

In this study, Meng et al. describe an antagonism between the conserved POPS and exonuclease domains of the yeast Pol2 polymerase and its importance in processive DNA synthesis and sister fork symmetry. They show that multiple defects caused by POPS mutations were rescued by exonuclease inactivation, including impaired growth and DNA synthesis, genome instability, and reliance on other genome maintenance factors, and single-molecule data revealed that the rescue stemmed from allowing sister replication forks to progress at equal rates.
Keywords: Pol2, replication elongation, sister fork asymmetry, EXO domain
Abstract
Pol2 is the leading-strand DNA polymerase in budding yeast. Here we describe an antagonism between its conserved POPS (Pol2 family-specific catalytic core peripheral subdomain) and exonuclease domain and the importance of this antagonism in genome replication. We show that multiple defects caused by POPS mutations, including impaired growth and DNA synthesis, genome instability, and reliance on other genome maintenance factors, were rescued by exonuclease inactivation. Single-molecule data revealed that the rescue stemmed from allowing sister replication forks to progress at equal rates. Our data suggest that balanced activity of Pol2's POPS and exonuclease domains is vital for genome replication and stability.
The eukaryotic genome is replicated through numerous sequence units referred to as replicons. Within each replicon, a pair of replisomes assemble at the replication initiation site (origin) to establish twin replication forks that travel outward until merging with neighboring replicons (Burgers and Kunkel 2017). Within the replisome, the leading and the lagging strand DNA polymerase complexes Polε and Polδ, respectively, are primarily responsible for copying the two parental strands (Pursell et al. 2007; Daigaku et al. 2015). While Polδ acts in a discontinuous manner, Polε carries out continuous synthesis that extends many kilobases and is a primary driver of replisome progression by promoting the rapid movement of the replicative helicase (Gan et al. 2017; Taylor and Yeeles 2019; Devbhandari and Remus 2020). The observation that sister replication forks emanating from the same replication origin progress at highly similar rates suggests that Polε is capable of continuous DNA synthesis through a complex chromatin environment (Claussin et al. 2022). Robust synthesis by Polε stems from both its intrinsic attributes and the assistance of extrinsic factors. For example, multiple replication-promoting factors can remove template barriers that impede Polε progression (Hizume and Araki 2019). In addition, unlike Polδ, the catalytic subunit of Polε binds directly to the replicative helicase, which encircles the leading strand template, thus stabilizing Polε association with the template strand (Langston et al. 2014; Goswami et al. 2018).
Besides being tethered to the replicative helicase, Polε has other features that contribute to the enzyme's ability to continuously synthesize DNA. In particular, the catalytic core of Polε possesses several conserved domains not found in Polδ or other DNA polymerases (Hogg et al. 2014). Thus far, only two of these domains have been examined in detail using the yeast catalytic subunit of Polε, Pol2, as the prototype of this family of enzymes. In one study, it was shown that the P domain of Pol2 enhances Polε–template association and processive leading strand synthesis (Hogg et al. 2014). Recently, we characterized another Pol2 family domain, referred to as POPS (Pol2 family-specific catalytic core peripheral subdomain), which is located at the periphery of the Pol2 catalytic (CAT) core (Supplemental Fig. S1A; Meng et al. 2020). This 68-residue region shows a high degree of sequence homology between the yeast Pol2 and its human counterpart, POLE (Meng et al. 2020). Modeling three recurrent cancer-associated mutations found in this region of the POLE enzyme in yeast generated the pol2-R567C,E611K,L621F (pol2-REL) allele, which led to increased gross chromosomal rearrangement (GCR) but normal mutation rates, distinguishing the POPS mutations from other POLE cancer mutants that cause a hypermutation phenotype (Meng et al. 2020). Replication progression defects were seen for pol2-REL cells, and the purified PolεREL complex reduced DNA synthesis in vitro, showing that POPS plays an important role in leading strand synthesis (Meng et al. 2020). However, how POPS contributes to Pol2-mediated replication elongation at the molecular level is unclear.
In this work, we addressed the molecular function of the POPS domain, aiming to gain insight into the intrinsic Polε features that promote processive DNA synthesis. To this end, we looked for suppressive mutations that could rescue pol2-REL defects. Surprisingly, we found that abolishing the activity of the Pol2 exonuclease (EXO) domain improved growth, S-phase progression, and genomic stability, as well as reduced reliance on other genome maintenance pathways of pol2-REL cells. Single-molecule analysis using Replicon-seq demonstrated that the pol2-REL mutant caused fork slowing or stalling, resulting in asymmetric progression of sister replication forks genome-wide. Importantly, replication fork progression was improved by abolishing the EXO activity of Pol2. Although the Pol2 EXO domain is best known for editing misincorporated nucleotides (Pursell and Kunkel 2008), our data provide evidence for another role for this domain in reducing fork movement and that such a role is balanced by POPS to permit processive leading strand synthesis and reduce the risk of chromosomal rearrangements.
Results and Discussion
Slow growth of a pol2 POPS mutant was rescued by abolishing the Pol2 exonuclease activity
We investigated whether the slow growth caused by the pol2-REL allele, which contains mutations in Pol2's POPS (Meng et al. 2020), could be suppressed by mutating other Pol2 regions implicated in the negative regulation of the enzyme. A recent study showed that mutating Pol2's PCNA binding motif (PIPm, F1119A, and F1200A) rescued the poor growth of cells carrying a pol2 allele lacking its polymerase domain, presumably because this mutation allowed Polδ to replace Polε for leading strand synthesis (Devbhandari and Remus 2020). However, PIPm did not improve the growth of pol2-REL at its semipermissive temperature (34°C); rather, it worsened pol2-REL growth, suggesting that when POPS was defective, weakening PCNA binding further reduced DNA synthesis (Fig. 1A). We confirmed that the slow growth of pol2-REL or pol2-REL-PIPm mutant cells was not caused by abnormal Pol2 protein levels (Supplemental Fig. S1B). As PCNA binding enhances Polε processivity (Chilkova et al. 2007), the negative genetic interaction between REL and PIPm mutations is consistent with the idea that POPS is also involved in processive DNA synthesis.
Figure 1.

Inactivating the Pol2 exonuclease suppressed the growth defect and genomic instability of a POPS mutant. (A) The growth of three pol2 mutant strains. In each, the endogenous POL2 locus was replaced by the indicated pol2 mutation. While pol2-REL cells grew poorly at the semipermissive temperature (34°C), further mutating two catalytic residues of the Pol2 exonuclease domain (D290A/E292A) led to better growth. In contrast, mutating the PCNA binding motif (PIPm) worsened pol2-REL growth. Cells were examined in threefold series of dilution on plates. (B) The growth of pol2-EXOcd-REL versus pol2-REL spore clones on dissection plates. Two representative tetrads are shown for the diploid cells carrying the indicated pol2 mutants. (C) Inactivating the Pol2 exonuclease rescued the sensitivity of pol2-REL cells to the loss of the genes encoding Rad52, Rnh201, or Rrm3. Cells were examined in threefold series of dilution on plates. (D) The growth of pol2-REL cells was improved by Rnh1 overexpression, which was driven by a galactose-inducible promoter. (E) Inactivating the Pol2 exonuclease reduced the GCR levels in pol2-REL cells.
Another study found that abolishing the Pol2 exonuclease activity by mutating two catalytic residues (EXOcd, as in the pol2-4 allele) improved the growth of a pol2 EXO domain mutant that is defective in DNA damage checkpoint-induced phosphorylation (Pellicanò et al. 2021). Although pol2-REL did not contain mutations in the EXO domain, its growth defect was also suppressed by the EXOcd mutation, since spore clones of pol2-EXOcd-REL grew better than those of pol2-REL (Fig. 1A,B). We confirmed that pol2-EXOcd-REL supported normal protein levels (Supplemental Fig. S1B). Thus, we concluded that abolishing the exonuclease activity of Pol2 could improve the growth of a POPS mutant.
The exonuclease domain of Pol2 is best studied for its proofreading ability, which corrects misincorporated nucleotides at the replication fork (Pursell and Kunkel 2008). However, a recent study suggested that the EXO domain could promote nascent strand degradation under conditions of replication stress, similar to the effect exerted by the Exo1 nuclease (Pellicanò et al. 2021). Given this similarity, we queried whether exo1Δ could also suppress the pol2-REL growth defect. In contrast to the lack of exonuclease activity of Pol2, Exo1 loss did not improve pol2-REL growth or affect the growth of pol2-EXOcd-REL cells (Supplemental Fig. S1C). Thus, the suppression of pol2-REL was specifically achieved by the EXO domain mutation and not by exo1Δ, unlike the situation reported in replication stress (Pellicanò et al. 2021).
Suppression specificity of Pol2 exonuclease inactivation toward the POPS mutant
Our genetic findings described above suggested a direct functional interplay between the POPS and EXO domains of Pol2. Since pol2-REL exhibited a wild-type mutation rate (Meng et al. 2020), such functional interplay is unlikely to be related to the mutation-editing function of Pol2's EXO domain. Consistent with this view, increasing cellular dNTP levels by deleting SML1, which is known to influence Pol2-mediated mutagenesis (Zhao et al. 1998; Williams et al. 2015), did not affect the growth of pol2-REL or pol2-REL-EXOcd (Supplemental Fig. S2A). Furthermore, Pol2 exonuclease inactivation did not serve as a general suppressor toward reduced leading strand synthesis caused by other mutants. First, pol2-EXOcd did not improve the growth of cells lacking the Polε subunit Dpb3, which promotes Polε–template association and DNA synthesis (Supplemental Fig. S2B; Hizume and Araki 2019). Second, pol2-EXOcd did not rescue the slow growth of cells lacking Mrc1, which maximizes leading strand synthesis speed, or another replisome component, Ctf4 (Supplemental Fig. S2C). Similarly, pol2-EXOcd did not affect the growth of cells showing increased pausing of replicative synthesis due to the lack of the Rrm3 helicase that removes protein barriers or the RNase H2 nuclease that removes RNA–DNA hybrids from the template (Supplemental Fig. S2C). Taken together, the specific suppression of the slow growth of pol2-REL by EXOcd provided evidence that POPS may uniquely counter a role of the EXO domain in nascent strand degradation in the absence of misincorporated nucleosides during normal growth that can slow DNA synthesis.
EXO inactivation reduced pol2-REL genome instability and its reliance on other genome maintenance factors
Next, we tested whether better growth of pol2-EXOcd-REL cells compared with pol2-REL cells correlated with an improvement of genome replication. The growth of cells suffering from defective replisome-mediated DNA synthesis increases reliance on replication-promoting factors, such as the Rrm3 helicase and RNase H2 (Hizume and Araki 2019). Consistent with compromised replication in pol2-REL cells, their growth strongly relies on the presence Rrm3 and the Rhn201 subunit of RNase H2 (Meng et al. 2020). Significantly, inactivating the Pol2 exonuclease improved the growth of pol2-REL in the absence of either Rrm3 or Rhn201 (Fig. 1C). Furthermore, simply overexpressing the gene encoding the RNase H1 protein capable of removing RNA–DNA hybrids improved the growth of pol2-REL (Fig. 1D). Taken together, these data provided evidence that pol2-REL had a reduced ability to replicate past template barriers, such as RNA–DNA hybrids, and this deficiency could be improved by inactivation of the Pol2 exonuclease.
A hallmark of defective DNA replication is a reliance on recombinational repair to fill unreplicated ssDNA gaps and on the DNA damage checkpoint that allows more time for DNA synthesis. We found that pol2-REL indeed showed synthetic sickness with mutations of the key recombination repair factor Rad52 and the DNA checkpoint mediator Rad9 (Fig. 1C; Supplemental Fig. S3A). Significantly, both negative genetic interactions were partially rescued by Pol2 exonuclease inactivation (Fig. 1C; Supplemental Fig. S3A).
Another hallmark of impaired genome replication is increased levels of GCR. We have previously shown that pol2-EXOcd did not change GCR rate as compared with wild type (Meng et al. 2020). However, EXOcd reduced the elevated GCR levels seen in pol2-REL cells (Fig. 1E). Collectively, our genetic analysis provided evidence that improving pol2-REL growth by Pol2 exonuclease inactivation correlated with enhanced genome replication and stability.
Abolishing Pol2 exonuclease activity improved genome replication of a POPS mutant that showed normal Mrc1 and Csm3 association with the replicative helicase
Next, we directly examined whether eliminating the exonuclease activity of Pol2 could correct genome-wide replication defects in pol2-REL cells. Flow cytometry analysis of G1-synchronized cells released into S phase confirmed slower DNA replication of pol2-REL cells at its semipermissive temperature (Fig. 2A; Meng et al. 2020). While wild-type cells mostly completed S phase 50 min after being released from G1 arrest, most pol2-REL cells remained in S phase at this time point. In contrast to the pol2-REL cells, DNA synthesis in pol2-EXOcd-REL cells was more advanced at 50 and 60 min, suggesting faster genome replication (Fig. 2A, red arrows; Supplemental Fig. S3B).
Figure 2.

pol2-REL replication defect was improved by inactivating the Pol2 exonuclease and was not associated with reduced levels of CMG-bound Mrc1 and Csm3. (A) Flow cytometry analysis of cell cycle progression of WT, pol2-REL, and pol2-EXOcd-REL cells at 34°C. Cells were arrested in G1 phase and then released into the cell cycle. Arrows highlight the different DNA contents between pol2-REL and pol2-EXOcd-REL at 50 and 60 min after G1 release. (B) CMG-bound Mrc1 or Csm3 in pol2-REL and wild-type cells. Cells were synchronized in G1 and then released into S phase at 24°C. At each examined time point, the Mcm4-FLAG protein was immunoprecipitated. Coimmunoprecipitation of the indicated proteins was examined by immunoblotting using the indicated antibodies. (C) Quantification of Mrc1 and Csm3 levels relative to Mcm4-bound Psf1 based on the coimmunoprecipitation experiments shown in B. Average values and standard deviations of two to three biological replicates are shown. (NS) Not statistically significant based on two-tailed Student's t-test.
Replication speed can be affected by the association of the replication progression factors Mrc1 and Csm3-Tof1 with the CMG replicative helicase, which is formed upon the association of the GINS complex and Cdc45 with the MCM helicase (Yeeles et al. 2017). We thus examined the association of Mrc1 and Csm3 with the GINS complex member Psf1 by coimmunoprecipitation. Since pol2-REL and wild-type cells progressed through S phase at different rates (Fig. 2A; Meng et al. 2020), averaging three S-phase time points reflected the overall S-phase situation of each genotype. Using this method, we found that the amounts of Mrc1 and Csm3 coimmunoprecipitated with the MCM-associated Psf1 were not reduced in pol2-REL cells compared with wild-type cells (Fig. 2B,C; Supplemental Fig. S3C). This observation provided evidence that the replication elongation defect caused by pol2-REL was not due to a lack of physical association of fork progression-promoting factors with the CMG complex.
Pol2 exonuclease activity partly accounts for genome-wide sister fork asymmetry and replisome progression defects of a POPS mutant
Our data thus far suggested that reduced DNA synthesis caused by a Pol2 POPS mutant allele could be partly due to Pol2 exonuclease activity impairing replisome movement. To test this model more directly, we used the Replicon-seq method to derive genome-wide, high-resolution maps of the movement of sister replication forks in pol2-REL, pol2-EXOcd-REL, pol2-EXOcd, and wild-type cells (Supplemental Fig. 4A; Claussin et al. 2022). Sister replication forks move at a constant rate through tens of kilobases of chromatin in wild-type cells, which gives rise to a symmetric “V”-shaped pattern when replicons are plotted according to their positions and lengths (Fig. 3; Claussin et al. 2022). In contrast, replicons in pol2-REL mutants showed extensive asymmetry, suggesting that sister replication forks moved at unequal rates due to frequent replisome stalling (Fig. 3; Supplemental Fig. S4C). Such defects appeared to be genome-wide, seen both at known fork barriers sites, such as tRNA genes, centromeres, and highly transcribed genes, and at other sites (Supplemental Figs. S4C, S5A). Significantly, progression of sister replisomes in pol2-EXOcd-REL was closer to that of the wild type than that of pol2-REL across the genome (Fig. 3). We note that the pol2-EXOcd mutant was essentially indistinguishable from wild type (Figs. 3, 4; Supplemental Figs. S4, S5). Read frequencies across a large range of read lengths were comparable between wild-type and all mutant data sets (Supplemental Fig. S4B).
Figure 3.
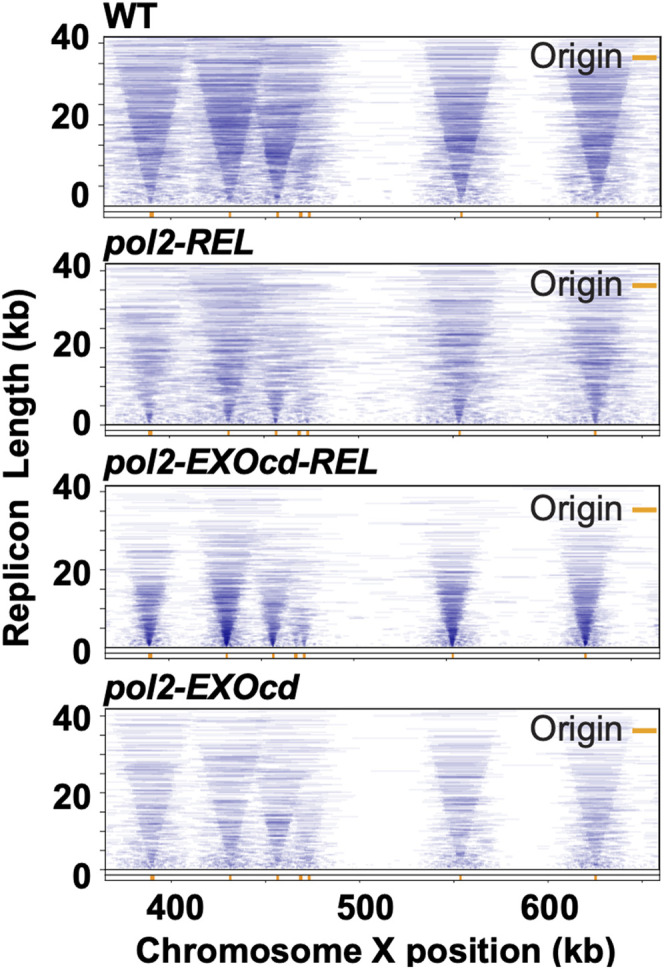
Replicon-seq analyses revealed genome-wide sister fork asymmetry of pol2-REL, which was improved by EXO inactivation. Examples of DNA replication mapped by Replicon-seq for the indicated strains are shown. The full lengths of BrdU-containing sequencing reads based on unfiltered data were plotted as lines according to their position (X-axis) and length (Y-axis) for chromosome X. Replication origins are indicated by orange bars.
Figure 4.

Sister replication fork asymmetry and leading strand DNA synthesis in pol2 mutants. (A) Genome-wide analysis of sister replication fork progression. All Replicon-seq data were filtered to select BrdU-containing reads that overlapped with defined ACSs (Eaton et al. 2010). Replicons were binned according to their length in 5-kb increments, and total read counts in each bin were normalized to allow comparison between bins. The positions of the midpoints of selected reads were plotted relative to the positions of the ACSs. The data are shown as heat maps for wild-type (WT), pol2-REL, pol2-EXOcd-REL, and pol2-EXOcd cells, as indicated. (B) Standard deviations of the data shown in A were calculated by fitting Gaussian distribution for WT, pol2-REL, pol2-EXOcd-REL, and pol2-EXOcd. Data were binned according to different sequencing read lengths indicated on the X-axis. (C) The relative differences in the lengths of leading versus lagging strands are shown as a heat map for WT, pol2-REL, pol2-EXOcd-REL, and pol2-EXOcd. The read orientation and the difference in position between the read midpoint and the ACS were used to define the fraction of each read that was synthesized by the leading or lagging strand polymerases. The relative difference in leading or lagging strand length was plotted on the X-axis; 0 indicates no difference, whereas signal to the left or right of 0 indicates that the leading or lagging strand is longer, respectively.
A quantitative analysis of replicons across the genome was performed as described in Claussin et al. (2022). The midpoints of replicon reads were first aligned to their respective ARS consensus sequences (ACSs), and then replicon reads were grouped by lengths. In all groups, read midpoints in wild-type or pol2-EXOcd cells aligned closely with the ACS sites, indicating that the left and right traveling sister replication forks and replisomes progressed at similar rates (Fig. 4A). In pol2-REL cells, the midpoints of the reads were broadly scattered, suggesting that the two replisomes were not progressing symmetrically from their origin (Fig. 4A). This asymmetry was partially corrected in pol2-EXOcd-REL cells (Fig. 4A). Calculation of the standard deviation of the data for replicons with different lengths confirmed that the pol2-EXOcd-REL partially rescued the broad increase in standard deviation seen in pol2-REL cells (Fig. 4B; Supplemental Fig. S5B). Collectively, these results indicated that a POPS mutation impeded replisome progression across all lengths of replicons, and this defect was partially corrected by disabling Pol2 exonuclease activity.
Replicon-seq data can also inform the coordination of leading and lagging strand synthesis at the replication fork. In wild-type cells, the leading strand is typically ∼300–400 nt longer than the contiguous lagging strand (Fig. 4C; Claussin et al. 2022). However, in pol2-REL cells, the two strands were more closely aligned and differed in position by ∼30 nt. Significantly, inactivating the Pol2 exonuclease activity in pol2-REL cells increased the relative length of the leading strand compared with the lagging strand to ∼80 nt, which more closely resembled wild type and pol2-EXOcd (Fig. 4C). This finding suggested that the POPS mutation interfered with leading strand synthesis likely by limiting Polε processivity.
The division of labor between the leading and lagging strand polymerases during genome replication is fundamentally important for the stable inheritance of genetic information. The striking difference in the lengths of nascent DNA synthesized by the two polymerases argues that they must adopt distinct strategies optimized for their specific tasks. In particular, the leading strand polymerase Polε faces the challenge of processive DNA synthesis over long stretches of template DNA that contain frequent barriers. Previous studies suggested that this challenge can be mitigated by several unique features of Polε that foster its association with template (Sengupta et al. 2013; Hogg et al. 2014; Langston et al. 2014; Goswami et al. 2018). Here we reveal a different strategy that also contributes to Polε-mediated leading strand synthesis. Our Replicon-seq data suggest that POPS aids processive synthesis in part by countering the Pol2 exonuclease function. This conclusion is supported by data in which several replication-related defects caused by a POPS mutant were improved by EXO inactivation, including slow growth and S-phase progression, increased GCR rates, and synthetic sickness with mutants affecting replication-promoting factors. Collectively, these data provide evidence for a functional antagonism between the POPs and EXO domains. We note that the suppression of pol2-REL defects by EXOcd was incomplete. This could be explained by a replication initiation defect seen for pol2-REL, which in principle should not be suppressed by EXOcd (Meng et al. 2020).
We show that the antagonism between POPs and EXO functions is specific and is unlikely to be explained by the role of the Pol2 EXO domain in mutation correction (Meng et al. 2020). Rather, our data suggest a model in which EXO-mediated polymerase backtracking impedes the forward movement of the enzyme when POPS is defective (Fig. 5). How POPS counterbalances such a role of EXO to favor forward polymerase movement is currently unclear. Our data suggest that POPS is unlikely to help by tethering Mrc1 and Csm3 to the replisome or promoting PCNA binding. One possibility to consider is that POPS may enable a Pol2 conformation that favors continued synthesis, such as by hindering the transfer of the nascent primer to the EXO domain in the absence of misincorporated nucleotides. In support of this view, we found that EXOcd exerted growth suppression on a pol2 mutant with weakened binding of the nascent DNA primer (Supplemental Fig. S5C; Ganai et al. 2015). Our proposed model also agrees with recent evidence showing the importance of partitioning the nascent primer between the EXO and CAT domains (Parkash et al. 2019; Xing et al. 2019). Our data further suggest that the proposed role for POPS in this process may be particularly useful when Pol2 is paused by template barriers, since the pol2-REL growth was improved when the burden of template barriers was reduced through Rhn1 overexpression. Given that individual mutations studied in pol2-REL correspond to recurrent cancer-associated mutations in POLE and exhibit replication defects (Meng et al. 2020), the synthetic sickness profile uncovered for pol2-REL, such as with mutations inactivating checkpoint and DNA repair proteins, may be able to inform strategies of selectively killing cancer cells bearing POPS mutations.
Figure 5.

A model for the antagonistic action of Pol2's POPS and EXO domains. During normal growth, processive leading strand synthesis benefits from POPS's counterbalance of EXO-mediated backtracking of the polymerase and/or nascent strand degradation. In POPS mutants, such as pol2-REL examined here, EXO-mediated Pol2 backtracking can impair leading strand synthesis, generating sister fork asymmetry and multiple replication-associated defects. These defects can be rescued by inactivating Pol2's EXO function. See the text for details
Materials and methods
Yeast strains, procedures, and cell cycle experiments
Yeast strains used in this study are listed in Supplemental Table S1. At least two strains per genotype were examined in each experiment. Standard PCR-based methods were used to generate integrated alleles and add tags to proteins at endogenous loci, followed by DNA sequencing verification. Primer information is available on request. Cell cycle arrest and release were conducted following standard protocols and are described in detail in the Supplemental Material.
Coimmunoprecipitation experiments and Western blotting
Standard protocols were followed. In brief, cells were disrupted by glass bead beating in lysis buffer that contained 50 mM HEPES-KOH (pH 7.5), 100 mM KOAc, 1% Triton X-100, 2 mM MgOAc, 2 mM NaF, 2mM β-glycerophosphate, 10 mM β-mercaptoethanol, and EDTA-free protease inhibitors (Roche). Benzonase was added to digest nucleic acid before centrifugation at 20,000g for 30 min to obtain whole-cell extract (WCE). WCE was then incubated with anti-FLAG beads (Sigma-Aldrich) for 2 h at 4°C. After washing the beads, bead-bound proteins were eluted with 2× Laemmli buffer. Proteins were boiled for 5 min before being subjected to SDS-PAGE on 4%–20% gradient gels (Bio-Rad) and transferred to nitrocellulose membrane (GE Healthcare) for Western blotting. Antibodies used include anti-Psf1 (a gift from K. Labib), anti-FLAG (Sigma-Aldrich F1804), anti-Mrc1 (a gift from G. De Piccoli), anti-Csm3 (a gift from K. Labib), and anti-HA (3F10).
GCR and mutation rate assays
GCR rates were measured using a standard protocol (Putnam and Kolodner 2010; Wan et al. 2019). Seven or more cultures were examined for each genotype. Cells were plated on SC + 5-FOA + Can (FC) and SC plates to obtain numbers of colonies that lose the URA3-CAN1 cassette (inserted at YEL068c) and total viable colonies, respectively. GCR rates were calculated as m/NT as follows: m[1.24 + ln(m)] – NFC = 0, where m is mutational events, NFC is the number of colonies on FC plates, and NT is the number of colonies on SC plates. The upper and lower 95% confidence intervals were then derived.
Replicon-seq data acquisition and analyses
Log-phase cells were arrested in G1 with α factor for 150 min at 25°C. BrdU was added to a final concentration of 400 µg/mL 30 min before the end of the arrest. Arrested cells were then washed with prewarmed YPD media and released from arrest at 30°C in YPD containing 400 µg/mL BrdU. DNA preparation and detection of nascent DNA were performed as described by Claussin et al. (2022) and is described in detail in the Supplemental Material. Consistency between two biological replicates per genotype was inspected before data combination to increase read numbers.
Supplementary Material
Acknowledgments
We thank Dr. Erik Johannsson and Dr. Dirk Remus for helpful discussion, and Dr. Karim Labib and Dr. Giacomo De Piccoli for sharing antibodies. X.Z. is supported by National Institute of General Medical Sciences (NIGMS) grants R01GM131058 and R35GM145260. I.W. is supported by NIGMS grants R01RGM102253 and R01GM129058. C.C. is supported by a Francois Wallace Monahan Fellowship.
Author contributions: All authors were involved in research design and data analyses, X.M., G.R.-M., and C.C. performed research. G.R.-M., I.W., and X.Z. wrote the paper with all authors’ input.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.350054.122.
Competing interest statement
The authors declare no competing interests.
References
- Burgers PMJ, Kunkel TA. 2017. Eukaryotic DNA replication fork. Annu Rev Biochem 86: 417–438. 10.1146/annurev-biochem-061516-044709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilkova O, Stenlund P, Isoz I, Stith CM, Grabowski P, Lundstrom EB, Burgers PM, Johansson E. 2007. The eukaryotic leading and lagging strand DNA polymerases are loaded onto primer-ends via separate mechanisms but have comparable processivity in the presence of PCNA. Nucleic Acids Res 35: 6588–6597. 10.1093/nar/gkm741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claussin C, Vazquez J, Whitehouse I. 2022. Single-molecule mapping of replisome progression. Mol Cell 82: 1372–1382.e4. 10.1016/j.molcel.2022.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigaku Y, Keszthelyi A, Müller CA, Miyabe I, Brooks T, Retkute R, Hubank M, Nieduszynski CA, Carr AM. 2015. A global profile of replicative polymerase usage. Nat Struct Mol Biol 22: 192–198. 10.1038/nsmb.2962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devbhandari S, Remus D. 2020. Rad53 limits CMG helicase uncoupling from DNA synthesis at replication forks. Nat Struct Mol Biol 27: 461–471. 10.1038/s41594-020-0407-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton ML, Galani K, Kang S, Bell SP, MacAlpine DM. 2010. Conserved nucleosome positioning defines replication origins. Genes Dev 24: 748–753. 10.1101/gad.1913210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan H, Yu C, Devbhandari S, Sharma S, Han J, Chabes A, Remus D, Zhang Z. 2017. Checkpoint kinase Rad53 couples leading- and lagging-strand DNA synthesis under replication stress. Mol Cell 68: 446–455.e3. 10.1016/j.molcel.2017.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganai RA, Bylund GO, Johansson E. 2015. Switching between polymerase and exonuclease sites in DNA polymerase ε. Nucleic Acids Res 43: 932–942. 10.1093/nar/gku1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami P, Abid Ali F, Douglas ME, Locke J, Purkiss A, Janska A, Eickhoff P, Early A, Nans A, Cheung AMC, et al. 2018. Structure of DNA–CMG–Polε elucidates the roles of the non-catalytic polymerase modules in the eukaryotic replisome. Nat Commun 9: 5061. 10.1038/s41467-018-07417-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hizume K, Araki H. 2019. Replication fork pausing at protein barriers on chromosomes. FEBS Lett 593: 1449–1458. 10.1002/1873-3468.13481 [DOI] [PubMed] [Google Scholar]
- Hogg M, Osterman P, Bylund GO, Ganai RA, Lundström E-B, Sauer-Eriksson AE, Johansson E. 2014. Structural basis for processive DNA synthesis by yeast DNA polymerase ε. Nat Struct Mol Biol 21: 49–55. 10.1038/nsmb.2712 [DOI] [PubMed] [Google Scholar]
- Langston LD, Zhang D, Yurieva O, Georgescu RE, Finkelstein J, Yao NY, Indiani C, O'Donnell ME. 2014. CMG helicase and DNA polymerase ε form a functional 15-subunit holoenzyme for eukaryotic leading-strand DNA replication. Proc Natl Acad Sci 111: 15390–15395. 10.1073/pnas.1418334111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Wei L, Devbhandari S, Zhang T, Xiang J, Remus D, Zhao X. 2020. DNA polymerase ε relies on a unique domain for efficient replisome assembly and strand synthesis. Nat Commun 11: 2437. 10.1038/s41467-020-16095-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkash V, Kulkarni Y, Ter Beek J, Shcherbakova PV, Kamerlin SCL, Johansson E. 2019. Structural consequence of the most frequently recurring cancer-associated substitution in DNA polymerase ε. Nat Commun 10: 373. 10.1038/s41467-018-08114-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicanò G, Al Mamun M, Jurado-Santiago D, Villa-Hernández S, Yin X, Giannattasio M, Lanz MC, Smolka MB, Yeeles J, Shirahige K, et al. 2021. Checkpoint-mediated DNA polymerase ε exonuclease activity curbing counteracts resection-driven fork collapse. Mol Cell 81: 2778–2792.e4. 10.1016/j.molcel.2021.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pursell ZF, Kunkel TA. 2008. DNA polymerase: a polymerase of unusual size (and complexity). Prog Nucleic Acid Res Mol Biol 82: 101–145. 10.1016/S0079-6603(08)00004-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pursell ZF, Isoz I, Lundström EB, Johansson E, Kunkel TA. 2007. Yeast DNA polymerase ε participates in leading-strand DNA replication. Science 317: 127–130. 10.1126/science.1144067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam CD, Kolodner RD. 2010. Determination of gross chromosomal rearrangement rates. Cold Spring Harb Protoc 2010: pdb.prot5492. 10.1101/pdb.prot5492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, van Deursen F, de Piccoli G, Labib K. 2013. Dpb2 integrates the leading-strand DNA polymerase into the eukaryotic replisome. Curr Biol 23: 543–552. 10.1016/j.cub.2013.02.011 [DOI] [PubMed] [Google Scholar]
- Taylor MRG, Yeeles JTP. 2019. Dynamics of replication fork progression following helicase–polymerase uncoupling in eukaryotes. J Mol Biol 431: 2040–2049. 10.1016/j.jmb.2019.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan B, Wu J, Meng X, Lei M, Zhao X. 2019. Molecular basis for control of diverse genome stability factors by the multi-BRCT scaffold Rtt107. Mol Cell 75: 238–251.e5. 10.1016/j.molcel.2019.05.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams LN, Marjavaarab L, Knowels GM, Schultz EM, Fox EJ, Chabes A, Herra AJ. 2015. dNTP pool levels modulate mutator phenotypes of error-prone DNA polymerase ε variants. Proc Natl Acad Sci 112: E2457–E2466. 10.1073/pnas.1422948112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing X, Kane DP, Bulock CR, Moore EA, Sharma S, Chabes A, Shcherbakova PV. 2019. A recurrent cancer-associated substitution in DNA polymerase ε produces a hyperactive enzyme. Nat Commun 10: 374. 10.1038/s41467-018-08145-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeeles JTP, Janska A, Early A, Diffley JFX. 2017. How the eukaryotic replisome achieves rapid and efficient DNA replication. Mol Cell 65: 105–116. 10.1016/j.molcel.2016.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Muller EG, Rothstein R. 1998. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell 2: 329–340. 10.1016/s1097-2765(00)80277-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.