

Abstract
Vascular dysfunction: develops progressively with ageing; increases the risk of cardiovascular diseases (CVD); and is characterized by endothelial dysfunction and arterial stiffening, which are primarily mediated by superoxide-driven oxidative stress and consequently reduced nitric oxide (NO) bioavailability and arterial structural changes. Interventions initiated before vascular dysfunction manifests may have more promise for reducing CVD risk than interventions targeting established dysfunction. Gut microbiome-derived trimethylamine N-oxide (TMAO) induces vascular dysfunction, is associated with higher CV risk, and can be suppressed by 3,3-dimethyl-1-butanol (DMB). We investigated whether DMB supplementation could prevent age-related vascular dysfunction in C57BL/6N mice when initiated prior to development of dysfunction. Mice received drinking water with 1% DMB or normal drinking water (control) from midlife (18 months) until being studied at 21, 24 or 27 months of age, and were compared to young adult (5 month) mice. Endothelial function [carotid artery endothelium-dependent dilatation (EDD) to acetylcholine; pressure myography] progressively declined with age in control mice, which was fully prevented by DMB via higher NO-mediated EDD and lower superoxide-related suppression of EDD (normalization of EDD with the superoxide dismutase mimetic TEMPOL). In vivo aortic stiffness (pulse wave velocity) increased progressively with age in controls, but DMB attenuated stiffening by ~ 70%, probably due to preservation of endothelial function, as DMB did not affect aortic intrinsic mechanical (structural) stiffness (stress–strain testing) nor adventitial abundance of the arterial structural protein collagen. Our findings indicate that long-term DMB supplementation prevents/attenuates age-related vascular dysfunction, and therefore has potential for translation to humans for reducing CV risk with ageing.
Keywords: arterial stiffness, gut microbiome, oxidative stress, trimethylamine N-oxide, vascular dysfunction
Graphical Abstract
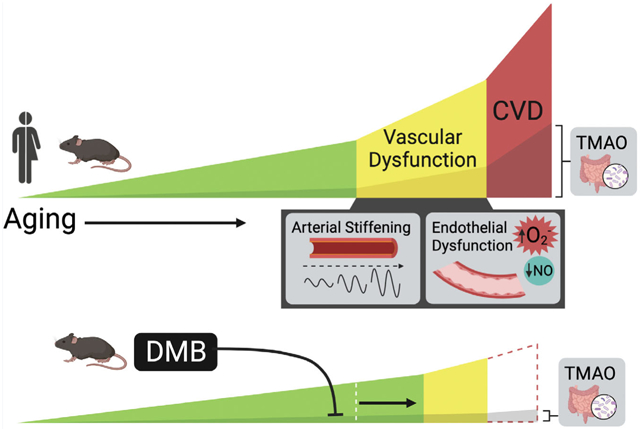
Age-related vascular dysfunction (arterial stiffening and endothelial dysfunction) contributes to the development of cardiovascular diseases (CVD). The gut microbiome-derived metabolite TMAO increases in circulation with age, is linked to vascular dysfunction and CVD in humans, causes vascular dysfunction in mice, and can be suppressed by DMB. We found that long-term supplementation with DMB prevents and delays vascular dysfunction when initiated in midlife, prior to the development of dysfunction, and continued into late life. DMB and similar gut microbiome-targeted interventions have potential for translation to humans for preventing/delaying vascular dysfunction and reducing CVD risk with ageing.
Introduction
Ageing is the primary risk factor for the development of cardiovascular diseases (CVD), the leading cause of death worldwide (Virani et al. 2021; World Health Organization, 2021). A key age-related change that contributes to the pathogenesis of CVD is the development of vascular dysfunction, which is characterized by impairments in endothelial function and stiffening of the large elastic arteries, e.g. the aorta (Lakatta & Levy, 2003). These adverse changes to arteries are largely driven by excess superoxide-associated oxidative stress, which reduces bioavailability of the vasodilatory molecule nitric oxide (NO) and induces arterial structural remodelling (e.g. increased collagen deposition) and consequent stiffening (Bachschmid et al., 2013; Fleenor et al., 2012; van der Loo et al., 2000).
Many interventions for improving vascular function have been conducted later in life with the goal of reversing established dysfunction (Hayashi et al., 2006; Jennings et al., 2019; Kaplon et al., 2016; Santos-Parker et al., 2017). However, the adverse underlying cellular and molecular processes that mediate vascular dysfunction develop progressively over the lifespan (Gioscia-Ryan et al., 2021; Lakatta, 2003; Sohal & Weindruch, 1996; Zieman et al., 2005). Thus, preventive interventions that target these processes before downstream dysfunction is detectable may have greater therapeutic potential (Lloyd-Jones et al., 2010). Conducting long-term intervention studies in humans is resource demanding and logistically challenging. Thus, to increase the likelihood of success for such interventions in clinical trials, conducting initial studies in relevant mouse models can provide insight into the efficacy of interventions and potential for translation to humans, as well as the mechanisms of action (Granger et al., 2012; Seals, 2013).
One intervention that shows promise for preventing age-related vascular dysfunction is supplementation with 3,3-dimethyl-1-butanol (DMB). DMB suppresses gut microbiota-dependent production of the metabolite trimethylamine N-oxide (TMAO) (Brunt et al., 2020; Chen et al., 2019; Li et al., 2017, 2018; Wang et al., 2015), a compound that increases in circulation with ageing (Brunt et al., 2019, 2020, 2021) and has been linked to age-associated vascular dysfunction in humans (Brunt et al., 2020, 2021). In mice, dietary supplementation with TMAO directly induces vascular dysfunction by increasing oxidative stress and reducing NO bioavailability (Brunt et al., 2020; Li et al., 2017), and increases arterial stiffness by altering the structural components of the arterial wall (Brunt et al., 2021). These findings support observations that higher circulating levels of TMAO increase CV risk in humans (Tang et al., 2013; Wang et al., 2011) and induce CVD in mice (Koeth et al., 2013; Wang et al., 2015). Importantly, DMB acts directly on certain intestinal bacteria but is non-lethal to these microbes (Wang et al., 2015), and has no known deleterious effects on host cells. Thus, there is strong interest in and potential for translation of long-term DMB supplementation to humans.
Therefore, the purpose of this study was to determine, for the first time, if long-term supplementation with DMB can prevent the development of age-related vascular dysfunction in mice when initiated in midlife, prior to the onset of dysfunction. We hypothesized that: (1) progressive impairments in endothelial function and increases in aortic stiffness with ageing in control mice would be attenuated by DMB supplementation; (2) preservation of endothelial function would be related to higher NO-mediated dilatation and lower superoxide production and superoxide-related suppression of endothelial function; and (3) DMB would ameliorate age-related increases in aortic stiffness by preventing age-related structural changes in the aorta.
Methods
Ethical approval
All procedures were approved by the University of Colorado Boulder Institutional Animal Care and Use Committee (protocol #2539) and were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory animals and the ethical guidelines set forth by The Journal of Physiology (Grundy, 2015).
Study design and experimental animals
We used a mouse model to provide a unique temporal and mechanistic assessment of the effects of long-term DMB supplementation on preventing vascular dysfunction when initiated in midlife and continued into late life. We conducted the study using male C57BL/6N mice, as mice of this strain and sex are an established model of human vascular ageing, i.e. these mice exhibit age-related impairments in endothelial function and aortic stiffening similarly to humans (Brunt et al., 2019; Ballak et al., 2020; Fleenor et al., 2012; Gioscia-Ryan et al., 2021), and such impairments develop progressively from late middle age onward (Gioscia-Ryan et al., 2021). Only male mice were used in the present study because, although human men and women develop vascular dysfunction at similar rates (Scuteri et al., 2014; Vlachopoulos et al., 2011), the development of vascular dysfunction is significantly delayed in female mice relative to males (DuPont et al., 2021), limiting their utility as an appropriate translational model of vascular ageing in women.
Mice were obtained at 2 months of age (young, n = 20 total; n = 7 for ex vivo intrinsic mechanical stiffness; n = 11–13 for all other measures) from the Charles River Laboratories (Wilmington, MA, USA) and at 13–17 months of age (middle-aged, n = 122) from the National Institute on Aging colony maintained by Charles River. Of the 122 mice obtained in middle age, 11 died prior to baseline testing and 27 (11 DMB, 16 control) died during the intervention, which was expected due to age-related attrition (Flurkey et al., 2007). Mice were housed in a conventional facility on a 12 h light/dark cycle and given ad libitum access to standard rodent chow containing 1890 mg kg−1 choline (Teklad 7917; Envigo, Indianapolis, IN, USA) and normal drinking water. Mice were group housed given the long duration of the intervention (3–9 months). All mice were acclimated to our facilities for >4 weeks prior to the intervention and/or any testing. Food and water intake were determined and reported as average intake per day in young mice over the course of 8 weeks, and older mice (18–27 months) over the course of the 3–9 months of intervention (described below; averaged across each 3 month interval). Mice were removed from the analysis as they were euthanized at each time point. As mice were group housed, food and water intake were determined as the total intake per cage divided by the number of mice within a given cage (Fig. 1).
Figure 1. Study overview.
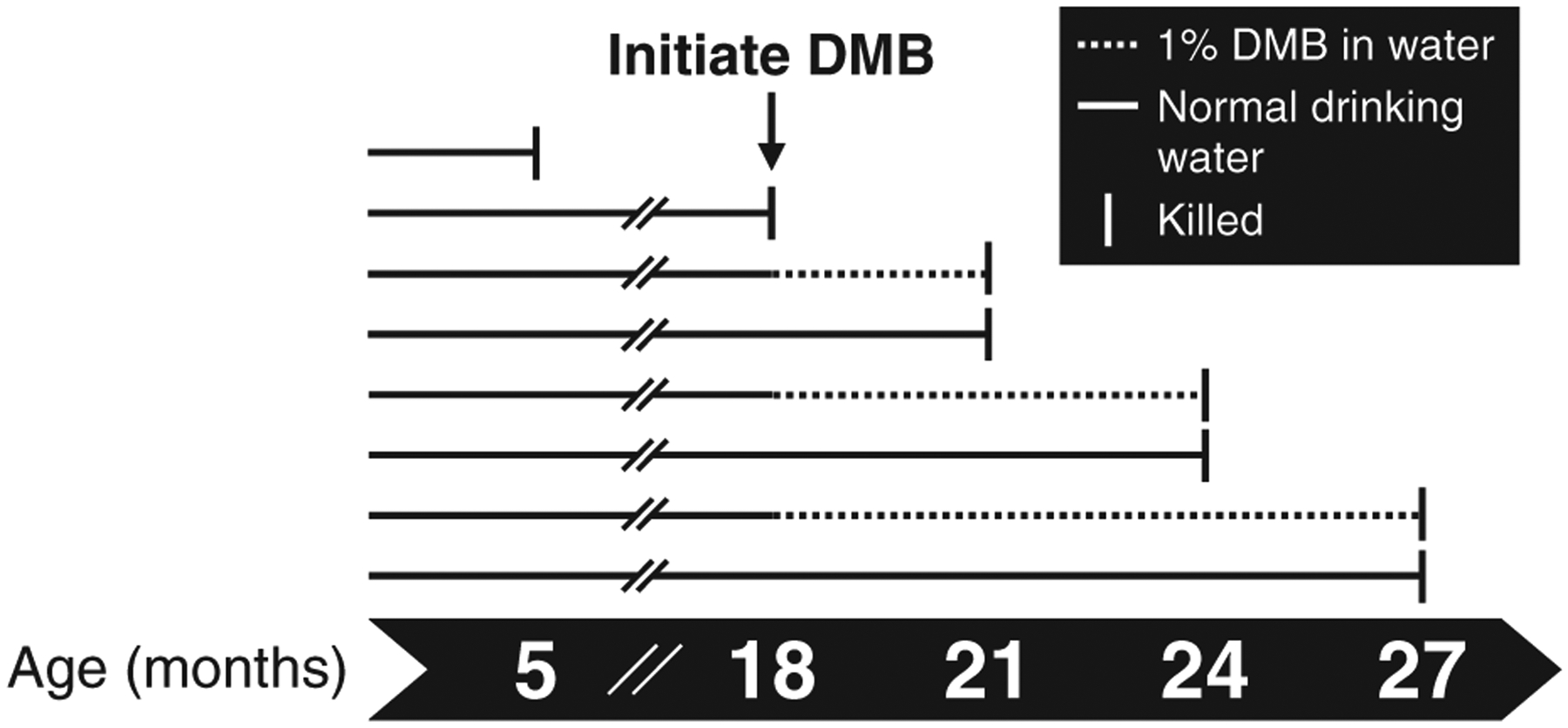
Male C57BL/6N mice were randomly assigned to receive normal drinking water (control) or water supplemented with 1% (v/v) 3,3-dimethyl-1-butanol (DMB) beginning in middle age (18 months) and continued into late life (up to 27 months). Mice were studied at 18, 21, 24 and 27 months of age and compared to a reference group of young adult (5 months) mice.
Intervention.
At 18 months of age (prior to baseline testing), mice were randomly assigned to either continue receiving normal drinking water (control) or begin receiving water supplemented with 1% (v/v) DMB (Sigma-Aldrich Corp., St. Louis, MO, USA). This dose and route of administration has previously been shown to be safe (i.e. no adverse effects on lipids, glucose, or liver and kidney function) (Wang et al., 2015), well tolerated and to reduce circulating levels of TMAO in mice (Brunt et al., 2020; Wang et al., 2015). A group of control mice was studied at 18 months of age (middle age, equivalent to ~55 years in humans) to obtain baseline measurements for terminal outcomes (n = 9), and mice in both the control and DMB-supplemented groups were studied at the following ages: 21, 24 and 27 months (equivalent to ~60, 68 and 75 years in humans; the median lifespan in these mice is 28 months) (Flurkey et al., 2007). In addition, a reference group of young adult control mice was studied at 5 months of age, the age at which these mice are physically mature (equivalent to ~25 years in humans) (Flurkey et al., 2007).
Experimental procedures
Investigators were blinded to treatment group for data collection related to endothelium-dependent dilatation (EDD) and biochemical assessments, and for all data analyses.
Aortic pulse wave velocity (aPWV).
We assessed aortic stiffness serially in vivo as aPWV (Brunt et al., 2021; Fleenor et al., 2012; Gioscia-Ryan et al., 2021). Mice were anaesthetized with 1–2.5% inhaled isoflurane in O2 for a maximum of 20 min. Blood velocity waves were detected at the transverse aortic arch and abdominal aorta using Doppler ultrasonography (Doppler Signal Processing Workstation, Indus Instruments, Webster, TX, USA). aPWV was calculated as the distance (cm) between the two sites divided by the difference in time delay (s) from the ECG R-wave to the onset of the blood velocity wave at each site.
Tissue collection.
All mice were studied for terminal outcomes in the morning and were not fasted overnight. Mice were euthanized using an approach consistent with the American Veterinary Medical Association guidelines (Underwood et al., 2020). Mice were anaesthetized with inhaled isoflurane (open-drop delivery in anaesthesia box) and were euthanized by exsanguination via cardiac puncture. Whole blood obtained from the cardiac puncture was heparinized, and plasma was separated by centrifugation and stored at −70°C for later analysis of TMAO and TMAO-related −metabolites (described below). The carotid arteries were excised, cleaned of surrounding tissue and used to assess endothelial function (described below). The thoracic aorta was removed and cleaned of surrounding perivascular adipose and connective tissue and sectioned/stored as described below. Additionally, we removed, cleaned and weighed the heart, brain, lung, liver, kidneys, spleen and visceral (epididymal) fat.
Plasma TMAO and related metabolites.
Plasma concentrations of TMAO, choline and betaine were assessed via liquid chromatography-tandem mass spectrometry using a stable isotope dilution method against internal standards, as previously described (Boutagy et al., 2015; Brunt et al., 2021; Wang et al., 2014).
Vascular endothelial function.
Vascular endothelial function was assessed in carotid arteries, an established model of age-related endothelial dysfunction, as previously described by our laboratory (Brunt et al., 2020; Fleenor et al., 2012; Gioscia-Ryan et al., 2021). Carotid arteries were excised at euthanasia, cannulated onto glass pipette tips in pressure myograph chambers (DMT, Inc., Arhaus, Denmark) in warm (37°C) physiological saline solution, and pressurized to 50 mmHg. Arteries were allowed to acclimate for 45 min prior to functional assessments. Resting luminal diameters were obtained, and arteries were preconstricted with phenylephrine (2 μmol L−1; Sigma-Aldrich Corp.) to simulate basal tone (~ 5–10% constriction) prior to each dose response. To determine EDD, arteries were treated with increasing doses of acetylcholine (ACh; 1 × 10−9 to 1 × 10−4 mol L−1; Sigma-Aldrich Corp.) for×2 min per dose, and luminal diameter was assessed at the end of each interval. Peak EDD was determined as the maximal luminal diameter obtained at any dose.
As EDD was similar between mice at 24 and 27 months of age within groups (control vs. DMB), the following mechanistic data are combined for these age groups where indicated.
To determine the extent to which NO contributes to the vasodilatory response to ACh (i.e. NO-mediated dilatation), we incubated a subset of carotid arteries from young (5 months) and older (24 and 27 months data combined) control and DMB-supplemented mice with the NO synthase inhibitor NG-nitro-l-arginine methyl ester (l-NAME; 0.1 mmol L−1; Sigma-Aldrich Corp.) for 30 min prior to the ACh dose response. NO-mediated dilatation was calculated as the difference in peak EDD to ACh in the presence vs. absence of l-NAME.
To determine the extent to which tonic excess superoxide-related oxidative stress impairs the vasodilatory response to ACh, we incubated a subset of carotid arteries from young (5 months) and older (24 and 27 months data combined) control and DMB-supplemented mice with the superoxide dismutase (SOD) mimetic 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPOL; 1 mmol L−1; Sigma-Aldrich Corp.) for 60 min prior to the ACh dose response.
To determine endothelium-independent dilatation (EID; i.e. smooth muscle sensitivity to NO), arteries were treated with increasing doses of the exogenous NO donor sodium nitroprusside (SNP; 1 × 10−9 to 1 × 10−3 mol L−1) for 2 min per dose. Luminal diameter was assessed at the end of each interval, and peak EID was determined as the maximal luminal diameter obtained at any dose.
Finally, arteries were incubated in Ca2+-free physiological saline solution for 20 min to ensure maximal luminal diameter was obtained. To account for anatomical differences in vessel diameter, all dose response data are reported as a percentage of maximal luminal diameter (Brunt et al., 2020; Fleenor et al., 2012; Gioscia-Ryan et al., 2021).
Aortic superoxide levels.
Aortic superoxide levels were assessed in a subset of young (5 months) and older (24 and 27 months data combined) control and DMB-supplemented mice in 1 mm segments of thoracic aorta via electron paramagnetic resonance (EPR) spectroscopy as previously described by our laboratory (Brunt et al., 2020; Fleenor et al., 2012; Gioscia-Ryan et al., 2021). Immediately after mice were euthanized, aortic rings were washed in warm physiological saline solution and incubated for 60 min in deoxygenated, via argon gas, Krebs/Hepes buffer [99 mm NaCl, 4.7 mm KCl, 1.87 mm CaCl2, 1.2 mm MgSO4, 25 mm NaHCO3, 1.03 mm KH2PO4, 20 mm Na-Hepes, 11.1 mm glucose, 0.1 mm diethylenetriaminepenta-acetic acid, 0.0035 mm sodium diethyldithiocarbamate and Chelex (Sigma-Aldrich Corp.)] containing 0.5 mmol L–1 of the spin probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (CMH; Enzo Life Sciences, Farmingdale, NY, USA). Samples were analysed using an MS300 X-band EPR spectrometer (Magnettech, Berlin, Germany). The instrument was set with the following parameters: BO-Field, 3350 G; sweep, 80 G; sweep time, 60 s; modulation, 3000 mG; MW atten, 7 dB; gain, 5 × eˆ2. Samples were collected and analysed in duplicate, and the values were averaged.
Aortic SOD abundance.
Protein abundance was determined in aortic lysates containing 0.4 μg μL−1 of protein via capillary electrophoresis Western detection using a WES instrument (Protein Simple, San Jose, CA, USA). The standard reagent kits and default settings for electrophoretic separation and immunodetection were used, except the primary antibody incubation phase was extended to 45 min. The chemiluminescent signal was analysed and displayed as an electro-pherogram/pseudo blot using Compass software (Protein Simple). Primary antibodies were anti-SOD1/Cu-Zn SOD (1:500; R&D, Minneapolis, MN, USA; Cat# AF3787), anti-SOD2/Mn-SOD (1:50; R&D; Cat# AF3419) and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:200, Cell Signaling Technology, Danvers, MA, USA; Cat # 14C10). The relative intensity of each SOD was normalized to the GAPDH signal. Antibodies were validated by the manufacturer.
Intrinsic mechanical stiffness.
Segments (~1 mm) of thoracic aorta were collected from a subset of young (5 months) and older (24 and 27 months) control and DMB-supplemented mice when they were euthanized and frozen and stored in physiological saline solution at −70°C. Intrinsic mechanical stiffness was later assessed in duplicate by incremental stress–strain testing using a pin (force) myograph (DMT) (Brunt et al., 2021; Gioscia-Ryan et al., 2021), and the values were averaged. These measures were conducted in aortic segments because our in vivo measure of stiffness (aPWV) was also conducted in the aorta. Aortic rings were mounted onto the pins in heated (37°C) phosphate-buffered saline (PBS) and pre-stretched three times by displacing the pins to 1 mm. The pins were then adjusted until a force of 1 mN was obtained. Force measurements were then recorded as the pins were displaced by 50 μm every 3 min until the vessel reached mechanical failure (i.e. a sudden drop in force). The pin displacement and corresponding force measurement were used to generate a stress–strain curve in which strain and stress were defined as:
where d is diameter and d(i) is the initial diameter, and:
where L is one-dimensional load, H is intima media thickness and D is vessel length.
The elastic modulus (EM) of the collagen-dominant (highest force) region of the stress–strain curve was defined as the slope of a linear equation fit to the final four points of the stress–strain curve. Stress–strain data for the entire curve were also fit with a seventh-order polynomial equation (r 2 > 0.99; RStudio, Boston, MA, USA) and the roots of the equation were calculated. The elastic modulus of the elastin-dominant region of the stress–strain curve (low force region where curvature is ~0) was defined as the slope of a linear equation fit to the stress–strain data between the first and second roots, which were considered the boundary between the very low-force region and the elastin-dominant region, and the boundary between the elastin-dominant region and the onset of the collagen-dominant region, respectively (Brunt et al., 2019; Gioscia-Ryan et al., 2018; Lammers et al., 2008).
To obtain measures of aortic intima-media thickness (IMT) and diameter, an ~1 mm segment of thoracic aorta was collected, placed in optimal cutting temperature compound, frozen in liquid nitrogen-cooled methylbutane and stored at −70°C. Samples were later sectioned (7 μm; Leica CM1520, Leica Biosystems, Wetzlar, Germany), plated on untreated microscope slides and imaged using a Nikon Eclipse TS100 (4 × magnification). Images were analysed using ImageJ software (National Institutes of Health, Bethesda, MA, USA). For analysis of IMT, the distinction between medial and adventitial layers was determined to be the point at which the regular banding patterns (media) abruptly shift to diffuse patterning (adventitia). Four separate images were used for analysis of each sample, and the values were averaged.
Aortic abundance of adventitial collagen.
Segments of thoracic aorta were obtained from a subset of young (5 months) and older (27 months) control and DMB-supplemented mice, stored, and later sectioned as described for IMT measures. Samples were plated on poly-L-lysine-coated slides, fixed in 2% paraformaldehyde, and stored at −70°C. Slides were later rehydrated (50 mmol L−1 glycine in PBS), permeabilized (0.1% Triton X-100) and incubated in 2.5% normal horse serum blocking buffer, with PBS washes between each step. The samples were then incubated with an anti-collagen I primary antibody (1:200; Abcam, Cambridge, MA, USA; Cat# ab21286; RRID:AB_446 161) for 60 min, as collagen I is the primary isoform found in arteries (Xu & Shi, 2014). Following washes, slides were then incubated with an alkaline phosphatase-conjugated secondary antibody according to the manufacturer’s instructions (Vector Laboratories, Inc., Burlingame, CA, USA; Cat# MP-7714). The samples were then mounted with Gelvatol mounting medium (The University of Queensland, n.d.) and allowed to cure overnight. The following day, samples were imaged using a Nikon Eclipse TS100 (4 × magnification) under identical conditions and analysed using the FIJI package on the ImageJ software (National Institutes of Health). Protein abundance was determined as the mean adventitial intensity normalized to the background intensity (Brunt et al., 2021; Fleenor et al., 2012).
Statistical methods
Power calculations were performed using G*power for our primary outcome variables, peak EDD and aPWV. Preliminary data comparing old control vs. short-term DMB-supplemented mice yielded pooled standard deviations of 17.5% and 82 cm s−1 and effect sizes of 0.57 and 0.61 for peak EDD and aPWV, respectively. N = 7 mice per group was determined to be sufficient to detect differences in peak EDD and aPWV with 90% statistical power (α = 0.05) using a two-way design. Given the long duration of the intervention and potential for age-related attrition and taste aversion to DMB, we randomized 122 middle-aged mice to either the control or DMB-supplemented groups, which resulted in group sizes of 7–13 at end-intervention.
Statistical analyses were conducted using GraphPad Prism version 9.2.0 (GraphPad Software, Inc., San Diego, CA, USA; RRID:SCR_0 02798). Statistical significance was set to α = 0.05. Data were assessed for outliers using the ROUT test (Q = 1%) prior to statistical analysis, and outliers were excluded from final analyses (Motulsky & Brown, 2006). Serial data were collected from some mice within these data sets, but non-repeated analyses were used as groups of mice were removed from the analyses at each time point that mice were euthanized.
Age-related changes in control mice.
To confirm expected changes in our outcomes with advancing age in control mice, we assessed the following variables over months of age (5, 18, 21, 24 and 27) using one-way ANOVA with Dunnett’s post hoc test: animal characteristics, circulating TMAO and related metabolites, peak EDD, area under the curve (AUC) of EDD and EID, aPWV, aortic EM, aortic IMT, and water and food intake.
Effect of DMB supplementation.
To determine whether there was an effect of DMB supplementation, we assessed the following variables between control and DMB mice at 21, 24 and 27 months of age using two-way ANOVA with Šídák’s post hoc test: animal characteristics, circulating metabolites, peak EDD, AUC of EDD and EID, aPWV, aortic intrinsic mechanical stiffness, aortic IMT, and water and food intake (assessed across 3 month intervals: 18–21, 21–24 and 24–27 months of age).
Carotid artery dilatory dose responses.
Differences in carotid artery dose responses were determined across treatment groups separately for each age relative to the young reference group using two-way mixed design ANOVA [between factor of group and repeated factor of dose (ACh or SNP)] with Tukey’s post hoc test.
Mechanisms of carotid artery endothelial function and aortic stiffening.
Differences in NO-mediated dilatation, aortic superoxide levels (EPR) and aortic protein abundance were determined between groups using one-way ANOVA with Tukey’s post hoc test. Differences in peak EDD in the presence vs. absence of TEMPOL were determined between groups using two-way mixed design ANOVA (between factor of group and repeated factor of ACh alone or ACh + TEMPOL) with Šídák’s post hoc test.
Results
Animal characteristics
Water and food intake are presented in Table 1, averaged across 3 month intervals. Relative to the reference group of young adult mice (5 months), water intake was slightly lower in control mice aged 18–21 months (P = 0.045) and 21–24 months (P = 0.010) but not different in control mice aged 24–27 months (P = 0.984). Mice supplemented with DMB in drinking water appeared to have a mild taste aversion to the water that subsided over time. Water intake was lowest in DMB-supplemented mice aged 18–21 months (i.e. during the first 3 months of the intervention; P < 0.001 vs. control), but then progressively increased over months 3–9 of the intervention (mice aged 21–24 and 24–27 months both P < 0.001 vs. mice aged 18–21 months, within the DMB group), such that water intake was only slightly lower than control mice during the last 3 month interval. Furthermore, mice were closely monitored by laboratory and veterinary staff and did not exhibit any signs of dehydration (e.g. sunken or recessed eyes, tight skin across the scapulae).
Table 1.
Water and food intake
| Age (months) | ||||
|---|---|---|---|---|
| 5 | 18–21 | 21–24 | 24–27 | |
| Time on intervention (months) | - | 0 to 3 | 3 to 6 | 6 to 9 |
| Water intake (mL day−1) | ||||
| Control | 3.8 ± 0.6 | 3.4 ± 0.7* | 3.5 ± 0.8* | 3.7 ± 0.7 |
| DMB | - | 2.3 ± 0.4† | 2.6 ± 0.6†,^ | 3.2 ± 0.8†,^ |
| Food intake (kcal day−1) | ||||
| Control | 12.4 ± 2.2 | 13.3 ± 2.8 | 11.8 ± 2.1 | 11.3 ± 2.4* |
| DMB | - | 11.9 ± 2.0† | 11.2 ± 2.7 | 11.3 ± 3.7 |
Data are mean ± SD. Food/water intake are reported as average intake per day in young mice (5 months; n = 12) over the course of 8 weeks, and older mice (18–27 months) over the course of the 3–9 months of intervention (averaged across each 3 month interval; mice were removed from the analysis as they were euthanized and studied at each time point; n = 12–41 mice per treatment group per 3 month interval). Mice were group housed, so food/water intake was determined as the total intake divided by the number of mice within a given cage. Statistics are one-way (effect of age in control mice only) ANOVA with Dunnett’s post hoc test and two-way mixed (age × treatment group) ANOVA with Šídák’s post hoc test.
P < 0.05 control vs. 5 month reference group.
P < 0.05 DMB vs. control within time point.
P < 0.01 vs. DMB aged 18–21 months. Abbreviations: DMB, 3,3-dimethyl-1-butanol.
In control mice, food intake did not differ between young mice and mice aged 18–21 (P = 0.062) and 21–24 months (P = 0.215) but was lower in mice aged 24–27 months (P = 0.011 vs. young). Food intake was lower in DMB-supplemented vs. control mice aged 18–21 months (i.e. the first 3 months of the intervention; P = 0.034), but was not different across groups in mice aged 21–27 months (i.e. during the last 6 months of the intervention; 21–24 months: P = 0.437; 24–27 months: P > 0.999). As such, there was no difference in caloric intake in older age between groups.
In control mice, body mass was highest in mice aged 18 months (i.e. middle age; P = 0.006 vs. young) but declined over time, such that there was no difference in body mass between young and older mice (21, 24 and 27 months vs. young: P = 0.167, P = 0.837 and P =0.920, respectively; Fig. 2). Relative to control mice, DMB-supplemented mice had lower body mass at 21 months of age (P 0.007; Fig. 2), possibly due to the lower water and/or food intake in DMB-supplemented mice during this interval of the intervention. However, there was no significant difference in body mass between groups at 24 months (P 0.055) or 27 months (P 0.085) of age (Fig. 2).
Figure 2. Body mass.
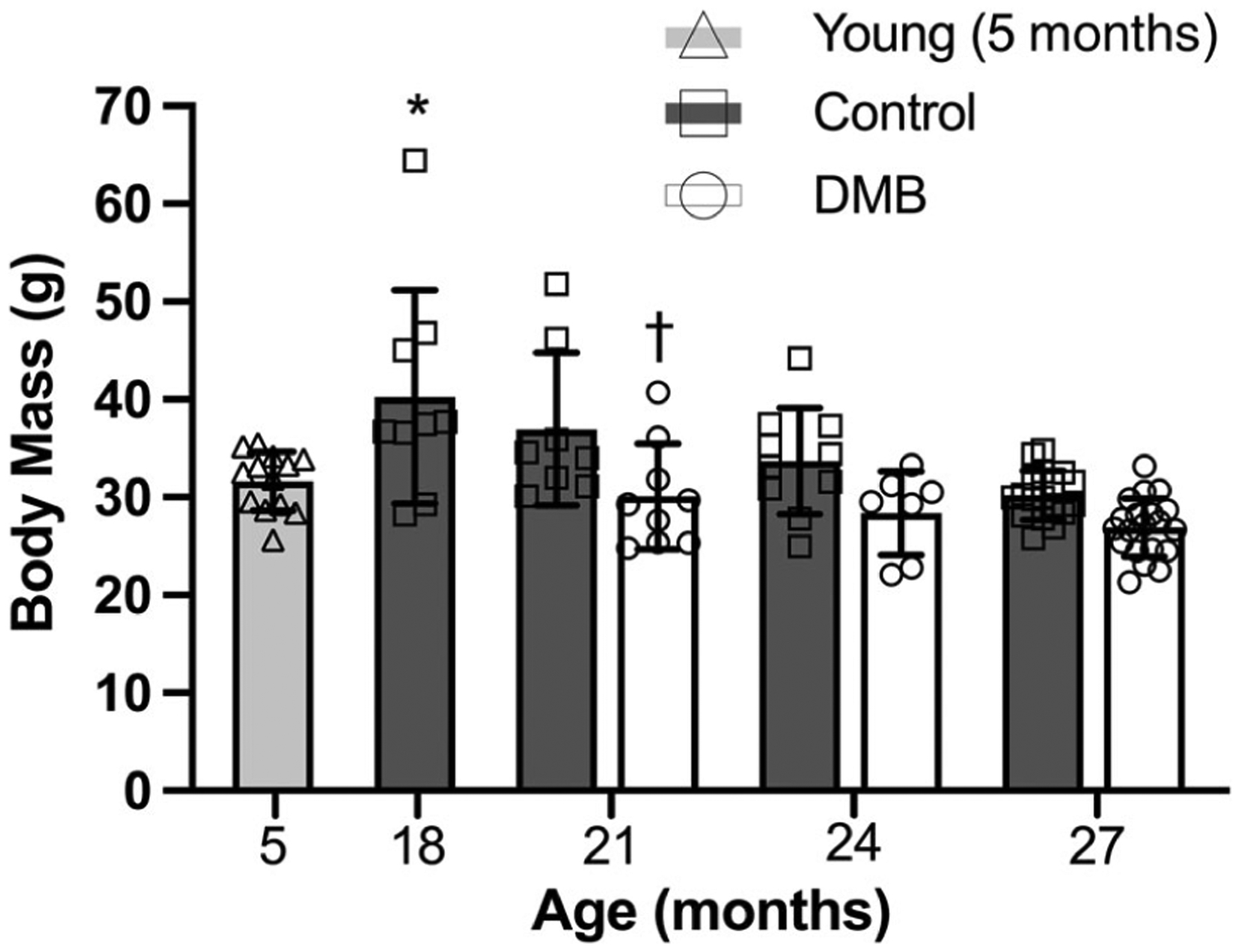
Body mass was highest at 18 months of age in control mice (P = 0.006 vs. young), and at 21 months of age, DMB-supplemented mice had lower body mass relative to control mice (P = 0.007). Body mass was obtained immediately prior to euthanasia. Data are mean ± SD n = 7–20 per age per group. Statistics are one-way (effect of age in control mice only) ANOVA with Dunnett’s post hoc test and two-way mixed (age × treatment group) ANOVA with Šídák’s post hoc test. *P < 0.05 control vs. 5 month reference group. †P < 0.05 DMB vs. control within time point.
Mass of key organs is given in Table 2. In control mice, heart (P = 0.0002), brain (P = 0.018) and lung (P < 0.0001) mass were increased with age, but there were no effects of age on liver (P = 0.238), kidney (P = 0.180) or spleen mass (P = 0.159). Importantly, there were no effects of DMB supplementation on mass of these organs (control vs. DMB within time point, all P ≥ 0.283). In control mice, visceral fat mass was highest in mice aged 18 months (i.e. middle age; P = 0.016 vs. young) and steadily declined into old age (young vs. 27 months: P = 0.039). Visceral fat mass similarly declined with age in DMB mice (age effect, P < 0.001), but was lower at each age time point relative to controls (treatment effect: P < 0.001; although no pairwise comparisons between groups reached statistical significance: 21, 24 and 27 months: P = 0.096, P = 0.072 and P = 0.391, respectively).
Table 2.
Mass of key organs and artery characteristics
| Age (months) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Control | DMB | |||||||
| 5 | 18 | 21 | 24 | 27 | 21 | 24 | 27 | |
| Mass of key organs | ||||||||
| Heart mass (mg) | 134 ± 9 | 174 ± 17* | 167 ± 24* | 166 ± 23* | 170 ± 28* | 150 ± 15 | 178 ± 22 | 171 ± 25 |
| Brain mass (mg) | 397 ± 47 | 429 ± 41 | 450 ± 73 | 468 ± 43* | 445 ± 46* | 398 ± 78 | 418 ± 41 | 434 ± 50 |
| Lung mass (mg) | 165 ± 19 | 192 ± 18 | 196 ± 32 | 209 ± 41* | 227 ± 25* | 183 ± 48 | 195 ± 30 | 214 ± 41 |
| Liver mass (g) | 1.56 ± 0.21 | 1.99 ± 0.64 | 1.77 ± 0.35 | 1.62 ± 0.44 | 1.59 ± 0.59 | 1.50 ± 0.20 | 1.44 ± 0.45 | 1.73 ± 0.70 |
| Kidney mass (mg) | 395 ± 66 | 452 ± 45 | 458 ± 70 | 432 ± 96 | 441 ± 47 | 446 ± 45 | 475 ± 54 | 461 ± 55 |
| Spleen mass (mg) | 72 ± 8 | 75 ± 13 | 84 ± 19 | 85 ± 38 | 106 ± 45 | 64 ± 9 | 92 ± 35 | 100 ± 52 |
| Visceral fat mass (g) | 0.98 ± 0.42 | 1.74 ± 0.95* | 1.17 ± 0.79 | 0.79 ± 0.57 | 0.42 ± 0.25* | 0.72 ± 0.58 | 0.30 ± 0.29 | 0.21 ± 0.15 |
| Artery characteristics (resting) | ||||||||
| Carotid artery | 447 ± 31 | 467 ± 18 | 486 ± 46 | 464 ± 79 | 488 ± 37 | 480 ± 28 | 491 ± 36 | 500 ± 53 |
| diameter (μm) | ||||||||
| Aorta diameter (μm) | 625 ± 20 | 611 ± 35 | 662 ± 45 | 653 ± 85 | 716 ± 55* | 631 ± 42 | 741 ± 58 | 706 ± 79 |
Data are mean ± SD. n = 7–20 per age per group. Statistics are one-way (effect of age in control mice only) ANOVA with Dunnett’s post hoc test and two-way mixed (age × treatment group) ANOVA with Šídák’s post hoc test.
P < 0.05 control vs. 5 month reference group.
P < 0.05 DMB vs. control within time point. Abbreviations: DMB, 3,3-dimethyl-1-butanol.
Diameters of the carotid artery and aorta are also presented in Table 2. There were no age-related differences in resting carotid artery luminal diameter in control mice (P = 0.122) and no effect of DMB supplementation (P = 0.392). Aorta luminal diameter increased with age (young vs. 27 months: P = 0.002), but there was no effect of DMB supplementation (P = 0.374).
Circulating TMAO and TMAO-related metabolites
We have previously shown that circulating plasma levels of TMAO are higher in old vs. young mice (Brunt et al., 2019, 2020), and we and others have found that short-term (~8 weeks) supplementation with DMB suppresses plasma TMAO (Brunt et al., 2020; Li et al., 2017; Wang et al., 2015). Here, we observed no differences in circulating TMAO from 5 to 24 months of age in control mice (all P > 0.999 vs. young). Consistent with previous findings, TMAO was higher at 27 months of age (P = 0.066 vs. young; Fig. 3), although the difference did not reach the P < 0.05 level because, as a secondary outcome, the comparison was underpowered (actual power = 68%). Importantly, plasma TMAO was lower in the DMB-supplemented mice vs. controls at 27 months of age (P = 0.040; Fig. 3).
Figure 3. Plasma trimethylamine N-oxide (TMAO).
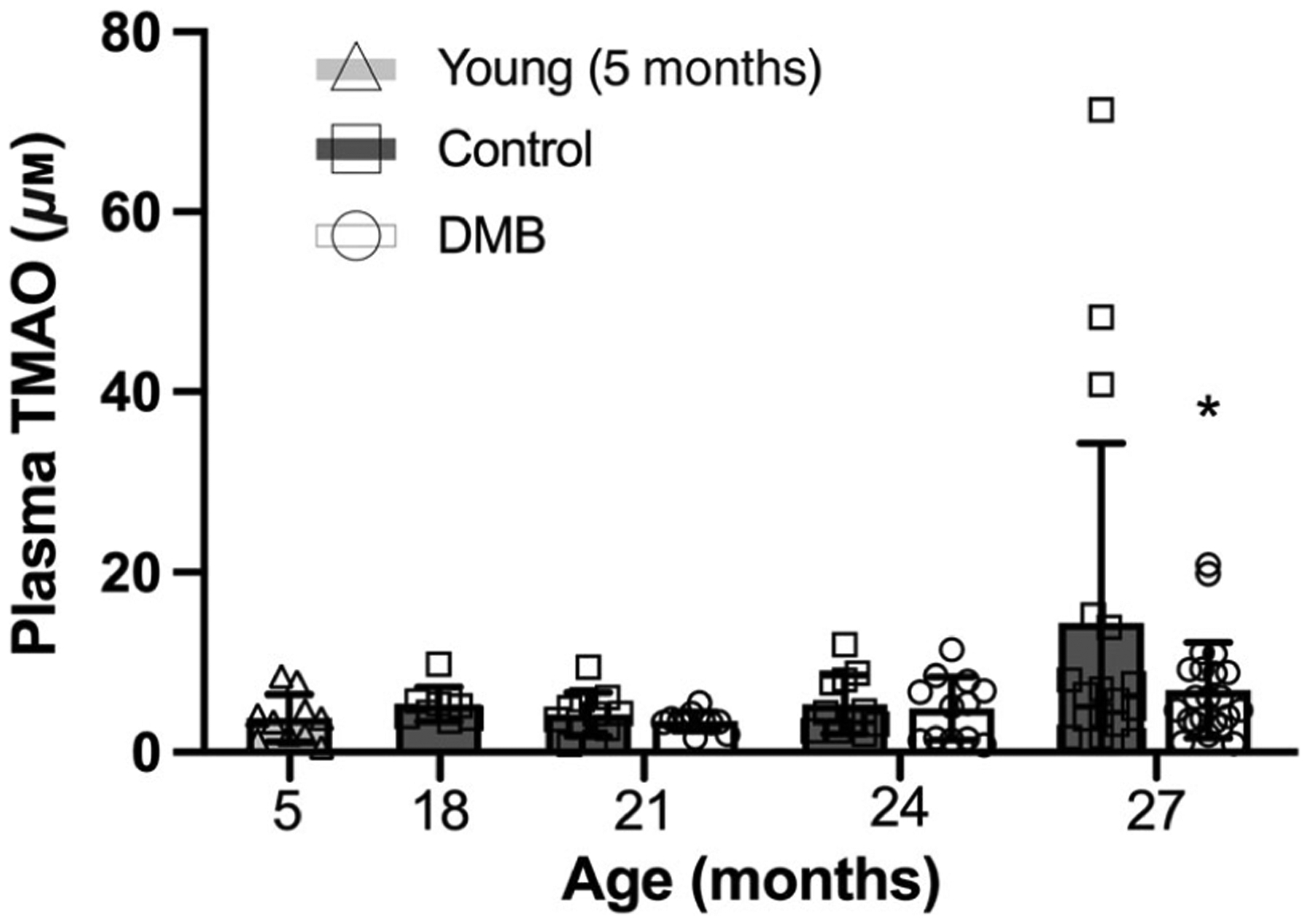
Plasma TMAO may increase with age in control mice (P = 0.066) but is lower in mice supplemented with 3,3-dimethyl-1-butanol (DMB) at 27 months of age. Plasma was collected from mice at the time the mice were euthanized and TMAO levels were determined via liquid chromatography-tandem mass spectrometry. Data are mean ± SD n = 9–22 per group. Statistics are one-way (effect of age in control mice only) ANOVA with Dunnett’s post hoc test and two-way mixed (age × treatment group) ANOVA with Šídák’s post hoc test. *P < 0.05 DMB vs. control within time point.
Circulating levels of TMAO precursors choline and betaine are presented in Table 3. Although l-carnitine is a key precursor of TMAO in humans, we did not include it as mice are vegetarians and standard rodent chow does not contain sources of l-carnitine (Teklad 7917). In control mice, plasma choline (P < 0.0001) and betaine were lower in mice aged 18–27 months vs. young mice (P = 0.0001). DMB supplementation had no effect on plasma choline levels (P = 0.580) but plasma betaine was higher in DMB-supplemented vs. control mice aged 27 months (P = 0.003). As DMB primarily suppresses conversion of choline to TMA (Wang et al., 2015), these data suggest there may have been some compensatory conversion of choline to betaine (Li & Vance, 2008)
Table 3.
Plasma concentrations of TMAO-related metabolites
| Age (months) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Control | DMB | |||||||
| 5 | 18 | 21 | 24 | 27 | 21 | 24 | 27 | |
| Choline (μM) | 39.8 ± 8.2 | 23.2 ± 8.0* | 17.1 ± 5.8* | 18.5 ± 6.6* | 21.5 ± 6.7* | 18.5 ± 7.1 | 20.5 ± 5.7 | 20.5 ± 7.4 |
| Betaine (μM) | 71.9 ± 36.4 | 29.0 ± 6.5* | 33.1 ± 11.2* | 40.5 ± 22.8* | 31.8 ± 9.8* | 41.2 ± 5.4 | 68.9 ± 55.1 | 69.3 ± 46.4† |
Data are mean ± SD. n = 9–22 per group. Statistics are one-way (effect of age in control mice only) ANOVA with Dunnett’s post hoc test and two-way mixed (age × treatment group) ANOVA with Šídák’s post hoc test.
P < 0.05 control vs. 5 month reference group.
P < 0.05 DMB vs. control within time point. Abbreviations: TMAO, trimethylamine N-oxide; DMB, 3,3-dimethyl-1-butanol.
Vascular endothelial function
Endothelial function was assessed by carotid artery EDD to increasing doses of ACh and compared to the reference group of young mice (Fig. 4A and B). EDD was not different from young mice in controls aged 18 (peak EDD: P = 0.999; EDD AUC: P = 0.999) and 21 months (peak EDD: P = 0.202; EDD AUC: P = 0.754). This finding is consistent with our previous observations (Gioscia-Ryan et al., 2021) and serves to validate our intervention study design by confirming that initiation of DMB treatment at 18 months occurred prior to the onset of age-associated endothelial dysfunction. As expected, endothelial dysfunction developed in control mice at 24 (peak EDD: P < 0.001; EDD AUC: P = 0.002) and 27 months of age (peak EDD: P < 0.001; EDD AUC: P < 0.001) relative to young mice, as indicated by reductions in EDD. These age-related impairments in EDD in the control mice were fully prevented in DMB-supplemented mice, such that there were no differences between young and DMB-supplemented mice at 24 (peak EDD: P = 0.998; EDD AUC: P = 0.998) and 27 months (peak EDD:P > 0.999; EDD AUC: P = 0.995).
Figure 4. Supplementation with 3,3-dimethyl-1-butanol (DMB) prevents age-related impairments in endothelium-dependent dilatation (EDD).
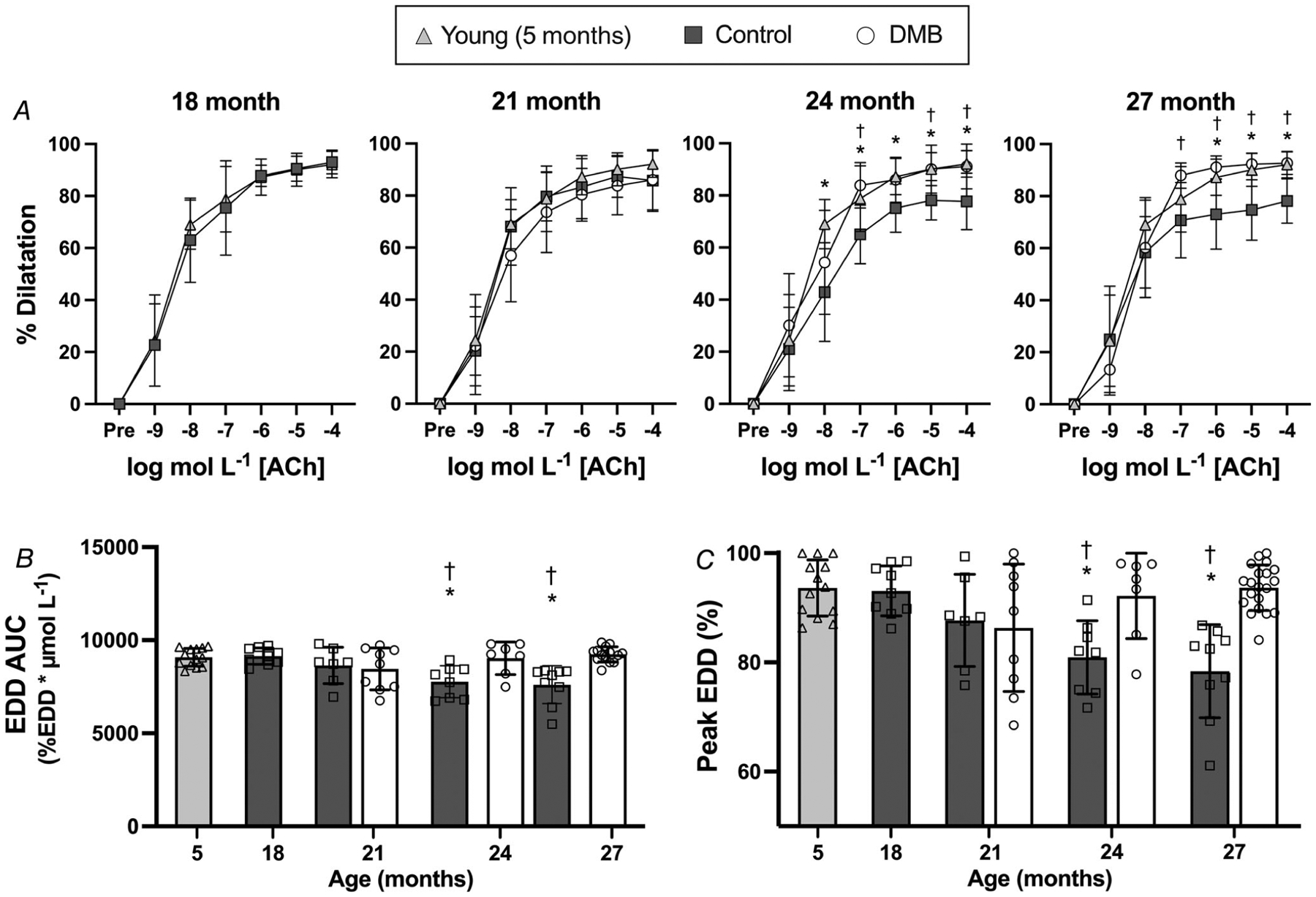
A, carotid artery EDD in response to increasing doses of acetylcholine (ACh). B, EDD to ACh area under the curve (AUC). C, peak EDD to ACh. Data are mean SD. Statistics are two-way mixed (group × dose) ANOVA with Tukey’s post hoc test (A), or one-way (effect of age in control mice only) ANOVA with Dunnett’s post hoc test and two-way mixed (age × treatment group) ANOVA with Šídák’s post hoc test (B and C). *P < 0.05 control vs. young 5 month reference group, within dose. †P < 0.05 DMB vs. control, within dose. n = 7–14 per group.
We next sought to determine the mechanisms underlying these functional differences. As impairments in endothelial function were observed at 24 and 27 months of age in control mice and declines in EDD were prevented with DMB-supplementation, the following mechanistic data are presented for each group with data from 24 and 27 months of age combined (old control vs. old DMB).
First, to determine if the maintenance of vasodilatory responses to ACh with ageing in the DMB-supplemented mice were endothelium-specific, we assessed endothelium-independent dilatation with increasing doses of the NO donor SNP, which assesses vascular smooth muscle sensitivity to NO (Fig. 5A). There were slight differences in dilatation across groups to submaximal doses of SNP, such that the SNP AUC was higher in DMB-supplemented vs. old control (P = 0.002) mice, but not different from young (P = 0.147). However, there were no differences in peak dilatation to SNP (P = 0.222). Thus, we conclude that the effects of DMB supplementation on improving EDD to ACh were largely endothelium-dependent, but that DMB may also improve smooth muscle sensitivity to lower concentrations of NO.
Figure 5. Supplementation with 3,3-dimethyl-1-butanol (DMB) improves endothelial function by enhancing nitric oxide (NO)-mediated dilatation and reducing superoxide-related suppression of endothelium-dependent dilatation (EDD).
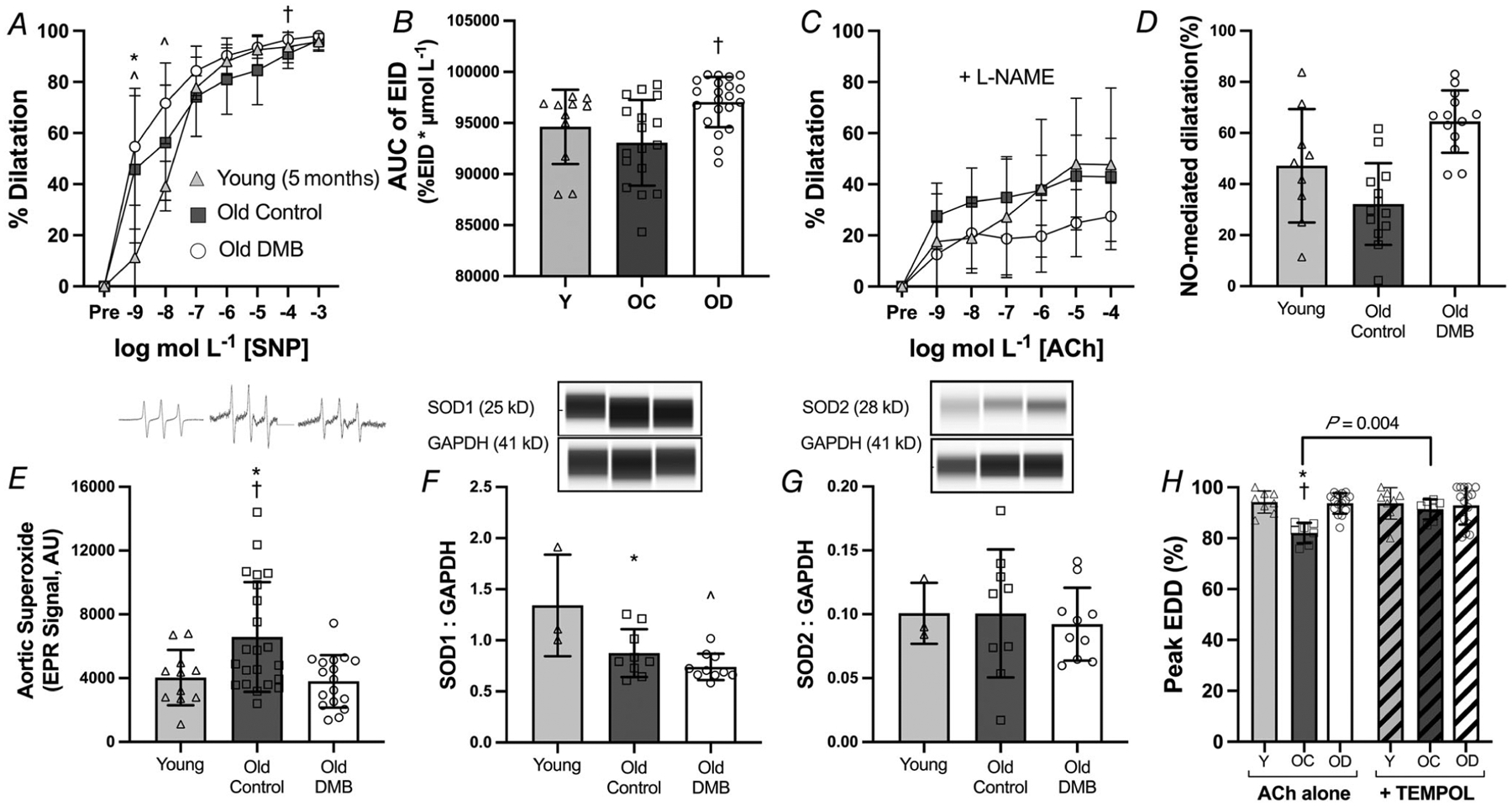
A, carotid artery endothelium-independent dilatation (EID) in response to increasing doses of the NO donor sodium nitroprusside (SNP); and B, EID area under the curve (AUC) (both n = 17–21 per group). C, EDD to acetylcholine (ACh) in the presence of the NO synthase inhibitor NG-nitro-l-arginine methyl ester (l-NAME); and D, NO-mediated dilatation, as assessed by the difference in peak EDD in the absence vs. presence of l-NAME (both n = 9–13 per group). E, superoxide production, measured in 1 mm aorta rings by electron paramagnetic resonance (EPR) spectroscopy, with representative tracings shown above (n = 11–22 per group). Aortic abundance of superoxide dismutase (SOD) 1 (F) and 2 (G) normalized to GAPDH with representative Western blot images generated from WES electropherograms shown above. H, peak EDD to ACh in the absence vs. presence of the superoxide dismutase mimetic 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPOL; n = 8–15 per group). Data are mean ± SD. Data are combined for mice aged 24 and 27 months. Statistics are two-way mixed (group dose) ANOVA with Tukey’s post hoc test (A and C), one-way ANOVA with Tukey’s post hoc test (B, D and E),×and two-way mixed (group × ACh alone or ACh + TEMPOL) with Šídák’s post hoc test (F). *P < 0.05 control vs. young 5 month reference group within dose/condition. †P < 0.05 control vs. DMB within dose/condition. ˆP < 0.01 DMB vs. young 5 month reference group within dose. Abbreviations: Y, young; OC, old control; OD, old DMB.
Next, to determine the contribution of NO to overall EDD, we repeated dose responses to ACh in the presence of the endothelial NO synthase inhibitor l-NAME and assessed NO-mediated dilatation as peak EDD in the presence vs. absence of l-NAME (Fig. 5C and D). There was a main effect of group, such that the DMB-supplemented mice had lower dilatation to ACh in the presence of l-NAME (P = 0.034); however, there were no pairwise differences across groups at any of the doses of ACh (all P ≥ 0.074). NO-mediated dilatation (difference in peak EDD in the presence vs. absence of the NO synthase inhibitor l-NAME) was higher in old DMB vs. old control mice (P < 0.001), indicating that DMB prevented the development of age-related endothelial dysfunction, at least in part, by preserving NO bioavailability.
Reductions in NO bioavailability with ageing occur primarily due to excess production of reactive oxygen species, particularly the free radical superoxide which readily scavenges NO, without a sufficient compensatory increase in antioxidant defences, i.e. SODs (van der Loo et al., 2000). To determine if superoxide production was increased with ageing and affected by DMB supplementation, we assessed superoxide levels in aorta rings using EPR spectroscopy (Fig. 5E). Aortic superoxide levels were higher in old control relative to young mice (P = 0.031), but there was no difference between old DMB-supplemented and young mice (P = 0.971). Next, we assessed aortic abundance of SOD1/Cu-Zn SOD and SOD2/Mn-SOD (Fig. 5F and G). Relative to young mice, SOD1 was lower in old control (P = 0.022) and old DMB-supplemented mice (P = 0.003), but there was no difference between old control and old DMB-supplemented mice (P = 0.451). There was no effect of age or DMB supplementation on abundance of SOD2 (P = 0.880). These findings demonstrate that DMB fully prevented the age-related increase in aortic superoxide levels without having effects on antioxidant defences; thus, DMB supplementation appears to prevent the production of superoxide.
To determine if these differences in superoxide production had a functional effect on EDD, we assessed peak EDD in the presence vs. absence of the superoxide dismutase mimetic TEMPOL (Fig. 5H). Impairments in peak EDD in old control mice were normalized to young levels with the addition of TEMPOL (P = 0.798 vs. young), whereas TEMPOL had no effect on peak EDD in young (P = 0.996 vs. peak EDD with Ach alone) or old DMB-supplemented mice (P = 0.959 vs. peak EDD with ACh alone). These data indicate that DMB improved EDD in old mice by ameliorating age-associated superoxide-driven suppression of EDD.
Together, our results suggest that functional declines in old control mice were due to excess tonic superoxide-associated suppression of EDD and reduced NO bioavailability, whereas DMB supplementation fully prevented these age-related effects.
Aortic stiffness
We assessed aortic stiffness in vivo by measuring aPWV (Fig. 6A). In control mice, aPWV remained at levels similar to those observed in young mice at 18 (P = 0.942 vs. young) and 21 months (P = 0.641 vs young), likewise validation our intervention study design by confirming that initiation of DMB treatment at 18 months occurred prior to the onset of age-associated aortic stiffening. However, there was a progressive, age-related increase in aPWV in control mice at 24 (P = 0.005 vs. young) and 27 months (P < 0.0001 vs. young).
Figure 6. Supplementation with 3,3-dimethyl-1-butanol (DMB) delays and attenuates age-related increases in in vivo aortic stiffness and aortic intima-media thickness but does not alter aortic intrinsic mechanical stiffness nor adventitial collagen abundance.
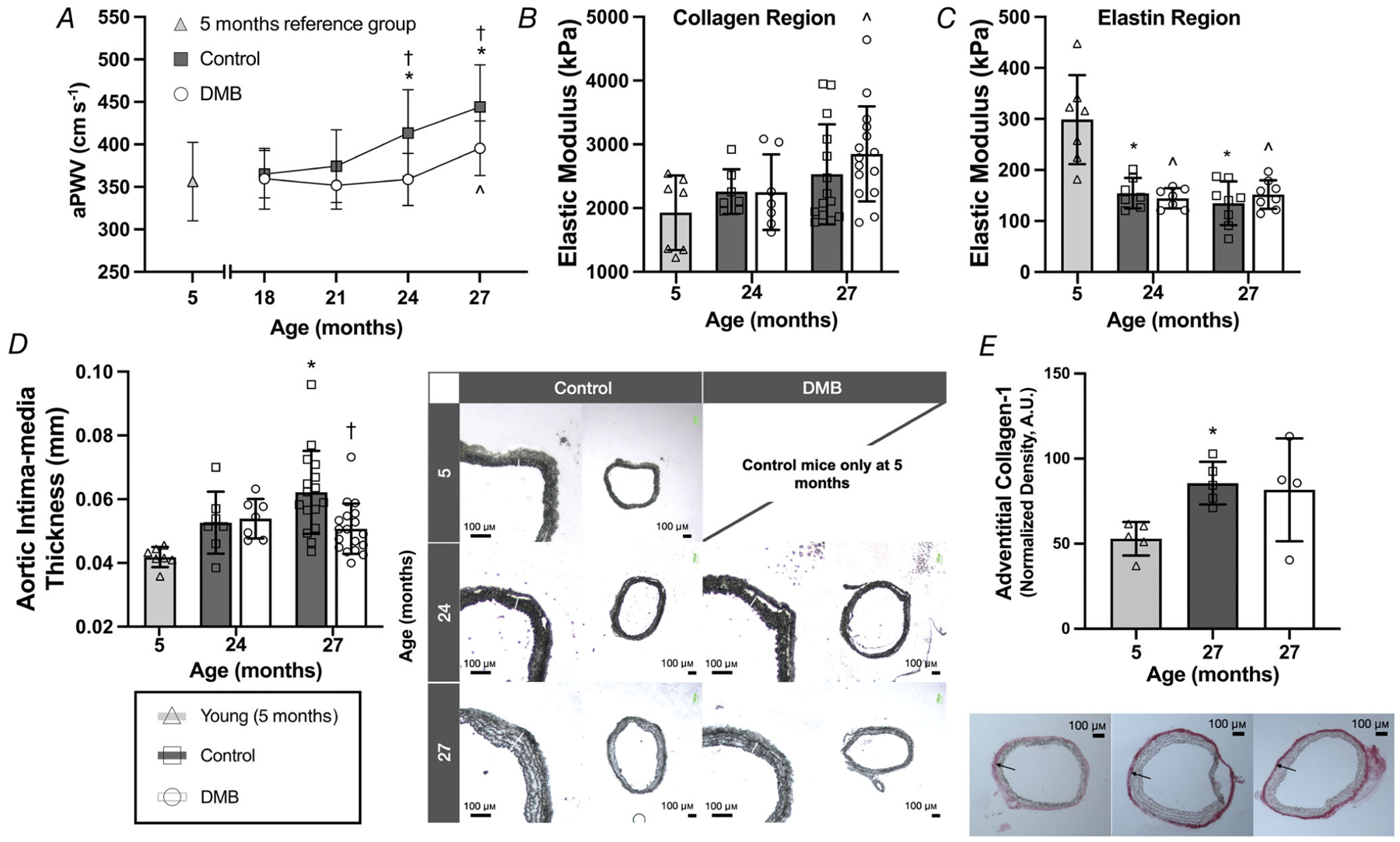
A, in vivo aortic pulse wave velocity (aPWV). B and C, elastic modulus of the (B) collagen-dominant (high force) region of the stress–strain curve and (C) elastin-dominant (low force) region, conducted in 1–2 mm segments of thoracic aorta. D, intima-media thickness measured in 7 μm segments of thoracic aorta, with representative images shown to the right (4× magnification). E, abundance of type-1 collagen measured via quantitative immunohistochemistry in the adventitia of 7 μm segments of thoracic aorta, with representative images shown below (4 × magnification). Data are mean ± SD. Statistics are one-way (effect of age in control mice only) ANOVA with Dunnett’s post hoc test and/or two-way mixed (age × treatment group; A–D) ANOVA with Šídák’s post hoc test.*P < 0.05 control vs. young 5 month reference group. †P < 0.05 DMB vs. control within age group. ˆP < 0.05 DMB vs. young 5 month reference group. n = 7–21 per group.
In DMB-supplemented mice, there was no age-related increase in aPWV in mice at 24 months of age (P = 0.999 vs. young), i.e. DMB supplementation prevented and/or delayed the development of aortic stiffening at this time point (P = 0.003 vs. 24 months control). DMB-supplemented mice experienced a slight age-related increase in aPWV at 27 months of age (P = 0.039 vs. young). However, the magnitude of increase was attenuated by 55% in DMB-supplemented relative to control mice (27 months: P = 0.016).
As we observed increases in aPWV in control mice at 24 and 27 months of age and, to a lesser extent, in DMB-supplemented mice at 27 months of age, we next sought to determine the mechanisms underlying these functional changes. In vivo aortic stiffening can occur due to both functional changes (e.g. impaired endothelial function) as well as structural (i.e. mechanical) changes to the artery (Lakatta et al., 1987; Lakatta & Levy, 2003). Changes in the structural component of aortic stiffness can be examined by assessing intrinsic mechanical (wall) stiffness and elasticity via stress–strain testing in segments of thoracic aorta. The EM of the high-force collagen-dominant region of the stress–strain curve (i.e. mechanical stiffness) increased with age in both control (P = 0.102, 27 months vs. young) and DMB-supplemented mice (P = 0.012, 27 months vs. young), and there was no difference in the collagen region EM between groups at 24 (P = 0.641) or 27 months of age (P = 0.979) (Fig. 6B). There was an age-related decline in the EM of the low-force elastin-dominant region of the stress–strain curve (i.e. decreased elasticity) in control mice at 24 and 27 months (both P < 0.001 vs. young), but there was no effect of DMB supplementation (P = 0.758) (Fig. 6C). Thus, we observed expected age-related changes in intrinsic stiffness and elasticity, but DMB supplementation did not appear to influence these measures of the structural component of aortic stiffness.
However, when calculating stress and strain, the stress component includes aortic IMT as a variable. Interestingly, we observed that aortic IMT increased with age in control mice (P < 0.001, 27 months vs. young), but was lower in DMB-supplemented mice at 27 months (P = 0.004 vs. controls) (Fig. 6D). A higher IMT results in a lower stress for any given amount of strain and, consequently, a lower EM value (see calculations in Methods). As such, it is possible that the observed lack of difference in aortic intrinsic stiffness between control and DMB-supplemented mice, despite differences in in vivo stiffness (aPWV), could be partly due to a greater IMT in control mice.
Increases in aortic intrinsic mechanical stiffness (collagen region EM) are primarily driven by increased abundance of the structural protein collagen (Zieman et al., 2005). Therefore, we measured protein abundance of type-1 collagen in the aortic adventitia via quantitative immunohistochemistry in a subset of mice (n = 4–5 per group). We observed that aortic collagen abundance was increased with age in control mice (P = 0.043, 27 months vs. young), but there was no obvious effect of DMB supplementation (P = 0.948, control vs. DMB; Fig. 6E).
Together, these data confirm that DMB supplementation did not alter the structural component of aortic stiffness, despite mitigating age-related increases in IMT. Therefore, attenuation of age-related aortic stiffening in vivo with DMB was probably mediated by functional rather than structural changes to the artery.
Discussion
Vascular endothelial dysfunction and arterial stiffening develop progressively over the lifespan (Gioscia-Ryan et al., 2021; Lakatta & Levy, 2003; Machin et al., 2020; Sohal & Weindruch, 1996; Zieman et al., 2005), are key contributors to the development of CVD (Lakatta & Levy, 2003) and independently predict risk of future CVD in humans (Boutouyrie et al., 2002; Mattace-Raso et al., 2006; Shechter et al., 2009; Shokawa et al., 2005; Sutton-Tyrrell et al., 2005; Yeboah et al., 2007). Strategies that target the underlying/initiating mechanisms of these processes and therefore prevent or delay dysfunction from manifesting may have a greater effect on reducing the risk of CVD than treating established dysfunction. One such effective intervention could be long-term supplementation with the gut microbiome-targeted compound DMB. In the present study, we found that DMB supplementation initiated at midlife prevents the development of endothelial dysfunction and both delays and attenuates in vivo arterial stiffening with ageing in mice. DMB acts locally on the gut microbiome with no known direct effects on the host (Wang et al., 2015). Thus, there is high potential for translation of DMB or other similar gut microbiome-targeted interventions to humans.
Primordial prevention of CVD
Much of clinical practice is focused on reducing the risk of CV events in individuals with existing CV risk factors (primary prevention) or reducing the risk of recurrent CV events in individuals with established disease (secondary prevention) (O’Keefe et al., 2009). Although interventions that favourably influence CV risk factors from suboptimal to optimal reduce the risk of CV events (primary prevention), the risk remains greater in these individuals compared with peers who have not yet developed risk factors by midlife (Liu et al., 2015). This elevated risk is probably due to cumulative exposure to risk factors prior to the initiation of treatment (Liu et al., 2015; Reges et al., 2021) and may not be fully reversed with treatment (Gioscia-Ryan et al., 2021; Lakatta & Levy, 2003; Sohal & Weindruch, 1996; Zieman et al., 2005). Thus, preservation of a low-risk status by reducing or halting the development of risk factors, i.e. primordial prevention, probably confers the most protection against development of CVD (Lloyd-Jones et al., 2021; Vaduganathan et al., 2015).
Our laboratory and others have shown that male C57BL/6N mice are an appropriate model of vascular ageing, as they develop progressive declines in vascular function in mid- to late life through similar mechanisms to those that drive vascular dysfunction in humans, such as excess vascular oxidative stress (Bachschmid et al., 2013; Brunt et al., 2019; Fleenor et al., 2012; Gioscia-Ryan et al., 2021; Lakatta & Levy, 2003). Furthermore, interventions that reverse age-related vascular dysfunction in this mouse model (Fleenor et al., 2013; Gioscia-Ryan et al., 2014, 2018) have been successfully translated to humans to improve vascular function (Rossman et al., 2018; Santos-Parker et al., 2017), demonstrating that this model is in fact representative of human vascular ageing.
Thus, we combined the primordial prevention paradigm and this model of vascular ageing to determine whether long-term supplementation with DMB could prevent vascular dysfunction with ageing, with the goal of providing a rationale for long-term use of DMB or other TMAO-suppressing compounds in future clinical trials. With this model, we were able to use unique temporal assessments to: (1) provide insight into the time course of development of age-related vascular dysfunction in control mice; (2) confirm that we initiated our intervention prior to the development of vascular dysfunction (i.e. conducted primordial prevention); and (3) demonstrate that our intervention was effective for preventing and delaying/attenuating vascular dysfunction with ageing.
DMB for preventing age-related endothelial dysfunction
In this study, we observed that, in control mice, endothelial function is maintained at young levels in early middle age (18–19 months), consistent with our previous report (Gioscia-Ryan et al., 2021). We did not observe consistent age-related endothelial dysfunction at 21 months of age, whereas dysfunction was apparent at 24 and 27 months of age, suggesting endothelial dysfunction began to manifest in most mice between 21 and 24 months of age. Our results provide novel insight regarding the temporal development of endothelial dysfunction throughout later life in this mouse model.
The declines in EDD that we observed in control mice with ageing were fully prevented with DMB supplementation due to lower arterial superoxide-associated suppression of endothelial function, higher NO bioavailability, and, at least at lower concentrations of ACh, improved smooth muscle sensitivity to NO. Our results align with previous findings demonstrating that DMB supplementation reverses established endothelial dysfunction in old mice by reducing superoxide-associated suppression of EDD and improving NO bioavailability (Brunt et al., 2020; Li et al., 2017), and show for the first time that DMB can also prevent the development of endothelial dysfunction. DMB also improves EDD in models of chronic kidney disease and pre-eclampsia (Chen et al., 2019; Li et al., 2018), suggesting that DMB or other TMAO-suppressing compounds may be effective for preventing endothelial dysfunction not only in the context of ageing, but also in other age-related diseases or diseases/conditions in which the aetiology is similar to ageing, i.e. where endothelial dysfunction is largely mediated by excess oxidative stress.
DMB for delaying and attenuating in vivo arterial stiffening with ageing
In control mice, we observed similar progressive impairments in in vivo aortic stiffness (aPWV), such that there were no impairments at 18 and 21 months of age, but aPWV was elevated in later life at 24 and 27 months of age. There is probably some variability in the exact age of onset of in vivo aortic stiffening, as a previous study from our laboratory reported detectable increases in aPWV at 18 months of age (Gioscia-Ryan et al., 2021). Regardless, aortic stiffening is a progressive phenomenon, with clearly established dysfunction observed by 24 months of age (Chen & Sun, 2018; Gioscia-Ryan et al., 2021; Machin et al., 2020).
Prior to this study, whether DMB has any effect on preventing or reversing arterial stiffening was unknown – to our knowledge, no studies have investigated the effects of DMB on arterial stiffness in any context. We observed that DMB supplementation delayed in vivo aortic stiffening such that aPWV remained at young levels through 24 months of age. aPWV was increased at 27 months of age in DMB-supplemented mice, but this increase was largely attenuated relative to control mice.
The rate of maturation/ageing in mice versus humans is such that a delay in stiffening of ~3 months in old mice is equivalent to nearly a decade in humans (Flurkey et al., 2007). PWV increases by ~1–2 m s−1 per decade in middle-aged to older adult humans (Reference Values for Arterial Stiffness’ Collaboration, 2010) which equates to an ~15% increase in risk of CV events and mortality (Vlachopoulos et al., 2010). Thus, DMB supplementation could confer a significant clinical benefit if it were similarly effective in delaying/attenuating arterial stiffening in humans.
Increases in in vivo aortic stiffness with ageing are mediated by both structural and functional changes to the arteries (Lakatta et al., 1987; Lakatta & Levy, 2003). Functional changes include impairments in endothelial function and effects on vascular tone (Lakatta & Levy, 2003). To isolate the structural component of aortic stiffening, we measured intrinsic mechanical stiffness, as well as elasticity, which are typically altered with ageing (increased stiffness and reduced elasticity) and contribute to increases in aPWV (Lakatta & Levy, 2003). Given that both dietary supplementation and acute ex vivo incubation with TMAO induce intrinsic mechanical stiffening in mouse aortas (Brunt et al., 2021), we postulated that attenuation of in vivo stiffening with long-term DMB supplementation would also be due to prevention of increases in the structural component of arterial stiffness. We observed that elasticity was reduced in control mice at 24 and 27 months of age, and intrinsic stiffness tended to increase at 27 months of age along with an increase in adventitial abundance of the arterial structural protein collagen. Thus, our findings align with previous reports (Brunt et al., 2019; Fleenor et al., 2012; Gioscia-Ryan et al., 2021) demonstrating that age-related increases in in vivo aortic stiffening are accompanied by structural changes to the arteries. However, we found that DMB had no effect on intrinsic mechanical stiffness nor aortic adventitial collagen abundance. This discrepancy could be due to lower circulating levels of TMAO in the current study (~15 μm on average in 27-month-old control mice) relative to the levels achieved when mice were supplemented/arteries were incubated with TMAO (~30 μm on average) (Brunt et al., 2021). In other words, the magnitude of increases in circulating TMAO that typically occur with ageing appears to be sufficient to induce functional changes in the arteries, but higher levels may be needed to induce structural changes. For these reasons, we speculate that the attenuation of age-related in vivo stiffening with DMB is probably mediated primarily by modulating the functional component of arterial stiffness, i.e. changes in endothelial function and effects on vascular tone.
Experimental considerations and limitations
Other studies have reported higher TMAO in old vs. young mice or rats (Brunt et al., 2019; Li et al., 2017). Contrary to these reports, we did not observe any age-related increase in plasma TMAO at 18–24 months of age and were unable to detect a statistically significant increase at 27 months, although we were underpowered for these comparisons. In addition, this variability and difference from previous reports could be related to the timing of our measurements. Plasma TMAO levels peak ~12 h after consumption of an oral choline bolus in humans (Mödinger et al., 2019) and ~5 h after an oral gavage of phosphatidylcholine-containing foods in mice (Gao et al., 2016). We did not control food intake in mice prior to plasma collection and only measured plasma TMAO levels at the time of assessing terminal measures to not induce additional stress on the mice. Thus, it is likely that we did not capture peak TMAO levels in all mice. However, no mice that received DMB had elevated TMAO levels relative to the young reference group, suggesting that DMB still acted as expected. Moreover, DMB supplementation may have preserved vascular function by reducing regular (daily) exposure to high postprandial levels of TMAO. Despite the apparent efficacy of DMB in the present study, there are some more recently developed second-generation TMA lyase inhibitors that suppress TMA production to a greater extent and could be considered for future studies (Roberts et al., 2018).
Mice supplemented with DMB had lower food and water consumption between 18 and 21 months of age, and lower body mass at 21 months of age. Thus, it is possible that improvements in vascular function could have been due in part to lower body mass or food intake. However, we do not believe this is the case because we observed no functional differences in EDD and aPWV between control and DMB-supplemented mice at 21 months of age, whereas functional differences were apparent at 24 and 27 months of age when there were no group differences in food intake and body mass, suggesting that improvements in vascular function were specific to DMB supplementation.
Conclusions
Long-term supplementation with DMB prevents age-related impairments in endothelial function by preserving NO bioavailability and reducing excess superoxide-related suppression of endothelial function. Additionally, DMB delays and attenuates progressive in vivo aortic stiffening with ageing, probably due in part to preservation of endothelial function. DMB or other TMAO-suppressing interventions have translational potential for preventing age-related vascular dysfunction in humans and thereby for reducing the risk of CVD.
Translational perspective.
We tested the hypothesis that long-term supplementation with the gut microbiome-targeted compound DMB could prevent the development of age-related vascular dysfunction when initiated in midlife, prior to the development of dysfunction. We observed that DMB fully prevented progressive declines in vascular endothelial function and both delayed and attenuated increases in in vivo aortic stiffness. Interventions aimed at preventing the development of cardiovascular (CV) risk factors have more potential for preventing CV diseases (CVD) relative to those aimed at reversing established dysfunction. Indeed, individuals who receive treatment to bring CV risk factors to optimal ranges still have elevated CV risk relative to those who did not yet develop risk factors in midlife. The gut microbiome has emerged as a promising target for preventing the development of vascular dysfunction. As such, DMB or other TMAO-suppressing and gut microbiome-targeted interventions initiated in midlife have potential for reducing CVD risk in humans by preventing the development of vascular dysfunction. Future research should explore and develop additional gut microbiome-targeted interventions using preclinical animal models and subsequently translate the findings of the present study and others to humans.
Key points.
Vascular dysfunction, characterized by endothelial dysfunction and arterial stiffening, develops progressively with ageing and increases the risk of cardiovascular diseases (CVD).
Interventions aimed at preventing the development of CV risk factors have more potential for preventing CVD relative to those aimed at reversing established dysfunction.
The gut microbiome-derived metabolite trimethylamine N-oxide (TMAO) induces vascular dysfunction, is associated with higher CV risk and can be suppressed by supplementation with 3,3-dimethyl-1-butanol (DMB).
In mice, DMB prevented the development of endothelial dysfunction and delayed and attenuated in vivo arterial stiffening with ageing when supplementation was initiated in midlife, prior to the development of dysfunction.
DMB supplementation or other TMAO-suppressing interventions have potential for translation to humans for reducing CV risk with ageing.
Acknowledgements
The authors thank Melanie Zigler, Jill Miyamoto-Ditmon, Zachary Cook and Danijel Djukovic for assistance with data collection.
Funding
This work was supported by R01 HL1348870-02S1 and R21 AG060884 (D.R.S.); F32 HL140875 (V.E.B.); and T32 AG000279 (A.G.C.).
Biography

Abigail G. Casso is a PhD Candidate in Dr D. R. Seals’ Integrative Physiology of Aging Laboratory at the University of Colorado Boulder. The studies described in the present paper represent work carried out as part of an NIH T32 fellowship through the Division of Geriatrics at the University of Colorado Denver. Abigail’s long-term research goals are to investigate novel mechanisms contributing to age-related vascular dysfunction and interventions for preserving function with ageing with the goal of preventing or delaying the progression of cardiovascular diseases.
Footnotes
Competing interests
None.
Supporting information
Additional supporting information can be found online in the Supporting Information section at the end of the HTML view of the article. Supporting information files available:
Data availability statement
All data underlying this article will be shared upon reasonable request to the corresponding author.
References
- Bachschmid MM, Schildknecht S, Matsui R, Zee R, Haeussler D, Cohen RA, Pimental D, & van der Loo B (2013). Vascular aging: Chronic oxidative stress and impairment of redox signaling-consequences for vascular homeostasis and disease. Annals of Medicine, 45(1), 17–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballak DB, Brunt VE, Sapinsley ZJ, Ziemba BP, Richey JJ, Zigler MC, Johnson LC, Gioscia-Ryan RA, Culp-Hill R, Eisenmesser EZ, D’Alessandro A, Dinarello CA, & Seals DR (2020). Short-term interleukin-37 treatment improves vascular endothelial function, endurance exercise capacity, and whole-body glucose metabolism in old mice. Aging Cell, 19(1), e13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutagy NE, Neilson AP, Osterberg KL, Smithson AT, Englund TR, Davy BM, Hulver MW, & Davy KP (2015). Short-term high-fat diet increases postprandial trimethylamine-N-oxide in humans. Nutrition Research, 35(10), 858–864. [DOI] [PubMed] [Google Scholar]
- Boutouyrie P, Tropeano AI, Asmar R, Gautier I, Benetos A, Lacolley P, & Laurent S (2002). Aortic stiffness is an independent predictor of primary coronary events in hypertensive patients: A longitudinal study. Hypertension, 39(1), 10–15. [DOI] [PubMed] [Google Scholar]
- Brunt VE, Casso AG, Gioscia-Ryan RA, Sapinsley ZJ, Ziemba BP, Clayton ZS, Bazzoni AE, VanDongen NS, Richey JJ, Hutton DA, Zigler MC, Neilson AP, Davy KP, & Seals DR (2021). Gut microbiome-derived metabolite trimethylamine n-oxide induces aortic stiffening and increases systolic blood pressure with aging in mice and humans. Hypertension, 78(2), 499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunt VE, Gioscia-Ryan RA, Casso AG, VanDongen NS, Ziemba BP, Sapinsley ZJ, Richey JJ, Zigler MC, Neilson AP, Davy KP, & Seals DR (2020). Trimethylamine-n-oxide promotes age-related vascular oxidative stress and endothelial dysfunction in mice and healthy humans. Hypertension, 76(1), 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunt VE, Gioscia-Ryan RA, Richey JJ, Zigler MC, Cuevas LM, Gonzalez A, Vázquez-Baeza Y, Battson ML, Smithson AT, Gilley AD, Ackermann G, Neilson AP, Weir T, Davy KP, Knight R, & Seals DR (2019). Suppression of the gut microbiome ameliorates age-related arterial dysfunction and oxidative stress in mice. The Journal of Physiology, 597(9), 2361–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Li J, Li N, Liu H, & Tang J (2019). Increased circulating trimethylamine N-oxide plays a contributory role in the development of endothelial dysfunction and hypertension in the RUPP rat model of preeclampsia. Hypertension in Pregnancy, 38(2), 96–104. [DOI] [PubMed] [Google Scholar]
- Chen K, & Sun Z (2018). Activation of DNA demethylases attenuates aging-associated arterial stiffening and hypertension. Aging Cell, 17(4), e12762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPont JJ, Kim SK, Kenney RM, & Jaffe IZ (2021). Sex differences in the time course and mechanisms of vascular and cardiac aging in mice: Role of the smooth muscle cell mineralocorticoid receptor. American Journal of Physiology Heart and Circulatory Physiology, 320(1), H169–H180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleenor BS, Seals DR, Zigler ML, & Sindler AL (2012). Superoxide-lowering therapy with TEMPOL reverses arterial dysfunction with aging in mice. Aging Cell, 11(2), 269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleenor BS, Sindler AL, Marvi NK, Howell KL, Zigler ML, Yoshizawa M, & Seals DR (2013). Curcumin ameliorates arterial dysfunction and oxidative stress with aging. Experimental Gerontology, 48(2), 269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flurkey K, Currer JM, & Harrison DE (2007). The mouse in aging research. In Fox JG et al. , (Ed.). The mouse in biomedical research (2nd ed.), American College Laboratory Animal Medicine (Elsevier), Burlington, MA. [Google Scholar]
- Gao X, Jiang C, Xu J, Yanagita T, Xue C, & Wang Y (2016). Serum pharmacokinetics of choline, trimethylamine, and trimethylamine-N-oxide after oral gavage of phosphatidylcholines with different fatty acid compositions in mice. Bioscience, Biotechnology, and Biochemistry, 80(11), 2217–2223. [DOI] [PubMed] [Google Scholar]
- Gioscia-Ryan RA, Battson ML, Cuevas LM, Eng JS, Murphy MP, & Seals DR (2018). Mitochondria-targeted antioxidant therapy with MitoQ ameliorates aortic stiffening in old mice. Journal of Applied Physiology (Bethesda, Md : 1985), 124(5), 1194–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gioscia-Ryan RA, Clayton ZS, Zigler MC, Richey JJ, Cuevas LM, Rossman MJ, Battson ML, Ziemba BP, Hutton DA, VanDongen NS, & Seals DR (2021). Lifelong voluntary aerobic exercise prevents age- and Western diet- induced vascular dysfunction, mitochondrial oxidative stress and inflammation in mice. Journal of Physiology, 599(3), 911–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gioscia-Ryan RA, LaRocca TJ, Sindler AL, Zigler MC, Murphy MP, & Seals DR (2014). Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. The Journal of Physiology, 592(12), 2549–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger JP, Barman SM, & Barrett KE (2012). Promoting physiology as an essential element in translational research. Physiology (Bethesda, Md), 27, 326. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. Journal of Physiology, 593(12), 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Miyagawa K, Sato K, Ueda R, & Dohi Y (2006). Temocapril, an Angiotensin converting enzyme inhibitor, ameliorates age-related increase in carotid arterial stiffness in normotensive subjects. Cardiology, 106(3), 190–194. [DOI] [PubMed] [Google Scholar]
- Jennings A, Berendsen AM, de Groot L, Feskens EJM, Brzozowska A, Sicinska E, Pietruszka B, Meunier N, Caumon E, Malpuech-Brugère C, Santoro A, Ostan R, Franceschi C, Gillings R, O’ Neill CM, Fairweather-Tait SJ, Minihane A-M, & Cassidy A (2019). Mediterranean-style diet improves systolic blood pressure and arterial stiffness in older adults. Hypertension, 73(3), 578–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplon RE, Hill SD, Bispham NZ, Santos-Parker JR, Nowlan MJ, Snyder LL, Chonchol M, LaRocca TJ, McQueen MB, & Seals DR (2016). Oral trehalose supplementation improves resistance artery endothelial function in healthy middle-aged and older adults. Aging (Albany NY), 8(6), 1167–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, DiDonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, … Hazen SL (2013). Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nature Medicine, 19(5), 576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakatta EG (2003). Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part III: cellular and molecular clues to heart and arterial aging. Circulation, 107(3), 490–497. [DOI] [PubMed] [Google Scholar]
- Lakatta EG, & Levy D (2003). Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: A “set up” for vascular disease. Circulation, 107(1), 139–146. [DOI] [PubMed] [Google Scholar]
- Lakatta EG, Mitchell JH, Pomerance A, & Rowe GG (1987). Human aging: Changes in structure and function. Journal of the American College of Cardiology, 10(2), 42A–47A. [DOI] [PubMed] [Google Scholar]
- Lammers SR, Kao PH, Qi HJ, Hunter K, Lanning C, Albietz J, Hofmeister S, Mecham R, Stenmark KR, & Shandas R (2008). Changes in the structure-function relationship of elastin and its impact on the proximal pulmonary arterial mechanics of hypertensive calves. American Journal of Physiology Heart and Circulatory Physiology, 295(4), H1451–H1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Chen Y, Gua C, & Li X (2017). Elevated circulating trimethylamine n-oxide levels contribute to endothelial dysfunction in aged rats through vascular inflammation and oxidative stress. Frontiers in Physiology, 8, 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Gua C, Wu B, & Chen Y (2018). Increased circulating trimethylamine N-oxide contributes to endothelial dysfunction in a rat model of chronic kidney disease. Biochemical and Biophysical Research Communications, 495(2), 2071–2077. [DOI] [PubMed] [Google Scholar]
- Li Z, & Vance DE (2008). Thematic review series: Glycerolipids. Phosphatidylcholine and choline homeostasis. Journal of Lipid Research, 49(6), 1187–1194. [DOI] [PubMed] [Google Scholar]
- Liu K, Colangelo LA, Daviglus ML, Goff DC, Pletcher M, Schreiner PJ, Sibley CT, Burke GL, Post WS, Michos ED, & Lloyd-Jones DM (2015). Can antihypertensive treatment restore the risk of cardiovascular disease to ideal levels?: The Coronary Artery Risk Development in Young Adults (CARDIA) Study and the Multi-Ethnic Study of Atherosclerosis (MESA). Journal of the American Heart Association, 4(9), e002275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Jones DM, Hong Y, Labarthe D, Mozaffarian D, Appel LJ, Van Horn L, Greenlund K, Daniels S, Nichol G, Tomaselli GF, Arnett DK, Fonarow GC, Ho PM, Lauer MS, Masoudi FA, Robertson RM, Roger V, Schwamm LH, Sorlie P, … Rosamond WD (2010). Defining and setting national goals for cardiovascular health promotion and disease reduction: The American Heart Association’s strategic Impact Goal through 2020 and beyond. Circulation, 121(4), 586–613. [DOI] [PubMed] [Google Scholar]
- Lloyd-Jones DM, Albert MA, & Elkind M (2021). The American Heart Association’s focus on primordial prevention. Circulation, 144(15), e233–e235. [DOI] [PubMed] [Google Scholar]
- van der Loo B, Labugger R, Skepper JN, Bachschmid M, Kilo J, Powell JM, Palacios-Callender M, Erusalimsky JD, Quaschning T, Malinski T, Gygi D, Ullrich V, & Lüscher TF (2000). Enhanced peroxynitrite formation is associated with vascular aging. Journal of Experimental Medicine, 192(12), 1731–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machin DR, Auduong Y, Gogulamudi VR, Liu Y, Islam MT, Lesniewski LA, & Donato AJ (2020). Lifelong SIRT-1 overexpression attenuates large artery stiffening with advancing age. Aging (Albany NY), 12(12), 11314–11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattace-Raso FUS, van der Cammen TJM, Hofman A, van Popele NM, Bos ML, Schalekamp M, Asmar R, Reneman RS, Hoeks APG, Breteler MMB, & Witteman JCM (2006). Arterial stiffness and risk of coronary heart disease and stroke: The Rotterdam Study. Circulation, 113(5), 657–663. [DOI] [PubMed] [Google Scholar]
- Mödinger Y, Schön C, Wilhelm M, & Hals P-A (2019). Plasma kinetics of choline and choline metabolites after a single dose of SuperbaBoostTM krill oil or choline bitartrate in healthy volunteers. Nutrients, 11(10), 2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motulsky HJ, & Brown RE (2006). Detecting outliers when fitting data with nonlinear regression - a new method based on robust nonlinear regression and the false discovery rate. Bmc Bioinformatics [Electronic Resource], 7(1), 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Keefe JH, Carter MD, & Lavie CJ (2009). Primary and secondary prevention of cardiovascular diseases: A practical evidence-based approach. Mayo Clinic Proceedings, 84(8), 741–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reference Values for Arterial Stiffness’ Collaboration (2010). Determinants of pulse wave velocity in healthy people and in the presence of cardiovascular risk factors: “establishing normal and reference values”. European Heart Journal, 31(19), 2338–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reges O, Ning H, Wilkins JT, Wu CO, Tian X, Domanski MJ, Lloyd-Jones DM, & Allen NB (2021). Association of cumulative systolic blood pressure with long-term risk of cardiovascular disease and healthy longevity: Findings from the lifetime risk pooling project cohorts. Hypertension, 77(2), 347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AB, Gu X, Buffa JA, Hurd AG, Wang Z, Zhu W, Gupta N, Skye SM, Cody DB, Levison BS, Barrington WT, Russell MW, Reed JM, Duzan A, Lang JM, Fu X, Li L, Myers AJ, Rachakonda S, … Hazen SL (2018). Development of a gut microbe-targeted nonlethal therapeutic to inhibit thrombosis potential. Nature Medicine, 24(9), 1407–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman MJ, Santos-Parker JR, Steward CAC, Bispham NZ, Cuevas LM, Rosenberg HL, Woodward KA, Chonchol M, Gioscia-Ryan RA, Murphy MP, & Seals DR (2018). Chronic supplementation with a mitochondrial antioxidant (MitoQ) improves vascular function in healthy older adults. Hypertension, 71(6), 1056–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Parker JR, Strahler TR, Bassett CJ, Bispham NZ, Chonchol MB, & Seals DR (2017). Curcumin supplementation improves vascular endothelial function in healthy middle-aged and older adults by increasing nitric oxide bioavailability and reducing oxidative stress. Aging (Albany NY), 9(1), 187–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scuteri A, Morrell CH, Orrù M, Strait JB, Tarasov KV, Ferreli LAP, Loi F, Pilia MG, Delitala A, Spurgeon H, Najjar SS, AlGhatrif M, & Lakatta EG (2014). Longitudinal perspective on the conundrum of central arterial stiffness, blood pressure, and aging. Hypertension, 64(6), 1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seals DR (2013). Translational physiology: From molecules to public health. Journal of Physiology, 591(14), 3457–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter M, Issachar A, Marai I, Koren-Morag N, Freinark D, Shahar Y, Shechter A, & Feinberg MS (2009). Long-term association of brachial artery flow-mediated vasodilatation and cardiovascular events in middle-aged subjects with no apparent heart disease. International Journal of Cardiology, 134(1), 52–58. [DOI] [PubMed] [Google Scholar]
- Shokawa T, Imazu M, Yamamoto H, Toyofuku M, Tasaki N, Okimoto T, Yamane K, & Kohno N (2005). Pulse wave velocity predicts cardiovascular mortality: Findings from the Hawaii-Los Angeles-Hiroshima study. Circulation Journal, 69(3), 259–264. [DOI] [PubMed] [Google Scholar]
- Sohal RS, & Weindruch R (1996). Oxidative stress, caloric restriction, and aging. Science, 273(5271), 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton-Tyrrell K, Najjar SS, Boudreau RM, Venkitachalam L, Kupelian V, Simonsick EM, Havlik R, Lakatta EG, Spurgeon H, Kritchevsky S, Pahor M, Bauer D, & Newman A, & Health ABC Study (2005). Elevated aortic pulse wave velocity, a marker of arterial stiffness, predicts cardiovascular events in well-functioning older adults. Circulation, 111(25), 3384–3390. [DOI] [PubMed] [Google Scholar]
- Tang WHW, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, & Hazen SL (2013). Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. New England Journal of Medicine, 368(17), 1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The University of Queensland (n.d.). IMBiol Mounting Media Recipe - Institute for Molecular Bioscience - University of Queensland. Available at: https://imb.uq.edu.au/facilities/microscopy/2020-microscopy-resources/sample-prep/imbiol-mounting-media-recipe [Accessed December 13, 2021].
- Vaduganathan M, Venkataramani AS, & Bhatt DL (2015). Moving toward global primordial prevention in cardiovascular disease: The heart of the matter. Journal of the American College of Cardiology, 66(14), 1535–1537. [DOI] [PubMed] [Google Scholar]
- Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Cheng S, Delling FN, Elkind MSV, Evenson KR, Ferguson JF, Gupta DK, Khan SS, Kissela BM, Knutson KL, Lee CD, … Lewis TT (2021). Heart disease and stroke statistics-2021 update: A report from the American Heart Association. Circulation, 143(8), e254–e743. [DOI] [PubMed] [Google Scholar]
- Vlachopoulos C, Aznaouridis K, & Stefanadis C (2010). Prediction of cardiovascular events and all-cause mortality with arterial stiffness: A systematic review and meta-analysis. Journal of the American College of Cardiology, 55(13), 1318–1327. [DOI] [PubMed] [Google Scholar]
- Vlachopoulos C, O’Rourke MF, & Nichols WW (2011). McDonald’s blood flow in arteries: Theoretical, experimental and clinical principles, th. CRC Press. Underwood W et al. (2020). AVMA Guidelines for the Euthanasia of Animals: 2020 Edition. [Google Scholar]
- Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung Y-M, Wu Y, Schauer P, Smith JD, Allayee H, Tang WHW, DiDonato JA, Lusis AJ, & Hazen SL (2011). Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature, 472(7341), 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Levison BS, Hazen JE, Donahue L, Li X-M, & Hazen SL (2014). Measurement of trimethylamine-N-oxide by stable isotope dilution liquid chromatography tandem mass spectrometry. Analytical Biochemistry, 455, 35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, Gu X, Huang Y, Zamanian-Daryoush M, Culley MK, DiDonato AJ, Fu X, Hazen JE, Krajcik D, DiDonato JA, Lusis AJ, & Hazen SL (2015). Non-lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell, 163(7), 1585–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2021). World Health Statistics. Available at: https://www.who.int/data/gho/publications/world-health-statistics [Accessed December 13, 2021].
- Xu J, & Shi G-P (2014). Vascular wall extracellular matrix proteins and vascular diseases. Biochimica Et Biophysica Acta, 1842(11), 2106–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeboah J, Crouse JR, Hsu F-C, Burke GL, & Herrington DM (2007). Brachial flow-mediated dilatation predicts incident cardiovascular events in older adults: The Cardiovascular Health Study. Circulation, 115(18), 2390–2397. [DOI] [PubMed] [Google Scholar]
- Zieman SJ, Melenovsky V, & Kass DA (2005). Mechanisms, pathophysiology, and therapy of arterial stiffness. Arteriosclerosis, Thrombosis, and Vascular Biology, 25(5), 932–943. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data underlying this article will be shared upon reasonable request to the corresponding author.