceted immunodeficiency with immune dysregulation. Haematologica. 2020;105:e488. doi: 10.3324/haematol.2019.231399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stoyanov B., Volinia S., Hanck T., Rubio I., Loubtchenkov M., Malek D., Stoyanova S., Vanhaesebroeck B., Dhand R., Nürnberg B. Cloning and characterization of a G protein-activated human phosphoinositide-3 kinase. Science. 1995;269:690–693. doi: 10.1126/science.7624799. [DOI] [PubMed] [Google Scholar]
- 25.Schmid M.C., Avraamides C.J., Dippold H.C., Franco I., Foubert P., Ellies L.G., Acevedo L.M., Manglicmot J.R.E., Song X., Wrasidlo W., et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kγ, a single convergent point promoting tumor inflammation and progression. Cancer Cell. 2011;19:715–727. doi: 10.1016/j.ccr.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo L., Wall A.A., Yeo J.C., Condon N.D., Norwood S.J., Schoenwaelder S., Chen K.W., Jackson S., Jenkins B.J., Hartland E.L., et al. Rab8a interacts directly with PI3Kγ to modulate TLR4-driven PI3K and mTOR signalling. Nat. Commun. 2014;5:4407. doi: 10.1038/ncomms5407. [DOI] [PubMed] [Google Scholar]
- 27.Deladeriere A., Gambardella L., Pan D., Anderson K.E., Hawkins P.T., Stephens L.R. The regulatory subunits of PI3Kγ control distinct neutrophil responses. Sci. Signal. 2015;8:ra8. doi: 10.1126/scisignal.2005564. [DOI] [PubMed] [Google Scholar]
- 28.Rynkiewicz N.K., Anderson K.E., Suire S., Collins D.M., Karanasios E., Vadas O., Williams R., Oxley D., Clark J., Stephens L.R., Hawkins P.T. Gβγ is a direct regulator of endogenous p101/p110γ and p84/p110γ PI3Kγ complexes in mouse neutrophils. Sci. Signal. 2020;13:eaaz4003. doi: 10.1126/scisignal.aaz4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bohnacker T., Marone R., Collmann E., Calvez R., Hirsch E., Wymann M.P. PI3Kgamma adaptor subunits define coupling to degranulation and cell motility by distinct PtdIns(3,4,5)P3 pools in mast cells. Sci. Signal. 2009;2:ra27. doi: 10.1126/scisignal.2000259. [DOI] [PubMed] [Google Scholar]
- 30.Vadas O., Dbouk H.A., Shymanets A., Perisic O., Burke J.E., Abi Saab W.F., Khalil B.D., Harteneck C., Bresnick A.R., Nürnberg B., et al. Molecular determinants of PI3Kγ-mediated activation downstream of G-protein-coupled receptors (GPCRs) Proc. Natl. Acad. Sci. USA. 2013;110:18862–18867. doi: 10.1073/pnas.1304801110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maier U., Babich A., Nürnberg B. Roles of non-catalytic subunits in gbetagamma-induced activation of class I phosphoinositide 3-kinase isoforms beta and gamma. J. Biol. Chem. 1999;274:29311–29317. doi: 10.1074/jbc.274.41.29311. [DOI] [PubMed] [Google Scholar]
- 32.Rathinaswamy M.K., Dalwadi U., Fleming K.D., Adams C., Stariha J.T.B., Pardon E., Baek M., Vadas O., DiMaio F., Steyaert J., et al. Structure of the phosphoinositide 3-kinase (PI3K) p110γ-p101 complex reveals molecular mechanism of GPCR activation. Sci. Adv. 2021;7:eabj4282. doi: 10.1126/sciadv.abj4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shymanets A., Prajwal P., Bucher K., Beer-Hammer S., Harteneck C., Nürnberg B. p87 and p101 subunits are distinct regulators determining class IB phosphoinositide 3-kinase (PI3K) specificity. J. Biol. Chem. 2013;288:31059–31068. doi: 10.1074/jbc.M113.508234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kurig B., Shymanets A., Bohnacker T., Prajwal, Brock C., Ahmadian M.R., Schaefer M., Gohla A., Harteneck C., Wymann M.P., et al. Ras is an indispensable coregulator of the class IB phosphoinositide 3-kinase p87/p110gamma. Proc. Natl. Acad. Sci. USA. 2009;106:20312–20317. doi: 10.1073/pnas.0905506106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walker E.H., Perisic O., Ried C., Stephens L., Williams R.L. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature. 1999;402:313–320. doi: 10.1038/46319. [DOI] [PubMed] [Google Scholar]
- 36.Rathinaswamy M.K., Gaieb Z., Fleming K.D., Borsari C., Harris N.J., Moeller B.E., Wymann M.P., Amaro R.E., Burke J.E. Disease-related mutations in PI3Kγ disrupt regulatory C-terminal dynamics and reveal a path to selective inhibitors. Elife. 2021;10:e64691. doi: 10.7554/eLife.64691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pacold M.E., Suire S., Perisic O., Lara-Gonzalez S., Davis C.T., Walker E.H., Hawkins P.T., Stephens L., Eccleston J.F., Williams R.L. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell. 2000;103:931–943. doi: 10.1016/s0092-8674(00)00196-3. [DOI] [PubMed] [Google Scholar]
- 38.Shymanets A., Ahmadian M.R., Kössmeier K.T., Wetzker R., Harteneck C., Nürnberg B. The p101 subunit of PI3Kγ restores activation by Gβ mutants deficient in stimulating p110γ. Biochem. J. 2012;441:851–858. doi: 10.1042/BJ20111664. [DOI] [PubMed] [Google Scholar]
- 39.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Evans R., O’Neill M., Pritzel A., Antropova N., Senior A., Green T., Žídek A., Bates R., Blackwell S., Yim J., et al. Protein complex prediction with AlphaFold-multimer. bioRxiv. 2021 doi: 10.1101/2021.10.04.463034. Preprint at. [DOI] [Google Scholar]
- 41.Gangadhara G., Dahl G., Bohnacker T., Rae R., Gunnarsson J., Blaho S., Öster L., Lindmark H., Karabelas K., Pemberton N., et al. A class of highly selective inhibitors bind to an active state of PI3Kγ. Nat. Chem. Biol. 2019;15:348–357. doi: 10.1038/s41589-018-0215-0. [DOI] [PubMed] [Google Scholar]
- 42.Lodowski D.T., Pitcher J.A., Capel W.D., Lefkowitz R.J., Tesmer J.J.G. Keeping G proteins at bay: a complex between G protein-coupled receptor kinase 2 and Gßγ. Science. 2003;300:1256–1262. doi: 10.1126/science.1082348. [DOI] [PubMed] [Google Scholar]
- 43.Okkenhaug K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu. Rev. Immunol. 2013;31:675–704. doi: 10.1146/annurev-immunol-032712-095946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shymanets A., Prajwal, Vadas O., Czupalla C., LoPiccolo J., Brenowitz M., Ghigo A., Hirsch E., Krause E., Wetzker R., et al. Different inhibition of Gβγ-stimulated class IB phosphoinositide 3-kinase (PI3K) variants by a monoclonal antibody. Specific function of p101 as a Gβγ-dependent regulator of PI3Kγ enzymatic activity. Biochem. J. 2015;469:59–69. doi: 10.1042/BJ20150099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walser R., Burke J.E., Gogvadze E., Bohnacker T., Zhang X., Hess D., Küenzi P., Leitges M., Hirsch E., Williams R.L., et al. PKCβ phosphorylates PI3Kγ to activate it and release it from GPCR control. PLoS Biol. 2013;11:e1001587. doi: 10.1371/journal.pbio.1001587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jin J.R., Gogvadze E., Xavier A.R., Bohnacker T., Voelzmann J., Wymann M.P. PI3Kγ regulatory protein p84 determines mast cell sensitivity to Ras inhibition-moving towards cell specific PI3K targeting? Front. Immunol. 2020;11:585070. doi: 10.3389/fimmu.2020.585070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burke J.E., Vadas O., Berndt A., Finegan T., Perisic O., Williams R.L. Dynamics of the phosphoinositide 3-kinase p110δ interaction with p85α and membranes reveals aspects of regulation distinct from p110α. Structure. 2011;19:1127–1137. doi: 10.1016/j.str.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burke J.E., Perisic O., Masson G.R., Vadas O., Williams R.L. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110α (PIK3CA) Proc. Natl. Acad. Sci. USA. 2012;109:15259–15264. doi: 10.1073/pnas.1205508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu J., Zhang Y., McIlroy J., Rordorf-Nikolic T., Orr G.A., Backer J.M. Regulation of the p85/p110 phosphatidylinositol 3’-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol. Cell Biol. 1998;18:1379–1387. doi: 10.1128/mcb.18.3.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brock C., Schaefer M., Reusch H.P., Czupalla C., Michalke M., Spicher K., Schultz G., Nürnberg B. Roles of G beta gamma in membrane recruitment and activation of p110 gamma/p101 phosphoinositide 3-kinase gamma. J. Cell Biol. 2003;160:89–99. doi: 10.1083/jcb.200210115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dbouk H.A., Vadas O., Shymanets A., Burke J.E., Salamon R.S., Khalil B.D., Barrett M.O., Waldo G.L., Surve C., Hsueh C., et al. G protein-coupled receptor-mediated activation of p110β by Gβγ is required for cellular transformation and invasiveness. Sci. Signal. 2012;5:ra89. doi: 10.1126/scisignal.2003264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rathinaswamy M.K., Fleming K.D., Dalwadi U., Pardon E., Harris N.J., Yip C.K., Steyaert J., Burke J.E. HDX-MS-optimized approach to characterize nanobodies as tools for biochemical and structural studies of class IB phosphoinositide 3-kinases. Structure. 2021;29:1371–1381.e6. doi: 10.1016/j.str.2021.07.002. [DOI] [PubMed] [Google Scholar]
- 53.Szwed A., Kim E., Jacinto E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol. Rev. 2021;101:1371–1426. doi: 10.1152/physrev.00026.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Siempelkamp B.D., Rathinaswamy M.K., Jenkins M.L., Burke J.E. Molecular mechanism of activation of class IA phosphoinositide 3-kinases (PI3Ks) by membrane-localized HRas. J. Biol. Chem. 2017;292:12256–12266. doi: 10.1074/jbc.M117.789263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Robert X., Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucl. Acids Res. 2014;42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kozasa T. Purification of G protein subunits from Sf9 insect cells using hexahistidine-tagged alpha and beta gamma subunits. Methods Mol. Biol. 2004;237:21–38. doi: 10.1385/1-59259-430-1:21. [DOI] [PubMed] [Google Scholar]
- 57.Jaitner B.K., Becker J., Linnemann T., Herrmann C., Wittinghofer A., Block C. Discrimination of amino acids mediating Ras binding from noninteracting residues affecting Raf activation by double mutant analysis. J. Biol. Chem. 1997;272:29927–29933. doi: 10.1074/jbc.272.47.29927. [DOI] [PubMed] [Google Scholar]
- 58.Huang W.Y.C., Alvarez S., Kondo Y., Lee Y.K., Chung J.K., Lam H.Y.M., Biswas K.H., Kuriyan J., Groves J.T. A molecular assembly phase transition and kinetic proofreading modulate Ras activation by SOS. Science. 2019;363:1098–1103. doi: 10.1126/science.aau5721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yin J., Lin A.J., Golan D.E., Walsh C.T. Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nat. Protoc. 2006;1:280–285. doi: 10.1038/nprot.2006.43. [DOI] [PubMed] [Google Scholar]
- 60.Ziemba B.P., Swisher G.H., Masson G., Burke J.E., Williams R.L., Falke J.J. Regulation of a coupled MARCKS-PI3K lipid kinase circuit by calmodulin: single-molecule analysis of a membrane-bound signaling module. Biochemistry. 2016;55:6395–6405. doi: 10.1021/acs.biochem.6b00908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hansen S.D., Huang W.Y.C., Lee Y.K., Bieling P., Christensen S.M., Groves J.T. Stochastic geometry sensing and polarization in a lipid kinase-phosphatase competitive reaction. Proc. Natl. Acad. Sci. USA. 2019;116:15013–15022. doi: 10.1073/pnas.1901744116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mirdita M., Schütze K., Moriwaki Y., Heo L., Ovchinnikov S., Steinegger M. ColabFold: making protein folding accessible to all. Nat. Methods. 2022;19:679–682. doi: 10.1038/s41592-022-01488-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gorrec F., Löwe J. Automated protocols for macromolecular crystallization at the MRC laboratory of molecular Biology. J. Vis. Exp. 2018:55790. doi: 10.3791/55790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stock D., Perisic O., Löwe J. Robotic nanolitre protein crystallisation at the MRC laboratory of molecular Biology. Prog. Biophys. Mol. Biol. 2005;88:311–327. doi: 10.1016/j.pbiomolbio.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 65.Gorrec F. The MORPHEUS protein crystallization screen. J. Appl. Crystallogr. 2009;42:1035–1042. doi: 10.1107/S0021889809042022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Powell H.R., Battye T.G.G., Kontogiannis L., Johnson O., Leslie A.G.W. Integrating macromolecular X-ray diffraction data with the graphical user interface iMosflm. Nat. Protoc. 2017;12:1310–1325. doi: 10.1038/nprot.2017.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Evans P. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 68.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dobbs J.M., Jenkins M.L., Burke J.E. Escherichia coli and Sf9 contaminant databases to increase efficiency of tandem mass spectrometry peptide identification in structural mass spectrometry experiments. J. Am. Soc. Mass Spectrom. 2020;31:2202–2209. doi: 10.1021/jasms.0c00283. [DOI] [PubMed] [Google Scholar]
- 72.Jaqaman K., Loerke D., Mettlen M., Kuwata H., Grinstein S., Schmid S.L., Danuser G. Robust single-particle tracking in live-cell time-lapse sequences. Nat. Methods. 2008;5:695–702. doi: 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Masson G.R., Burke J.E., Ahn N.G., Anand G.S., Borchers C., Brier S., Bou-Assaf G.M., Engen J.R., Englander S.W., Faber J., et al. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat. Methods. 2019;16:595–602. doi: 10.1038/s41592-019-0459-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Membrane composition: 96% DOPC, 2% PI(4,5)P2, 2% MCC-PE. Video associated with Figure 5E and 5G. Video plays at 25 frames per second. smTIRF-M data was collected with 52 ms time intervals (i.e. 19 fps). Scale bar is 2 μm.
Membrane composition: 96% DOPC, 2% PI(4,5)P2, 2% MCC-PE. Video associated with Figure 5F and 5H. Video plays at 25 frames per second. smTIRF-M data was collected with 22 ms time intervals (i.e. 45 fps). Scale bar is 2 μm.
The highest ranked alphafold models underlying the Gβγ interfaces in both p110γ and p101 are included in a zip file, with the b-factor column in the PDB file indicating the pLDDT score.
Data Availability Statement
-
•
The data underlying this manuscript are available at the following databases: The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository1 with the dataset identifier PXD035723, the X-ray crystallography data and model for the p110γ-p84 structure was deposited in the Protein DataBank (PDB), with the identifier PDB: 8AJ8, the EM density for the p110γ-p84 complex was deposited in the Electron Microscopy DataBank with the identifier 27738, and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.